

Abstract
The dystrobrevins (αDB and βDB) bind directly to dystrophin and are components of a transmembrane dystrophin–glycoprotein complex (DGC) that links the cytoskeleton to extracellular proteins in many tissues. We show here that αDB, βDB, and dystrophin are all concentrated at a discrete subset of inhibitory synapses on the somata and dendrites of cerebellar Purkinje cells. Dystrophin is depleted from these synapses in mice lacking both αDB and βDB, and DBs are depleted from these synapses in mice lacking dystrophin. In dystrophin mutants and αDB,βDB double mutants, the size and number of GABA receptor clusters are decreased at cerebellar inhibitory synapses, and sensorimotor behaviors that reflect cerebellar function are perturbed. Synaptic and behavioral abnormalities are minimal in mice lacking either αDB or βDB. Together, our results show that the DGC is required for proper maturation and function of a subset of inhibitory synapses, that DB is a key component of this DGC, and that interference with this DGC leads to behavioral abnormalities. We suggest that motor deficits in muscular dystrophy patients, which are their cardinal symptoms, may reflect not only peripheral derangements but also CNS defects.
Keywords: dystrophin, ERG, GABA receptor, muscular dystrophy, retina, behavior
Introduction
The integrity of skeletal muscle fibers is maintained in part by a multi-molecular, transmembrane protein complex called the dystrophin–glycoprotein complex (DGC) that links the cytoskeleton to the extracellular matrix (Ervasti and Campbell, 1991) (for review, see Blake et al., 2002). The best studied component of the DGC is dystrophin itself, a large (>400 kDa) protein associated with the inner face of the plasma membrane of the muscle cell. The dystrophin gene is mutated in patients with Duchenne muscular dystrophy, the most common congenital human neuromuscular disease (Hoffman et al., 1987). Likewise, mutations in genes encoding any of 10 other DGC components or DGC-related genes lead to muscular dystrophies in humans or animal models [α-, β-, γ-, and δ-sarcoglycan, α-dystrobrevin (αDB), laminin α2, and four enzymes that selectively glycosylate dystroglycan] (for review, see Dalkilic and Kunkel, 2003; Muntoni et al., 2004).
Duchenne’s original description of the disease that bears his name noted that many patients suffer from cognitive impairments as well as muscle disease (Duchenne, 1868). This finding has been amply confirmed (for review, see Blake and Kroger, 2000; Anderson et al., 2002) and extended to include defects in the neural retina (Pillers et al., 1993) (for review, see Schmitz and Drenckhahn, 1997). Likewise, neural defects have been documented in dystrophin-deficient mutant mice, including behavioral and visual abnormalities and alterations in the structure and function of inhibitory cerebellar and retinal photoreceptor synapses (Pillers et al., 1995; Knuesel et al., 1999; Anderson et al., 2003; Green et al., 2004; Vaillend et al., 2004).
How does dystrophin affect neuronal structure and function? One obvious possibility is that dystrophin acts as part of a DGC in brain as it does in muscle. Indeed, several DGC proteins are present in central neurons and appear to interact in a manner similar to that seen in muscle (Peters et al., 1997; Blake et al., 1998, 1999; Moukhles and Carbonetto, 2001; Neely et al., 2001; Zaccaria et al., 2001; Brunig et al., 2002; Levi et al., 2002). However, cerebral phenotypes of mutants in which the DGC is disrupted primarily reflect abnormalities in non-neuronal cells. Loss or abnormal glycosylation of brain dystroglycan, a core component of muscle DGC, results in cerebral malformations secondary to disruption of the developing pia (Michele et al., 2002; Moore et al., 2002). Mice lacking the cytoplasmic DGC component, α-syntrophin, have reduced levels of the water channel protein aquaporin-4 in perivascular astrocytes, resulting in impaired ion balance and susceptibility to epilepsy (Amiry-Moghaddam et al., 2003). Thus, despite growing evidence that the DGC is important for cerebral development and function, roles of the neuronal DGC remain unclear.
Here we have used mice lacking the DGC components αDB and βDB to reexamine this issue. αDB is a soluble cytoplasmic protein that binds directly to dystrophin and syntrophin. We showed previously that mice lacking αDB (adbn−/−) exhibit a mild muscular dystrophy as well as defects in their neuromuscular junction (NMJs) and myotendinous junctions (Grady et al., 1999, 2000, 2003; Akaaboune et al., 2002). αDB is abundant in brain (Blake et al., 1996, 1999), but adbn−/− mice showed no obvious CNS defects. We speculated that this might reflect the coexpression in brain of a homolog, βDB, which is predominantly expressed in the nervous system (Peters et al., 1997; Blake et al., 1998, 1999). We therefore generated βDB mutants (bdbn−/−) and double mutants lacking both αDB and βDB [double knock-outs (DKOs)]. We show here that dystrophin is lost from a subset of inhibitory synapses in DKO brain and that DKOs exhibit synaptic and behavioral defects similar, although not identical, to those seen in dystrophin-deficient (mdx) mice. Our results provide the first evidence for a role of βDB in any tissue, the first evidence that DGC components other than dystrophin affect synaptic structure in the brain, and indirect evidence that some but not all of the effects of dystrophin on neurons reflect its association with the DGC.
Materials and Methods
Mice
To generate bdbn−/− mice, embryonic stem cells in which the bdbn gene had been insertionally mutated (see Fig. 1A,B) were obtained from Lexicon Genetics (Woodlands, TX) [Omnibank sequence tag 109050 (described by Zambrowicz et al., 1998)], expanded, and injected into C57BL/6 mice. Generation of adbn−/− mice was described previously (Grady et al., 1999). mdx mice (C57BL/10ScSn-Dmdmdx/J) were purchased from The Jackson Laboratory (Bar Harbor, ME). All DB mutants were maintained on a C57BL/6 background. For Southern analysis, genomic DNA was digested with BamH1 and probed with the fragment of the bdbn gene shown in Figure 1B.
Figure 1.

Generation of bdbn−/− mice. A, Organization of the mouse bdbn gene, based on Loh et al. (1998). Exons are numbered 1–18, and the four subject to alternative splicing are indicated by * (Peters et al., 1997; Blake et al., 1998). UTR, Untranslated region. B, Location of gene trap vector that disrupted the bdbn gene. BamH1 sites and probe used for Southern blot analysis are indicated. C, Southern blot analysis of BamH1-digested genomic DNA from bdbn−/− mice and littermates. A ∼5.0 kb fragment present in the wild-type (+/+) allele is replaced by a ∼4.0 kb fragment in the mutant allele. D, Immunoblots of protein extracts from liver and kidney of bdbn+/+ and bdbn−/− mice using an antibody that recognizes both αDB (α) and βDB (β). The ∼70 kDa βDB band is completely absent from mutants, but the level of the ∼80 kDa αDB band is unchanged.
Immunochemistry
Antibodies used in this study were as follows. A rabbit polyclonal antibody to DB was generated in our laboratory to a recombinant fragment of αDB that is shared by all reported αDB isoforms (Peters et al., 1998; Grady et al., 1999). We also generated monoclonal antibodies to a recombinant fragment of βDB; several antibodies were obtained that reacted with βDB, but all of them also recognized αDB. One antibody, 1E1, of the IgG1 subclass, was used in this study. Mouse monoclonal anti-dystrophin (DYS2) was obtained from Novocastra (Newcastle, UK), rabbit polyclonal anti-GABAAα1 receptor subunit was from Alomone Labs (Jerusalem, Israel), mouse monoclonal anti-gephyrin (mAb7a) was from Synaptic Systems (Goettingen, Germany), mouse monoclonal anti-postsynaptic density-95 (PSD-95) was from Affinity BioReagents (Golden, CO), fluorescein- and peroxidase-conjugated goat anti-mouse IgG1 were from Roche Diagnostics (Indianapolis, IN), Alexa Fluor 488-conjugated goat anti-mouse IgG2a was from Invitrogen (Carlsbad, CA), and cyanine 3-conjugated goat anti-rabbit was from Jackson ImmunoResearch (West Grove, PA).
Immunoblotting was performed as described by Grady et al. (2003). Aliquots of whole-tissue extracts were resolved on 7.5% SDS-polyacrylamide gels, incubated with antibody, and detected with a peroxidase-conjugated secondary antibody.
For immunohistochemistry, tissue was frozen in liquid nitrogen-cooled isopentane and sectioned in a cryostat at 5–10 μm. Sections were stained with primary antibody for 2 h, rinsed, incubated 1 h with secondary antibody, mounted using 0.1% p-phenylenediamine in glycerol/PBS, and viewed with a microscope equipped for epifluorescence microscopy (Axioskop; Zeiss, Oberkochen, Germany) or with a confocal microscope (Olympus Optical, Melville, NY) (see Figs. 3G, 5A). In most cases, unfixed tissue was used; for GABAA receptor staining, sections were fixed in 2% paraformaldehyde/PBS before staining. Longitudinal sections of muscle were prepared and stained as described by Grady et al. (2000, 2003).
Figure 3.
