

Abstract
Prostaglandin (PG) D2 is well known as a mediator of inflammation. Hematopoietic PGD synthase (HPGDS) is responsible for the production of PGD2 involved in inflammatory responses. Microglial activation and astrogliosis are commonly observed during neuroinflammation, including that which occurs during demyelination. Using the genetic demyelination mouse twitcher, a model of human Krabbe’s disease, we discovered that activated microglia expressed HPGDS and activated astrocytes expressed the DP1 receptor for PGD2 in the brain of these mice. Cultured microglia actively produced PGD2 by the action of HPGDS. Cultured astrocytes expressed two types of PGD2 receptor, DP1 and DP2, and showed enhanced GFAP production after stimulation of either receptor with its respective agonist. These results suggest that PGD2 plays an important role in microglia/astrocyte interaction. We demonstrated that the blockade of the HPGDS/PGD2/DP signaling pathway using HPGDS- or DP1-null twitcher mice, and twitcher mice treated with an HPGDS inhibitor, HQL-79 (4-benzhydryloxy-1-[3-(1H-tetrazol-5-yl)-propyl]piperidine), resulted in remarkable suppression of astrogliosis and demyelination, as well as a reduction in twitching and spasticity. Furthermore, we found that the degree of oligodendroglial apoptosis was also reduced in HPGDS-null and HQL-79-treated twitcher mice. These results suggest that PGD2 is the key neuroinflammatory molecule that heightens the pathological response to demyelination in twitcher mice.
Introduction
Prostaglandin (PG) D2 is a well known mediator of inflammation. It augments vascular permeability (Flower et al., 1976), chemotaxis (Hirai et al., 2001), and antigen presentation (Herve et al., 2003), inhibits platelet aggregation (Whittle et al., 1978), and induces vasodilatation and bronchoconstriction (Wasserman et al., 1980). Hematopoietic PGD synthase (HPGDS) is responsible for the production of PGD2, including that linked to immune and inflammatory responses (Kanaoka and Urade, 2003). HPGDS is expressed in human Th2 lymphocytes (Tanaka et al., 2000), antigen-presenting cells (Urade et al., 1989), mast cells (Urade et al., 1990), and microglia (Mohri et al., 2003).
Microglial activation and astrogliosis are commonly observed during the neuroinflammation associated with brain injury, infection, and neurodegenerative diseases. However, the molecular mechanism underlying microglial activation and astrogliosis is still poorly understood. The twitcher mouse (C57BL/6J-GALCtwi: GALCtwi/twi) is an authentic animal model of human globoid cell leukodystrophy (Krabbe’s disease) resulting from the deficiency of galactosylceramidase (Duchen et al., 1980; Kobayashi et al., 1980). In this model, initial myelination progresses normally (Nagara et al., 1982), but then demyelination begins as a result of the apoptosis of oligodendrocytes (OLs) after postnatal day 30 (P30) (Taniike et al., 1999). Demyelination in twitcher progresses in an orderly manner (Taniike and Suzuki, 1994) and is always accompanied by both microglial activation and astrogliosis throughout the brain (Ohno et al., 1993b). Interestingly, the pathological features of GALCtwi/twi have many characteristics in common with multiple sclerosis (MS). For example, in both conditions, major histocompatibility complex (MHC) molecules contribute to disease progression (Ohno et al., 1993a; Matsushima et al., 1994, Taniike et al., 1997). The absence of MHC class II molecules reduces CNS demyelination and microglial/macrophage infiltration in the twitcher brain (Matsushima et al., 1994). It has also been reported that there is expression of proinflammatory molecules, such as interleukin-6 (IL-6), tumor necrosis factor α (TNFα) (LeVine and Brown, 1997), monocyte chemoattractant protein-1, interferon-induced protein-10, macrophage inflammatory protein-1α (MIP-1α), MIP-1β, and RANTES (regulated on activation normal T-cell expressed and secreted) (Wu et al., 2000) in twitcher. Furthermore, we recently reported that TNFα expression increased with the progression of demyelination in twitcher and that a TNFα inhibitor suppresses such demyelination (Kagitani-Shimono et al., 2005). These correlations make the twitcher mice a model system for investigating the molecular events of neuroinflammation in demyelinating diseases.
In the present study, we found that HPGDS expression was progressively upregulated in activated microglia in the GALCtwi/twi brain. This upregulation was accompanied by the expression of the PGD2 receptor DP1 in hypertrophic astrocytes located close to HPGDS-positive (HPGDS+) microglia. Using glial cells in primary culture, we demonstrated that activated microglia produced a large amount of PGD2 synthesized by HPGDS and that astrocytes expressed both DP1 and DP2 receptors and were activated by PGD2. Using HPGDS- or DP1-null GALCtwi/twi mice and GALCtwi/twi mice treated with an HPGDS-inhibitor, we also found that blockade of the HPGDS/PGD2/DP1 signaling pathway resulted in the suppression of astrogliosis and apoptosis of OLs as well as in that of demyelination. This is the first example of a PGD2-mediated microglia/astrocyte interaction that enhances neuroinflammation and demyelination, and our findings may cast new light on strategies for the treatment of Krabbe’s disease.
Materials and Methods
Mice.
All animal experiments were performed in accordance with the Japanese Law for the Protection of Experimental Animals and conformed to the regulations issued by the National Institutes of Health and the Society for Neuroscience. GALCtwi/+ mice were originally purchased from The Jackson Laboratory (Bar Harbor, ME), and the mutation was maintained by interbreeding of known heterozygous mice. DNA was extracted from clipped tails for the detection of wild-type or mutant alleles of GALC, which was done with primers gtwf2 (5′-CAGTCATTCAGAAAGTCTTCC-3′) and gtwr3 (5′-GCCCACTGTCTTCAGGTGATA-3′; mismatch primer). The PCR fragment of 237 bp of the normal allele was not digested by EcoRV, whereas that of the mutant allele was, resulting in a 217 bp fragment.
HPGDS−/− and DP1−/− mice were generated by standard gene-targeting technology with mouse embryonic stem cells derived from the 129 strain and backcrossed to the inbred C57BL/6J strain 15 times. The former strain was created by the Osaka Bioscience Institute and Japan Tobacco Inc. Pharmaceutical Frontier Research Laboratories, and the latter was created at Kyoto University (Matsuoka et al., 2000). Neither strain exhibits any adverse neurological symptomatology. For genotyping of HPGDS, primer FW1 (5′-GAGTTGCTGCATCTGACCTTTC-3′) and RV1 (5′-TAGCGAATAATTTCGGCTCTTCC-3′) were used to detect the wild-type allele, and H-FW1 (AAGATCTGTCTTGTTGCGTACGCT-3′) and NeoR8 (5′-GTCCAGATCATCCTGATCGAC-5′) were used for that of the targeted allele. For the detection of wild-type or mutant alleles of DP1, primer DP1 (5′-TCGGTCTTTTATGTGCTCGTG-3′) or Neo1 (5′-CCCGTGATATTGCTGAAGAGC-3′) was used together with DP2 (5′-GAATCATCTGGATGAAACACC-3′).
Double-mutant mice (HPGDS−/−GALCtwi/twi or DP1−/−GALCtwi/twi) were generated by mating HPGDS−/− C57BL/6J (HPGDS−/−GALC+/+) or DP1−/− C57BL/6J (DP1−/−GALC+/+) offspring with HPGDS+/+DP1+/+GALCtwi/+. The F1 litters (HPGDS+/−GALCtwi/+ and DP1+/−GALCtwi/+) were mated with each other to produce HPGDS−/−GALCtwi/twi, DP1−/−GALCtwi/twi, and HPGDS+/+DP1+/+GALCtwi/twi. These three types of GALCtwi/twi male mice were used for analysis. The frequency of each genotype in all generations conformed to Mendelian inheritance patterns.
HQL-79 (4-benzhydryloxy-1-[3-(1H-tetrazol-5-yl)-propyl]piperi-dine) (Aritake et al., 2006), an inhibitor of HPGDS, was purchased from Cayman Chemical (Ann Arbor, MI). GALCtwi/twi received a daily subcutaneous injection of HQL-79 (suspended in 0.5% methylcellulose; 50 mg · kg−1 · d−1) from P25 to P45. As a control, the same volume of vehicle was injected into GALCtwi/twi during the same period.
Materials.
Rabbit polyclonal (0.1 μg/ml) and rat monoclonal (0.2 μg/ml) anti-mouse HPGDS antibodies were raised and purified at Osaka Bioscience Institute (Mohri et al., 2003). The preabsorbed antibody was prepared by incubation of the polyclonal antibody with an excess amount of purified mouse HPGDS recombinantly expressed in Escherichia coli (Kanaoka et al., 2000). Guinea pig anti-DP1 antiserum (1:30) was also raised at Osaka Bioscience Institute (Mizoguchi et al., 2001). The other antibodies used were as follow: rabbit polyclonal antibodies against π-class glutathione S-transferase (π-GST) (1:1000; Biotrin International, Dublin, Ireland), NG2 (1:200; Chemicon, Temecula, CA), myelin basic protein (MBP) (1:5000; DakoCytomation, Glostrup, Denmark), cow glial fibrillary acidic protein (GFAP) (1:5000; DakoCytomation), and rat monoclonal anti-mouse F4/80 antibody (1:500; Serotec, Oxford, UK). Biotinylated Ricinus communis-agglutinin-1 (RCA-1) was purchased from Vector Laboratories (Burlingame, CA) (50 μg/ml).
Measurement of HPGDS activity and PGD2 content in the brain.
HPGDS activity in the brains of GALC+/+ and GALCtwi/twi mice at P39 (n = 4 each) was determined by using 40 μm [1-14C]PGH2 in the presence of 1 mm glutathione, as described previously (Urade et al., 1985). Protein concentrations were determined with bicinchoninic acid reagent (Pierce, Rockford, IL), using bovine serum albumin as a standard as per the protocol of the manufacturer. Quantities of PGD2, PGE2, and PGF2α in fresh-frozen brains of GALC+/+ and GALCtwi/twi mice decapitated at P39 (n = 6 each) were determined by enzyme immunoassays as described previously (Ram et al., 1997).
Northern blotting and quantitative PCR.
