

Abstract
In human addicts, craving and relapse are frequently evoked by the recall of memories connected to a drug experience. Established memories can become labile if recalled and can then be disrupted by several interfering events and pharmacological treatments, including inhibition of protein synthesis. Thus, reactivation of mnemonic traces provides an opportunity for disrupting memories that contribute to pathological states. Here, we tested whether the memory of a drug experience can be weakened by inhibiting protein synthesis after the reactivation of its trace. We found that an established morphine conditioned place preference (mCPP) was persistently disrupted if protein synthesis was blocked by either anisomycin or cycloheximide after the representation of a conditioning session. Unlike other types of memories, an established mCPP did not become labile after contextual recall, but required the concomitant re-experience of both the conditioning context and the drug. An established mCPP was disrupted after the conditioning session if protein synthesis was blocked selectively in the hippocampus, basolateral amygdala, or nucleus accumbens but not in the ventral tegmental area. This disruption seems to be permanent, because the preference did not return after further conditioning. Thus, established memories induced by a drug of abuse can be persistently disrupted after reactivation of the conditioning experience.
Keywords: morphine, conditioned place preference, memory, reconsolidation, protein synthesis inhibitor, addiction
Introduction
In human addicts, exposure to environmental cues previously associated with a drug strongly increases the risk of relapse (Ludwig and Stark, 1974; O’Brien et al., 1977). This conditioned response can occur despite years of abstinence from drug use (O’Brien et al., 1992) and represents a major challenge for the treatment of addiction. Several recent studies indicate that memory mechanisms and systems play a critical role in addiction (Landauer, 1969; White, 1996; Hyman and Malenka, 2001; Nestler, 2002; Robbins and Everitt, 2002). Indeed, because the experience of drugs of abuse creates new memories, these drugs can be viewed as memory modulators or enhancers (Landauer, 1969; White, 1996). Moreover, a growing body of evidence indicates that memory and addiction share both neural circuitry and molecular mechanisms (Fuchs et al., 2000; Vorel et al., 2001; Nestler, 2002; Kelley, 2004). Thus, given that several aspects of addiction depend on mnemonic processes induced by the drug experience, it is reasonable to hypothesize that disrupting these memories will help to diminish addictive behavior and the risk of relapse.
The stabilization of a new memory occurs through a process known as consolidation, which requires gene expression. In fact, disrupting protein synthesis immediately after learning prevents memory formation (Davis and Squire, 1984). Once consolidated, memory can again become transiently labile and sensitive to protein synthesis inhibitors if reactivated (Nader et al., 2000; Sara, 2000; Dudai and Eisenberg, 2004; Alberini, 2005). Therefore, it has been proposed that, to be maintained, a reactivated memory needs to undergo a process of reconsolidation (Lewis, 1979; Nader et al., 2000; Sara, 2000). This finding has important clinical implications because it shows that by blocking protein synthesis after reactivation, it is possible to selectively reduce or even eliminate long-lasting memories, including memories linked to pathological states.
Here, we investigated whether an established, drug-induced memory, similar to other types of memories, can be disrupted by inhibiting protein synthesis after its reactivation. Toward this end, we used a typical morphine conditioned place preference (mCPP) paradigm, in which the animal learns to associate a context with the rewarding effects of morphine. This paradigm models the formation of contextual/cued associations to drugs of abuse. Notably, the recall of these types of memories is known to play a critical role in several aspects of human addiction behavior, including relapse, craving, drug-seeking behavior, and withdrawal (Childress et al., 1993; O’Brien et al., 1993; Bardo and Bevins, 2000). Our results show that it is possible to persistently disrupt an established mCPP.
Materials and Methods
Animals.
Adult male, Long–Evans rats weighing between 200 and 250 g served as subjects in all the experiments. Animals were individually housed and maintained on a 12 h light/dark cycle. All experiments were performed during the light cycle. All rats were allowed ad libitum access to food and water. All protocols were complied with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the Mount Sinai School of Medicine Animal Care Committees.
Morphine conditioned place preference.
The protocol used was modified from Romieu et al. (2002). All experiments took place in a dimly lit room. On day 1, the rats received a single pre-exposure test in a Plexiglas shuttle box (Med Associates, St. Albans, VT) composed of two compartments of equal size (20.3 × 15.9 × 21.3 cm) separated by a sliding door. One side consisted of white walls, a grid floor, and a 60 W light, which was on throughout the experiments. The other side had black walls, a smooth plastic-covered floor, and was unlit throughout the experiments. The procedure consisted of three different phases: preconditioning (day 1), conditioning (days 2–5), and postconditioning days, as indicated in Results. For the preconditioning phase, each rat was placed in the white compartment with the door open. The animal was allowed to freely explore the apparatus for 10 min. The time spent in each chamber was recorded (unconditioned preference), and then the animals were returned to their home cages. Most animals spent approximately one-half of the time in each chamber (see Results). Animals showing a strong unconditioned preference (>540 s), which were <2% of the total number, were discarded. On the subsequent 4 d, place preference conditioning was conducted using a counterbalanced procedure, such that one-half of the animals in each experimental group were conditioned to the spontaneously preferred side and the other half to the spontaneously nonpreferred side. During conditioning, the animals received a subcutaneous injection of either morphine (10 mg/kg) or saline (vehicle solution) and were confined for 30 min in the assigned compartment. The postconditioning testing consisted of placing the animal into the white compartment and allowing it to freely access both chambers for 10 min. The amount of time that the animals spend in the conditioned chamber was recorded (CPP score). Because the aim of this study was to determine whether the reactivation of the memory trace causes the CPP to become labile again, we chose not to administer vehicle solution and confine the animals to the alternative compartment. In fact, exposure to a second compartment not paired with the drug but with vehicle also results in learning the contextual representation of that compartment (Pavlov, 1927; Bouton, 1993). Thus, to avoid any interference of this second learning trace on the mCPP and its reactivation, we performed only pairings with morphine. The potential drawback of omitting the vehicle pairing procedure is that CPP memory impairments could be confused with novelty seeking. However, we addressed the issue of novelty with control experiments described in Results, showing that no motivational effects of novelty were seen in our experiments. Our results are in agreement with previous data and conclusions on CPP, indicating that rats do not form a place preference for a novel side over one experienced previously (Mucha et al., 1982; Mucha and Iversen, 1984; van der Kooy, 1987; Bardo and Bevins, 2000).
The re-experience of a conditioning trial (reinforced trial) and a reminder conditioning consisted of a single morphine conditioning session.
Protein synthesis inhibitors.
For systemic treatments, anisomycin or cycloheximide were injected subcutaneously at 210 mg/kg (Milekic and Alberini, 2002) and 2.8 mg/kg (Squire et al. 1980), respectively. For double injection of cycloheximide, 2.2 mg/kg was used. These doses have been shown previously to disrupt memory consolidation (Davis et al., 1980; Squire et al., 1980; Milekic and Alberini, 2002). For local injections, animals were injected bilaterally into the basolateral amygdala (BLA), ventral tegmental area (VTA), and nucleus accumbens (NAc) with 0.5 μl of anisomycin at 125 μg/μl (Quevedo et al., 1999; Schafe and LeDoux, 2000; Taubenfeld et al., 2001) delivered over 1.25 min. Bilateral injections into the hippocampus (HC) consisted of 1 μl delivered over 2.5 min. Anisomycin was dissolved as described previously (Milekic and Alberini, 2002). Cycloheximide was dissolved in DMSO and finally diluted to 1%DMSO in saline. Vehicle solutions were prepared accordingly.
Inhibition of protein synthesis.
Groups of rats were injected intraperitoneally with 125 μCi l-[35S]methionine. Immediately after, they received an injection of either protein synthesis inhibitors (cycloheximide or anisomycin) or vehicle solution. One-half of these rats also received a second injection of either vehicle or inhibitor 5 h later. The rats that received a single injection were divided into two groups; one was killed 1 h later, and the other was killed 6 h later. The rats that received two injections were killed 1 h after the last injection. The brains were homogenized in 5 vol of cold lysis buffer (1% SDS, 60 mm Tris-Cl, pH 7.4, 62.4 mm imidazole, 40 mm DTT, 10% glycerol). The homogenates were boiled for 5 min and centrifuged at 18,000 × g for 20 min at room temperature. Equal amount of proteins were precipitated with 20% TCA and the radioactivity was counted. The inhibition of protein synthesis was calculated as follows: [1 − (CPMinhibitor/CPMvehicle)] × 100.
Surgical procedures.
Rats were anesthetized and implanted with cannulas (22 gauge; Plastics One, Roanoke, VA) positioned 1.5 mm just above each target areas using the following coordinates, according to Paxinos and Watson (1998): HC: anteroposterior (AP), −4.0 mm and mediolateral (ML), ±2.60 mm from bregma; dorsoventral (DV), −2.0 mm from the skull surface; BLA: AP, −2.8 mm; ML, ±5.3 mm; DV, −6.25 mm from the skull surface; NAc: AP, +1.4 mm; ML, ±1.7 mm; DV, −6.30 mm; VTA: AP, −5.2 mm; ML, ±1.80 mm with a 10° angle; DV, −6.45 mm. Rats were given at least 5 d to recover before undergoing any experimental procedures.
Histology.
At the end of the behavioral experiments, rats were anesthetized with ketamine (60 mg/kg, i.p.) and xylazine (7.5 mg/kg, i.p.) and perfused intracardially with 10% PBS-buffered formalin. The brains were removed, postfixed overnight in a 30% sucrose/formalin solution, and then cryoprotected in 30% sucrose/saline. To verify the location of the cannula tips, 40 μm coronal sections were cut through the targeted area, stained with cresyl violet, and examined under a light microscope. Rats with incorrect cannula placements were excluded from the study. Overall, four rats received improper cannula placements and were therefore excluded from the analyses.
