

Abstract
Endomorphins (EMs) are endogenous selective μ-opioid receptor agonists. Their role in inflammatory pain has not been fully elucidated. Here we examine peripheral antinociception elicited by exogenously applied EM-1 and EM-2 and the contribution of EM-containing leukocytes to stress- and corticotropin-releasing factor (CRF)-induced antinociception. To this end, we applied behavioral (paw pressure) testing, radioligand binding, immunohistochemistry, and flow cytometry in rats with unilateral hindpaw inflammation induced with Freund’s adjuvant. EMs injected directly into both hindpaws produced antinociception exclusively in inflamed paws. This was blocked by locally applied μ-receptor-selective (d-Phe-Cys-Tyr-d-Trp-Orn-Thr-Pen-Thr-NH2) but not κ-receptor-selective (nor-binaltorphimine) antagonists. δ-Receptor antagonists (naltrindole and N,N-diallyl-Tyr-Aib-Aib-Phe-Leu) did not influence EM-1-induced but dose-dependently decreased EM-2-induced antinociception. Antibodies against β-endorphin, methionine-enkephalin, or leucine-enkephalin did not significantly change EM-2-induced antinociception. Both EMs displaced binding of [3H]-[d-Ala2,N-Me-Phe4,Gly5-ol]enkephalin to μ-receptors in dorsal root ganglia (DRG). Using [3H]-naltrindole or [125I]-[d-Pen2,5]-enkephalin, no detectable δ-binding was found in DRG of inflamed hindlimbs. Numerous β-endorphin-containing and fewer EM-1- and EM-2-containing leukocytes were detected in subcutaneous tissue of inflamed paws. Leukocyte-depleting serum decreased the number of immigrating opioid-containing immune cells and attenuated swim stress- and CRF-induced antinociception in inflamed paws. Both forms of antinociception were strongly attenuated by anti-β-endorphin and to a lesser degree by anti-EM-1 and anti-EM-2 antibodies injected into inflamed paws. Together, exogenously applied and immune cell-derived EMs alleviate prolonged inflammatory pain through selective activation of peripheral opioid receptors. Exogenous EM-2 in addition to μ-receptors also activates peripheral δ-receptors, which does not involve actions via other opioid peptides.
Keywords: endomorphin, immune cells, leukocytes, antinociception, inflammation, pain
Introduction
Inflammatory pain can be effectively decreased by activation of opioid receptors on peripheral terminals of sensory neurons. This can be achieved with exogenous compounds and with opioid peptides derived from immune cells (Stein et al., 2003). Opioid-containing leukocytes, via activation of adhesion molecules and chemokines, migrate and accumulate in inflamed tissue (Machelska et al., 1998, 2002, 2004; Brack et al., 2004b). During stressful stimulation (swim stress, surgery) or local injection of corticotropin-releasing factor (CRF), leukocytes secrete opioids, which bind to their receptors on peripheral sensory nerve endings (Stein et al., 1990b, 1993, 1996; Schäfer et al., 1994). Selective activation of peripheral opioid receptors has the important advantage of providing effective analgesia without eliciting side effects typically associated with centrally acting opioids or with cyclooxygenase inhibitors (Stein et al., 2003).
The tetrapeptides endomorphin (EM)-1 and EM-2 are considered to have the highest specificity and affinity for the μ-opioid receptors of endogenous opioids (Zadina et al., 1997; Monory et al., 2000). Interestingly, preliminary results on the cloning of the EM precursor have been reported recently (Leff et al., 2005). EMs are distributed in regions of the CNS rich in μ-receptors and associated with pain perception (Martin-Schild et al., 1997, 1999; Schreff et al., 1998; Pierce and Wessendorf, 2000). In the spinal cord and brain, endogenous EMs can mediate electroacupuncture-induced antinociception (Han et al., 1999; Huang et al., 2000), and antinociception elicited by exogenously applied EMs is abolished in μ-receptor knock-out mice and by selective μ-receptor antagonists (Horvath, 2000; Przewlocki and Przewlocka, 2001). In addition, reasonable affinities of EMs for κ3-opioid receptors were reported (Goldberg et al., 1998), and some studies suggested that exogenous EM-2-induced antinociception was mediated by the release of dynorphin A(1-17) and methionine (Met)-enkephalin, which activated κ- and δ-receptors, respectively (Tseng, 2002). EMs are also expressed in primary sensory neurons and in immune cells (Jessop et al., 2002; Mousa et al., 2002), suggesting that EMs can be ligands at peripheral opioid receptors. Recently, EM-induced antinociception mediated by peripheral opioid receptors was reported in acute and neuropathic pain models (Spampinato et al., 2003; Obara et al., 2004).
In this study, we examined whether exogenous and endogenous immune cell-derived EMs can inhibit prolonged inflammatory pain and, if so, which opioid receptor types are involved. Because we had shown previously that β-endorphin is the most prominent opioid peptide mediating such effects (Stein et al., 2003), we also undertook a direct comparison to this peptide. To this end, we examined the following: (1) opioid receptor selectivity of exogenous EM-induced antinociception; (2) possible indirect effects of EM-2 through other opioid peptides; (3) binding of EMs to peripheral μ- and δ-receptors; (4) expression of EMs versus β-endorphin in inflamed tissue; (5) the impact of leukocyte depletion on the number of immune cells containing these peptides and on swim stress- and CRF-induced antinociception; and (6) the contribution of endogenous EMs in inflamed tissue to swim stress- and CRF-induced antinociception.
Materials and Methods
Animals
Experiments were conducted in male Wistar rats (200–250 g) (bred at the Charité-Universitätsmedizin Berlin, Campus Benjamin Franklin, Berlin, Germany) housed individually in cages lined with ground corncob bedding. Rats were kept in climate- and light-controlled rooms (22 ± 0.5°C; relative humidity, 60–65%; 12 h light/dark cycle) with standard rodent food pellets and water ad libitum. Experiments were performed according to the Policy on Ethics approved by the Society for Neuroscience and were approved by the local animal care committee (Landesamt für Arbeitsschutz, Gesundheitsschutz und Technische Sicherheit, Berlin, Germany). All efforts were made to minimize the number of animals used and their suffering.
Inflammation
Rats received 0.15 ml of complete Freund’s adjuvant (Calbiochem, La Jolla, CA) into the right hindpaw under brief isoflurane anesthesia (Abbott, Wiesbaden, Germany). The paw volume was monitored using a plethysmometer (Ugo Basile, Comerio, Italy) and determined by averaging two consecutive trials. The volume of displacement, which is equal to the paw volume, was indicated on a digital display. Experiments were performed at 4-6 d after induction of inflammation.
Assessment of nociceptive thresholds
We used the paw-pressure test (modified Randall-Selitto method). Rats (n = 6–8 per group) were gently restrained under paper wadding, and incremental pressure was applied via a wedge-shaped, blunt piston onto the dorsal surface of the hindpaw by means of an automated gauge (Ugo Basile). The paw-pressure threshold (PPT) (cutoff at 250 g) required to elicit paw withdrawal was determined by averaging three consecutive trials separated by 15 s intervals (Stein et al., 1990a; Machelska et al., 2003). PPTs were measured before (baseline) and after drug or antibody (Ab) treatments at the time points stated below. The contralateral paws were tested in the same way. The sequence of paws was alternated between animals to avoid “order” effects.
Drugs and antibodies for in vivo experiments
The following substances were used: EM-1, EM-2, β-endorphin, and rabbit IgG (Sigma, Taufkirchen, Germany); the following rabbit polyclonal Abs against opioid peptides anti-β-endorphin and anti-Met-enkephalin, (Peninsula Laboratories, Merseyside, UK), and anti-leucine (Leu)-enkephalin, anti-EM-1, and anti-EM-2 (Phoenix Pharmaceuticals, Belmont, CA); the opioid receptor antagonists d-Phe-Cys-Tyr-d-Trp-Orn-Thr-Pen-Thr-NH2 (CTOP) (selective at μ-receptors), naltrindole hydrochloride (NTI) and N,N-diallyl-Tyr-Aib-Aib-Phe-Leu (ICI 174,864) (both selective at δ-receptors), and nor-binaltorphimine dihydrochloride (norBNI) (selective at κ-receptors), CRF, and rabbit serum (all from Sigma), and rabbit anti-rat polymorphonuclear cell serum (anti-PMN) (Accurate Chemical and Scientific Corporation, Westbury, NY). Control and anti-PMN sera were injected intravenously following previously established protocols using 80 μl of serum diluted in 420 μl of 0.9% NaCl (Brack et al., 2004b). All other compounds were dissolved in sterile water and were injected into hindpaws [intraplantarly (i.pl.), in a volume of 0.1 ml] under brief isoflurane anesthesia or subcutaneously at the neck (in a volume of 0.2 ml).
