

Abstract
Developing neuronal networks acquire the ability to precisely time events, a key feature required for information processing. In the barrel cortex, encoding of information requires a high-precision temporal code with a resolution of ∼5 ms; however, it is not known what process drives the maturation in timing precision. Here, we report that long-term potentiation (LTP) at thalamocortical synapses in the neonatal layer IV barrel cortex produces a dramatic improvement in the timing of neuronal output and synaptic input. LTP strongly reduces the latency and variability of synaptically evoked action potentials, improving the fidelity of timing to within that predicted to be required for adult sensory processing. Such changes in timing also occur during development in the neonate. LTP also reduces the summation of EPSPs shortening the window for coincidence detection for synaptic input. In contrast to these reliable effects, LTP produced only a modest and variable change in synaptic efficacy. Thus, our findings suggest that the primary role of this form of neonatal LTP is for the acquisition of timing precision and the refinement of coincidence detection, rather than an increase in synaptic strength. Therefore, neonatal thalamocortical LTP may be a critical prerequisite for the maturation of information processing in the barrel cortex.
Keywords: development, glutamate, kainate receptor, AMPA receptor, long-term potentiation, spike timing
Introduction
A key feature of neuronal networks is their ability to precisely time events, a property critical for information processing (Singer, 1999; Koch and Segev, 2000; Fricker and Miles, 2001). For the ascending input to the mature rodent somatosensory cortex, evidence indicates that the first spike initiated by sensory activation encodes the majority of the information, and key to this is a high precision of the temporal code of 2–6 ms resolution (Ghazanfar et al., 2000; Panzeri et al., 2001; Celikel et al., 2004; Foffani et al., 2004). Developmentally, the emergence of timing precision correlates with a maturation in intrinsic neuronal excitability and in the properties of fast synaptic transmission (McCormick and Prince, 1987; Ben-Ari et al., 1997; Brenowitz and Trussell, 2001; Futai et al., 2001; Cathala et al., 2003). This coincides with a developmental period in which NMDA receptor (NMDAR)-dependent synaptic plasticity plays a critical role (Katz and Shatz, 1996; Zhang and Poo, 2001; Foeller and Feldman, 2004).
Cells in layer IV, the input layer, of the S1 somatosensory cortex are organized into structures termed barrels. These barrels are the anatomical correlates of a precise topographical map of the rodent whiskers (Woolsey and Van der Loos, 1970; Simons, 1978). Evidence suggests that NMDAR-dependent long-term potentiation (LTP) occurring in vivo during the first postnatal week plays a critical role in experience-dependent plasticity underlying the development of the cortical map. Activity and NMDAR activation are required for normal development of receptive fields in the barrel cortex and for experience-dependent plasticity during the first postnatal week (Schlaggar et al., 1993; Fox et al., 1996; Foeller and Feldman, 2004). LTP at thalamocortical synapses in the barrel cortex is NMDAR dependent and developmentally restricted to the first postnatal week (Crair and Malenka, 1995). LTP at this synapse also results in a switch from slow kainate receptor-mediated synaptic transmission to fast AMPA receptor (AMPAR)-mediated transmission, a change also occurring developmentally during this period (Kidd and Isaac, 1999).
Here, we investigated the role of LTP at neonatal thalamocortical synapses in the layer IV barrel cortex. We show that this form of LTP has a number of novel properties: it produces a change in the shape of the EPSP but only a relatively modest and variable increase in EPSP amplitude. The consequences of this are that LTP produces a rapid and robust improvement in the timing precision for synaptically evoked action potentials but a relatively small and inconsistent effect on synaptic efficacy. Moreover, the change in the EPSP shape with LTP also reduces postsynaptic summation of subthreshold input, thus reducing the window for coincidence detection of synaptic events. Developmentally, similar changes in EPSP kinetics and in timing precision occur during the first postnatal week, suggesting that this process may occur in vivo. Thus, our findings indicate that this form of neonatal LTP represents a novel mechanism for the activity-dependent acquisition of neuronal timing precision that may be the primary role of this form plasticity.
Materials and Methods
Thalamocortical slices were prepared from postnatal day 3 (P3) to P6 (P0 is designated as the day of birth) CD1 mouse pups as described previously (Kidd and Isaac, 1999, 2001; Kidd et al., 2002). Hippocampal slices were prepared from P12–P15 CD1 mouse pups as described previously (Daw et al., 2000). Slices were perfused with an extracellular solution as follows (in mm): 119 NaCl, 2.5 KCl, 1 NaH2PO4, 26.2 NaHCO3, 11 glucose, 1.3 MgSO4, and 2.5 CaCl2, saturated with 95% O2/5%CO2, pH 7.4, at 33–35°C. Visualized perforated-patch recordings were made under infrared illumination from neurons with stellate cell morphology (for layer IV recordings) and pyramidal cells in the CA1 region (for hippocampal recordings). Recordings were made with patch electrodes (4–7 MΩ) tip-filled with (in mm) 130 potassium methane sulfate, 8.5 NaCl, 5 HEPES, 0.5 EGTA, 0.5 Na-GTP, and 4 Mg-GTP, pH 7.3, 285 mOsm, and back-filled with the same solution but including 6 mg/ml amphotericin B (Isaac et al., 1996) or 1 mg/ml gramicidin D. EPSCs and EPSPs were evoked by electrical stimulation at a frequency of 0.1 Hz by a bipolar stimulating electrode placed in the stratum radiatum for hippocampal experiments and in the ventrobasal (VB) thalamus for thalamocortical experiments (Agmon and Connors, 1991; Kidd and Isaac, 1999, 2001; Kidd et al., 2002). LTP was induced at thalamocortical synapses by pairing 50 stimuli at 0.2 Hz (245 s) with a holding potential of 0 mV. Recent studies have shown that NMDAR-dependent LTP is induced by sensory-evoked activity in rodent primary sensory cortex [e.g., in the visual cortex (Froemke and Dan, 2002) and barrel cortex (Takahashi et al., 2003)]. Although pairing is an artificial protocol for induction of LTP, it fulfils the requirements of providing strong postsynaptic depolarization coincident with afferent activity that allows sufficient NMDAR activation to induce LTP. Therefore, this induction protocol is useful in studying the mechanisms and role of LTP in the barrel cortex.
For the experiments studying two thalamocortical inputs onto the same neuron (see Figs. 6, 7), two stimulating electrodes were placed in neighboring positions in the VB thalamus, and field potential recordings were used to establish that they activated neurons in the same barrel, before making perforated-patch recordings. Pathway specificity was confirmed by the induction of pathway-specific LTP. Recordings were made using a Multiclamp 700A (Molecular Devices, Palo Alto, CA), and signals were filtered at 4 kHz, digitized at 10 kHz, and stored on computer using CED Signal 2 software. Series resistance (30–60 MΩ) was analyzed in voltage clamp throughout the experiments as described previously (Daw et al., 2000) and displayed on-line. Input resistance was monitored throughout the experiments and was stable during recordings. In experiments testing changes in excitability, voltage ramps of 100 ms duration were applied to layer IV neurons in current-clamp mode before and after LTP induction. For experiments in which EPSC waveforms were injected into layer IV neurons (see Fig. 2), whole-cell patch-clamp recordings were made at 33–35°C using the same intracellular solution as above, except with no perforant. Recordings were made using an NPI SEC-05XL amplifier in current-clamp mode, and EPSC waveforms from 4 randomly chosen experiments were injected into neurons in 13 experiments. Data for each set of waveforms were pooled across experiments.
Figure 6.
