

Abstract
Working memory (WM), the ability to transiently hold information in mind, is essential for high-level cognitive functions that are often impaired in brain-injured patients. The cellular and molecular mechanisms contributing to WM deficits, which can manifest in the absence of overt damage, in these patients are unknown. The function of the dorsolateral prefrontal cortex in humans and monkeys, and the medial prefrontal cortex (mPFC), in rodents is critical for WM. We demonstrate that controlled cortical impact injury of rats causes a long-lasting WM impairment that is associated with increased levels of the GABA-synthesizing enzyme glutamic acid decarboxylase 67 (GAD67) in the mPFC for up to 1 month after injury. A single administration of dopamine D1 antagonists at 14 d after injury is sufficient to decrease GAD67 levels and restore WM for at least 1 week. These findings indicate that inhibition of prefrontal neuronal activity contributes to WM deficits and that strategies to reduce GAD67 expression can offer prolonged WM improvement in brain-injured patients.
Keywords: brain injury, executive function, dorsolateral prefrontal cortex, medial prefrontal cortex, delay cells, GABAergic inhibition, D1 antagonist
Introduction
Most brain-injured patients report some deficits in complex behaviors and high-level cognitive functions (e.g., comprehension, reasoning, planning, decision making) attributable to problems with working memory (Milner et al., 1998; Anderson et al., 1999). These deficits can manifest in the absence of overt brain damage, suggesting that neuronal dysfunction may be an underlying mechanism (Levin et al., 2002). However, the cellular and molecular mechanisms that contribute to neuronal dysfunction and working memory impairments are not known. Working memory, the ability to transiently hold information in mind and manipulate that information to guide subsequent behavior, is essential for many prefrontal functions (Baddeley, 1992). Imaging studies and electrophysiological recordings have identified the dorsolateral prefrontal cortex (DLPFC) in monkeys and humans, and the medial prefrontal cortex (mPFC) in rats, as a key structure for working memory function (Watanabe, 1986; Shaw and Aggleton, 1993; Broersen et al., 1994; D’Esposito et al., 2000). In vivo recordings from the DLPFC in monkeys during delay response tasks have identified neurons that show an increase in firing rate that is sustained throughout the delay period (Fuster and Alexander, 1971; Miller et al., 1996).
It is thought that dopamine is involved in the maintenance of delay neuronal activity in the PFC that subserves working memory. Based on these and other studies, it has been proposed that an optimal level of D1 signaling is required for proper working memory function, giving rise to an inverted U relationship between dopamine D1 receptor (D1R) stimulation and working memory (Williams and Goldman-Rakic, 1995). Although dopamine and other modulatory neurotransmitter systems (e.g., norepinephrine and acetylcholine) have been shown to be important, the inhibitory neurotransmitter GABA also appears to be critical for pyramidal cell activity in the DLPFC and working memory (Arnsten, 2000; Robbins, 2000; Lewis et al., 2005). For instance, intra-DLPFC administration of the GABAA receptor antagonist bicuculline impairs working memory (Sawaguchi et al., 1989). It has been proposed that GABA neuron activity may control which, and when, DLPFC pyramidal neurons are active during a working memory task (Constantinidis et al., 2002; Wang et al., 2002). It has been suggested that a reduction in the levels of the GABA synthesizing enzyme [glutamic acid decarboxylase (GAD)] in prefrontal inhibitory neurons, specifically parvalbumin-positive fast-spiking inhibitory neurons, may contribute to abnormal delayed cell activity and impaired working memory in schizophrenia patients (Akbarian et al., 1995; Volk et al., 2000), suggesting that prefrontal function may require an optimal range of inhibitory input.
In the present study, we examined whether brain injury in rats alters GAD levels in the prelimbic/infralimbic (PL/IL) cortices of the mPFC and whether these changes contribute to working memory dysfunction. Using a controlled cortical impact model of brain trauma in which the contusion site is located posterior to the PL/IL cortices, we report that traumatic brain injury (TBI) causes spatial working memory deficits in the absence of overt PL/IL cortical neuronal loss. This deficit is associated with increased expression of GAD67 in the PL/IL cortices. A single administration of the D1 antagonist SCH23390 [R(+)-7-chloro-8-hydroxy-3-methyl-1-phenyl-2,3,4,5-tetrahydro-1H-3-benzazepine hydrochloride], either systemically or targeted to the PL/IL cortices, results in a reversal of PL/IL GAD expression and a lasting improvement in working memory function.
Materials and Methods
Materials.
D1R, D2R, and D4R polyclonal antibodies were purchased from Alpha Diagnostic International (San Antonio, TX). GAD67 monoclonal, GAD65 polyclonal, and neuron-specific nuclear protein (NeuN) monoclonal antibodies were obtained from Chemicon (Temecula, CA). Parvalbumin (PV) polyclonal antibody was from Affinity BioReagents (Golden, CO). Calretinin (CR) polyclonal and calbindin (CB) polyclonal antibodies were purchased from Sigma (St. Louis, MO). SCH23390 and SKF83566 (8-bromo-2,3,4,5-tetrahydro-3-methyl-5-phenyl-1H-3-benzazepin-7-ol hydrobromide) were obtained from Tocris Cookson (Ellisville, MO).
Controlled cortical impact injury.
All protocols involving the use of animals were in compliance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee. Male Sprague Dawley rats (260–300 g) were purchased from Harlan Sprague Dawley (Indianapolis, IN). A controlled cortical impact (CCI) device was used to administer unilateral brain injury as described previously (Dixon et al., 1991; Smith et al., 1995). Rats were anesthetized with 4% isoflurane and a 2:1 mixture of N2O/O2 and then mounted in a stereotaxic frame. The head was held in a horizontal plane, and a 7 mm craniectomy was performed on the right cranial vault. The center of the craniectomy was placed at the midpoint between bregma and the lambda and 3.5 mm lateral to the midline. Animals received a single impact of 1.7 mm deformation with an impact velocity of 6 m/s at an angle of 10° from the vertical plane using a 6-mm-diameter impactor tip, making the impact orthogonal to the surface of the cortex. The impact was delivered onto the parietal association cortex. The body temperature was maintained at 37°C by the use of a heating pad.
Intra-mPFC drug infusion.
After CCI, rats were bilaterally implanted under isoflurane anesthesia with sterile stainless-steel guide cannulas aimed at the dorsal border of the prelimbic area using a stereotaxic device (bregma, 3.2 mm; lateral, ±0.75 mm, and depth, −2.5 mm). Animals were allowed to recover from the surgery for 14 d before drug infusion experiments. For drug administration, the infusion needles were inserted into awake, moving animals. The infusion needles extended 1.5 mm beyond the end of the guide cannulas, giving a total depth of 4.0 mm. Drugs were dissolved in saline, and infusions were performed at a rate of 0.25 μl/min for 4 min. After infusion, the needles were left in place for 2 min to allow for diffusion of the drug. Dosages of SCH23390 were based on previously published reports, and the specificity of the drug has been characterized (Seamans et al., 1998). Cannula placement was assessed in a representative group of animals. All infusion needle tracks examined terminated within the PL/IL cortices.
Spatial working memory testing.
All behavioral tests were performed by an experimenter blind to the treatment groups. Spatial working memory was assessed as described previously (Hamm et al., 1996; Steele and Morris, 1999; Kline et al., 2002). Briefly, 24 h before testing, animals were given five training trials in a working memory version of the Morris water maze task (delay match-to-place task) to familiarize them with the task (Dash et al., 2004). Each testing session consisted of a location trial, a 5 s delay, and a matching trial. Each location trial was initiated by placing the rat in a random start location. Once the animal found the platform, it was allowed to remain there for 10 s. The animal was then removed from the water tank for a 5 s delay period. The rat was then placed back into the maze at the same start position as used in the locating trial and allowed to search for the hidden escape platform (the matching trial). If the animal failed to locate the platform within 60 s on any given trial, it was led there by the experimenter. After each location–match pair, animals were given a 4 min intertrial interval. The influence of each drug on working memory performance was assessed in four location–match trials, with each pair having a unique start position and platform location.
Western blotting.
Animals were killed, and brains were dissected and submerged under ice-cold artificial CSF (in mm: 10 HEPES, pH 7.2, 1.3 NaH2PO4, 3 KCl, 124 NaCl, 10 dextrose, 26 NaHCO3, and 2 MgCl2). The PL/IL cortices and hippocampus ipsilateral to the side of impact were quickly removed and snap-frozen on dry ice. Sham-operated animals were killed at 24 h, 14 d, or 28 d after surgery and used as controls. The mPFC brain tissue was homogenized in a lysis buffer containing 10 mm Tris, pH 7.4, 1 mm EGTA, 1 mm EDTA, 0.5 μm DTT, 10 μg/ml leupeptin, 10 μg/ml aprotinin, 1 mm PMSF, and 0.1 μm okadaic acid, followed by centrifugation at 10,000 × g for 10 min. The supernatant solutions were used as cytosolic fraction samples to detect protein levels. The pelletized material was rinsed twice with the lysis buffer and then resuspended in lysis buffer containing 0.5% Triton X-100. After sonication, the Triton X-100-soluble fraction was collected and used for the detection of the dopamine receptors. The protein concentration was measured using a NanoOrange protein quantification kit (Invitrogen, Carlsbad, CA).