In vivo experimental protocols
Exogenous endomorphin antinociception.
Rats received intraplantar EM-1 (0.625 μg) or EM-2 (0.625 μg) into both hindpaws, and PPTs were reevaluated at 5–40 min after injection. Dose–response relationships of intraplantar EM-1 (0.3–1.25 μg), EM-2 (0.3–1.25 μg), and β-endorphin (0.125–2 μg) were assessed at 5 min after injections (i.e., at the time of maximum elevation of PPT). To confirm a peripheral site of action, separate groups of animals received EM-1 or EM-2 in a dose of 1.25 μg subcutaneously (the total amount of EM injected intraplantarly in the most effective dose, i.e., 0.625 μg per each hindpaw) and were tested 5–40 min later. The peripheral selectivity of intraplantar β-endorphin-induced antinociception had been demonstrated previously (Stein et al., 1990a). Because none of these compounds produced antinociception in noninflamed paws, only inflamed paws were examined in the subsequent experiments.
To evaluate opioid receptor selectivity, EM-1 (0.625 μg) or EM-2 (0.625 μg) was coadministered intraplantarly with CTOP (0.0625–0.5 μg), ICI 174,864 (0.075–2.5 μg), or norBNI (0.1–100 μg). Because EM-2-induced antinociception was reversed by ICI 174,864, we further examined the involvement of δ-receptors by testing another δ-selective antagonist (naltrindole; 10–80 μg), as well as Abs against Met-enkephalin (0.1–20 μg), Leu-enkephalin (0.1–8 μg), or β-endorphin (0.1–2 μg), which were coadministered intraplantarly with EM-2. In a separate experiment, the Ab against Met-enkephalin (100 μg) was injected intraplantarly either concomitantly or 1 h before EM-2, as in the study by Ohsawa et al. (2000). To confirm a peripheral site of action, the most effective intraplantar doses of antagonists were injected subcutaneously immediately before intraplantar EMs. PPTs were measured before and 5 min after injections.
Immune cell-derived endomorphin antinociception.
To activate endogenous opioidergic pathways of pain inhibition in inflamed tissue, we used the cold-water swim stress test. At 4 d after induction of inflammation, baseline PPTs were measured and animals were subjected to swimming for 1 min in a metal container filled with cold water (2–4°C). Thereafter, rats were dried and PPTs were reevaluated at 1 min after swimming (i.e., at the time of maximum elevation of PPT), as described previously (Stein et al., 1990a; Machelska et al., 2003). An endogenous trigger of swim stress-induced antinociception is CRF (Schäfer et al., 1996). To mimic this effect, CRF in the most effective dose determined previously (1.5 ng) (Schäfer et al., 1994), was injected into inflamed paws at 4 d after induction of inflammation. PPTs were measured before and 5 min after CRF injection (i.e., at the time of maximum elevation of PPT), as described previously (Schäfer et al., 1994; Machelska et al., 2003). Previously, we had shown that swim stress and CRF did not produce changes in PPT in noninflamed contralateral paws (Stein et al., 1990a; Schäfer et al., 1994; Machelska et al., 2003).
To examine the impact of general leukocyte depletion (see below, Immunohistochemistry and Flow cytometry) on swim stress- and CRF-induced antinociception, separate groups of animals were treated with anti-PMN serum. In contrast to single administration (Brack et al., 2004b), repeated anti-PNM treatment leads to a substantial reduction in the numbers not only of granulocytes but also of mononuclear cells (Savov et al., 2002; Eliason et al., 2005). Therefore, anti-PMN and control sera were injected intravenously immediately before and 1 and 3 d after induction of inflammation. Four days after induction of inflammation (i.e., 1 d after the last injection of sera), antinociception induced by swim stress and CRF was assessed.
To evaluate the participation of endogenous opioid peptides in stress-induced antinociception, Abs against β-endorphin (0.25–4 μg), EM-1 (0.75–3 μg), or EM-2 (0.25–0.75 μg) were injected intraplantarly into inflamed paws 5 min before swim stress. In separate experiments, Abs against EM-1 (2 μg) and EM-2 (0.5 μg) were injected simultaneously intraplantarly. PPTs were determined before Ab injection and were reevaluated at 1 min after swim stress (see above). To assess the contribution of endogenous opioid peptides to CRF-induced antinociception, Abs against β-endorphin (0.025–0.8 μg), EM-1 (2–6 μg), or EM-2 (0.25–0.75 μg) were injected intraplantarly concomitantly with CRF (1.5 ng). In separate experiments, Abs against EM-1 (4 μg) and EM-2 (0.5 μg) were intraplantarly coinjected with CRF (1.5 ng). PPTs were determined before and 5 min after injections (see above). Control groups were injected with equivalent doses of control rabbit IgG and were tested in the same way as groups treated with Abs. The experimenter was blinded to the treatments in all experiments.
Opioid receptor binding
Four days after intraplantar injections of complete Freund’s adjuvant or 0.9% NaCl (0.15 ml), animals were killed by an overdose of halothane, and lumbar (L3–L5) dorsal root ganglia (DRGs) ipsilateral to intraplantar injections were removed. Membranes were obtained from DRGs as described previously (Zöllner et al., 2003). Briefly, the tissue was placed immediately on ice in cold assay buffer (50 mm Tris-HCl and 5 mm EDTA, pH 7.4), homogenized with a Polytron homogenizer (Kinematica, Littau, Switzerland), and centrifuged at 48,000 × g at 4°C for 20 min. The pellet was resuspended in assay buffer followed by a 10 min incubation at 37°C to remove endogenous ligands. The homogenate was centrifuged again at 48,000 × g and resuspended in assay buffer. Membranes were aliquoted and stored at −80°C for 30 min.
Binding studies using [3H] ligands were performed according to Zöllner et al. (2003). μ- and δ-receptor binding sites were examined by incubating 100 μg of membrane protein with 2 nm [3H]-[d-Ala2,N-Me-Phe4,Gly5-ol]enkephalin ([3H]DAMGO) (65 Ci/mmol; selective μ-ligand) (Amersham Biosciences, Little Chalfont, UK) or with 1 nm [3H]-naltrindole (35 Ci/mmol; selective δ-ligand) (Amersham Biosciences) and fixed concentrations of EM-1 (10 μm) and EM-2 (10 μm). Nonspecific binding was defined using 10 μm naloxone hydrochloride (Sigma). Membranes were incubated for 1 h at 30°C in a final volume of 1 ml assay buffer.
Binding studies using a [125I] ligand were performed according to Knapp et al. (1991). Maximum numbers of δ-binding sites (Bmax) in DRG neurons were determined in competition binding experiments by incubating 20 μg of membrane protein with [125I]-[d-Pen2,5]-enkephalin ([125I]-DPDPE) (3.7 MBq, 100 μCi; selective δ-ligand) (Amersham Biosciences) in duplicate samples at six different concentrations (0.001–2 nm) in the presence or absence of unlabeled naloxone hydrochloride (10 μm). Membranes were incubated for 1 h at 30°C in a final volume of 800 μl assay buffer. In these experiments, we evaluated Bmax because [125I] ligands are more sensitive than [3H] ligands, and performing competition binding experiments with the latter ligands would require pooling of many DRGs from large numbers of animals.
In both cases, the reactions were terminated by rapid filtration under vacuum through Whatman GF/B glass fiber filters, followed by four washes with cold buffer (50 mm Tris-HCl, pH 7.4). Bound radioactivity was determined by liquid scintillation spectrophotometry after overnight extraction of the filters in 3 ml of scintillation fluid (EG&G Wallac, Turku, Finland) in the case of [3H] ligands or using a Packard Auto-Gamma 5650 scintillation counter in case of the [125I] ligand.
