

Abstract
The cellular and molecular mechanisms mediating the activity-dependent development of brain circuitry are still incompletely understood. Here, we examine the role of cAMP-dependent protein kinase [protein kinase A (PKA)] signaling in cortical development and plasticity, focusing on its role in thalamocortical synapse and barrel map development. We provide direct evidence that PKA activity mediates barrel map formation using knock-out mice that lack type IIβ regulatory subunits of PKA (PKARIIβ). We show that PKARIIβ-mediated PKA function is required for proper dendritogenesis and the organization of cortical layer IV neurons into barrels, but not for the development and plasticity of thalamocortical afferent clustering into a barrel pattern. We localize PKARIIβ function to postsynaptic processes in barrel cortex and show that postsynaptic PKA targets, but not presynaptic PKA targets, have decreased phosphorylation in pkar2b knock-out (PKARIIβ−/−) mice. We also show that long-term potentiation at TC synapses and the associated developmental increase in AMPA receptor function at these synapses, which normally occurs as barrels form, is absent in PKARIIβ−/− mice. Together, these experiments support an activity-dependent model for barrel map development in which the selective addition and elimination of thalamocortical synapses based on Hebbian mechanisms for synapse formation is mediated by a cAMP/PKA-dependent pathway that relies on PKARIIβ function.
Keywords: somatosensory cortex, activity dependent, glutamate, PKA, barrel, NMDA receptor
Introduction
A significant remaining challenge in developmental neuroscience is to understand the cellular and molecular mechanisms responsible for the organization of the intricate pattern of neural connections typical of the mammalian forebrain. Topographic maps of the sensory periphery are useful for this purpose, because even their gross form reflects the organization of precise underlying circuitry. Evidence suggests that the development of crude topography in these maps requires molecular cues, and map refinement depends, at least in part, on neural activity (Erzurumlu and Kind, 2001; Kind and Neumann, 2001; Cline, 2003; Lopez-Bendito and Molnar, 2003). A powerful model to study this process is the representation of facial whiskers in the primary somatosensory cortex of mice, known as “barrel” cortex. Each cortical barrel is composed of a barrel “wall” of densely packed layer IV cortical neurons that orient their dendrites into a cell-sparse barrel “hollow” occupied by clustered thalamocortical (TC) axons (TCAs) that relay input from a single whisker through the ventroposteromedial (VPM) thalamus. Evidence from a variety of sources, including studies of activity-dependent thalamocortical synapse development (Crair and Malenka, 1995; Feldman and Knudsen, 1998) and examination of various mutant mice with barrel map deficits (Erzurumlu and Kind, 2001; Kind and Neumann, 2001), suggests that barrel formation relies on an activity dependent process.
The cellular signaling events triggered by neuronal activity that influence barrel development are just now being examined in detail. Studies with “barrelless” (brl) mice, a loss of function mutant in the gene coding for adenylyl cyclase type I (Adcy1) (Welker et al., 1996; Abdel-Majid et al., 1998), have focused attention on the role of protein kinase A (PKA) signaling in barrel map development. Adcy1 is a Ca2+/calmodulin-stimulated adenylyl cyclase that catalyzes the formation of cAMP when Ca2+ concentrations increase, which occurs as a result of activation of NMDA receptors (NMDARs) (Wang and Storm, 2003). The main target of cAMP is PKA, a kinase strongly implicated in activity dependent changes in synaptic function related to learning and memory (Sweatt, 2001; Bauman et al., 2004) and developmental plasticity (Beaver et al., 2001, 2002; Yasuda et al., 2003; Fischer et al., 2004; Rao et al., 2004). We previously described deficits in thalamocortical long-term potentiation (LTP) and PKA-dependent phosphorylation of AMPA receptors (AMPARs) in brl mice, which are likely to play an important role in activity-dependent barrel map formation (Lu et al., 2003).