Total RNA was extracted from mouse cerebrum and cerebellum by the guanidinium thiocyanate-phenol-chloroform method using ISOGEN (Nippon Gene, Tokyo, Japan). Total RNA (10 μg) was electrophoresed in an agarose gel, transferred to Zeta Probe nylon membranes (Bio-Rad, Hercules, CA), and hybridized with 32P-labeled cDNA probes specific for mouse GFAP and glyceraldehyde-3-phosphate dehydrogenase (G3PDH). The blots were visualized by autoradiography with Eastman Kodak (Rochester, NY) XAR-5 film and an intensifying screen. The relative amount of each transcript was estimated by quantifying the associated radioactivity with a BAS-2000 (Fuji Film, Tokyo, Japan).
Quantitative PCR analysis of the amounts of mRNAs for HPGDS, DP1, DP2, peroxisome proliferator activated receptor γ (PPARγ), GFAP, and G3PDH was performed by using a LightCycler amplification and detection system (Roche Diagnostics, Indianapolis, IN) as described below. Each sample of total RNA (2 μg) was reverse transcribed at 50°C for 30 min into single-stranded cDNA in a reaction mixture containing 20 U of RNase inhibitor, 1× RNA PCR buffer, 1 mm dNTP mixture, 2.5 μm random primer, and 5 U of avian myeloblastosis virus reverse transcriptase (Takara Biomedicals, Kyoto, Japan). The sequence-specific primers used were as follow: HPGDS forward primer, 5′-GAATAGAACAAGCTGACTGGC-3′; HPGDS reverse primer, 5′-AGCCAAATCTGTGTTTTTGG-3′; DP1 forward primer, 5′-TTTGGGAAGTTCGTGCAGTACT-3′; DP1 reverse primer, 5′-GCCATGAGGCTGGAGTAGA-3′; DP2 forward primer, 5′-TGGCCTTCTTCAACAGCGT-3′; DP2 reverse primer, 5′-ACGCAGTTGGGGAATTCG-3′; PPARγ forward primer, 5′-GGAGATCTCCAGTGATATCGACCA-3′; PPARγ reverse primer, 5′-ACGGCTTCTACGGATCGAAACT-3′; GFAP forward primer, 5′-TTCCTGGAACAGCAAAACAAGGCGCT-3′; GFAP reverse primer, 5′-CTGTCTATACGCAGCCAGGTTGTTCTC-3′; inducible nitric oxide synthase (iNOS) forward primer, 5′-CAGCAATATAGGCTCATCCAGAG-3′; iNOS reverse primer, 5′-GTGGTGTAGGACAATCCACAACT-5′; G3PDH forward primer, 5′-TGAACGGGAAGCTCACTGG-3′; and G3PDH reverse primer, 5′-TCCACCACCCTGTTGCT-3′. The constructs used to create a standard curve were made by cloning each amplified fragment into the HindIII site of a pGEM vector (Promega, Madison, WI). The number of copies was calculated by plotting a dilution series on this standard curve in each PCR experiment. The PCR mixture contained Taq polymerase, X LightCycle DNA master SYBR Green (Roche Diagnostics) reaction buffer, 3 mm MgCl2, and 12.5 pmol of each primer. After PCR had been performed for 40 cycles of denaturation (95°C for 1 s), annealing (57°C for 5 s), and enzymatic chain extension (72°C for 10 s), the products for HPGDS, DP1, DP2, PPARγ, GFAP, and G3PDH were detected at 87, 83, 83, 91, 87, and 84°C, respectively. All PCR products were visualized under UV light after electrophoresis in an agarose gel containing ethidium bromide and were subsequently sequenced to verify that only the specific polymerization from the intended mRNA had occurred.
Western blotting analysis.
The brains were homogenized in 3 vol by weight of PBS. After centrifugation at 16,000 × g for 20 min, the supernatant and the precipitate were used in Western blotting for HPGDS and MBP, respectively. Protein was electrophoresed in 15/25% SDS-polyacrylamide gradient gel (Daiichi, Tokyo, Japan), and, subsequently, the separated proteins were electrophoretically transferred to an Immobilon polyvinylidene difluoride membrane (Millipore, Bedford, MA). The membrane was reacted with the rabbit polyclonal anti-HPGDS antibody (0.5 μg/ml) or anti-β-actin polyclonal antibody (1:1000; Abcam, Cambridge, UK) at 4°C overnight. After alkaline phosphatase-conjugated anti-rabbit IgG (1:3000) had been applied, HPGDS and MBP were detected by using a 5-bromo-4-chlor-indolyl-phosphate/nitroblue-tetrazolium-chloride developing kit (Amersham Bioscience, Uppsala, Sweden).
Immunocytochemistry.
GALCtwi/twi and GALC+/+ mice at P20, P30, P40, and P45 were used for immunocytochemical analysis. Under deep ether anesthesia, the mice were perfused via the heart with physiological saline, followed by 4% paraformaldehyde in 0.1 m sodium phosphate, pH 7.4, for 10 min. The brains were removed and postfixed in the same fixative overnight. After a coronal slice had been taken, they were routinely embedded in paraffin. Additionally, two each of the GALCtwi/twi and GALC+/+ mice at P45 were perfused with physiological saline only and processed for the preparation of fresh-frozen sections. Both paraffin and frozen sections (5 μm thick) were mounted on 3-aminopropyl-triethoxysilane-coated slides.
HPGDS immunostaining was performed as described previously (Mohri et al., 2003). Rabbit anti-mouse HPGDS antibody was applied followed by biotinylated anti-rabbit IgG and avidin-biotin complex (ABC) from an ABC Elite kit (Vector Laboratories). Immunoreactivity was visualized by the addition of diaminobenzidine hydrochloride (DAB) (Dotite, Kumamoto, Japan).
The procedures for double labeling were followed as described previously (Mohri et al., 2003). Briefly, in the case of double immunocytochemistry for HPGDS and F4/80, after the primary antibodies had been applied, the sections were sequentially incubated with Texas Red-conjugated anti-rat IgG antibody (20 μg/ml; ICN, Aurora, OH) and biotinylated anti-rabbit IgG antibody (20 μg/ml; Vector Laboratories), followed by FITC-conjugated avidin D (20 μg/ml; Vector Laboratories). In the case of double labeling for HPGDS and biotinylated RCA-1, after incubation with the primary antibody or lectin, the sections were sequentially incubated with the Texas Red-conjugated anti-rabbit IgG antibody (ICN), followed by FITC-conjugated avidin D. The fluorescence was examined with an Axiovert 100M microscope connected to a Zeiss (Oberkochen, Germany) laser-scanning microscope. The frozen sections, optimal for immunostaining of DP1, were fixed with 100% ethanol at −20°C for 30 min and then with acetone at room temperature for 5 min. After the sections had been incubated for 5 min with 0.1 m sodium phosphate, pH 7.4, containing 0.005% saponin (Nakarai Tesque, Kyoto, Japan) and 20 μm (4-amidino-phenyl)-methane-sulfonyl fluoride (Wako, Osaka, Japan), they were incubated with guinea pig anti-mouse DP1 antibody. They were then serially reacted with biotinylated anti-guinea pig antibody (dilution 1:400; Southern Biotechnology, Birmingham, AL) and ABC. The reaction using DAB as the chromogen was enhanced by adding 5 mg/ml nickel (II) ammonium sulfate hexahydrate (Nakarai Tesque) to the reaction solution. The double immunostaining for DP1 and F4/80 was performed on unfixed frozen sections as explained in the paragraph describing the double immunostaining of paraffin sections.
Terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end-labeling (TUNEL) histochemistry combined with immunocytochemistry for π-GST was performed as described previously (Taniike et al., 1999). π-GST-positive TUNEL-positive cells were counted in the cerebral parenchyma of paraffin sections prepared from mice at P45 (n = 4 each).
Primary cultures of mouse microglia and astrocytes.
We prepared primary cultures of microglia and astrocytes from wild-type mouse brains at P1 and P7, respectively. Cerebral cortices were dissected, and the leptomeninges were completely removed. The tissues were minced, suspended in PBS containing 0.05% trypsin (Invitrogen, Grand Island, NY) and 0.01% DNase I (Sigma, St. Louis, MO), and then incubated for 10 min at 37°C. After incubation and centrifugation, the cell pellets were washed three times with PBS. The cells were then filtered through a 75 μm nylon mesh, centrifuged, suspended in DMEM (Nakalai Tesque) containing 10% fetal bovine serum (FBS) (JRH Bioscience, Lenexa, KS), 100 IU/ml penicillin, and 100 μg/ml streptomycin (Invitrogen), and transferred to culture dishes. For microglial cultures, the medium was changed to DMEM containing 10% FBS after a 24 h culture period. This medium was exchanged for fresh medium twice weekly thereafter. The supernatant including microglial cells was collected and subcultured at 1 × 105 cells per well (six-well plate). After incubation in DMEM without FBS for 6 h, the microglia were treated with A23187 (5 μm; Sigma) for 30 min. When the microglial cells were treated with HQL-79, the inhibitor was added 15 min before A23187 treatment. The medium after treatment of the cells with these compounds was measured for its content of PGs.
For astrocyte cultures, the medium was changed to DMEM containing 20% FBS after the initial 24 h culture period. Then, 2 d later, the medium was changed back to that containing 10% FBS. Once the astrocytes in the primary culture reached confluence, the cells were rinsed with PBS, suspended in trypsin-containing PBS, and subcultured at 5 × 105 cells per well (six-well plate). After a 24 h incubation in DMEM containing 1% FBS, the astrocytes were incubated with BW245C (0.1–100 nm; Cayman Chemical) or 13, 14-dihydro-15-keto-PGD2 (DK-PGD2) (0.1–100 nm; Cayman Chemical) for 6 h at 37°C. After the cells had been washed with PBS, RNA was extracted from them and subjected to quantitative reverse transcription (RT)-PCR. We confirmed these cells to be either microglia or astrocytes by their positive immunoreaction with anti-RCA-1 and F4/80 or GFAP, respectively.
Statistical analysis.
Values were expressed as the mean ± SE. Data were analyzed by using the two-tailed t test; values of p < 0.05 were considered to be significant.
