

Abstract
Increasing evidence links intraneuronal β-amyloid (Aβ42) accumulation with the pathogenesis of Alzheimer’s disease (AD). In Aβ precursor protein (APP) mutant transgenic mice and in human AD brain, progressive intraneuronal accumulation of Aβ42 occurs especially in multivesicular bodies (MVBs). We hypothesized that this impairs the MVB sorting pathway. We used the trafficking of the epidermal growth factor receptor (EGFR) and TrkB receptor to investigate the MVB sorting pathway in cultured neurons. We report that, during EGF stimulation, APP mutant neurons demonstrated impaired inactivation, degradation, and ubiquitination of EGFR. EGFR degradation is dependent on translocation from MVB outer to inner membranes, which is regulated by the ubiquitin-proteasome system (UPS). We provide evidence that Aβ accumulation in APP mutant neurons inhibits the activities of the proteasome and deubiquitinating enzymes. These data suggest a mechanism whereby Aβ accumulation in neurons impairs the MVB sorting pathway via the UPS in AD.
Keywords: Alzheimer’s disease, endocytosis, neuron, EGFR, TrkB, endosome
Introduction
The mechanism by which β-amyloid (Aβ) is involved in the pathogenesis of Alzheimer’s disease (AD) is a central question in the field. Over the past few years, increasing evidence supports that intraneuronal Aβ accumulation is involved early on in AD pathogenesis (Gouras et al., 2005). Aβ can be generated after endocytosis of Aβ precursor protein (APP) from the plasma membrane (Koo and Squazzo, 1994). Internalized APP traffics from the plasma membrane to multivesicular bodies (MVBs) (Yamazaki et al., 1996), a sorting compartment within the endocytic pathway. β-Site APP cleaving enzyme (BACE) (Vassar et al., 1999) localizes to endosomal vesicles (Huse et al., 2000), which have the required low pH for BACE cleavage of APP. Subsequent cleavage of APP C-terminal fragments by γ-secretase to generate Aβ can also occur in late endosomes (Vetrivel et al., 2005). Indeed, the first immunoelectron microscopy study of presenilin 1 in brain localized this critical γ-secretase component prominently to MVBs of presynaptic and especially postsynaptic compartments (Lah et al., 1997). Abnormalities of neuronal endosomes are among the earliest known pathological changes in AD (Cataldo et al., 2000; Nixon, 2005).
Although the precise location(s) of Aβ production in neurons remains to be established, the subcellular site of Aβ42 accumulation in AD has become clearer. Aβ42 accumulation in the brains of APP mutant transgenic mice and human AD occurs especially in MVBs (Takahashi et al., 2002; Langui et al., 2004). MVBs are vesicular organelles morphologically defined by a limiting membrane and characteristic inner vesicles. MVBs are considered to be late endosomes, formed from early endosomes by membrane invaginations that generate inner vesicles with lower pH (∼5.5) (Gruenberg, 2001). MVBs are involved in many cellular functions, including the regulated trafficking of several proteins and membrane receptors (Gruenberg and Stenmark, 2004). The trafficking of the epidermal growth factor receptor (EGFR) to MVBs has been the most extensively studied. During endosomal maturation, inward invagination of the outer MVB membrane is thought to trap receptors in the MVB interior, terminate signaling and promote their subsequent degradation in the more acidic environment of inner vesicles of MVBs and lysosomes (Sorkin and von Zastrow, 2002; Matsuo et al., 2004). In neurons, the functional role of MVBs is less well understood. MVBs were described as important vesicles in retrograde transport, appearing to carry cargo from neuronal terminals to the cell body for signaling and/or degradation in lysosomes (Parton and Dotti, 1993).
We set out to study the MVB sorting pathway in cultured neurons derived from familial AD Swedish mutant APP transgenic mice, which we demonstrated previously parallel the subcellular accumulation of Aβ42 that occurs in vivo (Takahashi et al., 2004). Remarkably, we observed delayed dephosphorylation and degradation of EGFR during EGF stimulation in APP mutant neurons. EGFR receptor trafficking through the MVB sorting pathway has been described to be regulated by the ubiquitin-proteasome system (UPS) (Longva et al., 2002). We now provide evidence that Aβ accumulation in cultured APP mutant neurons impairs the MVB sorting pathway by inhibiting the UPS.
Materials and Methods
Antibodies.
We used the well characterized affinity-purified polyclonal Aβ42 antibody [immunofluorescence (IF) 1:200; Chemicon, Temecula, CA] (Kamal et al., 2001). We additionally used the following antibodies: early endosomal antigen-1 (EEA1), IF 1:100 (BD Transduction Laboratories, Lexington, KY); EGFR, Western blot (WB) 1:1000 (Upstate Biotechnology, Lake Placid, NY); EGFR, brain IF 1:200 (Santa Cruz Biotechnology, Santa Cruz, CA); p1173 EGFR, WB 1:1000 (Upstate Biotechnology); TrkB, WB 1:1000 (BD Transduction Laboratories); tumor susceptibility gene 101 (Tsg101), IF 1:100 (GeneTex, San Antonio, TX); microtubule associated protein 2 (MAP2), IF 1:1000 (Sigma, St. Louis, MO); ubiquitin, IF 1:200 (Chemicon); ubiquitin, WB 1:100 (Sigma); and α-tubulin, WB 1:10000 (Sigma). We applied secondary antibodies conjugated to Alexa Fluor-488 or -546 (IF 1:500; Invitrogen) or to HRP (WB 1:2000; Amersham Biosciences, Arlington Heights, IL).
Cell culture.
Primary neuronal cultures were prepared from cortices and hippocampi of embryonic day 16 (E16) Tg2576 and wild-type littermate mice (Almeida et al., 2005). One embryo corresponds to one set of cultures, and genotyping was performed using the cerebellum. We also prepared primary neurons from E16 APP−/− embryos (Zheng et al., 1995); a set of embryos correspond to one set of cultures. E16 brain tissue was dissociated by trypsinization and trituration in DMEM with 10% fetal bovine serum. Dissociated neurons were cultured on poly-d-lysine (Sigma) coated plates (2 ± 1 × 103 cells/cm2) and glass coverslips (5 ± 3 × 102 cells/cm2) and were maintained in neurobasal medium, B27 supplement, glutamine (Invitrogen), and antibiotics (Invitrogen). All animal experiments were performed in accordance with the NIH Guide for the Care and Use of Laboratory Animals.
Primary neuron immunofluorescence.
Cultured neurons at 12 and 19 d in vitro (DIV) were fixed in 4% formaldehyde in PBS with 0.12 m sucrose for 20 min, permeabilized, blocked in PBS containing 2% normal goat serum (NGS), 1% bovine serum albumin (BSA), and 0.1% saponin (room temperature, 1 h), and then immunolabeled in 2% NGS in PBS overnight at 4°C. After appropriate washing, coverslips were mounted with Fluoromount-G (Southern Biotechnology, Birmingham, AL). In multiple label experiments, channels were imaged sequentially to avoid bleed-through. Immunofluorescence was examined by confocal laser scanning microscopy (LSM510; Zeiss, Oberkochen, Germany) using a 63×, 1.4 numerical aperture (NA) objective, 488 nm argon and 543 nm helium–neon lasers, and long-pass filtered at 560 Hz and bandpass filtered at 505–530 Hz. For colocalization, optical sections were no thicker than 0.8 μm. We also performed wide-field fluorescence microscopy using an Olympus Optical (Thornwood, NY) IX-70 microscope equipped with an ORCA-ER CCD camera (Hamamatsu Photonics, Bridgewater, NJ) and a 60×, 1.4 NA plan apochromat objective.
For quantification of colocalization, we used a colocalization algorithm (MetaMorph 6.1; Universal Imaging Corporation, West Chester, PA) on thresholded images. We calculated percentage of area of overlap in 30 μm segments of neurite. The colocalization was presented as percentage area overlap of the Aβ42 total area. Images were prepared with Adobe Photoshop 7.0 (Adobe Systems, San Jose, CA).
Fluorescent-labeled ligand endocytosis.
For labeled EGF endocytosis experiments, cultured neurons (12 DIV) were incubated with rhodamine-EGF (rhod-EGF) (500 ng/ml) (Invitrogen), for 10 or 60 min at 37°C in neurobasal medium. For transferrin (Tf) endocytosis experiments, neurons (12 DIV) were preincubated in culture medium in the absence of B27 supplement for 15 min, and then Alexa 488–Tf (20 μg/ml; a gift from Dr. F. Maxfield, Weill Medical College, New York, NY) was added for 10 or 60 min at 37°C. Neurons were washed in PBS and fixed in 4% formaldehyde in PBS with 0.12 m sucrose for 20 min. For the proteasome inhibition experiments, wild-type neurons (12 DIV) were treated with MG132 (carbobenzoxy-Leu-Leu-leucinal, 50 μm; Calbiochem, La Jolla, CA), an irreversible proteasome inhibitor, for 1 or 2 h before 1 h incubation with rhod-EGF.
For quantification of EGF staining, puncta were defined by setting the threshold for staining intensity at least twice that of the background of the neurite. Puncta were delineated, and average intensities, number, and size of puncta per 30 μm of neurite were automatically quantified by image morphometric analysis using MetaMorph 6.1 on thresholded images. Images were prepared with Adobe Photoshop 7.0.
Immunoperoxidase electron microscopy.
Cryoprotected brain sections derived from adult male Sprague Dawley rats (250–300 g), fixed previously by perfusion with 3.75% acrolein (Polysciences, Warrington, PA) and 2% paraformaldehyde in 0.1 m phosphate buffer (PB), pH 7.4, and cut (40 μm thick) on a vibrating microtome, were treated with 1% sodium borohydride, as described previously (Takahashi et al., 2002). For labeling, free-floating sections were incubated with an antibody against the β-subunit of the 20S proteasome (Calbiochem) at a dilution of 1:400. Sections were then incubated in secondary Ig conjugated to biotin (Vector Laboratories, Burlingame, CA) in 0.1% BSA in 0.1 m Tris, followed by peroxidase–avidin complex (Vectastain Elite kit; Vector Laboratories) (Milner et al., 1998). The ABC reaction product was visualized after incubation of the tissues with 3,3-diaminobenzidine (Aldrich Chemical, Milwaukee, WI) and hydrogen peroxide. Sections were fixed in 2% osmium tetroxide in PB, embedded in EMBed 812, sectioned (65–76 nm thick), and counterstained with uranyl acetate and Reynolds’ lead citrate. Final preparations were examined with a Philips CM10 electron microscope. The EM studies were performed at the Division of Neurobiology, Weill Medical College of Cornell University and were partially supported by National Institutes of Health Program Project Grant HL 18974 (C. Iadecola, principle investigator). Final illustrations were generated from a high-resolution digital imaging CCD camera system (Advanced Microscopy Techniques, Danvers, MA) and processed using Adobe Photoshop 7.0.