Samples were resolved in a SDS-PAGE and transferred to an Immobilon-P membrane (Millipore, Bedford, MA), followed by blocking overnight in TBST (10 mm Tris, pH 7.5, 150 mm NaCl, and 0.05% Tween-20) plus 5% BSA. Membranes were then incubated with primary antibody (1.0, 0.5, 0.125, and 0.2 μg/ml for D1R, D2R, D4R, and GAD67 antibodies, respectively; 1:5000 dilution for GAD65 antibody) for 3 h at room temperature. After incubation with the primary antibody, membranes were washed three times, and immunoreactivity was assessed by an alkaline phosphatase-conjugated secondary antibody and a CDP-star chemiluminescent substrate (Cell Signaling Technology, Beverly, MA). The optical density of the immunoreactive bands was measured using NIH ImageJ software (http://rsb.info.nih.gov/ij/index.html). Before reprobing, blots were stripped by two 10 min washes in 50 mm NaOH at room temperature. The membranes were then washed extensively with TBST and reblocked for 1 h in 2% BSA before immunodetection.
Immunohistochemical staining.
Animals were anesthetized by an intraperitoneal injection of 0.7% chloral hydrate, followed by perfusion fixation using transcardiac infusion of 0.01 m PBS plus 1000 U/ml heparin sulfate, followed by 0.1 m phosphate buffer (PB) containing 4% paraformaldehyde and 0.2% glutaraldehyde. Brain tissue was removed and postfixed with 4% paraformaldehyde in 0.1 m PB at 4°C overnight. After cryoprotection in sucrose, 30-μm-thick frozen sections of the mPFC region were prepared. Sections were incubated in 2% normal goat serum and 0.2% Triton X-100 in 0.1 m PBS at room temperature for 2 h, followed by incubation with the primary antibody (2.5 μg/ml for anti-D1R, 0.5 μg/ml for anti-PV and anti-CB, 0.8 μg/ml for anti-CR, 0.25 μg/ml for anti-GAD67, and 1:2000 dilution for anti-NeuN antibodies) at 4°C overnight. After overnight incubations, sections were washed three times in TBS and incubated with Alexa Fluor 568-conjugated anti-rabbit-IgG or Alexa Fluor 488-conjugated anti-mouse-IgG, as suggested by the vendor. For cell counting, colorimetric staining was performed essentially as described above. Of exception, after the primary antibody incubation and washing, sections were incubated with a biotinylated secondary antibody that was detected using ABC and DAB kits (Vector Laboratories, Burlingame, CA) as directed by the manufacturer. The resultant images were adjusted for size and labeled using Photoshop 6.0 (Adobe Systems, San Jose, CA).
Cell counts.
Brain sections corresponding to 3.2 mm anterior to bregma were selected for cell counts that was performed using the optical dissector technique (Coggeshall and Lekan, 1996; Dash et al., 2001). Every third section was immunostained as described above (see Immunohistochemistry). Cells were quantified using Stereo Investigator, Optical Disector Probe (MicroBrightField, Colchester, VT) within a defined boundary of 2.15 × 10−2 mm3 encompassing the PL or IL at 40× magnification. The results obtained from five independent sections per animal were averaged. Cell counts were performed in both the PL and the IL by two independent researchers who were blind to the treatment groups. The corpus callosum was used as a marker to locate the medial prefrontal cortex.
Statistical analysis.
Student’s t test for unpaired variables or a one-way ANOVA was used for the analysis of cell counts and Western blot data, when appropriate. A two-way ANOVA was used for evaluating working memory performance and treatment effects. Results were considered significant at p < 0.05. Data are presented as the mean ± SEM.
Results
Lateral cortical impact injury causes spatial working memory deficits in the absence of overt mPFC (PL/IL) neuronal loss
The delay match-to-place task has been extensively used to study working memory and its dysfunction in pathological conditions (Hamm et al., 1996; Steele and Morris, 1999; Dash et al., 2004). In this task, animals are required to locate a hidden escape platform (location trial), removed from the maze, and then, after a few second delay, placed back into the training context and allowed to relocate the platform (match trial). The decrease in latency between the location (or first) and the match (or second) trials is used as a measure of working memory performance. The representative traces shown in Figure 1a illustrate the swimming paths taken by sham and injured animals during the location and match trials. Because the platform is moved before each location trial, rats must randomly search the water tank to find the platform. During the match trial, however, sham animals demonstrate a more constrained search within the immediate vicinity of the platform. In contrast, injured animals still use a random search strategy to solve the task. Figure 1b shows that sham animals had a significant decrease in latency during the matching trial at 14 and 28 d after injury (p < 0.001 in both time points), indicating functional working memory (n = 8 per each time point). By comparison, injured animals (when tested on either day 14 or day 28 after injury; n = 8 per each time point) did not show a significant decrease in latency between the location and the matching trials, indicating a working memory deficit (p = 0.558 and 0.069, respectively) (Fig. 1b). Comparison of the performance curves between injured and sham-operated animals revealed a significant interaction between trial number and injury status at both time points (14 d, F(1,14) = 14.02; 28 d, F(1,14) = 5.299).
Figure 1.
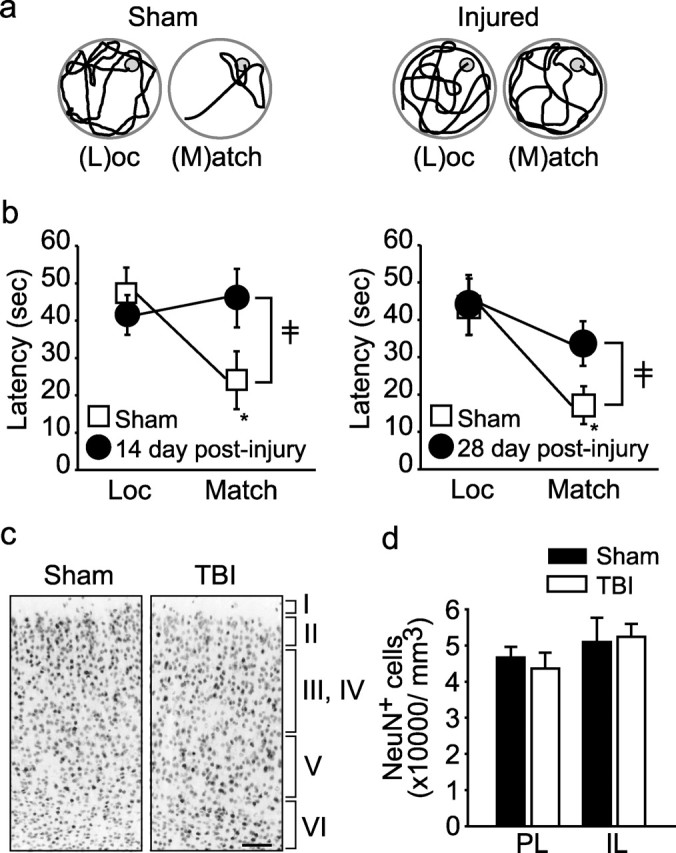
Cortical impact injury impairs spatial working memory performance. a, Representative traces of the swimming paths of a sham and an injured rat (14 d after injury) during the location (Loc) and match trials of the delay match-to-place task. b, Summary data displaying average latency to the hidden platform (in seconds) for location trials and match trials during the working memory task for sham and injured rats at 14 d (left) and 28 d (right) after injury (n = 8 per each time point). ‡Significant interaction between trial number and injury status (14 d sham vs injured, F(1,14) = 14.02; 28 d sham vs injured, F(1,14) = 5.299); *significant difference in latency between location and match trial (p < 0.001 in both time points). c, Representative photomicrographs for NeuN-positive neurons in PL/IL region of the mPFC of a sham and a 28 d post-injury animals. Neuronal layers in the mPFC are indicated by Roman numerals. Scale bar, 100 μm. d, Unbiased cell counts from sham (n = 5) and 28 d post-injury (n = 5) animals.
Lesion experiments, electrophysiological recordings, and neurochemical manipulations have demonstrated that the medial prefrontal cortex, especially the PL/IL cortices in rodents, is critical for working memory performance (Kolb, 1984; Runyan and Dash, 2005). Because loss of neurons in the PL/IL cortices as a result of CCI can give rise to the observed working memory deficits, we performed neuronal cell counts within the PL/IL cortices. After the completion of working memory testing (28 d after injury), animals were killed and coronal prefrontal sections (every 400 μm, five sections per animal) from sham and injured animals were stained using the neuron-specific antibody anti-NeuN (n = 5 per group). Figure 1c shows photomicrographs of representative NeuN-immunostained sections from a sham and an injured animal. NeuN-positive neurons in the PL/IL cortices were counted by an experimenter who was kept blind with respect to the injury condition using the optical dissector technique (counting frame, 250 × 180 μm; cap zones, 5 μm). The summary results shown in Figure 1d indicate that the cortical impact injury model used in the present study did not result in a significant loss of neurons within the PL/IL cortices (p = 0.806).
D1R, but not D2R or D4R, levels were increased after TBI
Previous studies have linked decreased levels of PFC dopamine D1 receptors to working memory deficits observed in schizophrenia patients (Okubo et al., 1997). To examine whether similar molecular changes occur after CCI, PL/IL tissues were dissected ipsilateral to the injury and analyzed by Western blots using D1 receptor- and D2 receptor-specific antibodies. The migration of the protein was identified based on its reported molecular weight. Samples obtained from animals 24 h after sham operation were used as controls. Optical density (n = 2 per each time point) measurements revealed that D1 immunoreactivity increases 3 h after injury and remains elevated for at least 3 d (Fig. 2a). The levels of D2 and D4 (data not shown) receptors did not change at any of the time points examined. Quantification of immunoreactivity at 24 h (n = 5) (Fig. 2b) and at 28 d (data not shown) revealed a significant increase in dopamine D1 receptor levels at 24 h (sham, 1.00 ± 0.118 vs TBI, 1.65 ± 0.132; p = 0.006) but not at 28 d (n = 5; sham, 1.00 ± 0.127 vs TBI, 1.09 ± 0.125; p = 0.622) after injury, time points when significant impairments in working memory were observed.
