

Abstract
Glutamate receptor (GluR) ion channels mediate fast synaptic transmission in the mammalian CNS. Numerous crystallographic studies, the majority on the GluR2-subtype AMPA receptor, have revealed the structural basis for binding of subtype-specific agonists. In contrast, because there are far fewer antagonist-bound structures, the mechanisms for antagonist binding are much less well understood, particularly for kainate receptors that exist as multiple subtypes with a distinct biology encoded by the GluR5–7, KA1, and KA2 genes. We describe here high-resolution crystal structures for the GluR5 ligand-binding core complex with UBP302 and UBP310, novel GluR5-selective antagonists. The crystal structures reveal the structural basis for the high selectivity for GluR5 observed in radiolabel displacement assays for the isolated ligand binding cores of the GluR2, GluR5, and GluR6 subunits and during inhibition of glutamate-activated currents in studies on full-length ion channels. The antagonists bind via a novel mechanism and do not form direct contacts with the E723 side chain as occurs in all previously solved AMPA and kainate receptor agonist and antagonist complexes. This results from a hyperextension of the ligand binding core compared with previously solved structures. As a result, in dimer assemblies, there is a 22 Å extension of the ion channel linkers in the transition from antagonist- to glutamate-bound forms. This large conformational change is substantially different from that described for AMPA receptors, was not possible to predict from previous work, and suggests that glutamate receptors are capable of much larger movements than previously thought.
Keywords: glutamate receptor, kainate, structure, gating, antagonist, crystallography
Introduction
At excitatory synapses, receptors for the neurotransmitter glutamate are the major mediators of information transfer in the mammalian CNS (Dingledine et al., 1999). The AMPA, kainate, NMDA, and δ ionotropic glutamate receptor (iGluR) families are encoded by 18 genes (Hollmann, 1999). This diversity underlies the specialized roles that each receptor species plays at individual synapses in the CNS, and its molecular basis is becoming increasingly well understood (Mayer, 2005a). Crystal structures for numerous agonist complexes have been solved for the ligand binding cores of GluR2-subtype AMPA receptors (Armstrong and Gouaux, 2000; Mayer and Armstrong, 2004), for GluR5- and GluR6-subtype kainate receptors (Mayer, 2005b; Nanao et al., 2005; Naur et al., 2005), and for the NR1 (Furukawa and Gouaux, 2003) and NR2 (Furukawa et al., 2005) subtypes of NMDA receptors as well as for a prokaryotic GluR homolog (Mayer et al., 2001). These structures, in conjunction with physical biochemical and electrophysiological studies, have revealed in broad terms the molecular mechanisms for ion channel gating, partial agonist activity, desensitization, and allosteric modulation (Sun et al., 2002; Jin et al., 2003, 2005; Inanobe et al., 2005).
The protein data bank (PDB) currently contains the structures of >60 iGluR agonist complexes, but strikingly, despite the vast number of antagonists that have been synthesized and that are widely used in neuroscience research, only four antagonist structures have been solved, two for the glycine-binding NR1 subunit of NMDA receptors (Furukawa and Gouaux, 2003; Inanobe et al., 2005) and two for the AMPA receptor GluR2 subunit (Armstrong and Gouaux, 2000; Hogner et al., 2003). This imbalance reflects the greater difficulty of crystallizing antagonist-bound forms of iGluRs. Although the available agonist and antagonist structures provided a starting point for modeling additional receptor subtypes and ligand complexes (Foucaud et al., 2003; Pentikainen et al., 2003; Moretti et al., 2004), they are limited in their usefulness by the conformational flexibility of iGluR ligand binding cores, for which domain closure varies with both ligand and iGluR subtype (Mayer, 2005b), and also by the complex network of solvent molecules that plays a key role in ligand binding but that it is difficult to model without previous structural knowledge (Armstrong and Gouaux, 2000).
Here, we report crystal structures of dimer assemblies of the GluR5 ligand-binding cores with two novel GluR5-selective antagonists solved at 1.74 and 1.87 Å resolution. These structures reveal an expanded cleft, more open than any previously solved iGluR structures. Surprisingly, despite the large difference in separation of the two domains that form the ligand binding site, there is substantial conservation of bound water molecules in the GluR5 agonist and antagonist complexes. The conformation of Glu723, a key side chain in the ligand binding site, is distinct from that found in the functionally related AMPA receptor antagonist complexes and suggests that GluR5 is trapped in the resting state rather than the partially activated structure observed for the GluR2 DNQX and 2-amino-3-[5-tert-butyl-3-(phosphonomethoxy)-4-isoxazolyl]propionic acid (ATPO) antagonist complexes (Armstrong and Gouaux, 2000; Hogner et al., 2003).
Materials and Methods
Protein purification and crystallization.
The rat GluR5 S1S2 ligand-binding core was overexpressed in OrigamiB (DE3) Escherichia coli and purified to homogeneity using Ni2+ NTA and ion exchange chromatography as described previously (Mayer, 2005b). The construct consisted of residues N416–K529, preceded by an 18 amino acid peptide encoding an NTA affinity tag and thrombin site, and was linked via a GT dipeptide to residues P652–E791. In the present experiments, we used the E791S mutant, which facilitated crystallization. The wild-type rat GluR6 and GluR2 ligand-binding cores were purified using similar procedures. The affinity tags were removed by proteolysis before ligand binding studies and crystallization. The GluR5 S1S2 selenomethionine derivative was prepared by growing cells in LB at 37°C to an OD of 1.2 at 600 nm; the cells were then collected by centrifugation and rinsed twice with PBS before resuspension in minimal medium at 16°C containing 100 mg/L each of (S)-lysine, (S)-threonine, (S)-isoleucine, (S)-phenylalanine, (S)-valine, and (S)-selenomethionine, followed by overnight induction of protein expression with 30 μm isopropyl β-d-1-thiogalactopyranoside. Incorporation of selenomethionine was established by electrospray ionization mass analysis, which revealed a major peak of Mr 29,596 and two minor peaks of Mr 29,549 and Mr 29,174, corresponding to species with 9, 8 and 0 Se, respectively. All crystals were grown in hanging drops at a temperature of 20°C with a 1:1 dilution of protein with reservoir. For complexes with UBP302 and UBP310, the protein was exhaustively dialyzed against a 2× crystallization buffer containing 40 mm NaCl, 20 mm HEPES, pH 7.0, 2 mm EDTA, and 10–20 μm ligand, for a total volume exchange of >107; the protein was then diluted by 50% with 10 mm ligand dissolved in water adjusted to pH 7.0 with NaOH and concentrated to between 5 and 10 mg/ml. For crystallization of complexes with glutamate, the protein was dialyzed against 1× crystallization buffer containing 10 mm (S)-glutamate and concentrated to 10–20 mg/ml. The reservoir solution for UBP302 and UBP310 complexes contained 17–22% PEG 1000 (polyethylene glycol) and 100 mm Tris chloride, pH 8.4–9.0. The reservoir solution for glutamate complexes contained 1.6–1.75 m (NH4) 2SO4 and 100 mm HEPES, pH 7.0–7.2. For the SeMet UBP310 complex, 10 mm β-mercaptoethanol was added to buffers and crystallization solutions. For both the glutamate and UBP complexes, seeding was required to obtain diffraction quality crystals. Cryopreservation was achieved by rapid serial transfers to mother liquor supplemented with increasing amounts of glycerol to a maximum concentration of 18–20%, followed by flash cooling in liquid N2.
Radiolabel displacement assays.
Ligand binding assays were performed as described previously (Mayer, 2005b), using exhaustively dialyzed protein and 12.5 nm [3H]S-glutamate (50 Ci/mmol; PerkinElmer Life Sciences, Norwalk, CT) for GluR5, 5 nm [3H]2S,4R-4-methylglutamate (47.9 Ci/mmol; Tocris Cookson, Ellisville, MO) for GluR6, or 10 nm [3H]AMPA (45.5 Ci/mmol; PerkinElmer Life Sciences) for GluR2. Kd values are mean ± SEM calculated from single binding site fits to three to six displacement curves per ligand corrected for the binding of [3H] ligands using experimentally determined Kd values of 57.2 nm for GluR5–glutamate, 35.6 nm for GluR6–2S,4R-4-methylglutamate, and 24.8 nm for GluR2–AMPA.
Electrophysiological assays.