Brain immunofluorescence.
For Tg2576 mouse brain immunofluorescence microscopy, floating sections, prepared as described above, were incubated in primary antibodies (ubiquitin, 1:250; EGFR, 1:200) for 24 h at room temperature and 40–48 h at 4°C and then incubated with fluorescent secondary antibodies: Alexa Fluor-488 goat anti-mouse and Alexa Fluor-546 goat anti-rabbit (1:500) for 30 min. Immunofluorescence was examined by confocal microscopy using a 63× oil-immersion lens, 1.4 NA; optical slices were 2 μm thick. For quantification of brain immunofluorescence, total fluorescence per field was measured using MetaMorph on thresholded images; three images per section were used. Total fluorescence quantified from staining in the absence of primary antibodies was subtracted from total fluorescence quantified from staining with primary antibodies. Averages of three measurements per section per mice were considered as an individual measurement. Images were prepared with Adobe Photoshop 7.0.
EGFR and TrkB endocytosis.
Wild-type and APP mutant neurons (12 DIV) were incubated with EGF (500 ng/ml; Calbiochem), TGFα (500 ng/ml; Calbiochem), and brain-derived neurotrophic factor (BDNF) (50 ng/ml; PreproTech, Rocky Hill, NJ) for the indicated times at 37°C.
Western blots.
Lysates were prepared using modified radioimmunoprecipitation assay (RIPA) buffer containing 50 mm Tris-HCl, pH 7.4, 1% NP-40, 0.25% sodium deoxycholate, 150 mm NaCl, 1 mm EGTA, 1 tablet per 50 ml complete protease inhibitor cocktail (Roche Diagnostics, Indianapolis, IN), and phosphatase inhibitor cocktail set II (1:100; Calbiochem). Protein concentrations were determined by the Bradford technique (Bio-Rad, Hercules, CA), and equal amounts of protein from each sample were analyzed by NuPage 4–12% Bis-Tris SDS-PAGE (Invitrogen), followed by immunoblotting.
Immunoprecipitation.
Cells were lysed in modified RIPA buffer and protein concentration was determined, as above. Lysates were incubated with protein G-coupled Sepharose beads (Amersham Biosciences) and anti-EGFR overnight at 4°C. Beads were washed three times with ice-cold PBS. After boiling in NuPage sample buffer, samples were analyzed by Western blot and immunoblotted with anti-EGFR or anti-ubiquitin.
Deubiquitinating enzyme activity assay.
The ubiquitin-aminomethylcoumarin (Ub-AMC) assay (Boston Biochem, Cambridge, MA) was performed essentially as described previously (Dang et al., 1998). Briefly, the reaction progress was monitored by the increase of fluorescence intensity at 425 nm (excitation at 340 nm), an indication of hydrolysis of Ub-AMC. Neuronal lysates were incubated in proteolysis buffer (in mm: 10 HEPES, pH 7.4, 5 ATP, 0.5 DTT, and 0.5 MgCl2) at 37°C in a cuvette and placed in a F-4500 Hitachi (Tokyo, Japan) spectrophotometer, and readings were initiated. After ∼60 s, Ub-AMC (250 nm) was added to the reaction that followed for a period of 200 s. Protein concentration was then determined, and, for each sample, proteasome activity was expressed as relative fluorescence units per milligram of protein per second.
Proteasome activity assay.
The proteasome activity assay was performed as described previously (Ehlers, 2003), with slight modifications. Briefly, neuronal lysates were incubated with 100 μm fluorogenic proteasome peptide substrate Suc-LLVY-AMC (Calbiochem) in proteolysis buffer (in mm: 10 HEPES, pH 7.4, 5 ATP, 0.5 DTT, and 0.5 MgCl2) for 30 min at 37°C with shaking. In control experiments, proteasome was inhibited by addition of 50 μm MG132 to lysates 10 min before the fluorogenic substrate. The assay was performed in a 96-well white plate, and fluorescence was measured every 5 min for 2 h after incubation on an HTS 7000 Plus fluorescent plate reader (PerkinElmer, Boston, MA) using excitation and emission wavelengths of 360 and 465 nm, respectively. Protein concentration was then determined, and, for each sample, proteasome activity was expressed as relative fluorescence units per milligram of protein per minute.
Statistical analysis.
Statistical comparisons were made using unpaired t tests or ANOVA with the automated Tukey–Cramer post hoc tests for multiple comparisons, with significance placed at p < 0.05. A set of cultures prepared from one mouse (Tg2576 or wild-type) embryo was considered as one independent experiment (n = 1). A set of cultures prepared from embryos of one pregnant APP−/− mouse was considered as one independent experiment (n = 1). One section prepared from one Tg2576 or wild-type mouse was considered as one independent experiment (n = 1). Data were expressed as mean ± SEM. Statistical analysis was performed using GraphPad Prism 3.0 software (GraphPad Software, San Diego, CA).
Results
In neurites of cultured APP mutant neurons, Aβ42 localizes to late endosomes/MVBs
To characterize the distribution of Aβ accumulation in cultured neurons from APP mutant transgenic mice harboring the human AD Swedish mutation (Tg2576) (Hsiao et al., 1996), we coimmunostained Aβ with a dendritic marker, MAP2 (Fig. 1A). Using a well characterized antibody specific to the C terminus of Aβ42 (Kamal et al., 2001), we observed a punctate pattern of Aβ42 immunofluorescence throughout cultured APP mutant neurons, consistent with the pattern observed using three other specific antibodies against Aβ42 (Runz et al., 2002; Takahashi et al., 2004; Jin et al., 2004). Aβ42 staining localized to both MAP2-positive dendrites and MAP2-negative axons (Fig. 1Aa–Ac). In contrast to Aβ42, immunocytochemistry of APP mutant neurons with an antibody specific for the C terminus of Aβ40 only showed modest fluorescence in neuronal soma, in which it colocalized with a Golgi marker, TGN38, and slight staining in neurites (supplemental Fig. S1, available at www.jneurosci.org as supplemental material). We confirmed the specificity of the Aβ42 immunofluorescence by staining neurons cultured from APP knock-out mice. Despite a low level of background staining in the cell body, there was no Aβ42 immunoreactivity in neurites (Fig. 1Ad–Af).
Figure 1.
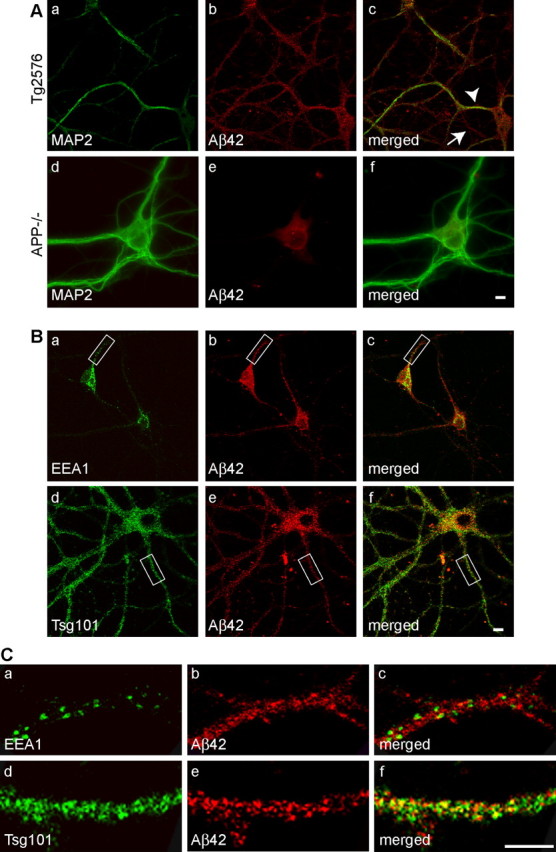
Late endocytic localization of Aβ42 in neurites of cultured APP mutant neurons. Neurons from APP mutant transgenic mice harboring the human AD Swedish mutation (Tg2576) were immunostained with an antibody specific for the C terminus of Aβ42 (Chemicon) and other subcellular markers at 19 DIV. Aa–Ac, Aβ42 localized to both dendrites, identified by MAP2-positive staining (arrowhead), and axons (arrow), identified by the absence of MAP2 staining. Ad–Af, Absence of the punctate Aβ42 immunostaining in neurites of APP−/− neurons; there was low level of background fluorescence in the cell body. Ba–Bc, Aβ42 did not colocalize with EEA1, a marker of early endosomes. Bd–Bf, Aβ42 overlap was evident with Tsg101, a marker of MVBs. C, Detail of neurites indicated in B, demonstrating Aβ42 colocalization with Tsg101 (d–f) but not with EEA1 (a–c). Scale bars, 10 μm.
We compared Aβ42 staining with that of established markers of the endocytic pathway in cultured APP mutant neurons at 19 DIV (Fig. 1B). EEA1 is a protein that is involved in the fusion of vesicles and early endosomes (Christoforidis et al., 1999). EEA1 mainly had a somatodendritic pattern of staining that did not reveal appreciable colocalization with Aβ42-positive vesicles (Fig. 1Ba–Bc). The lack of colocalization was especially evident in neurites (Fig. 1Ca–Cc). Tsg101 is a protein that mediates the sorting of ubiquitinated proteins into and localizes to MVBs (Katzmann et al., 2002). We observed that Tsg101 had a punctate pattern of staining with a more similar cellular distribution to Aβ42 (Fig. 1Bd–Bf). To better assess the extent of colocalization, we compared Aβ42 with EEA1 and Tsg101 staining of neurites at higher magnification, which more clearly delineated increased Aβ42 colocalization with Tsg101 than with EEA1 (Fig. 1Cd–Cf). Quantification of Aβ42 colocalization with EEA1 and Tsg101 revealed 8.4 ± 2.6% (23 neurites; n = 2) costaining with EEA1 compared with 37.3 ± 5.1% costaining with Tsg101 (34 neurites; n = 2). These data indicate that Aβ42 is present in both dendrites and axons and support previous work in which Aβ42 localized especially with markers of late endosomes/MVBs (Runz et al., 2002; Takahashi et al., 2004; Jin et al., 2004).
To further characterize the site of Aβ42 accumulation in APP mutant neurons at 12 DIV, at which time we previously reported early synaptic alterations (Almeida et al., 2005; Snyder et al., 2005), we used a dynamic assay for the endocytic pathway. We compared labeled compartments at different time points after endocytosis of fluorescent-labeled Tf and EGF with endogenous Aβ42 staining. The Tf receptor pathway is a well characterized pathway via the recycling endosome (Mukherjee et al., 1997). In contrast, EGF, after binding to EGFR and internalization, follows the MVB sorting pathway, in which it is sorted into the lumenal vesicles of MVBs (Katzmann et al., 2002).