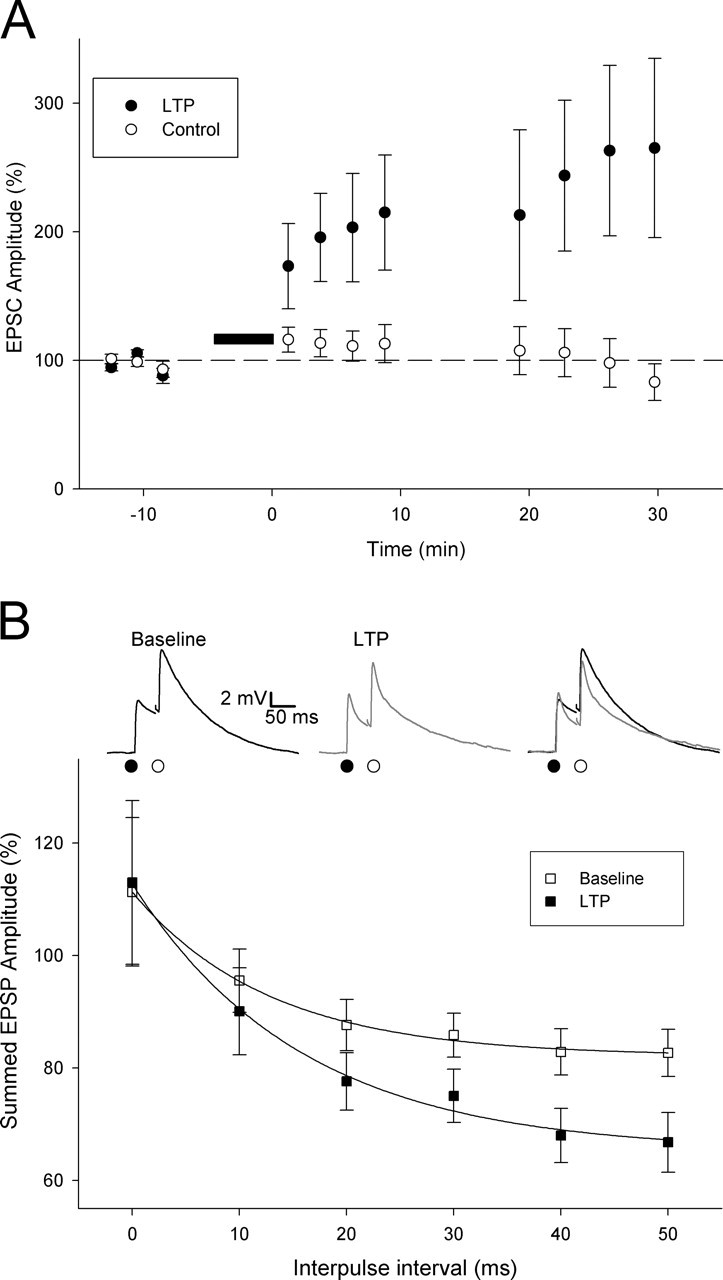
Neonatal LTP alters the time window for temporal summation of thalamocortical input. A, EPSC amplitude versus time for LTP experiments in which two thalamocortical inputs onto the same cell were monitored (LTP induction protocol applied to the pathway is represented by filled circles). The dashed line represents 100%. B, Temporal summation (peak of summed response as a percentage of arithmetic sum of the peaks of the individual contributing EPSPs) at different time intervals before (open squares) and after (filled squares) LTP induction. Inset, Example EPSPs evoked by paired-pulse stimulation (interpulse interval, 50 ms) during baseline (left; black), after LTP induction (middle; gray), and superimposed (right).
Figure 7.

The reduction of temporal summation with neonatal LTP reduces the ability of summed EPSPs to evoke action potentials. A, EPSC amplitude versus time for an example LTP experiment in which two thalamocortical inputs onto the same cell were monitored (LTP induction protocol applied to pathway is represented by filled circles). Inset, Diagram of experimental configuration for the summation LTP–control protocol. B, Current-clamp records taken at the times indicated from the experiment in A in which a stimulus was applied to the LTP path 30 ms before the control path to assess the amount of temporal summation (summation LTP–control protocol) before and after LTP induction. C, Pooled data for EPSC amplitude versus time for all two-pathway LTP experiments (n = 12; filled symbols represent LTP pathway). D, Percentage of second EPSPs that evoke an action potential (AP) baseline versus LTP for each cell (filled symbols) and the pooled data (open symbols). The squares represent summation LTP–control data (n = 6; stimulus interval, 30 ms), and the triangles represent summation control–LTP data (n = 6; stimulus interval, 30 ms). Insets, Diagrams of the two summation protocols. The dashed line is the line of unity. CTRL, Control.
Figure 2.

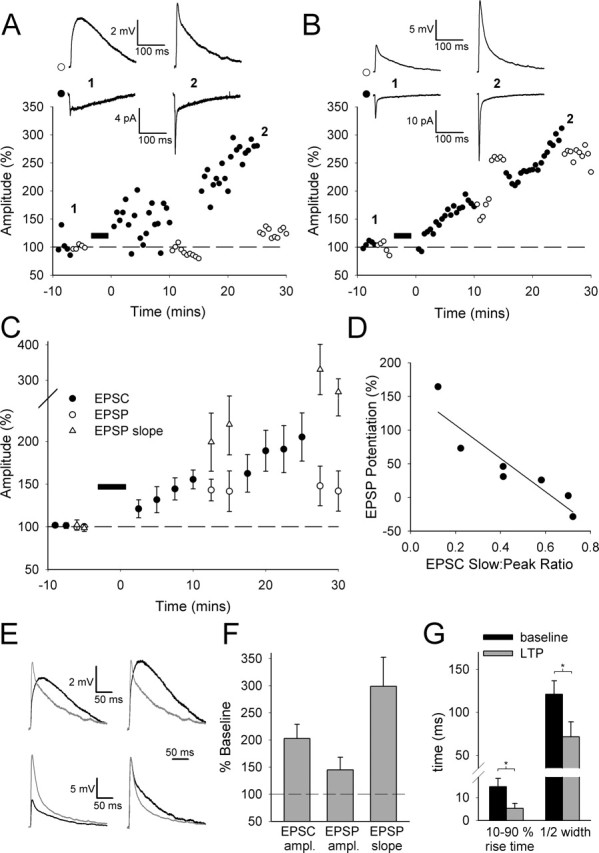
The properties of LTP in neonatal layer IV neurons are a consequence of the change in shape of the underlying EPSC and the membrane properties of the neurons. A, Superimposed voltage responses (EPSP waveforms; top) produced by somatically injected EPSC waveforms (bottom) for an example set of EPSC waveforms. Baseline EPSC (EPSCbaseline; black) and LTP EPSC (EPSCLTP, gray) pairs (left) are shown. Baseline EPSC (EPSCbaseline; black) and baseline EPSC scaled to the peak of the LTP EPSC (EPSCscaled; gray) pairs (right) are shown. B, Resulting EPSP waveforms for the experiment in A, scaled and superimposed, are the EPSCbaseline/EPSCLTP pair (top) and EPSCbaseline/EPSCscaled pair (bottom). C, Pooled data for changes in EPSC amplitude (ampl.) with LTP for the four data sets used and the change in EPSP waveform amplitude and slope for EPSCLTP (left) and EPSCscaled (right). D, Ratio of EPSC slow component: EPSC peak plotted versus mean EPSP peak amplitude change for EPSCLTP. The line is a linear regression (r2 = 0.41; p < 0.001). ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.005.
Statistical analyses were performed using the Student’s t test, and p < 0.05 considered significant.
