

Abstract
Accumulation of the microtubule-associated protein tau into neurofibrillary lesions is a pathological consequence of several neurodegenerative diseases, including Parkinson's disease and Alzheimer's disease. Hereditary mutations in the MAPT gene were shown to promote the formation of structurally distinct tau aggregates in patients that had a parkinsonian-like clinical presentation. Whether tau aggregates themselves or the soluble intermediate species that precede their aggregation are neurotoxic entities in these disorders has yet to be resolved; however, recent in vivo evidence supports the latter. We hypothesized that depletion of CHIP, a tau ubiquitin ligase, would lead to an increase in abnormal tau. Here, we show that deletion of CHIP in mice leads to the accumulation of non-aggregated, ubiquitin-negative, hyperphosphorylated tau species. CHIP−/− mice also have increased neuronal caspase-3 levels and activity, as well as caspase-cleaved tau immunoreactivity. Overexpression of mutant (P301L) human tau in CHIP −/− mice is insufficient to promote either argyrophilic or “pre-tangle” structures, despite marked phospho-tau accumulation throughout the brain. These observations are supported in postdevelopmental studies using RNA interference for CHIP (chn-1) in Caenorhabditis elegans and cell culture systems. Our results demonstrate that CHIP is a primary component in the ubiquitin-dependent degradation of tau. We also show that hyperphosphorylation and caspase-3 cleavage of tau both occur before aggregate formation. Based on these findings, we propose that polyubiquitination of tau by CHIP may facilitate the formation of insoluble filamentous tau lesions.
Keywords: Parkinson's disease, CHIP, synuclein, Alzheimer's disease, ubiquitination, proteasome degradation, neuronal cell death
Introduction
The most prevalent form of age-associated dementia, Alzheimer's disease (AD), is histologically characterized by the formation of neurofibrillary tangles (NFTs) composed of the microtubule-associated protein tau, accompanied by extracellular formation of amyloid plaques composed of the amyloid β peptide (Aβ); however, it is the tau pathology in AD that most closely correlates with neuronal loss (Gomez-Isla et al., 1997) and clinical dementia (Grundke-Iqbal et al., 1986; Ihara et al., 1986; Crystal et al., 1988). The discovery that mutations in the gene for tau (MAPT) cause FTDP-17 (frontotemporal dementia with parkinsonism-linked to chromosome 17) (Hutton et al., 1998; Spillantini et al., 1998), which is associated with tau aggregates within neurons and glia, provided conclusive evidence that abnormalities in tau were sufficient to produce neurodegeneration independent of Aβ. The accumulation of aberrant tau species is also the primary histopathological hallmark of several neurodegenerative disorders, including corticobasal degeneration, progressive supranuclear palsy, and argyrophilic grain disease (Ishizawa et al., 2003; Wszolek et al., 2004; Zimprich et al., 2004).
Recent work demonstrated that suppression of mutant tau expression in transgenic mice at an age when NFT pathology and memory deficits were manifest resulted in rescue of cognitive defects and neuronal loss without altering neurofibrillary pathology (Santacruz et al., 2005). This suggested that formation of tau aggregates might not be the primary mechanism linking tau pathology to neuronal death, but rather the soluble intermediates that culminate in NFT formation. Perhaps over-burdened or malfunctioning protein degradative systems, which would normally remove these toxic intermediate tau species (An et al., 2003; Layfield et al., 2003), are forced to partition tau into aggregates as an adaptive cellular response in terminally differentiated neurons to prevent their ill effects.
A role for the ubiquitin proteasome system (UPS) in the removal of aberrant tau species was suggested by studies demonstrating that proteasome-dependent degradation of constitutive heat shock protein 70 (Hsc70)-bound phospho-tau was specifically facilitated by the cochaperone CHIP (C terminus of the Hsc70-interacting protein) through direct ubiquitin ligase activity (Petrucelli et al., 2004; Shimura et al., 2004). Very recently, it was demonstrated that aggregated tau species isolated from AD brain tissue by immunoprecipitation with a conformation-specific antibody were, in fact, polyubiquitinated at lysine residues near or within the microtubule binding region (Cripps et al., 2006), suggesting that these sites are not only critical for tau degradation but perhaps have a role in microtubule dynamics. These findings correlate well with the recent finding that CHIP levels were elevated in AD samples relative to control tissue (Sahara et al., 2005), further suggesting that CHIP plays an important role in mature tangle formation. Additionally, a recent proteomic study identified HSP70, HSP90, and tau as CHIP binding partners (Bomar et al., 2003; Grelle et al., 2006). Here, we have characterized the effect of CHIP deletion on endogenous mouse tau as well as overexpressed mutant (P301L) human tau (Lewis et al., 2000; Dai et al., 2003). We have determined that CHIP is critical for degradation of phosphorylated tau species and may play a role in the formation of neurofibrillary pathology.
Materials and Methods
Antibodies for Western blotting.
The antibodies used were as follows: rabbit polyclonal CHIP antibody (1:1000) (Petrucelli et al., 2004); rabbit polyclonal total tau and ubiquitin antibodies (1:1000; Dako, Carpinteria, CA); Tau5 and Tau-C3 [mouse monoclonal total tau antibody (1:1000) and a monoclonal antibody recognizing tau cleaved by caspase-3 at Asp 421, respectively; provided by L. Binder, Northwestern University School of Medicine, Chicago, IL]; CP13 phospho-tau S202/T205 monoclonal antibody and PHF-1 phospho-tau S396/S404 monoclonal antibody (1:100; provided by P. Davies, Albert Einstein College of Medicine, Bronx, NY); 12E8 phospho-tau S262/356 monoclonal antibody (1:1000; provided by P. Seubert, Elan Pharmaceuticals, San Francisco, CA) (Seubert et al., 1995; Sun et al., 2003); glyceraldehyde-3-phosphate dehydrogenase (GAPDH) monoclonal antibody (1:5000; Biodesign International, Kennebunkport, ME); HSP70 monoclonal antibody (1:1000; StressGen Biotechnologies, Victoria, British Columbia, Canada); capase-3 and cleaved caspase-3 monoclonal antibodies (1:1000; Cell Signaling, Beverly, MA); β-adrenergic receptor monoclonal antibody (1:1000; Abcam, Cambridge, MA); α-synuclein (1:500; LB509; Zymed, San Francisco, CA); and HRP-conjugated secondary antibodies (1:5000; Jackson ImmunoResearch, West Grove, PA).
Cell culture and small interfering RNA transfections.
The mouse Neuro-2a neuroblastoma cell line (N2A) was grown in Opti-Mem plus 10% FBS and passaged every 3–5 d based on 90% confluence. For CHIP small interfering RNA (siRNA) transfections, siRNA was predesigned by Ambion (Austin, TX) for accession number NM_019719; the sense sequence was 5′-GGAGCAGCGACUCAACUUUtt-3′. The final siRNA (CHIP or a validated negative control siRNA; Ambion) concentration per well was 30 nm in Opti-Mem with 2 μl of siPORT Amine transfection reagent (Ambion) used per well. This mixture was incubated in a final volume of 200 μl for 15 min and added to 40–50% confluent N2A cells in 6-well dishes for a final in-well volume of 2.5 ml. For optimal knockdown of protein levels, the cells were incubated in the presence of the transfection reaction for 24 h and replaced with fresh media for an additional 48 h. Cells were harvested for subsequent Western blot analysis in lysis buffer containing 50 mm Tris-HCl, pH 7.4, 1 m NaCl, 0.1% Triton-X, 5 mm EDTA plus 1% SDS, PMSF, and both a protease and phosphatase inhibitor mixture.
Caenorhabditis elegans plasmid construction.
The human tau::EGFP fusion plasmid for expression in C. elegans was constructed using Gateway technology (Invitrogen, San Diego). Specifically, the unc-54 promoter plasmid pPD30.38 (a gift from A. Fire, Delft University of Technology, Delft, The Netherlands) was converted into a Gateway destination vector, pDEST-UNC-54, using Gateway technology. Additionally, a Gateway entry vector for tau::EGFP was generated by BP reaction with pDONR221 using PCR-amplified cDNA fragments from vector GFP-4R0N-WT tau. After this, the tau::EGFP fusion was cloned into the pDEST-UNC-54 vector via an LR reaction.
C. elegans vector delivery, imaging, and quantitation.
The plasmid encoding the wild-type human tau::EGFP fusion was injected into the gonads of early-adult hermaphrodites (strain N2, Bristol) at a concentration of 0.1 μg/ml. The injection mixture contained vector Punc-54::tau::EGFP as well as pRF4 rol-6 (su1006) as a dominant marker for transformation selection [as was green fluorescent protein (GFP) fluorescence] (Mello et al., 1991). Multiple worm lines expressing stable extrachromosomal arrays were obtained. After stable transmission of the arrays, multiple stable lines were compared and analyzed before a consensus line was integrated into the genome. Integration was performed using gamma irradiation with 3500–4000 rads from a Cobalt 60 source (Inoue and Thomas, 2000). The integrated transgenic lines were named according to appropriate C. elegans nomenclature as strains UA38 (baIn38) and UA39 (baIn39).
C. elegans strains UA38 and UA39 were cultured under standard conditions (Brenner, 1974). RNA interference (RNAi) was used to disrupt the expression of chn-1, the C. elegans ortholog of CHIP, using the bacterial feeding method (Timmons et al., 2001). The chn-1-containing fragment cloned into L4440 was obtained from the genomic library of RNAi targeting bacterial strains from the MRC GeneService (Kamath and Ahringer, 2003). Controls for RNAi specificity were determined on basis of unique Wormbase-predicted targets. Transformed HT115 (DE3) bacteria were grown for 14 h at 37°C in Luria broth supplemented with 50 μg/ml ampicillin. These cultures were plated onto standard [nematode growth medium (NGM)] worm plates containing 1 mm isopropyl β-d-1-thiogalactopyranoside to induce double-stranded RNA (dsRNA) production and were allowed to grow overnight at room temperature. Transgenic worms at the L4 stage were then placed onto these RNAi plates. The F1 generation offspring were subsequently transferred to fresh chn-1 RNAi plates, and their progeny (F2 generation) were examined as young adults for evidence of tau::EGFP expression. Worms were examined with a Nikon (Tokyo, Japan) E800 epifluorescence microscope using an Endow GFP HYQ filter cube. Images were captured with a Cool Snap HQ CCD camera (Photometrics, Tucson, AZ) driven by MetaMorph software (Universal Imaging Corporation, West Chester, PA).