Results
HPGDS expression is increased with progression of demyelination in GALCtwi/twi
First, we measured the levels of PGD2, PGE2, and PGF2α in the brain of 39-d-old GALCtwi/twi mice (Fig. 1A), in which demyelination was in progress. The PGD2 content was increased 10-fold in the GALCtwi/twi brain (35.0 ± 9.0 in GALC+/+ vs 390.0 ± 32.0 pg/brain in GALCtwi/twi, mean ± SE; n = 6), whereas the other PGs did not show such a drastic increase. That is, the PGE2 content remained the same (20.0 ± 6.0 vs 19.0 ± 12.0), and that of PGF2α increased only twofold (71.0 ± 13.0 vs 160.0 ± 35.0). At P39, the HPGDS enzymatic activity was threefold higher in the GALCtwi/twi cerebrum (0.62 ± 0.05 vs 2.10 ± 0.07 nmol · min−1 · mg−1 protein; n = 4) and fivefold higher in the GALCtwi/twi cerebellum (0.32 ± 0.10 vs 2.30 ± 0.28; n = 4) (Fig. 1B). Western blot analysis also revealed that the level of HPGDS protein in the cerebrum and cerebellum of the GALCtwi/twi brain at P40 was 2- and 10-fold higher, respectively, than that in the same brain regions of the GALC+/+ mice (Fig. 1C).
Figure 1.
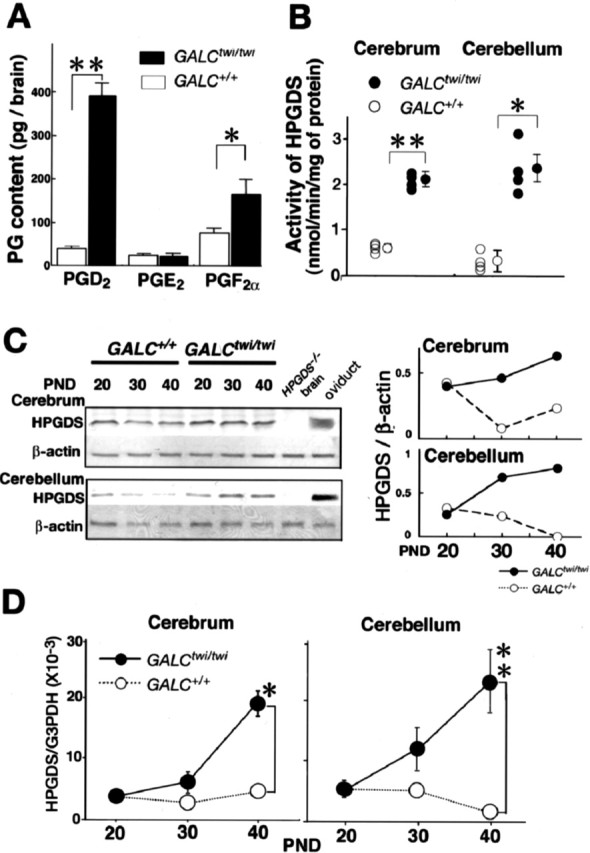
Increase in PGD2 in GALCtwi/twi brains together with upregulation of HPGDS. A, Quantities of PGD2, PGE2, and PGF2α in fresh-frozen brains of decapitated GALC+/+ (white columns) and GALCtwi/twi (black columns) mice at P39 were determined by enzyme immunoassays. n = 6. *p < 0.05, **p < 0.01. B, HPGDS activity in the cerebrum and cerebellum of GALC+/+ (white circles) and GALCtwi/twi (black circles) mice at P39 was determined by using 40 μm [1-14C]PGH2 in the presence of 1 mm glutathione. n = 4. *p < 0.05, **p < 0.01. C, Western blot analysis of HPGDS and β-actin in the brain from GALC+/+ and GALCtwi/twi. The top and bottom lanes contained the homogenate (5 μg of protein per lane) from the cerebrum and the cerebellum, respectively. The homogenates of the brain of HPGDS−/− mice and of the oviduct of the wild-type mice were used as the negative and positive control, respectively, of the HPGDS detection. The graphs on the right show the profiles of HPGDS/β-actin amount by morphometrical analysis of Western blots of GALC+/+ and GALCtwi/twi at P20, P30, and P40. D, Quantitative PCR analysis of the contents of mRNAs for HPGDS and G3PDH in GALC+/+ and GALCtwi/twi brains was performed by using a LightCycler amplification and detection system. n = 4. Data are mean ± SE. *p < 0.05, **p < 0.01. PND, Postnatal day.
The level of HPGDS mRNA in the GALC+/+ brains remained constant in the cerebrum and gradually decreased in the cerebellum, whereas it gradually increased in GALCtwi/twi brains after P30. By P40, the mRNA level in GALCtwi/twi was 4.5-fold higher in the cerebrum and 16.5-fold higher in the cerebellum than in the corresponding GALC+/+ brain regions (Fig. 1D).
Activated microglia/macrophages in GALCtwi/twi strongly express HPGDS
Immunocytochemical analysis at P40 showed that HPGDS was localized in resting (ramified) microglia in the GALC+/+ cerebellum (Fig. 2A, arrows and inset). In the GALCtwi/twi, the level of HPGDS protein was remarkably increased in activated microglia (Fig. 2B), which possessed irregular thick processes in the region of demyelination. Confocal double immunostaining revealed that ∼100% of the HPGDS+ cells were positive for RCA-1 (Fig. 2C), a marker for microglia and endothelial cells (Szumanska et al., 1987). RCA+ endothelial cells were negative for HPGDS (Fig. 2C, arrowhead). All cells positive for F4/80, a marker for activated microglia (Austyn and Gordon, 1981), were also positive for HPGDS (Fig. 2D, arrows). HPGDS+ cells were positive for neither GFAP, a marker for astrocytes, π-GST, a marker for OLs, nor NG2, a marker for OL precursor (data not shown).
Figure 2.
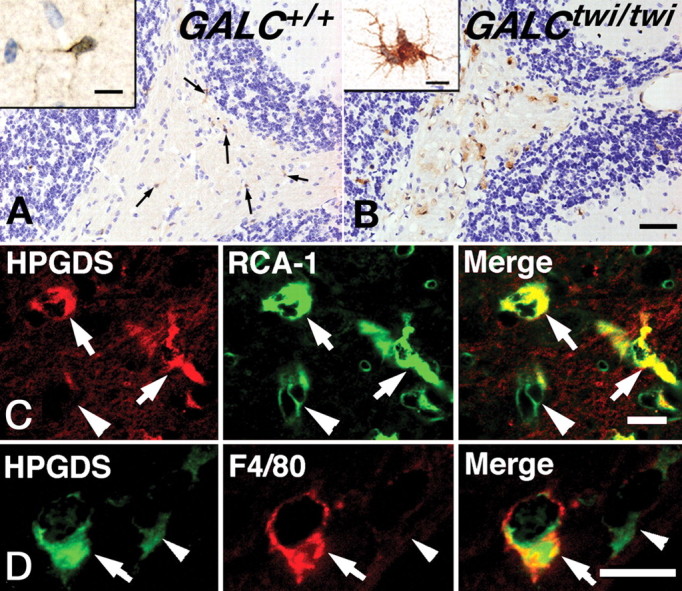
Upregulation of HPGDS in microglia of GALCtwi/twi brains at P45. A, B, HPGDS immunostaining visualized by using DAB in the cerebellum of GALC+/+ (A) and GALCtwi/twi (B). The Inset and arrows in A indicate resting HPGDS+ microglia. Scale bars: B, 50 μm; insets, 10 μm. C, D, Confocal images for HPGDS (red in C and green in D) and microglial markers, i.e., RCA-1 (green in C) and F4/80 (red in D). Arrows in C and D point to HPGDS+ microglia, and arrowheads in C point to RCA+ HPGDS− endothelial cells, judged from the morphology. Arrowheads in D indicate HPGDS-positive/F4/80-negative resting microglia. Scale bars: C, D, 10 μm.
DP1 receptor is induced in hypertrophic astrocytes of GALCtwi/twi
We next quantified the mRNAs of two types of membrane-bound receptors for PGD2 (i.e., DP1 and DP2 receptors), as well as the mRNA of PPARγ, a nuclear receptor for PGD2 metabolites (Forman et al., 1995; Kliewer et al., 1995). The DP1 mRNA level remained constant in the cerebrum and the cerebellum of the GALC+/+ mice from P20 through P40 but increased twofold in the cerebrum of the GALCtwi/twi brains and fourfold in their cerebellum at P40 (Fig. 3A). The expression profile of DP1 mRNA in the cerebrum and cerebellum in the GALCtwi/twi brain during development was thus similar to that of HPGDS (Fig. 1C,D). The contents of DP2 and PPARγ mRNAs in the brain were ∼1/10 and 1/100, respectively, of the content of DP1 mRNA and did not differ significantly between GALC+/+ and GALCtwi/twi mice at any of the ages examined (Fig. 3A).
Figure 3.