Results
Neonatal LTP produces a change in the shape of the thalamocortical EPSP
To investigate the role of neonatal LTP, we studied the properties of LTP at developing thalamocortical synapses in layer IV cells of the neonatal (P3–P6) mouse barrel cortex. We made perforated-patch-clamp recordings from layer IV neurons and evoked synaptic responses at thalamocortical synapses by electrical stimulation of thalamocortical axons in the VB thalamus (Agmon and Connors, 1991; Crair and Malenka, 1995; Isaac et al., 1997; Kidd and Isaac, 1999; Kidd et al., 2002). Although the properties of neonatal LTP at this input have been described in some detail under voltage clamp (Isaac et al., 1997; Kidd and Isaac, 1999), little is known about the consequences of LTP induction under current-clamp conditions, the physiological mode of operation. Therefore, we sequentially monitored EPSCs in voltage clamp and EPSPs in current clamp in the same neurons before and after LTP induction (Fig. 1A,B). After collection of a baseline, delivery of a pairing protocol induced a slowly developing LTP (Crair and Malenka, 1995; Isaac et al., 1997; Kidd and Isaac, 1999), measured as an increase in peak EPSC amplitude (average EPSC amplitude 20–25 min after pairing was 203 ± 26% of baseline; n = 7) (Fig. 1A–C). LTP induction, however, only had a variable effect on EPSP peak amplitude in the same cells. On average, a smaller potentiation in EPSP amplitude was observed (145 ± 23%; n = 7; p < 0.05 vs EPSC potentiation) (Fig. 1C,F). Indeed, the amount of EPSP potentiation varied considerably: in some cells there was little amplitude change despite a large increase in EPSC amplitude (Fig. 1A), whereas in others, a similar change in both EPSC and EPSP amplitude was observed (Fig. 1B). No change in input resistance occurred in these recordings (baseline, 479 ± 38 MΩ; LTP, 435 ± 28 MΩ; p = 0.2; n = 7), and the change in EPSP amplitude with LTP did not show any correlation with input resistance (p = 0.5; n = 7). We have described previously that the EPSC at the thalamocortical input in the neonatal barrel cortex is composed of fast AMPAR (AMPAR)-mediated and slow kainate receptor-mediated components (Kidd and Isaac, 1999, 2001). Interestingly, we found that the magnitude of change in EPSP amplitude with LTP was strongly correlated with the relative contribution of the fast and slow components of the baseline EPSC (p < 0.005) (Fig. 1D).
Figure 1.
LTP at thalamocortical synapses on layer IV neurons in the neonatal barrel cortex reliably alters the kinetics of the EPSP but has a variable effect on EPSP amplitude. A, Peak amplitude of EPSCs (filled circles) and EPSPs (open circles) versus time for an example experiment with a prominent slow component to the EPSC during baseline. In A and B, each point indicates an average of three consecutive responses, the filled bar indicates the time of LTP induction, and the insets show traces taken at the times indicated. B, Example LTP experiment for an input with a prominent fast component to the EPSC during baseline. C, Pooled data for all LTP experiments of this type (n = 7) including EPSP slope (triangles). D, Ratio of EPSC slow component: EPSC peak plotted versus EPSP peak amplitude change with LTP (0 = no change from baseline). The line is the linear regression through points (r2 = 0.91; p < 0.005). E, EPSPs before (black) and after (gray) LTP induction from example experiments in A (top) and B (bottom), superimposed (left) and scaled and superimposed (right). F, Changes in EPSC amplitude (ampl.), EPSP amplitude, and EPSP slope with LTP (n = 7). G, EPSP 10–90% rise time (left) and half-width (right) before (black bars) and after (gray bars) LTP induction (n = 7). ∗p < 0.05. For this and subsequent figures, error bars represent SEM.
Because LTP does not cause a reliable change in EPSP amplitude, we sought to determine what reliable change occurs in the synaptic response during LTP under current-clamp conditions. In contrast to EPSP amplitude changes, LTP consistently caused a change in the kinetics of the EPSP (Fig. 1E). There was a large increase in EPSP slope (298 ± 54%; p < 0.005; n = 7) (Fig. 1C,F), a decrease in rise time (10–90% rise time: baseline, 14.8 ± 3.6 ms; LTP, 5.3 ± 2.1 ms; p < 0.05; n = 7), and a speeding of the decay (half-width: baseline, 121.0 ± 15.5 ms; LTP, 71.5 ± 17.5 ms; p < 0.05; n = 7) (Fig. 1G). Our previous work demonstrates that LTP at this input is characterized by a simultaneous rapid increase in the fast AMPAR-mediated component and a decrease in the slow kainate receptor-mediated component to the EPSC (Kidd and Isaac, 1999). The present data indicate that this change consistently speeds up the kinetics of the EPSP but does not reliably increase EPSP amplitude.
The effects of neonatal LTP on the EPSP are attributable to the change in the shape of the EPSC
We wanted to understand whether the unusual features of this form of LTP result simply from a combination of the membrane properties of neonatal layer IV neurons and the properties of the thalamocortical synaptic response, or whether additional mechanisms are required. To investigate this, we randomly chose four LTP experiments and injected the averaged EPSCs from each experiment [the baseline EPSC (EPSCbaseline) and the EPSC after LTP induction (EPSCLTP)] as waveforms into neonatal layer IV neurons during somatic recordings. We recorded the resulting voltage responses (“EPSP waveforms”) in current-clamp mode (Fig. 2A,B). To address the role of the change in EPSC shape observed with LTP, we also injected an EPSC waveform that was the baseline EPSC scaled to the peak of the EPSC after LTP (EPSCscaled). Injection of the EPSCbaseline and EPSCLTP waveforms reproduced the features of LTP we observed experimentally. The increased amplitude of the fast component combined with the decrease in the slow component in EPSCLTP reliably produced little increase in EPSP amplitude (112 ± 23%; n = 4) but a robust increase in slope (240 ± 44%; n = 4; p < 0.01) (Fig. 2A–C). In contrast, EPSCscaled, in which there no is no change in EPSC shape, did not reproduce the properties of LTP because it produced a large increase in EPSP amplitude (195 ± 23%; n = 4; p < 0.05) (Fig. 2A–C). Injection of the EPSCLTP waveform also reproduced the finding that the increase in EPSP amplitude after LTP is correlated with the relative contribution of the slow and fast components to baseline transmission (r2 = 0.41; p < 0.0001) (Fig. 2D). Thus, the features of LTP in neonatal layer IV cells can be reproduced by somatic injections of the EPSC waveforms. This indicates that the properties of this form of LTP primarily result from a combination of the membrane properties of the neonatal layer IV cells combined with the change in shape of the synaptic response observed during LTP.
Neonatal LTP causes a rapid improvement in timing precision of synaptically evoked action potentials
Because LTP is commonly thought of as a mechanism for a long-lasting increase in synaptic strength, what then is the function of neonatal LTP at the thalamocortical input if an increase in EPSP amplitude does not consistently occur? We observe a robust change in the kinetics of the EPSP with LTP, and the ability of EPSPs to precisely time action potentials is dependent on their kinetics (Fricker and Miles, 2000, 2001; Galarreta and Hestrin, 2001; Cathala et al., 2003). Therefore, we wondered whether it is action potential timing that is altered by LTP induction. To address this, we studied the timing of action potentials evoked in layer IV neurons by thalamocortical EPSPs. We sequentially collected data in voltage clamp and current clamp using a stimulation intensity that evoked action potentials in ∼50% of current-clamp trials. The first thing we noticed with these experiments was that naive inputs exhibited remarkably poor timing precision. The latency from EPSP onset to the action potential (“E-S latency”) was very long (23.0 ± 2.2 ms; n = 13), and the timing precision (or jitter), as determined by calculating the SD in E-S latency for each cell (“E-S SD”), was very poor (9.3 ± 1.9; n = 13) compared with previous reports for more mature cortical neurons (Fricker and Miles, 2000, 2001; Jonas et al., 2004).
To investigate the effect of LTP on action potential timing, we measured the latency and variability of synaptically evoked action potentials after LTP induction. LTP (EPSC amplitude, 220 ± 27%; n = 13) (Fig. 3A,C) produced a reliable and pronounced shortening of the subthreshold EPSPs (10–90% rise time: baseline, 16.2 ± 2.5 ms; LTP, 8.4 ± 1.8 ms; p < 0.05; n = 13; half-width: baseline, 81.7 ± 8.3 ms; LTP, 60.3 ± 8.4 ms; p < 0.005; n = 13) and was associated with a robust decrease in E-S latency (baseline: 23.0 ± 2.2 ms, n = 13; LTP: 9.0 ± 1.1 ms, n = 13; p < 0.00005) (Fig. 3A,B,D,E) and in E-S SD (baseline: 9.3 ± 1.9, n = 13; LTP: 3.7 ± 0.7, n = 13; p < 0.005) (Fig. 3A,B,F). However, there was a variable and, on average, only a modest increase in the fraction of EPSPs that evoked action potentials (percentage of EPSPs that evoked an action potential: baseline: 41 ± 5%, n = 13; LTP: 70 ± 9%, n = 13; p < 0.005) (Fig. 3G). Indeed, in some cells there was no change or a decrease in the percentage of EPSPs evoking an action potential, despite a robust potentiation of the EPSC. Moreover, consistent with the variable effects of LTP on EPSP amplitude as shown in Figure 1, there was a correlation between the relative contribution of the fast and slow components to the baseline EPSC and the change in fraction of EPSPs that evoked an action potential (r2 = 0.63; p < 0.01; n = 13; data not shown).