Preparation of C. elegans extracts for immunoblotting.
Extracts were prepared after growth of strain UA38 to near confluence on two 100 mm NGM plates with and without bacteria expressing dsRNA targeting chn-1. Worms were collected by washing with M9 buffer and concentrated by centrifugation in a 1.5 ml microcentrifuge tube at 5000 × g for 1 min. The worm pellet was resuspended and lysed in buffer containing 50 mm Tris, 274 mm NaCl, 5 mm KCl, 2% SDS, and a protease and phosphatase inhibitor mixture, pH 8.0. After homogenization and sonication, 20 μg of protein (as determined by BCA assay) were loaded with equivalent volumes of SDS-PAGE sample buffer and subjected to SDS-PAGE Western blot analysis.
Breeding of CHIP−/−, CHIP−/− × Tau P301L, and twitcher mice colonies and tissue harvesting.
Mice lacking CHIP expression (CHIP−/−) were generated as described previously (Dai et al., 2003). Briefly, a neo-cassette was inserted into the CHIP gene, replacing exons 1–3. Mice were maintained on a mixed SvEv129/C57BL6 background and housed in a barrier facility. Heterozygote or homozygote mice lacking one or both alleles, respectively, were genotyped by PCR from tail-clip digestions (Dai et al., 2003). Tau P301L mice were as described previously (Lewis et al., 2000). CHIP+/− mice were bred with JNPL3 on a Swiss Webster background to generate CHIP+/− × JNPL3 mice. These mice were backcrossed to CHIP+/− mice to generate CHIP−/− × Tau P301L (CHT) mice. Breeding pairs of twitcher heterozygotes and C57BL/6J mice were purchased from The Jackson Laboratory (Bar Harbor, ME). The genotypes of pups born from the twitcher heterozygotes were determined by PCR (Sakai et al., 1996). CHIP−/−, CHIP+/+, JNPL3, CHT, and twitcher mice were humanely killed at postnatal day 25 (P25) to P30, and brains were quickly removed for dissection. The left cerebral hemisphere was immediately fixed in 10% formalin, and the other half was frozen on dry ice for subsequent biochemical and RNA analyses.
Quantitative real-time PCR.
RNA was isolated from tissue samples and analyzed by quantitative real-time PCR (qRT-PCR) as described previously (Dickey et al., 2003). Briefly, total RNA samples from the ventral half of four CHIP−/− and four CHIP+/+ mice right brain hemispheres were isolated using RNeasy technology from Qiagen (Valencia, CA) and reverse transcribed with Moloney murine leukemia virus reverse transcriptase and 1 m betaine. RNA was isolated after growth of strain UA38 to near confluence on 12 100 mm NGM plates with chn-1 siRNA bacteria or siRNA bacteria only. Worms were collected by washing with M9 buffer and concentrated by centrifugation in a 1.5 ml microcentrifuge tube at 1600 × g for 1 min. Worms were washed in M9 three times. Worm lysis was achieved via vortexing Trizol-treated worms with 150–212 μm glass beads. A standard curve was established within the reverse transcription reaction by adding total RNA (template) from an intra-experimental mouse RNA pool covering 3 logs. Five nanograms of total RNA from all samples were added to individual wells within the reverse transcription reaction for comparison to the standard curve. Primers for SYBR Green analysis by PCR were generated to as described previously (Dickey et al., 2004). To ensure that DNA contamination would not contribute to the detected signal, primers for CHIP were designed to span intron 1 (forward, 5′-CCAGCTGGAGATGGAGAGTT-3′; reverse, 5′-GCCGTTCCTCGATACTGTTC-3′). The primers for C. elegans were chn-1 forward 5′-CACTGGAACTGAGCCCAAAC-3′ and reverse 5′-GAGCGTTCGTGATAACAGCA-3′. Experimental wells containing 12 μl PCRs were run in quadruplicate in 384-well plates. PCR was run on the ABI 7900 as follows: 1 cycle of 95°C for 15 min, followed by 40 cycles of 95°C for 15 s and 60–65°C for 1 min. None of the primer pairs demonstrated more than one peak of fluorescence from melt-curve analysis. The standard curve was calculated by plotting the threshold cycle (Ct) against the log nanogram quantity of RNA added to the RT reactions. A linear regression was performed, and the slope, relating Ct to log nanogram RNA, was calculated and converted to a mass quantity of standard RNA. These mass values for the genes of interest were then normalized to 28S ribosomal RNA mass values and divided by GAPDH mRNA to determine fold change in expression relative to the standard RNA pool. For C. elegans, values were normalized to the GAPDH ortholog gpd3. These fold change values for samples in the CHIP−/− and CHIP+/+ groups were analyzed for significance using Student's t test.
Sarkosyl-soluble and insoluble tissue preparation.
After brain tissue was harvested as described above, each sample was homogenized in buffer containing 50 mm Tris, 274 mm NaCl, 5 mm KCl, 100 mm PMSF, and a protease and phosphatase inhibitor mixture, pH 8.0. Homogenates were centrifuged at 60,000 rpm for 15 min, and supernatant was saved as a “soluble” (S1) fraction. The remaining pellet was homogenized in buffer containing 10 mm Tris HCl, 0.8 m NaCl, 10% sucrose, 1 mm EGTA, and 1 mm PMSF, pH 7.4, and recentrifuged at 60,000 rpm for 15 min. The supernatant from this spin was incubated with 1% Sarkosyl at 37°C for 1 h. This mixture was then centrifuged at 60,000 rpm for 30 min, and the supernatant was removed as the “Sarkosyl-soluble” (S2) fraction. The remaining pellet was suspended in Tris–EDTA buffer and kept as the “Sarkosyl-insoluble” (P3) fraction.
Western blotting.
Cell lysates were sonicated, and tissue samples were homogenized and processed as described above. Protein concentrations were measured by a standard BCA assay (Pierce, Rockford, IL). Cell lysate samples and fractionated tissue homogenates were then heated in Laemmli's buffer, and equal amounts of protein were loaded into 10- or 15-well 10% Tris-glycine gels (Novex, San Diego, CA). After transfer, blots were blocked with Blotto (5% nonfat dry milk in TBS plus 0.1% Triton X-100) for 2 h, and antibodies were applied at appropriate dilutions in Blotto overnight at 4°C. Membranes were washed three times for 10 min in TBS with 0.05% TBS plus 0.1% Triton X-100 and incubated with secondary antibody for 1 h. Membranes were then washed three times for 10 min, and protein expression was visualized by ECL treatment and exposure to film. Bands were quantified using Scion (Frederick, MD) Image by analyzing pixel density. A semiquantitative analysis was performed by densitometry, correcting protein levels for GAPDH. For studies using the Odyssey scanner (Li-Cor, Lincoln, NE), conditions were similar to those described above; however, Li-Cor Odyssey blocking buffer was used in place of Blotto, and fluorescent-labeled [AlexaFluor 680 and Rockland (Gilbertsville, PA) IRDye800CW] secondary antibodies were used for detection.
Coimmunoprecipitation with PHF1 tau and Tau5 monoclonal antibodies.
Coimmunoprecipitation (co-IP) to assess tau ubiquitination levels in CHIP−/−, CHT, and JNPL3 mice was performed as described previously (Petrucelli et al., 2004). Briefly, 750 μg of total protein from CHIP−/−, CHIP+/+, CHT, and 4R0N P301L tau transgenic mouse tissue homogenates were suspended in 250 μl of co-IP buffer, sonicated, and incubated with 20 μl of protein G to pre-clear lysates for 1 h. After centrifugation, supernatants were incubated with anti-tau Tau5 antibody, anti-pS396/S404 tau PHF1 antibody, and 50 μl of protein G overnight at 4°C. Supernatants were washed three times in co-IP buffer, resuspended in sample buffer, and subjected to Western blot analyses after SDS-PAGE without controlling for protein concentration. Blots were probed with ubiquitin and total tau antibodies.
Alkaline phosphatase assay.
Tissue was homogenized in buffer containing 50 mm Tris-HCl, pH 7.4, 1 m NaCl, 0.1% Triton-X, 5 mm EDTA plus 1% SDS, PMSF, and a protease inhibitor mixture. One hundred micrograms of protein were suspended in 100 μl of 50 mm Tris HCl and 1 mm MgCl2 buffer and incubated at 30°C for 10 min. 30 U of calf intestine alkaline phosphatase were added to the samples and incubated at 30°C for 15 min. An equal volume of SDS-PAGE sample buffer was added to stop reactions, and samples were subjected to SDS-PAGE.
Immunohistochemistry.
Immunohistochemistry was performed on paraffin-embedded 5-μm-thick sections mounted on glass slides. The sections were deparaffinized and rehydrated in xylene and a graded series of alcohol and subjected to antigen retrieval with steaming in distilled H2O for 30 min. The tissue sections, along with positive and negative controls, were processed in a batch to assure uniformity, in a Dako Autostainer. The detection system used was the EnVision HRP system (Dako), and antibodies were diluted in Dako Antibody Diluent with background reducing components. The Liquid DAB Substrate-Chromogen system (Dako) was used as the chromogen. The stained slides were lightly counterstained in hematoxylin, dehydrated, and coverslipped. Terminal deoxnucleotidyl transferase-mediated dUTP nick end labeling (TUNEL) was performed using the Apoptag peroxidase kit (Chemicon, Temecula, CA) according to the manufacturer's instructions, as described previously (Zehr et al., 2004). The chromogen 3′,3′-diaminobenzidine was used to visualize staining. Sections were counterstained with methyl green.
Electron microscopy.
For routine ultrastructural studies, brains were removed and fixed overnight at 4°C in 2.5% glutaraldehyde in 0.1 m cacodylate buffer, pH 7.4. After washing in cacodylate buffer, the tissues were postfixed in 1% OsO4, en bloc stained in 1% uranyl acetate–50% ethanol, dehydrated in ethanols and propylene oxide, infiltrated, and embedded in Epon 812 (Polysciences, Warrington, PA). Ultrathin sections were counterstained with uranyl acetate and lead citrate and examined with a Philips 208S electron microscope.
Immunoelectron microscopy.