The PKA holoenzyme is a tetramer composed of a homodimer of two regulatory subunits (RIα, RIβ, RIIα, or RIIβ), each of which binds to one of three catalytic subunits (Cα, Cβ, or Cγ). Binding of cAMP to the regulatory subunit triggers its dissociation from the catalytic subunit, which becomes enzymatically active. In the last decade, mice with null mutations of the genes encoding the various PKA subunits have been generated in an effort to distinguish the role of different subunits in PKA activity (Brandon et al., 1997). Although the loss of a subunit is often compensated for by an increase in expression of other subunits, specific functions are perturbed in various PKA subunit knock-outs (Huang et al., 1995; Qi et al., 1996; Amieux et al., 1997; Brandon et al., 1998; Thiele et al., 2000; Howe et al., 2002; Fischer et al., 2004; Rao et al., 2004). Histological studies show that all PKA subunits are expressed in neurons, but PKARIIβ is a good candidate for providing specificity for PKA activity in neurons because of its low cAMP affinity and restricted subcellular expression pattern (Cadd and McKnight, 1989; Brandon et al., 1997).
Using pkar2b knock-out (PKARIIβ−/−) mice, we provide evidence for a direct role of PKA activity in the morphological and cytoarchitectonic development of cortical layer IV neurons during barrel map formation. Our findings also reveal that PKARIIβ-mediated PKA activity is not required for the gross patterning of TCA development and plasticity. Finally, we show that long-term potentiation and the associated development of AMPAR-mediated function at thalamocortical synapses is impaired in PKARIIβ−/− mice, lending further evidence in support of the hypothesis that activity-dependent regulation of thalamocortical synapse development by PKA plays an important role in barrel map formation.
Materials and Methods
Animals and PCR analysis
PKARIIβ homozygous knock-out mice of the sixth backcross generation of the incipient C57BL/6-pkar2b−/− 98% congenic inbred strain were originally generated by Dr. Stanley McKnight (University of Washington, Seattle, WA) (Brandon et al., 1998) and obtained commercially from Taconic Farms (Germantown, NY). PKARIIβ−/− mice were generated by a neomycin cassette insertion into the first exon of the pkar2b gene. Experiments were performed blind to genotype, with littermate controls. With the exception of the Golgi data, n in the text refers to the number of animals, because only one cell or section per animal was used for analysis. DNA for genotyping was purified from tails using the DNeasy kit from Qiagen (Valencia, CA). Wild-type PCR primers (5′-GCAGGATGAGCATCGAG-3′, 5′-TTCGAGAGTGAGGCGGA-3′) produced a 330 base pair (bp) band, and mutant primers (5′-GCAGGATGAGCATCGAG-3′, 5′-TTCGAGAGTGAGGCGGA-3′) produced a 244 bp band. Sixteen to 20 μl volumes were used for PCR prepared using the PCR kit from Qiagen. The PCR was performed with an initial denaturation step at 95°C for 5 min and 40 cycles at 94°C for 1 min, 57°C for 1 min, 72°C for 45 s, and a single final cycle at 72°C for 10 min. PCR products were resolved on a 2% agarose gel and detected with ethidium bromide.
Histology
Cytochrome oxidase staining.
Cytochrome oxidase (CO) is an enzyme of the mitochondrial electron transport chain that is responsible for most of the ATP production in cells. Its relative activity in tissue sections, which can be easily demonstrated by following a simple histochemical procedure (Seligman et al., 1968; Wong-Riley et al., 1978), reveals the pattern of thalamic afferent clustering into barrels in S1 cortex of rodents (Wong-Riley and Welt, 1980). It can also be used to detect changes in the pattern of thalamic afferent clustering into cortical barrels after manipulation of the sensory periphery (Wong-Riley et al., 1980; Lu et al., 2001). Briefly, barrel cortex was removed following the methods described by Strominger and Woolsey (1987), fixed and flattened for 2–4 h in 4% paraformaldehyde (PFA) at room temperature, and cut tangentially (parallel to layer IV) on a vibratome (VT1000S; Leica, Nussloch, Germany) into 100 μm sections and subject to CO staining. Sections were incubated with CO reaction solution free-floating for 2 h at room temperature or 10–14 h at 4°C. After visual detection of stain, sections were washed with PBS three times and mounted with Fluoromount-G (Electron Microscopy Sciences, Hatfield, PA).
Nissl staining and analysis of barrel cytoarchitecture.