Figure 2.
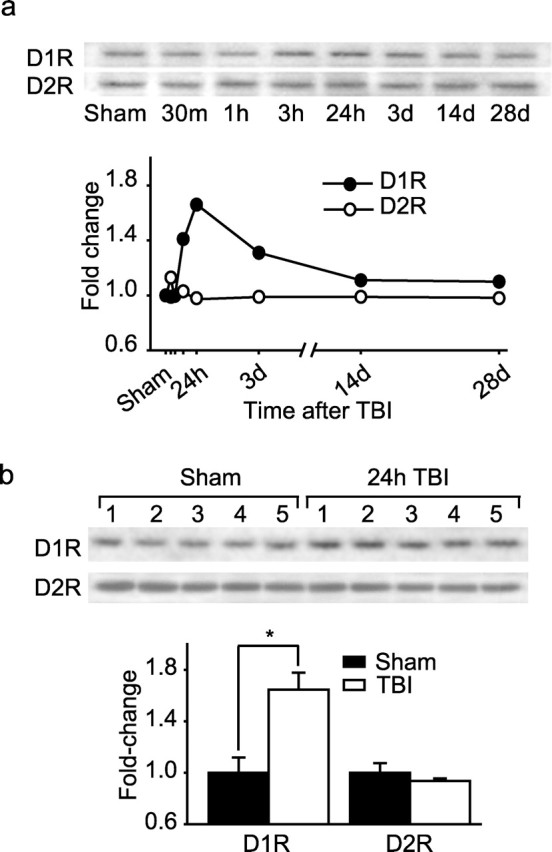
Cortical impact injury transiently increases dopamine D1, but not D2, receptor immunoreactivity in the PL/IL cortices. a, Representative Western blots and optical density measurement showing temporal changes in D1 and D2 receptor immunoreactivities in PL/IL protein extracts of sham (24 h) and injured animals at various time points after injury (n = 2 per each time point). b, Representative Western blots and optical density measurements of D1R and D2R immunoreactivities in mPFC of sham (n = 5) and 24 h post-injury (n = 5) animals. ∗p = 0.006.
TBI increases GAD67 protein and the number of GAD67-positive neurons
Because changes in GAD protein levels can alter GABAergic neurotransmission and impair working memory, Western blots using tissues taken from PL/IL cortices were performed. Figure 3a shows pictures of representative Western blots indicating the time courses for GAD67 and GAD65 protein levels after injury. The migrations of the GAD65- and GAD67-immunoreactive bands were identified based on their molecular weights. Although GAD65 levels did not change appreciably at the time points examined, GAD67 level increased within hours of TBI and remained elevated for at least 28 d after injury (Fig. 3a). Quantification of GAD67 immunoreactivity at the 24 h (Fig. 3b) and 28 d (data not shown) time points (n = 5 per each time point) revealed a significant increase in the PL/IL cortices as a result of injury at both time points (24 h, sham, 1.00 ± 0.132 vs TBI, 1.83 ± 0.176, p = 0.019; 28 d, sham, 1.00 ± 0.059 vs TBI, 1.34 ± 0.060, p = 0.011).
Figure 3.
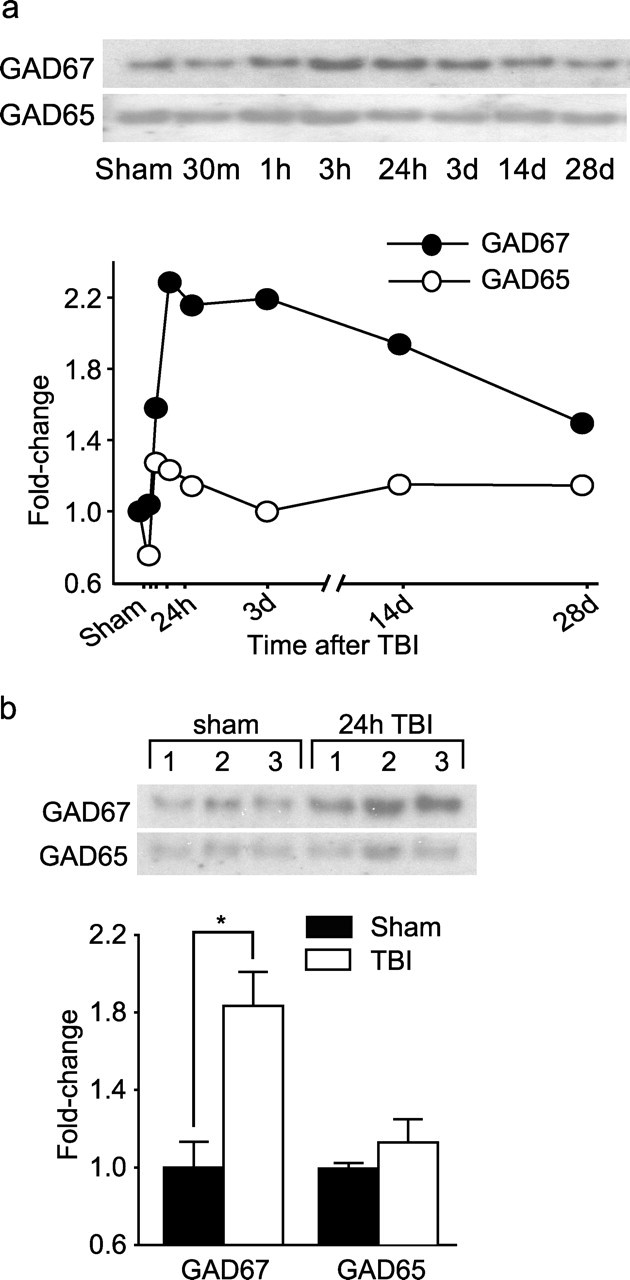
Cortical impact injury increases GAD67, but not GAD65, immunoreactivities in the PL/IL cortices. a, Representative Western blots and optical density measurements of sham (24 h) and injured animals at various time points after injury (n = 2 per each time point). b, Representative Western blots and optical density measurement of GAD67 and GAD65 immunoreactivity in the PL/IL cortices of sham (n = 5) and 24 h post-injury (n = 5) animals. ∗p = 0.019.
To determine whether the observed increase in GAD67 expression resulted in an increase in GAD67-immunopositive neuron number, mPFC tissue sections obtained from animals 14 d after injury or sham were immunostained using the GAD67 antibody. GAD67 immunoreactivity was detected using both fluorescent (Alexa Fluor 488) and colorimetric (DAB) methodologies, and the number of detectable cells were counted by an experimenter blind to the treatment groups. The CCI caused a significant increase in GAD67-positive cell number (n = 6 per group; sham, 6.10 ± 0.358 vs TBI, 7.64 ± 0.510 × 1000 cells/mm3; p = 0.032). This increase in GAD67-positive cell numbers as a result of CCI was independently confirmed by counting of the sections visualized using the fluorochrome-conjugated secondary antibody as shown in the representative pictures in Figure 4a.
Figure 4.
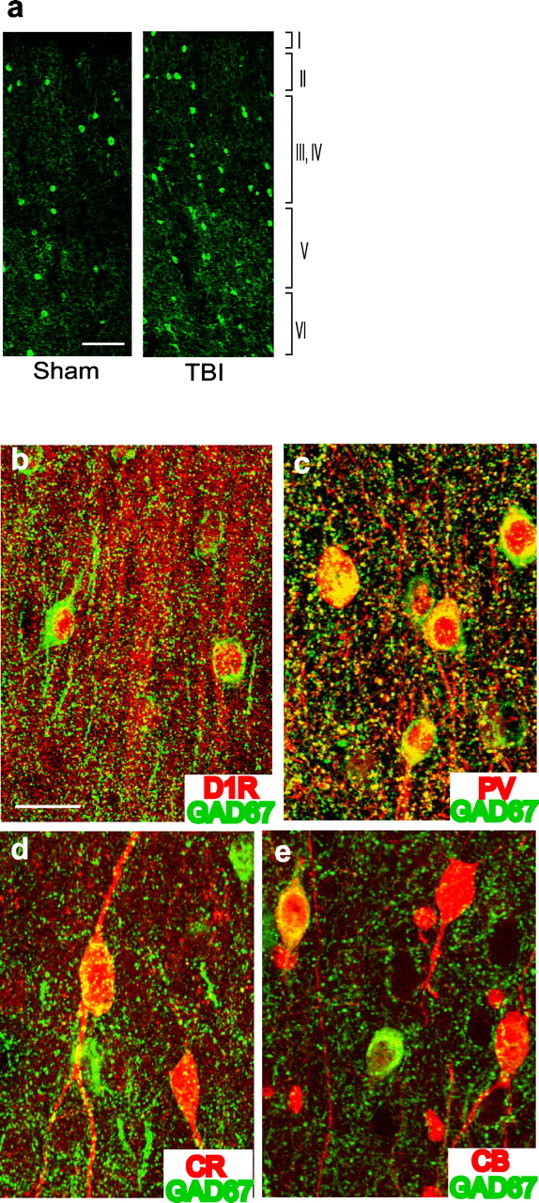
Increased number of GAD67-positive neurons in the mPFC after brain injury. a, Representative photomicrographs for GAD67-positive cells in mPFC tissue sections of sham and 14 d post-injury animals. Scale bar, 100 μm. Confocal micrographs showing double immunostaining of mPFC tissue section at 14 d after injury with GAD67 and D1R (b), GAD67 and PV (c), GAD67 and CR (d), and GAD67 and CB (e). Scale bar, 20 μm.