Cultured neurons were incubated with fluorescent-labeled Tf or EGF for 10 or 60 min (Fig. 2A). After 10 min incubation with rhod-EGF, which is sufficient for EGF–EGFR internalization into early endosomes (Sorkin and von Zastrow, 2002), we did not observe significant colocalization with Aβ42 (10.8 ± 2.1%; 28 neurites; n = 3) (Fig. 2Aa–Ac,B). However, after 60 min uptake, when internalized EGF–EGFR complexes are expected to traffic to MVBs (Longva et al., 2002), 23.1 ± 1.9% of EGF-labeled compartments overlapped with Aβ42 (106 neurites; n = 8) (Fig. 2Ad–Af). After 10 min incubation with Alexa 488–Tf, uptake was observed with no significant colocalization with Aβ42 (supplemental Fig. S2A, available at www.jneurosci.org as supplemental material). After 60 min uptake, Tf labeling was distributed throughout neurons, but the majority of labeled structures (87.4 ± 1.0%; 96 neurites; n = 6) were not stained by Aβ42 (Fig. 2Ag–Ai). These results support that Aβ42 shares more compartments with EGF in the late part of the endocytic pathway when EGF has reached MVBs than with Tf or EGF in the early part of the pathway and further confirms Aβ42 accumulation in MVBs of APP mutant neurons.
Figure 2.
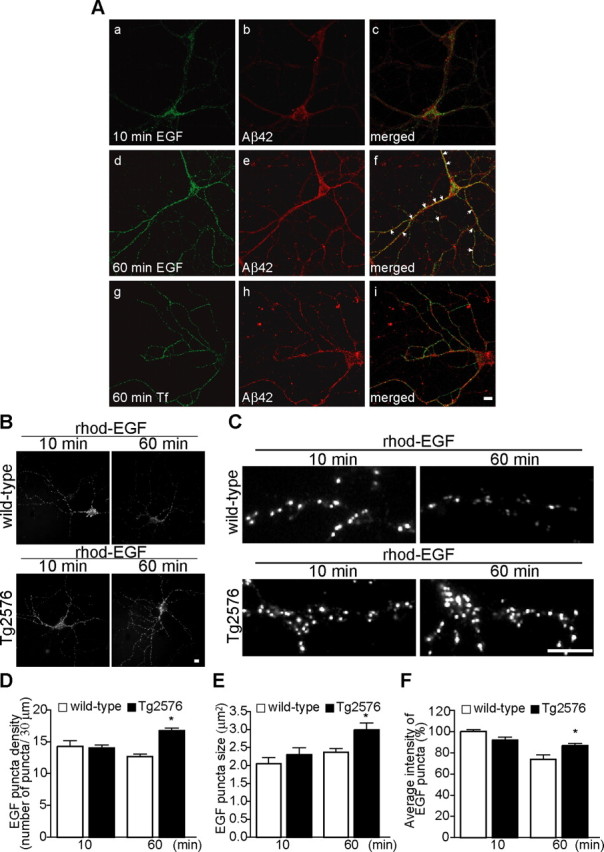
EGF endocytosis to the MVB sorting pathway is altered in cultured APP mutant neurons. Wild-type and APP mutant neurons (12 DIV) were incubated with rhod-EGF (500 ng/ml) or Alexa 488–Tf (20 μg/ml) for the indicated times at 37°C. Aa–Ac, rhod-EGF uptake after 10 min incubation followed by Aβ42 staining. Aβ42 did not share compartments with EGF after 10 min internalization, at which time EGF is in the early endocytic pathway. Ad–Af, rhod-EGF uptake after 60 min incubation followed by Aβ42 staining. Aβ42 shared more labeled compartments with EGF after 60 min internalization, at which time EGF traffics to the MVB sorting pathway. Ag–Ai, Alexa 488–Tf uptake after 60 min incubation followed by Aβ42 staining. Aβ42 did not share compartments with Tf after 60 min of uptake, at which time Tf localizes to the recycling pathway. B, Comparison of rhod-EGF endocytosis at 10 and 60 min in neurites of wild-type and APP mutant neurons (representative images). C, Representative higher-magnification images of neurites from wild-type and APP mutant neurons after 10 and 60 min internalization of rhod-EGF (segments of 30 μm). D, Quantification of the number of EGF puncta per 30 μm segment of neurite at 10 and 60 min after rhod-EGF internalization in wild-type (white bar) and APP mutant (red bar) neurons. At 10 min, there was no difference in the number of EGF puncta between wild-type (56 neurites; n = 3) and APP mutant (56 neurites; n = 3) neurons. At 60 min, there was a 33.4 ± 4.4% increase in rhod-EGF puncta density in APP mutant (290 neurites; n = 11) compared with wild-type (408 neurites; n = 15) neurons (∗p < 0.05 relative to wild-type neurons at 60 min). E, Quantification of the size of EGF puncta at 10 and 60 min after rhod-EGF internalization. At 10 min, there was no difference between wild-type (56 neurites; n = 3) and APP mutant (56 neurites; n = 3) neurons. At 60 min, there was a 26.2 ± 8.9% increase in rhod-EGF puncta size in APP mutant (290 neurites; n = 11) compared with wild-type (408 neurites; n = 15) neurons (∗p < 0.05 relative to wild-type neurons at 60 min). F, Quantification of the intensity of EGF puncta at 10 and 60 min after rhod-EGF internalization. In wild-type neurons (138 neurites; n = 3), there was a 25.9 ± 4.4% reduction in intensity of rhod-EGF puncta from 10 to 60 min (∗p < 0.05 relative to wild-type neurons at 10 min), whereas in APP mutant neurons (141 neurites; n = 3), there was no statistically significant reduction in EGF signal between the two time points. Error bars indicate SEM. Scale bars, 10 μm.
The MVB sorting pathway is altered in APP mutant neurons
To investigate whether Aβ42 accumulation in MVBs has consequences on the function of the endocytic pathway in neurons, we investigated the trafficking of rhod-EGF in APP mutant compared with wild-type neurons at 12 DIV (Fig. 2B–F). We compared EGF-labeled compartments at two time points: after 10 min, the rhod-EGF labeled compartments are predominantly early endosomes, whereas after 60 min, they are mainly MVBs (Longva et al., 2002). In wild-type neurons, there was a decrease from 10 to 60 min in rhod-EGF labeling. In contrast, in APP mutant neurons, there was no decrease in rhod-EGF from 10 to 60 min (Fig. 2B). At 10 min of EGF treatment, there was no difference in fluorescent EGF staining between wild-type and APP mutant neurons, seen especially clearly in segments of neurites (Fig. 2C). In contrast, after 60 min, rhod-EGF labeling was increased in APP mutant compared with wild-type neurons (Fig. 2C). We quantified both the number of rhod-EGF puncta per segment of neurite and measured the size and average fluorescence of rhod-EGF puncta. At 10 min, we found no statistical difference in number, size, or intensity of internalized rhod-EGF in APP mutant compared with wild-type neurons (Fig. 2D–F). This is consistent with an unaltered early endocytic pathway in APP mutant neurons. In addition, we observed no difference in Alexa 488–Tf endocytosis at 60 min of treatment in APP mutant compared with wild-type neurons (supplemental Fig. S2C,D, available at www.jneurosci.org as supplemental material), consistent with an unaltered recycling pathway. At 60 min of rhod-EGF treatment, the number of vesicles per 30 μm neurite segment was increased by 33.4 ± 4.4% in APP mutant (16.9 ± 0.4 vesicles/30 μm) compared with wild-type (12.7 ± 0.4 vesicles/30 μm) neurons (Fig. 2D, ∗p < 0.05). We also observed that rhod-EGF puncta were 26.2 ± 8.9% larger in APP mutant (3.0 ± 0.2 μm) compared with wild-type (2.4 ± 0.1 μm) neurons (Fig. 2E, ∗p < 0.05). In wild-type neurons, the average intensity of rhod-EGF puncta decreased by 25.9 ± 4.4% (∗p < 0.05) at 60 min compared with at 10 min, whereas there was no significant change in rhod-EGF puncta in APP mutant neurons (Fig. 2F). Possible explanations for increased rhod-EGF in MVBs of APP mutant neurons at 60 min included increased internalization from the plasma membrane or reduced EGF–EGFR degradation.
MVB-mediated receptor degradation is impaired in APP mutant neurons
To address these two hypotheses, we assessed for alterations in the internalization and/or degradation of EGF–EGFR in APP mutant neurons by biochemical analysis. We performed biotinylation assays of surface EGFR after EGF binding, which revealed no difference in the internalization of EGFR from the plasma membrane in APP mutant compared with wild-type neurons after 1 h treatment with EGF (supplemental Fig. S3, available at www.jneurosci.org as supplemental material). These data and the fluorescence results showing no difference in Tf endocytosis at 60 min (supplemental Fig. S2C,D, available at www.jneurosci.org as supplemental material) or in EGF endocytosis at 10 min (Fig. 2B–F) argue against altered receptor-mediated internalization in APP mutant neurons.
We next characterized the degradation of EGFR by Western blot after treatment with EGF and TGFα. EGFR can be stimulated by two different ligands, EGF and TGFα, that both lead to receptor clustering and internalization into early endosomes, membrane compartments with tubular–vesicular morphology with a mildly acidic pH. From early endosomes, receptors can be sorted to be recycled to the plasma membrane (recycling pathway) or into MVBs (MVB sorting pathway) for degradation in the lysosome. TGFα, more sensitive than EGF to lower pH, is released from EGFR in early endosomes, leading to deactivation and recycling of the receptor back to the plasma membrane. In contrast, the EGF–EGFR complex is stable at the pH of early endosomes, and therefore the activated receptor is retained on endosomal membranes. During endosomal maturation, inward invagination of the outer MVB membrane is thought to trap receptors in MVB inner vesicles, terminate signaling, and promote their subsequent degradation in lysosomes (Sorkin and von Zastrow, 2002; Matsuo et al., 2004). Although there was no difference in the total levels of EGFR in APP mutant compared with wild-type neurons at baseline, after 1 h activation with EGF, there was a significant increase in degradation of EGFR in wild-type (Fig. 3A,C) compared with APP mutant neurons, which persisted for as long as 7 h after treatment. In contrast, when TGFα was used as a ligand instead of EGF, no difference in EGFR degradation was observed between wild-type and APP mutant neurons (Fig. 3B,D). These results suggest that the endocytic pathway is specifically altered in APP mutant neurons at the level of MVB sorting and not in the internalization or recycling pathway.
Figure 3.