Nissl stain, which marks neuronal nuclei, was used to reveal cortical barrel cytoarchitecture. Briefly, animals were perfused transcardially with ice-cold PBS followed by 4% PFA in PBS. Barrel cortex was then removed and flattened for 4 h in 4% PFA at room temperature and cut tangentially (parallel to layer IV) on a vibratome (VT1000S; Leica) into 50-μm-thick tangential sections. Sections were then mounted and dried for 1 d on a slide warmer at 37°C. Slides were dehydrated and rehydrated in graded alcohol, then fixed in 10% formalin (Sigma, St. Louis, MO) and stained with 0.2% cresyl violet solution for 15 min. After dehydration with graded alcohol and xylene, slides were mounted with Cytoseal (Richard-Allen Scientific, Kalamazoo, MI). The density of neurons in barrel walls and hollows was determined in three representative barrels (b2, c2, and d2) by marking the cells within a closed fixed contour drawn using Neurolucida software (MicroBrightField, Colchester, VT). The average (Avg) cell density of these three barrels was calculated for each animal and compared between genotypes. All counts were done blind to the genotype.
Hematoxylin and eosin staining.
Whisker pads of the lesioned (right) and control (left) side were removed from the snout and fixed with 4% PFA overnight at 4°C. PFA (4%) was exchanged with 30% sucrose for cryoprotection for 2 d at 4°C. Serial tangential sections of 50 μm were collected using a cryostat and subjected to standard hematoxylin and eosin (H&E) staining to examine hair follicle cells.
Golgi staining and analysis of dendrite asymmetry.
Golgi stained neurons were obtained using the FD Rapid Golgi Stain kit (FD Neurotechnologies, Ellicott City, MD). Tissue was prepared according to the user manual, and 50 μm serial coronal sections were cut using a cryostat. Golgi staining was performed following kit instructions, and Nissl counterstaining, used to determine the boundaries of layer IV, was performed right after Golgi staining and before dehydration, as explained in the kit. Randomly selected layer IV spiny stellate neurons were examined under a light microscope at 10×, and dendrites were reconstructed in three dimensions at 100× with an oil immersion lens using Neurolucida software (MicroBrightField) blind to the genotype. Polar graphs that represent the length of dendritic processes binned every 10° (Fig. 3E,F) were plotted for each neuron using Neuroexplorer (MicroBrightField). Dendritic asymmetry was evaluated from the polar plots by calculating the dendritic length in the hemisphere (180°) with the greatest density of dendrites relative to the total dendritic length in a given neuron. Spiny stellate neurons with a dendritic asymmetry of 0.75 and above were accepted to have a dendritic orientation bias. Other parameters such as total dendritic length, spine density, and dendritic field span were also determined using Neuroexplorer (MicroBrightField).