Two-electrode voltage clamp was performed as described previously (Chen et al., 1999). Oocytes were injected with mRNA for rat GluR6 (50 pg), GluR5 (2 ng), or GluR2 (2 ng) 48 h before analysis by two-electrode voltage clamp at a holding potential of −60 mV. The extracellular solution contained (in mm) 100 NaCl, 1 KCl, 1 MgCl2, 0.5 BaCl2, and 5 HEPES, pH 7.5. The custom-built recording chamber had a volume of 5 μl; solutions were applied at 250 μl min−1 to allow rapid solution exchange.
Crystallographic data collection, phasing, and refinement.
Data sets for native and SeMet GluR5 S1S2 UBP310 complexes and for a native GluR5 glutamate complex were collected at APS beamline ID22 at 100°K using MAR 300 and MAR 225 CCD detectors, respectively; for the UBP302 complex, data were collected using CuKα radiation from a microfocus sealed tube with confocal optics (Micromax 002; Rigaku, The Woodlands, TX) and a MAR 345 image plate detector (Table 1). Data were indexed, scaled, and merged using HKL2000 (Otwinowski and Minor, 2001). The UBP310 and UBP302 complexes were solved initially by molecular replacement in the P41212 space group with one molecule in the asymmetric unit, using a GluR5 glutamate complex monomer (PDB code 1TXF) without ligand or solvent as the search probe. Because it was anticipated that there would be a large conformational change in the UBP310 structure compared with the search probe, domains 1 and 2 were located independently using the program Phaser-1.3.1 (McCoy et al., 2005), omitting Met106 through Leu108 and Ser213 through Gly215 in the interdomain β-strand linkers from the search model. After one round of simulated annealing, followed by positional and individual B-factor refinement using the CNS (Brunger et al., 1998), there was unambiguous density for the ligand and the six amino acids in the β-strands linking domains 1 and 2 that were omitted from the search model. Model building into Fo-Fc maps was performed using O (Jones and Kjeldgaard, 1997). Subsequent crystallographic refinement, including the addition of side chains in alternate conformations, ligand, ions, solvent, and a well ordered segment of a PEG molecule, combined with rounds of manual rebuilding, was performed with REFMAC (Winn et al., 2001) and resulted in values of Rwork and Rfree of 22.0 and 25.8%, respectively.
Table 1.
Data collection statistics
| Data set | UBP310 | UBP302 | Glutamate |
|---|---|---|---|
| Space group | C2221 | C2221 | P21 |
| Unit cell dimensions (Å) | |||
| a | 97.70 | 97.88 | 62.69 |
| b | 97.95 | 97.91 | 73.78 |
| c | 129.1 | 128.35 | 115.92 |
| α, β, γ (degrees) | 90, 90, 90 | 90, 90, 90 | 90, 99.74, 90 |
| Number per a.u. | 2 | 2 | 4 |
| Wavelength (Å) | 1.0000 | 1.54178 | 0.99997 |
| Resolution (Å)a | 40–1.74 (1.80) | 40–1.86 (1.93) | 30–2.1 (2.18) |
| Unique observations | 64,151 | 52,211 | 59,238 |
| Mosaicity | 0.60 | 0.65 | 0.58 |
| Mean redundancyb | 4.9 (4.9) | 7.1 (6.4) | 3.4 (3.4) |
| Completeness (%)b | 99.9 (99.7) | 98.6 (85.7) | 99.8 (100) |
| Rmerge (%)bc | 0.038 (0.441) | 0.057 (0.434) | 0.084 (0.344) |
| I/σ(I)b | 14.5 (3.38) | 16.3 (4.4) | 12.3 (3.62) |
a.u., Asymmetric unit.
aValues in parentheses indicate the low-resolution limit for the last shell of data.
bValues in parentheses indicate statistics for the last shell of data.
cRmerge = (Σ II − <II>)/ΣI II, where <II> is the mean II over symmetry–equivalent reflections.
After refinement in P41212 was completed, a subsequent analysis of a three-wavelength selenomethionine MAD data set was performed using SOLVE (Terwilliger and Berendzen, 1999) as a test to confirm the location of Met residues in domain 2 for which the overall B-factor was greater than twice the value for domain 1. Unexpectedly, the solution failed, and only two of nine SE atoms were located. A reinspection of the intensities in the 00l and h00 planes for data scaled in P41212 revealed some weak reflections that were incorrectly interpreted as systematic absences, and the data were reprocessed in the space group C2221, with two molecules in the asymmetric unit. The MAD solution now located 18 Se atoms in the asymmetric unit of the C2221 cell. For both space groups, the cumulative intensity distribution and moments matched those expected for untwinned data. After reprocessing in C2221, the a and b cell axes increased by √2 from their value of 69.07 Å in the tetragonal cell to 97.70 and 97.95 Å, with no change in the dimension of the c axis, and Rmerge decreased from 0.053 to 0.038. Although, in part, the change in Rmerge reflects a difference in the total number of reflections observed for P41212 and C2221 (we used data collected over a 120° rotation angle for C2221, giving an overall redundancy of 4.9-fold, whereas for P41212, a rotation angle of only 98° was used, giving an overall redundancy of 7.8-fold), the improved statistics in C2221 are likely to be genuine.
Two copies of the refined coordinates from the P41212 solution minus ligand and solvent were placed in the C2221 asymmetric unit by molecular replacement, and the analysis continued with positional and individual B-factor refinement using REFMAC, initially using tight NCS restraints, which were subsequently relaxed, before the building of ligand, a well ordered part of a PEG molecule, side chains with alternative conformations, ions, and solvent, guided by inspection of Fo-Fc maps coupled with iterative rounds of model building and refinement. A TLS motion determination analysis (Painter and Merritt, 2005) suggested that domain 2 was considerably more mobile than domain 1, consistent with the observed crystal packing in which domain 2 makes few intermolecular contacts. When refinement of the UBP310 complex was continued using four TLS groups per monomer that were defined based on motion analysis, values for Rwork and Rfree decreased to 17.8 and 20.8%, respectively (Table 2). A similar procedure was used to refine the UBP302 complex, which was solved by Fourier difference techniques. The occupancies of Met722 and E723 were refined manually by a combined procedure involving inspection of multiple rounds of Fo-Fc maps after refinement with occupancies incremented by steps of 0.05, combined with inspection of main-chain B-values; the final values chosen gave similar main-chain B-values for both conformations and the smallest residual density in Fo-Fc maps. Other side chains modeled in alternate conformations were refined at equal occupancy. The structure of a new crystal form of the GluR5 S1S2 glutamate complex, with four molecules in the asymmetric unit of a P21 cell, which are arranged as two pairs of dimers, was also solved by molecular replacement at 2.1 Å using the GluR5 monomer (PDB code 1TXF) as a search probe. Refinement proceeded without difficulty, and the final model, using a separate TLS group for each subunit, gave values for Rwork and Rfree that converged at 18.6 and 24.1%, respectively (Table 2). Additional crystallographic calculations were performed using CCP4 (1994) and the USF suite (Kleywegt et al., 2001). Domain closure was calculated using the program FIT after least-squares superposition of domain 1 using Cα atoms as described previously (Mayer, 2005b). Buried surface-area calculations were performed using the CCP4 program AREAIMOL with a probe radius of 1.4 Å and a point density of 20. Figures were prepared using BOBSCRIPT (Esnouf, 1997), MOLSCRIPT and RASTER3D (Kraulis, 1991; Merritt and Bacon, 1997), and PyMol (Delano, 2002). Coordinates and structure factors for the UBP310, UBP302, and glutamate complexes have been deposited in the PDB with accession codes 2F34, 2F35, and 2F36, respectively.
Table 2.
Refinement statistics
| Data set | UBP310 | UBP302 | Glutamate |
|---|---|---|---|
| Resolution (Å) | 33.41–1.74 | 39.92–1.87 | 29.9–2.11 |
| Protein atoms (alt conf) | 4355 (339) | 4313 (228) | 8065 (55) |
| Ligand atoms | 48 | 48 | 40 |
| Chloride/sulfate atoms | 2/0 | 2/0 | 0/4 |
| PEG atoms | 32 | 32 | 0 |
| Water atoms | 482 | 416 | 439 |
| Rwork/Rfree (%)a | 18.10/21.14 | 19.12/22.04 | 18.56/24.11 |
| rms deviations | |||
| Bond lengths (Å) | 0.014 | 0.012 | 0.016 |
| Bond angles (degrees) | 1.66 | 1.46 | 1.84 |
| Bonds B values (MC/SC) | 1.12/1.33 | 1.03/1.16 | 1.53/1.51 |
| Angles B values (MC/SC) | 1.72/1.88 | 1.55/1.57 | 2.32/2.21 |
| Mean B values (Å2) | |||
| Protein overall | 16.76 | 16.35 | 29.38 |
| Main chain | 16.00 | 15.60 | 28.19 |
| Side chain | 17.50 | 17.09 | 30.57 |
| Ligand | 30.48 | 35.14 | 20.72 |
| Ions | 23.39 | 25.24 | 48.25 |
| PEG | 22.43 | 23.56 | NA |
| Water | 40.78 | 40.52 | 31.26 |
| Ramachandran statisticsb | 93.2/6.8/0/0 | 92.8/7.2/0/0 | 92.0/7.9/0.1/0 |
alt conf, Alternative conformation; MC/SC, main chain/side chain; NA, not applicable.
aRwork = (Σ ‖Fo − Fc‖)/Σ Fo, where Fo and Fc denote observed and calculated structure factors, respectively; 5% of the reflections were set aside for the calculation of the Rfree value.
bPercentage of residues in most favored/allowed/generous/disallowed regions.