D1 receptors are expressed on GABAergic interneurons, as well as pyramidal neurons, in the prefrontal cortex of monkeys (Muly et al., 1998). To examine whether interneurons in the rat mPFC also express D1 receptors, double-labeled immunohistochemistry with D1 receptor and GAD67 was performed. D1 receptor-specific antibody showed strong staining in the apical dendrite of pyramidal neurons as well as cell bodies of nonpyramidal neurons. Most of the D1 receptor-positive nonpyramidal neurons were also positive for GAD67 (Fig. 4b). D1 receptor staining in the GAD67-positive cells showed punctated staining surrounding the nuclear membrane. This staining pattern is consistent with previous EM findings that indicate that interneurons of the macaque prefrontal cortex express D1 receptors that are localized in the Golgi apparatus and endoplasmic reticulum and in the cytoplasmic membrane of processes (Muly et al., 1998).
Cortical GABA neurons can be distinguished based on their expression of PV (basket and chandelier type), CR (double-bouquet type), and CB (double-bouquet type). PV-positive GABA neurons make synapses at sites proximal to the soma of pyramidal neurons and exert profound inhibition of prefrontal activity (Lewis et al., 2005). Double-labeled immunohistochemistry was performed to examine which inhibitory neuron subtype has increased GAD67 levels. Figure 4c–e shows representative photomicrographs of PL/IL tissue sections from an injured animal double-stained for GAD67 and PV, CR, or CB. Most of the PV-positive cells and fibers were strongly immunoreactive for GAD67 (Fig. 4c). In contrast, many CR- and CB-positive neurons were not labeled by the GAD67 antibody. Of the CR- and CB-immunopositive cells that were also immunoreactive for GAD67, the intensity of GAD67 staining was noticeably weaker than the surrounding GAD67-positive cells (Fig. 4d,e). These results suggest that the increase in GAD67-immunopositive cells likely reflects increased GAD67 expression within PV-positive inhibitory interneurons.
GABA receptor antagonists improve spatial working memory in injured rats
The above findings suggest that TBI-induced GAD67 expression could lead to inhibition of prefrontal activity and working memory deficits, and that GABA receptor antagonist should alleviate these deficits. Because complete blockade of GABA receptors is likely to cause unwanted side effects, a dose–response study was performed for each of three GABA receptor antagonists to determine the amount that can be administered without causing detectable working memory disturbances in normal animals. Once the dose for each compound was determined, its influence on working memory dysfunction was tested between days 10 and 14, time points at which GAD67 levels were found to be elevated in injured animals. Working memory testing was performed by an investigator blind to the treatment groups and unaware of the hypothesis being tested. Figure 5a shows that administration of the GABAA antagonist bicuculline (0.5 mg/kg; n = 10) improved working memory, as indicated by a significant interaction of treatment by trial (F(1,18) = 6.244). Similarly, the benzodiazepine inverse agonist ethyl-β-carboline-3-carboxylate (0.5 mg/kg carboline; n = 10), which acts as a negative regulator of GABAA receptor function, also improved working memory performance compared with vehicle-injected controls (F(1,18) = 8.765) (Fig. 5b). In contrast to the effects of GABAA antagonism, the GABAB receptor antagonist phaclofen (0.5 mg/kg; n = 10) did not improve working memory performance (F(1,18) = 0.104) (Fig. 5c). These results are consistent with the hypothesis that increased GABAergic activity, possibly as a result of enhanced GAD67 expression, contributes to the working memory deficits observed after brain injury.
Figure 5.

Systemic administration of GABAA receptor antagonists to injured animals on day 14 improves working memory performance. Summary data showing the latency to locate the hidden platform in the location (Loc) and match trials 30 min after systemic injection of the GABAA antagonist bicuculline (0.5 mg/kg) (a), the benzodiazepine inverse agonist carboline (0.5 mg/kg) (b), and the GABAB antagonist phaclofen (0.5 mg/kg) (c) compared with vehicle-treated injured animals (n = 10 per each drug). ‡Significant interaction between trial number and treatment (F(1,18) = 6.244 for bicuculline; F(1,18) = 8.765 for carboline). ∗Significant difference in latency between location and match trial (p < 0.001 for both bicuculline and carboline).
TBI-associated enhanced GAD67 expression can be attenuated by administration of a D1 receptor antagonist
Either electrolytic lesion of dopaminergic neurons in the ventral tegmental area (VTA) or chemical lesion using 6-hydroxy dopamine can alter the levels of GAD67 mRNA (Soghomonian and Chesselet, 1992; Billings and Marshall, 2004). To test whether modulation of dopamine receptors can be used to reverse GAD67 expression, the D1R antagonist SCH23390 (0.5 mg/kg) was administered systemically on day 14 after injury (Sawaguchi and Goldman-Rakic, 1991; Huang et al., 2004). The PL/IL tissues were taken 16 h later to examine GAD67 levels by Western blots (n = 10 per group) (Fig. 6a). A single administration of SCH23390 effectively reduces post-CCI GAD67 levels in the PL/IL cortices (p < 0.001). This effect was specific to injured animals because normal animals injected with SCH23390 did not show any statistically significant difference in GAD67 levels compared with vehicle-injected, uninjured controls (n = 5 per group; p = 0.098) (Fig. 6b).
Figure 6.
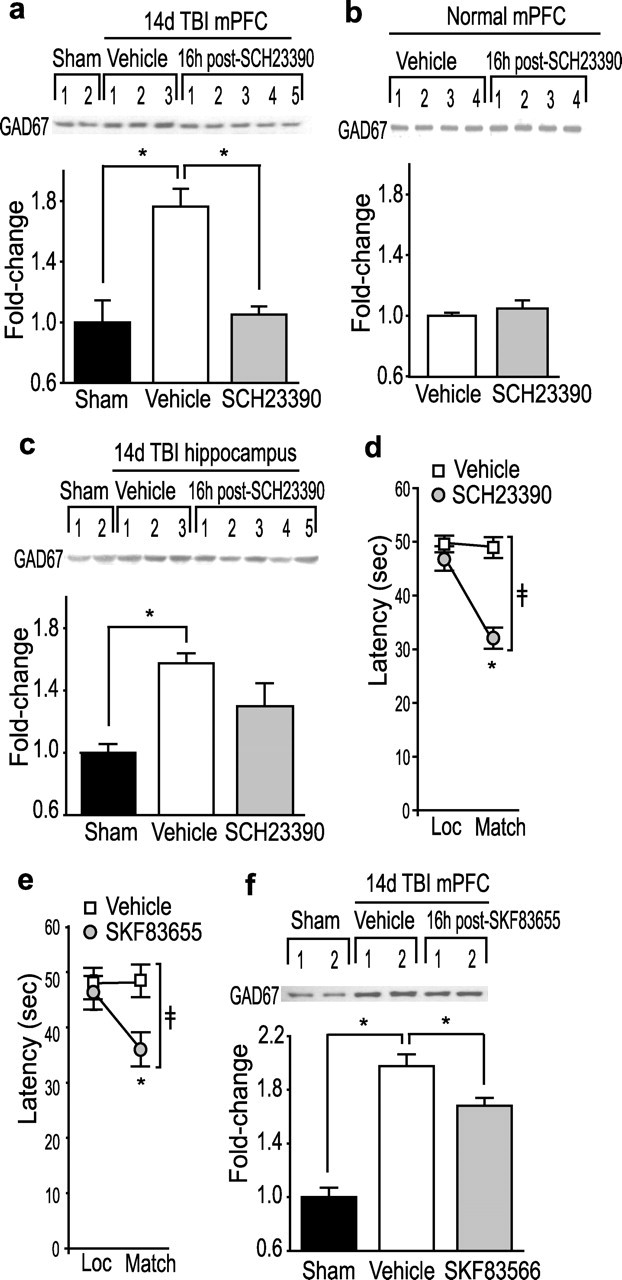
A single administration of D1-selective antagonists decreases mPFC GAD67 immunoreactivity and improves working memory performance in injured animals. a, A representative Western blot and summary results for GAD67 immunoreactivity in mPFC, measured 16 h after a single administration of the D1 antagonist SCH23390 (0.5 mg/kg) or vehicle (n = 10 per group) to injured or sham animals on day 14. ∗p = 0.006 for sham versus TBI vehicle and p < 0.001 for TBI vehicle versus TBI SCH23390. b, A representative Western blot and summary results for GAD67 immunoreactivity in mPFC of the uninjured animals at 16 h after an injection of SCH23390 (0.5 mg/kg) or vehicle (n = 5 per group). c, A representative Western blot and optical density measurement of GAD67 immunoreactivity in the hippocampus, measured 16 h after a single systemic injection of SCH23390 (0.5 mg/kg) or vehicle (n = 5 per group) to injured animals on day 14 after injury. ∗p < 0.001 for sham versus TBI vehicle. d, Summary data show the latency for the injured animals to find the hidden platform in the location (Loc) and match trials at 16 h after systemic injection of SCH23390 (0.5 mg/kg) compared with vehicle treatment (n = 10 per group). ‡Significant interaction between trial number and treatment (F(1,18) = 21.93). ∗Significant difference in latency between location and match trial (p < 0.001). e, Summary data show the latency for the injured animals to find the hidden platform in the location and match trials at 16 h after systemic injection of SKF83655 (50 μg/kg) compared with vehicle treatment (n = 11 per group). ‡Significant interaction between trial number and treatment (F(1,20) = 4.445). ∗Significant difference in latency between location and match trial (p = 0.021). f, A representative Western blot and summary results for GAD67 immunoreactivity in mPFC, measured 18–20 h after a single administration of the D1 antagonist SKF83655 (50 μg/kg) or vehicle (n = 11 per group) to injured or sham animals on day 14. ∗p = 0.011.