Figure 3.
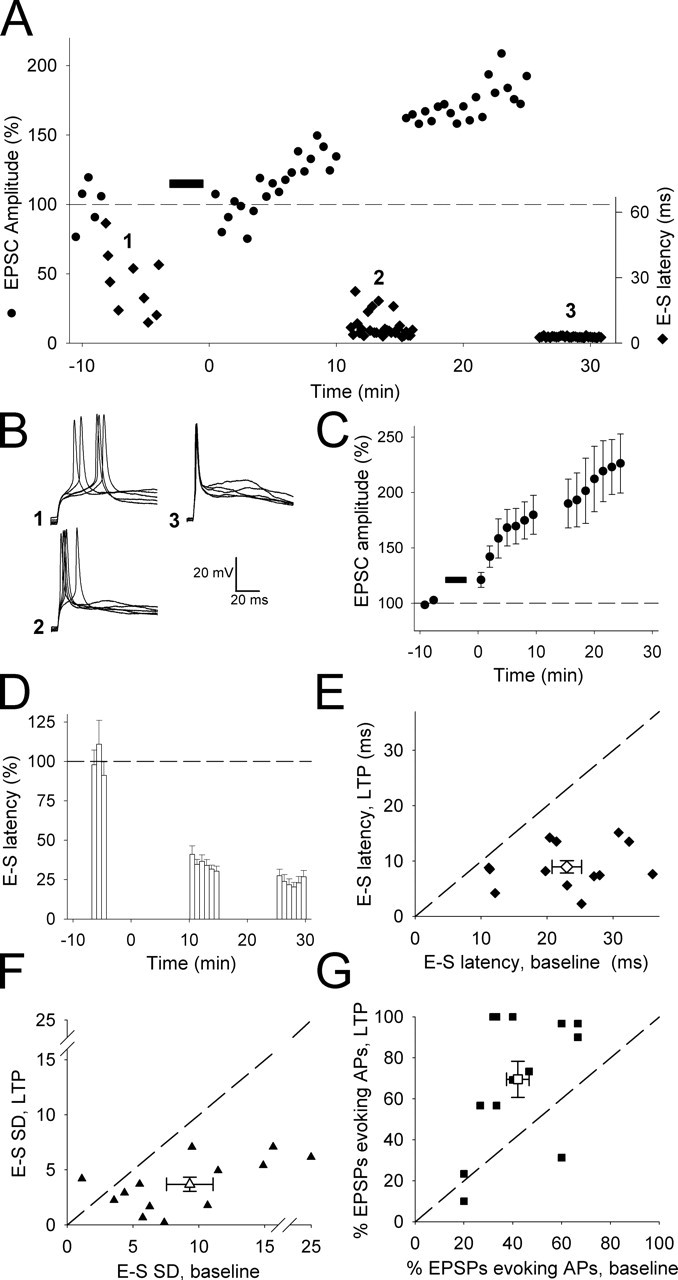
LTP at thalamocortical inputs onto neonatal layer IV neurons reduces the latency and increases the timing precision of synaptically evoked action potentials. A, EPSC amplitude (circles) and E-S latency (diamonds) versus time from an example LTP experiment. B, Representative superimposed current-clamp traces taken at the times indicated in A. C, Pooled data showing normalized EPSC versus time for all LTP experiments (n = 13). D, Pooled data showing normalized E-S latency versus time from these experiments. E, E-S latency during baseline plotted versus E-S latency after LTP induction in individual cells (filled diamonds) and for pooled data (open diamond). F, The E-S SD for baseline plotted versus E-S SD after LTP induction in individual cells (filled triangles) and for the pooled data (open triangle). G, Percentage of EPSPs evoking an action potential (AP) baseline plotted versus LTP in individual cells (filled squares) and for pooled data (open square). The dashed line is the line of unity.
There was no change in input resistance in these LTP experiments (baseline, 577 ± 119 MΩ; LTP, 536 ± 86 MΩ; p = 0.5; n = 13). The change in action potential timing was dependent on LTP induction, because in control experiments in which a sham LTP induction protocol was applied (cell was held at 0 mV, but no synaptic stimulation was applied) and no potentiation occurred (EPSC amplitude, 99 ± 14%; n = 6), there was no change in E-S latency (baseline, 20.1 ± 1.8 ms; n = 6; 20–25 min after sham induction protocol, 19 ± 4 ms) or E-S SD (baseline, 6.1 ± 1.4; n = 6; 20–25 min after sham induction protocol, 6.4 ± 1.6). In a separate set of experiments, we also investigated whether there was a change in excitability during LTP that contributed to the changes in action potential timing precision. We evoked action potentials using somatic injections of depolarizing current ramps during baseline and after LTP induction and measured action potential threshold and latency to action potential initiation. Neither of these parameters changed, demonstrating that LTP did not cause any change in cell excitability [EPSC amplitude: 203 ± 28%, n = 7; action potential threshold: baseline, −31.3 ± 1.3 mV; LTP, −31.6 ± 1.6 mV; change in latency to action potential (LTP baseline): 2.9 ± 2.8 ms, n = 7].
Feedforward activation of GABAergic interneurons by thalamocortical inputs has been shown to affect EPSP kinetics and play a critical role in determining the temporal precision of cortical responses to whisker stimulation in the barrel cortex of P14–P25 rats (Gabernet et al., 2005). We therefore investigated whether feedforward GABAergic transmission contributes to the effects of LTP on action potential timing precision in the neonate. We first determined the reversal potential for pharmacologically isolated GABAA receptor-mediated synaptic currents using gramicidin perforated-patch recordings and local electrical stimulation in layer IV of GABAergic afferents in the presence of NBQX (2,3-dioxo-6-nitro-1,2,3,4-tetrahydrobenzo[f]quinoxaline-7-sulfonoamide; 20 μm) and d-AP-5 (50 μm). In these recordings, the reversal potential for the GABAA receptor-mediated synaptic currents was −59.1 ± 2.8 mV (n = 20). A relatively depolarized reversal potential is typical for the neonatal brain, including the barrel cortex (Agmon et al., 1996). We next used the GABAA receptor antagonist bicuculline (10 μm) to determine the contribution of feedforward GABAergic transmission to the synaptic current evoked by thalamic stimulation. Acute application of bicuculline caused a small reduction in the size of the EPSC (at a holding potential of −70 mV) in some cells but had no effect in others. On average, no significant effect of bicuculline on EPSC amplitude was observed [EPSC integral in bicuculline (percentage of baseline), 97 ± 9%; n = 7]. In the same cells, we also investigated the effect of bicuculline on the timing precision of action potentials evoked by thalamocortical EPSPs. Bicuculline had no significant effect on E-S latency (baseline, 14.7 ± 2.5 ms; bicuculline, 12.1 ± 1.8 ms; n = 5), E-S SD (baseline, 10.3 ± 4.0 ms; bicuculline, 7.9 ± 2.9 ms; n = 5), or percentage of EPSPs evoking an action potential (baseline, 34 ± 5%; bicuculline, 34 ± 11%; n = 7). From these data, we conclude that feedforward GABAergic transmission at this developmental stage makes, at most, only a minor contribution to the synaptic input onto layer IV stellate cells evoked by thalamic stimulation and does not determine action potential timing precision.