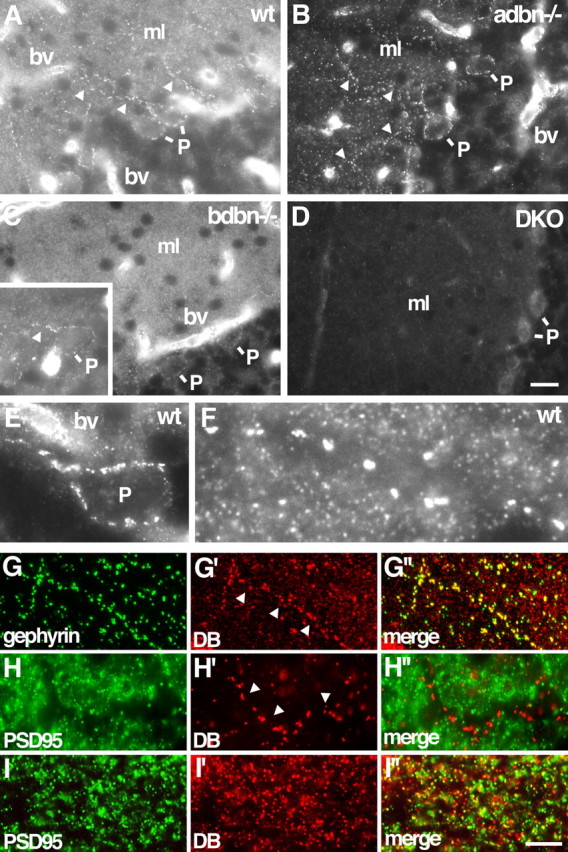
Localization of DB in cerebellum. A–D, Sections of wild-type (wt), adbn−/−, bdbn−/−, and DKO cerebellum were stained with an antibody that recognizes both αDB and βDB. Small puncta in the molecular layer (ml) contain αDB but not βDB. Walls of blood vessels (bv) and large puncta along Purkinje cell bodies (P) and dendrites (arrowheads) contain both αDB and βDB. Inset in C shows the large puncta in bdbn−/− tissue in another section. E, F, Large and small puncta in wild-type tissue at higher magnification. G–I, Sections of wild-type (G), adbn−/− (H), and bdbn−/− (I) cerebellum costained with antibodies to gephyrin (G), a component of inhibitory synapses, or PSD-95 (H, I), a component of excitatory synapses. Large DB-positive puncta (arrows) colocalize with gephyrin but not PSD-95. Few small DB-positive puncta colocalize with either gephyrin or PSD-95. Scale bars: (in D) A–D, 25 μm, E, 7 μm, F, 4 μm; (in I″) G, 14 μm, H, 10 μm, I, 5 μm.
Figure 5.
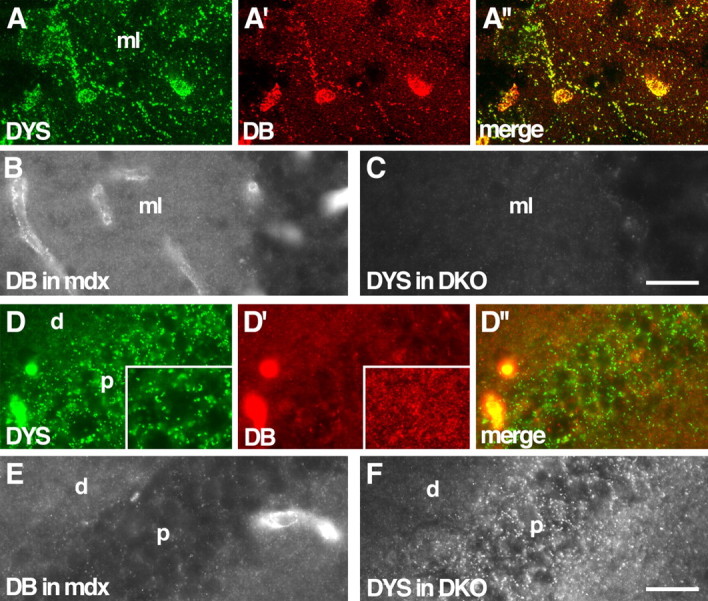
Localization of DB in dystrophin mutants and of dystrophin in DB mutants. A, Wild-type cerebellum stained with antibodies to dystrophin (DYS; A) and DB (A′). Large DB-positive puncta on Purkinje cell bodies and their dendrites in the molecular layer (ml) are also dystrophin positive. Both proteins are also present in blood vessels. B, Large DB-positive puncta are lost, but small puncta persist in dystrophin mutant (mdx) molecular layer. C, Large dystrophin-positive puncta are lost in DB mutant (DKO) molecular layer. D, Wild-type hippocampus stained with antibodies to dystrophin (D) and DB (D′). Large dystrophin-positive puncta, located primarily in the pyramidal cell soma layer (p), are DB negative. Small DB-positive puncta distribute primarily to dendritic layers (d). Insets show puncta at higher magnification. E, F, In hippocampus, distribution of DB is unaffected by loss of dystrophin, and distribution of dystrophin is unaffected by loss of DB. Scale bars: (in C) A, 20 μm, B, C, 40 μm; (in F) D–F, 30 μm.
For analysis of GABAA receptor clusters, whole-field digital micrographs of control, DKO, and mdx cerebellar sections, stained simultaneously, were taken at equivalent locations and exposure times using a 63× oil objective (Zeiss). A total of 21–25 micrographs from two mice per genotype were obtained. Images were imported into Photoshop 6.0 (Adobe Systems, San Jose, CA). Using the Levels command, intensity values of synaptic puncta for each micrograph were maintained while background intensity was adjusted to black. Puncta were then counted and sized using automated analysis functions (MetaMorph; Universal Imaging Corporation, Downington, PA). Statistical analysis was done using unpaired t test.
In situ hybridization
Riboprobes specific for αDB and βDB were synthesized and labeled with digoxigenin by standard protocols and hybridized overnight to 20 μm cryostat sections. Sections were washed 3 h in 0.2× SSC at 65°C, incubated with alkaline phosphatase-conjugated anti-digoxigenin antibody, and detected using nitroblue-tetrazolium-chloride/5-bromo-4-chlor-indolyl-phosphate solution (Roche Diagnostics). Nuclei were stained before mounting with 4′,6′-diamidino-2-phenylindole (Invitrogen).
Behavioral testing
Control (C57BL/6) and mutant mice were age (range of 3–9 months) and sex matched, housed in standard cages, and maintained ad libitum on food and water and a 12 h light/dark cycle. C57BL/6 served as control for mdx mice because previous work has shown no significant differences between C57BL/6 and C57BL/10 in a large battery of behavioral and motor analyses (Rafael et al., 2000).
Forelimb grip strength
Forelimb grip strength was evaluated using a grip strength meter (Stoelting, Wood Dale, IL) as described by Connolly et al. (2001). The procedure involved teaching a mouse to grab a “grasping trapeze” (a force transducer connected to a peak amplifier) when pulled by the tail. A peak pull force was measured when the pulling force overcame the mouse’s grip strength. Our protocol involved 3 d of habituation in which each mouse was given a number of trials until it performed five “solid” pulls. Testing involved two five-trial sessions, with each session being conducted on consecutive days. The dependent variable was mean pulling force (grams), which was computed across the total 10 trials of testing for each animal.
Sensorimotor tests
Sensorimotor tests were conducted as described previously (Wozniak et al., 2004).
Platform test.
Each mouse was timed for how long it remained on an elevated, circular platform (1.0 cm thick; 3.0 cm in diameter) with rounded edges. A maximum score of 60 s was assigned if the mouse remained on the platform for the entire test trial or if it could climb down on a very thin pole that supported the platform without falling.
Inverted screen test.
Each mouse was placed in the middle of wire mesh grid (16 squares per 10 cm) that was inverted to 180°. A mouse was timed for how long it remained upside down on the screen, with a maximum score of 60 s being given if the animal did not fall. Two trials were administered for each test with 2 h intervening between trials, and means were calculated across the trials for each mouse.
Motor coordination and balance
Motor coordination and balance were evaluated using the rotorod (Rotamex-5; Columbus Instruments, Columbus, OH) test, which involved three conditions: a stationary rod (60 s maximum); a rotating rod with a constant speed (5 rpm for 60 s maximum); and a rod that had an accelerating rotational speed (5–20 rpm over 0–180 s). The present protocol differed somewhat from our previously published methodology (Ho et al., 2000) in that it was designed to minimize learning. This included three training sessions, each session being separated by 4 d. Each session included one trial on the stationary rod, two trials on the constant-speed rotorod, and two trials on the accelerating rotorod. Time spent on the rod in each condition was used as the dependent variable.
Statistical analyses
ANOVA models were used to analyze the behavioral data. Typically, the statistical models included one between-subjects variable (genotype) and sometimes one within-subjects (repeated measures) variable, such as trials. When ANOVAs with repeated measures were conducted, the Huynh–Feldt adjustment of α levels was used for all within-subjects effects containing more than two levels to protect against violations of the sphericity/compound symmetry assumptions underlying this ANOVA model. Bonferroni’s correction was used to maintain α levels at 0.05 when multiple comparisons were made.