For postembedding immunoelectron microscopy, 4% paraformaldehyde-fixed brain tissue was dehydrated in graded ethanol, infiltrated in LR White resin (Polysciences, Warrington, PA), and polymerized in a vacuum oven at 50°C for 2 d. Ultrathin sections were collected on Formvar-coated nickel grids and used for immunocytochemistry as reported previously (Lin et al., 2003). The antibodies used from immunoelectron microscopy were CP13 (mouse monoclonal antibody to phosphorylated tau, pS202/T205, neat; from Dr. P. Davies) (Weaver et al., 2000) and E1 (polyclonal antibody to human tau; dilution, 1:200) (Crowe et al., 1991).
Results
CHIP−/− mice have reduced neuronal HSP70 RNA and protein levels and accumulate phospho-tau protein without affecting its transcription or other synaptic proteins
We demonstrated recently that CHIP and HSP70 were critical for the removal of tau and that CHIP-mediated ubiquitination of tau lead to increased accumulation of tau in an insoluble fraction (Petrucelli and Dawson, 2004). HSP70 overexpression had opposing effects. To better understand the role of CHIP in tau processing, we used mice deficient in CHIP expression to determine whether it is indeed essential for removal of phosphorylated tau species or involved in tau aggregation through its effects on ubiquitination. Founders used for previously described work by Dai et al. (2003) were used to generate a CHIP−/− in-house breeding colony. We found that ∼25% of the mice from our colony died at P30–P35, and these animals were distinctly smaller and feebler than their littermates (Fig. 1A), developing motor deficits around P20. Genotype analysis showed that these animals were indeed CHIP−/− mice; however, there were additional mice (25%) that were identified as CHIP deficient by PCR but lacked the small phenotype and possessed similar survival traits to wild-type siblings. Therefore, we decided to analyze tau levels in the small, symptomatic (sx) CHIP−/− mice and age-matched asymptomatic (asx) CHIP−/− mice, treating them as separate cohorts. Postmortem biochemical analyses of these mice with an antibody against total tau revealed a substantial increase in tau burden in sx CHIP−/− mice, particularly higher molecular weight tau species (subsequently shown to be phospho-tau), that was not present in age-matched CHIP+/+, CHIP+/−, or the remaining asx CHIP−/− littermates, nor was there any indication after 4.5 months of age that tau was accumulating (Fig. 1B). Further aging of these mice to 8 months did not precipitate aberrant tau accumulation (data not shown). This disparity in the CHIP−/− mice was most likely attributable to the use of non-isogenic founders to establish our line (Suzuki and Nakayama, 2003), bringing about allelic differences that resulted in the split phenotype. Therefore, given the unique and robust accumulation of tau demonstrated by the sx CHIP−/− mice, we focused our efforts on characterizing the neuropathology of these animals. For simplicity, sx CHIP−/− will be referred to as CHIP−/− mice unless noted otherwise.
Figure 1.

A large subset of CHIP−/− mice demonstrates P30–P35 morbidity and marked accumulation of cerebral phospho-tau levels. A, Symptomatic CHIP−/− mice are substantially smaller than either age-matched asymptomatic CHIP−/− or wild-type littermates. B, Whole brain was extracted from 30-d-old symptomatic CHIP−/− mice and 30-d-old and 4.5-month-old asymptomatic CHIP−/− mice, with age-matched wild-type littermates (n = 2), and homogenized for SDS-PAGE. Western blot analysis was performed using near-infrared fluorescent detection for total tau and GAPDH levels (in kilodaltons). No difference in total tau levels was observed in the asymptomatic CHIP−/− mice at either age compared with wild-type controls. SX, Symptomatic; ASX, asymptomatic; mo., month.
Using qRT-PCR, we validated that CHIP mRNA expression was indeed absent in these mice and determined that neither tau nor microtubule-associated protein 2 (MAP2) mRNA expression was changed compared with wild-type littermates (Fig. 2A). We also demonstrated that HSP70 mRNA expression was decreased, confirming that heat shock factor-1 (HSF1) activity was decreased because of CHIP deficiency in brain tissue, similar to previous findings in the periphery by Dai et al. (2003) (Fig. 2A). Interestingly, HSP27 mRNA levels were unaltered in these mice (data not shown), suggesting that the primary transcriptional machinery for this gene may not be HSF1. Western blot analyses demonstrated that CHIP protein was undetectable and HSP70 levels were decreased in CHIP−/− mice, consistent with the reductions in mRNA expression (Fig. 2B). Using a larger cohort of these mice, we confirmed that phospho-tau and total tau levels were dramatically increased. Given that tau mRNA levels were unchanged, this result was consistent with the role of CHIP in posttranslational tau protein degradation (Petrucelli et al., 2004; Shimura et al., 2004). To better establish the specificity of CHIP for tau, MAP2 protein levels were assessed and found to be unchanged (Fig. 2B). Immunohistochemical analyses comparing CHIP−/− and CHIP+/+ mice showed S202/T205 phospho-tau immunoreactivity in neuropil of forebrain and hindbrain gray matter in CHIP−/− mice that was not detected in age-matched CHIP+/+ mice (Fig. 2C).
Figure 2.
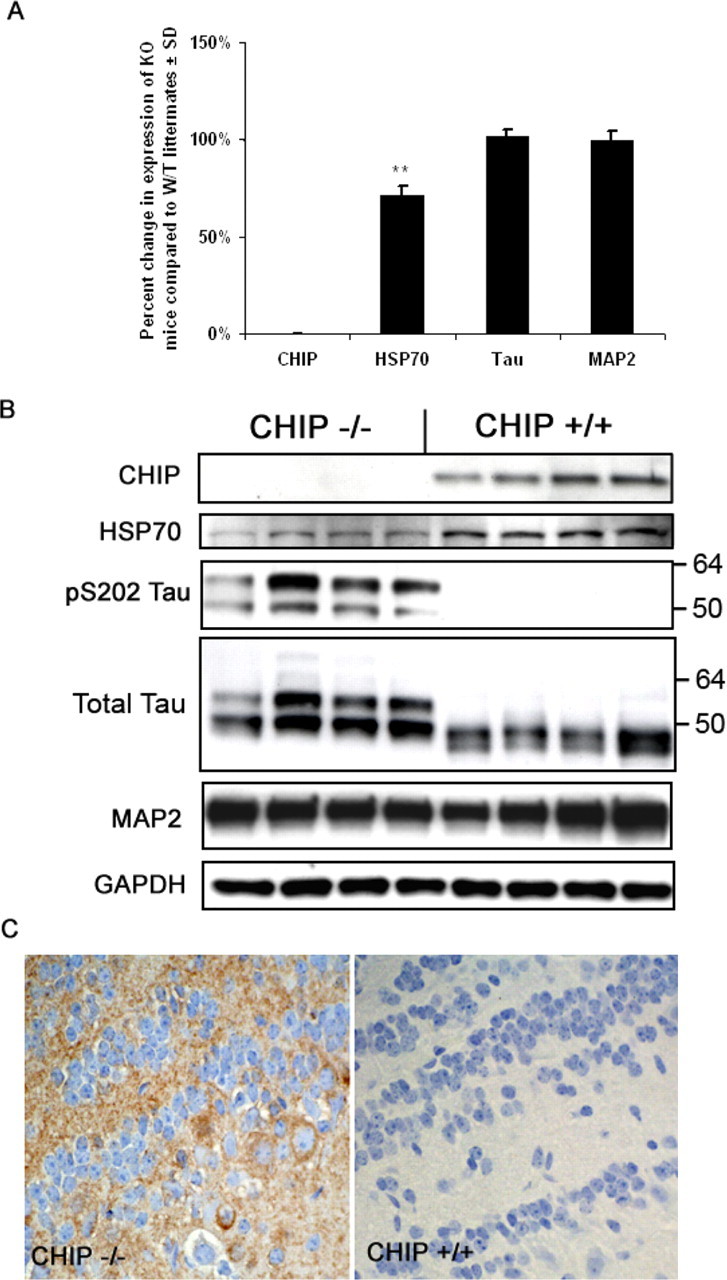
CHIP−/− mice have reduced neuronal HSP70 RNA and protein levels and accumulate phospho-tau protein without affecting its transcription or other microtubule-associated proteins. Total RNA from 30-d-old CHIP−/− mice (n = 4) and non-transgenic littermate (n = 4) brain tissue was extracted, and expression was analyzed by real-time PCR. Tau and MAP2 mRNA levels were unchanged in CHIP−/− mice relative to non-transgenic littermates after normalizing for GAPDH. A, CHIP was confirmed to be absent, and HSP70 levels were reduced, consistent with the previously described relationship between CHIP and HSF1 activity (∗∗p < 0.01). KO, Knock-out; W/T, wild type. Error bars represent ± SD. B, Brain protein levels (in kilodaltons) were assessed by Western blot analysis, confirming the absence of CHIP and reductions in HSP70 protein levels in CHIP−/− mice; however, pS202/T205 (CP13) tau levels were only observed in CHIP−/− mice. Elevations in total tau were also demonstrated. MAP2 and GAPDH levels remained unchanged, suggesting selectivity of CHIP for phospho-tau degradation. C, Levels of pS202/T205 (CP13) tau were also assessed by immunohistological methods, which revealed increases in phospho-tau species throughout the neuropil of forebrain and hindbrain gray matter in CHIP−/− mice compared with 30-d-old age-matched littermates as illustrated in the olfactory bulb.
CHIP−/− mice develop marked accumulation of soluble, ubiquitin-negative, phosphorylated tau species
Additional biochemical analyses of the CHIP−/− mice revealed a marked elevation in multiple phospho-tau species within the Sarkosyl-soluble protein fraction compared with age-matched CHIP+/+ littermates, including positive immunoreactivity with antibodies generated against the pS396/S404 (p = 0.0003), pS202/T205 (p = 0.014), and pS262/S356 (p = 0.031) epitopes on human tau (Fig. 3A). No abnormal accumulation of 60–72 kDa tau was seen in the Sarkosyl-insoluble protein fraction (data not shown). Treatment of homogenates with alkaline phosphatase (PPase) produced a collapse of the predominant ∼60 kDa molecular weight tau species in the CHIP−/− tissue into lower molecular weight species near the molecular weight of nonphosphorylated tau (∼55 kDa; detected with the Tau5 total tau antibody) (Fig. 3C), and immunoreactivity for pS396/S404 tau was mostly eliminated. Nonphosphorylated tau at S199/S202, recognized by the Tau 1 antibody, was significantly decreased (p = 0.0008) in the CHIP−/− homogenate compared with CHIP+/+ littermates (Fig. 3A). Other proteins that were probed for, including α-synuclein and MAP2 (Fig. 3A), along with three other putative CHIP substrates (androgen receptor, Smad3, and nNOS) based on previous in vitro studies remained unchanged in the CHIP−/− mice (data not shown). Although total tau levels were elevated significantly (p = 0.035) as measured by chemiluminescent Western analysis, we endeavored to definitively establish that tau was accumulating because of impaired degradation resulting from CHIP deletion, rather than simply enhanced kinase or decreased phosphatase activities. Thus, using a fluorescent-based Western blot approach for a more quantitative assessment of tau levels, we were able to further validate that the levels of total tau protein in the CHIP−/− mice were twice that in wild-type littermates (p = 0.0012) (Fig. 3B).