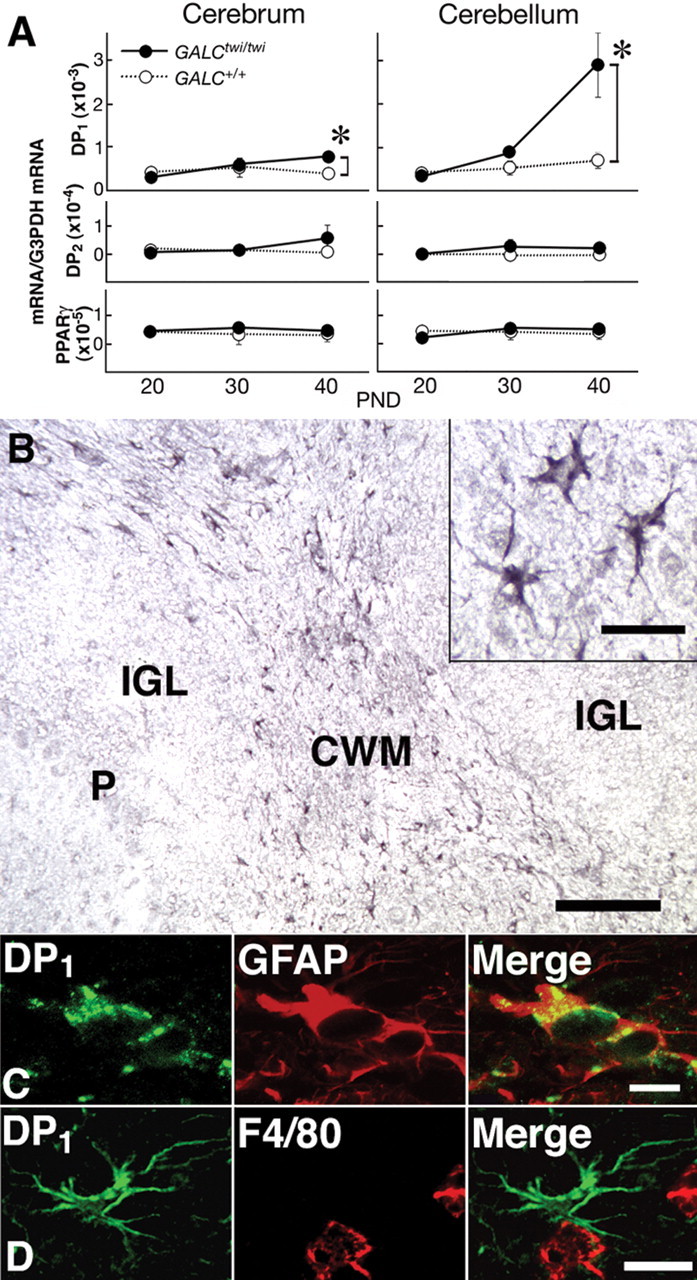
Induction of DP1 in GALCtwi/twi hypertrophic astrocytes. A, Quantitative PCR analysis of the contents of mRNAs for DP1, DP2, PPARγ, and G3PDH mRNA in GALC+/+ (white circles) and GALCtwi/twi (black circles) brains was performed by using a LightCycler amplification and detection system. n = 4. *p < 0.05. PND, Postnatal day. B, DP1 immunostaining in the GALCtwi/twi cerebellum at P45. DP1+ cells are recognized in the CWM but not in the internal granular layer (IGL) or Purkinje cell layer (P). Scale bar, 100 μm. Inset, High-power view of DP1+ cells. Scale bar, 10 μm. C, Double immunostaining for DP1 (green) and GFAP (red) in hypertrophied astrocytes. Scale bar, 5 μm. D, DP1+ astrocytes (green) and F4/80+ microglia (red) are intermingled in the CWM, although DP1 and F4/80 are not detectable in the same cells. Scale bar, 10 μm.
Previous studies have revealed that, in the normal mouse, DP1 receptor immunoreactivity was not detected in the brain except in the arachnoid trabecular cells of the basal forebrain (Mizoguchi et al., 2001). In GALCtwi/twi mice, ∼100% of DP1-positive cells in brain parenchyma were GFAP-positive astrocytes, and numerous DP1+ cells were recognized in the HPGDS-rich areas such as the corpus callosum and cerebellar white matter (CWM) (Fig. 3B) at P40–P45. The regional distribution profile was almost the same as that for HPGDS+ microglia (Fig. 2B). These DP1 receptor-positive cells were identified as hypertrophied astrocytes because they possessed large soma with thick-branched processes (Fig. 3B, inset) and expressed GFAP as judged by double immunostaining (Fig. 3C). DP1 receptor-positive cells were not identical to, but were intermingled with, F4/80+ microglia in the CWM (Fig. 3D), thus suggesting that DP1 receptor-positive astrocytes were located close to HPGDS+ activated microglia. These results suggest that activated microglia interact with hypertrophied astrocytes via PGD2/DP1 signaling in GALCtwi/twi mice.
Cultured microglia produce PGD2 by HPGDS action and cultured astrocytes upregulate GFAP expression when stimulated by DP
Using primary cultures of microglia, we investigated whether these cells produced PGD2 by HPGDS catalysis. As shown in Figure 4A, after treatment with 5 μm A23187, a Ca2+ ionophore, PGD2 release was increased ∼23-fold compared with that of controls, whereas PGE2 release was increased only 3.8-fold, and that of PGF2α remained unchanged. PGD2 production was suppressed dose dependently by treatment with HQL-79, a specific inhibitor of HPGDS (Matsushita et al., 1998).
Figure 4.
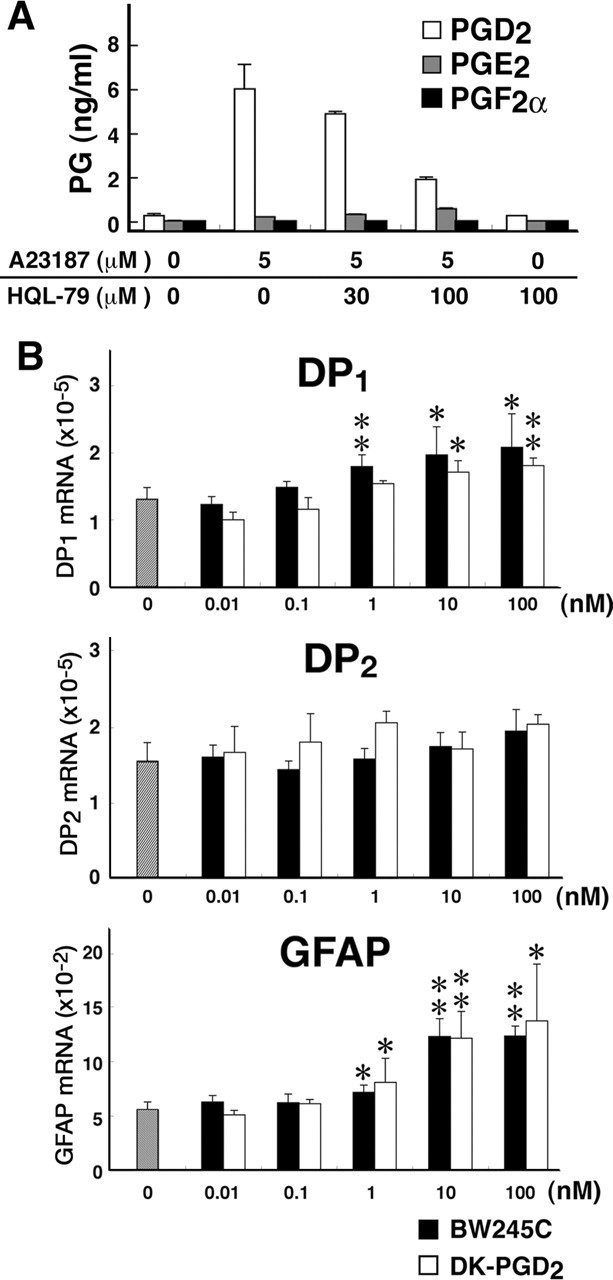
A, PG production by primary cultures of microglia 30 min after treatment with A23187 in the absence or presence of HQL-79. After incubation in DMEM without FBS for 6 h, the microglia were treated with 5 mm A23187 for 30 min. HQL-79 was added 15 min before A23187 treatment. PG contents in the medium were quantified by enzyme immunoassays. White columns, PGD2; gray columns, PGE2; black columns, PGF2α. n = 3. B, mRNA levels of DP1, DP2, and GFAP before (gray column) and after stimulation of primary cultures of astrocytes with BW245C (black columns) or DK-PGD2 (white columns). Once the astrocytes in the primary culture reached confluence, the cells were rinsed with PBS and subcultured at 5 × 105 cells per well (6-well plate). After a 24 h incubation in DMEM containing 1% FBS, the astrocytes were incubated with BW245C (0.1–100 nm) or DK-PGD2 (0.1–100 nm) for 6 h at 37°C. n = 3. *p < 0.05, **p < 0.01.
To investigate the effect of PGD2 on astrocyte activation, we determined the contents of DP1 and DP2 receptor mRNAs in cultured mouse astrocytes before and after stimulation with selective agonists for DP1 [BW245C (Sharif et al., 2000)] and DP2 [DK-PGD2 (Gervais et al., 2001)] (Fig. 4B). Astrocytes in primary culture contained mRNAs of both DP1 and DP2 receptors in almost equal amounts. The DP1 receptor mRNA content was increased after stimulation with either agonist in a dose-dependent manner from 1 to 100 nm and reached a level ∼1.5-fold higher than that without stimulation, whereas the DP2 receptor mRNA content remained almost unchanged after stimulation with either agonist. The GFAP mRNA content was also increased in a dose-dependent manner from 1 to 100 nm after stimulation with either agonist, becoming twofold higher than that in the untreated control cultures.
Inhibition of HPGDS/PGD2/DP1 receptor signaling alleviates astrogliosis of GALCtwi/twi
To assess the contribution of HPGDS and the DP1 receptor to astrogliosis in GALCtwi/twi mice, we generated HPGDS−/−GALCtwi/twi (supplemental Fig. 1A, available at www.jneurosci.org as supplemental material) and DP1−/−GALCtwi/twi (supplemental Fig. 1B, available at www.jneurosci.org as supplemental material) mice and then compared the mRNA content and the immunocytochemical profile of GFAP in the brain of these double-mutant mice at P45 with those of GALCtwi/twi mice. The content of psychosine, a potent neurotoxin and an accumulated substrate of the missing enzyme galactosylceramidase in GALCtwi/twi, was not significantly different among GALCtwi/twi, HPGDS−/−GALCtwi/twi, and DP1−/−GALCtwi/twi in the cerebrum (8.86 ± 0.45, 8.65 ± 2.47, and 9.07 ± 3.56 ng/mg protein, respectively; n = 4) or cerebellum (25.12 ± 2.21, 29.59 ± 4.94, and 35.78 ± 8.98 ng/mg protein, respectively; n = 4). Therefore, we consider the gliotic factors, with respect to the primary metabolic defect, to be uniform in all strains.