Immunohistochemistry
We used this procedure to compare the expression of EM-1 and EM-2 with β-endorphin and to evaluate the effect of anti-PMN serum on the number of leukocytes containing these opioids in the paw tissue. Rats (n = 5) were injected intravenously with anti-PMN or control sera, as described above. In separate experiments, we also evaluated expression of Leu-enkephalin in paw tissue (n = 3) as a possible peptide involved in EM-2-induced activation of δ-receptors (see Results). In all cases, rats were deeply anesthetized with isoflurane and perfused transcardially with 0.1 m PBS, pH 7.4, and with cold PBS containing 4% paraformaldehyde and 0.2% picric acid, pH 6.9 (fixative solution). The skin with adjacent subcutaneous tissue was dissected from plantar surfaces of both hindpaws, postfixed in the fixative solution, and cryoprotected in 15% sucrose solution at 4°C overnight, embedded in OCT compound (Miles, Elkhart, IN), and frozen. Seven-micrometer-thick sections were prepared on cryostat and mounted on gelatin-coated slides. These were incubated for 45 min in PBS with 0.3% H2O2 and 10% methanol to block endogenous peroxidase. To prevent nonspecific binding, the sections were incubated for 60 min in PBS containing 0.3% Triton X-100, 1% BSA, 4% goat serum, and 4% horse serum. The sections were then incubated overnight with rabbit anti-rat polyclonal Abs against Leu-enkephalin (4 μg/ml; Phoenix Pharmaceuticals), β-endorphin (1:1000; Peninsula Laboratories), EM-1, or EM-2 (7 μg/ml; Chemicon, Hampshire, UK). Additional staining was performed with a Vectastain avidin–biotin peroxidase complex according to the instructions of the manufacturer using goat anti-rabbit biotinylated secondary Ab and avidin–biotin peroxidase (Vectastain Elite kit; Vector Laboratories, Burlingame, CA). Finally, the sections were washed and stained with 3′,3′-diaminobenzidine tetrahydrochloride (Sigma) containing 0.01% H2O2 in 0.05 m Tris-PBS, pH 7.6, for 3–5 min. After the enzyme reaction, the sections were washed in tap water, counterstained with thionin, dehydrated in alcohol, cleared in xylene, and mounted in DPX (Merck, Darmstadt, Germany). Control experiments for staining specificity included the following: (1) preabsorption of Ab against Leu-enkephalin with Leu-enkephalin (8 μg/ml; Phoenix Pharmaceuticals) and of Abs against β-endorphin, EM-1, or EM-2 with their respective antigenic peptides (12 μg/ml; Sigma); (2) omission of either the primary or secondary Abs or avidin–biotin complex. These control experiments did not show staining for any of the peptides. These procedures were performed according to Mousa et al. (2002) and Machelska et al. (2003).
β-Endorphin-, EM-1-, and EM-2-positive leukocytes were quantified by an observer blinded to the experimental protocol, using a Zeiss (Jena, Germany) microscope (objective, 20×; eyepiece, 10×). The mean number of the respective peptide-expressing cells in four sections per animal and 15 squares (384 μm2 each) per section was calculated, as described previously (Mousa et al., 2002; Machelska et al., 2003).
Flow cytometry
This procedure was used to examine the effect of anti-PMN serum on the leukocyte subpopulations in inflamed paws. Rats (n = 8) were injected intravenously with anti-PMN or control sera, as described above. At 4 d after induction of inflammation, rats were killed with an overdose of isoflurane, and plantar subcutaneous paw tissue from inflamed paws was collected. To obtain a single-cell suspension, the tissue was cut into 1–2 mm pieces, enzymatically digested with collagenase and hyaluronidase (Sigma), and pressed through a 70 μm nylon filter (BD Biosciences, Heidelberg, Germany), as described previously (Rittner et al., 2001; Brack et al., 2004a,b).
Cell surface staining was performed by incubation with mouse anti-rat CD45 conjugated with CyChrome monoclonal Ab (mAb) (4 μg/ml; identifies all hematopoetic cells) and with mouse anti-rat CD3 conjugated with phytoerythrin mAb (4 μg/ml; identifies T cells) (both from BD Biosciences). Other leukocyte subpopulations were identified by intracellular staining after fixation with 1% paraformaldehyde and permeabilization with saponin buffer (Rittner et al., 2001; Brack et al., 2004a,b). Cell suspensions were incubated with CD45 conjugated with CyChrome mAb, mouse anti-rat RP-1 conjugated with phytoerythrin mAb (12 μg/ml; identifies PMN; BD Biosciences), and mouse anti-rat ED1 conjugated with fluorescein isothiocyanate mAb (2 μg/ml; identifies monocytes/macrophages; Serotec, Oxford, UK). In separate aliquots, the specificity of the staining was verified by incubation with appropriate species and isotype-matched control Abs. Cells obtained from a single inflamed paw were quantified by mixing the stained single-cell suspension with fluorescent TruCOUNT beads (BD Biosciences). A total of 70,000 events were acquired per paw. Cell numbers were calculated in relation to the known number of TruCOUNT beads. Data were analyzed using CellQuest Pro software (BD Biosciences).
Statistical analysis
Data are presented as means ± SEM and are expressed in raw values. In behavioral experiments, two-sample comparisons were made using the t test for independent data and paired t test for dependent data. Dose–response curves were analyzed by one-way ANOVA, followed by linear regression. In experiments using opioid receptor antagonists and Abs against opioid peptides, effects of their most effective doses were tested against their own baseline PPTs and against PPTs of respective control groups. For clarity of graphs, effects of antagonists and Abs on EM-induced antinociception are compared with baseline PPTs of a respective representative group (there were no significant differences in baseline PPTs between groups; p > 0.05, one-way ANOVA). Changes in PPTs over time were evaluated by two-way repeated-measures ANOVA followed by the Bonferroni’s test, and multiple comparisons at one time point were performed with one-way ANOVA followed by the Bonferroni’s test. Differences were considered significant if p < 0.05.
Ligand binding data represent values of at least three independent experiments, each performed in duplicate. [125I]-DPDPE saturation binding experiments were fitted to a one-site binding hyperbola using GraphPad Prism software (GraphPad, San Diego, CA) to determine Bmax values.
Results
Exogenous endomorphin antinociception
Antinociceptive effects of EM-1, EM-2, and β-endorphin
At 4–6 d after intraplantar injection of complete Freund’s adjuvant, inflammation was confined to inoculated paws and resulted in swelling (1.9 ± 0.07 vs 0.97 ± 0.01 ml, inflamed vs noninflamed paws) and hyperalgesia (decreased PPT, 38 ± 2.5 vs 70 ± 2.4 g, inflamed vs noninflamed paws) (p < 0.001, paired t test). At 5 min after intraplantar injection of EM-1 (0.3–1.25 μg), EM-2 (0.3–1.25 μg), or β-endorphin (0.125–2 μg) into both hindpaws, PPT increased dose dependently in inflamed (p < 0.001, ANOVA, linear regression) (Fig. 1, left) but not in contralateral noninflamed paws (p > 0.05, ANOVA) (Table 1). There were no significant differences among PPT elevations produced by the most effective doses of EM-1 (0.625 μg), EM-2 (0.625 μg), and β-endorphin (1 μg) (p > 0.05, ANOVA) (Fig. 1, left). EM-1-induced (0.625 μg) and EM-2-induced (0.625 μg) antinociception was maximal at 5 min, remained significantly elevated at 10 min, and returned to the baseline level by 20 min after injection (p < 0.001, Bonferroni’s test) (Fig. 1, middle). We have shown previously that exogenous β-endorphin produced a similar time course of antinociception (Stein et al., 1990a). Systemic injections (subcutaneous) of either EM at the dose of 1.25 μg (the total amount of EM injected intraplantarly in the most effective dose, i.e., 0.625 μg per each hindpaw) did not significantly change PPTs of inflamed (Fig. 1, right) or noninflamed paws (data not shown) (p > 0.05, two-way repeated-measures ANOVA). Peripheral selectivity of intraplantar β-endorphin (in the dose range used here) was demonstrated previously (Stein et al., 1990a).
Figure 1.

Antinociception elicited by exogenous EM-1, EM-2, and β-endorphin (END) in inflamed paws. Left, Dose-dependent antinociceptive effects at 5 min after intraplantar injections (p < 0.001, ANOVA, linear regression). Middle, Time course of EM-1- and EM-2-induced antinociception. *p < 0.001, Bonferroni’s test. Right, Lack of EM-1- and EM-2-induced antinociception after systemic (subcutaneous) injection at the neck (p > 0.05, two-way repeated-measures ANOVA). Data are expressed as means ± SEM.
Table 1.