Results
Ligand binding assays
The first potent antagonist identified for AMPA and kainate receptors, the quinoxalinedione CNQX (Honore et al., 1988; Brauner-Osborne et al., 2000), showed little discrimination between AMPA and kainate receptor subtypes (Fig. 1). UPB302 and UBP310, in contrast, are representative members of a new class of selective antagonist in which the parent molecule is based on willardiine (More et al., 2004; Dolman et al., 2005). The agonist members of the willardine family have already proven to be useful tools for determining the structural basis of partial agonist activity for AMPA receptors (Jin et al., 2003), and willardiine-based antagonists such as UBP302 have helped to establish a role for kainate receptors in synaptic plasticity (More et al., 2004; Dolman et al., 2005). In this study, we report a novel antagonist, UBP310, which binds to GluR5-subtype kainate receptors with nanomolar affinity. To characterize the affinity and selectivity of UBP310 for iGluR subtypes, we performed displacement assays using highly purified preparations of the GluR2, GluR5, and GluR6 S1S2 ligand-binding cores and appropriate radioligands (Fig. 1). The results reveal that UBP310 binds to GluR5 S1S2 with an affinity of 130 nm and shows 12,700-fold selectivity for GluR5 versus GluR6 and 830-fold selectivity for GluR5 versus GluR2. UBP302, which lacks the 5-methyl group of UBP310 and has a phenyl ring substitution for the thiophene group of UBP310, retains high selectivity for GluR5 versus GluR6 and AMPA receptors, but with a 30-fold reduction in affinity. In contrast, NBQX, a second-generation quinoxalinedione, shows little discrimination between GluR5 and GluR6 but has a >100-fold selectivity for AMPA versus kainate receptors, consistent with its use as a selective AMPA receptor antagonist in studies on native kainate receptors expressed in hippocampal interneurons (Mulle et al., 2000).
Figure 1.

Binding experiments with AMPA and kainate receptor S1S2 ligand-binding cores. A, Chemical structures of UBP310, UBP302, and the quinoxalinediones NBQX and CNQX. B, Summary of the results of radioligand displacement assays for the S1S2 constructs of GluR5, GluR6, and GluR2; Kd values are the mean ± SEM of three to six experiments per ligand. Selectivity was calculated from the ratio of the indicated Kd pairs. C, Displacement curves for binding of willardiine derivatives to GluR5 and GluR6. D, Displacement curves for binding of quinoxalinediones to GluR5 and GluR6; data points for C and D show the mean ± SEM.
Electrophysiological measurements of antagonist selectivity
Functional assays of antagonist action using two-electrode voltage-clamp recording from Xenopus oocytes expressing GluR2, GluR5, or GluR6 confirmed the high selectivity of UBP310 binding observed in radiolabel displacement assays for the S1S2 constructs (Fig. 2). When applied at 3.25 μm, a concentration 25 times its Kd value for binding to GluR5, UBP310 produced 100% inhibition of responses to 45 μm glutamate for GluR5 but <1% inhibition of responses for GluR2 (0.98 ± 0.31%; n = 4) and GluR6 (0.28 ± 0.12%; n = 5). When applied at a 10 times higher concentration, 250 times the Kd value for GluR5, UBP310 produced only 8.8 ± 0.73% and 9.9 ± 0.53% inhibition of GluR2 and GluR6 responses to glutamate (Fig. 2). These experiments likely underrepresent the selectivity of UBP310, because the 45 μm glutamate concentration used to activate ion channel gating is ∼800 times the Kd value for binding of glutamate to GluR5 S1S2 but only 60 and 30 times the Kd value for binding of glutamate to GluR2 S1S2 and GluR6 S1S2, respectively. Consistent with its nanomolar affinity for GluR5, UBP310 showed extremely slow washoff with sigmoidal kinetics (Fig. 2E). As a result, in experiments on the synaptic activation of kainate receptors, the binding of glutamate and UBP310 is unlikely to come to equilibrium. The sigmoidal washoff kinetics for UBP310 are reminiscent of those observed for antagonists in studies on AMPA receptor gating mechanisms and likely reflect the need for agonist binding to multiple subunits to efficiently activate ion channel gating (Rosenmund et al., 1998).
Figure 2.

Selective antagonist action of UBP310. A–C, Inhibition of glutamate (Glu)-activated currents is shown for GluR2 (A), GluR5 (B), and GluR6 (C); UBP310 was applied at 3.25 μm for GluR5 and 32.5 μm for GluR2 and GluR6. D, Mean ± SD of responses from four to five cells per GluR subtype recorded as shown in A–C. E, Kinetics of recovery from inhibition by 3.25 μm UBP310 of the GluR5 response to glutamate.
The UBP GluR5 complexes have a hyperextended conformation
Crystal structures of the GluR5 S1S2 complex with UBP310, UBP302, and glutamate were solved at resolutions of 1.74, 1.87, and 2.1 Å, respectively (Tables 1, 2). Figure 3A shows a superposition of the GluR5 glutamate, GluR5 UBP310, and GluR2 ATPO complexes and reveals substantial differences in domain closure that follows the sequence Glu > ATPO > UBP310. To quantify differences in the extent of domain closure, we superimposed domain 1 of each protomer in the GluR5 UBP310 and UBP302 structures on domain 1 of monomer A from the GluR5 glutamate complex. This analysis was repeated for the previously solved AMPA receptor DNQX and ATPO antagonist complexes (Armstrong and Gouaux, 2000; Hogner et al., 2003) and for the NMDA receptor NR1 5,7-dichlorokynurenic acid (DCKA) and cycloleucine (cLeu) antagonist complexes (Furukawa and Gouaux, 2003; Inanobe et al., 2005), using the appropriate parent agonist complex as the reference structure. Domain closure was quantified by calculating the rotation angle required to superimpose domain 2 of the individual complexes on their parent agonist structures after previous superposition of domain 1. Figure 3B shows the results of this analysis and reveals that the UBP302 complex is the most open structure (30.1°), with UPB310 only slightly more closed (29.3°). The AMPA receptor GluR2 complexes with DNQX (16.6°) and ATPO (18.9°) and the NR1 complex with cLeu (17.6 °) are substantially more closed in comparison, whereas the NR1 complex with DCKA (24.9°) is intermediate between these extremes. Because the extent of domain closure for iGluR agonist complexes varies for AMPA, kainate, and NMDA receptor subtypes, with the GluR5 glutamate complex 6° more closed than the GluR2 glutamate complex (Armstrong and Gouaux, 2000; Mayer, 2005a), these results reflect two variables: differences in domain closure for agonist complexes and differences in domain closure for antagonist complexes. To measure differences in domain closure that arise solely from the antagonist contribution, the above analysis was repeated, using the UBP310 complex as the reference structure. The results, shown in Figure 3C, reveal that the DNQX (15.3°), ATPO (14.4°), DCKA (9.6°), and cLeu (12.5°) complexes are all more closed, by the amount indicated in parentheses, than the UBP310 complex.
Figure 3.

Ligand-induced domain closure for iGluR ligand binding cores. A, Stereoview of the GluR5 UBP310 (red), GluR2 ATPO (PDB code 1NOT; blue), and GluR5 glutamate (green) complexes superimposed using domain 1 Cα coordinates; the labels indicate the position of helices F, G, H, and I in domain 2. B, Analysis of domain closure defined as the rotation that is required to superimpose domain 2 for the indicated antagonist complexes following superimposition of domain 1 onto the GluR2, GluR5, or NR1 agonist complexes with glutamate or glycine as appropriate. C, Analysis of the rotation that is required to superimpose domain 2 for the indicated antagonist complexes following superimposition of domain 1 on the GluR5 UBP310 complex.