Statistical analyses.
Statistical analyses were performed using one- and two-way ANOVAs followed by Newman–Keul or Bonferroni’s post hoc comparisons. For pair-wise comparisons, Student’s t test was used.
Results
The consolidation of mCPP requires protein synthesis
In the first set of experiments, we determined whether a 4 d conditioning protocol requires a protein synthesis-dependent process of consolidation to establish mCPP. Toward this end, we assessed the effect of two widely used protein synthesis inhibitors, anisomycin and cycloheximide, which block protein synthesis by distinct mechanisms, to confirm that the results were specifically related to protein synthesis inhibition and not to unspecific effects. As detailed below, both inhibitors produced similar outcomes.
Rats were conditioned to morphine or vehicle once a day for 4 d, and at the end of a 4 d conditioning session, one-half of the morphine-conditioned rats received a single subcutaneous dose of anisomycin or cycloheximide, whereas the other half received vehicle solution. CPP was tested 24 h later. To determine whether the effect was stable, the animals were retested 1 week later.
The test at 24 h showed that both inhibitors completely blocked mCPP (Fig. 1A). A one-way ANOVA that compared the CPP scores across vehicle, morphine, and morphine-with-inhibitor-injected groups revealed that there was a significant group effect (F(2,22) = 7.6; p < 0.01). A Newman–Keul post hoc test showed that morphine-conditioned animals treated with anisomycin or cycloheximide had a significantly lower conditioning score (280.1 ± 17.5 s; p < 0.01) compared with morphine-conditioned, vehicle-injected controls (390.3 ± 31.5 s). Moreover, their postconditioning score was not significantly different from that of the vehicle-conditioned animals (262.0 ± 73.8 s) or their preconditioning score (243.3 ± 36.5 s). Because both anisomycin and cycloheximide produced a similar effect, the CPP scores of the two groups were combined. When the same animals were retested 1 week later, they showed a partial, but significant, recovery of the place preference (358.9 ± 28.0 s; p < 0.05; t test) compared with their 24 h conditioning score (Fig. 1A). This suggested that inhibition of protein synthesis at the end of conditioning impaired CPP only transiently.
Figure 1.
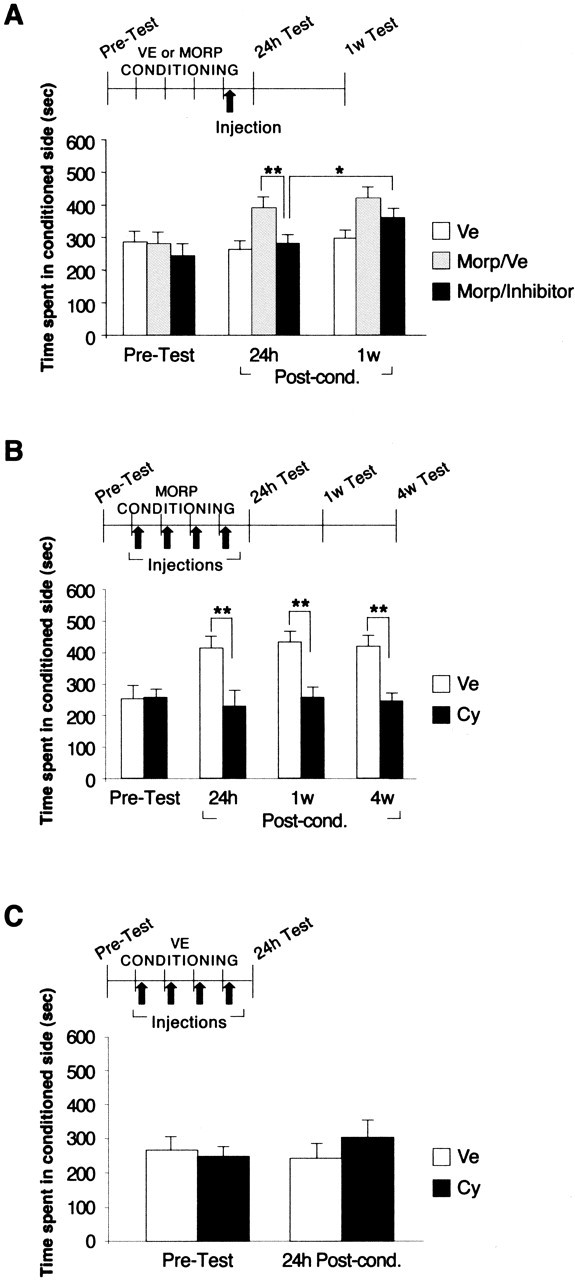
Protein synthesis is required for the induction of mCPP. A–C, Values are expressed as mean ± SEM time spent in the drug-conditioned chamber. *p < 0.05; **p < 0.01. A, Groups of rats were conditioned for 4 d to either vehicle (n = 8; white bars) or morphine (n = 15). At the end of conditioning, the morphine-conditioned rats were injected subcutaneously with either vehicle (n = 8; gray bars) or inhibitor (n = 7, of which n = 4 with cycloheximide and n = 3 with anisomycin; black bars). All rats were tested 24 h and 1 week later. B, Rats received daily administration of either cycloheximide (n = 8; black bars) or vehicle (n = 8; white bars) immediately after each conditioning session. CPP was tested 24 h, 1 week, and 4 weeks later. C, Rats were conditioned to vehicle instead of morphine and administered cycloheximide (n = 9; black bars) or vehicle (n = 11; white bars) immediately after each conditioning session. CPP was tested after 24 h. VE or Ve, Vehicle; MORP or morp, morphine; w, week; Post-cond., postconditioning; Cy, cycloheximide.
One possible explanation for this recovery is that a single dose of inhibitor does not completely block the protein synthesis (Flood et al., 1975) required for CPP consolidation. If this is the case, a partial inhibition might only delay but not sufficiently disrupt the consolidation process. A similar hypothesis has been proposed by Lattal and Abel (2004) to explain transient effects of protein synthesis on contextual fear conditioning memory reconsolidation. A second possibility is that each conditioning event induces a protein synthesis-dependent phase and that blocking protein synthesis only during the last day of conditioning is not sufficient to disrupt the consolidation of mCPP.
We first tested whether a more extended inhibition of protein synthesis at the end of the 4 d conditioning results in a persistent blockade of mCPP. Groups of rats were conditioned as described above but received two injections of cycloheximide, one immediately after the last conditioning session and the second 5 h later. In parallel, the rates of protein synthesis inhibition, over time, after a single or double injection were established. Rats were injected with 35S-methionine and divided into two groups: one was challenged by one injection of either cycloheximide or vehicle solution, whereas the other received two injections of the each solution. The single injection and the first of the two injections were administered immediately after the 35S-methionine; the second injection was applied 5 h after the first. The groups that received one injection were divided into two subgroups: one was killed 1 h after injection (n = 4 per group) and the other at 6 h after injection (n = 4 per group). The groups that received two injections were killed 6 h after the first injection (n = 4 per group). The rat brains were dissected and homogenized. TCA precipitates of these homogenates were used to determine the amount of protein synthesis inhibition, as described in Materials and Methods. One hour after a single injection, 70% of protein synthesis was inhibited, whereas 6 h after a single injection, only 23% of protein synthesis was blocked. With the double injection, the rate of protein synthesis inhibition at 6 h was maintained at 71%. Similar degrees of protein synthesis inhibition have been described previously (Flood et al., 1977, 1978).
When we applied the double injection protocol, which led to a more extended inhibition of protein synthesis at the end of conditioning, the behavioral results were similar to those found after a single injection; CPP was significantly inhibited at 24 h but recovered 1 week later (data not shown).
We then tested the second hypothesis outlined above by measuring the effect of blocking protein synthesis after each daily conditioning. Groups of rats were injected with one dose of either cycloheximide or vehicle every day at the end of each conditioning trial and tested 24 h, 1 week, and 4 weeks later. As depicted in Figure 1B, a two-way ANOVA that compared the mCPP score across treatment and test revealed a significant effect of treatment (F1,42 = 15.58; p < 0.05) and a significant treatment-by-test interaction (F2,42 = 4.29; p < 0.001). Bonferroni’s post hoc test showed that cycloheximide significantly inhibited mCPP at the 24 h test (cycloheximide, 226.6 ± 47.2 s; vehicle, 410.9 ± 36.3 s; p < 0.01). This inhibition persisted at 1 week (254.8 ± 32.8 and 429.4 ± 33.1 s, respectively; p < 0.01) and 4 weeks after conditioning (243.3 ± 21.8 and 416.8 ± 31.4 s, respectively; p < 0.01) (Fig. 1B). Thus, blocking protein synthesis after each conditioning session persistently disrupts the consolidation of mCPP.
To exclude the possibility that systemically injected protein synthesis inhibitors cause nonspecific behavioral effects, we tested the CPP scores of rats that underwent 4 d conditioning with vehicle instead of morphine and received either cycloheximide or vehicle injection immediately after each vehicle conditioning. CPP was tested 24 h later. As depicted in Figure 1C and revealed by Student’s t test, no significant difference was found between vehicle- and cycloheximide-treated animals after vehicle conditioning (240.3 ± 42.8 and 301.7 ± 50.4 s, respectively). These CPP scores were similar to their preconditioning scores (265.6 ± 38.7 and 247.1 ± 28.5 s, respectively). We concluded that cycloheximide per se does not cause any change in place preference.