Our data therefore suggest that a change in GABAergic transmission is unlikely to contribute to the alteration in spike timing precision observed during LTP. However, to directly test this, we also investigated whether LTP causes the improvement in action potential timing precision in the absence of GABAA receptor-mediated transmission, by sequentially recording in voltage and current clamp and using a stimulation intensity that evoked action potentials in ∼50% of current-clamp trials. LTP was reliably induced in the presence of the GABAA receptor blocker picrotoxin (100 μm) and produced a robust potentiation of EPSC amplitude (184 ± 21%; n = 7). Similar to experiments with GABAA receptor-mediated transmission intact, LTP produced a significant shortening of EPSPs measured from the subthreshold responses (rise time: baseline, 14.8 ± 4.5 ms; LTP, 7.3 ± 1.8 ms; p < 0.05; n = 7; half-width: baseline, 70.2 ± 8.3 ms; LTP, 52.9 ± 7.2 ms; p < 0.05; n = 7). LTP also caused an increase in action potential timing precision: E-S latency was reduced (baseline, 16.0 ± 1.3 ms; LTP, 10.2 ± 1.0 ms; p < 0.0001; n = 7) and became less variable (E-S SD: baseline, 6.0 ± 1.1 ms; LTP, 3.0 ± 1.1 ms; p < 0.005; n = 7), but there was no change in efficacy (percentage of EPSPs evoking an action potential: baseline, 44 ± 8%; LTP, 44 ± 14%; n = 7). Therefore, the LTP-induced improvement in action potential timing precision occurs in the absence of GABAA receptor-mediated responses, suggesting that changes in feedforward GABAergic transmission do not significantly contribute to the effects of LTP we observe.
Little change in timing of synaptically evoked action potentials with hippocampal CA1 LTP
Our data suggest that this form of neonatal LTP primarily affects action potential timing precision and that this is attributable to the change in EPSP shape caused by the switch from kainate- to AMPAR-mediated transmission observed at thalamocortical synapses. To further test this, we studied the properties of LTP at CA1 synapses in adolescent (P12–P15) mice, a form that does not involve such a switch in the type of receptors mediating transmission (Bliss and Collingridge, 1993; Collingridge et al., 2004). The timing of synaptically evoked action potentials in hippocampal CA1 pyramidal neurons at naive inputs was much more precise than in neonatal layer IV neurons (E-S latency: 9.6 ± 1.5 ms, n = 11, p < 0.0005 vs layer IV neurons; E-S SD: 3.9 ± 1.1, n = 11, p < 0.05 vs layer IV neurons) (Fig. 4A) and was very similar to that reported previously for this cell type (Fricker and Miles, 2000). Induction of LTP produced a small change in E-S latency (baseline: 9.6 ± 1.6, n = 10; LTP: 4.6 ± 0.6, n = 10; p < 0.05) (Fig. 4A–C) and E-S SD (baseline: 3.9 ± 1.4, n = 10; LTP: 1.4 ± 0.6, n = 10; p < 0.05) (Fig. 4A,D). In contrast to the variable effect of LTP at thalamocortical synapses, there was a large and much more consistent increase in the fraction of EPSPs that evoked action potentials (baseline: 45 ± 7%, n = 10; LTP: 93 ± 4%, n = 10; p < 0.0005) (Fig. 4E) such that after LTP in most cells, all trials resulted in a postsynaptic action potential. This increase in efficacy with CA1 LTP was significantly larger than that observed with LTP at thalamocortical synapses in the neonatal barrel cortex (p < 0.05).
Figure 4.

LTP at CA1 synapses increases efficacy with only a small effect on action potential timing. A, EPSC amplitude (circles) versus time from pooled LTP experiments (n = 11) in CA1 pyramidal cells and E-S latency (diamonds) from an example cell. B, Representative superimposed current-clamp traces for the example experiment in A, taken at the times indicated. C, E-S latency during baseline plotted versus E-S latency after LTP induction in individual cells and for pooled data. D, E-S SD for baseline plotted versus E-S SD after LTP induction in individual cells and for the pooled data. E, Percentage of EPSPs evoking an action potential baseline plotted versus LTP in individual cells and for pooled data. The dashed line is the line of unity.
Developmental improvement in timing precision
Together, these findings show that neonatal LTP at the thalamocortical input produces a reliable and dramatic improvement in action potential latency and timing precision. The finding that there are no changes in excitability associated with this form of LTP strongly suggests that it is the change in EPSP shape that is responsible for the improvement in timing precision. Given the developmental decrease in the kainate receptor-mediated component of the EPSC at this synapse (Kidd and Isaac, 1999; Bannister et al., 2005) and the evidence that an NMDAR-dependent LTP-like process is driven by experience during early postnatal development (Fox, 2002; Foeller and Feldman, 2004), we investigated whether there was a developmental change in EPSP shape and improvement in timing precision for the thalamocortical input onto layer IV cells by collecting data from cells in slices prepared from animals aged between P3 and P6. Over this age range, we found that there is a reduction in EPSP 10–90% rise time and half-width (10–90% rise time significantly negatively correlated with age: r2 = 0.81, p < 0.05, n = 9; half-width significantly negatively correlated with age: r2 = 0.91, p < 0.05, n = 9) (Fig. 5A). Moreover, we found a strong negative correlation of both E-S latency and E-S SD with age (E-S latency: r2 = 0.96, p < 0.005, n = 12; E-S SD: r2 = 0.81, p < 0.05, n = 12) (Fig. 5B,C). This developmental change in timing was associated with the developmental change in EPSP kinetics because there is a strong co-correlation between these parameters with age (E-S latency vs 10–90% rise time, r2 = 0.997; E-S latency vs half width, r2 = 0.993; E-S SD vs 10–90% rise time, r2 = 0.953; E-S SD vs half-width, r2 = 0.979) (Fig. 5D). We found no developmental correlation of action potential timing with other parameters such as input resistance (Fig. 5E), action potential threshold (Fig. 5F), or EPSC amplitude, ruling out a contribution of these factors to the improvement in timing precision.
Figure 5.
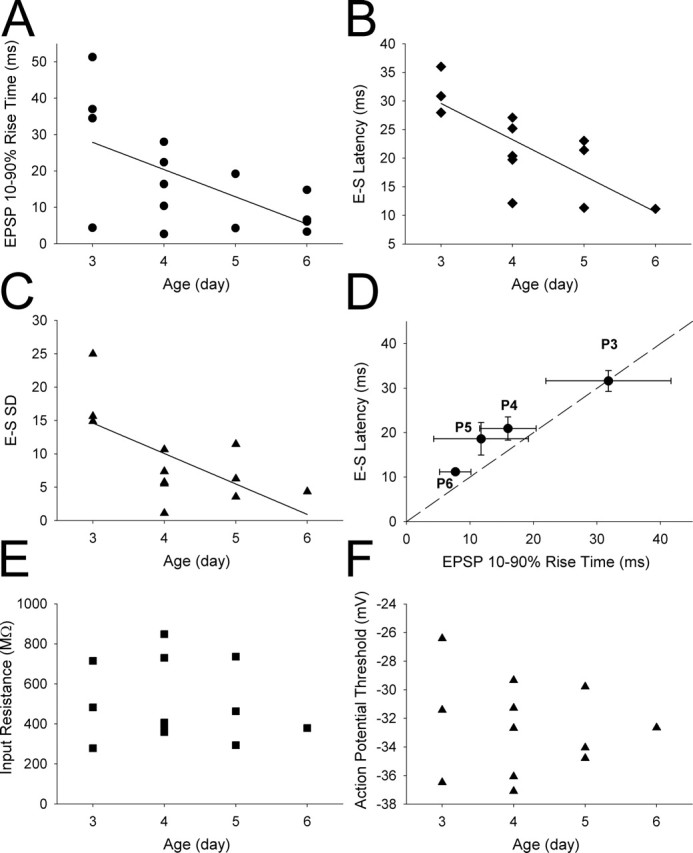
EPSP kinetics and timing of synaptically evoked action potentials in the layer IV neonatal barrel cortex are developmentally regulated. A, EPSP 10–90% rise time plotted versus age of animal from which the slices were made; the line is a linear regression through points (r2 = 0.81; p < 0.05). B, E-S latency plotted versus age; the line is a linear regression through points (r2 = 0.98; p < 0.0001). C, E-S SD plotted versus age; the line is a linear regression through points (r2 = 0.89; p < 0.01). D, Mean E-S latency plotted versus mean EPSP rise time at different postnatal ages; the dashed line is the line of unity. E, Input resistance plotted versus age; there was no significant correlation. F, Action potential threshold plotted versus age; there was no significant correlation.