GFAP immunostaining at P45 revealed that only weakly GFAP+ astrocytes with thin and delicate processes (Fig. 5A, inset) were present in both the cerebrum and cerebellum (Fig. 5A,E) of GALC+/+ mice. In GALCtwi/twi mice, intensely GFAP+ astrocytes with enlarged soma and many thick processes (Fig. 5B, inset) were numerous and diffusely distributed in both cerebrum and cerebellum (Fig. 5B,F). In HPGDS−/−GALCtwi/twi (Fig. 5C,G) and DP1−/−GALCtwi/twi (Fig. 5D,H) mice, the GFAP immunoreactivity was slightly more intense than that in GALC+/+ mice but remarkably weaker than that in GALCtwi/twi mice, and the processes of GFAP+ astrocytes were clearly thinner than those in the GALCtwi/twi mice. The suppression of astrogliosis was more remarkable in HPGDS−/−GALCtwi/twi than in DP1−/−GALCtwi/twi mice. Quantitative RT-PCR and Northern blot analyses revealed that the GFAP mRNA content in the cerebellum at P40 was decreased to 15 and 43% of that of GALCtwi/twi mice in HPGD−/−GALCtwi/twi and DP1−/− GALCtwi/twi mice, respectively (Fig. 5I).
Figure 5.
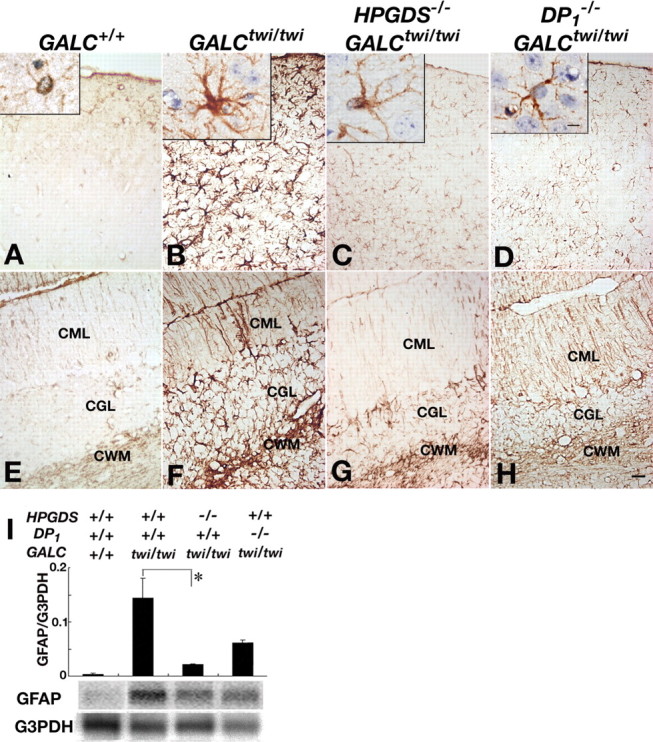
Alleviated astrogliosis in the mutant twitcher lacking HPGDS or DP1 gene. A–H, GFAP immunostaining in the cerebrum (A–D) and the cerebellum (E–H) of GALC+/+ (A, E), GALCtwi/twi (B, F), HPGDS−/−GALCtwi/twi (C, G), and DP1−/−GALCtwi/twi (D, H) mice. Insets show higher-magnification view of GFAP-immunoreactive astrocytes for each genotype. Scale bars: H, 50 μm; insets, 5 μm. CGL, Cerebellar granular layer; CML, cerebellar molecular layer. I, Quantitative RT-PCR (top) and Northern blot analyses (bottom) for GFAP and G3PDH in GALC+/+, GALCtwi/twi, HPGDS−/−GALCtwi/twi, and DP1−/−GALCtwi/twi. The quantitative PCR analysis of the contents of mRNAs for GFAP and G3PDH was performed by using a LightCycler amplification and detection system. For Northern blotting, total RNA (10 μg) was electrophoresed in an agarose gel, transferred to nylon membranes, and hybridized with 32P-labeled cDNA probes specific for mouse GFAP and G3PDH. n = 3–5. *p < 0.05.
Furthermore, when we treated GALCtwi/twi mice with HQL-79, the astrogliosis of GALCtwi/twi mice (Fig. 6A,B) was markedly suppressed in both the cerebrum (Fig. 6D) and cerebellum (Fig. 6E). DP1 receptor-positive astrocytes in the GALCtwi/twi mice (Fig. 6C) were also decreased in number after the HQL-79 treatment (Fig. 6F). Quantitative RT-PCR and Northern blot analyses (Fig. 6G) revealed that the GFAP mRNA content in the GALCtwi/twi cerebellum at P45 was decreased to 29% of the control by the inhibitor treatment.
Figure 6.

HPGDS-inhibitor alleviated astrogliosis via inhibition of DP1 upregulation in GALCtwi/twi. GALCtwi/twi received a daily subcutaneous injection of 50 mg · kg−1 · d−1 HQL-79 from P25 to P45. As a control, the same volume of vehicle was injected into GALCtwi/twi during the same period. A–C, Vehicle-treated GALCtwi/twi. D–F, HQL-79-treated GALCtwi/twi. A, B, D, E, GFAP immunostaining. C, F, DP1 immunostaining. Scale bar, 50 μm. G, Quantitative RT-PCR (top) and Northern blot analyses (bottom) for vehicle-treated and HQL-79-treated GALCtwi/twi. n = 3. *p < 0.05. CGL, Cerebellar granular layer; CML, cerebellar molecular layer.
Inhibition of HPGDS/PGD2/DP1 receptor signaling suppresses apoptosis of OLs and demyelination of GALCtwi/twi
MBP immunostaining revealed that demyelination was severe in the GALCtwi/twi brain (Fig. 7A), yet myelin was well conserved in the brains of HPGDS−/−GALCtwi/twi, DP1−/−GALCtwi/twi, and HQL-79-treated GALCtwi/twi (Fig. 7B–D). Western blotting analysis for MBP of these mice confirmed the immunocytochemical observation (Fig. 7E,F). As shown in Figure 7F, relative amounts of MBP were significantly retained in the brains of HPGDS−/−GALCtwi/twi, DP1−/−GALCtwi/twi (n = 5), and HQL-79-treated GALCtwi/twi (n = 7) mice. Especially MBPs of 18.5 and 14 kDa, which are expressed in the early phase of myelination, were better preserved in HPGDS−/−GALCtwi/twi, DP1−/−GALCtwi/twi and HQL-79-treated GALCtwi/twi than those of 21.5 and 17 kDa MBPs, expressed in the later phase. As shown in Figure 7G, the number of π-GST-positive TUNEL-positive cells, which indicate apoptotic OLs, was significantly decreased in HPGDS−/−GALCtwi/twi and HQL-79-treated GALCtwi/twi (Fig. 7G). To determine whether proinflammatory molecule levels were changed in those mice, we measured the mRNAs for several proinflammatory molecules, such as iNOS, IL-1β, IL-6, and TNFα. The level of iNOS, but not that of IL-1β, IL-6, or TNFα, was significantly reduced in HPGDS−/−GALCtwi/twi and HQL-79-treated GALCtwi/twi animals (n = 4) (Fig. 7H).
Figure 7.
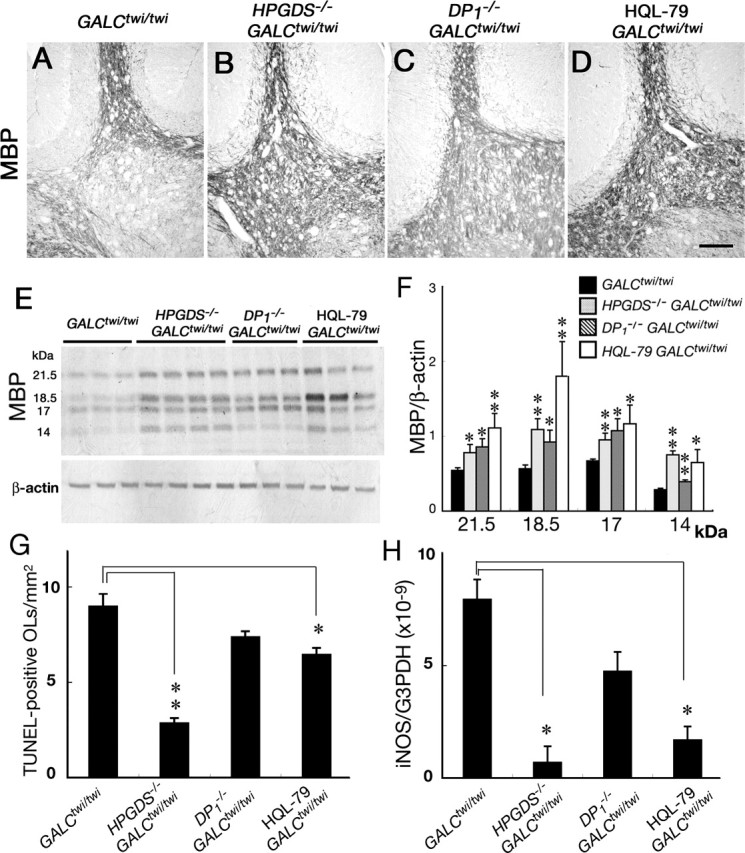
Demyelination suppressed via inhibition of neuroinflammation in GALCtwi/twi. A–D, MBP immunostaining. Cerebellum of mice at P45. Scale bar, 100 μm. E, Western blot analysis of MBP (top) and β-actin (bottom). The top and bottom lanes contained the homogenate (2 μg of protein per lane) from the cerebrum. F, Morphometrical analysis of MBP protein content in the brain of GALCtwi/twi (black columns; n = 4), HPGDS−/−GALCtwi/twi (gray columns; n = 5), DP1−/−GALCtwi/twi (hatched columns; n = 5), and HQL-79-treated GALCtwi/twi (white columns; n = 7). *p < 0.05, **p < 0.01. G, TUNEL histochemistry combined with immunocytochemistry for π-GST was performed, and π-GST-positive TUNEL-positive cells were counted in the basal ganglia of the brains of paraffin sections prepared from mice at P45 (n = 4 each). *p < 0.05, **p < 0.01. H, Quantitative analysis of iNOS mRNA in the cerebellum at P45. n = 4. *p < 0.05.
These results indicate that the genetic or pharmacological blockade of the HPGDS/PGD2/DP1 signaling pathway not only suppressed the hypertrophic change in astrocytes or astrogliosis in GALCtwi/twi mice but also suppressed iNOS expression and subsequent enhancement of demyelination.