Effects of opioid peptide injections into inflamed paws on paw-pressure thresholds in contralateral noninflamed paws
| Treatment (μg) | Paw-pressure threshold (g) |
|---|---|
| Control | 63 ± 2.3 |
| Endomorphin-1 | |
| 0.3 | 67 ± 2.6 |
| 0.45 | 74 ± 6.8 |
| 0.625 | 71 ± 3.7 |
| 1.25 | 74 ± 3.1 |
| Control | 70 ± 2.9 |
| Endomorphin-2 | |
| 0.3 | 65 ± 3.1 |
| 0.625 | 66 ± 2.8 |
| 1.25 | 63 ± 3.0 |
| Control | 63 ± 1.4 |
| β-Endorphin | |
| 0.125 | 67 ± 2.3 |
| 0.5 | 62 ± 2.2 |
| 1 | 61 ± 2.2 |
| 2 | 63 ± 3.1 |
Data are means ± SEM.
Opioid receptor selectivity of EM-1-induced antinociception
EM-1 (0.625 μg, i.pl.)-induced antinociception was dose dependently blocked by CTOP (0.125–0.5 μg, i.pl.) (p < 0.001, ANOVA, linear regression) (Fig. 2, left). At the highest dose, CTOP (0.5 μg) reduced PPTs to the baseline level (p > 0.05, paired t test). ICI 174,864 (1.25 μg, i.pl.) also significantly attenuated EM-1-induced antinociception (p < 0.05, t test). However, the effect was small and not dependent on the dose of ICI 174,864 (0.625–2.5 μg; p > 0.05, ANOVA) (Fig. 2, left). NorBNI (0.1–100 μg, i.pl.) had no significant effect (p > 0.05, ANOVA) (Fig. 2, left). Systemic injection (subcutaneous) of the most effective intraplantar dose of CTOP did not significantly change intraplantar EM-1 (0.625 μg)-induced antinociception (p > 0.05, t test; data not shown).
Figure 2.

Opioid receptor selectivity of exogenous EM-1- and EM-2-induced antinociception. Left, EM-1-induced antinociception was dose dependently blocked by μ-receptor (CTOP) but not δ-receptor (ICI 174,864) or κ-receptor (norBNI) selective antagonists (p < 0.001, ANOVA, linear regression; p > 0.05, ANOVA, respectively). Middle, EM-2-induced antinociception was dose dependently blocked by μ-receptor (CTOP) and attenuated by δ-receptor (ICI 174,864 and naltrindole) selective antagonists (p < 0.001, ANOVA, linear regression). EM-2-induced antinociception was not significantly changed by a κ-receptor (norBNI) selective antagonist (p > 0.05, ANOVA). Right, EM-2-induced antinociception was not significantly changed by antibodies against Met-enkephalin (anti-Met-ENK), Leu-enkephalin (anti-Leu-ENK), and β-endorphin (anti-END) (p > 0.05, ANOVA). Receptor antagonists and peptide antibodies were coadministered intraplantarly into inflamed paws with either EM-1 or EM-2. PPTs were measured 5 min after injections. Dashed lines represent baseline paw-pressure thresholds of representative groups and are 40 ± 1.2 g. Data are expressed as means ± SEM.
Opioid receptor selectivity of EM-2-induced antinociception
EM-2 (0.625 μg, i.pl.)-induced antinociception was dose dependently blocked by CTOP (0.0625–0.25 μg, i.pl.) (p < 0.001, ANOVA, linear regression) (Fig. 2, middle). The effect of CTOP (0.25 μg, i.pl) was not significantly different from baseline PPT (p > 0.05, paired t test). Both ICI 174,864 (0.075–0.3 μg, i.pl.) and naltrindole (10–40 μg, i.pl) dose dependently decreased EM-2-induced antinociception (p < 0.001, ANOVA, linear regression) (Fig. 2, middle). The most effective doses of ICI 174,864 (0.15 μg, i.pl.) and naltrindole (40 μg, i.pl.) did not completely abolish this antinociception, i.e., the remaining PPTs were significantly higher than baseline PPTs (p < 0.05, paired t test). Higher doses of ICI 174,864 (0.3 μg, i.pl.) and naltrindole (80 μg, i.pl) did not produce stronger inhibition, and norBNI (0.1–100 μg, i.pl.) had no significant effect on EM-2-induced antinociception (p > 0.05, ANOVA) (Fig. 2, middle). Systemic (subcutaneous) injection of the most effective intraplantar doses of CTOP, ICI 174,864, or naltrindole did not significantly change intraplantar EM-2 (0.625 μg, i.pl.)-induced antinociception (p > 0.05, t test; data not shown).
As a possible mechanism of EM-2-induced activation of δ-receptors, we examined the hypothesis that this effect is indirect after a release of other opioid peptides acting at δ-receptors, such as Met-enkephalin, Leu-enkephalin, or β-endorphin. We reported previously the expression of Met-enkephalin and β-endorphin in inflamed subcutaneous tissue (Stein et al., 1990b; Rittner et al., 2001; Machelska et al., 2003). In the present study, we found specific staining for Leu-enkephalin in mononuclear and polymorphonuclear leukocytes (data not shown). Intraplantar injections of Abs against Met-enkephalin (0.1–20 μg), Leu-enkephalin (0.1–8 μg), or β-endorphin (0.1–2 μg) did not significantly change EM-2-induced antinociception (p > 0.05, ANOVA) (Fig. 2, right). Also, intraplantar Ab against Met-enkephalin (100 μg), given simultaneously or 1 h before EM-2, did not significantly change EM-2-induced antinociception (88 ± 3.6 vs 87 ± 4.8 and 88 ± 3.6 vs 81 ± 6.3 g, respectively; p > 0.05, t test).
EM-1 and EM-2 binding to μ- and δ-receptors in DRG
In DRG cell membranes, we detected specific [3H]-DAMGO binding at μ-receptors with a significant increase in the number of μ-binding sites in DRG ipsilateral to inflamed paws (42.5 ± 2.3 fmol/mg protein) compared with DRG from noninflamed hindlimbs (23.4 ± 2.4 fmol/mg protein) (p < 0.001, t test). EM-1 and EM-2 displaced [3H]-DAMGO binding in DRG from noninflamed (EM-1, 92 ± 1.9%; EM-2, 94.3 ± 1.6%) and inflamed (EM-1, 92.7 ± 2.9%; EM-2, 95.8 ± 1.5%) hindlimbs compared with naloxone displacement (100%).
Because EM-2 produced antinociception also via δ-receptors, we attempted to examine whether EMs bind to peripheral δ-receptors. However, using [3H]-naltrindole, we could not detect any binding in DRG of noninflamed or inflamed hindlimbs. Using [125I]-DPDPE, we found little binding in DRG from untreated animals (Bmax of specific [125I]-DPDPE binding, 7 ± 2.4 fmol/mg protein) and no detectable binding in DRG from inflamed hindlimbs, which made it impossible to examine EM binding to δ-receptors.
Immune cell-derived endomorphin antinociception
Comparative expression of EM-1, EM-2, and β-endorphin
Immunohistochemistry revealed strong immunoreactivities for β-endorphin, EM-1, and EM-2 in leukocytes of inflamed subcutaneous paw tissue at 4 d after intraplantar injection of complete Freund’s adjuvant. These leukocytes had morphological appearances consistent with mononuclear cells and granulocytes (Fig. 3). The number of β-endorphin-positive cells (8.3 ± 0.5) was significantly higher than the number of cells expressing either EM-1 (4.2 ± 0.2) or EM-2 (4.9 ± 0.2) (p < 0.001, t test). The number of EM-1-stained cells was not significantly different from the number of EM-2-positive cells (p > 0.05, t test). A few scattered nerve fibers were positive for EM-2 in both noninflamed and inflamed subcutaneous tissue (data not shown), as reported previously (Mousa et al., 2002). There were no opioid-containing cells in noninflamed contralateral paws (data not shown).
Figure 3.