Transmembrane linker separation in GluR5 ligand-binding core dimers
In the multiple structures that have been solved for the ligand binding cores of iGluRs, the protein molecules crystallize in a number of different configurations, with both crystallographic and noncrystallographic symmetry. There is substantial experimental evidence that the twofold symmetric dimer formed by the agonist-bound complexes of wild-type GluR2 and the nondesensitizing L483Y mutant corresponds to the active state of AMPA receptors (Sun et al., 2002; Horning and Mayer, 2004). In contrast, in previous work on GluR5- and GluR6-subtype kainate receptors (Mayer, 2005b), dimers were observed that are extremely unlikely to have any functional significance and arise simply to satisfy crystal packing requirements. The GluR5 glutamate complex described here contained four molecules in the asymmetric unit, whereas the antagonist complexes contained two molecules (Table 1). Analysis of crystal packing in the GluR5 UBP310 and UBP302 complexes revealed two essentially identical dimers, both generated by crystallographic symmetry. These dimers are strikingly similar to the dimers reported previously for numerous AMPA receptor agonist and antagonist complexes (Armstrong and Gouaux, 2000), for a GluR6 domoate complex (Nanao et al., 2005), for the NR1 cLeu complex (Inanobe et al., 2005), and for the bacterial homolog GluR0 (Mayer et al., 2001). In the GluR5 glutamate complex reported here, there are also two dimers, but these are generated by noncrystallographic symmetry. The structure of these dimers is also closely related to that observed previously for GluR2 and GluR6 but markedly different from that of a GluR5 glutamate complex dimer that was suggested to correspond to a desensitized state (Naur et al., 2005), but which alternatively could have been generated by crystal packing and have no functional significance.
In AMPA receptor dimer assemblies, differences in domain closure for agonist- and antagonist-bound complexes have been suggested to underlie gating of the ion channel (Armstrong and Gouaux, 2000; Jin et al., 2002; Sun et al., 2002), and it is likely that the same principle applies to other iGluR subtypes. In the pair of GluR5 UBP310 complex dimers, the linkers leading to the transmembrane ion channel segments are separated by 18.6 and 19.0 Å, as defined by the position of the Cα atoms of Ile653, with similar values of 19.3 and 19.5 Å for the pair of GluR5 UBP302 complex dimers. In contrast, in the pair of GluR5 glutamate complex dimers, the linkers are separated by 40.9 and 41.0 Å. Superposition of the GluR5 glutamate AB dimer and each of the pair of UBP310 complex dimers, using domain 1 Cα coordinates (Fig. 4), revealed an increase in separation of the ion channel linkers by a mean value of 22.1 Å after agonist binding with only minimal changes in the structure of the individual domains, as judged by rms values of 0.52 and 0.44 Å when domains 1 and 2 were fit independently. In comparison, when the wild-type and nondesensitizing GluR2 L483Y mutant dimer complexes with AMPA and DNQX are superimposed, the linkers separate by only 7.1 and 8 Å (Armstrong and Gouaux, 2000; Sun et al., 2002). It seems plausible that bulkier AMPA receptor antagonists could produce further linker separation, because for GluR2, the DNQX structures are closed by 2.5–6° compared with the apo protein structure. Despite considerable effort, our attempts to crystallize the apo state of kainate receptors have not been successful, and at present, we do not know whether the UBP302 and UBP310 complexes are hyperextended compared with the apo state of GluR5 or whether the apo state of kainate receptors is more open than that of AMPA receptors.
Figure 4.
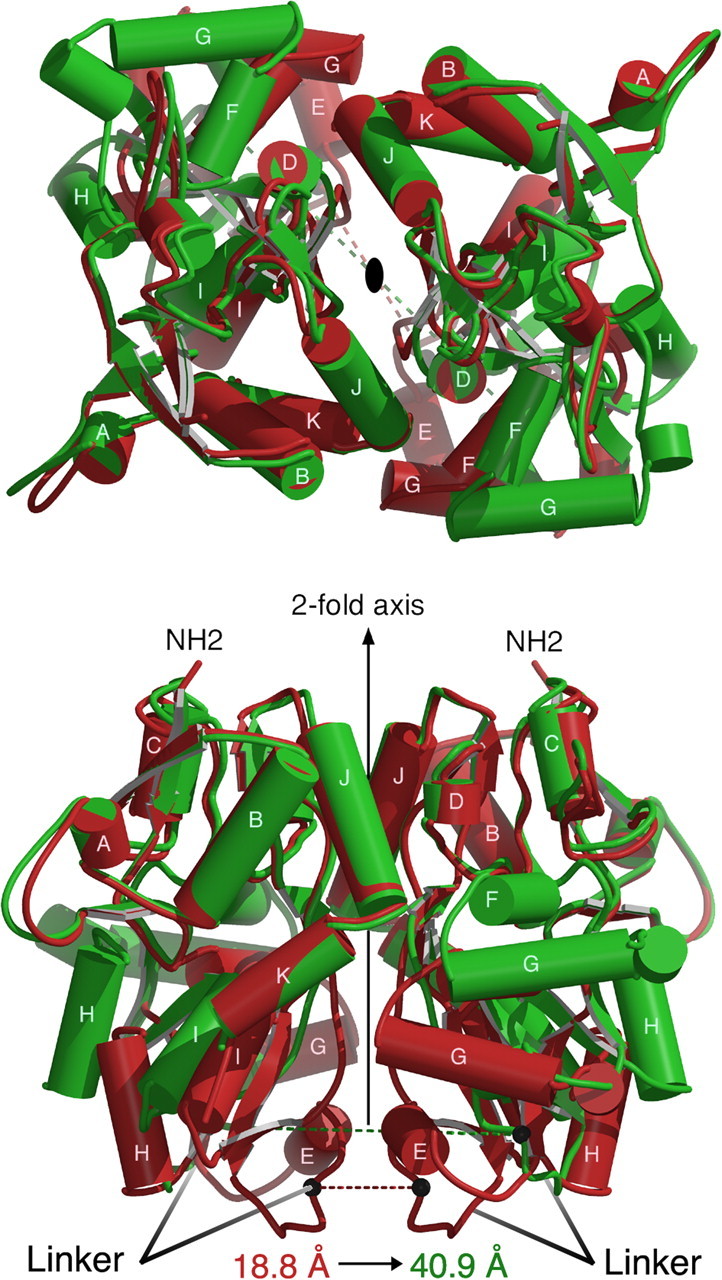
Agonist-induced conformational changes in the GluR5 dimer assembly illustrated by superposition using domain 1 Cα coordinates of the UBP310 (red) and the glutamate (green) complexes. The top panel is viewed from the N terminus looking down the twofold axis; the bottom panel shows a view from the side after rotation by 90°. The relative movement of the linker regions, which connect domain 2 to the ion channel pore, is represented by black spheres connected by red and green dashed lines, which show the difference in position of the Cα coordinates of Ile653 in the two structures. This distance increases from 18.8 Å in the UBP310 complex to 40.9 Å in the glutamate complex.
Mechanism of binding of GluR5 antagonists
A detailed inspection of the UBP310 model after TLS refinement revealed that, despite similar mean isotropic B-values for domain 1 (17.0 Å2) and domain 2 (16.4 Å2), helix E, helix H, and the top of helix G are less well ordered than the rest of the domain 2, whereas, with the exception of loop 2, domain 1 is well ordered throughout. This is illustrated by a ribbon diagram painted by the B-factor shown in Figure 5A. The GluR5 S1S2 side chains in contact with ligand are in well ordered parts of the structure for both domains and omit density for UBP310, and some key water molecules involved in the binding of antagonist were of high quality (Fig. 5B); the UBP302 complex showed similar structural features. Before TLS refinement, there was a large difference in B-values for domain 1 (23.8 Å2) and domain 2 (58.0 Å2), which complicated model building. To increase confidence in the final model, a MAD experiment was performed for a UBP310 complex crystal grown from selenomethionine-labeled protein. The GluR5 S1S2 construct contains a total of nine methionine residues, five in domain 1 and four in domain 2, all of which were unambiguously identified in anomalous difference electron density maps for data collected at the peak energy for Se (Fig. 5A). In addition, the anomalous difference maps confirmed the location of methionine side chains built in alternate conformations including Met722 in the ligand binding pocket.
Figure 5.