The magnitude of the developmental change in EPSP kinetics and action potential timing precision was similar to that observed acutely during LTP. At P3–P4, the EPSP rise time was 23.0 ± 2.9 ms (n = 8), the half-width was 144.4 ± 22.9 ms (n = 8), the E-S latency was 27.4 ± 3.4 ms (n = 6), and the E-S SD was 16.7 ± 3.7 (n = 6). By P5–P6, these parameter values were reduced to an EPSP rise time of 9.1 ± 2.4 ms (n = 6), a half-width of 94.7 ± 9.9 ms (n = 6), an E-S latency of 11.2 ± 4.0 ms (n = 4), and an E-S SD of 6.4 ± 2.0 (n = 4). Thus, although P5–P6 EPSP kinetics were still somewhat slower and spike timing was not quite as precise as that observed after LTP, the magnitude of the developmental change was at least as large as observed acutely during LTP. This is explained by the fact that at P3–P4 EPSP kinetics were slower and spike timing was less precise than observed during baseline before LTP induction (the LTP experiments were performed primarily on slices from P4 and P5 animals). Therefore, although it is possible that other developmental processes could also contribute, our findings are consistent with neonatal LTP in vivo, driving a change in EPSP kinetics that produces the developmental improvement in timing precision.
Neonatal LTP alters the time window for coincidence detection precision for synaptic input
Another key feature of mature cortical networks is coincidence detection; a property required for the induction of certain forms of synaptic plasticity, for the generation and detection of synchrony, and for information processing (Jeffress, 1948; Hebb, 1949; Abeles, 1982; Singer, 1999; Bi and Poo, 2001; Fricker and Miles, 2001; Galarreta and Hestrin, 2001). Because a primary effect of neonatal LTP is to speed the decay of the EPSP, we tested the idea that this alters the window for coincidence detection for synaptic input. We investigated this by studying whether the temporal summation of EPSPs was altered after LTP induction. To do this, we evoked responses at two independent pathways onto the same neuron with two stimulating electrodes placed in the thalamus. We tested heterosynaptic summation between subthreshold EPSPs evoked sequentially at each input at a range of intervals from 0 to 50 ms. This heterosynaptic comparison was used to avoid the confounding effect of homosynaptic short-term depression that is prominent at this developmental stage when using paired-pulse stimulation of the same pathway (Kidd and Isaac, 2001; Kidd et al., 2002). The extent of summation was estimated by comparing the peak amplitude of the summed EPSPs with the arithmetic sum of the peaks of the two contributing EPSPs (evoked in isolation in interleaved trials). In baseline conditions, as expected, the peak of the summed response decreased with the increasing interpulse interval (interpulse interval of: 0 ms, 111 ± 13% of arithmetic sum of EPSPs; 10 ms, 96 ± 6%; 20 ms, 88 ± 5%; 30 ms, 86 ± 4%; 40 ms, 83 ± 4%; 50 ms, 83 ± 4%; n = 9) (Fig. 6B). An LTP induction protocol was applied to one input, which resulted in a pathway-specific LTP (EPSC amplitude: LTP path, 265 ± 70%; control path, 83 ± 14%; n = 9) (Fig. 6A): this pathway-specific LTP confirmed the independence of the two pathways in these experiments. The induction of LTP resulted in a speeding of the decay of the EPSP (consistent with our other data), and this produced a decrease in summation at intervals of ≥30 ms (0 ms, 113 ± 15%; 10 ms, 90 ± 8%; 20 ms, 78 ± 5%; 30 ms, 75 ± 5%; 40 ms, 68 ± 5%; 50 ms, 67 ± 5%; n = 9; EPSP summation relationship with an interpulse interval significantly different for baseline vs LTP, p < 0.05, two-way ANOVA with replication) (Fig. 6B). In addition to a reduction in the peak amplitude of the summed EPSP, there was also a pronounced reduction in the duration of the summed EPSP after LTP induction (Fig. 6B).
To investigate whether the reduction in EPSP summation after LTP has a functional consequence for spike output, we tested whether it reduces the ability of summation of EPSPs to evoke action potentials. We therefore repeated the summation experiment using a stimulation intensity sufficient to evoke an action potential by the summed EPSP in ∼50% of trials. We investigated this for a time interval for which the subthreshold experiments indicated that LTP causes a robust decrease in summation: 30 ms. Application of the LTP induction protocol to one input caused a pathway-specific LTP (EPSC amplitude: LTP path: 204 ± 20%, p < 0.0005; control path: 115 ± 14%, p = 0.3, n = 12) (Fig. 7A,C). In six of these experiments, EPSP summation was tested when the LTP path EPSP preceded the control path EPSP, as for subthreshold experiments (“summation LTP-CTRL”) (Fig. 7A,B). In these experiments, LTP caused a reduction in summation as indicated by a reduction in the number of second EPSPs evoking an action potential (percentage of second EPSPs evoking an action potential: baseline: 73 ± 5%, n = 6; LTP: 24 ± 8%, n = 6; p < 0.005) (Fig. 7D, squares). Thus, LTP reduces the probability of evoking an action potential by summed synaptic input when the LTP path precedes the control path. To test whether this decrease in action potential firing is a result of the change in EPSP kinetics with LTP rather than other changes not specifically related to EPSP summation, in the other six experiments we investigated the ability of the summed EPSPs to evoke action potentials when the control path EPSP preceded the LTP path EPSP (“summation CTRL-LTP”). In these experiments, LTP had the opposite effect in that it increased the proportion of second EPSPs evoking an action potential (percentage of second EPSPs evoking an action potential: baseline: 29 ± 6%, n = 6; LTP: 62 ± 12%, n = 6; p < 0.05) (Fig. 7D, triangles). Thus, the probability of the summed EPSPs to evoke an action potential is increased when the LTP path follows the control path. These summation experiments therefore indicate that neonatal LTP, in addition to changing the timing precision of synaptically evoked action potentials, shortens the window for coincidence detection of synaptic input and that this affects the temporal integration of EPSPs and their ability to evoke action potentials.
Discussion
A critical step in the development of neural circuits is the emergence of the ability to precisely time events. During the same developmental period, another prominent feature of neuronal circuits is their ability to undergo activity- and NMDAR-dependent long-term synaptic plasticity, a process that is critically important for the appropriate development of the brain (Katz and Shatz, 1996; Bi and Poo, 2001; Zhang and Poo, 2001; Foeller and Feldman, 2004). Here, we bring together these two important themes of development by demonstrating that NMDAR-dependent LTP at neonatal thalamocortical synapses dramatically improves the precision by which layer IV neurons in the barrel cortex can time events. LTP expression at this input is characterized by a rapid, simultaneous increase in the fast AMPAR-mediated component and a reduction in the slow kainate receptor-mediated component to transmission (Kidd and Isaac, 1999). We show that such a change speeds the rise and shortens the decay of the EPSP, resulting in a robust reduction in the latency and variability of synaptically evoked action potentials. Moreover, the shortening of the EPSP alters the window for coincidence detection, thereby also improving the precision by which synaptic input is timed.
Properties of thalamocortical LTP
Our findings show that the increase in the peak EPSC amplitude that occurs during neonatal LTP in the layer IV barrel cortex does not necessarily result in an increase in EPSP amplitude. Instead, the only reliable change in this form of LTP is a decrease in both the rise time and half-width of the EPSP. This suggests that an increase in synaptic transmission may not be the primary function of this form of LTP. The discrepancy between changes in peak amplitude of EPSPs and EPSCs could simply be attributable to the change in EPSC waveform combined with the membrane properties of the cell, or may involve other processes. One possibility is a reduction in input resistance with LTP; however, we found no change in this parameter occurred during LTP, ruling out this possibility. Experiments in which EPSC waveforms recorded before and after LTP induction were injected into layer IV cells demonstrate that the LTP phenomenon could be reproduced simply by the shape change in the underlying EPSC. Thus, the properties of LTP at thalamocortical inputs onto neonatal layer IV cells are likely to be a consequence of the unique change in the shape of the EPSC observed at these synapses combined with the membrane properties of the neonatal layer IV cells.