Figure 3.
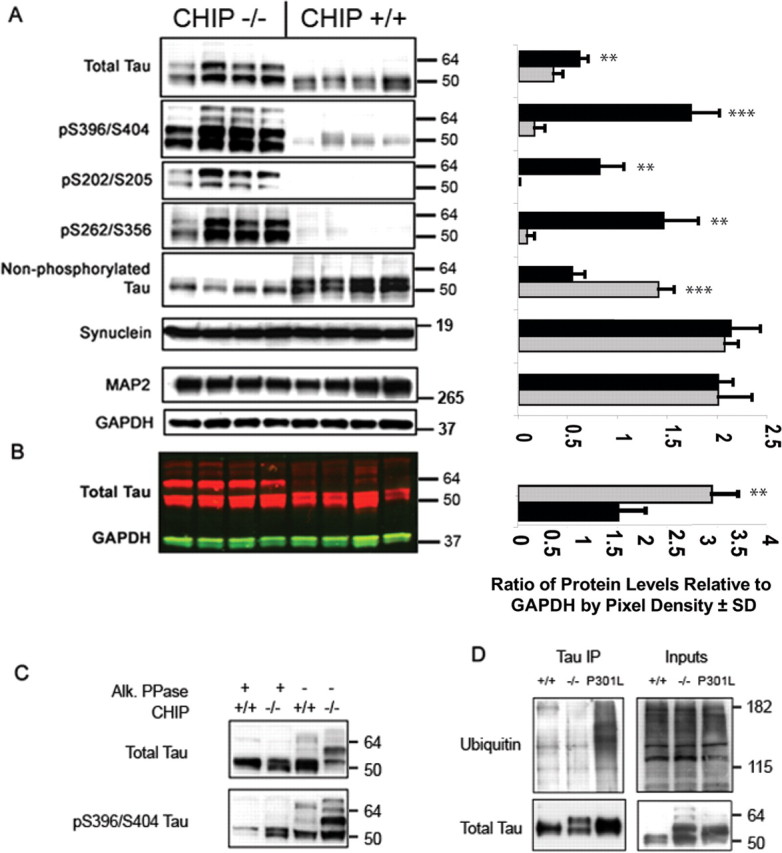
CHIP−/− mice develop marked accumulation of soluble phosphorylated tau species. A, Whole brain was extracted from 30-d-old CHIP−/− mice (n = 4; gray bars) and non-transgenic littermates (n = 4; black bars) and homogenized for SDS-PAGE and Western blot analyses (left), and bands were quantitated by densitometry (right). Probing for three distinct phospho-tau epitopes at S396/S404 (PHF1), S202/T205 (CP13), and S262/S356 (12E8) demonstrated a marked elevation in these tau species in CHIP−/− mice. Total tau levels were also elevated with a distinct upward shift in the molecular weight of nearly 50% of the total protein levels. Tau dephosphorylated at S199/S202 (detected by the Tau 1 antibody) was decreased in CHIP−/− mice, indicating a conversion of total tau to a phosphorylated state. Synuclein levels, which are also affected by ubiquitination, remained unaffected because of CHIP deletion, thus further suggesting selectivity of CHIP for tau. No differences were detected in MAP2 or GAPDH levels. ∗∗p < 0.01; ∗∗∗p < 0.001. B, Western blot analysis of the same mice in A using near-infrared fluorescent detection for tau and GAPDH levels. ∗∗p < 0.01. Error bars represent ± SD. C, Homogenates were also subjected to alkaline phosphatase (Alk. PPase) treatment, and tau levels were assessed by Western blot to determine which species of total tau were phosphorylated. To confirm dephosphorylation efficiency by alkaline phosphatase, we first probed with the pS396/S404 tau antibody (PHF1) and demonstrated that nearly all tau becomes dephosphorylated after treatment. Subsequent probing for total levels showed that the upper molecular weight species seen in the CHIP−/− brain homogenate are primarily hyperphosphorylated forms of tau. D, Coimmunoprecipitation (IP) for total tau and subsequent probing for ubiquitin and total tau demonstrated that 4-week-old CHIP−/− mice, which have a substantial phospho-tau burden comparable with, if not greater than, 34-week-old transgenic mice overexpressing mutant human tau, are unable to ubiquitinate tau species. Even CHIP+/+ mice that lack any type of tau pathology have slightly increased levels of ubiquitinated tau compared with CHIP−/− mice, further suggesting that endogenous mouse tau is indeed a target of CHIP activity. For blots, values shown on the right are in kilodaltons.
We then performed a coimmunoprecipitation experiment on brain homogenates from CHIP−/−, CHIP+/+, and JNPL3 mutant P301L tau mice [as a phospho-tau positive control (Lewis et al., 2000; Sahara et al., 2005)]. Although accumulation of ubiquitinated tau at 1 month of age in mice is normally very low, there were slight indications that more ubiquitin-positive tau was present in the CHIP+/+ mice compared with CHIP−/− mice; however, more striking was the absence of polyubiquitinated tau in CHIP−/− mice in contrast to the large amount of polyubiquitinated tau in JNPL3 mice (Fig. 3D) despite both models having substantial phospho-tau burden (Lewis et al., 2000). Therefore, only the JNPL3 mice demonstrated polyubiquitination of phospho-tau species, presumably mediated by endogenous CHIP activity that is absent in CHIP−/− mice.
Presynaptic and perikaryal accumulation of phospho-tau but not Alzheimer type tau conformational epitopes in CHIP−/− mice
Immunohistochemical studies of CHIP−/− mice (Fig. 4) showed increase in phospho-tau recognized by the pS202/T205 antibody (CP13) in the cerebral cortex, hippocampus, olfactory bulb, midbrain tectum, and cerebellar deep nuclei (Fig. 4A–D). Immunoreactivity was more robust for pS202/T205 (Fig. 4E,F, CP13) than pS396/S404 (Fig. 4G,H, PHF-1), as demonstrated in hippocampal mossy fibers and the cerebellar molecular layer. Mossy fibers were less intensely immunoreactive for pS262/S356 tau (Fig. 4I) and normal tau with Tau5 (Fig. 4K), whereas both antibodies showed Purkinje cell perikaryal staining (12E8 more than Tau5) rather than molecular layer neuropil. The paucity of immunoreactivity for total normal tau is not unexpected because it is difficult to detect normal tau in tissue that has not been perfused, presumably as a result of rapid breakdown of microtubules during brain removal and fixation. The ability to detect any normal tau immunoreactivity is remarkable and fits with the increased expression detected in biochemical analyses (Fig. 3A). There was no evidence of either perikaryal or neuropil immunoreactivity with MC1, a monoclonal antibody that recognizes conformational changes in tau seen in neurofibrillary degeneration (Weaver et al., 2000) (Fig. 4M,N). The phospho-tau immunoreactivity of CHIP−/− mouse brain was suggestive of presynaptic distribution, and localization in mossy fibers in the hippocampus and cerebellum is consistent with this observation, which was later confirmed by immunoelectron microscopy studies (see Fig. 10).
Figure 4.
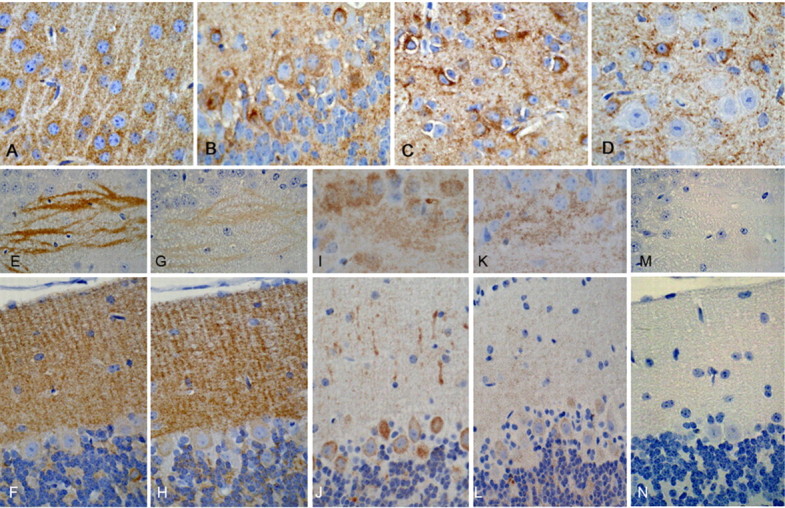
Presynaptic and perikaryal accumulation of phospho-tau but not Alzheimer-type tau conformational epitopes in CHIP−/− mice. A, Neocortical sections show diffuse neuropil immunoreactivity with CP13 (pS202/T205) revealing a negative outline of apical dendrites of pyramidal neurons. B–D, CP13 immunoreactivity was present in occasional neuronal perikarya in the olfactory bulb (B), midbrain tectum (C), and cerebellar deep nuclei (D), regions that also had increased neuronal apoptosis (see Fig. 7D,E). E, G, I, K, M, Adjacent sections of the hippocampal CA3 sector of a CHIP−/− mouse show phospho-tau immunoreactivity of mossy fibers with CP13 (pS202/T205; E), PHF1 (pS396/S404; G), 12E8 (pS262/S356; I), and tau 5 (total normal tau; K) but not MC1 (Alzheimer-type conformational tau epitope; M). F, H, In the cerebellar molecular layer, there is diffuse neuropil staining of the molecular layer, consistent with a presynaptic distribution with antibodies to phospho-tau, CP13 (F), and PHF1 (H). J, L, N, There is more perikaryal staining with 12E8 (J) and tau 5 (L) but neither neuropil nor perikaryal immunoreactivity with MC1 (N).