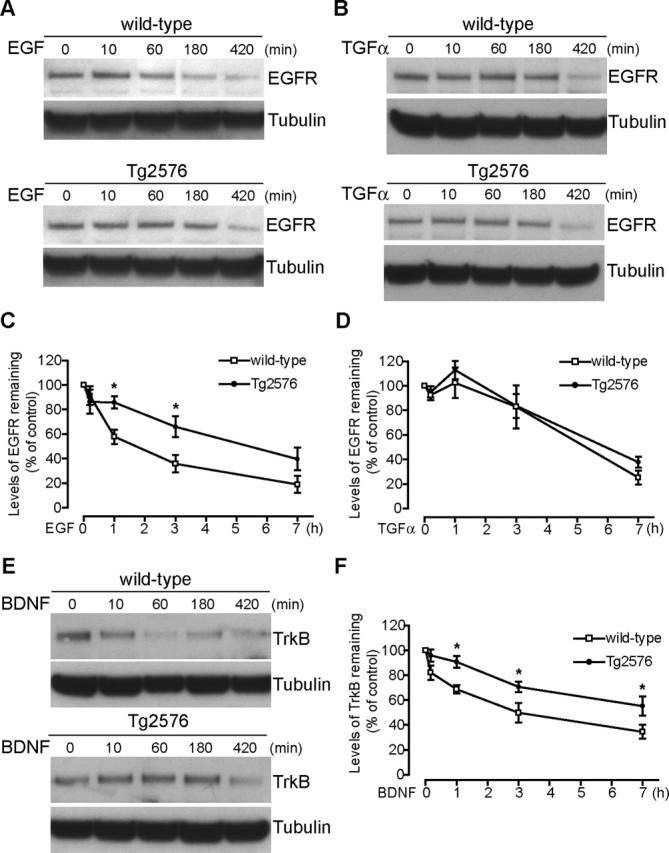
Delayed degradation of EGFR after EGF, but not TGFα, and of TrkB after BDNF activation, in APP mutant neurons. Wild-type and APP mutant neurons (12 DIV) were incubated with EGF (500 ng/ml), TGFα (500 ng/ml), or BDNF (50 ng/ml) for the indicated times at 37°C, and then cell lysates were subjected to SDS-PAGE and immunoblotted for EGFR or TrkB and for α-tubulin, as a loading control. A, During treatment with EGF, preserved EGFR signal, consistent with reduced degradation, was evident in APP mutant compared with wild-type cell lysates at and after 60 min. B, During treatment with TGFα, no difference in EGFR signal was evident in APP mutant compared with wild-type cell lysates at and after 60 min, consistent with an unaltered recycling pathway. C, Quantification of remaining levels of EGFR relative to unstimulated conditions revealed that EGF activation induced greater downregulation of EGFR in wild-type (open squares; n = 6–16 for each time point) compared with APP mutant (filled circles; n = 6–16 for each time point) neurons, which was statistically significant at 1 and 3 h (∗p < 0.05). D, In contrast, TGFα activation induced a similar downregulation of EGFR in wild-type (open squares; n = 7) and APP mutant (filled circles; n = 7) neurons, consistent with an unaltered TGFα–EGFR recycling pathway. E, Similar to EGFR after EGF treatment, the decrease in levels of TrkB was delayed in APP mutant neurons, consistent with reduced degradation in APP mutant compared with wild-type neurons at and after 60 min of BDNF treatment. F, Quantification of TrkB levels revealed that BDNF activation induced greater downregulation of TrkB in wild-type (open squares; n = 6–10 for each time point) compared with APP mutant (filled circles; n = 6–12 for each time point) neurons, which was statistically significant at 1, 3, and 7 h (∗p < 0.05). Error bars indicate SEM.
TrkB is a neuronal receptor tyrosine kinase involved in learning and memory that, similarly to EGFR, is internalized during ligand activation. Although less is known about the TrkB endocytic pathway, it was reported recently that, after internalization of TrkB, the formed macroendosomes are quickly processed to MVBs (Valdez et al., 2005). Indeed, MVBs are hypothesized to be an important structure in mediating Trk retrograde signaling (Weible and Hendry, 2004). We therefore treated APP mutant and wild-type neurons (12 DIV) with BDNF and measured total levels of TrkB (Fig. 3E). There was a trend for decreased total levels of TrkB in nonstimulated APP mutant compared with wild-type neurons, although this did not reach significance. Treatment with BDNF, already after 1 h, significantly reduced the relative degradation of TrkB in APP mutant compared with wild-type neurons, and this difference persisted even at 7 h of treatment (Fig. 3E,F).
Delayed dephosphorylation of EGFR after EGF treatment in APP mutant neurons
Because EGFR dephosphorylation is thought to occur at the lower pH of the inner vesicles of MVBs, we next assessed whether this step was also altered in APP mutant neurons. Therefore, we measured levels of EGFR phosphorylated at residue Y1173 by Western blot. Indeed, EGFR dephosphorylation after EGF treatment was decreased in APP mutant compared with wild-type neurons at 12 DIV (Fig. 4A,B). After 60 min of EGF treatment, the levels of phosphorylated EGFR (P-EGFR) were increased by 89.6 ± 35.8% (p < 0.05) in APP mutant compared with wild-type neurons. After 180 min, there still was a trend for increased levels of P-EGFR in APP mutant neurons that did not reach statistical significance. These data are consistent with slower dephosphorylation and/or degradation of EGFR in APP mutant neurons during treatment with EGF. Of note, there was no difference between the levels of P-EGFR at 10 min of treatment with EGF or treatment with TGFα (Fig. 4C,D) in APP mutant compared with wild-type neurons. These results support the scenario of normal internalization, activation, and initial trafficking steps of EGFR, followed by alterations on reaching the MVB sorting pathway. Because Aβ42 accumulation, given the transmembrane orientation of APP, should be occurring at the inner aspect of, or within, the outer membrane of MVBs, this could be slowing the downregulation of EGF–EGFR, which requires transport from the outer limiting membranes to the inner vesicles of MVBs (Futter et al., 1996; Katzmann et al., 2002; Sorkin and von Zastrow, 2002).
Figure 4.
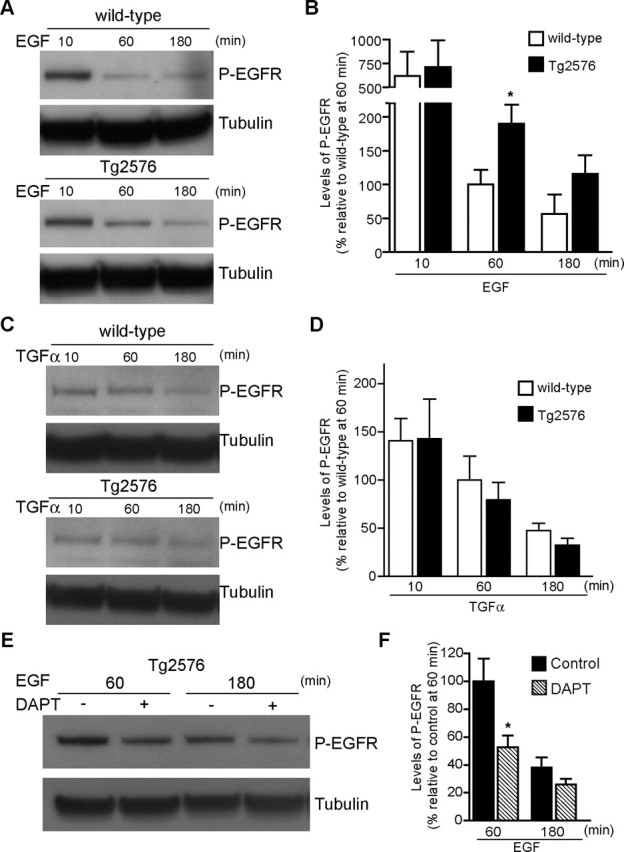
Delayed dephosphorylation of EGFR after EGF, but not TGFα, treatment in APP mutant neurons. A, After EGF treatment, cell lysates (12 DIV) were subjected to SDS-PAGE and immunoblotted for phospho-EGFR (pY1173-EGFR) and for α-tubulin, as a loading control. The preserved P-EGFR signal in APP mutant compared with wild-type neurons at 60 min is consistent with reduced deactivation of the receptor. B, EGFR deactivation, quantified by reduction in levels of P-EGFR after EGF treatment for 60 and 180 min, was greater in wild-type (white bars; n = 5–9 for each time point) compared with APP mutant (black bars; n = 6–9 for each time point) neurons. Deactivation of EGFR was significantly reduced by 89.6 ± 35.8% in APP mutant neurons after 60 min treatment with EGF (∗p < 0.05). C, In contrast, after TGFα treatment, no difference was detected in P-EGFR signal in APP mutant compared with wild-type neurons. D, Quantification of EGFR deactivation revealed no difference in APP mutant compared with wild-type neurons after TGFα treatment. E, Treatment of APP mutant neurons with the γ-secretase inhibitor DAPT (250 nm, 17 h), before the indicated time points of EGF treatment, reduced the levels of P-EGFR. F, Quantification of EGFR deactivation in APP mutant neurons with or without DAPT treatment. There was a 47.2 ± 17.9% increase in EGFR deactivation (hatched bars; n = 9) in DAPT-treated compared with untreated APP mutant (black bars; n = 8) neurons at 60 min (∗p < 0.05). Error bars indicate SEM.
Aβ is responsible for delaying the dephosphorylation of EGFR
To assess whether Aβ is required for slowing down the dephosphorylation of EGFR in APP mutant neurons, we treated neurons with the γ-secretase inhibitor N-[N-(3,5-difluorophenacetyl)-l-alanyl]-S-phenylglycine-t-butylester (DAPT) (250 nm). We confirmed that DAPT treatment reduced levels of Aβ in cultured APP mutant neurons (supplemental Fig. S4, available at www.jneurosci.org as supplemental material). Seventeen hours of DAPT treatment accelerated the dephosphorylation of EGFR in APP mutant neurons, which was especially evident during 1 h of treatment with EGF (Fig. 4E,F). DAPT treatment of APP mutant neurons also accelerated the degradation of TrkB, restoring TrkB levels to those of wild-type neurons (supplemental Fig. S5, available at www.jneurosci.org as supplemental material). This suggests that Aβ42 accumulation in MVBs affects normal trafficking within MVBs.
Increased levels of EGF-induced ubiquitinated EGFR in APP mutant neurons
Ubiquitination, in particular monoubiquitination, is a sorting signal that marks receptors for entry into the MVB pathway (Longva et al., 2002; Haglund et al., 2003). Ubiquitination of EGFR is known to be necessary for EGFR degradation in the lysosome. Ubiquitination of EGFR is the signal that recruits endosomal sorting complexes required for transport (ESCRT) via hepatocyte growth factor-regulated tyrosine kinase substrate and Tsg101, after the complex is formed at the outer membrane of MVBs (Babst, 2005). Subsequently, invagination of the MVB membrane occurs and EGFR is translocated into the inner vesicles of MVBs, in which it is thought that dephosphorylation/deactivation of the receptor occurs (Sorkin and von Zastrow, 2002; Longva et al., 2002).