Figure 3.
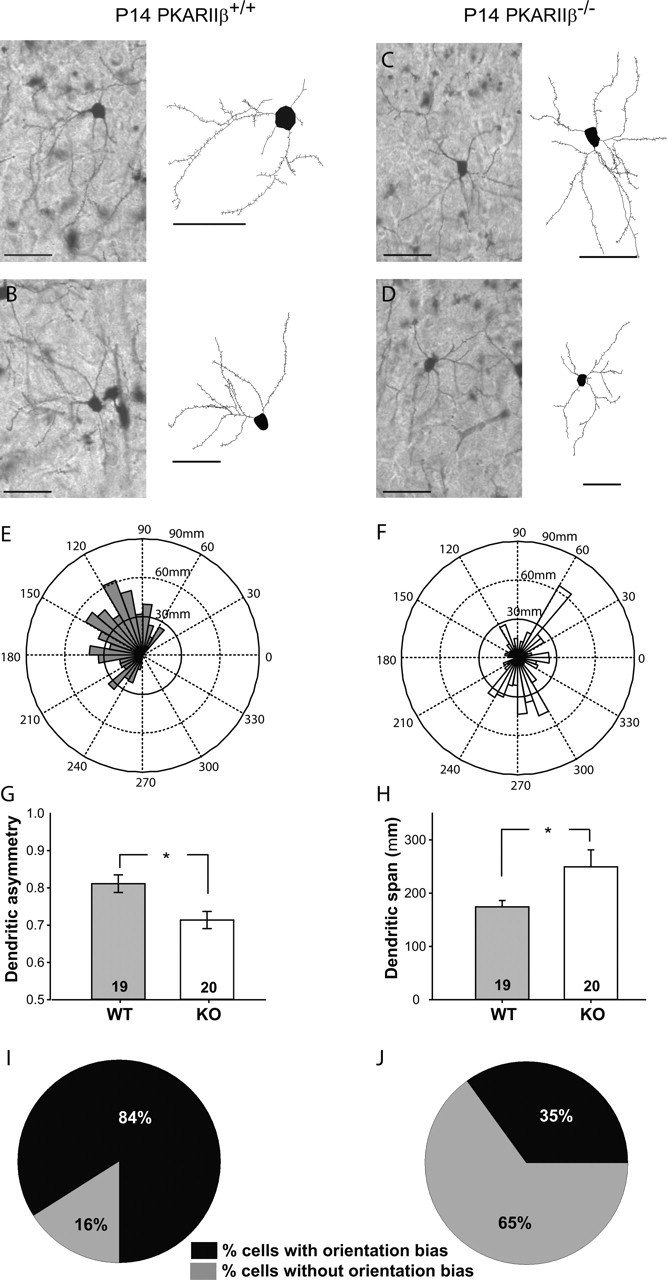
Layer IV spiny stellate neurons in PKARIIβ−/− mice have less orientation bias than wild-type littermate controls. A–D, Examples of Golgi-stained layer IV spiny stellate neurons at low magnification (left) and their computer-aided reconstructions (right) in wild-type (A, B) and PKARIIβ−/− (C, D) mice at P14. E, F, Polar graphs of the dendrites of neurons illustrated in B and D (see Materials and Methods). G, PKARIIβ−/− (KO) mice (0.71 ± 0.02; 20 neurons from 4 animals) have significantly lower (*p < 0.01; t test) dendritic asymmetry compared with wild-type (WT) littermate controls (0.81 ± 0.02; 19 neurons from 4 animals). Dendritic asymmetry, which must be ≥0.5, is defined as the ratio of the dendritic length in the hemisphere with the greatest density of dendrites relative to the total dendritic length. H, Dendritic field span, the greatest distance between the most distal dendrite tips of a particular layer IV spiny stellate neuron, is significantly higher (*p < 0.05; t test) in PKARIIβ−/− (KO) mice (249.43 ± 31.77 μm) compared with wild-type (WT) littermate controls (174.24 ± 11.96 μm). I, J, Pie charts showing the percentage of cells with and without orientation bias in PKARIIβ−/− (J) and wild-type littermate control mice (I). Layer IV spiny stellate neurons are defined to have an orientation bias if their dendritic asymmetry is ≥0.75 (see Materials and Methods). Scale bar, 50 μm. Error bars indicate SEM.
Sensory manipulations
C row of whiskers and whisker follicles from postnatal day 1 (P1) and P5 animals were cauterized with a surgical cautery device (Malis Bipolar Coagulator & Cutter; model CMC II; Codman, Piscataway, NJ). Pups were anesthetized on ice, and whiskers on the right side were visualized under a light microscope. After cauterization, the pups were revived in a warmed, oxygenated chamber and returned to their mother. The animals were then killed at P12–P14 for CO and H&E staining (see above). Animals with lesions that spread beyond row C, or did not include at least the first four whiskers in row C, as revealed by the H&E staining, were excluded from analysis.
For quantification of barrel map plasticity, the width of CO-stained whisker barrels corresponding to the b1, b2, b3, c1, c2, c3, d1, d2, and d3 whiskers were measured using ImageJ Software (W. S. Rasband, National Institutes of Health, Bethesda, MD). The ratio of the average width of C-row whisker barrels (c1–c3) relative to the B- and D-row whisker barrels (b1–b3 and d1–d3) was used to quantify a map plasticity index (MPI) defined as follows: MPI = 2 × [Avg(c1, c2, c3)]/[Avg(b1, b2, b3) + Avg(d1 + d2 + d3)].