Figure 10.

Presynaptic accumulation of human tau in CHT mice. A, Pontine nuclei from CHT mice immunostained for pS202/T205 (CP13) tau shows diffuse parenchymal immunoreactivity similar to that seen in the CHIP−/− mice. B, There was also no argyrophilia with Gallyas silver stain in the CHT mice, consistent with CHIP−/− pathology. C, Mossy fibers in the cerebellar internal granular layer of CHT mice show immunoreactivity for human-specific tau polyclonal antibody E1. D, Electron micrograph of the cerebellar granular layer in CHT mice fixed with glutaraldehyde and embedded in epon resin shows well preserved synaptic vesicles (arrows), synaptic densities (double arrows), and mitochondria (M), but no abnormal tau filaments. E, Low-power electron micrograph of the cerebellar granular layer of CHT mouse fixed in paraformaldehyde and embedded in LR White resin shows granule cells (arrows) and glomeruli (arrowheads). The boxed area is enlarged in F. Immunoelectron microscopy of CHT mouse with E1 and 10 nm gold particles (arrows) shows nonfilamentous tau immunoreactivity in axonal terminal of mossy fibers. Mitochondria (M) and synaptic densities (double arrows) are not immunolabeled.
Stress state alone does not precipitate phospho-tau accumulation
To validate that this marked accumulation of phospho-tau was indeed attributable to the absence of CHIP and not simply a result of aberrant kinase activation or stress-associated activities of the sx CHIP−/− mice, we examined levels of tau in another diseased mouse model that has a very similar phenotype caused by a naturally occurring nonsense mutation in the GALC (galactosylceramidase) gene (Duchen et al., 1980). The twitcher mice can be identified phenotypically by their failure to thrive and hindlimb paralysis (Kobayashi et al., 1980), which coincides quite similarly with the phenotypic progression of the sx CHIP−/− mice. Therefore, these mice provided an appropriate disease control model to test whether agonal state alone can facilitate tau accumulation. Both total tau and phospho-tau levels were assessed in P30 sx CHIP−/− and twitcher mice by Western blot, and as demonstrated in previous figures, CHIP null mice have a marked accumulation of phospho-tau species in the soluble fraction; however, twitcher mice showed no evidence of tau accumulation despite their moribund condition (Fig. 5). Therefore, these results powerfully demonstrate that it is the absence of CHIP activity promoting the degradation of phospho-tau species that is provoking its abnormal accumulation, not the overall debilitated condition of these mice.
Figure 5.

Stress state alone does not precipitate phospho-tau accumulation. Whole brain was extracted from 30-d-old CHIP−/− mice, twitcher (Galc−/−) mice, and age-matched wild-type littermates (n = 2) and homogenized for SDS-PAGE and Western blot analysis. Membrane was probed for total tau, CP13 (pS202/T205), CHIP, and GAPDH levels (in kilodaltons). Tau accumulation was present only in the CHIP−/− mice compared with the twitcher mice or wild-type controls.
RNAi for CHIP and the C. elegans ortholog chn-1 promotes accumulation of abnormally phosphorylated tau in both a mouse neuroblastoma cell line and a transgenic nematode line
To further investigate the impact of CHIP on the degradation of tau protein, isogenic strains of transgenic C. elegans expressing a protein fusion between wild-type human tau and enhanced GFP (EGFP) in the bodywall muscles were generated to facilitate direct comparison of the effect of CHIP gene knockdown on tau protein levels by RNAi. The C. elegans ortholog of CHIP, chn-1 [see sequence alignment using the Clustal program (Chenna et al., 2003)] (supplemental Fig. 1, available at www.jneurosci.org as supplemental material), is ubiquitously expressed in many tissues including the muscles of the bodywall. As depicted in Figure 6, worms fed bacteria producing dsRNA targeting the C. elegans chn-1 exhibited a dramatic increase in tau::EGFP fluorescence in the bodywall muscles (C, D) compared with control animals fed RNAi bacteria only (A, B). Specifically, in the case of isogenic strain UA38, tau::EGFP worms exposed to chn-1 dsRNA displayed robust fluorescence throughout the bodywall muscle tissue in 96.4% of animals tested. This is in contrast to the 9.9% of transgenic worms observed with high levels of tau::EGFP fluorescence in the absence of chn1 RNAi treatment (p = 0.003). A similar significant increase in tau levels was observed using a separate transgenic line (UA39; p = 0.001). Western blot analyses of the soluble (S1) fractions from these worms demonstrated that tau levels in transgenic worms exposed to chn-1 RNAi realized dramatic increases in both total and phosphorylated GFP-tagged tau protein levels (Fig. 6E). Worms grown on the RNAi bacterial control strain did not exhibit an increase in tau::EGFP levels over uninduced chn-1 RNAi animals (Fig. 6E). Real-time PCR analysis of chn1 mRNA levels demonstrated ∼50% reductions in worms fed chn1 RNAi bacteria compared with those fed RNAi bacteria alone (Fig. 6F). We then went on to generate siRNA to specifically knockdown CHIP expression in a mouse neuroblastoma cell line (N2A). When CHIP protein was effectively absent because of CHIP RNAi, phospho-tau recognized by a pS396/S404-specific antibody was significantly increased by 40–45%, and total tau levels were increased by 20–25% compared with cells exposed to an siRNA control (n = 4) (Fig. 6G,H). Statistical analysis comparing the increases in phospho-tau compared with total tau levels demonstrated that phospho-tau was preferentially accumulating compared with overall tau levels (∼20% greater increase) (Fig. 6I).
Figure 6.

RNAi for CHIP and the C. elegans ortholog chn-1 promotes accumulation of abnormally phosphorylated tau in both a mouse neuroblastoma cell line and a transgenic nematode line. An isogenic C. elegans line transgenic for tau::EGFP was generated and fed bacteria containing a chn-1 short hairpin RNA construct. A, C, Although Tau::EGFP worms had fluorescence (A), those receiving the chn-1 RNAi had robust increases in tau::EGFP protein in the tail region (C). The boxed areas are shown at higher magnification in B and D, respectively. Scale bars: A, C, 100 μm; B, D, 20 μm. E, Western blot analyses of the worms demonstrate increases in GFP, total tau, and pS396/S404 tau levels when exposed to chn-1 siRNA compared with tau::EGFP RNAi bacteria-fed control worms. F, Real-time PCR demonstrated that chn1 mRNA expression was reduced by ∼50% in worms fed chn1 RNAi bacteria compared with worms fed control RNAi bacteria. G, Four-well replicates of N2A cells transfected with CHIP siRNA or a scrambled siRNA for 72 h were harvested for Western blot analyses. H, Those cells exposed to the CHIP siRNA had a more highly significant increase in pS396/S404 tau (p < 0.0009) compared with total tau immunoreactivity (p < 0.016). Quantitation of bands by densitometry revealed an ∼40–45% increase in pS396/S404 tau levels and an ∼20–25% increase in total tau levels in cells exposed to CHIP siRNA (gray bars) compared with scrambled siRNA (black bars). ∗p < 0.05; ∗∗p < 0.001. I, Additional analysis demonstrated a significant 20% increase in pS396/S404 tau compared with total tau levels, suggesting selectivity of CHIP for aberrant tau species. ∗p < 0.05. For blots, values shown on the right are in kilodaltons. Ctrl, Control. Error bars represent SD.
Caspase-3 activation is increased in CHIP−/− mice and is associated with increased apoptosis and elevated levels of caspase-3-cleavage of tau
Although it has been demonstrated previously that caspase-3 activity is increased in peripheral tissues in CHIP−/− mice (Dai et al., 2003), it remained to be determined (1) whether this apoptotic marker was a developmental consequence of CHIP deletion, (2) whether it was similarly elevated in terminally differentiated neurons, and (3) whether there was a functional link with tau. To address the first issue, RNAi was used to knockdown CHIP RNA expression in N2A cells. RNAi of CHIP caused increases not only in total caspase-3 levels but also in the cleavage product of caspase-3 (cleaved caspase-3) compared with cells treated with a scrambled siRNA, indicating apoptotic activation (Fig. 7A). Tau cleaved by caspase-3 was also slightly elevated in these cells. This in vitro result demonstrated that caspase activation occurring in the CHIP−/− mice was likely not attributable to a developmental compensation but rather was a direct result of the loss of CHIP activity. Western blots for both caspase-3 and cleaved caspase-3 in CHIP−/− brain homogenate also revealed elevated caspase-3 expression and activity in these mice compared with CHIP+/+ littermates (Fig. 7B), and qRT-PCR demonstrated a 70% increase in caspase-3 mRNA levels, thus answering the second question as to whether caspase-3 was elevated in the CNS. Finally, to address the third issue regarding caspase-3 activity in CHIP−/− mice, we used an antibody that selectively detects tau after it has been cleaved by caspase-3 (Tau-C3) to demonstrate that caspase-3-mediated tau cleavage was significantly increased in CHIP−/− mice compared with CHIP+/+ littermates (Fig. 7B). Immunohistochemical analyses further demonstrated weak Tau-C3 immunoreactivity in the neuropil but not aggregated in perikarya of CHIP−/− mice (Fig. 7C). Immunostaining for cleaved caspase-3 demonstrated abundant apoptotic bodies in regions with high levels of phospho-tau immunoreactivity particularly within the deep cerebellar nuclei (Fig. 7D), cerebellar internal granular layer, olfactory bulb, midbrain tectum, and basal layers of the hippocampal dentate fascia. Apoptotic activation was also assessed by TUNEL. TUNEL-positive cells were detected throughout the brain, but particularly in regions that also showed high phospho-tau levels, including the midbrain tectum (Fig. 7E). The cytologic profile of both cleaved caspase-3 and TUNEL-labeled cells was consistent with apoptosis, with nuclear condensation and fragmentation. Not all apoptotic bodies detected with nuclear counterstains (hematoxylin or methyl green) were labeled by both methods. In age-matched CHIP+/+ littermates, apoptotic bodies were limited to sparse cells in the olfactory bulb, subventricular zone, and basal layers of the hippocampal dentate fascia (data not shown).
Figure 7.