Inhibition of HPGDS/PGD2/DP1 receptor signaling diminishes symptomatology of GALCtwi/twi
The lifespan of HPGDS−/−GALCtwi/twi and DP1−/−GALCtwi/twi mice (47.3 ± 4.5 and 46.2 ± 0.5 d, respectively) was slightly longer than that of GALCtwi/twi mice (45.5 ± 0.4 d) despite the fact that the severe peripheral neuropathy, which primarily determines the lifespan of GALCtwi/twi mice, was observed across all three groups of mice. The lifespan of HQL-79-treated GALCtwi/twi mice was significantly longer than that of the vehicle-treated mice (47.8 ± 1.3 and 44.0 ± 0.7 d, respectively; n = 5; p < 0.03).
Spasticity and ataxic symptoms of HPGDS−/−GALCtwi/twi and DP1−/−GALCtwi/twi mice at P45 were remarkably milder than those of GALCtwi/twi mice. GALCtwi/twi mice barely staggered with remarkable intentional tremor, whereas HPGDS−/−GALCtwi/twi mice moved about noticeably faster at P45. When suspended by the tail, HPGDS−/−GALCtwi/twi animals retained the righting reflex, exhibiting a milder tremor (supplemental movie A, available at www.jneurosci.org as supplemental material). Clinical symptoms including spasticity and ataxic symptoms were also milder in HQL-79-treated GALCtwi/twi than in vehicle-treated GALCtwi/twi mice. HQL-79-treated GALCtwi/twi moved around smoothly at P45 and retained the righting reflex with milder tremor when suspended by the tail (supplemental movie B, available at www.jneurosci.org as supplemental material).
The main cause of the death of GALCtwi/twi mice is peripheral and cranial nerve palsy, which leads to difficulty in eating and limits the lifespan. MBP immunostaining (Fig. 7A–D) and electron microscopy (data not shown) revealed that demyelination was attenuated in the CNS, but not in the sciatic nerve, of HPGDS−/−GALCtwi/twi, DP1−/−GALCtwi/twi, and HQL-79-treated GALCtwi/twi mice. A similar discrepancy between CNS and PNS has been reported previously in the case of GALCtwi/twi mice receiving bone-marrow transplantation (Toyoshima et al., 1986; Ichioka et al., 1987). Furthermore, Escolar et al. (2005) recently reported that transplantation of umbilical-cord blood into babies with infantile Krabbe’s disease resulted in improvement in the CNS but not in the PNS.
Using immunocytochemistry, we found that DP1 receptor expression was upregulated in the CNS but not detected in the sciatic nerve of GALCtwi/twi (data not shown), suggesting that PGD2 may not be involved in progression of the pathology of the PNS of GALCtwi/twi. This coincides with a distinction between the CNS and PNS pathologies in twitcher in which Schwann cells, unlike OLs, proliferate in response to demyelination. Furthermore, apoptotic death does not appear to occur in the peripheral nerves (Komiyama and Suzuki, 1992). The demyelination mechanism, therefore, appears to be distinct between CNS and PNS, particularly regarding the contribution of support cells as well as PGD2 to the demyelination.
Discussion
PGD2-mediated enhancement of microglial activation and astrogliosis during neuroinflammation
Microglia are believed to act primarily as a line of defense for the brain, because they express MHC molecules, secrete proinflammatory cytokines (Woodroofe et al., 1991), and produce NO (Wood et al., 1994) and PGs (Gebicke-Haerter et al., 1989; Minghetti and Levi, 1995; Bauer et al., 1997). Cyclooxygenases (COXs) are the key enzymes for the production of PGs and are upregulated in the CNS in various neurodegenerative diseases, including Alzheimer’s disease (Yermakova et al., 1999), Parkinson’s disease (Teismann et al., 2003), prion disease (Walsh et al., 2000; Deininger et al., 2003), spinal cord injury (Schwab et al., 2000), and ischemia (Govoni et al., 2001). Several investigators have reported selective COX inhibitors to be neuroprotective (McGeer and McGeer, 1995; Stewart et al., 1997; Pasinetti, 1998; Lim et al., 2000; Govoni et al., 2001), suggesting that some classes of PGs may play important roles in neurodegeneration. Positive immunoreactivities for COX-1 and COX-2 are mostly observed in microglia (Pasinetti and Aisen, 1998; Yermakova et al., 1999; Walsh et al., 2000). Several reports have demonstrated that microglia produce PGD2 and PGE2 in vitro (Gebicke-Haerter et al., 1989; Minghetti and Levi, 1995; Bauer et al., 1997). Between these two prostanoids, PGE2 has been proposed to act as a mediator in inflammatory processes (Petrova et al., 1999; Kim et al., 2001) of neurodegenerative diseases such as amyotrophic lateral sclerosis (Ilzecka, 2003), whereas the direct mechanism of PGD2 involvement in neurodegenerative diseases has not been well studied.
The pathological phenotypes within the CNS of HPGDS- or DP1-null twitcher mice are quite different from those seen in their peripheral immune responses. We reported previously that DP1−/− mice used in the ovalbumin-induced asthma model exhibited a remarkably decreased accumulation of lymphocytes and proinflammatory cytokines in their lungs compared with wild-type mice (Matsuoka et al., 2000). In contrast, HPGDS−/− mice used in this same model did not show any changes in the lymphocyte and cytokine components in their lungs (K. Aritake and Y. Urade, unpublished results). Together, these results suggest that the above functional peripheral defect of DP1−/− mice may not be simply caused by the deficiency of DP1 signaling but may be attributable to the shunting of PGD2 signaling to the DP2 pathway. Based on these results, we believe that the mild gliosis and demyelination in the HPGDS- and DP1- null twitcher mice are mainly caused by the suppression of neuroinflammation around the HPGDS-expressing microglia and DP1/DP2-possessing astrocytes, although it is possible that decreased cell infiltration into the CNS in these mice leads to the suppression of secondary gliosis.
We demonstrated here that PGD2 was actively produced in the GALCtwi/twi brain (Fig. 1A) by HPGDS upregulated in activated microglia (Figs. 2, 4A). In contrast, the level of PGE2 was unchanged in the GALCtwi/twi brain when compared with that in the GALC+/+ brain (Fig. 1A), and microsomal PGE synthase was not induced in the GALCtwi/twi brain, as judged from the results of quantitative RT-PCR and immunocytochemistry (data not shown). Furthermore, only a small amount of PGE2 release was observed in cultured microglia after activation (Fig. 4A). Therefore, PGD2 is thought to play more important roles than PGE2 in the processes of neuroinflammation that induce microglia activation and astrogliosis.
In addition, we demonstrated that astrocytes had DP receptors and that PGD2 stimulation accelerated astrogliosis (Figs. 3, 4B). We also showed that inhibition of HPGDS/PGD2/DP1 receptor signaling by genetic depletion of HPGDS or DP1, or administration of HQL-79, reduced astrogliosis and ameliorated the clinical symptoms of GALCtwi/twi. As shown in Figure 4B, cultured astrocytes expressed both DP1 and DP2 receptors and increased GFAP production when stimulated by either a DP1 or DP2 agonist. This may explain the finding that the astrogliosis of HPGDS−/−GALCtwi/twi mice was milder than that of DP1−/−GALCtwi/twi mice, in which PGD2-induced gliosis progressed in a DP2 receptor-mediated manner. Conversely, in HPGDS−/−GALCtwi/twi mice, PGD2 was not produced in the neuroinflammatory loci, signal transduction through both DP1 and DP2 receptors was blocked, and astrocytic activation was minimal.
In this study, we unexpectedly found that blockade of the HPGDS/PGD2 signaling resulted in suppressed apoptosis of OLs (Fig. 7G). This suppression may have contributed to the amelioration of demyelination in HPGDS−/−GALCtwi/twi and HQL-79-treated GALCtwi/twi mice. However, DP1−/−GALCtwi/twi mice exhibited milder symptoms and their demyelination was clearly suppressed, as judged from the results of the immunohistochemical and Western blot analyses (Fig. 7C,E,F) compared with the GALCtwi/twi mice, despite the fact that the number of TUNEL-positive cells was not significantly decreased (Fig. 7G). Therefore, secondary demyelination caused by neuroinflammation might also have been suppressed in the DP1−/−GALCtwi/twi mice, leading to decreased spasticity and improved ataxic symptoms.
Predicted mechanism of the HPGDS/PGD2/DP1 signaling pathway leading to enhanced astrogliosis and demyelination
Based on the findings described in this present study, we hypothesize the molecular events leading to neuroinflammation in the GALCtwi/twi brains to be the following (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). First, myelin and/or OL debris produced by the apoptotic death of OLs in the GALCtwi/twi brain (Taniike et al., 1999) activates microglia to synthesize PGD2 through the action of HPGDS. The PGD2 produced by these activated microglia then stimulates both DP1 and DP2 receptors on neighboring astrocytes in a paracrine manner and upregulates the expression of DP1, but not that of DP2, receptors on these astrocytes. The activation of DP1, a Gs-coupled receptor (Hirata et al., 1994), increases the intracellular cAMP level in the astrocytes to induce hypertrophy of these cells. DP2 stimulation also reportedly elevates the intracellular Ca2+ level. Engagement of DP2, a Gαi-coupled receptor (Hirai et al., 2001), also increases GFAP expression, presumably through elevation of the intracellular Ca2+ level.
We reported previously that OLs express lipocalin-type PGD synthase (L-PGDS) but not HPGDS (Taniike et al., 2002). Although L-PGDS and HPGDS catalyze the same reaction to produce PGD2, these two enzymes have quite different roles within the context of demyelination. In L-PGDS−/−GALCtwi/twi, both OL apoptosis and gliosis were enhanced (Taniike et al., 2002), unlike the case of HPGDS−/−GALCtwi/twi, in which gliosis was ameliorated. Therefore, we hypothesize that L-PGDS localized in OLs plays a protective role in twitcher mice and that HPGDS, upregulated in activated microglia, is involved in the secondary pathology in this disease model.