Some authors indicate that one concern regarding a disruptive effect on CPP is the potential confounding influence of novelty-seeking behavior on the test day (Bardo and Bevins, 2000). Our results depicted in Figure 1C show that no effects of novelty were seen in the place conditioning paradigm used. Indeed, vehicle-conditioned rats did not change their place preference in favor of the novel context on the test day, because the preference that the animals showed on the test day remained similar to the spontaneous preference recorded during the preconditioning test. Furthermore, the additional treatment with the protein synthesis inhibitor cycloheximide also did not affect their place preference.
Reactivation with a conditioning trial but not with contextual recall destabilizes an established mCPP
We then asked whether mCPP becomes labile if the rats recall this memory by being re-exposed to the conditioning context. As shown in Figure 2A, groups of rats were conditioned for 4 d. One week after the end of conditioning, the rats were tested for mCPP. This test recalled the experience (test 1) or, in other words, reactivated mCPP. Immediately after test 1, one-half of the rats received an injection of cycloheximide or anisomycin and the other half was injected with vehicle. We then retested the animals 24 h later (test 2). We found no change between the mCPP scores at test 2 compared with their test 1 preference; in fact, all rats maintained a strong preference for the conditioned side. Moreover, the mCPP of anisomycin- or cycloheximide-injected rats were similar to that of vehicle-injected controls (test 1: vehicle, 394.4 ± 42.6 s; inhibitor, 387.8 ± 25.8 s; Test 2: vehicle, 413.3 ± 35.9 s; inhibitor, 425.9 ± 25.3 s). Because both cycloheximide and anisomycin had similar effects, their scores were combined.
Figure 2.
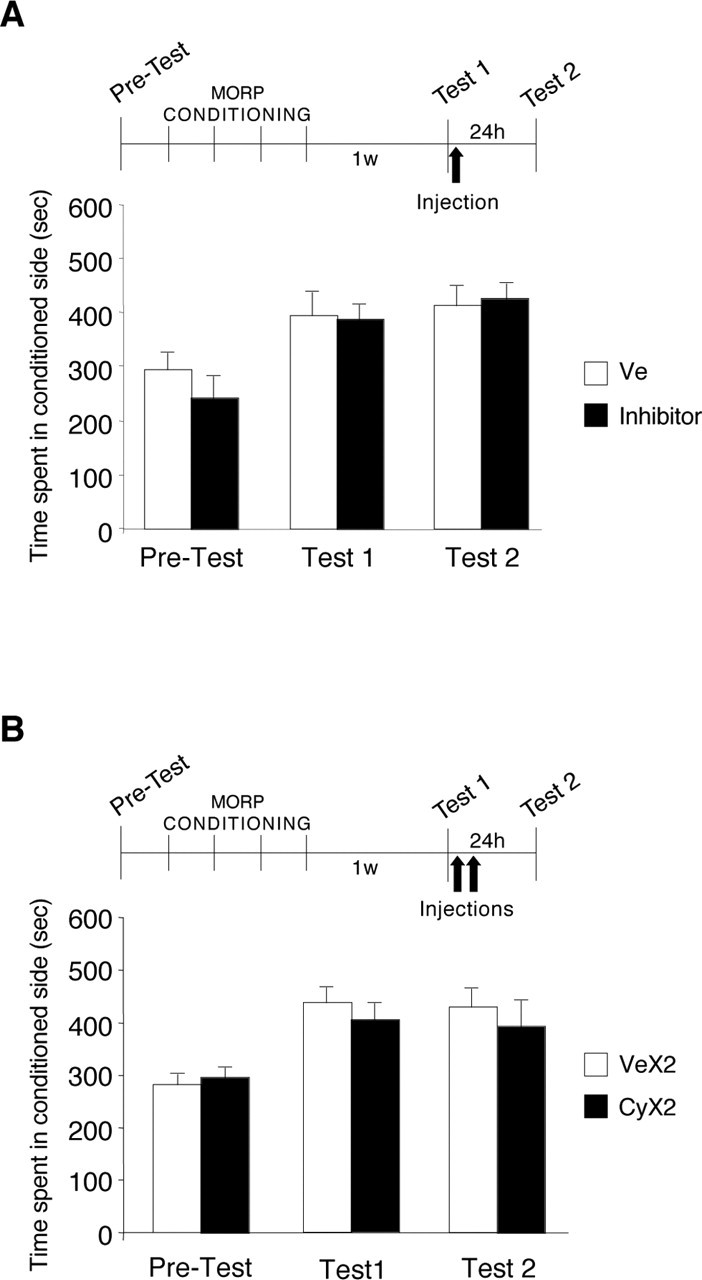
Protein synthesis inhibitors administered after contextual reactivation do not affect the stability of mCPP. All values are expressed as mean ± SEM time spent in the drug-conditioned chamber. A, Rats were conditioned to morphine and tested after 1 week (test 1) to reactivate mCPP. Immediately after test 1, one-half of the rats received one injection of cycloheximide or anisomycin (inhibitor; n = 8, of which n = 4 with cycloheximide and n = 4 with anisomycin; black bars), and the other half of vehicle solution (n = 7; white bars). When retested 24 h later (test 2), all rats maintained their mCPP scores. B, Rats were conditioned and tested as described in A. Immediately after test 1, rats received two injections of either cycloheximide (CyX2; n = 8) or vehicle (VeX2; n = 7), 5 h apart. When retested 24 h later (test 2), all rats maintained their mCPP scores. MORP, Morphine; w, week; Ve, vehicle.
Although no disruption (not even a transient one) of mCPP was observed after a single injection of protein synthesis inhibitor after contextual recall, in line with the hypothesis described above, we tested the effect of blocking protein synthesis for a more extended period of time. As shown in Figure 2B, groups of rats were treated as above but received two subcutaneous injections of cycloheximide, one immediately after contextual recall (test 1) and the second 5 h later. When the animals were retested 24 h later (test 2), again no effect of the treatment was observed, and all rats maintained a strong preference for the conditioned side (test 1: vehicle, 436.4 ± 24.9 s; cycloheximide, 403.5 ± 32.3 s; test 2: vehicle, 428.7 ± 27.4 s; cycloheximide, 393.4 ± 45.6 s).
These data showed that, unlike other types of contextual memories (Nader et al., 2000; Sara, 2000; Dudai and Eisenberg, 2004; Alberini, 2005), mCPP does not become sensitive to protein synthesis inhibitors after contextual recall.
Because the drug may serve as an internal reinforcer, we then hypothesized that mCPP may become labile if the associations formed between the effects of the drug and the context were reactivated. Thus, we tested the effect of protein synthesis inhibitors on mCPP after the re-experience of a single reinforced trial (i.e., a conditioning session). As depicted in Figure 3A, rats were conditioned to morphine for 4 d and, 1 week later, underwent one conditioning session. Immediately after this session, one-half of the animals received a systemic injection of inhibitor (cycloheximide or anisomycin) and the other half received vehicle. All animals were then retested 24 h and 1 week later. A two-way ANOVA, which compared the CPP scores of the two groups across treatment and test, revealed a significant effect of treatment (F(1,48) = 11.97; p < 0.01) and test (F(2,48) = 5.42; p < 0.01). Bonferroni’s post hoc test showed that the inhibitor caused a significant disruption of mCPP at 24 h after reconditioning (inhibitor, 261.5 ± 33.9 s; vehicle, 461.6 ± 42.5 s; p < 0.01), but this disruption did not persist 1 week later (inhibitor, 365.4 ± 45.1 s; vehicle, 442.3 ± 29.1 s) (Fig. 3A).
Figure 3.
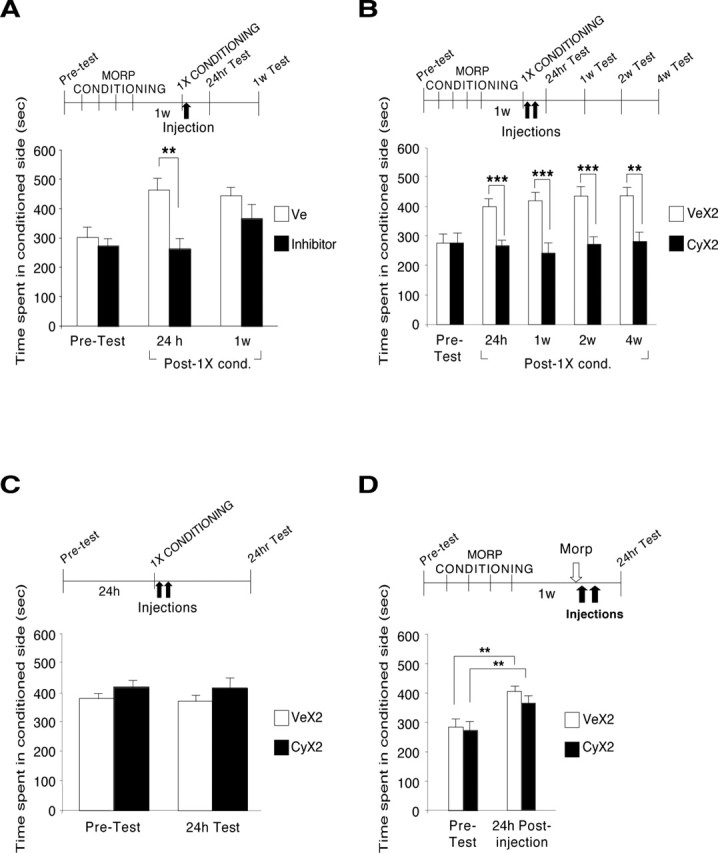
An established mCPP is disrupted by protein synthesis inhibitors administered after a single conditioning session. A–D, Values are expressed as mean ± SEM time spent in the drug-conditioned chamber. **p < 0.01; ***p < 0.001. A, Rats were conditioned to morphine and 1 week later received one additional conditioning session (1X CONDITIONING). Immediately after, one-half of the animals were injected with cycloheximide or anisomycin (inhibitors; n = 10, of which n = 6 with cycloheximide and n = 4 with anisomycin; black bars), and the other half were injected with vehicle (n = 8; white bars). Rats were tested 24 h and 1 week later (Post-1X cond.). B, Animals were conditioned and 1 week later received one additional conditioning (1X CONDITIONING) as described in A, followed by two injections of cycloheximide (CyX2; n = 10; n = 7 in 4 week test; black bars) or vehicle (VeX2; n = 11; white bars) 5 h apart. Rats were tested 24 h and 1, 2, and 4 weeks later. C, Rats received one morphine conditioning in the spontaneously preferred context followed by two injections of either cycloheximide (CyX2; black bars; n = 8) or vehicle (VeX2; white bars; n = 8) and were tested 24 h later. D, The conditioning context was omitted. Rats were conditioned for 4 d and 1 week later received one morphine treatment in their home cage. Thirty minutes later, they received two injections of either cycloheximide (CyX2; black bars; n = 13) or vehicle (VeX2; white bars; n = 12). CPP was tested 24 h after injection. MORP, Morphine; w, week; Ve, vehicle.