Caspase-3 activation is increased in CHIP−/− mice and is associated with increased apoptosis and elevated levels of caspase-3-cleavage of tau. A, Mouse neuroblastoma cells were transfected with CHIP or negative control (Ctrl) siRNAs, and homogenates were subjected to Western blot analysis. Both caspase-3 levels and its activity (measured by cleaved caspase-3 levels) were elevated in response to knockdown of CHIP expression. Tau cleaved at Asp421, a site at which caspase-3 is known to cleave tau in humans (recognized by monoclonal antibody C3), was modestly increased in these cells as well. B, Similarly, in CHIP−/− mice, both caspase-3 and cleaved caspase-3 levels were elevated compared with CHIP+/+ littermates. Caspase-3-cleaved tau was also elevated in CHIP−/− mice, demonstrating an interaction of these two proteins in this model of tau accumulation. C, The cerebellar molecular layer shows neuropil immunoreactivity for C3 in CHIP−/− mice but not in CHIP+/+ littermates. D, Caspase-3 immunoreactivity in paraffin sections of CHIP−/− mice demonstrates many apoptotic bodies (arrows and inset at higher magnification) in the cerebellar deep nuclei, a region with increased tau immunoreactivity in cell processes and neuronal perikarya (see Fig. 4D). E, TUNEL labeling shows similar profiles in other brain regions, such as the midbrain tectum. Insets, Note that the dense and fragmented nuclear profiles (hematoxylin in D and methyl green in E) are consistent with apoptosis in both cleaved caspase-3-immunoreactive cells and in TUNEL-labeled cells.
CHIP-deficient mice overexpressing a mutant form of human tau convert all human tau into a higher molecular weight species but still fail to accumulate insoluble tau species or develop pre-tangle-like structures compared with littermates
To determine the effect that mutant human tau may have on the phenotype and pathology of CHIP−/− mice, we bred the JNPL3 mutant tau strain with CHIP−/− mice to generate CHT mice. Although Western blot analyses of the CHT mice revealed that phospho-tau levels (pS396/S404) were identically elevated between CHIP−/− and CHT mice compared with JNPL3 and CHIP−/− littermates, there was an upward shift in the molecular weight of human tau-immunoreactive species by ∼5 kDa in CHT mice compared with JNPL3 littermates (Fig. 8A). These species collapsed to normal tau molecular weight species after phosphatase treatment (as described above). Immunoprecipitation of tau from both CHT and age-matched JNPL3 littermates confirmed that the tau accumulating in these mice was not polyubiquitinated, whereas the tau present in age-matched JNPL3 mice was (Fig. 8B).
Figure 8.

CHIP-deficient mice overexpressing a mutant form of human tau convert all human tau into a higher molecular weight species but still fail to accumulate insoluble tau species. Whole brain was extracted from 21-d-old CHIP+/+, CHIP−/−, CHT, and JNPL3 mice (n = 3 per group, 2 shown per group), and homogenates were fractionated using Sarkosyl solubilization for SDS-PAGE and Western blot analyses. A, Membranes were probed with the following tau antibodies: pS396/S404-tau (PHF1), total tau (Tau5), dephosphorylated at S199/S202 (Tau 1), and human-specific total tau (E1). CHT mice had a similar tau profile to CHIP−/− mice in that phospho-tau and total tau levels were elevated and dephosphorylated tau was reduced. Note the absence of phospho-tau species in age-matched JNPL3 mice compared with CHT mice. CHT mice probed with the human-specific total tau antibody detected similar expression levels compared with JNPL3 mice; however, the molecular weight of tau was higher in the CHT mice, which collapsed with alkaline phosphatase (PPase) treatment. GAPDH levels were unchanged. B, Coimmunoprecipitation (IP) of tau, followed by immuno-probing for polyubiquitinated species, demonstrated that the tau accumulating in the CHT mice is ubiquitin negative, whereas age-matched JNPL3 littermates did exhibit some polyubiquitination of tau. For blots, values shown on the right are in kilodaltons.
Immunohistochemical analyses for human tau immunoreactivity using the E1 antibody comparing CHT mice with age-matched JNPL3 littermates revealed that JNPL3 mice had marked perikaryal staining (“pre-tangles”) in the CA3 and CA1 pyramidal neurons of the hippocampus, as well as neurons in the brainstem, whereas CHT mice had mostly neuropil (Fig. 9D) or neuritic (axonal/presynaptic) staining, for example in mossy fibers in CA3 (Fig. 9B) or in brainstem (Fig. 9F).
Figure 9.

Human tau fails to form pre-tangles in JNPL3 mice on the CHIP−/− background. Immunohistochemistry with the human tau-specific antibody E1 was used to compare CHT mice (bottom row) with age-matched littermate JNPL3 mice (top row). A, B, E1 immunoreactivity was present in mossy fibers in CA3 of the hippocampus (A, B), but only JNPL3 mice had perikaryal staining in CA3 neurons (A). C, D, In the CA1 sector of the hippocampus, both mice had neuropil staining (C, D), but only JNPL3 had pyramidal neuronal perikaryal staining (C). E, F, Although a few neurons had perikaryal staining in CHIP−/− mice in the brainstem (F), they were more numerous in the brainstem of JNPL3 mice (E). Neuritic staining was present in the brainstem in both mice.
Additional immunohistochemical analysis in CHT mice confirmed the presence of diffuse neuropil phospho-tau (S202/T205) immunoreactivity in the presynaptic neuronal compartment (Fig. 10A) that showed no staining with the Gallyas silver stain (Fig. 10B). Immunostaining for E1 revealed neuritic staining in cerebellar internal granular cell layer within glomeruli (Fig. 10C). Electron microscopy was used to investigate the nature of the cerebellar glomerular immunoreactivity, which had the morphology of presynaptic mossy fiber terminals. In tissue fixed in the absence of glutaraldehyde for immunoelectron microscopy (Fig. 10E,F) immunogold was concentrated in the glomeruli in structures that were rich in synaptic densities and mitochondria consistent with mossy fiber termini. In tissue that had been fixed with glutaraldehyde to preserve ultrastructural morphology (Fig. 10D), there was no evidence of tau filaments in mossy fiber termini (Fig. 10D).
Discussion
Recent progress toward understanding the involvement of the UPS in disorders associated with protein aggregation has implicated a subset of chaperones in removal of abnormal proteins in a number of neurodegenerative diseases (Korhonen and Lindholm, 2004). Several recent studies have implicated the unique U-box ubiquitin ligase CHIP as a necessary component for the sequestration and removal of aberrant proteins prone to aggregation in neurological disorders (Sahara et al., 2002; Urushitani et al., 2004; Miller et al., 2005). Despite these findings, a definitive link between ubiquitination of tau and its aggregation has yet to be established; however, several lines of correlative evidence do exist. Recently, our group implicated CHIP as the primary ubiquitin ligase for tau, and, as such, tau lesions in AD consistently stain positive for CHIP (Petrucelli et al., 2004). Moreover, levels of CHIP were found to be elevated in AD, further suggesting the role of CHIP in tangle maturation (Sahara et al., 2005). Immunoelectron microscopy of filaments isolated from AD and other tauopathies shows direct incorporation of ubiquitin in tau filaments (Yang and Ksiezak-Reding, 1998), whereas immunohistochemical studies of AD conducted over a decade ago suggested that ubiquitination may be associated with a relatively late stage of NFT formation, preceding accumulation of phospho-tau species (Bancher et al., 1991). Indeed, the study by Sahara et al. (2005), in which an 18-month-old CHIP−/− mouse was analyzed, demonstrated that there was a slight increase in SDS-insoluble tau in this animal compared with an age-matched littermate. The current study challenges the notion that polyubiquitination of tau occurs only after NFT formation has initiated. We suggest that ubiquitination of excess phospho-tau facilitates tau aggregation and/or Sarkosyl insolubility as an adaptive neuronal response when the degradative machinery is overwhelmed, as in a diseased state or in models of overexpression.
Previously, our group and others demonstrated that CHIP specifically bound to tau within the microtubule binding region, and other reports revealed that tau is indeed polyubiquitinated within and around this functional domain of the tau protein (Morishima-Kawashima et al., 1993; Cripps et al., 2006). Here, using CHIP (chn-1 in C. elegans) RNAi and CHIP−/− mice, we demonstrate that CHIP is essential for the in vivo degradation of phospho-tau species, perhaps indicating that tau must first be released from the microtubule to become a CHIP substrate. The finding by Nishimura et al. (2004) suggests that phosphorylation of tau within this microtubule binding domain by PAR1, the Drosophila ortholog of the microtubule affinity regulating kinase 2 (MARK2), causes the initial release of tau from the microtubule, and only then are other tau kinases able to recognize tau as a substrate. This finding would support the notion that CHIP specifically recognizes phospho-tau species after their release from the microtubule (Nishimura et al., 2004).
We therefore endeavored to more thoroughly examine the relationship between CHIP activity and tau accumulation in a mammalian system. The present results indicate that loss of functional activity of CHIP is associated with rapid accumulation of ubiquitin-negative phospho-tau species that accumulate in the presynaptic compartment of mice that are 25–30 d old. When these CHIP−/− mice were crossbred with mice expressing mutant human tau (JNPL3), human tau was converted to a higher molecular weight species. Dephosphorylation experiments proved that the high molecular weight tau present in CHT and CHIP−/− mice was phosphorylated (Figs. 3D, 8), suggesting that tau is continually and rapidly phosphorylated as a consequence of normal neuronal plasticity. Thus, ubiquitination of phospho-tau species by CHIP is critical to prevent their accumulation. If the delicate balance of phospho-tau turnover is altered by impaired proteasomal function, tau overexpression, or enhanced tau phosphorylation, aggregation of these hyperphosphorylated tau species into NFTs may be a CHIP/chaperone-mediated protective mechanism rather than a direct route to neuronal cell death. Indeed, the histochemical analyses for human tau in CHT mice further supports this notion, because age-matched JNPL3 littermates already develop pre-tangle perikaryal structures, whereas human tau diffusely accumulates primarily in presynaptic compartments of JNPL3 mice when crossed onto a CHIP null background (CHT) (Figs. 9, 10).