An activity-dependent improvement in timing precision
The ability to precisely time inputs and outputs enables neural circuits to detect and generate synchrony, processes widely believed to be critical for information processing (Singer, 1999; Koch and Segev, 2000; Fricker and Miles, 2001; Galarreta and Hestrin, 2001). Moreover, precise timing allows coincidence detection that is required for spike timing-dependent plasticity, a prominent form of synaptic plasticity at more mature hippocampal (Bi and Poo, 2001) and primary sensory cortical (Feldman, 2000; Sjostrom et al., 2001; Froemke and Dan, 2002; Celikel et al., 2004) synapses. Our present findings show that the slow EPSPs observed at naive thalamocortical inputs to layer IV cells result in a very poor temporal precision in the synaptically evoked action potentials. This is in contrast to CA1 pyramidal cells in adolescent animals in which EPSPs evoke action potentials with relatively high precision. LTP results in little change in action potential timing precision in CA1 pyramidal cells, whereas in neonatal layer IV cells, a very prominent decrease in latency and variability of action potential timing is observed. In addition, although LTP at CA1 synapses results in a reliable and large (in most cases maximal) increase in action potential firing probability, the effect of LTP in neonatal layer IV cells on firing probability is highly variable and, on average, significantly smaller.
An additional factor that has a strong influence on the timing of action potentials evoked by EPSPs is feedforward GABAergic transmission. In the adolescent and adult cortex, the hyperpolarization produced by feedforward activation of GABergic interneurons prevents the activation of long-latency action potentials and alters coincidence detection (Pouille and Scanziani, 2001; Wehr and Zador, 2003). In the present study, GABAergic transmission was intact. However, at this developmental stage, we find that the feedforward GABAergic postsynaptic potential is depolarizing (Agmon et al., 1996) and therefore unlikely to play a role in enforcing temporal fidelity. Plasticity of this GABAergic input might be expected to affect the timing of postsynaptic action potentials; however, because we induce LTP by a pairing protocol in individual neurons in our study, there is unlikely to be any plasticity of the feedforward glutamatergic input onto the GABAergic interneurons (because these cells did not experience the pairing protocol). Moreover, an activity-dependent plasticity of the GABAergic synapses onto the recorded neurons, as has been described for CA1 pyramidal neurons (Woodin et al., 2003), is very unlikely to be induced by our pairing protocol. This form of plasticity requires postsynaptic action potentials and is dependent on L-type voltage-gated calcium channel activation, both of which are prevented by our pairing protocol (voltage clamp at 0 mV). Indeed, we show that feedforward GABAA receptor-mediated transmission makes little contribution to the thalamocortical input at this neonatal developmental period, and robust changes in EPSP kinetics and action potential timing precision are observed with LTP in the presence of a GABAA receptor antagonist. Thus, changes in GABAergic transmission are very unlikely to contribute to the effects of LTP on action potential timing.
In addition to a role in the acquisition of output timing precision, neonatal LTP also improves the timing of synaptic input by virtue of its effect on temporal summation of EPSPs. EPSPs at naive thalamocortical inputs in layer IV show pronounced temporal summation. This may be required early in development to provide a large coincidence detection window for induction of synaptic plasticity. However, it is desirable for mature information processing to have a narrow window for coincidence detection (Singer, 1999; Fricker and Miles, 2001). Neonatal LTP contributes to this requirement by shortening the EPSP that reduces summation and thus increases the temporal precision for coincidence detection.
Developmental changes in timing precision
A number of lines of evidence suggest that NMDAR-dependent LTP at the thalamocortical input, occurring in vivo during development in the first postnatal week, plays a critical role in development and experience-dependent plasticity in the barrel cortex (Fox, 2002; Foeller and Feldman, 2004). Previous work has shown that LTP at thalamocortical synapses in the barrel cortex is NMDAR dependent and developmentally restricted to the first postnatal week (Crair and Malenka, 1995). Consistent with an in vivo role for this, activity and NMDAR activation are required for normal development of receptive fields in the barrel cortex and for experience-dependent plasticity during the first postnatal week (Schlaggar et al., 1993; Fox et al., 1996; Foeller and Feldman, 2004). Furthermore, the present study shows that the acute changes in synaptic function and timing we describe for LTP also occur developmentally in vivo, suggesting that an LTP-like mechanism may contribute to the improvement in timing precision during development in layer IV of the barrel cortex. However, maturation of feedforward inhibition and/or changes in neuronal active membrane properties likely also contributes to this developmental phenomenon. Thus, together, our data provide evidence that the primary role LTP at the neonatal thalamocortical input in the barrel cortex is not to cause an increase in synaptic strength, but rather to produce a rapid and long-lasting increase in the temporal precision for timing both inputs and outputs.
Conclusion
A number of studies indicate that for ascending input to mature rodent somatosensory cortex, the first spike initiated by sensory activation encodes the majority of the information, and key to this is a high precision of the temporal code of 2–6 ms resolution (Ghazanfar et al., 2000; Panzeri et al., 2001; Celikel et al., 2004; Foffani et al., 2004). It is noteworthy that, in the present study, we find that naive thalamocortical inputs to layer IV cells evoke action potentials with much poorer resolution than this. However, neonatal LTP induction rapidly improves action potential timing precision to within this required level of precision. Therefore, this form of LTP may be an important developmental mechanism by which activity leads to the acquisition of high-precision timing observed in the adult. Layer IV receives the majority of the ascending input from the sensory periphery via the thalamocortical input. Thus, an early activity-dependent mechanism to produce high-precision timing at this input in layer IV neurons would be an important early step to the development of mature information processing in the barrel cortex. Moreover, there is good evidence that precise timing in layer IV neurons is required for subsequent forms of spike timing-dependent plasticity in layer II/III that are observed later in development (Feldman, 2000; Celikel et al., 2004). Thus, neonatal LTP at the thalamocortical input may be a critical prerequisite for the subsequent development of sensory processing.
Footnotes
This work was supported by the Wellcome Trust (J.T.R.I.) and the National Institute of Neurological Disorders and Stroke Intramural Program (J.T.R.I.).