Because there was a transient disruption, we tested whether blocking protein synthesis for a more extended period of time after the re-experience of a single conditioning session results in a sustained disruption of mCPP. As depicted in Figure 3B, groups of rats were treated as above but received two injections of either cycloheximide or vehicle, one immediately after the re-experience of a conditioning session and the second 5 h later. As described above, this double injection of cycloheximide blocks 70% of protein synthesis for at least 6 h. Results showed that this cycloheximide treatment after the re-experience of the conditioning session completely and persistently blocked mCPP (cycloheximide: 24 h, 264.1 ± 13.3 s; 1 week, 239.9 ± 34.4 s; 2 weeks, 270.1 ± 27.2 s; 4 weeks, 278.4 ± 32.5 s; vehicle: 24 h, 396.5 ± 26.7 s; 1 week, 416.8 ± 26.8 s; 2 weeks, 432.2 ± 27.6 s; 4 weeks, 432.5 ± 29.6 s). A two-way ANOVA, which compared the CPP scores of the two groups across treatment and test, revealed a significant effect of treatment (F(1,95) = 49.04; p < 0.0001), test (F(4,95) = 2.69; p < 0.05), and a treatment-by-test interaction (F(4,95) = 3.26; p < 0.05). Bonferroni’s post hoc test showed that cycloheximide significantly and persistently disrupted CPP (p < 0.001 for 24 h, 1 week, 2 weeks; p < 0.01 for 4 weeks) compared with vehicle treatment. Moreover, at all time points of testing, the CPP scores of cycloheximide-treated rats were similar to their preconditioning score (273.7 ± 30.6 s).
To exclude that the disruption of mCPP by cycloheximide reflects an avoidance of the conditioning context induced, for example, by malaise caused by the treatment, groups of rats were tested for their preconditioning preference and the following day conditioned with morphine in the spontaneously preferred context. We chose to condition the animals in the spontaneously preferred context to exclude any confound caused by their natural avoidance. Immediately after conditioning, one-half of the rats received a subcutaneous double injection of cycloheximide, whereas the other half received vehicle. As depicted in Figure 3C, the test at 24 h showed that cycloheximide-injected rats had mCPP scores similar to those of vehicle-injected rats. These scores were also similar to their respective preconditioning scores (before conditioning: cycloheximide, 413.9 ± 25.6 s; vehicle, 376.6 ± 16.2 s; after conditioning: cycloheximide, 411.3 ± 33.0 s; vehicle, 367.0 ± 18.2 s). This showed that morphine followed by a prolonged inhibition of protein synthesis does not cause avoidance of the conditioned context.
Finally, to determine whether the blocking effect of the inhibitors on mCPP after reactivation by conditioning was context dependent, the conditioning context was omitted. Rats were conditioned for 4 d and 1 week later received an injection of morphine in their home cage, followed by a double injection of either cycloheximide or vehicle 30 min later. As shown in Figure 3D, testing at 24 h showed that cycloheximide did not decrease mCPP (before conditioning, 271.5 ± 24.8 s; after conditioning, 364.8 ± 16.4 s) compared with vehicle-injected controls (before conditioning, 284.0 ± 28.1 s; after conditioning, 404.8 ± 24.8 s). These results show that, once consolidated, CPP does not become labile after the re-experience of the drug in a different context. The same results were obtained using another alternative context, which consisted of a smooth and clear plastic box (width, 18.9 cm; length, 29.7 cm; height, 12.8 cm) with an opaque lid placed in a different room, for the reactivation by conditioning (data not shown).
Together, these data led us to conclude that an established mCPP can be persistently disrupted by inhibiting protein synthesis after the concomitant reactivation of both the contextual cues and the experience of the drug.
An established mCPP is disrupted by inhibition of protein synthesis in the HC, BLA, or NAc, but not in the VTA, after the re-experience of a conditioning trial
To determine in which brain regions protein synthesis is required to maintain or reconsolidate an established mCPP reactivated by a reinforced trial, we tested the effect of blocking protein synthesis in selected brain areas. We targeted the HC, BLA, NAc, and VTA, which are known to mediate drug addiction and/or memory formation (Tzschentke, 1998; McGaugh, 2004). Whereas the HC and BLA are required for spatial– and contextual–emotional associative types of memories (Ambrogi-Lorenzini et al., 1995; McGaugh, 2004; Richter-Levin, 2004), the NAc and VTA are part of the mesolimbic dopaminergic circuit, which mediates drug reward. Moreover, the HC, BLA, and NAc have all been found to play a critical role in both memory and addiction (Ambrogi-Lorenzini et al., 1999; Fuchs et al., 2000; Di Chiara, 2002; Meyers et al., 2003), and, interestingly, the NAc seems to represent a site of convergence, because it receives direct projection from both HC and BLA (Groenewegen et al., 1987; Wright et al., 1996). Finally, the VTA, an area of synaptic plasticity for drug addiction (Kauer, 2004), is critical for the induction of mCPP and for behavioral sensitization to drugs of abuse (Kalivas and Weber, 1988; Vezina and Stewart, 1990; Harris et al., 2004).
Rats were bilaterally implanted with cannulas targeting HC, BLA, NAc, or VTA. After recovering from surgery, rats were pretested, conditioned for 4 d, and, a week later, received a reinforced trial as described above. Immediately after this session, animals were injected with either anisomycin or vehicle into the area of interest and tested for mCPP 24 h and 1 week later. Results showed that the HC, BLA, or NAc injections of anisomycin completely and persistently disrupted mCPP, whereas the same treatment in the VTA had no effect. Parallel experiments determined the rate of protein synthesis inhibition by a local injection of anisomycin. We investigated this in the HC, and 35S-methionine incorporation revealed that, 1 h after injection of 1 μl of anisomycin at 125 μg/μl (see Materials and Methods), protein synthesis was blocked by 82% compared with vehicle injection. This inhibition was maintained at 6 h after injection (85%), suggesting that new protein synthesis is strongly inhibited for an extended period of time.
Figure 4 shows the locations of injections targeting HC (Fig. 4A), BLA (Fig. 4B), NAc (Fig. 4C), and VTA (Fig. 4D) and examples of the injection sites. Specifically, the injections mainly targeted the dorsal HC, the BLA, the NAc core, and the anterior VTA. Figure 5A depicts the results obtained with HC injections. A two-way ANOVA, which compared CPP scores across treatment and test, revealed a significant effect of treatment (F(1,56) = 146.9; p < 0.0001), test (F(3,56) = 14.5; p < 0.0001), and a treatment-by-test interaction (F(3,56) = 15.4; p < 0.0001). Bonferroni’s post hoc test showed that hippocampal inhibition of protein synthesis caused a significant decrease of mCPP when tested at both 24 h (vehicle, 421.4 ± 20.7 s; anisomycin, 284.4 ± 42.8 s; p < 0.001) and 1 week (vehicle, 428. 6 ± 18.9 s; anisomycin, 294.4 ± 42.1 s; p < 0.001) after the re-experience of a conditioning trial.
Figure 4.
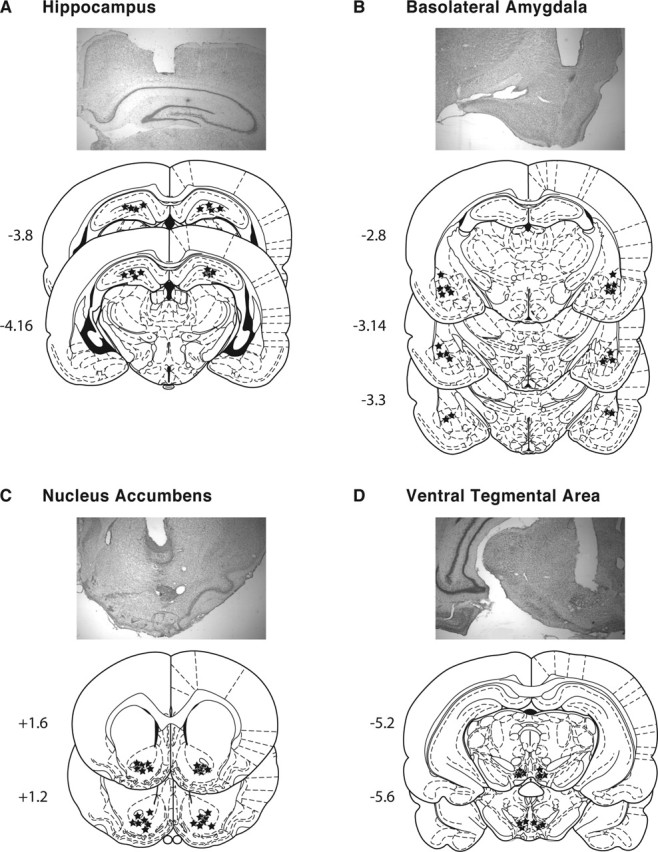
A–D, Photomicrographs of representative cannula placements and schematic representations of anisomycin injection sites in HC (A), BLA (B), NAc (C), and VTA (D) at the indicated rostrocaudal planes. The numbers represent the posterior (−) or anterior (+) coordinate from bregma (in millimeters) according to Paxinos and Watson (1998). Injection sites are represented by black stars.