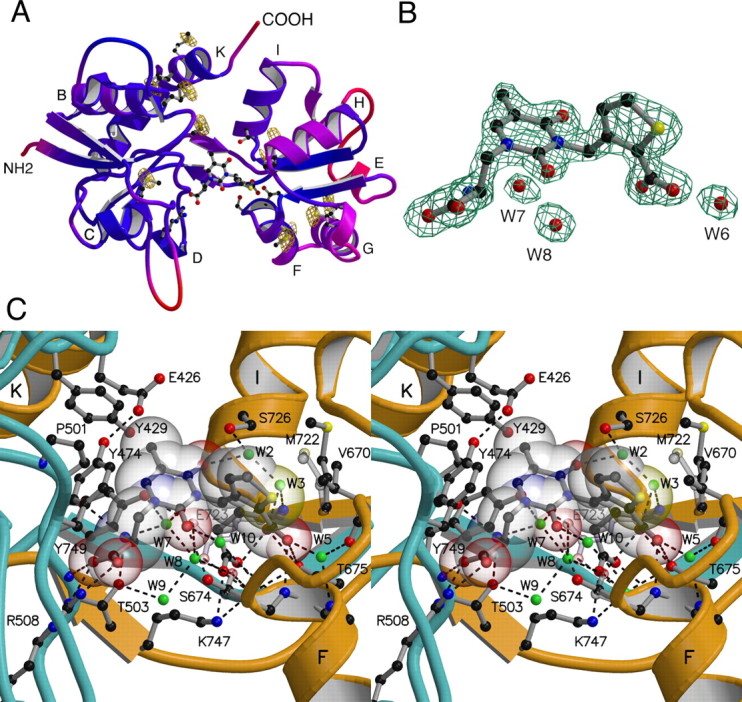
High-resolution crystal structure of the GluR5 UBP310 complex. A, Ribbon representation of the GluR5 S1S2 UBP310 complex colored by B-factor with a color ramp from blue to red over the B-value range 10–35; the ligand and side chains that interact with UBP310 are shown as ball-and-stick representations; the labels indicate the locations of α-helices; the Se peak energy anomalous difference electron density map contoured at 6 σ confirms the location of all nine methionine side chains, which are drawn as ball-and-stick representations. B, Fo-Fc omit electron density map calculated using data to 1.74 Å and contoured at 4.5 σ; atoms for UBP310 and the indicated waters were omitted from the Fc calculation. C, Stereoview of the ligand binding pocket in the UBP310 complex; the peptide chains from segments S1 and S2 are colored cyan and gold, respectively; UBP310 is shown as a transparent CPK model together with a ball-and-stick representation for the ligand and some side chains; water molecules are shown as green spheres; dashed lines indicate hydrogen bonds; alternative conformations for the side chains of Ser674, Met722 and Glu723 are shown as transparent objects.
UBP302 and UBP310 are large molecules (volume, 226 and 234 Å3) that, because of steric overlaps, cannot be accommodated by the conformation the GluR5-binding pocket adopts in the glutamate-bound complex. As a result of steric hindrance, the binding of UBP302 and UBP310 forces the ligand binding site to expand to an open cleft conformation; this occurs via rotation of domain 2 by 30°, which positions a pocket formed by the N terminus of helix F in contact with the thiophene and phenyl groups of UBP310 and UBP302. Both antagonists make extensive contacts with domain 2, principally with residues near the N terminus of helix F but also with helix I (Fig. 5C). The thiophene ring carboxyl group, which acts as a surrogate for the γ-carboxyl group of glutamate, makes direct hydrogen bond contacts with the side-chain hydroxyl group and the main-chain amide of Thr675. At the tip of helix F, the side chain of Ser674 was modeled in two conformations: one permits a hydrogen bond contact with the 2 position carbonyl oxygen of the uracil ring; the second conformation is similar to that found in the GluR5 glutamate complex and makes a weak hydrogen bond contact with the thiophene ring carboxyl group.
Solvent-mediated hydrogen bond contacts with UBP antagonists are made by the domain 2 side chains of Glu723, Ser726, and Tyr749; by the main-chain amides of Ser674, Met676, and Glu723; and by the main-chain carbonyl oxygen of Val670. The side chain of Val670 plays an important role by making van der Waals contact with the hydrophobic thiophene and phenyl groups of UBP310, partially shielding them from solvent. The side chain of Met722 was modeled in two conformations with occupancies of 0.65 and 0.35; the former conformation is similar to that found in the GluR5 glutamate complex, whereas the latter permits van der Waals contact with the UBP310 thiophene group. The larger size of the phenyl group in UBP302, which bumps up against the side chain of Val670, causes a slight expansion of the ligand binding core compared with the UBP310 complex (Fig. 3B).
Domain 1 contacts with the antagonists closely resemble those made by glutamate, reflecting common structural elements of these ligands. In fact, all known GluR ligands share a common mode of interaction with domain 1 in which the ligand α-carboxyl group makes an ion pair contact with the side chain of Arg508 and a hydrogen bond with the main-chain amide of Thr503. The ligand α-amino group makes hydrogen bond contacts with the side chain of Thr503 and the main-chain carbonyl oxygen of Pro501. In the UBP310 complex, the 5-methly group slots into a hydrophobic cavity in domain 1 formed by the methylene groups of Glu426, Tyr429, and Pro501. Desolvation of the methyl group would be expected to increase the binding energy for UBP310 by 1.2 kcal mol−1 compared with UBP302, which lacks the 5-methyl group (Anslyn and Dougherty, 2005). For both antagonists, the aromatic side chain of Tyr474, which acts as a lid in the closed cleft agonist complex (Mayer, 2005b), makes extensive van der Waals contacts with the willardiine uracil ring.
Subsite analysis and solvent structure
Previous work on AMPA and kainate receptors has revealed that solvent molecules play key structural roles in the binding sites of glutamate receptor ion channels (Armstrong and Gouaux, 2000; Mayer, 2005b). In addition to acting as surrogate ligand atoms, water molecules also form a hydrogen-bonded network that links domain 1 with domain 2, further stabilizing the closed cleft conformation. In the GluR5 glutamate complex, there are six water molecules trapped in the closed cleft conformation stabilized by glutamate; five of these occupy sites in domain 2, and several are displaced by GluR5-selective agonists (Mayer, 2005b). To compare solvent structure in the GluR5 glutamate and antagonist complexes, we constructed subsite maps, as has been done previously for AMPA receptors (Armstrong and Gouaux, 2000; Mayer and Armstrong, 2004). Although we expected the antagonists to displace many of the water molecules trapped in the GluR5 glutamate complex, the analysis instead revealed conservation of four of six water molecules in the antagonist complexes, suggesting that they may play a structural role (Fig. 6). A more detailed description of the chemistry of the subsite analysis for the GluR5 UBP310 and glutamate complexes is available as supplemental material (available at www.jneurosci.org).
Figure 6.

Subsite map of the GluR5 ligand-binding pocket. Schematic representations for the GluR5-binding pocket occupied by (S)-glutamate (A) and UBP310 (B). Hydrogen bond and ion pair sites generated by domains 1 and 2 are colored pink and blue, respectively; stripes indicate sites generated by both domains; sites of van der Waals contacts are indicated by hatched curved lines. In the glutamate complex, there are six trapped water molecules shown as numbered red spheres; four of these are maintained in the UBP310 complex. To make this figure, torsion angles in the UBP310 ligand were adjusted compared with the conformation found in the crystal structure to bring the heterocyclic rings into approximately the same plane for ease of illustration. A detailed explanation of the subsite maps is available as supplemental material (available at www.jneurosci.org).
In addition to revealing remarkable conservation of solvent molecules in the ligand binding site of the GluR5 agonist and antagonist complexes, these maps explain the selective affinity of UBP310 and UBP302 for GluR5 versus GluR6 and AMPA receptors. Surprisingly, the key determinants of GluR5- versus GluR6-subtype selectivity for agonists (Mayer, 2005b), the exchange of Ser706 for Asn690 and Leu720 for Phe704, play no major role in the binding of antagonists. Instead, there are two alanine substitutions in GluR6 that replace GluR5 side chains that make key contacts with the antagonists; these are the pairs Ala487, which replaces Thr503, and Ala658, which replaces Ser674. The loss of the direct hydrogen bond to the ligand α-amino group made by Thr503 is likely to be particularly significant because, in the antagonist complexes, the E723 side chain does not make the canonical interaction with the ligand α-amino group observed in all previously solved AMPA and kainate receptor complexes. The low affinity of UBP310 and UBP302 for AMPA receptors must arise in large part because of steric clashes imposed by the substitution of Leu650 for Val670 and Met708 for Ser726. Likewise, binding of willardiine antagonists to the KA1 and KA2 subtypes of kainate receptor is hindered by the replacement of Val670 and Ser726 by the much larger Ile and Met side chains, whereas for GluR7, the exchange of Ala for Ser674 will also lower affinity.