In addition to the PFC, working memory-related delay cell activities have been recorded in other brain structures (Miller et al., 1991; Egorov et al., 2002). However. delay cell activity in these areas appears to be coordinated and regulated in a top-down manner by the PFC to guide behavior (Fuster et al., 1985). We, therefore, examined whether the enhanced GAD67 expression after TBI, and the effect of the systemic D1R antagonist, is specific to the PL/IL cortices or whether these events also occur in another brain area. Because the spatial working memory task we used requires hippocampal function, we examined the post-injury and treatment-related levels of GAD67 in hippocampal extracts (n = 5). Figure 6c shows that, by 14 d after injury, GAD67 protein levels are also enhanced in the hippocampus after TBI (sham, 1.00 ± 0.057; TBI vehicle, 1.57 ± 0.063; p < 0.001). Although administration of SCH23390 decreased GAD67 levels in the hippocampus, this reduction did not achieve statistical significance (TBI vehicle, 1.57 ± 0.063; TBI SCH23390, 1.30 ± 0.15; p = 0.125).
D1R antagonist improves spatial working memory in injured rats
We next examined whether the decrease in GAD67 levels after SCH23390 administration translates to improvement of working memory performance. SCH23390 (0.5 mg/kg) or vehicle was systemically administered on day 14 after injury, and working memory was tested 16 h later (n = 10 per group). All behavioral tests involving the use of SCH23390 were performed by an experimenter blind to the treatment groups and unaware of the hypothesis being tested. Figure 6d shows that the SCH23390-treated injured animals performed significantly better in the spatial working memory task than did vehicle-treated injured animals. This is indicated by decreases in latency during the match trials compared with the location trials (location trial, 46.9 ± 2.29 s; matching trial, 32.1 ± 1.97 s; p < 0.001). In contrast, vehicle-treated animals displayed similar latencies in both the location and match trials (location trial, 49.6 ± 1.53 s; matching trial, 48.9 ± 1.96 s; p = 0.747), indicating an inability to perform the task. Comparison of the performance curves between vehicle- and SCH23390-treated injured animals revealed a significant interaction between trial number and treatment (vehicle vs SCH23390, F(1,18) = 21.93).
It has been reported that, at high concentrations, SCH23390 can act as a serotonin 1C/2C receptor agonist (Millan et al., 2001). We therefore tested whether a second D1 antagonist, SKF83566, can reduce post-injury GAD67 levels and improve working memory performance in a separate group of animals. SKF83566 (50 μg/kg) or vehicle was systemically administered on day 14 after injury, and working memory was tested 16 h later (n = 11 per group). Figure 6e shows that SKF83566-treated injured animals had significantly improved working memory performance (location trial, 46.3 ± 3.11 s; matching trial, 36.0 ± 3.09 s; p = 0.021) compared with vehicle-treated animals. Comparison of the performance curves between vehicle- and SKF83655-treated injured animals revealed a significant interaction between trial number and treatment (vehicle vs SKF83655, F(1,20) = 4.445). As seen for SCH23390, this improvement in working memory was associated with a decrease in GAD67 levels (n = 11 per group; TBI vehicle, 1.98 ± 0.089; TBI SCH23390, 1.68 ± 0.059; p = 0.011) (Fig. 6f). However, the reduction in GAD67 levels caused by SKF83655 treatment was smaller than that observed after SCH23390 treatment, a finding consistent with the modest improvement in working memory caused by SKF83655. These effects could be attributable to the low dose of SKF83655 used in this study.
Single administration of a D1R antagonist is sufficient to cause a lasting decrease in GAD67 levels and improvement in working memory
To determine whether the beneficial effect of the D1R antagonist on working memory after TBI can persist, we examined whether a single administration of SCH23390 can decrease GAD67 levels for longer than 16 h. Fourteen days after injury, groups of animals (n = 5 per group) were intraperitoneally injected with either 0.5 mg/kg SCH23390 or vehicle. Seven days after the injection, animals were killed and the PL/IL tissues were analyzed by Western blots to determine the levels of GAD67. A single administration of SCH23390 is sufficient to decrease GAD67 to a level comparable with that detected in sham animals (p < 0.001) (Fig. 7a). To explore whether this decrease in GAD67 levels detected at 7 d after SCH23390 administration translates to behavioral improvement, groups of animals received vehicle or drug on day 14 and were tested in the working memory task on day 21 after injury (n = 8 per group). Animals receiving SCH23390 exhibited a decrease in latency in the matching trials, giving rise to a significant interaction between treatment and trial (F(1,14) = 9.096) (Fig. 7b). These findings show that a single administration of the D1 antagonist SCH23390 to injured animals can provide a lasting improvement in working memory function, possibly by decreasing GAD67 expression.
Figure 7.
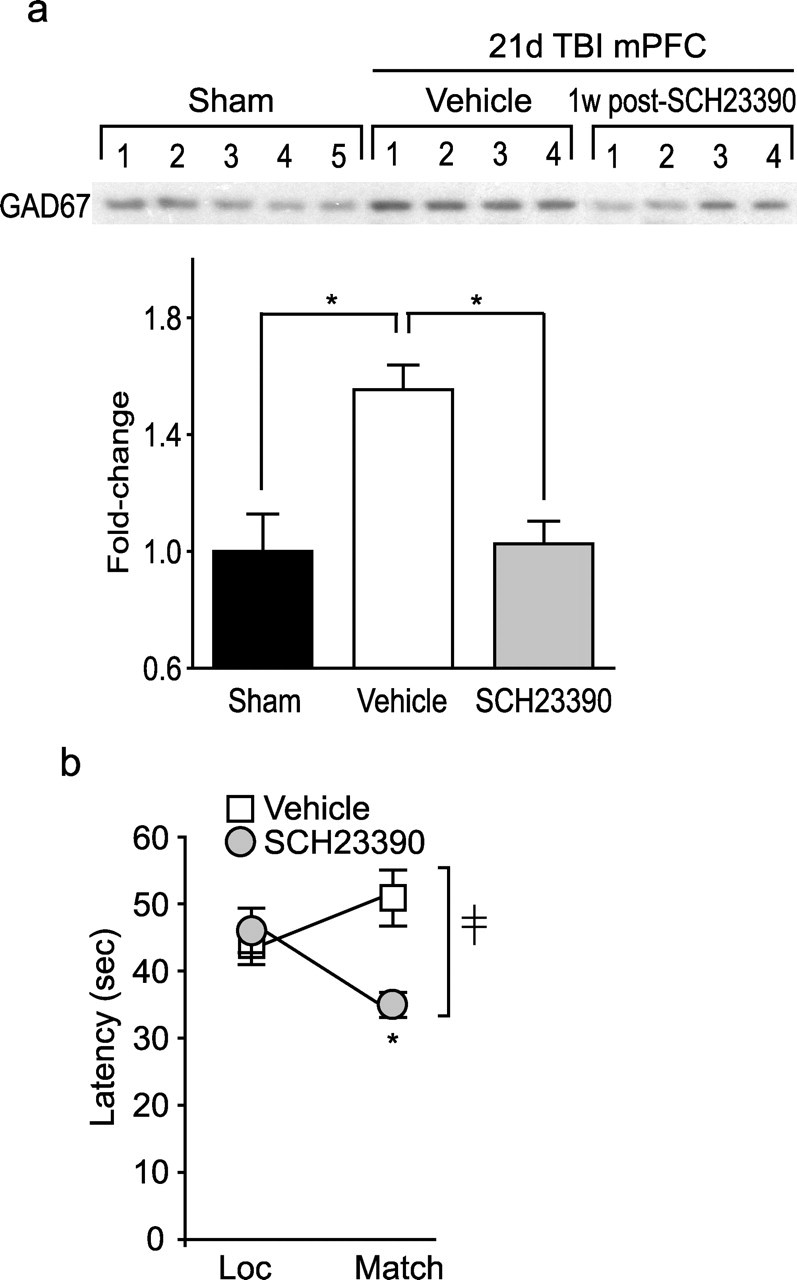
Lasting effect of D1 receptor antagonist on working memory performance and GAD67 levels. a, Representative Western blot and optical density measurement of GAD67 immunoreactivity in mPFC of sham and injured animals on day 21 after the completion of working memory testing (n = 5 per group). ∗p = 0.004 for sham versus TBI vehicle and p < 0.001 for TBI vehicle versus TBI SCH23390. b, Working memory performance (n = 8 per group) tested 7 d after a single administration of SCH23390 (0.5 mg/kg) on day 14 after injury. ‡Significant interaction between trial number and treatment (F(1,14) = 9.096). ∗Significant difference in latency between location (Loc) and match trial (p = 0.022).