The instigation of the pathological condition in twitcher is attributable to the accumulation of psychosine, yet it has been reported that inflammatory cells and many cytokines are also involved in the progression of the disease (Wu et al., 2000). Although the toxicity of psychosine plays a significant role in the apoptotic death of OLs and in preliminary demyelination, the ensuing pathology is intensified by cytokines and chemokines released by activated microglia and possibly by astrocytes as well. In this sense, the demyelinating process closely resembles that of pattern III of MS, in which the lesions contain an inflammatory cell infiltration, activated microglia, and OL apoptosis but without any deposition of Ig and complement (Lucchinetti et al., 2000). Considering this, along with reports that MHC class II-null twitcher mice show only mild pathology (Matsushima et al., 1994) and that inhibiting the proinflammatory cytokine TNFα suppresses demyelination and apoptosis of OLs in GALCtwi/twi (Kagitani-Shimono et al., 2005), we believe that secondary neuroinflammation is a critical component in the progression of the degenerative condition found in twitcher. Furthermore, because HPGDS inhibition in GALCtwi/twi significantly suppressed the disease process, including apoptosis of OLs, it is reasonable to surmise that neuroinflammation in twitcher mice exacerbates demyelination and functional deficits.
Germane to our contention is identifying the physiological role of astrogliosis within the disease model. Although still a subject not yet clarified despite intense discussion, we speculate that hypertrophic astrocytes may be related to neuroinflammation through their production of cytokines. This study, then, represents the first example of a PGD2-mediated microglia/astrocyte interaction in neuroinflammation. Here, PGD2 was shown to be an important mediator of neuroinflammation, and it was demonstrated that the HPGDS inhibitor HQL-79, an orally active compound remarkably effective at attenuating neuroinflammation-associated astrogliosis, was an able attenuating substance. Our present results encourage additional study into both the role of PGD2 in inflammatory and demyelinating diseases as well as the use of HPGDS inhibitors as novel strategies for anti-neuroinflammatory therapy.
Footnotes
*I.M. and M.T. contributed equally to this work.
This work was supported by the following: Ministry of Education, Culture, Sports, Science, and Technology of Japan Grants 09670806 (M.T.), 13557016 (N.E.), and 12558078 (Y.U.); the Japan Science and Technology Corporation (N.E., Y.U.); the Suntory Institute for Bioorganic Research (N.E.); the Osaka Medical Research Foundation for Incurable Diseases (M.T.); the Takeda Science Foundation (N.E., Y.U.); the Yamanouchi Foundation for Research on Metabolic Disorders (N.E.); the Mitsubishi Foundation (Y.U.); National Institutes of Health–United States Public Health Service Grants NS-24453 and HD-03110 (K.S.); and Osaka City. We thank Sigeko Matsumoto for conducting the immunocytochemistry and Yumiko Hoshikawa and Masumi Sakata for technical assistance. We also thank Dr. Osamu Hayaishi for general support of this study and Dr. Aya Jakobovits (Abgenix, Fremont, CA) for the donation of ES cells (E14-1).
References
- Aritake K, Kado Y, Inoue T, Miyano M, Urade Y (2006). Structural and functional characterization of HQL-79, an orally active, selective inhibitor for human hematopoietic prostaglandin D synthase. J Biol Chem in press. [DOI] [PubMed]
- Austyn JM, Gordon S (1981). F4/80, a monoclonal antibody directed specifically against the mouse macrophage. Eur J Immunol 11:805–815. [DOI] [PubMed] [Google Scholar]
- Bauer MK, Lieb K, Schulze-Osthoff K, Berger M, Gebicke-Haerter PJ, Bauer J, Fiebich BL (1997). Expression and regulation of cyclooxygenase-2 in rat microglia. Eur J Biochem 243:726–731. [DOI] [PubMed] [Google Scholar]
- Deininger MH, Bekure-Nemariam K, Trautmann K, Morgalla M, Meyermann R, Schluesener HJ (2003). Cyclooxygenase-1 and -2 in brains of patients who died with sporadic Creutzfeldt-Jakob disease. J Mol Neurosci 20:25–30. [DOI] [PubMed] [Google Scholar]
- Duchen LW, Eicher EM, Jacobs JM, Scaravilli F, Teixeira F (1980). Hereditary leucodystrophy in the mouse: the new mutant twitcher. Brain 103:695–710. [DOI] [PubMed] [Google Scholar]
- Escolar ML, Poe MD, Provenzale JM, Richards KC, Allison J, Wood S, Wenger DA, Pietryga D, Wall D, Champagne M, Morse R, Krivit W, Kurtzberg J (2005). Transplantation of umbilical-cord blood in babies with infantile Krabbe’s disease. N Engl J Med 352:2069–2081. [DOI] [PubMed] [Google Scholar]
- Flower RJ, Harvey EA, Kingston WP (1976). Inflammatory effects of prostaglandin D2 in rat and human skin. Br J Pharmacol 56:229–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forman BM, Tontonoz P, Chen J, Brun RP, Spiegelman BM, Evans RM (1995). 15-Deoxy-delta 12, 14-prostaglandin J2 is a ligand for the adipocyte determination factor PPAR gamma. Cell 83:803–812. [DOI] [PubMed] [Google Scholar]
- Gebicke-Haerter PJ, Bauer J, Schobert A, Northoff H (1989). Lipopolysaccharide-free conditions in primary astrocyte cultures allow growth and isolation of microglial cells. J Neurosci 9:183–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gervais FG, Cruz RP, Chateauneuf A, Gale S, Sawyer N, Nantel F, Metters KM, O’Neill GP (2001). Selective modulation of chemokinesis, degranulation, and apoptosis in eosinophils through the PGD2 receptors CRTH2 and DP. J Allergy Clin Immunol 108:982–988. [DOI] [PubMed] [Google Scholar]
- Govoni S, Masoero E, Favalli L, Rozza A, Scelsi R, Viappiani S, Buccellati C, Sala A, Folco G (2001). The cycloxygenase-2 inhibitor SC58236 is neuroprotective in an in vivo model of focal ischemia in the rat. Neurosci Lett 303:91–94. [DOI] [PubMed] [Google Scholar]
- Herve M, Angeli V, Pinzar E, Wintjens R, Faveeuw C, Narumiya S, Capron A, Urade Y, Capron M, Riveau G, Trottein F (2003). Pivotal roles of the parasite PGD2 synthase and of the host D prostanoid receptor 1 in schistosome immune evasion. Eur J Immunol 33:2764–2772. [DOI] [PubMed] [Google Scholar]
- Hirai H, Tanaka K, Yoshie O, Ogawa K, Kenmotsu K, Takamori Y, Ichimasa M, Sugamura K, Nakamura M, Takano S, Nagata K (2001). Prostaglandin D2 selectively induces chemotaxis in T helper type 2 cells, eosinophils, and basophils via seven-transmembrane receptor CRTH2. J Exp Med 193:255–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirata M, Kakizuka A, Aizawa M, Ushikubi F, Narumiya S (1994). Molecular characterization of a mouse prostaglandin D receptor and functional expression of the cloned gene. Proc Natl Acad Sci USA 91:11192–11196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichioka T, Kishimoto Y, Brennan S, Santos GW, Yeager AM (1987). Hematopoietic cell transplantation in murine globoid cell leukodystrophy (the twitcher mouse): effects on levels of galactosylceramidase, psychosine, galactocerebrosides. Proc Natl Acad Sci USA 84:4259–4263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilzecka J (2003). Prostaglandin E2 is increased in amyotrophic lateral sclerosis patients. Acta Neurol Scand 108:125–129. [DOI] [PubMed] [Google Scholar]
- Kagitani-Shimono K, Mohri I, Fujitani Y, Suzuki K, Ozono K, Urade Y, Taniike M (2005). Anti-inflammatory therapy by ibudilast, a phosphodiesterase inhibitor, in demyelination of twitcher, a genetic demyelination model. J Neuroinflamm 2:10–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanaoka Y, Urade Y (2003). Hematopoietic prostaglandin D synthase. Prostaglandins Leukot Essent Fatty Acids 69:163–167. [DOI] [PubMed] [Google Scholar]
- Kanaoka Y, Fujimori K, Kikuno R, Sakaguchi Y, Urade Y, Hayaishi O (2000). Structure and chromosomal localization of human and mouse genes for hematopoietic prostaglandin D synthase. Conservation of the ancestral genomic structure of sigma-class glutathione S-transferase. Eur J Biochem 267:3315–3322. [DOI] [PubMed] [Google Scholar]
- Kim EJ, Lee JE, Kwon KJ, Lee SH, Moon CH, Baik EJ (2001). Differential roles of cyclooxygenase isoforms after kainic acid-induced prostaglandin E(2) production and neurodegeneration in cortical and hippocampal cell cultures. Brain Res 908:1–9. [DOI] [PubMed] [Google Scholar]
- Kliewer SA, Lenhard JM, Willson TM, Patel I, Morris DC, Lehmann JM (1995). A prostaglandin J2 metabolite binds peroxisome proliferator-activated receptor gamma and promotes adipocyte differentiation. Cell 83:813–819. [DOI] [PubMed] [Google Scholar]
- Kobayashi T, Yamanaka T, Jacobs JM, Teixeira F, Suzuki K (1980). The Twitcher mouse: an enzymatically authentic model of human globoid cell leukodystrophy (Krabbe disease). Brain Res 202:479–483. [DOI] [PubMed] [Google Scholar]
- Komiyama A, Suzuki K (1992). Progressive impairment of Schwann cell proliferation in vitro in murine globoid cell leukodystrophy (twitcher). Brain Res 598:1–9. [DOI] [PubMed] [Google Scholar]
- LeVine SM, Brown DC (1997). IL-6 and TNFα expression in brains of twitcher, quaking and normal mice. J Neuroimmunol 73:47–56. [DOI] [PubMed] [Google Scholar]