Conformational rearrangements of Glu723 in the ligand binding site
In numerous structures solved for AMPA and kainate receptor agonist complexes, the ligand α-amino group is bound by a conserved glutamate side chain in domain 2 that makes a key ion pair contact with the ligand. There is substantial evidence that these structures correspond to the active state of the receptor (Armstrong and Gouaux, 2000; Sun et al., 2002; Armstrong et al., 2003). In the UBP302 and UBP310 antagonist complexes, the side chain of Glu723 was refined in two conformations with 65 and 35% occupancy, neither of which was capable of forming a contact with the antagonist α-amino group due to the extreme domain opening required to accommodate these bulky ligands. Figure 7 shows omit maps for the Glu723 side chain and residues in the second β-strand linking domains 1 and 2 for the UBP302 and glutamate complexes. In the UBP302 and UBP310 complexes, Chi2 of the Glu723 side-chain conformation refined with 65% occupancy has rotated by 115° compared with the conformation found in GluR5 agonist complexes, such that the carboxyl group projects toward the β-strand linking domain 2 to domain 1, where it makes a strong hydrogen bond contact with the side chain of Lys747. In this “high-occupancy” conformation of the GluR5 antagonist complexes, which resembles that found in the GluR2 apo state (Armstrong and Gouaux, 2000), a pair of well ordered water molecules with mean B-values of 22.6 and 29.8 Å2 is located close to the position where the carboxyl group of the Glu723 side chain is found in GluR5 agonist complexes; these water molecules (Fig. 5, W7 and W8) form a hydrogen-bonded network linking the antagonist α-amino group with domain 2 residues. This is reminiscent of the binding mechanism for the NMDA receptor NR2 subunit in which Asp731, the equivalent residue to Glu723, binds to the glutamate ligand α-amino group via a water molecule (Furukawa et al., 2005). In the second “low-occupancy” conformation observed in the UBP antagonist complexes, the Glu723 side chain projects toward the ligand, displacing W7 and W8, but the 4.6 and 4.2 Å distance from the oxygen atoms of the Glu723 carboxyl group to the UBP302 and UBP310 α-amino groups is too large for hydrogen bond formation; instead, in this conformation, the Glu723 side chain is stabilized by hydrogen bond formation with the hydroxyl group of Tyr749. These results are of interest in light of the gating model for iGluR activation proposed on the basis of differences between crystal structures of the apo- and agonist-bound ligand-binding cores of the AMPA receptor GluR2 subunit (Armstrong and Gouaux, 2000). They suggest that, in contrast to the previously solved AMPA receptor antagonist complexes with ATPO and DNQX, in which the Glu705 side chain switches to the active conformation found in agonist complexes, UBP302 and UBP310 stabilize the receptor in a conformation closer to the resting state.
Figure 7.
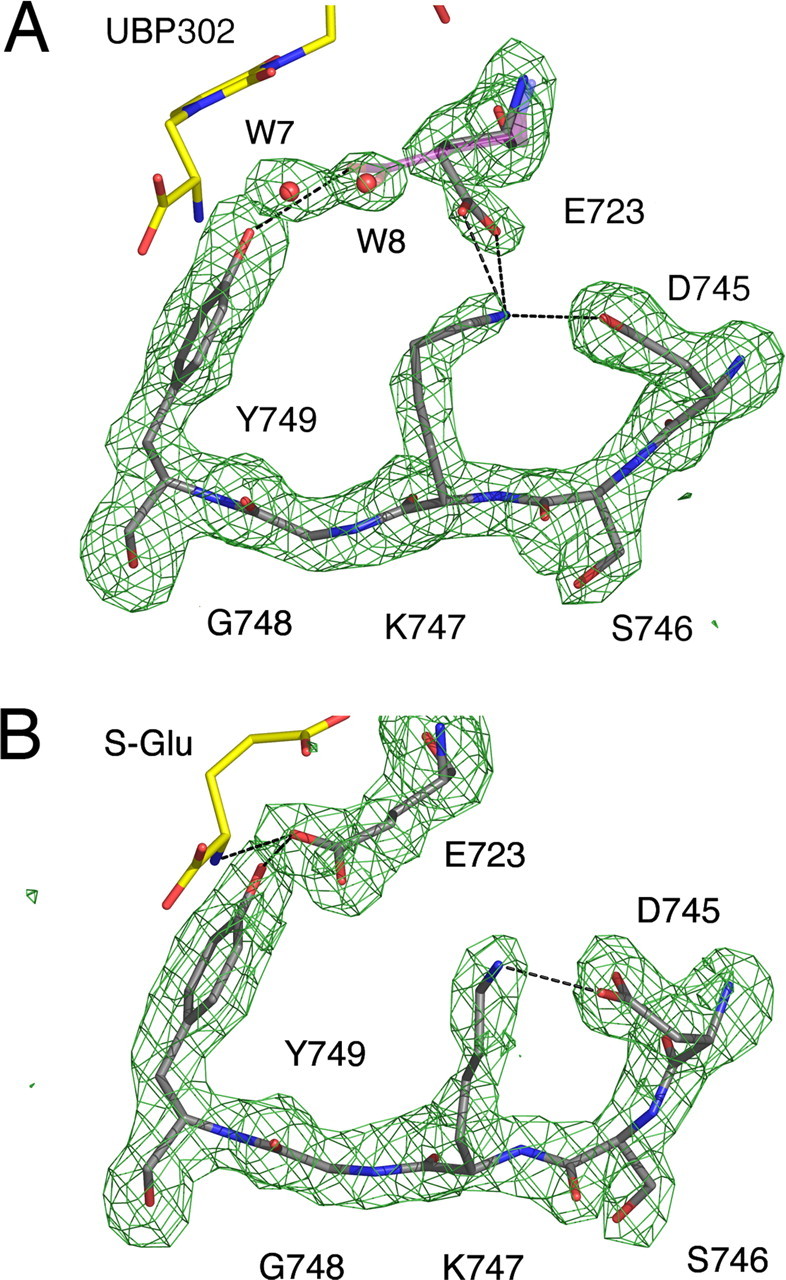
The Glu723 side chain undergoes a conformational switch. Fo-Fc omit electron density maps calculated using data to 1.87 and 2.11 Å for the GluR5 UBP302 (A) and the GluR5 glutamate (B) complexes contoured at 3.1 and 3.2 σ, respectively; atoms for Glu723, W7 and W8, and Asp745 through Tyr749 were omitted from the Fc calculation. Hydrogen bonds are shown by dashed lines. The UBP302 and glutamate ligands are drawn as yellow stick models. In the UBP302 structure, the Glu723 side chain was refined in two conformations: the first with 65% occupancy, makes a hydrogen bond with the side chain of Lys747, and the second, refined with 35% occupancy, is shown as a transparent stick figure and projects toward but does contact the ligand. In contrast, in the glutamate complex, the Glu723 side chain makes a hydrogen bond contact with the ligand α-amino group. Glu, Glutamate.
Discussion
Structural diversity of iGluR ligand binding core dimers
Although the numerous subtypes of glutamate receptor ion channels share a related structure, which for the ligand binding domain is similar to that of bacterial periplasmic binding proteins (Quiocho and Ledvina, 1996), high-resolution structural studies are beginning to reveal substantial diversity in their binding and gating mechanisms. The first structures of the AMPA receptor GluR2 ligand-binding core dimers have dominated our understanding of the molecular function of non-NMDA receptors (Armstrong and Gouaux, 2000; Sun et al., 2002; Jin et al., 2003). These studies suggested that the conformational change the ligand binding core undergoes during the transition from the apo- to agonist-bound conformation, which results in a 7–8 Å separation of domain 2, is substantially less than observed for periplasmic binding proteins. A key difference between iGluRs and the periplasmic proteins is the presence of the ion channel, which, because of its physical coupling to the ligand binding core, could reasonably be expected to restrict the range of motion available during the transition from resting to agonist-bound states. The results of the present study reveal that the GluR5 ligand-binding cores can separate by an additional 11–12 Å beyond that observed for AMPA receptors and raise the question of whether this is unique to kainate receptors, or a general feature of iGluR gating. Given the similar length and amino acid sequence of the linkers leading from the ligand binding core to the ion channel segments of AMPA and kainate receptors, the latter seems more likely and worth exploring in future experiments. The result will have profound implications for shaping our thinking about the gating mechanism of iGluRs, and the data presented here reveals that the coupling mechanism linking the ligand binding cores to the ion channel segments of kainate receptors must be capable of supporting much larger conformational changes than suggested by previous work on AMPA receptors.