Intra-PL/IL administration of SCH23390 improves working memory in injured animals
The spatial working memory enhancement seen after systemic administration of GABA and D1 antagonists could have arisen from the action of these drugs on brain structures other than the PL/IL cortices. To determine whether intra-PL/IL administration of SCH23390 is sufficient to improve working memory performance, injured animals were surgically implanted with guide cannulas aimed at PL/IL cortices (Fig. 8a). Fourteen days after injury, animals were bilaterally injected with either 6 ng/side SCH23390 or an equal volume (1 μl) of vehicle (n = 9 per group). Sixteen hours later, working memory testing was performed. Compared with vehicle-treated animals, animals that received intra-PL/IL infusion of SCH23390 showed significant improvement in working memory performance compared with vehicle-infused controls (F(1,16) = 8.325) (Fig. 8b). Because the drug was administered 16 h before testing and continuous washout of the drug by circulating CSF would have effectively removed the agent from the PL/IL cortices, it is unlikely that the observed improvement in working memory was attributable to the blockade of D1 receptors at the time of testing. To examine whether the drug was exerting its beneficial effect by decreasing GAD67 levels in PL/IL cortices, at the completion of behavioral training, animals were killed and PL/IL tissues were processed for Western blotting (n = 5 per group). Figure 8c (representative Western blot and summary data) shows that intra-PL/IL infusion of SCH23390 significantly decreases GAD67 expression in the PL/IL cortices (p = 0.006).
Figure 8.
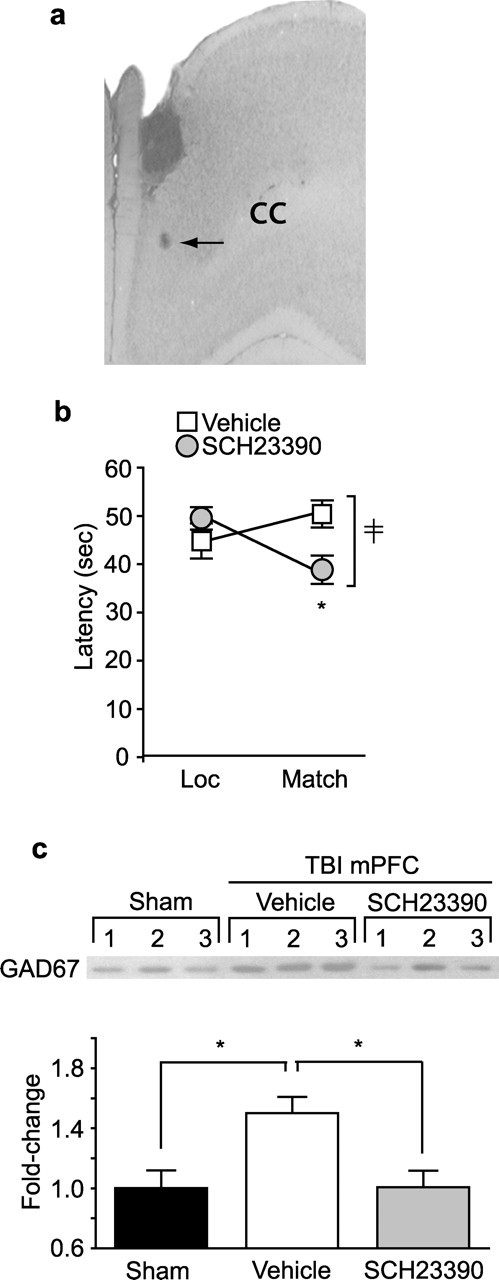
Intra-mPFC administration of SCH23390 improves working memory performance and decreases GAD67 level in injured animals. a, Photomicrograph showing the infusion needle track. Arrow, Dye injected through the infusion needle, showing the position of the needle tip. CC, Corpus callosum. b, Summary data of working memory performance 16 h after the targeted infusion of SCH23390 (6 ng/side) or vehicle (n = 9 per group). ‡Significant interaction between trial number and treatment (F(1,16) = 8.325). ∗Significant difference in latency between location (Loc) and match trial (p = 0.014). c, Representative Western blot and optical density measurement of GAD67 immunoreactivity in mPFC of sham and post-injury animals treated with targeted infusion of SCH23390 (6 ng/side) or vehicle (n = 5 per group) after behavioral testing. ∗p = 0.014 for sham versus TBI vehicle and p = 0.006 for TBI vehicle versus TBI SCH23390.
Discussion
Brain-injured patients often display cognitive deficits that can be attributable to dysfunction of the prefrontal cortex (Anderson et al., 1999; Levin and Hanten, 2005). These deficits include socially inappropriate behaviors, impairments in judgment, reduced attention, and working memory dysfunction. Working memory, the ability to hold information “on-line” for seconds and to manipulate that information to guide goal-directed behavior, lies at the core of many higher cognitive functions (Baddeley, 1992). Working memory is impaired in several neurological diseases (Germano and Kinsella, 2005; Lewis et al., 2005). For example, the degree of working memory deficits is the most reliable indicator of prognosis in schizophrenia patients (Lewis et al., 2005). Using a rodent CCI model of TBI, the present study examines cellular and molecular mechanisms that contribute to the impairment of working memory. The key findings of this study are as follows: (1) traumatic brain injury can cause enduring working memory deficits in the absence of overt prefrontal cell loss; (2) TBI enhances the expression of the GABA-synthesizing enzyme GAD67, and GABAA receptor antagonists improve working memory function in injured animals; and (3) a single administration of dopamine D1 receptor antagonists can suppress GAD67 expression in the PL/IL cortices of injured animals and improve working memory function for days.
Alterations in catecholamine signaling have been proposed to contribute to a variety of behavioral deficits after TBI. For example, using a cortical ablation model of injury, Feeney et al. (1981) showed that amphetamine, a monoamine uptake inhibitor, can restore tactile placing and beam walking, suggesting that catecholamine depletion may play a role in these motor deficits. Likewise, Zhu et al. (2000) have reported that central fluid percussion injury combined with entorhinal cortex lesion decreases dopamine β-hydroxylase-immunoreactive fibers in the hippocampus. The monoamine oxidase-B inhibitor l-deprenyl attenuated this loss, and performance in a spatial memory task improved as a result of the treatment. Although dopamine levels have not been specifically examined in the PL/IL cortices after injury, an increase in dopamine levels as early as 1 h after injury has been observed in the frontal cortex (Massucci et al., 2004).
Our results suggest that one potential consequence of increased dopamine levels in the prefrontal cortex is an increase in GAD67 levels. Our Western blot data using PL/IL tissue samples revealed that TBI causes a sustained increase in GAD67, but not GAD65, levels. GAD65 and GAD67 are the products of two separate genes whose expressions are independently regulated (Erlander et al., 1991). Although both enzymes are present in most GABA-containing neurons, GAD65 is a hydrophobic membrane-anchored form primarily found in axon terminals, whereas GAD67 is a soluble hydrophilic isoform distributed throughout the cell (Esclapez et al., 1994). Although both isoforms require the cofactor pyridoxal phosphate (PLP) for their enzymatic activity, GAD67 has a significantly higher binding affinity and lower dissociation constant for PLP than GAD65 (Chen et al., 2003). Therefore, most GAD67 exists as an active holoenzyme (bound with PLP), whereas GAD65 is primarily present as inactive apoenzymes (not bound with PLP) (Chen et al., 2003). Furthermore, brain GABA levels are normal in GAD65 knock-out mice (Asada et al., 1996), whereas GABA levels are reduced in adult heterozygotes of GAD67 knock-out mice (Asada et al., 1997). The change in GAD67 levels, as well as the increase in GAD67 cell number, independent of a change in GAD65, suggests that an upregulation of GAD67 within inhibitory neurons is more likely than generation of new GABAergic neurons via neurogenesis. This conclusion is further supported by the observation that the increase in GAD67-positive neurons could be detected as early as 24 h after injury (data not shown), a time period unlikely to be sufficient for the production, migration, and differentiation of newly proliferated neurons.
Our results demonstrating an increase in PL/IL GAD67 levels suggest that enhanced inhibition within this structure may impair delay activity and play a role in the working memory deficits seen after brain injury. The ability of prefrontal neurons to generate delay activity in the absence of stimulation is fundamentally important to working memory. The delay time (10–20 s) of sustained firing is orders of magnitude longer than the biophysical time constants (tens of milliseconds) of fast electrical signals (Fuster, 1995). For this reason, it is believed that delay activity is generated by the local neural network. This persistent activity relies on a recurrent excitatory network of pyramidal cells that is controlled by inhibitory interneurons (Constantinidis et al., 2002). Consistent with a role for excess GABA receptor-mediated inhibition of the PFC in injured animals, the GABAA receptor antagonist bicuculline and the benzodiazepine inverse agonist ethyl-β-carboline-3-carboxylate, but not the GABAB antagonist phaclofen, improved working memory performance in injured animals. This lack of effect by phaclofen is likely attributable to the relatively small number of GABAB receptors within the rat mPFC.