To evaluate whether the ubiquitin-regulated sorting of EGFR is altered in APP mutant neurons, we measured the levels of ubiquitin-conjugated EGFR after 1 h of EGF activation (Fig. 5A). Immunoprecipitation of EGFR followed by Western blotting for ubiquitin revealed an increase in ubiquitinated EGFR in APP mutant compared with wild-type neurons after 1 h of treatment with EGF (Fig. 5A). Because this difference could be solely attributable to a slower degradation rate of EGFR in APP mutant neurons, we measured the increase in EGFR ubiquitination by dividing the intensity of ubiquitin bands by the EGFR bands in corresponding blots, which indicated a 2.9 ± 0.8-fold (p < 0.05; n = 3) increase in the levels of ubiquitinated EGFR in APP mutant compared with wild-type neurons. These data suggest that the markedly elevated levels of ubiquitinated EGFR in APP mutant neurons are not primarily attributable to reduced degradation of EGFR.
Figure 5.
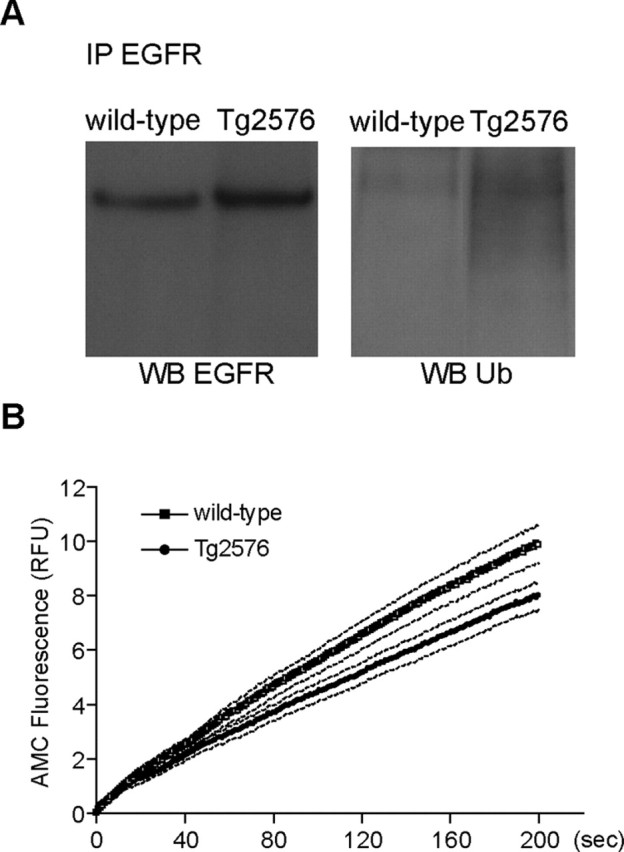
Decreased deubiquitination of EGFR after EGF stimulation in APP mutant neurons. A, Wild-type and APP mutant transgenic neurons (12 DIV) were incubated with EGF (500 ng/ml) for 1 h at 37°C. Cell lysates were subjected to immunoprecipitation (IP) with anti-EGFR, followed by SDS-PAGE and immunoblotting for EGFR or ubiquitin. Representative blots revealed elevated levels of ubiquitinated-EGFR in APP mutant compared with wild-type neurons. B, Kinetics of Ub-AMC hydrolysis, a measure of deubiquitinating enzyme activity, revealed decreased deubiquitination activity in cell lysates from APP mutant (filled circles; n = 9) compared with wild-type (open squares; n = 7) neurons. Data are expressed as mean (solid line) ± SEM (dotted lines). RFU, Relative fluorescence units.
Reduced deubiquitination in APP mutant neurons
The increased levels of ubiquitinated EGFR in APP mutant neurons during EGF treatment suggest that ubiquitination was not impaired, whereas the subsequent deubiquitination step, which occurs before and is necessary for the translocation into MVB inner membranes (Alwan et al., 2003), might be altered. Indeed, deubiquitination is increasingly recognized as an important regulatory step (d’Azzo et al., 2005). Deubiquitinating enzymes are cysteine proteases that specifically cleave off ubiquitin from its conjugates, thereby regenerating free ubiquitin (Chung and Baek, 1999). Therefore, we measured the activity of the deubiquitinating enzymes UCH-L1, UCH-L3, and isopeptidase T by incubating cell lysates of APP mutant and wild-type neurons with the ubiquitinated substrate Ub-AMC and measuring the accumulation of released fluorescence (Fig. 5B) (Dang et al., 1998; Russell and Wilkinson, 2005). The rate of ubiquitin hydrolysis was reduced by 20.3 ± 9.1% (p < 0.05) in APP mutant compared with wild-type neuronal lysates.
Increased ubiquitin and EGFR in APP mutant neurons
In the absence of EGF treatment, total levels of ubiquitin conjugates were unaltered in 12 DIV neurons by both Western blot (Fig. 6A,B) and immunofluorescence (Fig. 6C), suggesting that the increased ubiquitination of EGFR after EGF treatment in APP mutant neurons does not reflect a general alteration in cellular ubiquitination. However, after 19 d in culture, APP mutant neurons did reveal increased levels of total ubiquitin conjugates by Western blot (Fig. 6A,B) and by immunofluorescence (Fig. 6C). These data indicate that a modest but specific defect in deubiquitination at 12 DIV develops into a more general increase in ubiquitinated proteins at 19 DIV.
Figure 6.
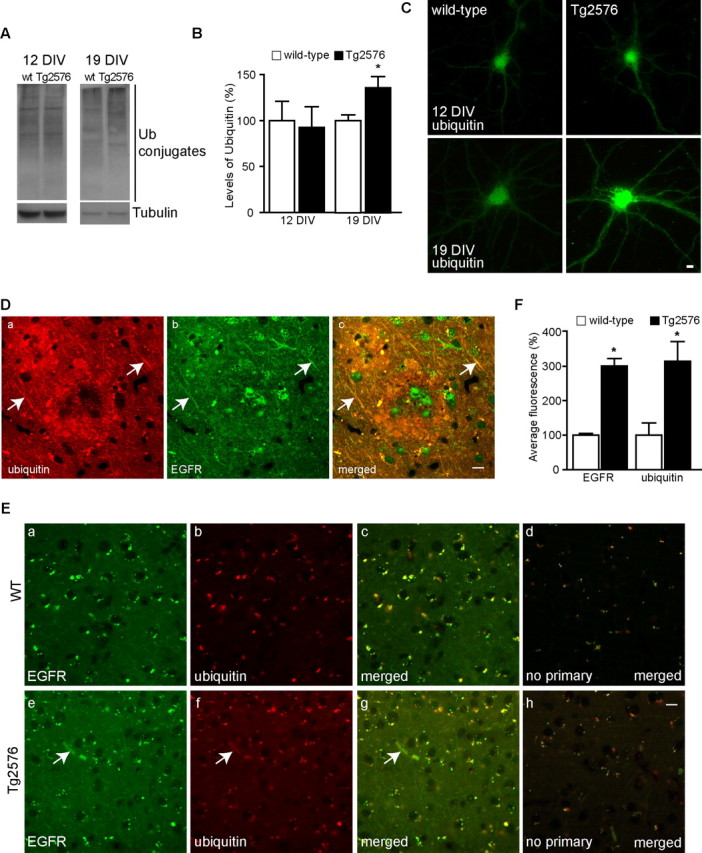
Increased ubiquitin and EGFR in APP mutant neurons. A, Cell lysates from 12 and 19 DIV wild-type (wt) and APP mutant neurons (both without EGF treatment) were subjected to SDS-PAGE and immunoblotted for ubiquitin and for α-tubulin, as a loading control. APP mutant neurons revealed a similar pattern of ubiquitin conjugates to wild-type neurons at 12 DIV and an increase in total ubiquitin conjugates at 19 DIV. B, Quantification of levels of ubiquitin conjugates indicated that there was no significant difference at 12 DIV, whereas at 19 DIV, there was a 35.9 ± 12.9% increase in ubiquitin conjugates in APP mutant (black bars; n = 9) compared with wild-type (white bars; n = 12) neurons (∗p < 0.05). C, By immunofluorescence, ubiquitin immunoreactivity was increased especially in processes of APP mutant compared with wild-type neurons at 19 DIV but not at 12 DIV. Da–Dc, Representative tissue section from a late stage (18 months) Tg2576 mouse brain double labeled with anti-EGFR (green) and anti-ubiquitin (red) revealed that increased EGFR and ubiquitin immunoreactivity colocalized in dystrophic neurites (arrows) around a plaque. E, Representative images from cortical areas without plaques from late stage (18 months) Tg2576 (e–h) and wild-type (a–d) mice. Anti-EGFR (a, e, green), anti-ubiquitin (b, f, red), merged image (c, g), and secondary antibodies (d, h) in the absence of primary antibodies (merged image). Tg2576 brain sections showed an increase in immunoreactivity for EGFR and ubiquitin when compared with wild-type. F, Quantification of total fluorescence after subtraction of background staining showed an almost threefold increase in EGFR and ubiquitin immunoreactivity in Tg2576 as compared with wild-type brain (∗p < 0.05; n = 3). Scale bars, 10 μm. Error bars indicate SEM.
We also assessed by immunofluorescence for alterations in EGFR and ubiquitin in Tg2576 compared with wild-type littermate mouse brains (18 months). In dystrophic neurites surrounding plaques, there was increased staining for ubiquitin and EGFR (Fig. 6D, arrows). We also observed a 200.4 ± 22.6% (p < 0.05) increase of EGFR immunostaining in Tg2576 neurons in cortical areas without plaques when compared with neurons in the wild-type brain (Fig. 6E,F). There was a similar increase (214.5 ± 67.1%; p < 0.05) in ubiquitin immunostaining in the same neurons (Fig. 6E,F), suggesting a correlation between the increase in ubiquitin and EGFR in APP mutant neurons.
Aβ accumulation in APP mutant neurons reduces proteasome activity
The 20% inhibition of deubiquitinating enzyme activity noted above seemed insufficient to account for the almost threefold increase in levels of ubiquitinated EGFR during treatment with EGF in APP mutant neurons. Although the proteasome is best known for degrading polyubiquitinated proteins in the cytoplasm, the proteasome is also involved in regulating the recycling to the plasma membrane and degradation via lysosomes of membrane-associated proteins, such as tyrosine kinase receptors, including EGFR. Proteasome inhibition was reported to decrease the deubiquitination of EGFR and the translocation of EGFR into the inner vesicles of MVBs, thereby slowing EGFR degradation (Longva et al., 2002; Alwan et al., 2003), paralleling the alterations that we demonstrated in EGFR during EGF activation in APP mutant neurons above. To evaluate whether proteasome inhibition contributes to the decreased EGFR degradation and increased levels of ubiquitinated EGFR in APP mutant neurons, we measured proteasome enzymatic activity in APP mutant compared with wild-type neurons.