Figure 5.
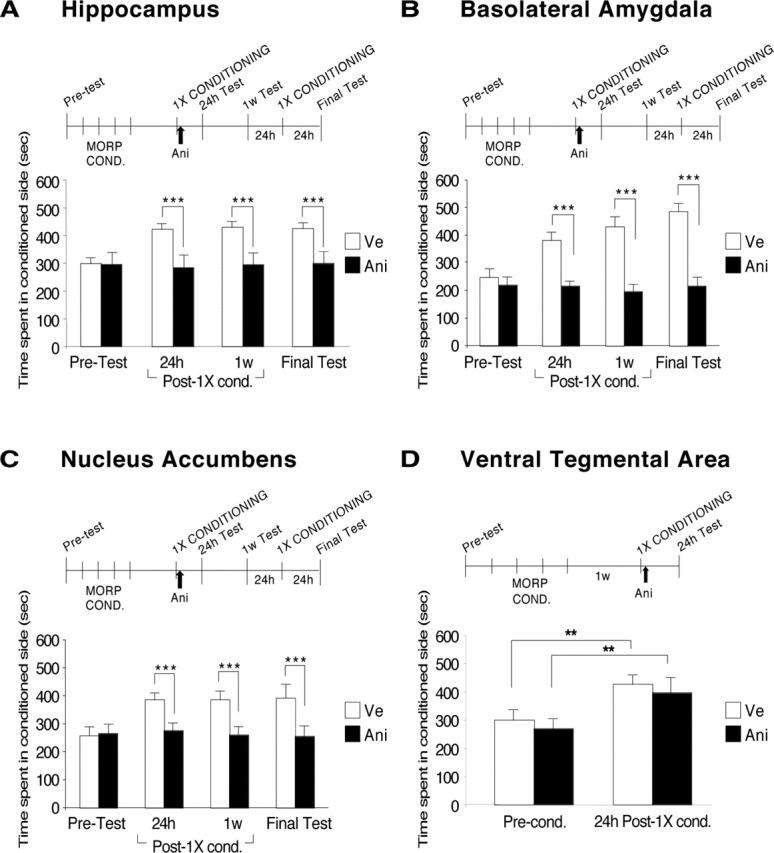
Inhibition of protein synthesis in the HC, BLA, or NAc, but not in the VTA, after conditioning, disrupts an established mCPP. Rats were conditioned to morphine and 1 week later received a single conditioning session (1× CONDITIONING). Immediately after, one-half of the animals were injected bilaterally into the targeted area with anisomycin (black bars) and the other half with vehicle (white bars). Rats were tested 24 h later and, if an effect was evident, retested 1 week later (1w Test). For the experiments in HC, BLA, and NAc, 1 d after the 1 week test, rats were again conditioned to morphine (1× CONDITIONING) and, finally, retested the next day (final test). Values are expressed as mean ± SEM time spent in the drug-conditioned chamber. **p < 0.01; *** p < 0.001. A, HC, Bar graphs representing the CPP scores of vehicle-injected (n = 8) and anisomycin-injected (n = 8) groups of rats. Pretest, 24 h, 1 week, and final test scores are shown. B, BLA, Bar graphs representing the CPP scores of vehicle-injected [n = 14, except final test (n = 11)] and anisomycin-injected (n = 11, n = 10 in 1 week and final tests) groups of rats. Pretest, 24 h, 1 week, and final test scores are shown. C, NAc, Bar graphs representing the CPP scores of vehicle-injected (n = 15) and anisomycin-injected (n = 14) groups of rats. Pretest, 24 h, 1 week, and final test scores are shown (Ve, n = 8; Ani, n = 8). D, VTA, Bar graphs representing the CPP scores of vehicle-injected (n = 7) and anisomycin-injected (n = 7) groups of rats. Pretest and 24 h test scores are shown. MORP COND., Morphine conditioning; Ani, anisomycin; Ve, vehicle; Post-1X cond., 1 week after conditioning; Pre-cond., preconditioning.
Is this memory deficit reflecting a suppression of the mCPP? The amnesia induced by the disruption of an established memory could be the result of a temporary suppression of the memory (Judge and Quartermain, 1982; Riccio and Richardson, 1984; Anokhin et al., 2002; Lattal and Abel, 2004). For example, amnesia induced by electroconvulsive shock after retrieval of a memory is restored by administration of a reminder shock (Miller and Kraus, 1977). Recently, Fischer et al. (2004) showed that the amnesia caused by inhibition of protein synthesis after contextual fear conditioning reactivation is not because of a permanent memory loss because, in fact, a subsequent exposure to a reminder shock fully reinstates the original memory. We therefore tested whether, similar to these memories, the disruption of mCPP caused by protein synthesis inhibition after the re-experience of a conditioning trial is caused by suppression of the response and whether mCPP re-emerges after conditioning reminders. The rats that underwent hippocampal anisomycin injection after the re-experience of a conditioning trial received another conditioning session 1 d after the 1 week test. This session acted as a strong reminder (reminder conditioning). When tested 24 h later, mCPP remained completely abolished in anisomycin-treated rats (vehicle, 424.3 ± 19.5 s; anisomycin, 298.9 ± 41.7 s). Bonferroni’s post hoc test confirmed that there was no significant change between the mCPP scores before or after the reminder conditioning (Fig. 5A).
Similar results were found when the BLA was targeted. A two-way ANOVA, which compared CPP scores across treatment and test, revealed a significant effect of drug treatment (F(1,87) = 905.0; p < 0.0001), test (F(3,87) = 72.12; p < 0.0001), and test-by-treatment interaction (F(3,87) = 85.51; p < 0.0001). Bonferroni’s post hoc test showed that BLA inhibition of protein synthesis caused a significant disruption of mCPP when tested both 24 h (vehicle, 381.6 ± 28.7 s; anisomycin, 219.2 ± 28.0 s; p < 0.001) and 1 week (vehicle, 430. 7 ± 32.9 s; anisomycin, 196.0 ± 23.2 s; p < 0.001) after the re-experience of a conditioning trial. Moreover, Bonferroni’s post hoc test confirmed that, similar to the HC-injected rats, BLA-injected animals had no significant recovery of mCPP after a reminder conditioning (vehicle, 485. 5 ± 26.4 s; anisomycin, 215.9 ± 30.3 s) (Fig. 5B).
NAc experiments also showed similar outcomes. A two-way ANOVA that compared CPP scores across treatment and test revealed a significant effect of treatment (F(1,95) = 206.6; p < 0.0001), test (F(3,95) = 26.8; p < 0.0001), and test-by-treatment interaction (F(3,95) = 28.0; p < 0.0001). Bonferroni’s post hoc test revealed that inhibition of protein synthesis in NAc caused a significant disruption of mCPP when tested both 24 h (vehicle, 385.8 ± 24.3 s; anisomycin, 275.3 ± 26.1 s; p < 0.001) and 1 week (vehicle, 385.9 ± 30.3 s; anisomycin, 260.3 ± 30.1 s; p < 0.001) after the re-experience of a conditioning trial. Moreover, similar to the HC- and BLA-injected rats, animals injected with anisomycin into the NAc did not regain the preference for the conditioning side after the reminder conditioning. Bonferroni’s post hoc test confirmed that there was no significant change between the CPP scores before or after the reminder conditioning (vehicle, 391.9 ± 47.2 s; anisomycin, 255.4 ± 35.0 s; p < 0.001) (Fig. 5C).
In contrast, when anisomycin was injected into the VTA, no effect on mCPP was observed. A two-way ANOVA comparing the groups across treatment and test revealed no significant difference for either factor. In fact, both groups performed similarly and had strong mCPP for the conditioning side when tested 24 h after re-experiencing the conditioning trial (vehicle, 426.0 ± 36.9 s; anisomycin, 396.3 ± 45.6 s) (Fig. 5D). As expected, the mCPPs of both groups of conditioned rats were significantly higher than their preconditioning scores (vehicle, 299.1 ± 63.0 s; anisomycin, 268.6 ± 49.4 s; p < 0.01 for both groups; t test).
Together, these results demonstrated that protein synthesis in the HC, BLA, or NAc, but not in the VTA, is required for maintaining or reconsolidating CPP after the re-experience of a conditioning trial.
Discussion
Our results show that both a new and an established mCPP can be persistently disrupted by inhibiting protein synthesis either during conditioning or after the re-experience of a conditioning trial, respectively. Specifically, with an established mCPP, a reinforced trial but not contextual recall or morphine treatment alone induces a protein synthesis-dependent reconsolidation process. Brain regions in which protein synthesis is required for this reconsolidation include the HC, BLA, or NAc but not the VTA. Notably, the disruption evoked by the protein synthesis inhibitors in HC, BLA, and NAc is persistent, because mCPP is not reinstated by further conditioning.
These results have important clinical implications, because they indicate that it is possible to persistently disrupt established memories induced by drugs of abuse by interfering with the reconsolidation process.