Electroretinograms
Control (C57BL/6; n = 7) and DKO (n = 5) mice, aged 7–10 months, were dark adapted overnight. Flash electroretinograms (ERGs) were recorded from ketamine/xylazine-anesthetized mice as described by Ohlemiller et al. (2000). Responses to five flashes from a Grass Instruments (Quincy, MA) PS33-Plus xenon white light flash source (integrated intensity of 2.3 cd/s/m2 at the eye), separated by 10 s, were bandpass filtered (0.1–1000 Hz), digitized at 3 kHz, and averaged. Amplitude and latency (time-to-peak) of both a-wave and b-wave were measured and analyzed by unpaired t test.
Results
Generation of DB mutant mice
We previously generated adbn−/− mice and showed that they were viable and fertile but had mild muscular dystrophy as well as defects in their neuromuscular and myotendinous junctions (Grady et al., 1999, 2000, 2003). To generate bdbn−/− mice, we used embryonic stem cells bearing a mutational insert between exons 3 and 4 (Fig. 1A,B). These exons are upstream of all four known sites of alternative splicing (Loh et al., 1998), and the insert contains sequences that promote aberrant splicing and that terminate translation and transcription of the mutated gene (Zambrowicz et al., 1998). Accordingly, the insert would be expected to block expression of all βDB isoforms. The location of the mutagenic insert was confirmed by Southern blot analysis (Fig. 1C). Immunoblotting of tissue extracts from bdbn−/− liver, kidney, and brain, using a monoclonal antibody that recognizes both αDB and βDB (1E1; see Materials and Methods) revealed complete loss of βDB with no obvious compensatory increase in αDB (Fig. 1D and data not shown). In addition, reverse transcription-PCR, using an upstream primer in exon 3 and a downstream primer in exon 4, generated no wild-type product from bdbn−/− brain, whereas abundant product was obtained from control brain (data not shown). Thus, we conclude that the allele was a functional null.
Heterozygous (bdbn+/−) mice appeared normal, and homozygous (bdbn−/−) mice were produced in expected numbers. The bdbn−/− mice were viable, fertile, and outwardly normal, consistent with results reported for an independently generated βDB mutant allele (Loh et al., 2001). The adbn−/− and bdbn−/− mice were crossed to generate adbn+/−, bdbn+/− and then adbn−/−,bdbn−/− DKOs. DKOs were produced in expected numbers from matings of adbn+/−,bdbn+/−, adbn−/−,bdbn+/−, or adbn+/−,bdbn−/− animals. Like adbn−/− and bdbn−/− mice, DKOs were viable, fertile, and overtly normal in appearance.
Localization of DB in muscle and forebrain
Antibodies to DB that we generated, purchased, or obtained from colleagues either recognized both αDB and βDB or failed to stain brain tissue satisfactorily, as judged by persistence of staining in mutants (data not shown). We therefore determined localization of the two orthologs by immunostaining tissue sections from wild-type, adbn−/−, bdbn−/−, and DKO mice with antisera that recognized both αDB and βDB and comparing the resultant staining patterns. Consistent with previous reports, DB was detected along the surface of skeletal muscle fibers, with increased levels at the neuromuscular junctions (Blake et al., 1996; Peters et al., 1998; Grady et al., 1999, 2000). No immunoreactivity persisted in adbn−/− muscle, whereas bdbn−/− muscle did not differ detectably from controls. We conclude that all DB present in muscle fibers is αDB, a finding consistent with other reports (Peters et al., 1997; Blake et al., 1998). Neither was DB was detected in the vasculature (Fig. 2A–D and data not shown).
Figure 2.
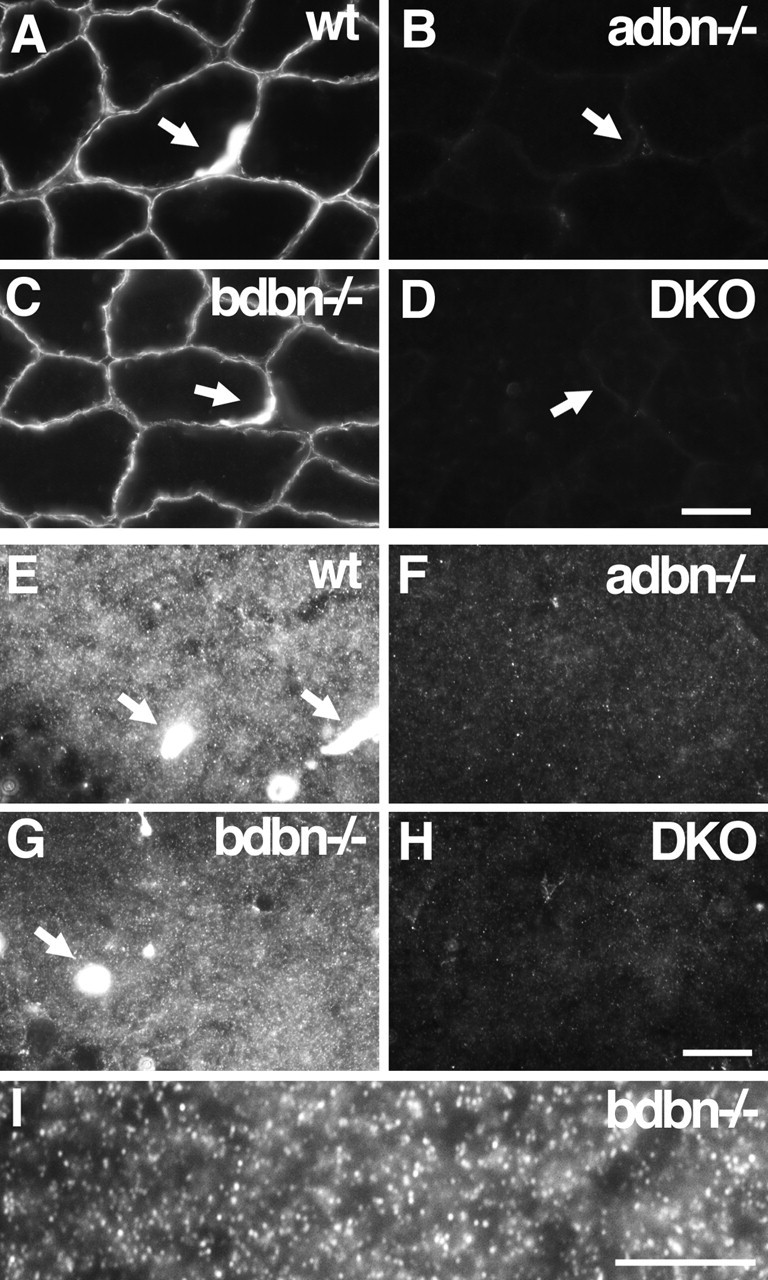
Localization of DB in skeletal muscle and hippocampus. Sections of wild-type (wt), adbn−/−, bdbn−/−, and DKO tissue were stained with an antibody that recognizes both αDB and βDB. A–D, In skeletal muscle, αDB is associated with the sarcolemma of muscle fibers; its levels are markedly increased at neuromuscular junctions. Synaptic sites (arrows) were identified by counterstaining with α-bungarotoxin (data not shown). No immunoreactivity is present in adbn−/− muscle, whereas bdbn−/− muscle is indistinguishable from that of controls, indicating that all DB in this tissue is αDB. E–H, In hippocampus, αDB is present in small puncta in the neuropil and in the walls of blood vessels (arrows). I, Higher magnification of αDB staining in bdbn−/− hippocampus. Scale bars: (in D) A–D, 20 μm; (in H) E–H, 10 μm; I, 10 μm.
In contrast, both αDB and βDB were present in the forebrain (cerebral cortex and hippocampus) (Fig. 2E–I and data not shown). Three types of staining were observed in wild-type brain. First, small, densely packed DB-positive puncta were present throughout most of the neuropil (gray matter) but were absent from myelinated (white matter) tracts. Second, DB was present in the walls of cerebral blood vessels (in contrast to its absence from vasculature in muscle). Third, DB was present along pial surfaces surrounding the brain. In adbn−/− brain, no punctate staining was observed, and staining of blood vessels and pia was greatly reduced. Staining of puncta, blood vessels, and pia in bdbn−/− brain was similar to that in controls. No DB immunoreactivity was detectable in sections from DKO brain. Together, these results indicate that the small puncta contain αDB but not βDB, whereas blood vessels and pia contain both αDB and βDB. Furthermore, it seems likely that levels of αDB are higher than levels of βDB in vessels and pia. We cannot completely rule out an alternative possibility, that loss of βDB in bdbn−/− mice led to a compensatory increase in αDB levels, but immunoblotting of bdbn−/− tissue provided no evidence for such compensation (Fig. 1D) (Loh et al., 2001).