The hypothesis that aggregation of hyperphosphorylated tau in neurofibrillary lesions may be a neuroprotective mechanism was recently proposed by Santacruz et al. (2005), who showed that repression of mutant tau expression in an inducible tau model, at an age when tangle pathology is present, rescued memory deficits and prevented additional neuronal loss. NFTs, however, persisted and even showed a tendency to increase, presumably because of redistribution of soluble intermediates into NFTs. Therefore, although ubiquitination of phospho-tau mediated by CHIP may target phosphorylated tau for proteasomal degradation, the excess of abnormally phosphorylated soluble tau in tauopathies or overexpressing models may exceed the capacity of the proteasome, with the excess ubiquitinated phospho-tau being shifted into stable aggregates.
In addition to interacting with Hsp70 and Hsp90 (Grelle et al., 2006), it was also demonstrated that CHIP was capable of facilitating HSF-1 activity, demonstrating the regulatory role that this protein may have in the inducible chaperone response. Thus, because reductions in Hsp70 have been shown to facilitate caspase-3 activation (Li et al., 2000), one could certainly argue that the deletion of CHIP could result in a number of consequences because of the number of Hsp70/Hsp90 client proteins that are also potential CHIP substrates. Here, however, the finding that all of the phospho-tau species examined were elevated, coupled with the fact that the classical tau kinases such as GSK3-β and MARK2 have not been identified as Hsp client proteins, suggests that the accumulation of tau in these mice is attributable to the absence of CHIP-mediated ubiquitination, not a secondary effect that is altering tau biology. Interestingly, 50% of the CHIP−/− and CHT mice were smaller and feebler compared with CHIP +/+ mice and JNPL3 littermates of similar age, whereas the remaining 50% were indistinguishable from aged wild-type littermates. This difference may be explained by allelic variations in the hybrid SvEv129/C57BL6 genetic backgrounds that gave rise to either a protective factor/variant in the unimpaired mice or a risk factor in the small mice. Although mRNA expression analyses are ongoing to identify which factor or factors are responsible for the dramatic differences observed in these mice, it is remarkable that only those mice with the small phenotype demonstrated marked accumulation of tau.
As discussed above briefly, the CHIP−/− and CHT mice exhibited increased caspase-3 activity in multiple brain regions. Increases in caspase-3, cleaved caspase-3, as well as caspase-3-cleaved tau were detected in brain homogenates from these mice but not littermate controls. Although it has been suggested that caspase cleavage of tau is an early event in NFT formation (Rissman et al., 2004), it was clearly insufficient to lead to tangle formation of either endogenous (CHIP−/−) or overexpressed mutant human tau (CHT). This provides additional evidence that ubiquitination is a critical component for classical tau aggregation.
Whereas it is possible that the inherent properties of mouse tau may make it resistant to filament formation, mice deficient in Pin1, an enzyme shown to be critical for dephosphorylation of tau, and mice engineered to have increased activation of Cdk5 both accumulate phosphorylated endogenous mouse tau and develop argyrophilic tau aggregates (Liou et al., 2003; Noble et al., 2003). Although ubiquitin immunoreactivity was not assessed in either of these models, our findings of robust phospho-tau accumulation in CHIP−/− and CHT mice without any detectable argyrophilic pathology implies that CHIP may function to relieve neurons of potentially toxic properties of soluble phospho-tau intermediates through facilitation of its degradation and/or perhaps promotion of aggregation.
In conclusion, neither hyperphosphorylation nor caspase-3 cleavage of tau, alone or in combination, are sufficient to induce aggregation of tau in the absence of CHIP, indicating that CHIP may function to not only facilitate proteasomal degradation of phospho-tau species, but that NFTs may actually be a protective mechanism of the neuron while an intermediate(s) is the cause of cognitive impairment and neurodegeneration in tauopathies. This also demonstrates that hyperphosphorylation and caspase-3-mediated tau cleavage are occurring before aggregation and that perhaps their effects on soluble tau species are the primary neurotoxic events in the pathogenic cascade. The CHIP−/− mice also demonstrate that tau phosphorylation may be a much more dynamic process than currently thought. The accumulation of endogenous phospho-tau in the CHIP−/− mice suggests that tau phosphorylation is an ongoing process under normal conditions and deletion of CHIP results in its accumulation. Thus, wild-type mice with an intact repertoire of degradative components are able to prevent tau accumulation, but not aggregation, whereas the CHIP−/− mice accumulate massive amounts of nonaggregated, ubiquitin-negative phospho-tau. Therefore, a very delicate balance must be maintained between de novo synthesis, phosphorylation, microtubule binding, and degradation of tau for proper neuronal function. Certainly, there would be redundancies in place to compensate for the loss of one of these critical factors involved, a phenomenon that may account for the CHIP−/− mice that lack a degenerate phenotype. In summary, CHIP reduces the accumulation of phospho-tau species within the brain but may promote its sequestration into aggregates under diseased conditions.
Footnotes
This work was supported by the Florida Alzheimer's Center Research Initiative, the Byrd Institute, the Institute for the Study of Aging, and National Institutes of Health Grants P50-NS40256 and P01-AG17216. Research in the Caldwell laboratory at the University of Alabama was supported by a Howard Hughes Medical Institute Undergraduate Science Education Program Grant. We thank Drs. Peter Seubert, Peter Davies, and Lester Binder for graciously providing the 12E8; CP13, MC1, and PHF-1; and Tau5, Tau 1, and TauC3 antibodies, respectively. We also thank Amy Shirley, Stephanie Brennan, and Michelle Norris for technical assistance with worm tau strain construction and maintenance.
References
- An WL, Cowburn RF, Li L, Braak H, Alafuzoff I, Iqbal K, Iqbal IG, Winblad B, Pei JJ (2003). Up-regulation of phosphorylated/activated p70 S6 kinase and its relationship to neurofibrillary pathology in Alzheimer's disease. Am J Pathol 163:591–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bancher C, Grundke-Iqbal I, Iqbal K, Fried VA, Smith HT, Wisniewski HM (1991). Abnormal phosphorylation of tau precedes ubiquitination in neurofibrillary pathology of Alzheimer disease. Brain Res 539:11–18. [DOI] [PubMed] [Google Scholar]
- Bomar JM, Benke PJ, Slattery EL, Puttagunta R, Taylor LP, Seong E, Nystuen A, Chen W, Albin RL, Patel PD, Kittles RA, Sheffield VC, Burmeister M (2003). Mutations in a novel gene encoding a CRAL-TRIO domain cause human Cayman ataxia and ataxia/dystonia in the jittery mouse. Nat Genet 35:264–269. [DOI] [PubMed] [Google Scholar]
- Brenner S (1974). The genetics of Caenorhabditis elegans Genetics 77:71–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chenna R, Sugawara H, Koike T, Lopez R, Gibson TJ, Higgins DG, Thompson JD (2003). Multiple sequence alignment with the Clustal series of programs. Nucleic Acids Res 31:3497–3500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cripps D, Thomas S, Jeng Y, Yang F, Davies P, Yang A (2006). Alzheimer's-disease-specific conformation of hyperphosphorylated phf-tau is polyubiquitinated through lys-48, lys-11, and lys-6 ubiquitin conjugation. J Biol Chem 281:10825–10838. [DOI] [PubMed] [Google Scholar]
- Crowe A, Ksiezak-Reding H, Liu WK, Dickson DW, Yen SH (1991). The N terminal region of human tau is present in Alzheimer's disease protein A68 and is incorporated into paired helical filaments. Am J Pathol 139:1463–1470. [PMC free article] [PubMed] [Google Scholar]
- Crystal H, Dickson D, Fuld P, Masur D, Scott R, Mehler M, Masdeu J, Kawas C, Aronson M, Wolfson L (1988). Clinico-pathologic studies in dementia: nondemented subjects with pathologically confirmed Alzheimer's disease. Neurology 38:1682–1687. [DOI] [PubMed] [Google Scholar]
- Dai Q, Zhang C, Wu Y, McDonough H, Whaley RA, Godfrey V, Li HH, Madamanchi N, Xu W, Neckers L, Cyr D, Patterson C (2003). CHIP activates HSF1 and confers protection against apoptosis and cellular stress. EMBO J 22:5446–5458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickey CA, Loring JF, Eastman PS, Montgomery JR, Gordon M, Morgan DG (2003). Selectively reduced expression of synaptic plasticity-related genes in APP+PS1 transgenic mice. J Neurosci 23:5219–5226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickey CA, Gordon MN, Mason JE, Wilson NJ, Diamond DM, Guzowski JF, Morgan D (2004). Amyloid suppresses induction of genes critical for memory consolidation in APP + PS1 transgenic mice. J Neurochem 88:434–442. [DOI] [PubMed] [Google Scholar]
- Duchen LW, Eicher EM, Jacobs JM, Scaravilli F, Teixeira F (1980). Hereditary leucodystrophy in the mouse: the new mutant twitcher. Brain 103:695–710. [DOI] [PubMed] [Google Scholar]
- Gomez-Isla T, Hollister R, West H, Mui S, Growdon JH, Petersen RC, Parisi JE, Hyman BT (1997). Neuronal loss correlates with but exceeds neurofibrillary tangles in Alzheimer's disease. Ann Neurol 41:17–24. [DOI] [PubMed] [Google Scholar]