Different types of memory reactivation
Previous studies have shown that several types of memories, including spatial, fear-conditioning, and appetitive memories, can be disrupted by blocking protein synthesis after their reactivation (Nader et al., 2000; Sara, 2000; Dudai and Eisenberg, 2004; Alberini, 2005). Here, we provide evidence that, in addition to these more conventional types of memories, associative memories induced by a drug of abuse can become labile and be disrupted by protein synthesis inhibitors after their reactivation. In our experiments, reactivation required the re-experience of a conditioning session and did not occur after contextual or drug exposure alone.
Recently, two studies have suggested similar conclusions (Lee et al., 2005; Miller and Marshall 2005). However, Miller and Marshall (2005) found that cocaine-induced CPP (COC-CPP) is disrupted by MEK (mitogen-activated protein kinase kinase) inhibitors injected into the core of the NAc after contextual recall alone. Several reasons may explain the differences between these results and ours. First, they used MEK inhibitors, which block a post-translational regulation event, whereas we investigated the requirement for protein synthesis. Indeed, in addition to influencing the activity of transcription and translation, ERK (extracellular signal-regulated kinase) activation affects membrane electrical properties and receptor activation via post-translational modifications (Sweatt, 2004). Second, both MEK inhibitors blocked COC-CPP memory retrieval, whereas protein synthesis inhibitors generally fail in this respect but inhibit the consolidation phase of memory (Davis and Squire, 1984). Furthermore, it is not clear whether MEK inhibitors injected into the NAc have a transient effect (like protein synthesis inhibitors) or cause a persistent biochemical inhibition. In fact, the levels of proteins (pErk, pCREB, pELK-1, and Fos) investigated by Miller and Marshall to monitor the effect of the MEK inhibitors were reported to be extremely low, and it is unknown whether these levels were below the control values. Notably, our data are in agreement with previous findings by Hernandez and Kelley (2004), who reported that reactivation by the conditioned stimulus (CS) alone of a well learned appetitive memory (lever-pressing task) was not impaired by the systemic administration of protein synthesis inhibitors. Finally, we cannot exclude that the discrepancy between the findings of Miller and Marshall and ours are attributable to differences in behavioral protocols (e.g., drugs used, number of conditioning trials, time elapsing between initial conditioning and reactivation, systemic vs intra-NAc core injections, etc.). Lee et al. (2005) showed that knocking down the expression of the transcription factor Zif268 after the reactivation of a cocaine-induced memory (nose-poke response paired with a light CS) by a CS presentation disrupted the acquisition of a new instrumental drug-seeking behavioral response (active lever pressing). Because the behavioral tasks used in this investigation are very different from ours, a comparison between the two studies is impractical. Moreover, this paradigm showed a CS reactivation-dependent disruption of the acquisition of a new response and not on the reconsolidation of the original association.
In conclusion, the works of both Miller and Marshall (2005) and Lee et al. (2005) indicate that it is possible to disrupt drug-induced associative memories by applying interfering treatments after the presentation of a CS alone. However, our studies show that, at least under certain conditions, the disruption of the protein synthesis-dependent phase underlying reconsolidation can be more efficiently obtained by the re-experience of a reinforced trial than by exposure to the CS alone.
In agreement with previous reports (Mactutus et al., 1979; Richardson et al., 1982; Sara, 2000; Duvarci and Nader, 2004; Eisenberg and Dudai, 2004; Inda et al., 2005), our results also demonstrate that a reinforced trial, in addition to a CS or US alone, can reactivate a memory trace and make it labile again. One may argue that the exposure to one conditioning trial leads to new learning. This hypothesis, however, posits that only the new learning should be blocked during reactivation and should spare the original conditioning. In contrast, our results, in agreement with previous studies (Duvarci and Nader, 2004), show that the administration of protein synthesis inhibitors after one reinforced trial completely blocks the original memory, suggesting that the trace created by the original learning has become labile.
Although, in our experiments, contextual reactivation alone was unable to destabilize mCPP, even when protein synthesis was substantially blocked for >6 h, we cannot exclude that, with other induction and reactivation protocols, mCPP may become sensitive to protein synthesis inhibitors after exposure to CS alone. Previous work on memory reconsolidation indicates that the strength of training and reactivation correlate with the degree of memory stability after reactivation. Suzuki et al. (2004) showed that both weaker memories and those that undergo stronger reactivations are more susceptible to becoming labile. Moreover, the degree of protein synthesis requirement may change according to the nature of the reactivation. For example, in agreement with our results on mCPP, Bozon et al. (2003) reported that an object recognition memory does not become labile after the exposure of the contextual information or the previously presented objects in a different context. Similar results had been found by DeVietti and Holliday with electroconvulsive shock (DeVietti and Holliday, 1972). Hence, the strength of initial learning and the strength, nature, and number of reactivation events all contribute to the degree of memory destabilization.
The reason why contextual reactivation or morphine treatment alone does not disrupt mCPP is unclear. One possibility is that conditioning reactivates a larger representation of the trace. Alternatively, reactivation by conditioning may be effective because the context–drug associations play a major role in the protein synthesis-dependent phase of reconsolidation. Another possibility is that contextual reactivation alone may induce extinction, which, if blocked by protein synthesis inhibitors, results in the preservation of the preference. This latter hypothesis is consistent with several findings showing that extinction of several types of memories, including CPP, is blocked if protein synthesis inhibitors are administered after the extinction trial (Berman and Dudai, 2001; Eisenberg et al., 2003; Sangha et al., 2003; Vianna et al., 2003; Fischer et al., 2004) (but see Lattal and Abel, 2001; Lien et al., 2004). Finally, another often discussed issue with CPP deficits is that they may result from effects of state dependence. Our results argue against the possibility that the impairment produced by protein synthesis inhibitors is because of a state dependence. In fact, systemic administration of protein synthesis inhibitors at the end of the 4 d conditioning period impairs mCPP at 24 h, but memory returns at 1 week. If the impairment was caused by the effect of a state induced by the protein synthesis inhibitor and/or morphine, then memory would not re-emerge.
The importance of the extent of protein synthesis inhibition
A persistent disruption of mCPP after reactivation by conditioning requires that protein synthesis is inhibited for a sufficiently long period of time. These results are in agreement with previous studies using systemic or local injections of protein synthesis inhibitors (Flood et al., 1975, 1977, 1978; Kesner et al., 1981; Rosenblum et al., 1993).
Our findings may provide an explanation for previous conflicting results on memory consolidation and reconsolidation, in which, in some cases, the amnesia produced by protein synthesis inhibitors appeared to be transient (Flexner et al., 1966; Roberts and Flexner, 1969; Judge and Quartermain, 1982; Anokhin et al., 2002; Lattal and Abel, 2004). It is possible that in these cases, the synthesis of proteins essential for the memory stabilization was blocked only transiently, and therefore memory re-emerged at a later time.
Circuitry required for the stabilization of mCPP after reactivation by conditioning
Given the role played by the HC, BLA, and NAc, but not the VTA, in processing information related to context, reward, and motivation, our results are not surprising. In fact, the HC is known to process contextual associations, the BLA is critical for mediating various forms of appetitive and aversive conditioning, and the NAc, particularly the core, plays a central role in maintaining cue-elicited drug-seeking behavior (Fuchs et al., 2004; Ito et al., 2004). Moreover, the NAc represents a convergent node that receives BLA, HC, and prefrontal cortex projections and sends outputs to the systems that are responsible for the execution of the behavior. Both HC and BLA projections to the NAc are important for learned association between stimuli and drug reward and appear to be involved in cue-elicited craving (Everitt et al., 2001). Notably, drugs of abuse have been found to induce long-lasting synaptic changes in all these areas (Thomas et al., 2001; Huang et al., 2003; Thompson et al., 2004).
On the other hand, the inability of anisomycin to disrupt mCPP when injected into the VTA demonstrates that the effect of protein synthesis inhibitors is selective for certain regions and not others. Consistent with previous evidence (Wolf, 1998; Vanderschuren and Kalivas, 2000; Everitt and Wolf, 2002), one explanation for VTA dispensability during reconsolidation is that the VTA is involved in the induction of a drug-conditioned behavior but not during its maintenance. This was argued also by Borgland et al. (2004), who, using behavioral sensitization, reported that the cocaine-induced synaptic plasticity at VTA excitatory synapses is transient. These authors suggested that the locus of the critical plasticity induced during cocaine sensitization shifts from its initiation site in the VTA (Wolf, 1998; Everitt and Wolf, 2002) to its sites of maintenance, one of which is believed to be the NAc (Wolf, 1998; Vanderschuren and Kalivas, 2000).
An anatomical shift may also explain why, in our study, protein synthesis inhibitors administered after the single conditioning event, 1 week after the end of conditioning, results in a permanent disruption of mCPP, whereas the same treatment applied on the last day of the 4 d conditioning protocol (induction phase) produces no stable effect. Perhaps, protein synthesis in the VTA is involved during the 4 d conditioning but not recruited 1 week later after a single conditioning event. If this is true, blocking protein synthesis during either the initial conditioning phase or at later times during mCPP maintenance would affect differentially activated circuits and therefore produce different effects.
Notably, because our injection site mainly targeted the dorsal HC, the BLA, the NAc core, and the anterior VTA, it should be kept in mind that targeting other subregions of HC, amygdala, NAc, and VTA may produce a different outcome. Identifying in more detail the circuitry (subregions and neuronal subpopulations) that underlies the mCPP reconsolidation process will be an important addition.
Reactivation and the question of persistence
Our results indicate that the disruption of mCPP by protein synthesis inhibitors after reactivation by conditioning is persistent. In fact, mCPP does not recover over time nor reinstate with further conditioning. Although we cannot exclude that more vigorous rescuing methods may recover the memory, our results imply that it is the synthesis of key proteins after reactivation by conditioning that mediates the reconsolidation of mCPP. These results have important clinical implications, because they show that memories induced by drugs of abuse can be persistently disrupted.