Association of DB with a subset of cerebellar inhibitory synapses
In cerebellar cortex as in cerebral cortex, αDB was present in small puncta and both αDB and βDB were present in blood vessels and pia (Fig. 3A–D and data not shown). In addition, however, a second, distinct set of DB-positive puncta was present in cerebellum but not cortex. These structures were readily distinguishable from the smaller puncta by both size (generally <0.7 μm compared with >1.5 μm in diameter) and brightness. They were confined to the molecular and Purkinje cell layers and appeared to localize to the somata and dendrites of Purkinje cells (Fig. 3E,F). They were completely absent from DKO mice and their intensity was greatly reduced in sections from bdbn−/− mice, but they remained prominent in sections from adbn−/− mice. [In fact, the large puncta were more prominent in adbn−/− than in control sections because of the absence of the smaller puncta in the latter (Fig. 3, compare A, B)]. Thus, the larger, Purkinje cell-associated puncta contain both αDB and βDB. Levels of βDB appear to be higher than those of αDB, although, as above, we cannot rule out the possibility that loss of αDB led to a compensatory increase in βDB.
The size and distribution of the large βDB-rich puncta in cerebellum suggested that they were synapses. To test this idea, we costained sections for DB plus gephyrin and PSD-95, which mark inhibitory and excitatory postsynaptic membranes, respectively. All large, βDB-rich puncta associated with Purkinje cell dendrites but not somata and were also gephyrin-rich, whereas few if any were PSD-95 positive (Fig. 3G,H). Thus, βDB associated with Purkinje cell dendrites localizes to inhibitory synapses.
The βDB associated with inhibitory synapses on Purkinje cells might be present in presynaptic or postsynaptic elements. To distinguish these possibilities, we performed in situ hybridization with a probe specific for βDB (Fig. 4B). Purkinje cells expressed βDB, supporting the idea that βDB is a postsynaptic component. The presynaptic elements at such synapses are formed by interneurons of the molecular layer: basket and stellate cells (Palay and Chan-Palay, 1974). We detected no βDB RNA in such cells but cannot exclude the possibility that βDB is present at low levels presynaptically as well as postsynaptically.
Figure 4.

Localization of αDB and βDB RNA in cerebellum. In situ hybridization to sections of wild-type cerebellum with probes specific for αDB (A) or βDB (B). A′ and B′ show high-magnification views of areas in A and B. αDB RNA is present at highest levels in Bergmann glial somata, which flank Purkinje cell (P) somata. βDB RNA is present at highest levels in Purkinje cell bodies. Two Purkinje cell somata are outlined by dotted lines. Scale bar: A, B, 20 μm; A′, B′, 10 μm.
We also asked whether the small, αDB-rich puncta in cerebellum were associated with synapses. In fact, none of these puncta colocalized with gephyrin and few with PSD-95 (Fig. 3H,I). Thus, the small, αDB puncta may not be synaptic. To determine their cellular source, we performed in situ hybridization with a probe specific for αDB (Fig. 4A). Although low levels of αDB RNA were present in Purkinje cells, the most prominent deposits of αDB RNA appeared to be within Bergmann glial cell bodies located between Purkinje cells (Palay and Chan-Palay, 1974). Processes of Bergmann glial cells form specialized envelopes around nerve terminals on Purkinje cells, in which they appear to modulate synaptic transmission (Grosche et al., 2002; Huang and Bordey, 2004). We therefore speculate that the small αDB-positive structures in the molecular layer represent sites of interaction between glial cells and surrounding neurons.
Our findings that αDB is present in astrocytes and radial glia are consistent with those of Blake et al. (1999). Their biochemical data, showing concentration of βDB in postsynaptic densities, is also consistent with our immunohistochemical finding that this isoform is concentrated at inhibitory synaptic sites. Conversely, their report of broadly distributed βDB-like immunoreactivity in neuronal somata and processes is inconsistent with our results. Our confidence in the distribution we report stems from the finding that immunoreactivity is absent from mutant tissue.
Interdependence of DB and dystrophin colocalization
Knuesel et al. (1999) showed previously that dystrophin is associated with a subset of inhibitory synapses in cerebellum. We asked whether the same synapses were DB positive. Indeed, DB and dystrophin completely colocalized to large synapses on Purkinje cells (Fig. 5A). This colocalization included clusters associated with Purkinje cell bodies as well as their dendrites.
To ask whether the localization of dystrophin and DB was interdependent, we used mdx and DKO mutants. Loss of dystrophin, in mdx mice, led to loss of DB immunoreactivity from large puncta (Fig. 5B). Likewise, loss of DB, in DKO mice, led to loss of dystrophin from large puncta (Fig. 5C). Therefore, DB and dystrophin not only colocalize within Purkinje cells but also depend on each other for maintaining that localization. The smaller, αDB-rich puncta neither colocalized with dystrophin nor were affected by its loss (Fig. 5A–C). Thus, in the absence of DB, there is a specific loss of dystrophin at Purkinje synapses.
We also examined the relationship between DB and dystrophin in the hippocampus. Dystrophin was present in large puncta concentrated near pyramidal cell bodies (Fig. 5D), as also shown previously by Knuesel et al. (1999). DB was absent from these puncta. Likewise, little if any dystrophin was associated with the small DB-rich puncta in the dendritic layers. Consistent with the lack of colocalization, the distribution of DB was not detectably perturbed in mdx hippocampus, nor was the distribution of dystrophin detectably altered in DKO hippocampus (Fig. 5E,F). Thus, the relationship between dystrophin and DB differs among structures in the brain.
Loss of DB disrupts GABA receptor clustering
To assess the role of DB in the formation or maintenance of cerebellar inhibitory synapses, we stained control and DKO brains with antibodies to the GABAAα1 receptor subunit, a prominent component of inhibitory postsynaptic sites on the somata and dendrites of cerebellar Purkinje cells (Fritschy and Mohler, 1995) (Fig. 6A). In DKO cerebellum, the number of GABAAα1-positive puncta was reduced by ∼33%, and the size of the puncta was reduced by ∼50% (Fig. 6B,D–F). Thus, DB plays a regulatory role in maintaining the integrity of Purkinje cell inhibitory synapses.
Figure 6.
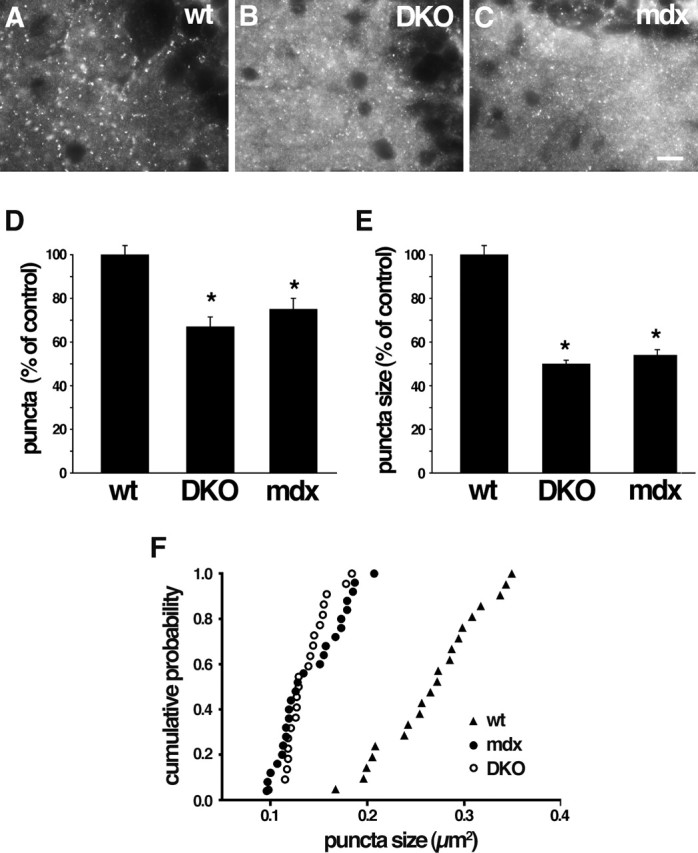
Disruption of GABAA receptor clusters in DKO and mdx cerebellum. A–C, Wild-type (wt), DB mutant (DKO), and dystrophin mutant (mdx) cerebella immunostained with antibody to GABAAα1 receptor subunit. GABAAα1-positive puncta are decreased in number and size in both mutants. Higher levels of diffuse staining in DKO and mdx (B, C) were seen consistently and may represent nonclustered GABAA receptors. D–F, Quantification of cluster number and size from micrographs such as those shown in A–C. Bars in D and E show mean ± SEM and are expressed as percentage of control. *p < 0.001, significantly different from wild type. F shows cumulative probability analysis of puncta size. Scale bar: A–C, 10 μm.
We used single mutants to ask whether the integrity of inhibitory receptor clusters required both αDB and βDB. Moderate reductions in the number and size of GABAAα1-positive puncta were observed in bdbn−/− mice, whereas no significant abnormalities were detected in adbn−/− mice (data not shown). This pattern is consistent with the distribution of DB isoforms at these synapses documented above: a predominance of βDB with lower amounts of αDB (Fig. 3).