The capacity of the proteasome to degrade the fluorogenic peptide substrate Suc-LLVY-AMC results in an increase in fluorescence with time attributable to accumulation of cleaved AMC. Remarkably, the rate of AMC accumulation was significantly reduced in APP mutant compared with wild-type neurons (Fig. 7A). As a control, we performed the assay in the presence of the proteasome inhibitor MG132, which as expected completely blocked the accumulation of AMC. By linear regression analysis, the slopes of the curves were 0.15 ± 0.03 for wild-type and 0.07 ± 0.01 for APP mutant neurons, which corresponds to a significant 51.4 ± 21.4% (p < 0.05) inhibition of the proteasome in APP mutant compared with wild-type neurons (Fig. 7B). This inhibition does not appear to be secondary to a reduction in proteasomes, because there was no difference in the levels of the α-subunit of the 20S subunit of the proteasome in APP mutant compared with wild-type neurons (Fig. 7C).
Figure 7.
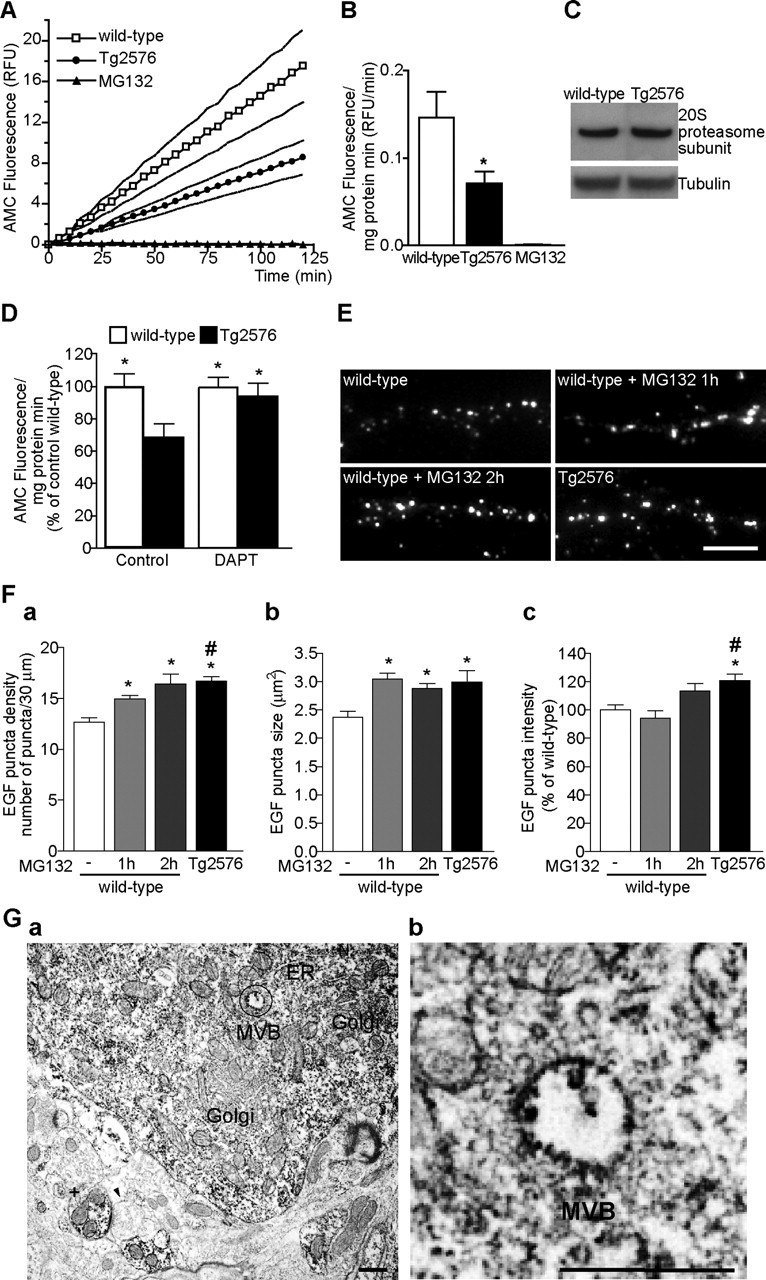
Aβ-dependent inhibition of the proteasome. A, Proteasome activity was assayed in lysates prepared from wild-type (open squares) and APP mutant (filled circles) neurons (12 DIV). The kinetics of fluorescence accumulation from the degradation of the proteasome substrate Suc-LLVY-AMC monitored for 120 min revealed markedly increased AMC accumulation in wild-type compared with APP mutant neurons. Addition of MG132 completely prevented AMC accumulation (filled triangles). Data are expressed as mean (solid line) ± SEM (dotted lines). B, Bar graph of the average slope obtained by linear regression analysis during the 120 min of Suc-LLVY-AMC degradation in wild-type (white bar; n = 14), APP mutant (black bar; n = 16), and MG132 (black bar; n = 2); there was a 51.4 ± 21.4% decrease in proteasome activity in APP mutant compared with wild-type neurons (∗p < 0.05). C, There was no appreciable difference in the levels of the 20S proteasome α-subunit in wild-type compared with APP mutant neurons (representative blots). D, Treatment of APP mutant neurons with DAPT (250 nm, 17 h; n = 11) normalized the levels of proteasome activity to wild-type (n = 11) levels (∗p < 0.05 relative to untreated APP mutant neurons; n = 9). Treatment with DAPT had no statistically significant effect on proteasome activity in wild-type neurons (n = 11). E, Representative images of neurites from wild-type and APP mutant neurons after 1 h internalization of rhod-EGF (segments of 30 μm). Wild-type neurons pretreated with MG132 for 1 or 2 h revealed similar EGF uptake to that in APP mutant neurons. Scale bar, 10 μm. Fa, The bar graph depicts increased number of rhod-EGF puncta in wild-type neurons treated with MG132 (112 neurites, n1 h = 7; 119 neurites, n2 h = 5), especially after 2 h (n = 5) to levels similar to that in APP mutant neurons (290 neurites, n = 11) [∗p < 0.05 relative to untreated wild-type neurons (408 neurites, n = 15); #p < 0.05 relative to wild-type neurons pretreated for 1 h with MG132 (112 neurites, n = 7)]. Fb, The size of EGF puncta in wild-type neurons treated with MG132 was significantly increased only after 1 h, reaching APP mutant levels (∗p < 0.05). Fc, Quantification of the intensity of rhod-EGF puncta revealed that 2 h of treatment with MG132 was necessary to increase rhod-EGF puncta intensity. Error bars indicate SEM. Ga, Immunoreactivity of the β-subunit of the 20S proteasome in normal rat brain was evident in the cytoplasm of pyramidal neurons and in dendrites (+). Immunoperoxidase labeling was distributed diffusely in neuronal perikaryon, with pronounced labeling especially associated with MVBs, endoplasmic reticulum, and small vesicles. In contrast, Golgi revealed little labeling. A presynaptic compartment (arrowhead) had no appreciable labeling, whereas the postsynaptic compartment to which it is connected by a synaptic density demonstrates marked staining. Gb, Higher-magnification image of circled area in a revealed labeling in the outer membrane and inner vesicle of a MVB. ER, Endoplasmic reticulum; Golgi, Golgi apparatus; RFU, relative fluorescence units. Scale bars, 500 nm.
To verify whether inhibition of proteasome activity was attributable to Aβ, we again inhibited Aβ generation by treating APP mutant neurons with the γ-secretase inhibitor DAPT for 17 h and measured proteasome activity. In APP mutant neurons, there was a nearly complete normalization in proteasome activity to wild-type levels, whereas there was no significant change in proteasome activity when treating wild-type neurons with DAPT (Fig. 7D). These data support the scenario that Aβ42 accumulation is altering the MVB sorting pathway via inhibition of the proteasome.
To test the hypothesis that proteasome inhibition by Aβ42 is responsible for the altered MVB sorting pathway in APP mutant neurons, we repeated the assay of rhod-EGF endocytosis in wild-type neurons pretreated with the proteasome inhibitor MG132. Remarkably, MG132 treatment reduced degradation of EGF–EGFR complexes after 1 h of rhod-EGF endocytosis in wild-type neurons, similar to levels observed in APP mutant neurons (Fig. 7E). The response to MG132 was time dependent and required at least a 2 h preincubation with MG132 to approach the increase in number and size of rhod-EGF puncta that was observed in APP mutant neurons (Fig. 7Fa,Fb). There was a trend for an increase in rhod-EGF puncta intensity that did not reach significance when compared with control conditions (Fig. 7Fc).
Localization of a proteasome subunit (XAPC7) to late endosomal membranes was recently described in BHK21 cells (Dong et al., 2004). To investigate whether proteasomes also localize to membrane compartments in neurons of the brain, we performed immuno-EM using an antibody against the 20S subunit of the proteasome (Fig. 7G). The 20S subunit labeling was present in multiple sites in neurons, including the outer membranes and inner vesicles of MVBs, confirming that proteasome subunits also localize to late endosomal membranes in neurons.
Inhibition of proteasome activity by accumulating Aβ42 provides a mechanism for the decrease in deubiquitination and subsequent degradation of EGFR in APP mutant neurons (supplemental Fig. S6, available at www.jneurosci.org as supplemental material).
Discussion
The endocytosis of EGFR is among the best understood UPS-regulated pathways. Work from several laboratories has shown that, during ligand (EGF, TGFα) binding, EGFR at the plasma membrane is activated by phosphorylation and then subsequently is ubiquitinated and trafficked to early endosomes, followed by transport either via recycling endosomes back to the cell surface (TGFα) or via the MVB sorting pathway for degradation (EGF). The ubiquitination machinery at the outer surface of MVB outer membranes and the proteasome coordinate the translocation of the EGF–EGFR complex into the inner vesicles of MVBs for subsequent deactivation/dephosphorylation and later degradation in lysosomes. Proteasome inhibition was demonstrated previously to block EGF–EGFR translocation into inner vesicles of MVBs and thereby to elevate levels of ubiquitinated EGFR and inhibit the degradation of the EGF–EGFR complex (Longva et al., 2002). Because AD-linked Aβ42 accumulates, given the topology of APP, on the inner leaflet or within MVB outer membranes, we hypothesized that this aberrant Aβ accumulation may alter MVB sorting. We have now demonstrated that Aβ-accumulating APP mutant neurons cultured from Tg2576 transgenic mice revealed normal internalization and recycling but altered MVB sorting of EGFR during EGF activation. The accumulation of EGF–EGFR in MVBs of APP mutant compared with wild-type neurons suggested reduced degradation of EGFR. We verified altered MVB sorting and degradation of another ligand–receptor complex, BDNF–TrkB, which provided similar results. To obtain more information on the altered trafficking of EGFR within MVBs in APP mutant neurons, we assessed the levels of phosphorylated EGFR, because dephosphorylation is thought to occur at the low pH of the inner vesicles of MVBs, and found elevated levels of P-EGFR.