A novel binding mechanism for UBP310
Our results also have implications for molecular modeling studies on iGluRs in general. Previous attempts to model the binding of UBP302 based on the GluR2 ATPO structure were hampered by the 14° difference in domain closure for the AMPA and kainate receptor antagonist structures and failed to correctly identify domain 2 contacts with the antagonist (More et al., 2004). Beyond the large differences in domain closure for individual iGluR antagonists, an unusual feature of the structures reported here is the surprising observation that Glu723, the conserved glutamate side chain responsible for binding the α amino group of glutamate, and its equivalent in heterocyclic amino acid derivatives, plays only a minor role in the binding of UBP310 and UBP302. This novel binding mechanism is a consequence of the hyperextension of the ligand binding core, which positions domain 2 and Glu723 too far away from the ligand to permit an energetically favorable contact. It is striking that the conformation adopted by Glu723 closely resembles that found in the GluR2 apo state, which to date gives our sole insight into the structure of a resting glutamate receptor. In contrast, for the GluR2 DNQX and ATPO structures, Glu705, the equivalent side chain, adopts the same conformation found in the agonist complexes. Relevant to this, recent studies on AMPA receptors reveal that DNQX and CNQX can weakly activate ion channel gating in the presence of the allosteric modulator CX614 (Li et al., 2005). Crystallographic and binding studies with CX614 reveal that the allosteric modulator binds at the base of the ligand binding core dimer and stabilizes dimer assembly without producing additional structural changes in agonist-bound complexes (Jin et al., 2005). This is consistent with the switch in conformation of the GluR2 Glu705 side from a resting to active conformation in the DNQX complex.
Chemistry of the ligand binding pocket
The structures of the UBP310 and UBP302 complexes reinforce the emerging picture that a key determinant of glutamate receptor subtype selectivity is generated by the surface of a pocket that lies immediately below the tip of helix F in domain 2. Both agonists and antagonists slot into this pocket, and amino acid substitutions in this region confer subtype selectivity in part by steric effects. The high affinity of UBP310 and UBP302 for GluR5 is likely to result in part from the close contact of the thiophene and phenyl groups with the side chain of Val670, which forms the cap of pocket. In contrast, the larger leucine side chain in AMPA receptors will hinder binding of these antagonists. Another feature of the GluR5 antagonist binding site that has precedent from previous work with AMPA receptors, albeit for different subsites in the ligand binding core, is the conformational mobility of methionine side chains. In GluR5, Met722 was refined in two conformations, only one of which makes contact with the ligand. In AMPA receptors, conformational mobility of the methionine residue at a different location, Met708, plays a key role in the binding of willardiine partial agonists (Jin et al., 2003). In the 5-iodo and 5-bromo willardiine complexes, Met708 rotates outward compared with the conformation found in the 5-fluoro complex, to accommodate the larger halogen atoms.
The water molecules observed in high-resolution crystal structures of the iGluR ligand binding cores play roles in receptor function and stability that are not well understood at present. In the glutamate complexes, water molecules act as surrogate ligand atoms, the binding of which is stabilized by the presence of agonists (Arinaminpathy et al., 2006). Displacement of some of these water molecules permits the binding of heterocyclic amino acids like AMPA, ATPA, and quisqualate (Armstrong and Gouaux, 2000; Jin et al., 2002; Mayer, 2005b). In contrast, in the GluR5 UBP310 and glutamate complexes, it is striking that the majority of water molecules in the ligand binding site are conserved, the exception being displacement of W1 to permit binding of the antagonist 5-methyl group. The antagonist crystal structures reveal strings of water molecules linking domains 1 and 2 in a complex hydrogen-bonded network, but the stability of these networks relative to that of bulk solvent and their role in stabilizing different conformational states of the receptor are not known. In this sense, the iGluR crystal structures remind us of how little we really understand about the complex chemistry of ligand binding. Recent nuclear magnetic resonance studies reinforce the point that glutamate receptors are not static structures and that the higher B-factors observed for domain 2 compared with domain 1 in crystallographic structures correspond to higher mobility for domain 2 in the ligand-bound complexes of receptors in solution (McFeeters and Oswald, 2002; Valentine and Palmer, 2005). However, at present, we have no experimental information about mobility at the level of individual side chains. The role of water molecules will be especially difficult to resolve, and, at present, the application of molecular dynamics simulations seems the most promising approach to address these issues (Mamonova et al., 2005; Arinaminpathy et al., 2006).
Footnotes
This work was supported by the intramural research program of the National Institute of Child Health and Human Development, National Institutes of Health, Department of Health and Human Services. We thank Eric Gouaux and Lothar Esser for advice and discussion; the Swartz laboratory, National Institute of Neurological Disorders and Stroke (NINDS), for providing oocytes; Carla Glasser for preparing plasmids and cRNA; and Neali Armstrong for critical review of this manuscript. Carla Glasser and Tatiana Molchanova performed ligand binding experiments. Min Zhang helped to prepare proteins and set up crystallization screens. Mass spectral analysis was performed by Howard Jaffe (Protein/Peptide Sequencing Facility, NINDS). [3H]2S,4R-4-methylglutamate was a generous gift from Tocris Cookson (Ellisville, MO). Synchrotron diffraction data were collected at Southeast Regional Collaborative Access Team (SER-CAT) 22-ID beamline at the Advanced Photon Source, Argonne National Laboratory. Supporting institutions may be found at www.ser-cat.org/members.html. Use of the Advanced Photon Source was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Contract W-31-109-Eng-38. Development of UBP302 and UBP310 was supported by the Biotechnology and Biological Sciences Research Council.
References
- Anslyn EV, Dougherty DA (2005). In: Modern physical organic chemistry Sausalito, CA: University Science Books.
- Arinaminpathy Y, Sansom MS, Biggin PC (2006). Binding site flexibility: molecular simulation of partial and full agonists with a glutamate receptor. Mol Pharmacol 69:5–12. [DOI] [PubMed] [Google Scholar]
- Armstrong N, Gouaux E (2000). Mechanisms for activation and antagonism of an AMPA-sensitive glutamate receptor: crystal structures of the GluR2 ligand binding core. Neuron 28:165–181. [DOI] [PubMed] [Google Scholar]
- Armstrong N, Mayer M, Gouaux E (2003). Tuning activation of the AMPA-sensitive GluR2 ion channel by genetic adjustment of agonist-induced conformational changes. Proc Natl Acad Sci USA 100:5736–5741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brauner-Osborne H, Egebjerg J, Nielsen EO, Madsen U, Krogsgaard-Larsen P (2000). Ligands for glutamate receptors: design and therapeutic prospects. J Med Chem 43:2609–2645. [DOI] [PubMed] [Google Scholar]
- Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL (1998). Crystallography and NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr D Biol Crystallogr 54:905–921. [DOI] [PubMed] [Google Scholar]
- Chen GQ, Cui C, Mayer ML, Gouaux E (1999). Functional characterization of a potassium-selective prokaryotic glutamate receptor. Nature 402:817–821. [DOI] [PubMed] [Google Scholar]
- Delano WL (2002). In: The PyMol molecular graphics system San Carlos, CA: DeLano Scientific.
- Dingledine R, Borges K, Bowie D, Traynelis SF (1999). The glutamate receptor ion channels. Pharmacol Rev 51:7–45. [PubMed] [Google Scholar]
- Dolman NP, Troop HM, More JC, Alt A, Knauss JL, Nistico R, Jack S, Morley RM, Bortolotto ZA, Roberts PJ, Bleakman D, Collingridge GL, Jane DE (2005). Synthesis and pharmacology of willardiine derivatives acting as antagonists of kainate receptors. J Med Chem 48:7867–7881. [DOI] [PubMed] [Google Scholar]
- Esnouf RM (1997). An extensively modified version of MolScript that includes greatly enhanced coloring capabilities. J Mol Graph Model 15:132–134112–133. [DOI] [PubMed] [Google Scholar]
- Foucaud B, Laube B, Schemm R, Kreimeyer A, Goeldner M, Betz H (2003). Structural model of the N-methyl-d-aspartate receptor glycine site probed by site-directed chemical coupling. J Biol Chem 278:24011–24017. [DOI] [PubMed] [Google Scholar]
- Furukawa H, Gouaux E (2003). Mechanisms of activation, inhibition and specificity: crystal structures of NR1 ligand-binding core. EMBO J 22:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furukawa H, Singh SK, Mancusso R, Gouaux E (2005). Subunit arrangement and function in NMDA receptors. Nature 438:185–192. [DOI] [PubMed] [Google Scholar]
- Hogner A, Greenwood JR, Liljefors T, Lunn ML, Egebjerg J, Larsen IK, Gouaux E, Kastrup JS (2003). Competitive antagonism of AMPA receptors by ligands of different classes: crystal structure of ATPO bound to the GluR2 ligand-binding core, in comparison with DNQX. J Med Chem 46:214–221. [DOI] [PubMed] [Google Scholar]
- Hollmann M (1999). Structure of ionotropic glutamate receptors. In: Ionotropic glutamate receptors in the CNS (Jonas P, Monyer H, eds) pp. 3–98. Berlin: Springer.