References
- Abeles M (1982). Role of the cortical neuron: integrator or coincidence detector? Isr J Med Sci 18:83–92. [PubMed] [Google Scholar]
- Agmon A, Connors BW (1991). Thalamocortical responses of mouse somatosensory (barrel) cortex in vitro. Neuroscience 41:365–379. [DOI] [PubMed] [Google Scholar]
- Agmon A, Hollrigel G, O’Dowd DK (1996). Functional GABAergic synaptic connection in neonatal mouse barrel cortex. J Neurosci 16:4684–4695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bannister NJ, Benke TA, Mellor J, Scott H, Gurdal E, Crabtree JW, Isaac JT (2005). Developmental changes in AMPA and kainate receptor-mediated quantal transmission at thalamocortical synapses in the barrel cortex. J Neurosci 25:5259–5271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Ari Y, Khazipov R, Leinekugel X, Caillard O, Gaiarsa JL (1997). GABAA, NMDA and AMPA receptors: a developmentally regulated “menage a trois.”. Trends Neurosci 20:523–529. [DOI] [PubMed] [Google Scholar]
- Bi G, Poo M (2001). Synaptic modification by correlated activity: Hebb’s postulate revisited. Annu Rev Neurosci 24:139–166. [DOI] [PubMed] [Google Scholar]
- Bliss TV, Collingridge GL (1993). A synaptic model of memory: long-term potentiation in the hippocampus. Nature 361:31–39. [DOI] [PubMed] [Google Scholar]
- Brenowitz S, Trussell LO (2001). Maturation of synaptic transmission at end-bulb synapses of the cochlear nucleus. J Neurosci 21:9487–9498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cathala L, Brickley S, Cull-Candy S, Farrant M (2003). Maturation of EPSCs and intrinsic membrane properties enhances precision at a cerebellar synapse. J Neurosci 23:6074–6085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celikel T, Szostak VA, Feldman DE (2004). Modulation of spike timing by sensory deprivation during induction of cortical map plasticity. Nat Neurosci 7:534–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collingridge GL, Isaac JT, Wang YT (2004). Receptor trafficking and synaptic plasticity. Nat Rev Neurosci 5:952–962. [DOI] [PubMed] [Google Scholar]
- Crair MC, Malenka RC (1995). A critical period for long-term potentiation at thalamocortical synapses. Nature 375:325–328. [DOI] [PubMed] [Google Scholar]
- Daw MI, Chittajallu R, Bortolotto ZA, Dev KK, Duprat F, Henley JM, Collingridge GL, Isaac JT (2000). PDZ proteins interacting with C-terminal GluR2/3 are involved in a PKC-dependent regulation of AMPA receptors at hippocampal synapses. Neuron 28:873–886. [DOI] [PubMed] [Google Scholar]
- Feldman DE (2000). Timing-based LTP and LTD at vertical inputs to layer II/III pyramidal cells in rat barrel cortex. Neuron 27:45–56. [DOI] [PubMed] [Google Scholar]
- Foeller E, Feldman DE (2004). Synaptic basis for developmental plasticity in somatosensory cortex. Curr Opin Neurobiol 14:89–95. [DOI] [PubMed] [Google Scholar]
- Foffani G, Tutunculer B, Moxon KA (2004). Role of spike timing in the forelimb somatosensory cortex of the rat. J Neurosci 24:9266–9271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox K (2002). Anatomical pathways and molecular mechanisms for plasticity in the barrel cortex. Neuroscience 111:799–814. [DOI] [PubMed] [Google Scholar]
- Fox K, Schlaggar BL, Glazewski S, O’Leary DD (1996). Glutamate receptor blockade at cortical synapses disrupts development of thalamocortical and columnar organization in somatosensory cortex. Proc Natl Acad Sci USA 93:5584–5589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fricker D, Miles R (2000). EPSP amplification and the precision of spike timing in hippocampal neurons. Neuron 28:559–569. [DOI] [PubMed] [Google Scholar]
- Fricker D, Miles R (2001). Interneurons, spike timing, and perception. Neuron 32:771–774. [DOI] [PubMed] [Google Scholar]
- Froemke RC, Dan Y (2002). Spike-timing-dependent synaptic modification induced by natural spike trains. Nature 416:433–438. [DOI] [PubMed] [Google Scholar]
- Futai K, Okada M, Matsuyama K, Takahashi T (2001). High-fidelity transmission acquired via a developmental decrease in NMDA receptor expression at an auditory synapse. J Neurosci 21:3342–3349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabernet L, Jadhav SP, Feldman DG, Carandini M, Scanziani M (2005). Somatosensory integration controlled by dynamic thalamocortical feed-forward inhibition. Neuron 48:315–327. [DOI] [PubMed] [Google Scholar]
- Galarreta M, Hestrin S (2001). Spike transmission and synchrony detection in networks of GABAergic interneurons. Science 292:2295–2299. [DOI] [PubMed] [Google Scholar]
- Ghazanfar AA, Stambaugh CR, Nicolelis MA (2000). Encoding of tactile stimulus location by somatosensory thalamocortical ensembles. J Neurosci 20:3761–3775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebb DO (1949). In: The organization of behavior: a neuropsychological theory New York: Wiley.
- Isaac JT, Hjelmstad GO, Nicoll RA, Malenka RC (1996). Long-term potentiation at single fiber inputs to hippocampal CA1 pyramidal cells. Proc Natl Acad Sci USA 93:8710–8715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isaac JT, Crair MC, Nicoll RA, Malenka RC (1997). Silent synapses during development of thalamocortical inputs. Neuron 18:269–280. [DOI] [PubMed] [Google Scholar]
- Jeffress LA (1948). A place theory of sound localization. J Comp Physiol Psychol 41:35–39. [DOI] [PubMed] [Google Scholar]
- Jonas P, Bischofberger J, Fricker D, Miles R (2004). Interneuron Diversity series: Fast in, fast out–temporal and spatial signal processing in hippocampal interneurons. Trends Neurosci 27:30–40. [DOI] [PubMed] [Google Scholar]
- Katz LC, Shatz CJ (1996). Synaptic activity and the construction of cortical circuits. Science 274:1133–1138. [DOI] [PubMed] [Google Scholar]
- Kidd FL, Isaac JT (1999). Developmental and activity-dependent regulation of kainate receptors at thalamocortical synapses. Nature 400:569–573. [DOI] [PubMed] [Google Scholar]
- Kidd FL, Isaac JT (2001). Kinetics and activation of postsynaptic kainate receptors at thalamocortical synapses: role of glutamate clearance. J Neurophysiol 86:1139–1148. [DOI] [PubMed] [Google Scholar]
- Kidd FL, Coumis U, Collingridge GL, Crabtree JW, Isaac JT (2002). A presynaptic kainate receptor is involved in regulating the dynamic properties of thalamocortical synapses during development. Neuron 34:635–646. [DOI] [PubMed] [Google Scholar]
- Koch C, Segev I (2000). The role of single neurons in information processing. Nat Neurosci 3:[Suppl], 1171–1177. [DOI] [PubMed] [Google Scholar]
- McCormick DA, Prince DA (1987). Post-natal development of electrophysiological properties of rat cerebral cortical pyramidal neurones. J Physiol (Lond) 393:743–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panzeri S, Petersen RS, Schultz SR, Lebedev M, Diamond ME (2001). The role of spike timing in the coding of stimulus location in rat somatosensory cortex. Neuron 29:769–777. [DOI] [PubMed] [Google Scholar]
- Pouille F, Scanziani M (2001). Enforcement of temporal fidelity in pyramidal cells by somatic feed-forward inhibition. Science 293:1159–1163. [DOI] [PubMed] [Google Scholar]
- Schlaggar BL, Fox K, O’Leary DD (1993). Postsynaptic control of plasticity in developing somatosensory cortex. Nature 364:623–626. [DOI] [PubMed] [Google Scholar]
- Simons DJ (1978). Response properties of vibrissa units in rat SI somatosensory neocortex. J Neurophysiol 41:798–820. [DOI] [PubMed] [Google Scholar]
- Singer W (1999). Neuronal synchrony: a versatile code for the definition of relations? Neuron 24:49–65. 111–125. [DOI] [PubMed] [Google Scholar]
- Sjostrom PJ, Turrigiano GG, Nelson SB (2001). Rate, timing, and cooperativity jointly determine cortical synaptic plasticity. Neuron 32:1149–1164. [DOI] [PubMed] [Google Scholar]
- Takahashi T, Svoboda K, Malinow R (2003). Experience strengthening transmission by driving AMPA receptors into synapses. Science 299:1585–1588. [DOI] [PubMed] [Google Scholar]
- Wehr M, Zador AM (2003). Balanced inhibition underlies tuning and sharpens spike timing in auditory cortex. Nature 426:442–446. [DOI] [PubMed] [Google Scholar]
- Woodin MA, Ganguly K, Poo MM (2003). Coincident pre- and postsynaptic activity modifies GABAergic synapses by postsynaptic changes in Cl- transporter activity. Neuron 39:807–820. [DOI] [PubMed] [Google Scholar]
- Woolsey TA, Van der Loos H (1970). The structural organization of layer IV in the somatosensory region (SI) of mouse cerebral cortex. The description of a cortical field composed of discrete cytoarchitectonic units. Brain Res 17:205–242. [DOI] [PubMed] [Google Scholar]
- Zhang LI, Poo MM (2001). Electrical activity and development of neural circuits. Nat Neurosci 4:[Suppl], 1207–1214. [DOI] [PubMed] [Google Scholar]