GABA interneurons have been classified into three subpopulations according to their electrophysiological characteristics and expression of calcium binding proteins PV, CR, or CB (Zaitsev et al., 2005). Our immunohistochemistry results suggest that the PV-expressing subtype is likely to be responsible for the increased GAD67 expression we observed. The PV-containing GABA neurons are physiologically termed fast-spiking neurons that are known to fire at an extremely high frequency. These neurons correspond morphologically to wide arbor (basket) and chandelier cells that make synapses on soma and axon initial segments of the pyramidal neurons, respectively, and strongly regulate cortical output activity (Gonzalez-Burgos et al., 2005). Most cortical PV-containing GABA neurons, compared with only 10% of the CB-expressing interneurons, express both D1 and D2 subtypes of dopamine receptor (Le Moine and Gaspar, 1998). Recently, it has been observed that dopaminergic input to GABA neurons not only regulates the excitability of these cells but also modulates GABA synthesis (Caboche et al., 1992; Martin and Rimvall, 1993). Interestingly, these manipulations primarily influence GAD67 mRNA, whereas GAD65 mRNA remains essentially unresponsive. For example, electrolytic lesion of the VTA, which results in dopaminergic denervation to the mPFC, leads to the downregulation of GAD67 mRNA (Retaux et al., 1994), suggesting that the loss of D1 receptor stimulation can decrease GAD67 expression.
The Western blot results we present indicate that TBI acutely increases the levels of dopamine D1 receptors in the PL/IL cortices. This increase could have contributed to the upregulation of PL/IL GAD67. Interestingly, we found that a single administration of the dopamine D1 receptor antagonist SCH23390 was sufficient to reduce GAD67 levels for up to 1 week after the injection. However, SCH23390 not only decreased GAD67 expression when administered at time points when D1 receptor levels were high (data not shown) but also at time points beyond this increase (e.g., 14 d after injury). The enhanced expression of GAD67 after TBI was not restricted to the PL/IL cortices but was also observed in the hippocampus. Because the working memory task used in the present study has a high demand for spatial information processing, hippocampal dysfunction could contribute to the observed behavioral deficits (Bramlett et al., 1997). To specifically examine the consequences of D1 receptor antagonism within the PL/IL cortices, targeted administration of SCH23390 was performed. This treatment paradigm also resulted in improved working memory and decreased GAD67 expression in the PL/IL cortices, suggesting that dopamine-mediated regulation of GAD67 in the PL/IL cortices is a major contributor to working memory deficits in injured animals.
At present, it is not clear how modulation of D1 receptor activity leads to alterations in GAD67 levels. Dopamine agonists, especially D1-selective agonists, are known to increase the levels of the intracellular second-messenger cAMP, leading to activation of the cAMP-dependent protein kinase A (PKA). Once activated by cAMP, the catalytic subunit of PKA can translocate into the nucleus in which it can phosphorylate specific transcription factors such as cAMP response element-binding protein (CREB), leading to enhanced transcription of genes containing the binding site for CREB in their promoter region, such as the immediate-early gene c-fos. Together with c-Jun, c-Fos stimulates the expression of genes carrying activator protein-1 (AP-1) binding sites. In addition to CREB, PKA has been shown to activate the heat-shock transcription factor (HSE) (Choi et al., 1991). A search for transcription factor motifs in the promoter region of rat gad67 revealed motifs with 90–100% homology to the binding sites for CREB, AP-1, and HSE. Thus, it is plausible that increased levels of dopamine after injury can lead to enhanced GAD67 levels via activation of one or more of these transcription factors. In addition, translational, as well as posttranslational, mechanisms may also contribute to enhanced GAD67 protein levels (Rimvall and Martin, 1994). Whereas dopamine D1 receptors increase cAMP-mediated signaling, engagement of D2 receptors decreases cAMP levels via activation of inhibitory Gi-proteins, suggesting that both D1 receptor antagonism and D2 receptor agonism may have similar behavioral consequences. Consistent with this notion, it has been demonstrated that chronic systemic administration of bromocriptine, a D2 receptor agonist, attenuates working memory deficits in rats after TBI (Kline et al., 2002). Furthermore, Castner et al. (2000) have shown that systemic administrations of the D1 agonist ABT 431 [(−)-trans 9,10-acetoxy-2-propyl-4,5,5a,6,7,11-b-hexahydro-3-thia-5-azacyclopent-1-ena[c]phenanthrene hydrochloride] can reverse the working memory impairments caused by D1 receptor downregulation after chronic antipsychotic drug exposure. This working memory improvement was found to last for up to 1 year. Although it has not been examined, it would be interesting to determine whether these effects are mediated via altered expression of GAD67.
Footnotes
We thank John Byrne and Anthony Moore for insightful comments and suggestions and Melanie Moody and Min Zhang for technical assistance. This work was supported by National Institutes of Health Grants NS35457 and NS049160, the Mike Hogg Foundation, and The Institute for Rehabilitation and Research/Mission Connect Consortium.
References
- Akbarian S, Kim JJ, Potkin SG, Hagman JO, Tafazzoli A, Bunney WE Jr, Jones EG (1995). Gene expression for glutamic acid decarboxylase is reduced without loss of neurons in prefrontal cortex of schizophrenics. Arch Gen Psychiatry 52:258–266. [DOI] [PubMed] [Google Scholar]
- Anderson SW, Bechara A, Damasio H, Tranel D, Damasio AR (1999). Impairment of social and moral behavior related to early damage in human prefrontal cortex. Nat Neurosci 2:1032–1037. [DOI] [PubMed] [Google Scholar]
- Arnsten AF (2000). Through the looking glass: differential noradenergic modulation of prefrontal cortical function. Neural Plast 7:133–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asada H, Kawamura Y, Maruyama K, Kume H, Ding R, Ji FY, Kanbara N, Kuzume H, Sanbo M, Yagi T, Obata K (1996). Mice lacking the 65 kDa isoform of glutamic acid decarboxylase (GAD65) maintain normal levels of GAD67 and GABA in their brains but are susceptible to seizures. Biochem Biophys Res Commun 229:891–895. [DOI] [PubMed] [Google Scholar]
- Asada H, Kawamura Y, Maruyama K, Kume H, Ding RG, Kanbara N, Kuzume H, Sanbo M, Yagi T, Obata K (1997). Cleft palate and decreased brain gamma-aminobutyric acid in mice lacking the 67-kDa isoform of glutamic acid decarboxylase. Proc Natl Acad Sci USA 94:6496–6499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baddeley A (1992). Working memory. Science 255:556–559. [DOI] [PubMed] [Google Scholar]
- Billings LM, Marshall JF (2004). Glutamic acid decarboxylase 67 mRNA regulation in two globus pallidus neuron populations by dopamine and the subthalamic nucleus. J Neurosci 24:3094–3103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bramlett HM, Green EJ, Dietrich WD (1997). Hippocampally dependent and independent chronic spatial navigational deficits following parasagittal fluid percussion brain injury in the rat. Brain Res 762:195–202. [DOI] [PubMed] [Google Scholar]
- Broersen LM, Heinsbroek RP, de Bruin JP, Joosten RN, van Hest A, Olivier B (1994). Effects of local application of dopaminergic drugs into the dorsal part of the medial prefrontal cortex of rats in a delayed matching to position task: comparison with local cholinergic blockade. Brain Res 645:113–122. [DOI] [PubMed] [Google Scholar]
- Caboche J, Vernier P, Rogard M, Julien JF, Mallet J, Besson MJ (1992). Role of dopaminergic D2 receptors in the regulation of glutamic acid decarboxylase messenger RNA in the striatum of the rat. Eur J Neurosci 4:438–447. [DOI] [PubMed] [Google Scholar]
- Castner SA, Williams GV, Goldman-Rakic PS (2000). Reversal of antipsychotic-induced working memory deficits by short-term dopamine D1 receptor stimulation. Science 287:2020–2022. [DOI] [PubMed] [Google Scholar]
- Chen CH, Battaglioli G, Martin DL, Hobart SA, Colon W (2003). Distinctive interactions in the holoenzyme formation for two isoforms of glutamate decarboxylase. Biochim Biophys Acta 1645:63–71. [DOI] [PubMed] [Google Scholar]
- Choi HS, Li B, Lin Z, Huang E, Liu AY (1991). cAMP and cAMP-dependent protein kinase regulate the human heat shock protein 70 gene promoter activity. J Biol Chem 266:11858–11865. [PubMed] [Google Scholar]
- Coggeshall RE, Lekan HA (1996). Methods for determining numbers of cells and synapses: a case for more uniform standards of review. J Comp Neurol 364:6–15. [DOI] [PubMed] [Google Scholar]
- Constantinidis C, Williams GV, Goldman-Rakic PS (2002). A role for inhibition in shaping the temporal flow of information in prefrontal cortex. Nat Neurosci 5:175–180. [DOI] [PubMed] [Google Scholar]
- Dash PK, Mach SA, Moore AN (2001). Enhanced neurogenesis in the rodent hippocampus following traumatic brain injury. J Neurosci Res 63:313–319. [DOI] [PubMed] [Google Scholar]