- Honore T, Davies SN, Drejer J, Fletcher EJ, Jacobsen P, Lodge D, Nielsen FE (1988). Quinoxalinediones: potent competitive non-NMDA glutamate receptor antagonists. Science 241:701–703. [DOI] [PubMed] [Google Scholar]
- Horning MS, Mayer ML (2004). Regulation of AMPA receptor gating by ligand binding core dimers. Neuron 41:379–388. [DOI] [PubMed] [Google Scholar]
- Inanobe A, Furukawa H, Gouaux E (2005). Mechanism of partial agonist action at the NR1 subunit of NMDA receptors. Neuron 47:71–84. [DOI] [PubMed] [Google Scholar]
- Jin R, Horning M, Mayer ML, Gouaux E (2002). Mechanism of activation and selectivity in a ligand-gated ion channel: structural and functional studies of GluR2 and quisqualate. Biochemistry 41:15635–15643. [DOI] [PubMed] [Google Scholar]
- Jin R, Banke TG, Mayer ML, Traynelis SF, Gouaux E (2003). Structural basis for partial agonist action at ionotropic glutamate receptors. Nat Neurosci 6:803–810. [DOI] [PubMed] [Google Scholar]
- Jin R, Clark S, Weeks AM, Dudman JT, Gouaux E, Partin KM (2005). Mechanism of positive allosteric modulators acting on AMPA receptors. J Neurosci 25:9027–9036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones TA, Kjeldgaard M (1997). Electron-density map interpretation. Methods Enzymol 277:173–208. [DOI] [PubMed] [Google Scholar]
- Kleywegt GJ, Zou JY, Kjeldgaard M, Jones TA (2001). Around O. In: International tables for crystallography, Vol F, Crystallography of biological macromolecules (Rossman MG, Arnold E, eds) pp. 353–356. Dordrecht: Kluwer Academic Publishers.
- Kraulis PJ (1991). MOLSCRIPT: a program to produce both detailed and schematic plots of protein structures. J Appl Crystallogr 24:946–950. [Google Scholar]
- Li Y, Nilsson L, Rogers GA (2005). Agonist activities of competitive AMPA receptor antagonists. Soc Neurosci Abstr 31:486.6. [Google Scholar]
- Mamonova T, Hespenheide B, Straub R, Thorpe MF, Kurnikova M (2005). Protein flexibility using constraints from molecular dynamics simulations. Phys Biol 2:S137–S147. [DOI] [PubMed] [Google Scholar]
- Mayer ML (2005a). Glutamate receptor ion channels. Curr Opin Neurobiol 15:282–288. [DOI] [PubMed] [Google Scholar]
- Mayer ML (2005b). Crystal structures of the GluR5 and GluR6 ligand binding cores: molecular mechanisms underlying kainate receptor selectivity. Neuron 45:539–552. [DOI] [PubMed] [Google Scholar]
- Mayer ML, Armstrong N (2004). Structure and function of glutamate receptor ion channels. Annu Rev Physiol 66:161–181. [DOI] [PubMed] [Google Scholar]
- Mayer ML, Olson R, Gouaux E (2001). Mechanisms for ligand binding to GluR0 ion channels: crystal structures of the glutamate and serine complexes and a closed apo state. J Mol Biol 311:815–836. [DOI] [PubMed] [Google Scholar]
- McCoy AJ, Grosse-Kunstleve RW, Storoni LC, Read RJ (2005). Likelihood-enhanced fast translation functions. Acta Crystallogr D Biol Crystallogr 61:458–464. [DOI] [PubMed] [Google Scholar]
- McFeeters RL, Oswald RE (2002). Structural mobility of the extracellular ligand-binding core of an ionotropic glutamate receptor. Analysis of NMR relaxation dynamics. Biochemistry 41:10472–10481. [DOI] [PubMed] [Google Scholar]
- Merritt EA, Bacon DJ (1997). Raster3D: photorealistic molecular graphics. Methods Enzymol 277:505–524. [DOI] [PubMed] [Google Scholar]
- More JC, Nistico R, Dolman NP, Clarke VR, Alt AJ, Ogden AM, Buelens FP, Troop HM, Kelland EE, Pilato F, Bleakman D, Bortolotto ZA, Collingridge GL, Jane DE (2004). Characterisation of UBP296: a novel, potent and selective kainate receptor antagonist. Neuropharmacology 47:46–64. [DOI] [PubMed] [Google Scholar]
- Moretti L, Pentikainen OT, Settimo L, Johnson MS (2004). Model structures of the N-methyl-d-aspartate receptor subunit NR1 explain the molecular recognition of agonist and antagonist ligands. J Struct Biol 145:205–215. [DOI] [PubMed] [Google Scholar]
- Mulle C, Sailer A, Swanson GT, Brana C, O’Gorman S, Bettler B, Heinemann SF (2000). Subunit composition of kainate receptors in hippocampal interneurons. Neuron 28:475–484. [DOI] [PubMed] [Google Scholar]
- Nanao MH, Green T, Stern-Bach Y, Heinemann SF, Choe S (2005). Structure of the kainate receptor subunit GluR6 agonist-binding domain complexed with domoic acid. Proc Natl Acad Sci USA 102:1708–1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naur P, Vestergaard B, Skov LK, Egebjerg J, Gajhede M, Kastrup JS (2005). Crystal structure of the kainate receptor GluR5 ligand-binding core in complex with (S)-glutamate. FEBS Lett 579:1154–1160. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z, Minor W (2001). Denzo and Scalepack. In: International tables for crystallography, Vol F, Crystallography of biological macromolecules (Rossman MG, Arnold E, eds) pp. 226–235. Dordrecht: Kluwer Academic Publishers.
- Painter J, Merritt EA (2005). A molecular viewer for the analysis of TLS rigid-body motion in macromolecules. Acta Crystallogr D Biol Crystallogr 61:465–471. [DOI] [PubMed] [Google Scholar]
- Pentikainen OT, Settimo L, Keinanen K, Johnson MS (2003). Selective agonist binding of (S)-2-amino-3-(3-hydroxy-5-methyl-4-isoxazolyl)propionic acid (AMPA) and 2S-(2alpha,3beta,4beta)-2-carboxy-4-(1-methylethenyl)-3-pyrrolidineacetic acid (kainate) receptors: a molecular modeling study. Biochem Pharmacol 66:2413–2425. [DOI] [PubMed] [Google Scholar]
- Quiocho FA, Ledvina PS (1996). Atomic structure and specificity of bacterial periplasmic receptors for active transport and chemotaxis: variation of common themes. Mol Microbiol 20:17–25. [DOI] [PubMed] [Google Scholar]
- Rosenmund C, Stern-Bach Y, Stevens CF (1998). The tetrameric structure of a glutamate receptor channel. Science 280:1596–1599. [DOI] [PubMed] [Google Scholar]
- Sun Y, Olson R, Horning M, Armstrong N, Mayer M, Gouaux E (2002). Mechanism of glutamate receptor desensitization. Nature 417:245–253. [DOI] [PubMed] [Google Scholar]
- Terwilliger TC, Berendzen J (1999). Automated MAD and MIR structure solution. Acta Crystallog sect D 55:849–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valentine ER, Palmer AGI (2005). Microsecond-to-millisecond conformational dynamics demarcate the GluR2 glutamate receptor bound to agonists glutamate, quisqualate, and AMPA. Biochemistry 44:3410–3417. [DOI] [PubMed] [Google Scholar]
- Winn MD, Isupov MN, Murshudov GN (2001). Use of TLS parameters to model anisotropic displacements in macromolecular refinement. Acta Crystallogr D Biol Crystallogr 57:122–133. [DOI] [PubMed] [Google Scholar]