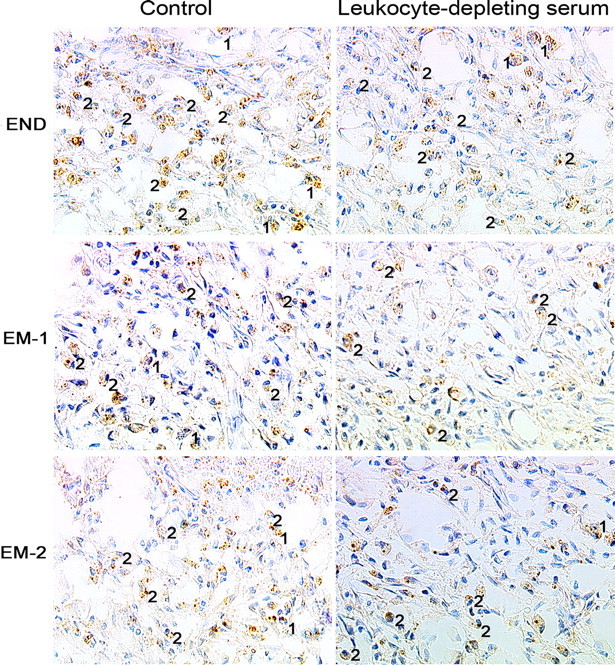
Representative immunohistochemical images showing the effects of leukocyte-depleting serum (intravenously) on the number of cells expressing β-endorphin (END), EM-1, and EM-2 in inflamed subcutaneous paw tissue. The control group was treated with nonimmune serum (intravenously). All three peptides are localized in granulocytes (1) and mononuclear cells (2). Scale bar, 20 μm.
Effects of leukocyte depletion on the number of immune cells containing EM-1, EM-2, or β-endorphin
Pretreatment with intravenous anti-PMN serum significantly reduced the total number of all hematopoetic (CD45+) cells in inflamed paws (Table 2). This treatment caused significant decreases of granulocytes (by 70%), monocytes/macrophages (by 33%), and T cells (by 39%) (p = 0.002, Mann–Whitney U test; p = 0.046, p = 0.009, t tests, respectively) (Table 2), in line with previous studies (Savov et al., 2002; Eliason et al., 2005).
Table 2.
Effect of intravenous leukocyte-depleting serum on immune cell subpopulations in inflamed subcutaneous paw tissue
| Treatment | Total cell number (× 103 per paw) |
|||
|---|---|---|---|---|
| CD45+ cells | Granulocytes | Monocytes/macrophages | T cells | |
| Control | 795 ± 98 | 136 ± 40 | 499 ± 63 | 22 ± 2.4 |
| Anti-PMN | 483 ± 63* | 35 ± 4.5* | 335 ± 37* | 13 ± 1.6* |
Effects were assessed with flow cytometry. Data are means ± SEM.
* indicates significant differences versus respective controls (treated with nonimmune serum, intravenously) (p = 0.006 for CD45+ cells; p = 0.002 for granulocytes; p = 0.046 for monocytes/macrophages; p = 0.009 for T cells, t test).
The pretreatment with intravenous anti-PMN serum significantly diminished the number of cells containing β-endorphin, EM-1, or EM-2 in inflamed paws (Fig. 3). The number of leukocytes containing β-endorphin, EM-1, or EM-2 was decreased by 53% (3.9 ± 0.4 cells), 45% (2.3 ± 0.3 cells), and 41% (2.9 ± 0.3 cells), respectively, compared with the respective control groups (8.3 ± 0.5, 4.2 ± 0.2, and 4.9 ± 0.2 cells; p < 0.001, t test).
Effects of leukocyte depletion on stress- and CRF-induced antinociception
Swim stress produced potent antinociception in inflamed paws of control rats (34 ± 0.8 vs 169 ± 11.9 g, baseline PPT vs PPT at 1 min after stress; p < 0.001, paired t test). This effect was significantly reduced by anti-PMN serum (169 ± 11.9 vs 82 ± 5.6 g, PPT of control vs PPT of anti-PMN; p < 0.001, t test). CRF (1.5 ng, i.pl.) produced antinociception in inflamed paws of control rats (34 ± 0.9 vs 108 ± 5.5 g, baseline PPT vs PPT at 5 min after CRF; p < 0.001, paired t test). Anti-PMN serum significantly decreased this CRF-induced antinociception (108 ± 5.5 vs 71 ± 6.7 g, PPT of control vs PPT of anti-PMN; p = 0.002, t test). In contralateral noninflamed paws, PPTs were not significantly changed by swim stress or by CRF (p > 0.05, paired t test; data not shown), consistent with our previous studies (Stein et al., 1990a; Schäfer et al., 1994; Machelska et al., 2003). PPTs in noninflamed paws were not significantly changed by anti-PMN serum (p > 0.05, t test; data not shown).
Relative contribution of EM-1 and EM-2 versus β-endorphin to stress-induced antinociception
The individual intraplantar application of Abs against β-endorphin (0.25–2 μg), EM-1 (0.75–2 μg), or EM-2 (0.25–0.5 μg) dose dependently attenuated swim stress-induced antinociception (p < 0.001, p = 0.007, and p = 0.002, respectively, ANOVA, linear regression) (Fig. 4, left). The most effective doses of Abs against β-endorphin (2 μg), EM-1 (2 μg), and EM-2 (0.5 μg) decreased this antinociception by 73, 30, and 36%, respectively. These maximum effects of Abs against the two EMs were not significantly different from each other (p > 0.05) but were significantly smaller compared with the effect of Ab against β-endorphin (p < 0.05, Bonferroni’s test) (Fig. 4, right). The effect of concomitant intraplantar Abs against EM-1 (2 μg) and EM-2 (0.5 μg) was not significantly different compared with Abs against each EM injected separately (p > 0.05), but it was significantly smaller than the effect of Ab against β-endorphin (2 μg; p < 0.05, Bonferroni’s test) (Fig. 4, right). None of the treatments significantly changed the PPTs in noninflamed contralateral paws (p > 0.05, ANOVA; data not shown).
Figure 4.

Effects of antibodies against EM-1 (anti-EM-1), EM-2 (anti-EM-2), and β-endorphin (anti-END) injected (intraplantarly) into inflamed paws on swim stress-induced antinociception. Left, Dose dependency of anti-opioid peptides injected separately (p = 0.007 for anti-EM-1; p = 0.002 for anti-EM-2; p < 0.001 for anti-END, ANOVA, linear regression). Right, Comparison of single injections of anti-END, anti-EM-1, and anti-EM-2 with concomitant injection of anti-EM-1 and anti-EM-2. * indicates significant differences versus control, and + indicates significant differences versus anti-END (p < 0.05, Bonferroni’s test). Data are means ± SEM.
Relative contribution of EM-1 and EM-2 versus β-endorphin to CRF-induced antinociception
Single intraplantar administrations of Abs against β-endorphin (0.025–0.4 μg), EM-1 (2–4 μg), or EM-2 (0.25–0.5 μg) dose dependently decreased CRF-induced antinociception in inflamed paws (p < 0.001, p = 0.02, and p = 0.016, respectively, ANOVA, linear regression) (Fig. 5, left). The most effective doses of Abs against β-endorphin (0.2 μg), EM-1 (4 μg), and EM-2 (0.5 μg) decreased this antinociception by 49, 21, and 28%, respectively. Effects of Abs against each EM injected separately were not significantly different compared with each other (p > 0.05) but were significantly smaller compared with the effect of Ab against β-endorphin (p < 0.05, Bonferroni’s test) (Fig. 5, right). The effect of concomitant injection of Abs against EM-1 (4 μg) and EM-2 (0.5 μg) was not significantly different compared with Ab against β-endorphin (0.2 μg) and EM-2 (0.5 μg) injected alone (p > 0.05), but it was significantly stronger than the effect of Ab against EM-1 (4 μg) injected alone (p < 0.05, Bonferroni’s test) (Fig. 5, right). None of the treatments significantly changed the PPTs in noninflamed contralateral paws (p > 0.05, ANOVA; data not shown).
Figure 5.

Effects of antibodies against EM-1 (anti-EM-1), EM-2 (anti-EM-2), and β-endorphin (anti-END) injected (intraplantarly) into inflamed paws on CRF-induced antinociception. Left, Dose dependency of anti-opioid peptides injected separately (p = 0.02 for anti-EM-1; p = 0.016 for anti-EM-2; p < 0.001 for anti-END, ANOVA, linear regression). Right, Comparison of single injections of anti-END, anti-EM-1, and anti-EM-2 with concomitant injection of anti-EM-1 and anti-EM-2. * indicates significant differences versus control, + indicates significant differences versus anti-END, and # indicates a significant difference versus anti-EM-1 (p < 0.05, Bonferroni’s test). Data are means ± SEM.