Because loss of dystrophin leads to loss of DB from cerebellar inhibitory synapses (Fig. 5), one might expect that these synapses would be defective in dystrophin-deficient mice. Indeed, Knuesel et al. (1999) showed that clustering of GABAA receptors was disrupted in mdx mice. We found that loss of dystrophin and DB led to effects of similar magnitude on GABAA synapses (Fig. 6). In contrast, neither loss of dystrophin (Knuesel et al., 1999) nor loss of DB (data not shown) detectably affected the association of gephyrin with these synapses. The role of gephyrin at cerebellar synapses has not been tested in vivo because of the neonatal lethality of gephyrin-deficient mutant mice (Feng et al., 1998). However, gephyrin is present at the DB- and dystrophin-rich synapses (Fig. 3) and is required for synaptic clustering of multiple types of inhibitory neurotransmitter receptors, including receptors containing the GABAAα1 subunit (Essrich et al., 1998; Kneussel et al., 1999, 2001). Together, these results suggest that two separate complexes are required for integrity of the inhibitory postsynaptic membrane, one containing DB and dystrophin and another containing gephyrin.
Abnormal motor behavior in mice lacking DB
Results presented so far show that inhibitory synaptic input to cerebellar Purkinje cells is perturbed in the absence of DB. Output from the cerebellum is therefore likely to be aberrant in DKOs. In light of the role of the cerebellum in motor coordination, one might expect that motor function would be affected in DKOs. To assess this possibility, we submitted DKO mice to a battery of sensorimotor behavior tests. The ability of DKO mice to balance on a stationary rotorod was comparable with that of age-, sex-, and strain-matched control mice (Fig. 7D), as was their stride length (data not shown). In contrast, the performance of DKOs was significantly worse than that of controls in five other tests: forelimb grip strength (Fig. 7A), ability to balance on a small platform (Fig. 7B), ability to hang from an inverted screen (Fig. 7C), ability to balance on a rotorod rotating at a constant speed (data not shown), and ability to balance on a rotorod rotating at accelerating speed (Fig. 7E). Thus, DB is important for maintaining normal motor function.
Figure 7.
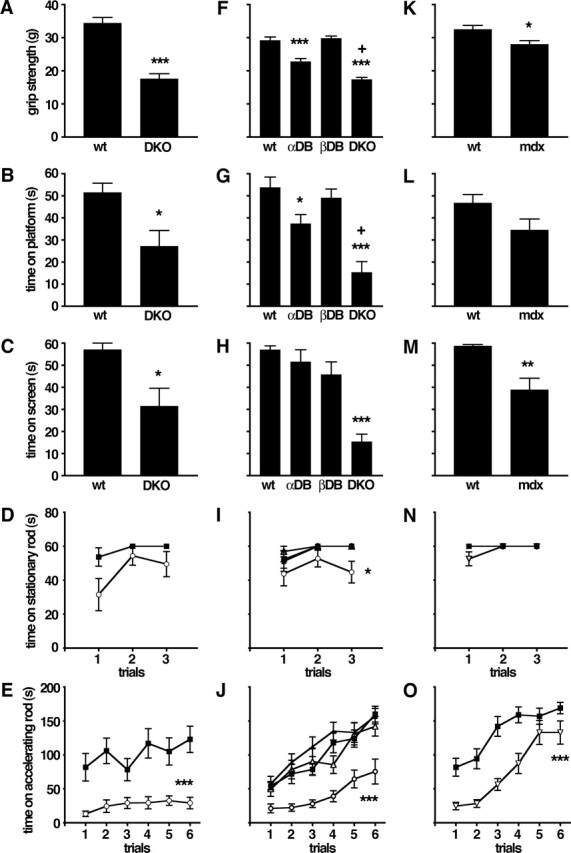
Defects in motor behavior in mice lacking DB or dystrophin. Results from five tests are shown: A, F, K, grip strength; B, G, L, time balanced on a small circular platform; C, H, M, time before falling from an inverted screen; D, I, N, time before falling from a stationary rotorod; and E, J, O, time before falling from an accelerating rotorod. A–E show results from wild-type (wt) and DKO mice. F–J show results from a second cohort of wild-type (wt) and DKO mice that were tested in parallel with adbn−/− (αDB) and bdbn−/− (βDB) mice. K–O show results from wild-type and mdx mice. DKO mice performed worse than wild-type, adbn−/−, and bdbn−/− mice on all tests; mdx mice performed worse than control in grip strength (K), time on screen (M), and time on accelerating rotorod (O). ▪, Wild type; ▵, αDB; •, βDB; ○, DKO; ▿, mdx. Graphs show mean ± SEM; n = 8–12 (mean, 10.3) animals per group. Significance of differences from wild type are as follows: *p < 0.05; **p < 0.005; ***p < 0.0005. In F and G, +p < 0.005, significantly different from αDB.
One complication in interpreting these results is that adbn−/− mice have peripheral abnormalities, including mild muscular dystrophy and defective neuromuscular synapses and myotendinous junctions (Grady et al., 1999, 2000, 2003). It was therefore possible that the defects observed in DKOs reflected peripheral rather than (or in addition to) central defects. To address this possibility, we compared the behavior of single DB mutants (adbn−/− and bdbn−/−) with that of a second, independent cohort of controls and DKOs. In two tests, forelimb grip strength and platform balance, adbn−/− mice performed significantly worse than controls, but in both cases they were significantly less impaired than DKOs (Fig. 7F,G). In all other tests, including the inverted screen test, adbn−/− mice were indistinguishable from controls (Fig. 7H–J). In all tests, defects in DKOs were similar to those observed in the first cohort, and in no case was the behavior of bdbn−/− single mutants distinguishable from that of controls. Thus, although neuromuscular defects attributable to the absence of αDB may contribute to the abnormal behavior observed in DKOs, the additive loss of βDB, which is present in brain but not muscle (Fig. 2A–D) (Peters et al. 1997; Blake et al., 1998), is required for full expression of the DKOs sensorimotor disabilities.
Another interpretive difficulty is that neuromuscular structure might be more severely affected in DKOs than in adbn−/− mice, with this difference contributing to the behavioral difference between genotypes. This seemed unlikely in view of the absence of detectable βDB from muscle (see above). We nonetheless tested this possibility directly by comparing the structure of neuromuscular junctions in adbn−/−, bdbn−/−, and DKO muscles. As reported previously, acetylcholine receptors (AChRs) smoothly outline each branch of the postsynaptic membrane at control neuromuscular junctions but are fragmented in adbn−/− mice. In addition, receptor density is lower in mutants than controls (Grady et al., 2000, 2003). No defects were observed in bdbn−/− muscles, and no differences were detected between adbn−/− and DKO muscles (Fig. 8). Similarly, there was no dystrophy (fibrosis or muscle fiber degeneration and regeneration) in bdbn−/− muscles, and the mild dystrophy observed in adbn−/− mice (Grady et al., 1999) was not exacerbated in DKOs (data not shown). These results support the conclusion that central synaptic defects underlie the behavioral defects in DKO mice.
Figure 8.

Neuromuscular structure in DB mutants. Longitudinal sections of muscles were stained with rhodamine–α-bungarotoxin to label acetylcholine receptors in the postsynaptic membrane of the neuromuscular junction. In wild-type mice (wt; A) and in adbn−/−, bdbn−/−, and DKO mice (data not shown), the mature synapse is composed of a pretzel-like array of branches. At higher magnification, receptors are regularly arrayed in wild-type muscles, with subtle striations indicative of junction folds (B). In adbn−/− mice, in contrast, receptors are broken into small irregular patches with radiating spicules (C) (Grady et al., 2000). No defects are detectable in bdbn−/− neuromuscular junctions (D), and no differences are detectable between adbn−/− and DKO junctions (E). Scale bar: A, 10 μm; B–E, 6 μm.
Abnormal motor behavior in mice lacking dystrophin
The observation that striking defects in inhibitory cerebellar synapses and in sensorimotor behavior are seen only in DKOs is consistent with the idea that the abnormalities result from cerebellar defects. Because mdx and DKO mice have similar cerebellar synaptic defects (Fig. 6) (Knuesel et al., 1999), we asked whether they have similar defects in sensorimotor behavior. Patterns of behavior in mdx mice were generally similar to those observed in DKO mice (Fig. 7K–O). In several tests, however, the severity of defects in mdx mice was less than that in DKO mice compared with wild type (Fig. 3K,L,N). This difference is noteworthy in that mdx mice display a more severe muscular dystrophy than that observed in adbn−/− or DKO mice (Grady et al., 1999 and data not shown). As exemplified by transgenic rescue experiments, muscular dystrophy in mdx and adbn−/− mice primarily reflects the respective loss of dystrophin and dystrobrevin within skeletal muscle (Cox et al., 1993; Grady et al., 2003). Nonetheless, our results support the idea that the sensorimotor impairments in mdx and DKO mice have a central component as well.