Ubiquitination is required for trafficking of EGFR to MVBs, subsequent translocation within MVBs, and later degradation of EGFR via the MVB sorting pathway. It was therefore possible that reduced ubiquitination of EGFR might be blocking EGFR from translocating to inner MVB membranes. Instead, we found increased ubiquitin conjugated EGFR in APP mutant neurons, suggesting that ubiquitination of EGFR was not impaired. Therefore, we next hypothesized that deubiquitination activity might be compromised. Analysis of deubiquitinating enzyme activity did reveal reduced deubiquitination in APP mutant neurons, although the modest change seemed insufficient for the more marked elevation in ubiquitinated EGFR.
Considerable evidence over the past few years has indicated that the UPS regulates the trafficking of EGFR through the MVB sorting pathway. Proteasome activity is necessary for deubiquitination to occur before EGFR translocation (Alwan et al., 2003). Moreover, proteasome inhibition is known to lead to decreased translocation of the EGF–EGFR complex from the outer membrane to the inner vesicles of MVBs and subsequent decreased degradation of EGFR (Longva et al., 2002). Therefore, we next examined proteasome activity in APP mutant compared with wild-type neurons, which revealed a marked reduction in proteasome activity in APP mutant neurons. Because proteasome inhibition also inhibits deubiquitination, reduced proteasome activity therefore could be responsible for the cellular alterations that we describe in the APP mutant neurons. The mechanism by which this occurs appears unclear, because the proteasome is generally considered to be cytosolic and Aβ42 accumulates in the inner side of, or within, the outer membrane of MVBs. However, Aβ42 was reported to inhibit proteasome function in vitro (Gregori et al., 1995, 1997) and a direct interaction between the α-subunit XAPC7 of the proteasome to MVB/late endosomes was recently described (Dong et al., 2004). Moreover, by immunoelectron microscopy, we observed that the β-subunit of the 20S proteasome localizes to MVBs in neurons of the brain.
That treatment with γ-secretase inhibitor reversed the alterations in the MVB sorting pathway and the UPS supports that Aβ rather than overexpression of human APP is responsible. Although others and we have observed especially accumulation of Aβ42 within neurons with AD pathogenesis (Gouras et al., 2000; D’Andrea et al., 2001; Gyure et al., 2001; Mori et al., 2002; Ohyagi et al., 2005), the results with the γ-secretase inhibitor cannot exclude a role also for the more soluble and abundantly secreted Aβ40 peptides in the cellular alterations that occur in APP mutant neurons. Because the UPS-regulated MVB sorting pathway is best characterized for EGFR, a better understanding of UPS-mediated MVB sorting of other neuronal receptors will be required to establish whether there is a general or more selective dysfunction in this pathway in APP mutant neurons.
Both the aberrant intracellular accumulation of disease-linked proteins and the UPS are increasingly being implicated in neurodegenerative diseases of aging. Intraneuronal accumulation of Aβ in AD therefore follows this common theme. The cellular mechanism by which aberrant accumulation is involved in the disease process is of major interest. In familial forms of Parkinson’s disease (PD), genetic studies have revealed that mutations in the UPS, specifically, mutations in the ubiquitin ligase Parkin and in the hydrolase UCH-L1, are linked with early onset recessive forms of PD (Bertram and Tanzi, 2005). Recently, mutations in CHMP2B, encoding a component of the MVB-associated ESCRTIII complex, were linked to frontotemporal dementia (Skibinski et al., 2005). A genetic association between variants of the gene encoding ubiquilin 1 (UBQLN1) and AD was also reported recently (Bertram et al., 2005). Although the cellular function of ubiquilin remains unclear, it is known to contain an ubiquitin-like domain in its N terminus and an ubiquitin-associated domain in its C terminus and therefore could interact both with ubiquitin ligases and the proteasome (Kleijnen et al., 2003). Ubiquitin is well known to be increased especially in dystrophic neurites of human AD and of mouse models of β-amyloidosis (Blanchard et al., 2003), an aberrant form of ubiquitin was reported to accumulate in human AD and Down syndrome brains (van Leeuwen et al., 1998), Aβ was reported to decrease proteasome activity in cultured neurons (Lopez Salon et al., 2003), and proteasome activity was described to be reduced in the brains of Tg2576 mice (Oh et al., 2005) and human AD (Keller et al., 2000).
We provide evidence that Aβ is responsible for the alterations in the MVB sorting pathway and the UPS in APP mutant neurons, because treatment with the γ-secretase inhibitor DAPT blocked the cellular alterations in APP mutant neurons, including impairment in proteasome activity. Moreover, in support of Aβ-induced proteasome inhibition occurring in APP mutant neurons, we found that proteasome inhibition of wild-type neurons produced alterations in the MVB sorting pathway paralleling those seen in APP mutant neurons. The findings that the disease-linked Aβ42 peptides localize and accumulate especially in MVBs and thereby alter the trafficking of membrane receptors by inhibiting the UPS suggest a mechanism whereby Aβ42 accumulation contributes to AD pathogenesis. The UPS is a complex system of interconnecting biochemical pathways that are increasingly being found to have major cellular functions even beyond the degradation of proteins. The UPS is being studied in the regulated recycling and degradation of membrane receptors and has been linked to the regulation of synaptic plasticity (Ehlers, 2003). Indeed, we hypothesize that the inhibition of the UPS by Aβ42 impairs the endocytic trafficking of neuronal receptors and thereby may be the cause of synaptic dysfunction in AD. Our data supports a novel mechanism whereby Aβ accumulation alters the cell biology of vulnerable neurons in AD.
Footnotes
This study was supported by grants from the Alzheimer’s Association (G.K.G), the American Health Assistance Foundation (G.K.G), National Institutes of Health Grant NS045677 (G.K.G.), and a doctoral fellowship from Fundação para a Ciência e a Tecnologia, Portugal (C.G.A.). We acknowledge the input from the trafficking club meetings, Department of Biochemistry, especially that from Francis Lee, Tim McGraw, Tim Ryan, and Fred Maxfield. We thank Michael Lin for helpful discussions and critical reading of this manuscript. We also thank Edgar Gomes for helpful discussions.
References
- Almeida CG, Tampellini D, Takahashi RH, Greengard P, Lin MT, Snyder EM, Gouras GK (2005). Beta-amyloid accumulation in APP mutant neurons reduces PSD-95 and GluR1 in synapses. Neurobiol Dis 20:187–198. [DOI] [PubMed] [Google Scholar]
- Alwan HA, van Zoelen EJ, van Leeuwen JE (2003). Ligand-induced lysosomal epidermal growth factor receptor (EGFR) degradation is preceded by proteasome-dependent EGFR de-ubiquitination. J Biol Chem 278:35781–35790. [DOI] [PubMed] [Google Scholar]
- Babst M (2005). A protein’s final ESCRT. Traffic 6:2–9. [DOI] [PubMed] [Google Scholar]
- Bertram L, Tanzi RE (2005). The genetic epidemiology of neurodegenerative disease. J Clin Invest 115:1449–1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertram L, Hiltunen M, Parkinson M, Ingelsson M, Lange C, Ramasamy K, Mullin K, Menon R, Sampson AJ, Hsiao MY, Elliott KJ, Velicelebi G, Moscarillo T, Hyman BT, Wagner SL, Becker KD, Blacker D, Tanzi RE (2005). Family-based association between Alzheimer’s disease and variants in UBQLN1. N Engl J Med 352:884–894. [DOI] [PubMed] [Google Scholar]
- Blanchard V, Moussaoui S, Czech C, Touchet N, Bonici B, Planche M, Canton T, Jedidi I, Gohin M, Wirths O, Bayer TA, Langui D, Duyckaerts C, Tremp G, Pradier L (2003). Time sequence of maturation of dystrophic neurites associated with Abeta deposits in APP/PS1 transgenic mice. Exp Neurol 184:247–263. [DOI] [PubMed] [Google Scholar]
- Cataldo AM, Peterhoff CM, Troncoso JC, Gomez-Isla T, Hyman BT, Nixon RA (2000). Endocytic pathway abnormalities precede amyloid beta deposition in sporadic Alzheimer’s disease and Down syndrome: differential effects of APOE genotype and presenilin mutations. Am J Pathol 157:277–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christoforidis S, McBride HM, Burgoyne RD, Zerial M (1999). The Rab5 effector EEA1 is a core component of endosome docking. Nature 397:621–625. [DOI] [PubMed] [Google Scholar]
- Chung CH, Baek SH (1999). Deubiquitinating enzymes: their diversity and emerging roles. Biochem Biophys Res Commun 266:633–640. [DOI] [PubMed] [Google Scholar]
- D’Andrea MR, Nagele RG, Wang HY, Peterson PA, Lee DH (2001). Evidence that neurones accumulating amyloid can undergo lysis to form amyloid plaques in Alzheimer’s disease. Histopathology 38:120–134. [DOI] [PubMed] [Google Scholar]
- Dang LC, Melandri FD, Stein RL (1998). Kinetic and mechanistic studies on the hydrolysis of ubiquitin C-terminal 7-amido-4-methylcoumarin by deubiquitinating enzymes. Biochemistry 37:1868–1879. [DOI] [PubMed] [Google Scholar]
- d’Azzo A, Bongiovanni A, Nastasi T (2005). E3 ubiquitin ligases as regulators of membrane protein trafficking and degradation. Traffic 6:429–441. [DOI] [PubMed] [Google Scholar]
- Dong J, Chen W, Welford A, Wandinger-Ness A (2004). The proteasome alpha-subunit XAPC7 interacts specifically with Rab7 and late endosomes. J Biol Chem 279:21334–21342. [DOI] [PubMed] [Google Scholar]
- Ehlers MD (2003). Activity level controls postsynaptic composition and signaling via the ubiquitin-proteasome system. Nat Neurosci 6:231–242. [DOI] [PubMed] [Google Scholar]