In conclusion, a combination of both behavioral and pharmacological interventions that target newly synthesized proteins may be used for developing novel treatments for weakening memories induced by drugs of abuse and subsequently reducing the risk of withdrawal and relapse.
Footnotes
This work was supported by National Institute on Drug Abuse Grant R21, Cutting-Edge Basic Research Awards Grant DA017672, and the Hirschl Foundation (C.M.A.). We thank Bob Blitzer, Gabriella Pollonini, Stephen Taubenfeld, Khatuna Gagnidze, Reginald Miller, and the Center for Comparative Medicine and Surgery facility of Mount Sinai for technical support.
References
- Alberini CM (2005). Mechanisms of memory stabilization: are consolidation and reconsolidation similar or distinct processes? Trends Neurosci 28:51–56. [DOI] [PubMed] [Google Scholar]
- Ambrogi-Lorenzini CG, Baldi E, Bucherelli C, Tassoni G (1995). Time-dependent deficits of rat’s memory consolidation induced by tetrodotoxin injections into the caudate-putamen, nucleus accumbens, and globus pallidus. Neurobiol Learn Mem 63:87–93. [DOI] [PubMed] [Google Scholar]
- Ambrogi-Lorenzini CG, Baldi E, Bucherelli C, Sacchetti B, Tassoni G (1999). Neural topography and chronology of memory consolidation: a review of functional inactivation findings. Neurobiol Learn Mem 71:1–18. [DOI] [PubMed] [Google Scholar]
- Anokhin KV, Tiunova AA, Rose SP (2002). Reminder effects–reconsolidation or retrieval deficit? Pharmacological dissection with protein synthesis inhibitors following reminder for a passive-avoidance task in young chicks. Eur J Neurosci 15:1759–1765. [DOI] [PubMed] [Google Scholar]
- Bardo MT, Bevins RA (2000). Conditioned place preference: what does it add to our preclinical understanding of drug reward? Psychopharmacology 153:31–43. [DOI] [PubMed] [Google Scholar]
- Berman DE, Dudai Y (2001). Memory extinction, learning anew, and learning the new: dissociations in the molecular machinery of learning in cortex. Science 291:2417–2419. [DOI] [PubMed] [Google Scholar]
- Borgland SL, Malenka RC, Bonci A (2004). Acute and chronic cocaine-induced potentiation of synaptic strength in the ventral tegmental area: electrophysiological and behavioral correlates in individual rats. J Neurosci 24:7482–7490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouton ME (1993). Context, time, and memory retrieval in the interference paradigms of Pavlovian learning. Psychol Bull 114:80–99. [DOI] [PubMed] [Google Scholar]
- Bozon B, Davis S, Laroche S (2003). A requirement for the immediate early gene zif268 in reconsolidation of recognition memory after retrieval. Neuron 40:695–701. [DOI] [PubMed] [Google Scholar]
- Childress AR, Hole AV, Ehrman RN, Robbins SJ, McLellan AT, O’Brien CP (1993). Cue reactivity and cue reactivity interventions in drug dependence. NIDA Res Monogr 137:73–95. [PubMed] [Google Scholar]
- Davis HP, Squire LR (1984). Protein synthesis and memory: a review. Psychol Bull 96:518–559. [PubMed] [Google Scholar]
- Davis HP, Rosenzweig MR, Bennet EL, Squire LR (1980). Inhibition of cerebral protein synthesis: dissociation of nonspecific effects and amnesic effects. Behav Neural Biol 28:99–104. [DOI] [PubMed] [Google Scholar]
- DeVietti TL, Holliday JH (1972). Retrograde amnesia produced by electroconvulsive shock after reactivation of a consolidated memory trace: a replication. Psychon Sci 29:137–138. [DOI] [PubMed] [Google Scholar]
- Di Chiara G (2002). Nucleus accumbens shell and core dopamine: differential role in behavior and addiction. Behav Brain Res 137:75–114. [DOI] [PubMed] [Google Scholar]
- Dudai Y, Eisenberg M (2004). Rites of passage of the engram: reconsolidation and the lingering consolidation hypothesis. Neuron 44:93–100. [DOI] [PubMed] [Google Scholar]
- Duvarci S, Nader K (2004). Characterization of fear memory reconsolidation. J Neurosci 24:9269–9275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenberg M, Dudai Y (2004). Reconsolidation of fresh, remote, and extinguished fear memory in Medaka: old fears don’t die. Eur J Neurosci 20:3397–3403. [DOI] [PubMed] [Google Scholar]
- Eisenberg M, Kobilo T, Berman DE, Dudai Y (2003). Stability of retrieved memory: inverse correlation with trace dominance. Science 301:1102–1104. [DOI] [PubMed] [Google Scholar]
- Everitt BJ, Wolf ME (2002). Psychomotor stimulant addiction: a neural systems perspective. J Neurosci 22:3312–3320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everitt BJ, Dickinson A, Robbins TW (2001). The neuropsychological basis of addictive behaviour. Brain Res Brain Res Rev 36:129–138. [DOI] [PubMed] [Google Scholar]
- Fischer A, Sananbenesi F, Schrick C, Spiess J, Radulovic J (2004). Distinct roles of hippocampal de novo protein synthesis and actin rearrangement in extinction of contextual fear. J Neurosci 24:1962–1966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flexner LB, Flexner JB, Roberts RB (1966). Stages of memory in mice treated with acetoxycycloheximide before or immediately after learning. Proc Natl Acad Sci USA 56:730–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flood JF, Jarvik ME, Bennett EL, Orme AE, Rosenzweig MR (1977). Protein synthesis inhibition and memory for pole jump active avoidance and extinction. Pharmacol Biochem Behav 7:71–77. [DOI] [PubMed] [Google Scholar]
- Flood JF, Bennett EL, Orme AE, Rosenzweig MR, Jarvik ME (1978). Memory: modification of anisomycin-induced amnesia by stimulants and depressants. Science 199:324–326. [DOI] [PubMed] [Google Scholar]
- Flood JFB, Bennet EL, Orme E, Rosenzweig MR (1975). Relation of memory formation to controlled amounts of brain protein synthesis. Physiol Behav 15:97–102. [DOI] [PubMed] [Google Scholar]
- Fuchs RA, Weber SM, Rice HJ, Neisewander JL (2000). Effects of excitotoxic lesions of the basolateral amygdala on cocaine-seeking behavior and cocaine conditioned place preference in rats. Brain Res 929:15–25. [DOI] [PubMed] [Google Scholar]
- Fuchs RA, Evans KA, Parker MC, See RE (2004). Differential involvement of the core and shell subregions of the nucleus accumbens in conditioned cue-induced reinstatement of cocaine seeking in rats. Psychopharmacology (Berl) 176:459–465. [DOI] [PubMed] [Google Scholar]
- Groenewegen HJ, Vermeulen-Van der Zee E, te Kortschot A, Witter MP (1987). Organization of the projections from the subiculum to the ventral striatum in the rat. A study using anterograde transport of Phaseolus vulgaris leucoagglutinin. Neuroscience 23:103–120. [DOI] [PubMed] [Google Scholar]
- Harris GC, Wimmer M, Byrne R, Aston-Jones G (2004). Glutamate-associated plasticity in the ventral tegmental area is necessary for conditioning environmental stimuli with morphine. Neuroscience 129:841–847. [DOI] [PubMed] [Google Scholar]
- Hernandez PJ, Kelley AE (2004). Long-term memory for instrumental responses does not undergo protein synthesis-dependent reconsolidation upon retrieval. Learn Mem 11:748–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang YC, Wang SJ, Chiou LC, Gean PW (2003). Mediation of amphetamine-induced long-term depression of synaptic transmission by CB1 cannabinoid receptors in the rat amygdala. J Neurosci 23:10311–10320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyman SE, Malenka RC (2001). Addiction and the brain: the neurobiology of compulsion and its persistence. Nat Rev Neurosci 2:695–703. [DOI] [PubMed] [Google Scholar]
- Inda MC, Delgado-Garcia JM, Carrion AM (2005). Acquisition, consolidation, reconsolidation, and extinction of eyelid conditioning responses require de novo protein synthesis. J Neurosci 25:2070–2080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito R, Robbins TW, Everitt BJ (2004). Differential control over cocaine-seeking behavior by nucleus accumbens core and shell. Nat Neurosci 7:389–397. [DOI] [PubMed] [Google Scholar]
- Judge ME, Quartermain D (1982). Characteristics of retrograde amnesia following reactivation of memory in mice. Physiol Behav 28:585–590. [DOI] [PubMed] [Google Scholar]
- Kalivas PW, Weber B (1988). Amphetamine injection into the ventral mesencephalon sensitizes rats to peripheral amphetamine and cocaine. J Pharmacol Exp Ther 245:1095–1102. [PubMed] [Google Scholar]
- Kauer JA (2004). Learning mechanisms in addiction: synaptic plasticity in the ventral tegmental area as a result of exposure to drugs of abuse. Annu Rev Physiol 66:447–475. [DOI] [PubMed] [Google Scholar]
- Kelley AE (2004). Memory and addiction: shared neural circuitry and molecular mechanisms. Neuron 44:161–179. [DOI] [PubMed] [Google Scholar]
- Kesner RP, Partlow LM, Bush LG, Berman RF (1981). A quantitative regional analysis of protein synthesis inhibition in the rat brain following localized injection of cycloheximide. Brain Res 209:159–176. [DOI] [PubMed] [Google Scholar]