Location and function of DB in retina
Most boys with Duchenne muscular dystrophy exhibit defects in the b-wave of the ERG, which reflects synaptic responses in the inner retina (Pillers et al., 1993, 1995; Schmitz and Drenckhahn, 1997). A short form of dystrophin (Dp260), which lacks the N-terminal domains of the muscle isoform, is concentrated in photoreceptor terminals of normal humans and mice but is lacking in most Duchenne patients. No retinal defects are apparent in mdx mice, reflecting the fact that the mutation in this allele does not prevent expression of Dp260 (D’Souza et al., 1995). However, other murine dystrophin mutants, in which Dp260 is missing (mdx3cv), do show defects in the ERG (D’Souza et al., 1995; Green et al., 2004).
DB colocalizes with Dp260 in photoreceptor terminals, but its form is unclear: Dalloz et al. (2001) reported that αDB is present in this location in mice, Blank et al. (2002) reported that βDB is concentrated at this site in chick retina, and Ueda et al. (2000a) used antibodies unable to distinguish the two isoforms in rats. Using sections from wild-type retina, we confirmed the presence of DB in the outer plexiform layer, in which photoreceptor terminals form synapses, as well as in the inner limiting membrane and blood vessels (Fig. 9A). Use of mutant sections showed that photoreceptor terminals contained βDB but not αDB, that the inner limiting membrane contained αDB but not βDB, and that both isoforms were associated with blood vessels (Fig. 9B–D). As noted previously, βDB is colocalized with dystrophin and dystroglycan in photoreceptor terminals (Fig. 9E and data not shown).
Figure 9.

Localization and function of DB in retina. A–D, Sections of wild-type (wt), adbn−/−, bdbn−/−, and DKO retina were stained with an antibody that recognizes both αDB and βDB. The inner limiting membrane (ILM) contains αDB only, the outer plexiform layer (OPL) contains βBD only, and blood vessels (bv) contain both proteins. E, Dystrophin (DYS) and DB are colocalized in the outer plexiform layer; blood vessels contain DB but not dystrophin. F, Concentration of dystrophin in the outer plexiform layer persists in the absence of βDB. G, Representative ERG records from wild-type and DKO mice show similar configurations. H, I, Amplitude and latency of b-wave are normal in the absence of DB. Bars show mean ± SEM from 10 wild-type and 7 DKO mice. Scale bars: (in D) A–D, 10 μm; (in F″) E, F, 10 μm.
Using antibodies that recognize all dystrophin isoforms, we found no alteration in retinal dystrophin distribution in bdbn−/− or DKO mice (Fig. 9F and data not shown). Thus, the interdependence of DB and dystrophin localization at photoreceptor synapses differs from that documented above for inhibitory cerebellar synapses. Likewise, DB persisted in mdx mice (data not shown), but this result is not informative because Dp260 also persists in photoreceptor terminals of this strain (see above).
Finally, we measured ERGs in DKO mice and controls. We found no differences between the two genotypes (Fig. 9G). In particular, the amplitude and latency of the b-wave, which are altered in mdx3cv mice, were normal in DKO mice (Fig. 9H,I).
Discussion
The two DBs, α and β, bind directly to dystrophin and are prominent components of the DGC that links the cytoskeleton to the extracellular milieu in many cell types (Peters et al., 1997; Blake et al., 1998, 2002). Previous work has shown that αDB is critical for the maturation or maintenance of skeletal muscle fibers and their neuromuscular and myotendinous junctions (Grady et al., 1999, 2000, 2003), but no roles have been reported for αDB in nonmuscle tissues or for βDB in any tissue. Here, we show that both αDB and βDB are required for the maturation or maintenance of a subset of inhibitory synapses in the cerebellum and for proper execution of motor behaviors that depend on cerebellar integrity. In addition, by comparing the localization of DBs and dystrophin, the interdependence of their localization, and the defects that occur in their absence, we have provided evidence that DB in some parts of the CNS acts as part of a DGC.
It is useful to consider our results in the context of previous studies on the NMJ, at which synaptic roles of the DGC have been best studied to date. A DGC underlies and traverses the postsynaptic membrane at this synapse. Components of this DGC include dystrophin and its homolog utrophin, as well as dystroglycan, α and β2 syntrophin, and αDB. Subtle defects in synaptic topology and neurotransmitter receptor density are evident at NMJs lacking either dystrophin or utrophin, with more dramatic defects at NMJs lacking both proteins (Deconinck et al., 1997; Grady et al., 1997, 2000). NMJs are also aberrant in muscles lacking dystroglycan, α-syntrophin, or αDB (Côté et al., 1999; Adams et al., 2000, 2004; Grady et al., 2000; Jacobson et al., 2001). For adbn−/− mice, postsynaptic defects include alterations in the density, metabolic stability, and mobility of AChRs and a decrease in the number of junctional folds (Grady et al., 2000, 2003; Akaaboune et al., 2002). Three lines of evidence indicate that a main role of dystrophin and utrophin at the NMJ is to hold in place a subcomplex containing αDB. First, synaptic αDB is lost in the absence of dystrophin and utrophin, whereas dystrophin and utrophin remain concentrated at synaptic sites in the absence of αDB. Second, defects in αDB mutant mice are as severe as those in mice lacking dystrophin plus utrophin. Third, synaptic defects in mice lacking all three components are no more severe than in mice lacking only αDB (Grady et al., 2000).
The role of the DGC at cerebellar inhibitory synapses resembles that at the NMJ in that it is required for complete postsynaptic differentiation at both sites. Loss of either DB or dystrophin results in a significant reduction in the number and size of cerebellar GABAA receptor clusters, reminiscent of the fragmentation of AChR clusters seen in myotubes lacking DB or both utrophin and dystrophin (Knuesel et al., 1999; Grady et al., 2000). Moreover, the DGC appears to act along with a primary scaffolding mechanism at both sites, with rapsyn and gephyrin playing a predominant and DGC-independent role at the NMJ and inhibitory synapses, respectively (Gautam et al., 1995; Feng et al., 1998). Previous work using cultured neurons suggested that the DGC is not essential for GABAergic synaptogenesis (Levi et al., 2002). Consistent with this view, gephyrin localization at inhibitory synapses is not dependent on DB or dystrophin (Knuesel et al., 1999 and this study). Thus, the DGC is dispensable for receptor clustering at NMJs and cerebellar inhibitory synapses but important for the subsequent maintenance and stabilization of both synaptic types. Conversely, assembly of the DGC may differ at these two sites: synaptic localization of DB and dystrophin in cerebellum depends on the presence of each other, whereas at the NMJ loss of DB does not perturb the localization of dystrophin or utrophin (Grady et al., 2000). It is interesting to note that the dystrophin expressed in Purkinje cells and skeletal muscle (as well as cerebral cortex) have distinct promoters, each giving rise to unique isoforms (for review, see Sadoulet-Puccio and Kunkel, 1996).
In contrast, the involvement of the DGC in photoreceptor synapses appears to be very different from that at NMJs and cerebellar inhibitory synapses. First, the DGC is most prominently associated with the presynaptic terminals of photoreceptors in retina (Ueda et al., 2000a) but with the postsynaptic membrane in muscle and cerebellum. Second, synaptic transmission by photoreceptors is compromised in the absence of dystrophin but not in the absence of DB (Fig. 8G–I) (Pillers et al., 1993; Dalloz et al., 2001), whereas NMJs and cerebellar synapses require both dystrophin (or dystrophin plus utrophin) and DB. Third, the localization of DB and dystrophin are more strikingly interdependent at the NMJ and in cerebellum than in photoreceptors (Fig. 8) (Dalloz et al., 2001). Interestingly, both NMJs and cerebellar inhibitory synapses bear full-length dystrophin (427 kDa), whereas a 260 kDa dystrophin isoform predominates in photoreceptors (D’Souza et al., 1995; Pillers et al., 1995; Schmitz and Drenckhahn, 1997).
In addition to their synaptic roles, DGC components are present in and required for the proper function of astrocytes whose foot processes directly appose pial and perivascular basal laminas (Ueda et al., 2000b; Neely et al., 2001; Zaccaria et al., 2001; Michele et al., 2002; Moore et al., 2002; Amiry-Moghaddam et al., 2003). This DGC contains Dp71, a short dystrophin isoform that can bind both αDB and βDB (Blake et al., 1999; Haenggi et al., 2004). αDB is present at these sites, as well as in Bergmann glia that cap nerve terminals in the cerebellum (Grosche et al., 2002; Huang and Bordey, 2004). Roles of DB at these sites remain to be determined. Loss of DB clearly does not result in the striking neuronal migration defects seen in mice lacking dystroglycan, which are believed to result from disruption of the pial basal lamina (Moore et al., 2002). It is possible, however, that DB affects functions more subtly, as shown in studies on the role of α-syntrophin in water transport (Amiry-Moghaddam et al., 2003). More generally, our results support the idea that the 427, 260, and 71 kDa forms of dystrophin interact with DB and other components of the DGC in different ways.