Discussion
The major finding of this study is that both exogenously applied and immune cell-derived EM-1 and EM-1 can alleviate prolonged inflammatory pain through selective activation of peripheral opioid receptors. In particular, we found the following: (1) that both EMs bind selectively to peripheral μ-receptors, resulting in antinociception; (2) κ-receptors are not involved in EM-1 and EM-2-induced effects; (3) exogenous EM-2, but not EM-1, can also activate peripheral δ-receptors, which is not mediated by other opioid peptides such as Met-enkephalin, Leu-enkephalin, or β-endorphin; and (4) both EMs are expressed in leukocytes accumulating in inflamed tissue and contribute to swim stress- and CRF-induced antinociception.
Peripheral exogenous endomorphin antinociception
Previous studies using spinal and supraspinal injections of EMs have reported antinociception in models of acute pain (without sustained tissue injury) or in short-lasting inflammation (1–3 h) (Horvath, 2000; Przewlocki and Przewlocka, 2001). We have now shown that both EMs can also produce antinociception in prolonged inflammatory pain and that this effect is mediated by peripheral opioid receptors. This is supported by our finding that the antinociceptive effects of both intraplantar EMs were blocked by intraplantar application of systemically ineffective doses of opioid receptor antagonists. Peripheral effects of EMs were also found in animals with neuropathic pain (Obara et al., 2004). In our study, both EMs were similarly effective and produced antinociception of comparable duration, in line with findings in other models (Stone et al., 1997; Tseng et al., 2000; Sakurada et al., 2001; Obara et al., 2004). Also, the antinociceptive efficacies of both EMs were similar to the effect of β-endorphin, and all of these effects seem to be less than those produced by morphine in our previous study (Stein et al., 1989). The selective effect of EMs in inflamed paws is in agreement with previous studies and likely related to upregulation and increased G-protein coupling of peripheral opioid receptors, to a disruption of the perineural barrier, and to an increased number of sensory nerve terminals in inflamed tissue (for review, see Stein et al., 2003).
Both EMs activated peripheral μ-receptors because their effects were fully and dose dependently blocked by the selective μ-receptor antagonist CTOP. Consistently, we found high-affinity binding of both peptides to μ-receptors in DRG, displacing [3H]-DAMGO binding by ∼92–96%. Our findings on μ-receptor-mediated effects of EMs are in accordance with other studies evaluating EM antinociception, with radioligand receptor binding and signaling in the CNS or cell cultures (Zadina et al., 1997; Goldberg et al., 1998; Horvath, 2000; Monory et al., 2000; Przewlocki and Przewlocka, 2001). Although in our study EM-1-induced antinociception was decreased by a δ-antagonist, this effect was small and not dose dependent, and a κ-antagonist had no effect. This indicates that EM-1 can reduce prolonged inflammatory pain acting exclusively at μ-receptors in peripheral tissue, similar to the CNS (Horvath, 2000; Przewlocki and Przewlocka, 2001).
That EM-2-induced peripheral antinociception was not mediated by κ-receptors in our experiments is in agreement with Goldberg et al. (1998) but in contrast to other studies evaluating supraspinal (Ohsawa et al., 2000; Tseng et al., 2000; Makulska-Nowak et al., 2001) or spinal (Ohsawa et al., 2001a; Sakurada et al., 2001) effects of EM-2. We also found that peripheral EM-2-induced antinociception was significantly and dose dependently attenuated by two antagonists selective at δ-receptors (naltrindole and ICI 174,864). Such effects were not observed by Goldberg et al. (1998) but were reported by Ohsawa et al. (2000) and Sakurada et al. (2001). In the latter two studies, the authors suggested that EM-2 releases Met-enkephalin, which activates δ-receptors to produce antinociception (Ohsawa et al., 2001b). Although opioid peptides with affinity at δ-receptors, such as enkephalins and β-endorphin, are expressed in inflammatory cells (Stein et al., 1990b; Machelska et al., 2003; present study) and in sensory neurons (Antunes Bras et al., 2001), intraplantar Abs against Met-enkephalin, Leu-enkephalin, and β-endorphin (in wide dose ranges) did not significantly alter intraplantar EM-2-induced antinociception in our study. It is possible that differences between our study and those by Sakurada et al. (2001) and Ohsawa et al. (2000) might be related to differences between sources of Abs against Met-enkephalin. However, the same Ab against Met-enkephalin used in our present experiments decreased swim stress-induced antinociception in our previous studies (Machelska et al., 2003). Also, the dose and time of Ab preinjection do not seem to account for these differences because administration of this Ab following the schedule used by Ohsawa et al. (2000) did not change EM-2-induced antinociception in our present study. Thus, in contrast to the CNS (Ohsawa et al., 2000; Sakurada et al., 2001), EM-2-triggered release of other endogenous opioid peptides does not seem to play a role in peripheral inflamed tissue in our model.
Finally, we tested the hypothesis that EM-2 binds directly to δ-receptors on sensory neurons. In previous studies, EM-2 did not bind or activate δ-receptors in membranes from δ-receptor-expressing cells or in brain of wild-type and μ-receptor knock-out mice without inflammation (Goldberg et al., 1998; Monory et al., 2000). Because inflammation elevates the number and efficacy of G-protein coupling of opioid receptors in DRGs (Zöllner et al., 2003), we performed binding experiments in animals with peripheral inflammation. Nonetheless, we only detected small amounts of δ-binding sites in DRGs from noninflamed and no detectable δ-binding in DRGs from inflamed hindlimbs. This may be attributed to a low expression of δ-receptor mRNA, inflammation-induced downregulation, or preferential cytoplasmic localization of δ-receptors in DRGs (Buzas and Cox, 1997; Zhang et al., 1998; Bao et al., 2003; Pühler et al., 2004). Conversely, δ-receptors have been demonstrated repeatedly on peripheral terminals of sensory neurons (Wenk and Honda 1999; Brack et al., 2004a), and the injection of selective δ-receptor agonists directly into inflamed tissue produces peripheral antinociception in our model (Stein et al., 1989). This might be explained by inflammation-induced enhanced axonal transport of opioid receptors toward peripheral nerve terminals (Mousa et al., 2001; Pühler et al., 2004). Thus, a possible direct action of EM-2 at δ-receptors on peripheral terminals of sensory nerves remains to be elucidated. Unfortunately, it is impossible to perform quantitative binding experiments in inflamed subcutaneous tissue because of its highly heterogeneous nature.
Immune cell-derived endomorphin antinociception
The expression of EMs in mononuclear cells and granulocytes is consistent with previous reports (Jessop et al., 2002, Mousa et al., 2002). Extending those observations, we now compare the expression of EMs with β-endorphin, the most extensively characterized opioid peptide in the control of inflammatory pain (Stein et al., 1990a; Mousa et al., 2004). Our quantitative analysis revealed similar numbers of leukocytes containing EM-1 and EM-2, but these cells were significantly less abundant (by approximately twofold) than cells containing β-endorphin. Furthermore, we found that depletion of hematopoietic (CD45+) leukocytes, including cells containing EM-1, EM-2, and β-endorphin, in inflamed tissue attenuates stress- and CRF-induced antinociception.
Consistent with the degree of depletion of CD45+ leukocytes and of immune cells containing β-endorphin, EM-1, or EM-2 (all by ∼50%), anti-PMN serum attenuated stress- and CRF-induced antinociception by ∼40–50%. This indicates that leukocytes are the source of opioids producing both forms of antinociception and is in line with previous studies (Schäfer et al., 1994; Hermanussen et al., 2004; Machelska et al., 2004). Importantly, both stress- and CRF-induced antinociception was dose dependently attenuated by pretreatment with Abs against EM-1 and EM-2. Based on data provided by the supplier, both Abs appear selective because they react in 100% with their respective peptides and their cross-reactivities do not exceed 1%. The calculated 1.7-fold higher affinity of anti-EM-2 versus anti-EM-1 for their respective peptides (Phoenix Pharmaceuticals) might account for lower potency of anti-EM-1 (0.75–4 μg) compared with anti-EM-2 (0.25–0.75 μg) to attenuate swim stress- and CRF-induced antinociception observed in our experiments. Thus, these data suggest that EM release is stimulated by CRF, as we showed previously for β-endorphin (Schäfer et al., 1994; Cabot et al., 1997). However, the effects of either single or combined Abs against EM-1 and EM-2 were less than that of anti-β-endorphin Ab. Thus, it appears that leukocyte-derived EMs contribute less than leukocyte-derived β-endorphin to the endogenous attenuation of inflammatory pain.