- Landauer TK (1969). Reinforcement as consolidation. Psych Rev 76:82–96. [DOI] [PubMed] [Google Scholar]
- Lattal KM, Abel T (2001). Different requirements for protein synthesis in acquisition and extinction of spatial preferences and context-evoked fear. J Neurosci 21:5773–5780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lattal KM, Abel T (2004). Behavioral impairments caused by injections of the protein synthesis inhibitor anisomycin after contextual retrieval reverse with time. Proc Natl Acad Sci USA 101:4667–4672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JL, Di Ciano P, Thomas KL, Everitt BJ (2005). Disrupting reconsolidation of drug memories reduces cocaine-seeking behavior. Neuron 47:795–801. [DOI] [PubMed] [Google Scholar]
- Lewis DJ (1979). Psychobiology of active and inactive memory. Psychol Bull 86:1054–1083. [PubMed] [Google Scholar]
- Lien WH, Yeh TL, Yang YK, Cherng CF, Chen HH, Chen PS, Yu L (2004). Cycloheximide enhances maintenance of methamphetamine-induced conditioned place preference. Chin J Physiol 47:23–30. [PubMed] [Google Scholar]
- Ludwig AM, Stark LH (1974). Alcohol craving. Subjective and situational aspects. Q J Stud Alcohol 35:899–905. [PubMed] [Google Scholar]
- Mactutus CF, Riccio DC, Ferek JM (1979). Retrograde amnesia for old reactivated memory: some anomalous characteristics. Science 204:1319–1320. [DOI] [PubMed] [Google Scholar]
- McGaugh JL (2004). The amygdala modulates the consolidation of memories of emotionally arousing experiences. Annu Rev Neurosci 27:1–28. [DOI] [PubMed] [Google Scholar]
- Meyers RA, Zavala AR, Neisewander JL (2003). Dorsal, but not ventral, hippocampal lesions disrupt cocaine place conditioning. NeuroReport 14:2127–2131. [DOI] [PubMed] [Google Scholar]
- Milekic MH, Alberini CM (2002). Temporally graded requirement for protein synthesis following memory reactivation. Neuron 36:521–525. [DOI] [PubMed] [Google Scholar]
- Miller CA, Marshall JF (2005). Molecular substrates for retrieval and reconsolidation of cocaine-associated contextual memory. Neuron 47:873–884. [DOI] [PubMed] [Google Scholar]
- Miller RR, Kraus JN (1977). Somatic and autonomic indexes of recovery from electroconvulsive shock-induced amnesia in rats. J Comp Physiol Psychol 91:434–442. [DOI] [PubMed] [Google Scholar]
- Mucha RF, Iversen SD (1984). Reinforcing properties of morphine and naloxone revealed by conditioned place preferences: a procedural examination. Psychopharmacology (Berl) 82:241–247. [DOI] [PubMed] [Google Scholar]
- Mucha RF, van der Kooy D, O’Shaughnessy M, Bucenieks P (1982). Drug reinforcement studied by the use of place conditioning in rat. Brain Res 243:91–105. [DOI] [PubMed] [Google Scholar]
- Nader K, Schafe GE, LeDoux JE (2000). The labile nature of consolidation theory. Nat Rev Neurosci 1:216–219. [DOI] [PubMed] [Google Scholar]
- Nestler EJ (2002). Common molecular and cellular substrates of addiction and memory. Neurobiol Learn Mem 78:637–647. [DOI] [PubMed] [Google Scholar]
- O’Brien CP, Testa T, O’Brien TJ, Brady JP, Wells B (1977). Conditioned narcotic withdrawal in humans. Science 195:1000–1002. [DOI] [PubMed] [Google Scholar]
- O’Brien CP, Childress AR, McLellan AT, Ehrman R (1992). Classical conditioning in drug-dependent humans. Ann NY Acad Sci 654:400–415. [DOI] [PubMed] [Google Scholar]
- O’Brien CP, Childress AR, McLellan AT, Ehrman R (1993). Developing treatments that address classical conditioning. NIDA Res Monogr 135:71–91. [PubMed] [Google Scholar]
- Pavlov IP (1927). In: Conditioned reflexes London: Oxford UP.
- Paxinos G, Watson C (1998). In: The rat brain in stereotaxic coordinates, Ed 4 London: Academic.
- Quevedo J, Vianna MR, Roesler R, de-Paris F, Izquierdo I, Rose SP (1999). Two time windows of anisomycin-induced amnesia for inhibitory avoidance training in rats: protection from amnesia by pretraining but not pre-exposure to the task apparatus. Learn Mem 6:600–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riccio DC, Richardson R (1984). The status of memory following experimentally induced amnesias: gone, but not forgotten. Physiol Psychol 12:59–72. [Google Scholar]
- Richardson R, Riccio DC, Mowrey H (1982). Retrograde amnesia for previously acquired Pavlovian conditioning: UCS exposure as a reactivation treatment. Physiol Psychol 10:384–390. [Google Scholar]
- Richter-Levin G (2004). The amygdala, the hippocampus, and emotional modulation of memory. The Neuroscientist 10:31–39. [DOI] [PubMed] [Google Scholar]
- Robbins TW, Everitt BJ (2002). Limbic-striatal memory systems and drug addiction. Neurobiol Learn Mem 78:625–636. [DOI] [PubMed] [Google Scholar]
- Roberts RB, Flexner LB (1969). The biochemical basis of long-term memory. Q Rev Biophys 2:135–173. [DOI] [PubMed] [Google Scholar]
- Romieu P, Phan VL, Martin-Fardon R, Maurice T (2002). Involvement of the sigma(1) receptor in cocaine-induced conditioned place preference: possible dependence on dopamine uptake blockade. Neuropsychopharmacology 26:444–455. [DOI] [PubMed] [Google Scholar]
- Rosenblum K, Meiri N, Dudai Y (1993). Taste memory: the role of protein synthesis in gustatory cortex. Behav Neural Biol 59:49–56. [DOI] [PubMed] [Google Scholar]
- Sangha S, Scheibenstock A, Morrow R, Lukowiak K (2003). Extinction requires new RNA and protein synthesis and the soma of the cell right pedal dorsal 1 in Lymnaea stagnalis. J Neurosci 23:9842–9851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sara SJ (2000). Retrieval and reconsolidation: toward a neurobiology of remembering. Learn Mem 7:73–84. [DOI] [PubMed] [Google Scholar]
- Schafe GE, LeDoux JE (2000). Memory consolidation of auditory pavlovian fear conditioning requires protein synthesis and protein kinase A in the amygdala. J Neurosci 20:1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Squire LR, Davis HP, Spanis CW, Gold PE, Sternberg DB (1980). Neurobiology of amnesia. Science 209:836–837. [DOI] [PubMed] [Google Scholar]
- Suzuki A, Josselyn SA, Frankland PW, Masushige S, Silva AJ, Kida S (2004). Memory reconsolidation and extinction have distinct temporal and biochemical signatures. J Neurosci 24:4787–4795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweatt JD (2004). Mitogen-activated protein kinases in synaptic plasticity and memory. Curr Opin Neurobiol 14:311–317. [DOI] [PubMed] [Google Scholar]
- Taubenfeld SM, Milekic MH, Monti B, Alberini CM (2001). The consolidation of new but not reactivated memory requires hippocampal C/EBPbeta. Nat Neurosci 4:813–818. [DOI] [PubMed] [Google Scholar]
- Thomas MJ, Beurrier C, Bonci A, Malenka RC (2001). Long-term depression in the nucleus accumbens: a neural correlate of behavioral sensitization to cocaine. Nat Neurosci 4:1217–1223. [DOI] [PubMed] [Google Scholar]
- Thompson AM, Swant J, Gosnell BA, Wagner JJ (2004). Modulation of long-term potentiation in the rat hippocampus following cocaine self-administration. Neuroscience 127:177–185. [DOI] [PubMed] [Google Scholar]
- Tzschentke TM (1998). Measuring reward with the conditioned place preference paradigm: a comprehensive review of drug effects, recent progress and new issues. Prog Neurobiol 56:613–672. [DOI] [PubMed] [Google Scholar]
- van der Kooy D (1987). Place conditioning: a simple and effective method for assessing the motivational properties of drugs. In: Methods of assessing the reinforcing properties of abused drugs (Bozarth MA, ed.) pp. 229–240. New York: Springer.
- Vanderschuren LJ, Kalivas PW (2000). Alterations in dopaminergic and glutamatergic transmission in the induction and expression of behavioral sensitization: a critical review of preclinical studies. Psychopharmacology 151:99–120. [DOI] [PubMed] [Google Scholar]
- Vezina P, Stewart J (1990). Amphetamine administered to the ventral tegmental area but not to the nucleus accumbens sensitizes rats to systemic morphine: lack of conditioned effects. Brain Res 516:99–106. [DOI] [PubMed] [Google Scholar]
- Vianna MR, Igaz LM, Coitinho AS, Medina JH, Izquierdo I (2003). Memory extinction requires gene expression in rat hippocampus. Neurobiol Learn Mem 79:199–203. [DOI] [PubMed] [Google Scholar]
- Vorel SR, Liu X, Hayes RJ, Spector JA, Gardner EL (2001). Relapse to cocaine-seeking after hippocampal theta burst stimulation. Science 292:1175–1178. [DOI] [PubMed] [Google Scholar]
- White NM (1996). Addictive drugs as reinforcers: multiple partial actions on memory systems. Addiction 91:921–965. [PubMed] [Google Scholar]
- Wolf ME (1998). The role of excitatory amino acids in behavioral sensitization to psychomotor stimulants. Prog Neurobiol 54:679–720. [DOI] [PubMed] [Google Scholar]
- Wright CI, Beijer AV, Groenewegen HJ (1996). Basal amygdaloid complex afferents to the rat nucleus accumbens are compartmentally organized. J Neurosci 16:1877–1893. [DOI] [PMC free article] [PubMed] [Google Scholar]