Mice lacking both αDB and βDB performed poorly on tests of sensorimotor behavior that depend on proper cerebellar function, whereas mice lacking either αDB or βDB showed few defects. The cellular basis of these defects are undoubtedly complex, but several lines of evidence suggest that cerebellar inhibitory synapses are involved. (1) Both αDB and βDB localize to inhibitory synapses on Purkinje cells, and both are required for maintenance of GABAA receptor clusters at these synapses. (2) Dystrophin is concentrated at the same synapses, and mice lacking dystrophin show both synaptic and sensorimotor defects similar to those of DKOs. (3) Dystrophin persists in the vasculature and pia of mdx mice (presumably because the dystrophin isoform Dp71 persists in mdx mice) (Haenggi et al., 2004). The similarity of behavioral disabilities in mdx and DKO mice thus argues that the defects are unlikely to primarily reflect DGC function at these non-neuronal sites. (4) Neuromuscular defects are similar in mice lacking αDB or both αDB and βDB, presumably because only αDB is expressed in muscle or at the NMJ. However, motor defects are dramatically greater in DKOs than in mice lacking only αDB, indicating that the behavioral defects are not secondary to neuromuscular abnormalities.
In summary, our results indicate that the DGC is required for proper maturation and function of a subset of central inhibitory synapses, that DB is a key component of this DGC, and that interference with this DGC leads to behavioral abnormalities. DKO mice may therefore be useful in exploring mechanisms that underlie cognitive deficits in patients with Duchenne muscular dystrophy (Duchenne, 1868; Blake and Kroger, 2000; Anderson et al., 2002). Moreover, our results suggest that motor deficits in muscular dystrophy patients, which are their cardinal symptoms, may reflect not only peripheral derangements but alterations within the CNS as well.
Footnotes
This work was supported by grants from the National Institutes of Health (NIH) (R.M.G., J.R.S.) and by NIH Core Grant DC04665 (K.K.O.). We thank Alex Cohen and Joshua Weiner for important contributions to monoclonal antibody characterization and in situ hybridization, respectively.
References
- Adams ME, Kramarcy N, Krall SP, Rossi SG, Rotundo RL, Sealock R, Froehner SC (2000). Absence of alpha-syntrophin leads to structurally aberrant neuromuscular synapses deficient in utrophin. J Cell Biol 150:1385–1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams ME, Kramarcy N, Fukuda T, Engel AG, Sealock R, Froehner SC (2004). Structural abnormalities at neuromuscular synapses lacking multiple syntrophin isoforms. J Neurosci 24:10302–10309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akaaboune M, Grady RM, Turney S, Sanes JR, Lichtman JW (2002). Neurotransmitter receptor dynamics studied in vivo by reversible photo-unbinding of fluorescent ligands. Neuron 34:865–876. [DOI] [PubMed] [Google Scholar]
- Amiry-Moghaddam M, Williamson A, Palomba M, Eid T, de Lanerolle NC, Nagelhus EA, Adams ME, Froehner SC, Agre P, Ottersen OP (2003). Delayed K+ clearance associated with aquaporin-4 mislocalization: phenotypic defects in brains of alpha-syntrophin-null mice. Proc Natl Acad Sci USA 100:13615–13620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson JL, Head SI, Rae C, Morley JW (2002). Brain function in Duchenne muscular dystrophy. Brain 125:4–13. [DOI] [PubMed] [Google Scholar]
- Anderson JL, Stewart I, Morley JW (2003). Altered inhibitory input to Purkinje cells of dystrophin-deficient mice. Brain Res 982:280–283. [DOI] [PubMed] [Google Scholar]
- Blake DJ, Kroger S (2000). The neurobiology of Duchenne muscular dystrophy: learning lessons from muscle? Trends Neurosci 23:92–99. [DOI] [PubMed] [Google Scholar]
- Blake DJ, Nawrotzki R, Peters MF, Froehner SC, Davies KE (1996). Isoform diversity of dystrobrevin, the murine 87-kDa postsynaptic protein. J Biol Chem 271:7802–7810. [DOI] [PubMed] [Google Scholar]
- Blake DJ, Nawrotzki R, Loh NY, Gorecki DC, Davies KE (1998). β-Dystrobrevin, a member of the dystrophin-related protein family. Proc Natl Acad Sci USA 95:241–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blake DJ, Hawkes R, Benson MA, Beesley PW (1999). Different dystrophin-like complexes are expressed in neurons and glia. J Cell Biol 147:645–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blake DJ, Weir A, Newey SE, Davies KE (2002). Function and genetics of dystrophin and dystrophin-related proteins in muscle. Physiol Rev 82:291–329. [DOI] [PubMed] [Google Scholar]
- Blank M, Blake DJ, Kroger S (2002). Molecular diversity of the dystrophin-like protein complex in the developing and adult avian retina. Neuroscience 111:259–273. [DOI] [PubMed] [Google Scholar]
- Brunig I, Suter A, Knuesel I, Fritschy JM (2002). GABAergic terminals are required for postsynaptic clustering of dystrophin but not of GABAA receptors and gephyrin. J Neurosci 22:4805–4813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connolly AM, Keeling RM, Mehta S, Pestronk A, Sanes JR (2001). Three mouse models of muscular dystrophy: the natural history of strength and fatigue in dystrophin-, dystrophin/utrophin-, and laminin alpha2-deficient mice. Neuromuscul Disord 11:703–712. [DOI] [PubMed] [Google Scholar]
- Côté PD, Moukhles H, Lindenbaum M, Carbonetto S (1999). Chimaeric mice deficient in dystroglycans develop muscular dystrophy and have disrupted myoneural synapses. Nat Genet 23:338–342. [DOI] [PubMed] [Google Scholar]
- Cox GA, Cole NM, Matsumura K, Phelps SF, Hauschka SD, Campbell KP, Faulkner JA, Chamberlain JS (1993). Overexpression of dystrophin in transgenic mdx mice eliminates dystrophic symptoms without toxicity. Nature 364:725–729. [DOI] [PubMed] [Google Scholar]
- Dalkilic I, Kunkel LM (2003). Muscular dystrophies: genes to pathogenesis. Curr Opin Genet Dev 13:231–238. [DOI] [PubMed] [Google Scholar]
- Dalloz C, Claudepierre T, Rodius F, Mornet D, Sahel J, Rendon A (2001). Differential distribution of the members of the dystrophin glycoprotein complex in mouse retina: effect of the mdx(3Cv) mutation. Mol Cell Neurosci 17:908–920. [DOI] [PubMed] [Google Scholar]
- Deconinck AE, Potter AC, Tinsley JM, Wood SJ, Vater R, Young C, Metzinger L, Vincent A, Slater CR, Davies KE (1997). Postsynaptic abnormalities at the neuromuscular junctions of utrophin-deficient mice. J Cell Biol 136:883–894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Souza VN, Nguyen TM, Morris GE, Karges W, Pillers DM, Ray PN (1995). A novel dystrophin isoform is required for normal retinal electrophysiology. Hum Mol Genet 4:837–842. [DOI] [PubMed] [Google Scholar]
- Duchenne GBA (1868). Recherches sur la paralysie musculaire pseudo-hypertrophique ou paralysie myo-sclerosique. Arch Gen Med 121:5–25. 179–209, 305–321, 421–443, 552–588. [Google Scholar]
- Ervasti JM, Campbell KP (1991). Membrane organization of the dystrophin-glycoprotein complex. Cell 66:1121–1131. [DOI] [PubMed] [Google Scholar]
- Essrich C, Lorez M, Benson JA, Fritschy JM, Luscher B (1998). Postsynaptic clustering of major GABAA receptor subtypes requires the gamma 2 subunit and gephyrin. Nat Neurosci 1:563–571. [DOI] [PubMed] [Google Scholar]
- Feng G, Tintrup H, Kirsch J, Nichol MC, Kuhse J, Betz H, Sanes JR (1998). Dual requirement for gephyrin in glycine receptor clustering and molybdoenzyme activity. Science 282:1321–1324. [DOI] [PubMed] [Google Scholar]
- Fritschy JM, Mohler H (1995). GABAA-receptor heterogeneity in the adult rat brain: differential regional and cellular distribution of seven major subunits. J Comp Neurol 359:154–194. [DOI] [PubMed] [Google Scholar]
- Gautam M, Noakes PG, Mudd J, Nichol M, Chu GC, Sanes JR, Merlie JP (1995). Failure of postsynaptic specialization to develop at neuromuscular junctions of rapsyn-deficient mice. Nature 377:232–236. [DOI] [PubMed] [Google Scholar]
- Grady RM, Merlie JP, Sanes JR (1997). Subtle neuromuscular defects in utrophin-deficient mice. J Cell Biol 136:871–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grady RM, Grange RW, Lau KS, Maimone MM, Nichol MC, Stull JT, Sanes JR (1999). Role for α-dystrobrevin in the pathogenesis of dystrophin-dependent muscular dystrophies. Nat Cell Biol 1:215–220. [DOI] [PubMed] [Google Scholar]