- Futter CE, Pearse A, Hewlett LJ, Hopkins CR (1996). Multivesicular endosomes containing internalized EGF-EGF receptor complexes mature and then fuse directly with lysosomes. J Cell Biol 132:1011–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouras GK, Tsai J, Naslund J, Vincent B, Edgar M, Checler F, Greenfield JP, Haroutunian V, Buxbaum JD, Xu H, Greengard P, Relkin NR (2000). Intraneuronal Abeta42 accumulation in human brain. Am J Pathol 156:15–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouras GK, Almeida CG, Takahashi RH (2005). Intraneuronal Abeta accumulation and origin of plaques in Alzheimer’s disease. Neurobiol Aging 26:1235–1244. [DOI] [PubMed] [Google Scholar]
- Gregori L, Fuchs C, Figueiredo-Pereira ME, Van Nostrand WE, Goldgaber D (1995). Amyloid beta-protein inhibits ubiquitin-dependent protein degradation in vitro. J Biol Chem 270:19702–19708. [DOI] [PubMed] [Google Scholar]
- Gregori L, Hainfeld JF, Simon MN, Goldgaber D (1997). Binding of amyloid beta protein to the 20 S proteasome. J Biol Chem 272:58–62. [DOI] [PubMed] [Google Scholar]
- Gruenberg J (2001). The endocytic pathway: a mosaic of domains. Nat Rev Mol Cell Biol 2:721–730. [DOI] [PubMed] [Google Scholar]
- Gruenberg J, Stenmark H (2004). The biogenesis of multivesicular endosomes. Nat Rev Mol Cell Biol 5:317–323. [DOI] [PubMed] [Google Scholar]
- Gyure KA, Durham R, Stewart WF, Smialek JE, Troncoso JC (2001). Intraneuronal Abeta-amyloid precedes development of amyloid plaques in Down syndrome. Arch Pathol Lab Med 125:489–492. [DOI] [PubMed] [Google Scholar]
- Haglund K, Di Fiore PP, Dikic I (2003). Distinct monoubiquitin signals in receptor endocytosis. Trends Biochem Sci 28:598–603. [DOI] [PubMed] [Google Scholar]
- Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G (1996). Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science 274:99–102. [DOI] [PubMed] [Google Scholar]
- Huse JT, Pijak DS, Leslie GJ, Lee VM, Doms RW (2000). Maturation and endosomal targeting of beta-site amyloid precursor protein-cleaving enzyme. The Alzheimer’s disease beta-secretase. J Biol Chem 275:33729–33737. [DOI] [PubMed] [Google Scholar]
- Jin LW, Maezawa I, Vincent I, Bird T (2004). Intracellular accumulation of amyloidogenic fragments of amyloid-beta precursor protein in neurons with Niemann-Pick type C defects is associated with endosomal abnormalities. Am J Pathol 164:975–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamal A, Almenar-Queralt A, LeBlanc JF, Roberts EA, Goldstein LSB (2001). Kinesin-mediated axonal transport of a membrane compartment containing beta-secretase and presenilin-1 requires APP. Nature 414:643–648. [DOI] [PubMed] [Google Scholar]
- Katzmann DJ, Odorizzi G, Emr SD (2002). Receptor downregulation and multivesicular-body sorting. Nat Rev Mol Cell Biol 3:893–905. [DOI] [PubMed] [Google Scholar]
- Keller JN, Hanni KB, Markesbery WR (2000). Impaired proteasome function in Alzheimer’s disease. J Neurochem 75:436–439. [DOI] [PubMed] [Google Scholar]
- Kleijnen MF, Alarcon RM, Howley PM (2003). The ubiquitin-associated domain of hPLIC-2 interacts with the proteasome. Mol Biol Cell 14:3868–3875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koo EH, Squazzo SL (1994). Evidence that production and release of amyloid beta-protein involves the endocytic pathway. J Biol Chem 269:17386–17389. [PubMed] [Google Scholar]
- Lah JJ, Heilman CJ, Nash NR, Rees HD, Yi H, Counts SE, Level AI (1997). Light and electron microscopic localization of presenilin-1 in primate brain. J Neurosci 17:1971–1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langui D, Girardot N, El Hachimi KH, Allinquant B, Blanchard V, Pradier L, Duyckaerts C (2004). Subcellular topography of neuronal Abeta peptide in APPxPS1 transgenic mice. Am J Pathol 165:1465–1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longva KE, Blystad FD, Stang E, Larsen AM, Johannessen LE, Madshus IH (2002). Ubiquitination and proteasomal activity is required for transport of the EGF receptor to inner membranes of multivesicular bodies. J Cell Biol 156:843–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez Salon M, Pasquini L, Besio MM, Pasquini JM, Soto E (2003). Relationship between beta-amyloid degradation and the 26S proteasome in neural cells. Exp Neurol 180:131–143. [DOI] [PubMed] [Google Scholar]
- Matsuo H, Chevallier J, Mayran N, Le Blanc I, Ferguson C, Faure J, Blanc NS, Matile S, Dubochet J, Sadoul R, Parton RG, Vilbois F, Gruenberg J (2004). Role of LBPA and Alix in multivesicular liposome formation and endosome organization. Science 303:531–534. [DOI] [PubMed] [Google Scholar]
- Milner TA, Lee A, Aicher SA, Rosin DL (1998). Hippocampal alpha2a-adrenergic receptors are located predominantly presynaptically but are also found postsynaptically and in selective astrocytes. J Comp Neurol 395:310–327. [PubMed] [Google Scholar]
- Mori C, Spooner ET, Wisniewsk KE, Wisniewski TM, Yamaguch H, Saido TC, Tolan DR, Selkoe DJ, Lemere CA (2002). Intraneuronal Abeta42 accumulation in Down syndrome brain. Amyloid 9:88–102. [PubMed] [Google Scholar]
- Mukherjee S, Ghosh RN, Maxfield FR (1997). Endocytosis. Physiol Rev 77:759–803. [DOI] [PubMed] [Google Scholar]
- Nixon RA (2005). Endosome function and dysfunction in Alzheimer’s disease and other neurodegenerative diseases. Neurobiol Aging 26:373–382. [DOI] [PubMed] [Google Scholar]
- Oh S, Hong HS, Hwang E, Sim HJ, Lee W, Shin SJ, Mook-Jung I (2005). Amyloid peptide attenuates the proteasome activity in neuronal cells. Mech Ageing Dev 126:1292–1299. [DOI] [PubMed] [Google Scholar]
- Ohyagi Y, Asahara H, Chui DH, Tsuruta Y, Sakae N, Miyoshi K, Yamada T, Kikuchi H, Taniwaki T, Murai H, Ikezoe K, Furuya H, Kawarabayashi T, Shoji M, Checler F, Iwaki T, Makifuchi T, Takeda K, Kira J, Tabira T (2005). Intracellular Abeta42 activates p53 promoter: a pathway to neurodegeneration in Alzheimer’s disease. FASEB J 19:255–257. [DOI] [PubMed] [Google Scholar]
- Parton RG, Dotti CG (1993). Cell biology of neuronal endocytosis. J Neurosci Res 36:1–9. [DOI] [PubMed] [Google Scholar]
- Runz H, Rietdorf J, Tomic I, de Bernard M, Beyreuther K, Pepperkok R, Hartmann T (2002). Inhibition of intracellular cholesterol transport alters presenilin localization and amyloid precursor protein processing in neuronal cells. J Neurosci 22:1679–1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell NS, Wilkinson KD (2005). Deubiquitinating enzyme purification, assay inhibitors, and characterization. Methods Mol Biol 301:207–219. [DOI] [PubMed] [Google Scholar]
- Skibinski G, Parkinson NJ, Brown JM, Chakrabarti L, Lloyd SL, Hummerich H, Nielsen JE, Hodges JR, Spillantini MG, Thusgaard T, Brandner S, Brun A, Rossor MN, Gade A, Johannsen P, Sorensen SA, Gydesen S, Fisher EM, Collinge J (2005). Mutations in the endosomal ESCRTIII-complex subunit CHMP2B in frontotemporal dementia. Nat Genet 37:806–808. [DOI] [PubMed] [Google Scholar]
- Snyder EM, Nong Y, Almeida CG, Paul S, Moran T, Choi EY, Nairn AC, Salter MW, Lombroso PJ, Gouras GK, Greengard P (2005). Regulation of NMDA receptor trafficking by amyloid-beta. Nat Neurosci 8:1051–1058. [DOI] [PubMed] [Google Scholar]
- Sorkin A, von Zastrow M (2002). Signal transduction and endocytosis: close encounters of many kinds. Nat Rev Mol Cell Biol 3:600–614. [DOI] [PubMed] [Google Scholar]
- Takahashi RH, Milner TA, Li F, Nam EE, Edgar MA, Yamaguchi H, Beal MF, Xu H, Greengard P, Gouras GK (2002). Intraneuronal Alzheimer Abeta42 accumulates in multivesicular bodies and is associated with synaptic pathology. Am J Pathol 161:1869–1879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi RH, Almeida CG, Kearney PF, Yu F, Lin MT, Milner TA, Gouras GK (2004). Oligomerization of Alzheimer’s β-amyloid within processes and synapses of cultured neurons and brain. J Neurosci 24:3592–3599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valdez G, Akmentin W, Philippidou P, Kuruvilla R, Ginty DD, Halegoua S (2005). Pincher-mediated macroendocytosis underlies retrograde signaling by neurotrophin receptors. J Neurosci 25:5236–5247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Leeuwen FW, de Kleijn DP, van den Hurk HH, Neubauer A, Sonnemans MA, Sluijs JA, Koycu S, Ramdjielal RD, Salehi A, Martens GJ, Grosveld FG, Peter J, Burbach H, Hol EM (1998). Frameshift mutants of beta amyloid precursor protein and ubiquitin-B in Alzheimer’s and Down patients. Science 279:242–247. [DOI] [PubMed] [Google Scholar]
- Vassar R, Bennett BD, Babu-Khan S, Kahn S, Mendiaz EA, Denis P, Teplow DB, Ross S, Amarante P, Loeloff R, Luo Y, Fisher S, Fuller J, Edenson S, Lile J, Jarosinski MA, Biere AL, Curran E, Burgess T, Louis JC, Collins F, Treanor J, Rogers G, Citron M (1999). Beta-secretase cleavage of Alzheimer’s amyloid precursor protein by the transmembrane aspartic protease BACE. Science 286:735–741. [DOI] [PubMed] [Google Scholar]
- Vetrivel KS, Cheng H, Kim SH, Chen Y, Barnes NY, Parent AT, Sisodia SS, Thinakaran G (2005). Spatial segregation of γ-secretase and substrates in distinct membrane domains. J Biol Chem 280:25892–25900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weible MW, Hendry IA (2004). What is the importance of multivesicular bodies in retrograde axonal transport in vivo? J Neurobiol 58:230–243. [DOI] [PubMed] [Google Scholar]
- Yamazaki T, Koo EH, Selkoe DJ (1996). Trafficking of cell-surface amyloid beta-protein precursor. II. Endocytosis, recycling and lysosomal targeting detected by immunolocalization. J Cell Sci 109:999–1008. [DOI] [PubMed] [Google Scholar]
- Zheng H, Jiang M, Trumbauer ME, Sirinathsinghji DJ, Hopkins R, Smith DW, Heavens RP, Dawson GR, Boyce S, Conner MW, Stevens KA, Slunt HH, Sisoda SS, Chen HY, Van Der Ploeg LH (1995). beta-Amyloid precursor protein-deficient mice show reactive gliosis and decreased locomotor activity. Cell 81:525–531. [DOI] [PubMed] [Google Scholar]