- Lim GP, Yang F, Chu T, Chen P, Beech W, Teter B, Tran T, Ubeda O, Ashe KH, Frautschy SA, Cole GM (2000). Ibuprofen suppresses plaque pathology and inflammation in a mouse model for Alzheimer’s disease. J Neurosci 20:5709–5714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucchinetti C, Bruck W, Parisi J, Scheithauer B, Rodriguez M, Lassmann H (2000). Heterogeneity of multiple sclerosis lesions: implications for the pathogenesis of demyelination. Ann Neurol 47:707–717. [DOI] [PubMed] [Google Scholar]
- Matsuoka T, Hirata M, Tanaka H, Takahashi Y, Murata T, Kabashima K, Sugimoto Y, Kobayashi T, Ushikubi F, Aze Y, Eguchi N, Urade Y, Yoshida N, Kimura K, Mizoguchi A, Honda Y, Nagai H, Narumiya S (2000). Prostaglandin D2 as a mediator of allergic asthma. Science 287:2013–2017. [DOI] [PubMed] [Google Scholar]
- Matsushima GK, Taniike M, Glimcher LH, Grusby MJ, Frelinger JA, Suzuki K, Ting JP (1994). Absence of MHC class II molecules reduces CNS demyelination, microglial/macrophage infiltration, and twitching in murine globoid cell leukodystrophy. Cell 78:645–656. [DOI] [PubMed] [Google Scholar]
- Matsushita N, Aritake K, Takada A, Hizue M, Hayashi K, Mitsui K, Hayashi M, Hirotsu I, Kimura Y, Tani T, Nakajima H (1998). Pharmacological studies on the novel antiallergic drug HQL-79. II. Elucidation of mechanisms for antiallergic and antiasthmatic effects. Jpn J Pharmacol 78:11–22. [DOI] [PubMed] [Google Scholar]
- McGeer PL, McGeer EG (1995). The inflammatory response system of brain: implications for therapy of Alzheimer and other neurodegenerative diseases. Brain Res Brain Res Rev 21:195–218. [DOI] [PubMed] [Google Scholar]
- Minghetti L, Levi G (1995). Induction of prostanoid biosynthesis by bacterial lipopolysaccharide and isoproterenol in rat microglial cultures. J Neurochem 65:2690–2698. [DOI] [PubMed] [Google Scholar]
- Mizoguchi A, Eguchi N, Kimura K, Kiyohara Y, Qu WM, Huang ZL, Mochizuki T, Lazarus M, Kobayashi T, Kaneko T, Narumiya S, Urade Y, Hayaishi O (2001). Dominant localization of prostaglandin D receptors on arachnoid trabecular cells in mouse basal forebrain and their involvement in the regulation of non-rapid eye movement sleep. Proc Natl Acad Sci USA 98:11674–11679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohri I, Eguchi N, Suzuki K, Urade Y, Taniike M (2003). Hematopoietic prostaglandin D synthase is expressed in microglia in the developing postnatal mouse brain. Glia 42:263–274. [DOI] [PubMed] [Google Scholar]
- Nagara H, Kobayashi T, Suzuki K, Suzuki K (1982). The twitcher mouse: normal pattern of early myelination in the spinal cord. Brain Res 244:289–294. [DOI] [PubMed] [Google Scholar]
- Ohno M, Komiyama A, Martin PM, Suzuki K (1993a). MHC class II antigen expression and T-cell infiltration in the demyelinating CNS and PNS of the twitcher mouse. Brain Res 602:186–196. [DOI] [PubMed] [Google Scholar]
- Ohno M, Komiyama A, Martin PM, Suzuki K (1993b). Proliferation of microglia/macrophages in the demyelinating CNS and PNS of twitcher mouse. Brain Res 602:268–274. [DOI] [PubMed] [Google Scholar]
- Pasinetti GM (1998). Cyclooxygenase and inflammation in Alzheimer’s disease: experimental approaches and clinical interventions. J Neurosci Res 54:1–6. [DOI] [PubMed] [Google Scholar]
- Pasinetti GM, Aisen PS (1998). Cyclooxygenase-2 expression is increased in frontal cortex of Alzheimer’s disease brain. Neuroscience 87:319–324. [DOI] [PubMed] [Google Scholar]
- Petrova TV, Akama KT, Van Eldik LJ (1999). Selective modulation of BV-2 microglial activation by prostaglandin E(2). Differential effects on endotoxin-stimulated cytokine induction. J Biol Chem 274:28823–28827. [DOI] [PubMed] [Google Scholar]
- Ram A, Pandey HP, Matsumura H, Kasahara-Orita K, Nakajima T, Takahata R, Satoh S, Terao A, Hayaishi O (1997). CSF levels of prostaglandins, especially the level of prostaglandin D2, are correlated with increasing propensity towards sleep in rats. Brain Res 751:81–89. [DOI] [PubMed] [Google Scholar]
- Schwab JM, Brechtel K, Nguyen TD, Schluesener HJ (2000). Persistent accumulation of cyclooxygenase-1 (COX-1) expressing microglia/macrophages and upregulation by endothelium following spinal cord injury. J Neuroimmunol 111:122–130. [DOI] [PubMed] [Google Scholar]
- Sharif NA, Crider JY, Xu SX, Williams GW (2000). Affinities, selectivities, potencies, and intrinsic activities of natural and synthetic prostanoids using endogenous receptors: focus on DP class prostanoids. J Pharmacol Exp Ther 293:321–328. [PubMed] [Google Scholar]
- Stewart WF, Kawas C, Corrada M, Metter EJ (1997). Risk of Alzheimer’s disease and duration of NSAID use. Neurology 48:626–632. [DOI] [PubMed] [Google Scholar]
- Szumanska G, Vorbrodt AW, Mandybur TI, Wisniewski HM (1987). Lectin histochemistry of plaques and tangles in Alzheimer’s disease. Acta Neuropathol (Berl) 73:1–11. [DOI] [PubMed] [Google Scholar]
- Tanaka K, Ogawa K, Sugamura K, Nakamura M, Takano S, Nagata K (2000). Cutting edge: differential production of prostaglandin D2 by human helper T cell subsets. J Immunol 164:2277–2280. [DOI] [PubMed] [Google Scholar]
- Taniike M, Suzuki K (1994). Spacio-temporal progression of demyelination in twitcher mouse: with clinico-pathological correlation. Acta Neuropathol (Berl) 88:228–236. [DOI] [PubMed] [Google Scholar]
- Taniike M, Marcus JR, Popko B, Suzuki K (1997). Expression of major histocompatibility complex class I antigens in the demyelinating twitcher CNS and PNS. J Neurosci Res 47:539–546. [PubMed] [Google Scholar]
- Taniike M, Marcus JR, Nishigaki T, Fujita N, Popko B, Suzuki K, Suzuki K (1998). Suppressed UDP-galactose: ceramide galactosyltransferase and myelin protein mRNA in twitcher mouse brain. J Neurosci Res 51:536–540. [DOI] [PubMed] [Google Scholar]
- Taniike M, Mohri I, Eguchi N, Irikura D, Urade Y, Okada S, Suzuki K (1999). An apoptotic depletion of oligodendrocytes in the twitcher, a murine model of globoid cell leukodystrophy. J Neuropathol Exp Neurol 58:644–653. [DOI] [PubMed] [Google Scholar]
- Taniike M, Mohri I, Eguchi N, Beuckmann CT, Suzuki K, Urade Y (2002). Perineuronal oligodendrocytes protect against neuronal apoptosis through the production of lipocalin-type prostaglandin D synthase in a genetic demyelinating model. J Neurosci 22:4885–4896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teismann P, Vila M, Choi DK, Tieu K, Wu DC, Jackson-Lewis V, Przedborski S (2003). COX-2 and neurodegeneration in Parkinson’s disease. Ann NY Acad Sci 991:272–277. [DOI] [PubMed] [Google Scholar]
- Toyoshima E, Yeager AM, Brennan S, Santos GW, Moser HW, Mayer RF (1986). Nerve conduction studies in the Twitcher mouse (murine globoid cell leukodystrophy). J Neurol Sci 74:307–318. [DOI] [PubMed] [Google Scholar]
- Urade Y, Fujimoto N, Hayaishi O (1985). Purification and characterization of rat brain prostaglandin D synthetase. J Biol Chem 260:12410–12415. [PubMed] [Google Scholar]
- Urade Y, Ujihara M, Horiguchi Y, Ikai K, Hayaishi O (1989). The major source of endogenous prostaglandin D2 production is likely antigen-presenting cells. Localization of glutathione-requiring prostaglandin D synthetase in histiocytes, dendritic, and Kupffer cells in various rat tissues. J Immunol 143:2982–2989. [PubMed] [Google Scholar]
- Urade Y, Ujihara M, Horiguchi Y, Igarashi M, Nagata A, Ikai K, Hayaishi O (1990). Mast cells contain spleen-type prostaglandin D synthetase. J Biol Chem 265:371–375. [PubMed] [Google Scholar]
- Walsh DT, Perry VH, Minghetti L (2000). Cyclooxygenase-2 is highly expressed in microglial-like cells in a murine model of prion disease. Glia 29:392–396. [PubMed] [Google Scholar]
- Wasserman MA, Griffin RL, Marsalisi FB (1980). Potent bronchoconstrictor effects of aerosolized prostaglandin D2 in dogs. Prostaglandins 20:703–715. [DOI] [PubMed] [Google Scholar]
- Whittle BJ, Moncada S, Vane JR (1978). Comparison of the effects of prostacyclin (PGI2), prostaglandin E1 and D2 on platelet aggregation in different species. Prostaglandins 16:373–388. [DOI] [PubMed] [Google Scholar]
- Wood PL, Choksi S, Bocchini V (1994). Inducible microglial nitric oxide synthase: a large membrane pool. NeuroReport 5:977–980. [DOI] [PubMed] [Google Scholar]
- Woodroofe MN, Sarna GS, Wadhwa M, Hayes GM, Loughlin AJ, Tinker A, Cuzner ML (1991). Detection of interleukin-1 and interleukin-6 in adult rat brain, following mechanical injury, by in vivo microdialysis: evidence of a role for microglia in cytokine production. J Neuroimmunol 33:227–236. [DOI] [PubMed] [Google Scholar]
- Wu YP, Matsuda J, Kubota A, Suzuki K, Suzuki K (2000). Infiltration of hematogenous lineage cells into the demyelinating central nervous system of twitcher mice. J Neuropathol Exp Neurol 59:628–639. [DOI] [PubMed] [Google Scholar]
- Yermakova AV, Rollins J, Callahan LM, Rogers J, O’Banion MK (1999). Cyclooxygenase-1 in human Alzheimer and control brain: quantitative analysis of expression by microglia and CA3 hippocampal neurons. J Neuropathol Exp Neurol 58:1135–1146. [DOI] [PubMed] [Google Scholar]