- Grady RM, Zhou H, Cunningham JM, Henry MD, Campbell KP, Sanes JR (2000). Maturation and maintenance of the neuromuscular synapse: Genetic evidence for the roles of the dystrophin-glycoprotein complex. Neuron 25:279–293. [DOI] [PubMed] [Google Scholar]
- Grady RM, Akaaboune M, Cohen AL, Maimone MM, Lichtman JW, Sanes JR (2003). Tyrosine-phosphorylated and nonphosphorylated isoforms of α-dystrobrevin: roles in skeletal muscle and its neuromuscular and myotendinous junctions. Neuron 160:741–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green DG, Guo H, Pillers DA (2004). Normal photoresponses and altered b-wave responses to APB in the mdx(Cv3) mouse isolated retina ERG supports role for dystrophin in synaptic transmission. Vis Neurosci 21:739–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grosche J, Kettenmann H, Reichenbach A (2002). Bergmann glial cells form distinct morphological structures to interact with cerebellar neurons. J Neurosci Res 68:138–149. [DOI] [PubMed] [Google Scholar]
- Haenggi T, Soontormalai A, Schaub MC, Fritschy JM (2004). The role of utrophin and Dp71 for assembly of different dystrophin-associated protein complexes (DPCs) in the choroids plexus and microvasculature of the brain. Neuroscience 129:403–413. [DOI] [PubMed] [Google Scholar]
- Ho N, Liauw JA, Blaeser F, Wei F, Hanissian S, Muglia LM, Wozniak DF, Nardi A, Arvin KL, Holtzman DM, Linden DJ, Zhuo M, Muglia LJ, Chatila TA (2000). Impaired synaptic plasticity and camp response element-binding protein activation in Ca2+/calmodulin-dependent protein kinase type IV/Gr-deficient mice. J Neurosci 20:6459–6472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman EP, Brown RH, Kunkel LM (1987). Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell 51:919–928. [DOI] [PubMed] [Google Scholar]
- Huang H, Bordey A (2004). Glial glutamate transporters limit spillover activation of presynaptic NMDA receptors and influence synaptic inhibition of Purkinje neurons. J Neurosci 24:5659–5669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson C, Cote PD, Rossi SG, Rotundo RL, Carbonetto S (2001). The dystroglycan complex is necessary for stabilization of acetylcholine receptor clusters at neuromuscular junctions and formation of the synaptic basement membrane. J Cell Biol 152:435–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kneussel M, Brandstatter JH, Laube B, Stahl S, Muller U, Betz H (1999). Loss of postsynaptic GABAA receptor clustering in gephyrin-deficient mice. J Neurosci 19:9289–9297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kneussel M, Brandstatter JH, Gasnier B, Feng G, Sanes JR, Betz H (2001). Gephyrin-independent clustering of postsynaptic GABAA receptor subtypes. Mol Cell Neurosci 17:973–982. [DOI] [PubMed] [Google Scholar]
- Knuesel I, Mastrocola M, Zuellig RA, Bornhauser B, Schaub MC, Fritschy JM (1999). Altered synaptic clustering of GABAA receptors in mice lacking dystrophin (mdx mice). Eur J Neurosci 11:4457–4462. [DOI] [PubMed] [Google Scholar]
- Levi S, Grady RM, Henry MD, Campbell KP, Sanes JR, Craig AM (2002). Dystroglycan is selectively associated with inhibitory GABAergic synapses but is dispensable for their differentiation. J Neurosci 22:4274–4285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loh NY, Ambrose HJ, Guay-Woodford LM, DasGupta S, Nawrotzki RA, Blake DJ, Davies KE (1998). Genomic organization and refined mapping of the mouse β-dystrobrevin gene. Mamm Genome 9:857–862. [DOI] [PubMed] [Google Scholar]
- Loh NY, Nebenius-Oosthuizen D, Blake DJ, Smith AJH, Davies KE (2001). Role of β-dystrobrevin in nonmuscle dystrophin-associated protein complex-like complexes in kidney and liver. Mol Cell Biol 21:7442–7448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michele DE, Barresi R, Kanagawa M, Saito F, Cohn RD, Satz JS, Dollar J, Nishino I, Kelley RI, Somer H, Straub V, Mathews KD, Moore SA, Campbell KP (2002). Post-translational disruption of dystroglycan-ligand interactions in congenital muscular dystrophies. Nature 418:417–422. [DOI] [PubMed] [Google Scholar]
- Moore SA, Saito F, Chen J, Michele DE, Henry MD, Messing A, Cohn RD, Ross-Barta SE, Westra S, Williamson RA, Hoshi T, Campbell KP (2002). Deletion of brain dystroglycan recapitulates aspects of congenital muscular dystrophy. Nature 418:422–425. [DOI] [PubMed] [Google Scholar]
- Moukhles H, Carbonetto S (2001). Dystroglycan contributes to the formation of multiple dystrophin-like complexes in brain. J Neurochem 78:824–834. [DOI] [PubMed] [Google Scholar]
- Muntoni F, Brockington M, Torelli S, Brown SC (2004). Defective glycosylation in congenital muscular dystrophies. Curr Opin Neurol 17:205–209. [DOI] [PubMed] [Google Scholar]
- Neely JD, Amiry-Moghaddam M, Ottersen OP, Froehner SC, Agre P, Adams ME (2001). Syntrophin-dependent expression and localization of aquaporin-4 water channel protein. Proc Natl Acad Sci USA 98:14108–14113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohlemiller KK, Vogler CA, Roberts M, Glavin N, Sands MS (2000). Retinal function is improved in a murine model of a lysosomal storage disease following bone marrow transplantation. Exp Eye Res 71:469–481. [DOI] [PubMed] [Google Scholar]
- Palay SL, Chan-Palay V (1974). In: Cerebellar cortex New York: Springer.
- Peters MF, O’Brien KF, Sadoulet-Puccio HM, Kunkel LM, Adams ME, Froehner SC (1997). β-Dystrobrevin, a new member of the dystrophin family. J Biol Chem 272:31561–31569. [DOI] [PubMed] [Google Scholar]
- Peters MF, Sadoulet-Puccio HM, Grady RM, Kramarcy NR, Kunkel LM, Sanes JR, Sealock R, Froehner SC (1998). Differential membrane localization and intermolecular associations of α-dystrobrevin isoforms in skeletal muscle. J Cell Biol 142:1269–1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pillers DM, Bulman DE, Weleber RG, Sigesmund DA, Musarella MA, Powell BR, Murphey WH, Westall C, Panton C, Becker LE, Worton RG, Ray PN (1993). Dystrophin expression in the human retina is required for normal function as defined by electroretinography. Nat Genet 4:82–86. [DOI] [PubMed] [Google Scholar]
- Pillers DM, Weleber RG, Woodward WR, Green DG, Chapman VM, Ray PN (1995). mdxCv3 Mouse is a model for electroretinography of Duchenne/Becker muscular dystrophy. Invest Ophthalmol Vis Sci 36:462–466. [PubMed] [Google Scholar]
- Rafael JA, Nitta Y, Peters J, Davies KE (2000). Testing of SHIRPA, a mouse phenotypic assessment protocol, on Dmdmdx and Dmdmdx3cv dystrophin-deficient mice. Mamm Genome 11:725–728. [DOI] [PubMed] [Google Scholar]
- Sadoulet-Puccio HM, Kunkel LM (1996). Dystrophin and its isoforms. Brain Pathol 6:25–35. [DOI] [PubMed] [Google Scholar]
- Schmitz F, Drenckhahn D (1997). Dystrophin in the retina. Prog Neurobiol 53:547–560. [DOI] [PubMed] [Google Scholar]
- Ueda H, Baba T, Kashiwagi K, Iijima H, Ohno S (2000a). Dystrobrevin localization in photoreceptor axon terminals and at blood-ocular barrier sites. Invest Ophthalmol Vis Sci 41:3908–3914. [PubMed] [Google Scholar]
- Ueda H, Baba T, Terada N, Kato Y, Fujii Y, Takayama I, Mei X, Ohno S (2000b). Immunolocalization of dystrobrevin in the astrocytes endfeet and endothelial cells in the rat cerebellum. Neurosci Lett 283:121–124. [DOI] [PubMed] [Google Scholar]
- Vaillend C, Billard JM, Laroche S (2004). Impaired long-term spatial and recognition memory and enhanced CA1 hippocampal LTP in the dystrophin-deficient Dmdmdx mouse. Neurobiol Dis 17:10–20. [DOI] [PubMed] [Google Scholar]
- Wozniak DF, Hartman RE, Boyle MP, Vogt SK, Brooks AR, Tenkova T, Young C, Olney JW, Muglia LJ (2004). Apoptotic neurodegeneration induced by ethanol in neonatal mice is associated with profound learning/memory deficits in juveniles followed by progressive functional recovery in adults. Neurobiol Dis 17:403–414. [DOI] [PubMed] [Google Scholar]
- Zaccaria ML, Di Tommaso F, Brancaccio A, Paggi P, Petrucci TC (2001). Dystroglycan distribution in adult mouse brain: a light and electron microscopy study. Neuroscience 104:311–324. [DOI] [PubMed] [Google Scholar]
- Zambrowicz BP, Friedrich GA, Buxton EC, Lilleberg SL, Person C, Sands AT (1998). Disruption and sequence identification of 2,000 genes in mouse embryonic stem cells. Nature 392:608–611. [DOI] [PubMed] [Google Scholar]