Finally, we found EM-2 but not EM-1 in sensory neurons in subcutaneous paw tissue (Mousa et al., 2002; present study). However, it seems unlikely that neuron-derived EM-2 contributes substantially to swim stress- and CRF-induced antinociception because (1) the degree of attenuation of stress- and CRF-induced antinociception by anti-PMN serum strongly correlated with the effect on leukocyte depletion (see above paragraph), and (2) stress- and CRF-induced antinociception were not significantly attenuated by anti-EM-2 compared with anti-EM-1, which has not been found in neurons (Mousa et al., 2002; present study). Together, it appears that leukocytes in peripheral inflamed tissue are the main source of EMs to produce local antinociception in response to swim stress or CRF application.
Conclusions
Exogenously applied EM-1 and EM-2 decrease pain in inflamed tissue. Both peptides bind to peripheral μ-receptors, and EM-1 selectively activates these receptors. EM-2 apparently also activates peripheral δ-receptors, which is not related to release of other opioid peptides. Furthermore, leukocyte-derived EM-1 and EM-2 contribute to the control of inflammatory pain, yet their participation seems smaller than that of the classical opioid peptide β-endorphin. Additional studies need to clarify whether leukocyte-derived EMs are generally less important or whether other stimuli are more efficient to elicit their release and increase their role in pain control.
Footnotes
This study was supported by Bundesminsterium für Bildung und Forschung Grant 01GZ0311 (H.M., C.S.) and State Committee for Scientific Research Grant K062/P05/2003 (B.P.).
References
- Antunes Bras J, Becker C, Bourgoin S, Lombard M, Cesselin F, Hamon M, Pohl M (2001). Met-enkephalin is preferentially transported into the peripheral processes of primary afferent fibres in both control and HSV1-driven proenkephalin A overexpressing rats. Neuroscience 103:1073–1083. [DOI] [PubMed] [Google Scholar]
- Bao L, Jin SX, Zhang C, Wang LH, Xu ZZ, Zhang FX, Wang LC, Ning FS, Cai HJ, Guan JS, Xiao HS, Xu ZQ, He C, Hökfelt T, Zhou Z, Zhang X (2003). Activation of delta opioid receptors induces receptor insertion and neuropeptide secretion. Neuron 37:121–133. [DOI] [PubMed] [Google Scholar]
- Brack A, Rittner HL, Machelska H, Shaqura M, Mousa SA, Labuz D, Zollner C, Schafer M, Stein C (2004a). Endogenous peripheral antinociception in early inflammation is not limited by the number of opioid-containing leukocytes but by opioid receptor expression. Pain 108:67–75. [DOI] [PubMed] [Google Scholar]
- Brack A, Rittner HL, Machelska H, Leder K, Mousa SA, Schafer M, Stein C (2004b). Control of inflammatory pain by chemokine-mediated recruitment of opioid-containing polymorphonuclear cells. Pain 112:229–238. [DOI] [PubMed] [Google Scholar]
- Buzas B, Cox BM (1997). Quantitative analysis of mu and delta opioid receptor gene expression in rat brain and peripheral ganglia using competitive polymerase chain reaction. Neuroscience 76:479–489. [DOI] [PubMed] [Google Scholar]
- Cabot PJ, Carter L, Gaiddon C, Zhang Q, Schäfer M, Loeffler JP, Stein C (1997). Immune cell-derived beta-endorphin. Production, release, and control of inflammatory pain in rats. J Clin Invest 100:142–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eliason JL, Hannawa KK, Ailawadi G, Sinha I, Ford JW, Deogracias MP, Roelofs KJ, Woodrum DT, Ennis TL, Henke PK, Stanley JC, Thompson RW, Upchurch GR Jr (2005). Neutrophil depletion inhibits experimental abdominal aortic aneurysm formation. Circulation 112:232–240. [DOI] [PubMed] [Google Scholar]
- Goldberg IE, Rossi GC, Letchworth SR, Mathis JP, Ryan-Moro J, Leventhal L, Su W, Emmel D, Bolan EA, Pasternak GW (1998). Pharmacological characterization of endomorphin-1 and endomorphin-2 in mouse brain. J Pharmacol Exp Ther 286:1007–1013. [PubMed] [Google Scholar]
- Han Z, Jiang YH, Wan Y, Wang Y, Chang JK, Han JS (1999). Endomorphin-1 mediates 2 Hz but not 100 Hz electroacupuncture analgesia in the rat. Neurosci Lett 274:75–78. [DOI] [PubMed] [Google Scholar]
- Hermanussen S, Do M, Cabot PJ (2004). Reduction of beta-endorphin-containing immune cells in inflamed paw tissue corresponds with a reduction in immune-derived antinociception: reversible by donor activated lymphocytes. Anesth Analg 98:723–729. [DOI] [PubMed] [Google Scholar]
- Horvath G (2000). Endomorphin-1 and endomorphin-2: pharmacology of the selective endogenous mu-opioid receptor agonists. Pharmacol Ther 88:437–463. [DOI] [PubMed] [Google Scholar]
- Huang C, Wang Y, Chang JK, Han JS (2000). Endomorphin and mu-opioid receptors in mouse brain mediate the analgesic effect induced by 2 Hz but not 100 Hz electroacupuncture stimulation. Neurosci Lett 294:159–162. [DOI] [PubMed] [Google Scholar]
- Jessop DS, Richards LJ, Harbuz MS (2002). Opioid peptides endomorphin-1 and endomorphin-2 in the immune system in humans and in a rodent model of inflammation. Ann NY Acad Sci 966:456–463. [DOI] [PubMed] [Google Scholar]
- Knapp RJ, Sharma SD, Toth G, Duong MT, Fang L, Bogert CL, Weber SJ, Hunt M, Davis TP, Wamsley JK, Hruby VJ, Yamamura HI (1991). d-Pen2,4′-125I-Phe4,d-Pen5]enkephalin: a selective high affinity radioligand for delta opioid receptors with exceptional specific activity. J Pharmacol Exp Ther 258:1077–1083. [PubMed] [Google Scholar]
- Leff P, Matus M, Calva JC, Salazar A, Acevedo R, De los Heros P, Gompfh H, Allen C, Peng B, Alagon A, Gamba G, Pintar J, Anton B (2005). In: Cloning and functional characterization of a novel opioid peptide system for the mu opioid receptor Presented at the annual International Narcotics Research Conference meeting, Annapolis, MD, July, abstract S48, http://www.inrcworld.org/2005meeting/INRC05Prog6_28_05LATE.pdf.