- Dash PK, Moore AN, Moody MR, Treadwell R, Felix JL, Clifton GL (2004). Post-trauma administration of caffeine plus ethanol reduces contusion volume and improves working memory in rats. J Neurotrauma 21:1573–1583. [DOI] [PubMed] [Google Scholar]
- D’Esposito M, Postle BR, Rypma B (2000). Prefrontal cortical contributions to working memory: evidence from event-related fMRI studies. Exp Brain Res 133:3–11. [DOI] [PubMed] [Google Scholar]
- Dixon CE, Clifton GL, Lighthall JW, Yaghmai AA, Hayes RL (1991). A controlled cortical impact model of traumatic brain injury in the rat. J Neurosci Methods 39:253–262. [DOI] [PubMed] [Google Scholar]
- Egorov AV, Hamam BN, Fransen E, Hasselmo ME, Alonso AA (2002). Graded persistent activity in entorhinal cortex neurons. Nature 420:173–178. [DOI] [PubMed] [Google Scholar]
- Erlander MG, Tillakaratne NJ, Feldblum S, Patel N, Tobin AJ (1991). Two genes encode distinct glutamate decarboxylases. Neuron 7:91–100. [DOI] [PubMed] [Google Scholar]
- Esclapez M, Tillakaratne NJ, Kaufman DL, Tobin AJ, Houser CR (1994). Comparative localization of two forms of glutamic acid decarboxylase and their mRNAs in rat brain supports the concept of functional differences between the forms. J Neurosci 14:1834–1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feeney DM, Gonzales A, Law WA (1981). Amphetamine restores locomotor function after motor cortex injury in the rat. Proc West Pharmacol Soc 24: pp. 15–17. [PubMed] [Google Scholar]
- Fuster JM (1995). Temporal processing. Ann NY Acad Sci 769:173–181. [DOI] [PubMed] [Google Scholar]
- Fuster JM, Alexander GE (1971). Neuron activity related to short-term memory. Science 173:652–654. [DOI] [PubMed] [Google Scholar]
- Fuster JM, Bauer RH, Jervey JP (1985). Functional interactions between inferotemporal and prefrontal cortex in a cognitive task. Brain Res 330:299–307. [DOI] [PubMed] [Google Scholar]
- Germano C, Kinsella GJ (2005). Working memory and learning in early Alzheimer’s disease. Neuropsychol Rev 15:1–10. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Burgos G, Krimer LS, Povysheva NV, Barrionuevo G, Lewis DA (2005). Functional properties of fast spiking interneurons and their synaptic connections with pyramidal cells in primate dorsolateral prefrontal cortex. J Neurophysiol 93:942–953. [DOI] [PubMed] [Google Scholar]
- Hamm RJ, Temple MD, Pike BR, O’Dell DM, Buck DL, Lyeth BG (1996). Working memory deficits following traumatic brain injury in the rat. J Neurotrauma 13:317–323. [DOI] [PubMed] [Google Scholar]
- Huang YY, Simpson E, Kellendonk C, Kandel ER (2004). Genetic evidence for the bidirectional modulation of synaptic plasticity in the prefrontal cortex by D1 receptors. Proc Natl Acad Sci USA 101:3236–3241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kline AE, Massucci JL, Marion DW, Dixon CE (2002). Attenuation of working memory and spatial acquisition deficits after a delayed and chronic bromocriptine treatment regimen in rats subjected to traumatic brain injury by controlled cortical impact. J Neurotrauma 19:415–425. [DOI] [PubMed] [Google Scholar]
- Kolb B (1984). Functions of the frontal cortex of the rat: a comparative review. Brain Res 320:65–98. [DOI] [PubMed] [Google Scholar]
- Le Moine C, Gaspar P (1998). Subpopulations of cortical GABAergic interneurons differ by their expression of D1 and D2 dopamine receptor subtypes. Brain Res Mol Brain Res 58:231–236. [DOI] [PubMed] [Google Scholar]
- Levin HS, Hanten G (2005). Executive functions after traumatic brain injury in children. Pediatr Neurol 33:79–93. [DOI] [PubMed] [Google Scholar]
- Levin HS, Hanten G, Chang CC, Zhang L, Schachar R, Ewing-Cobbs L, Max JE (2002). Working memory after traumatic brain injury in children. Ann Neurol 52:82–88. [DOI] [PubMed] [Google Scholar]
- Lewis DA, Hashimoto T, Volk DW (2005). Cortical inhibitory neurons and schizophrenia. Nat Rev Neurosci 6:312–324. [DOI] [PubMed] [Google Scholar]
- Martin DL, Rimvall K (1993). Regulation of gamma-aminobutyric acid synthesis in the brain. J Neurochem 60:395–407. [DOI] [PubMed] [Google Scholar]
- Massucci JL, Kline AE, Ma X, Zafonte RD, Dixon CE (2004). Time dependent alterations in dopamine tissue levels and metabolism after experimental traumatic brain injury in rats. Neurosci Lett 372:127–131. [DOI] [PubMed] [Google Scholar]
- Millan MJ, Newman-Tancredi A, Quentric Y, Cussac D (2001). The “selective” dopamine D1 receptor antagonist, SCH23390, is a potent and high efficacy agonist at cloned human serotonin2C receptors. Psychopharmacology (Berl) 156:58–62. [DOI] [PubMed] [Google Scholar]
- Miller EK, Li L, Desimone R (1991). A neural mechanism for working and recognition memory in inferior temporal cortex. Science 254:1377–1379. [DOI] [PubMed] [Google Scholar]
- Miller EK, Erickson CA, Desimone R (1996). Neural mechanisms of visual working memory in prefrontal cortex of the macaque. J Neurosci 16:5154–5167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milner B, Squire LR, Kandel ER (1998). Cognitive neuroscience and the study of memory. Neuron 20:445–468. [DOI] [PubMed] [Google Scholar]
- Muly EC III, Szigeti K, Goldman-Rakic PS (1998). D1 receptor in interneurons of macaque prefrontal cortex: distribution and subcellular localization. J Neurosci 18:10553–10565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okubo Y, Suhara T, Suzuki K, Kobayashi K, Inoue O, Terasaki O, Someya Y, Sassa T, Sudo Y, Matsushima E, Iyo M, Tateno Y, Toru M (1997). Decreased prefrontal dopamine D1 receptors in schizophrenia revealed by PET. Nature 385:634–636. [DOI] [PubMed] [Google Scholar]
- Retaux S, Trovero F, Besson MJ (1994). Role of dopamine in the plasticity of glutamic acid decarboxylase messenger RNA in the rat frontal cortex and the nucleus accumbens. Eur J Neurosci 6:1782–1791. [DOI] [PubMed] [Google Scholar]
- Rimvall K, Martin DL (1994). The level of GAD67 protein is highly sensitive to small increases in intraneuronal gamma-aminobutyric acid levels. J Neurochem 62:1375–1381. [DOI] [PubMed] [Google Scholar]
- Robbins TW (2000). Chemical neuromodulation of frontal-executive functions in humans and other animals. Exp Brain Res 133:130–138. [DOI] [PubMed] [Google Scholar]
- Runyan JD, Dash PK (2005). Distinct prefrontal molecular mechanisms for information storage lasting seconds versus minutes. Learn Mem 12:232–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawaguchi T, Goldman-Rakic PS (1991). D1 dopamine receptors in prefrontal cortex: involvement in working memory. Science 251:947–950. [DOI] [PubMed] [Google Scholar]
- Sawaguchi T, Matsumura M, Kubota K (1989). Delayed response deficits produced by local injection of bicuculline into the dorsolateral prefrontal cortex in Japanese macaque monkeys. Exp Brain Res 75:457–469. [DOI] [PubMed] [Google Scholar]
- Seamans JK, Floresco SB, Phillips AG (1998). D1 receptor modulation of hippocampal-prefrontal cortical circuits integrating spatial memory with executive functions in the rat. J Neurosci 18:1613–1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw C, Aggleton JP (1993). The effects of fornix and medial prefrontal lesions on delayed non-matching-to-sample by rats. Behav Brain Res 54:91–102. [DOI] [PubMed] [Google Scholar]
- Smith DH, Soares HD, Pierce JS, Perlman KG, Saatman KE, Meaney DF, Dixon CE, McIntosh TK (1995). A model of parasagittal controlled cortical impact in the mouse: cognitive and histopathologic effects. J Neurotrauma 12:169–178. [DOI] [PubMed] [Google Scholar]
- Soghomonian JJ, Chesselet MF (1992). Effects of nigrostriatal lesions on the levels of messenger RNAs encoding two isoforms of glutamate decarboxylase in the globus pallidus and entopeduncular nucleus of the rat. Synapse 11:124–133. [DOI] [PubMed] [Google Scholar]
- Steele RJ, Morris RG (1999). Delay-dependent impairment of a matching-to-place task with chronic and intrahippocampal infusion of the NMDA-antagonist d-AP5. Hippocampus 9:118–136. [DOI] [PubMed] [Google Scholar]
- Volk DW, Austin MC, Pierri JN, Sampson AR, Lewis DA (2000). Decreased glutamic acid decarboxylase67 messenger RNA expression in a subset of prefrontal cortical gamma-aminobutyric acid neurons in subjects with schizophrenia. Arch Gen Psychiatry 57:237–245. [DOI] [PubMed] [Google Scholar]
- Wang X, Zhong P, Yan Z (2002). Dopamine D4 receptors modulate GABAergic signaling in pyramidal neurons of prefrontal cortex. J Neurosci 22:9185–9193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe M (1986). Prefrontal unit activity during delayed conditional Go/No-Go discrimination in the monkey. I. Relation to the stimulus. Brain Res 382:1–14. [DOI] [PubMed] [Google Scholar]
- Williams GV, Goldman-Rakic PS (1995). Modulation of memory fields by dopamine D1 receptors in prefrontal cortex. Nature 376:572–575. [DOI] [PubMed] [Google Scholar]
- Zaitsev AV, Gonzalez-Burgos G, Povysheva NV, Kroner S, Lewis DA, Krimer LS (2005). Localization of calcium-binding proteins in physiologically and morphologically characterized interneurons of monkey dorsolateral prefrontal cortex. Cereb Cortex 15:1178–1186. [DOI] [PubMed] [Google Scholar]
- Zhu J, Hamm RJ, Reeves TM, Povlishock JT, Phillips LL (2000). Postinjury administration of l-deprenyl improves cognitive function and enhances neuroplasticity after traumatic brain injury. Exp Neurol 166:136–152. [DOI] [PubMed] [Google Scholar]