- Grelle G, Kostka S, Otto A, Kersten B, Genser KF, Muller EC, Walter S, Boddrich A, Stelzl U, Hanig C, Volkmer-Engert R, Landgraf C, Alberti S, Hohfeld J, Strodicke M, Wanker EE (2006). Identification of VCP/p97, carboxyl terminus of Hsp70-interacting protein (CHIP), and amphiphysin II interaction partners using membrane-based human proteome arrays. Mol Cell Proteomics 5:234–244. [DOI] [PubMed] [Google Scholar]
- Grundke-Iqbal I, Iqbal K, Tung YC, Quinlan M, Wisniewski HM, Binder LI (1986). Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc Natl Acad Sci USA 83:4913–4917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutton M, Lendon CL, Rizzu P, Baker M, Froelich S, Houlden H, Pickering-Brown S, Chakraverty S, Isaacs A, Grover A, Hackett J, Adamson J, Lincoln S, Dickson D, Davies P, Petersen RC, Stevens M, de Graaff E, Wauters E, van Baren J (1998). Association of missense and 5′-splice-site mutations in tau with the inherited dementia FTDP-17. Nature 393:702–705. [DOI] [PubMed] [Google Scholar]
- Ihara Y, Nukina N, Miura R, Ogawara M (1986). Phosphorylated tau protein is integrated into paired helical filaments in Alzheimer's disease. J Biochem (Tokyo) 99:1807–1810. [DOI] [PubMed] [Google Scholar]
- Inoue T, Thomas JH (2000). Targets of TGF-beta signaling in Caenorhabditis elegans dauer formation. Dev Biol 217:192–204. [DOI] [PubMed] [Google Scholar]
- Ishizawa T, Mattila P, Davies P, Wang D, Dickson DW (2003). Colocalization of tau and alpha-synuclein epitopes in Lewy bodies. J Neuropathol Exp Neurol 62:389–397. [DOI] [PubMed] [Google Scholar]
- Kamath RS, Ahringer J (2003). Genome-wide RNAi screening in Caenorhabditis elegans Methods 30:313–321. [DOI] [PubMed] [Google Scholar]
- Kobayashi T, Yamanaka T, Jacobs JM, Teixeira F, Suzuki K (1980). The Twitcher mouse: an enzymatically authentic model of human globoid cell leukodystrophy (Krabbe disease). Brain Res 202:479–483. [DOI] [PubMed] [Google Scholar]
- Korhonen L, Lindholm D (2004). The ubiquitin proteasome system in synaptic and axonal degeneration: a new twist to an old cycle. J Cell Biol 165:27–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Layfield R, Cavey JR, Lowe J (2003). Role of ubiquitin-mediated proteolysis in the pathogenesis of neurodegenerative disorders. Ageing Res Rev 2:343–356. [DOI] [PubMed] [Google Scholar]
- Lewis J, McGowan E, Rockwood J, Melrose H, Nacharaju P, Van Slegtenhorst M, Gwinn-Hardy K, Paul Murphy M, Baker M, Yu X, Duff K, Hardy J, Corral A, Lin WL, Yen SH, Dickson DW, Davies P, Hutton M (2000). Neurofibrillary tangles, amyotrophy and progressive motor disturbance in mice expressing mutant (P301L) tau protein. Nat Genet 25:402–405. [DOI] [PubMed] [Google Scholar]
- Li CY, Lee JS, Ko YG, Kim JI, Seo JS (2000). Heat shock protein 70 inhibits apoptosis downstream of cytochrome c release and upstream of caspase-3 activation. J Biol Chem 275:25665–25671. [DOI] [PubMed] [Google Scholar]
- Lin WL, Lewis J, Yen SH, Hutton M, Dickson DW (2003). Filamentous tau in oligodendrocytes and astrocytes of transgenic mice expressing the human tau isoform with the P301L mutation. Am J Pathol 162:213–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liou YC, Sun A, Ryo A, Zhou XZ, Yu ZX, Huang HK, Uchida T, Bronson R, Bing G, Li X, Hunter T, Lu KP (2003). Role of the prolyl isomerase Pin1 in protecting against age-dependent neurodegeneration. Nature 424:556–561. [DOI] [PubMed] [Google Scholar]
- Mello CC, Kramer JM, Stinchcomb D, Ambros V (1991). Efficient gene transfer in C. elegans: extrachromosomal maintenance and integration of transforming sequences. EMBO J 10:3959–3970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller VM, Nelson RF, Gouvion CM, Williams A, Rodriguez-Lebron E, Harper SQ, Davidson BL, Rebagliati MR, Paulson HL (2005). CHIP suppresses polyglutamine aggregation and toxicity in vitro and in vivo. J Neurosci 25:9152–9161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morishima-Kawashima M, Hasegawa M, Takio K, Suzuki M, Titani K, Ihara Y (1993). Ubiquitin is conjugated with amino-terminally processed tau in paired helical filaments. Neuron 10:1151–1160. [DOI] [PubMed] [Google Scholar]
- Nishimura I, Yang Y, Lu B (2004). PAR-1 kinase plays an initiator role in a temporally ordered phosphorylation process that confers tau toxicity in Drosophila Cell 116:671–682. [DOI] [PubMed] [Google Scholar]
- Noble W, Olm V, Takata K, Casey E, Mary O, Meyerson J, Gaynor K, LaFrancois J, Wang L, Kondo T, Davies P, Burns M, Veeranna M, Nixon R, Dickson D, Matsuoka Y, Ahlijanian M, Lau LF, Duff K (2003). Cdk5 is a key factor in tau aggregation and tangle formation in vivo. Neuron 38:555–565. [DOI] [PubMed] [Google Scholar]
- Petrucelli L, Dawson TM (2004). Mechanism of neurodegenerative disease: role of the ubiquitin proteasome system. Ann Med 36:315–320. [DOI] [PubMed] [Google Scholar]
- Petrucelli L, Dickson D, Kehoe K, Taylor J, Snyder H, Grover A, De Lucia M, McGowan E, Lewis J, Prihar G, Kim J, Dillmann WH, Browne SE, Hall A, Voellmy R, Tsuboi Y, Dawson TM, Wolozin B, Hardy J, Hutton M (2004). CHIP and Hsp70 regulate tau ubiquitination, degradation and aggregation. Hum Mol Genet 13:703–714. [DOI] [PubMed] [Google Scholar]
- Rissman RA, Poon WW, Blurton-Jones M, Oddo S, Torp R, Vitek MP, LaFerla FM, Rohn TT, Cotman CW (2004). Caspase-cleavage of tau is an early event in Alzheimer disease tangle pathology. J Clin Invest 114:121–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahara N, Lewis J, DeTure M, McGowan E, Dickson DW, Hutton M, Yen SH (2002). Assembly of tau in transgenic animals expressing P301L tau: alteration of phosphorylation and solubility. J Neurochem 83:1498–1508. [DOI] [PubMed] [Google Scholar]
- Sahara N, Murayama M, Mizoroki T, Urushitani M, Imai Y, Takahashi R, Murata S, Tanaka K, Takashima A (2005). In vivo evidence of CHIP up-regulation attenuating tau aggregation. J Neurochem 94:1254–1263. [DOI] [PubMed] [Google Scholar]
- Sakai N, Inui K, Tatsumi N, Fukushima H, Nishigaki T, Taniike M, Nishimoto J, Tsukamoto H, Yanagihara I, Ozono K, Okada S (1996). Molecular cloning and expression of cDNA for murine galactocerebrosidase and mutation analysis of the twitcher mouse, a model of Krabbe's disease. J Neurochem 66:1118–1124. [DOI] [PubMed] [Google Scholar]
- Santacruz K, Lewis J, Spires T, Paulson J, Kotilinek L, Ingelsson M, Guimaraes A, DeTure M, Ramsden M, McGowan E, Forster C, Yue M, Orne J, Janus C, Mariash A, Kuskowski M, Hyman B, Hutton M, Ashe KH (2005). Tau suppression in a neurodegenerative mouse model improves memory function. Science 309:476–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seubert P, Mawal-Dewan M, Barbour R, Jakes R, Goedert M, Johnson GV, Litersky JM, Schenk D, Lieberburg I, Trojanowski JQ, Lee VM (1995). Detection of phosphorylated Ser262 in fetal tau, adult tau, and paired helical filament tau. J Biol Chem 270:18917–18922. [DOI] [PubMed] [Google Scholar]
- Shimura H, Schwartz D, Gygi SP, Kosik KS (2004). CHIP-Hsc70 complex ubiquitinates phosphorylated tau and enhances cell survival. J Biol Chem 279:4869–4876. [DOI] [PubMed] [Google Scholar]
- Spillantini MG, Murrell JR, Goedert M, Farlow MR, Klug A, Ghetti B (1998). Mutation in the tau gene in familial multiple system tauopathy with presenile dementia. Proc Natl Acad Sci USA 95:7737–7741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L, Liu SY, Zhou XW, Wang XC, Liu R, Wang Q, Wang JZ (2003). Inhibition of protein phosphatase 2A- and protein phosphatase 1-induced tau hyperphosphorylation and impairment of spatial memory retention in rats. Neuroscience 118:1175–1182. [DOI] [PubMed] [Google Scholar]
- Suzuki Y, Nakayama M (2003). Differential profiles of genes expressed in neonatal brain of 129X1/SvJ and C57BL/6J mice: a database to aid in analyzing DNA microarrays using nonisogenic gene-targeted mice. DNA Res 10:263–275. [DOI] [PubMed] [Google Scholar]
- Timmons L, Court DL, Fire A (2001). Ingestion of bacterially expressed dsRNAs can produce specific and potent genetic interference in Caenorhabditis elegans Gene 263:103–112. [DOI] [PubMed] [Google Scholar]
- Urushitani M, Kurisu J, Tateno M, Hatakeyama S, Nakayama K, Kato S, Takahashi R (2004). CHIP promotes proteasomal degradation of familial ALS-linked mutant SOD1 by ubiquitinating Hsp/Hsc70. J Neurochem 90:231–244. [DOI] [PubMed] [Google Scholar]
- Weaver CL, Espinoza M, Kress Y, Davies P (2000). Conformational change as one of the earliest alterations of tau in Alzheimer's disease. Neurobiol Aging 21:719–727. [DOI] [PubMed] [Google Scholar]
- Wszolek ZK, Pfeiffer RF, Tsuboi Y, Uitti RJ, McComb RD, Stoessl AJ, Strongosky AJ, Zimprich A, Muller-Myhsok B, Farrer MJ, Gasser T, Calne DB, Dickson DW (2004). Autosomal dominant parkinsonism associated with variable synuclein and tau pathology. Neurology 62:1619–1622. [DOI] [PubMed] [Google Scholar]
- Yang L, Ksiezak-Reding H (1998). Ubiquitin immunoreactivity of paired helical filaments differs in Alzheimer's disease and corticobasal degeneration. Acta Neuropathol (Berl) 96:520–526. [DOI] [PubMed] [Google Scholar]
- Zehr C, Lewis J, McGowan E, Crook J, Lin WL, Godwin K, Knight J, Dickson DW, Hutton M (2004). Apoptosis in oligodendrocytes is associated with axonal degeneration in P301L tau mice. Neurobiol Dis 15:553–562. [DOI] [PubMed] [Google Scholar]
- Zimprich A, Biskup S, Leitner P, Lichtner P, Farrer M, Lincoln S, Kachergus J, Hulihan M, Uitti RJ, Calne DB, Stoessl AJ, Pfeiffer RF, Patenge N, Carbajal IC, Vieregge P, Asmus F, Muller-Myhsok B, Dickson DW, Meitinger T, Strom TM (2004). Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron 44:601–607. [DOI] [PubMed] [Google Scholar]