- Machelska H, Cabot PJ, Mousa SA, Zhang Q, Stein C (1998). Pain control in inflammation governed by selectins. Nat Med 4:1425–1428. [DOI] [PubMed] [Google Scholar]
- Machelska H, Mousa SA, Brack A, Schopohl JK, Rittner HL, Schäfer M, Stein C (2002). Opioid control of inflammatory pain regulated by intercellular adhesion molecule-1. J Neurosci 22:5588–5596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machelska H, Schopohl JK, Mousa SA, Labuz D, Schäfer M, Stein C (2003). Different mechanisms of intrinsic pain inhibition in early and late inflammation. J Neuroimmunol 141:30–39. [DOI] [PubMed] [Google Scholar]
- Machelska H, Brack A, Mousa SA, Schopohl JK, Rittner HL, Schäfer M, Stein C (2004). Selectins and integrins but not platelet-endothelial cell adhesion molecule-1 regulate opioid inhibition of inflammatory pain. Br J Pharmacol 142:772–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makulska-Nowak HE, Gumulka SW, Lipkowski AW, Rawa MA (2001). Effects of endomorphin-2 on arterial blood pressure and pain threshold in spontaneously hypertensive rats and modification of these effects by beta-funaltrexamine and nor-binaltorphimine. Life Sci 69:581–589. [DOI] [PubMed] [Google Scholar]
- Martin-Schild S, Zadina JE, Gerall AA, Vigh S, Kastin AJ (1997). Localization of endomorphin-2-like immunoreactivity in the rat medulla and spinal cord. Peptides 18:1641–1649. [DOI] [PubMed] [Google Scholar]
- Martin-Schild S, Gerall AA, Kastin AJ, Zadina JE (1999). Differential distribution of endomorphin 1- and endomorphin 2-like immunoreactivities in the CNS of the rodent. J Comp Neurol 405:450–471. [PubMed] [Google Scholar]
- Monory K, Bourin MC, Spetea M, Tomboly C, Toth G, Matthes HW, Kieffer BL, Hanoune J, Borsodi A (2000). Specific activation of the mu opioid receptor (MOR) by endomorphin 1 and endomorphin 2. Eur J Neurosci 12:577–584. [DOI] [PubMed] [Google Scholar]
- Mousa SA, Zhang Q, Sitte N, Ji R, Stein C (2001). Beta-endorphin-containing memory-cells and mu-opioid receptors undergo transport to peripheral inflamed tissue. J Neuroimmunol 115:71–78. [DOI] [PubMed] [Google Scholar]
- Mousa SA, Machelska H, Schäfer M, Stein C (2002). Immunohistochemical localization of endomorphin-1 and endomorphin-2 in immune cells and spinal cord in a model of inflammatory pain. J Neuroimmunol 126:5–15. [DOI] [PubMed] [Google Scholar]
- Mousa SA, Shakibaei M, Sitte N, Schafer M, Stein C (2004). Subcellular pathways of beta-endorphin synthesis, processing, and release from immunocytes in inflammatory pain. Endocrinology 145:1331–1341. [DOI] [PubMed] [Google Scholar]
- Obara I, Przewlocki R, Przewlocka B (2004). Local peripheral effects of mu-opioid receptor agonists in neuropathic pain in rats. Neurosci Lett 360:85–89. [DOI] [PubMed] [Google Scholar]
- Ohsawa M, Mizoguchi H, Narita M, Chu M, Nagase H, Tseng LF (2000). Differential mechanisms mediating descending pain controls for antinociception induced by supraspinally administered endomorphin-1 and endomorphin-2 in the mouse. J Pharmacol Exp Ther 294:1106–1111. [PubMed] [Google Scholar]
- Ohsawa M, Mizoguchi H, Narita M, Nagase H, Kampine JP, Tseng LF (2001a). Differential antinociception induced by spinally administered endomorphin-1 and endomorphin-2 in the mouse. J Pharmacol Exp Ther 298:592–597. [PubMed] [Google Scholar]
- Ohsawa M, Shiraki M, Mizoguchi H, Narita M, Kawai K, Nagase H, Cheng EY, Narita M, Tseng LF (2001b). Release of [Met5]enkephalin from the spinal cord by intraventricularly administered endomorphin-2, but not endomorphin-1 in the anesthetized rat. Neurosci Lett 316:1–4. [DOI] [PubMed] [Google Scholar]
- Pierce TL, Wessendorf MW (2000). Immunocytochemical mapping of endomorphin-2-immunoreactivity in rat brain. J Chem Neuroanat 18:181–207. [DOI] [PubMed] [Google Scholar]
- Przewlocki R, Przewlocka B (2001). Opioids in chronic pain. Eur J Pharmacol 429:79–91. [DOI] [PubMed] [Google Scholar]
- Pühler W, Zöllner C, Brack A, Shaqura MA, Krause H, Schäfer M, Stein C (2004). Rapid upregulation of mu opioid receptor mRNA in dorsal root ganglia in response to peripheral inflammation depends on neuronal conduction. Neuroscience 129:473–479. [DOI] [PubMed] [Google Scholar]
- Rittner HL, Brack A, Machelska H, Mousa SA, Bauer M, Schäfer M, Stein C (2001). Opioid peptide-expressing leukocytes: identification, recruitment, and simultaneously increasing inhibition of inflammatory pain. Anesthesiology 95:500–508. [DOI] [PubMed] [Google Scholar]
- Sakurada S, Hayashi T, Yuhki M, Orito T, Zadina JE, Kastin AJ, Fujimura T, Murayama K, Sakurada C, Sakurada T, Narita M, Suzuki T, Tan-no K, Tseng LF (2001). Differential antinociceptive effects induced by intrathecally administered endomorphin-1 and endomorphin-2 in the mouse. Eur J Pharmacol 427:203–210. [DOI] [PubMed] [Google Scholar]
- Savov JD, Gavett SH, Brass DM, Costa DL, Schwartz DA (2002). Neutrophils play a critical role in development of LPS-induced airway disease. Am J Physiol Lung Cell Mol Physiol 283:L952–L962. [DOI] [PubMed] [Google Scholar]
- Schäfer M, Carter L, Stein C (1994). Interleukin 1 beta and corticotropin-releasing factor inhibit pain by releasing opioids from immune cells in inflamed tissue. Proc Natl Acad Sci USA 91:4219–4223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schäfer M, Mousa SA, Zhang Q, Carter L, Stein C (1996). Expression of corticotropin-releasing factor in inflamed tissue is required for intrinsic peripheral opioid analgesia. Proc Natl Acad Sci USA 93:6096–6100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreff M, Schulz S, Wiborny D, Höllt V (1998). Immunofluorescent identification of endomorphin-2-containing nerve fibers and terminals in the rat brain and spinal cord. NeuroReport 9:1031–1034. [DOI] [PubMed] [Google Scholar]
- Spampinato S, Qasem AR, Calienni M, Murari G, Gentilucci L, Tolomelli A, Cardillo G (2003). Antinociception by a peripherally administered novel endomorphin-1 analogue containing beta-proline. Eur J Pharmacol 469:89–95. [DOI] [PubMed] [Google Scholar]
- Stein C, Millan MJ, Shippenberg TS, Peter K, Herz A (1989). Peripheral opioid receptors mediating antinociception in inflammation. Evidence for involvement of mu, delta and kappa receptors. J Pharmacol Exp Ther 248:1269–1275. [PubMed] [Google Scholar]
- Stein C, Gramsch C, Herz A (1990a). Intrinsic mechanisms of antinociception in inflammation: local opioid receptors and β-endorphin. J Neurosci 10:1292–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein C, Hassan AHS, Przewlocki R, Gramsch C, Peter K, Herz A (1990b). Opioids from immunocytes interact with receptors on sensory nerves to inhibit nociception in inflammation. Proc Natl Acad Sci USA 87:5935–5939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein C, Hassan AH, Lehrberger K, Giefing J, Yassouridis A (1993). Local analgesic effect of endogenous opioid peptides. Lancet 342:321–324. [DOI] [PubMed] [Google Scholar]
- Stein C, Pflüger M, Yassouridis A, Hoelzl J, Lehrberger K, Welte C, Hassan AHS (1996). No tolerance to peripheral morphine analgesia in presence of opioid expression in inflamed synovia. J Clin Invest 98:793–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein C, Schäfer M, Machelska H (2003). Attacking pain at its source: new perspectives on opioids. Nat Med 9:1003–1008. [DOI] [PubMed] [Google Scholar]
- Stone LS, Fairbanks CA, Laughlin TM, Nguyen HO, Bushy TM, Wessendorf MW, Wilcox GL (1997). Spinal analgesic actions of the new endogenous opioid peptides endomorphin-1 and -2. NeuroReport 8:3131–3135. [DOI] [PubMed] [Google Scholar]
- Tseng LF (2002). The antinociceptive properties of endomorphin-1 and endomorphin-2 in the mouse. Jpn J Pharmacol 89:216–220. [DOI] [PubMed] [Google Scholar]
- Tseng LF, Narita M, Suganuma C, Mizoguchi H, Ohsawa M, Nagase H, Kampine JP (2000). Differential antinociceptive effects of endomorphin-1 and endomorphin-2 in the mouse. J Pharmacol Exp Ther 292:576–583. [PubMed] [Google Scholar]
- Wenk HN, Honda CN (1999). Immunohistochemical localization of delta opioid receptors in peripheral tissues. J Comp Neurol 408:567–579. [DOI] [PubMed] [Google Scholar]
- Zadina JE, Hackler L, Ge LJ, Kastin AJ (1997). A potent and selective endogenous agonist for the mu-opiate receptor. Nature 386:499–502. [DOI] [PubMed] [Google Scholar]
- Zhang Q, Schäfer M, Elde R, Stein C (1998). Effects of neurotoxins and hindpaw inflammation on opioid receptor immunoreactivities in dorsal root ganglia. Neuroscience 85:281–291. [DOI] [PubMed] [Google Scholar]
- Zöllner C, Shaqura MA, Bopaiah CP, Mousa S, Stein C, Schäfer M (2003). Painful inflammation-induced increase in micro-opioid receptor binding and G-protein coupling in primary afferent neurons. Mol Pharmacol 64:202–210. [DOI] [PubMed] [Google Scholar]