PKA activity assay
PKA activity was measured in total tissue homogenates as a ratio in the presence and absence of cAMP using the SignaTECT cAMP-dependent protein kinase assay kit (Promega, Madison, WI). Briefly, somatosensory cortex was removed at P11 and homogenized in ice-cold homogenization buffer (0.32 m sucrose, 10 mm HEPES, pH 7.4) with protease and phosphatase inhibitor mixtures (Sigma) on the same day as the activity test. One gram of tissue was homogenized in 5 ml of homogenization buffer. Appropriate dilutions were prepared in 0.1 mg/ml BSA, and the reaction was performed following the steps clearly explained in the kit. A control reaction without substrate was also run for each sample to determine the background radioactivity, which was counted using a scintillation counter.
Protein analyses
Antibodies.
The following primary antibodies were used: PKARIIβ (at 1:1000 dilution for immunoblotting and 1:500 for immunohistochemistry) and PKAC (1:1000) from PharMingen (San Diego, CA); serotonin transporter (5-HTT; 1:250) from Immunostar (Hudson, WI); microtubule-associated protein (MAP2; 1:1000) from Sternberger (Lutherville, MD); actin (1:750) from Sigma; phospho-synapsin (Ser9) (1:1000) from Cell Signaling Technology (Beverly, MA); phospho-MAPK (mitogen-activated protein kinase; 1:1000) from Chemicon (Temecula, CA); phospho-Rim (1:1000) from Dr. G. Lonart (Eastern Virginia Medical School, Norfolk, VA); phospho-glutamate receptor 1 (phospho-GluR1; 1:1000), phospho-NMDAR subunit 1 (phospho-NR1; 1:1000), and GluR1 (1:1000) from Upstate Biotechnology (Lake Placid, NY); Rim (1:1000) and NR1 (1:5000) from Synaptic Systems (Göttingen, Germany). The following secondary antibodies were used: mouse IgG–HRP (1:20,000) and rabbit IgG–HRP (1:12,500) from Pierce (Rockford, IL); mouse IgG–Alexa 488 (1:500), mouse IgG–cyanine 3 (Cy3) (1:500), rabbit IgG–Alexa 488 (1:500) from Invitrogen (San Diego, CA).
Immunoblotting.
Barrel cortex was isolated from P11 mice and homogenized in homogenization buffer with protease and phosphatase inhibitor mixtures (Sigma). Subcellular fractions were acquired as described previously (Lu et al., 2003). Briefly, the homogenates were centrifuged at 800 × g for 10 min and the pellet was saved as P1, which is enriched with cell bodies. The supernatant was then centrifuged at 7100 × g for 15 min to yield pellet P2, the synaptosome-enriched fraction. Synaptosome enrichment was confirmed by probing different fractions with antibodies for synaptic marker proteins [e.g., PSD-95 (postsynaptic density-95)]. Protein concentrations were measured with Bradford assay using Bio-Rad (Hercules, CA) protein assay kit II. Twenty micrograms of total protein were loaded into 5–20% gradient SDS-PAGE gels and electrophoretically transferred to nitrocellulose membranes (Criterion system; Bio-Rad). Primary antibodies against the protein in question were applied and the signal detected with HRP-conjugated secondary antibodies. Immunoreactivity was quantified with ECL pico (Pierce), and densitometric quantification was performed with Optiquant software (Packard Bioscience, Meriden, CT) by background subtracting and normalizing band intensities to the wild-type band in each set. When using PKA site-specific antibodies, quantification was performed by comparing the level of phospho-specific antibody to the activity of antibodies against the total protein. Otherwise, quantification was performed relative to actin, which is expressed at consistent high levels throughout barrel development.
Immunofluorescence and colocalization analysis.
Mice were perfused as explained above, and the brain was removed and fixed overnight at 4°C. One hundred micrometer sections in the thalamocortical plane were cut with a vibratome, and sections remained free-floating for all incubations and washes. Briefly, tissues were permeabilized with 0.7% Triton X-100 in PBS for 20 min, incubated with 0.1 m glycine for 30 min, and then blocked with 1% normal goat serum (NGS) and 2 mg/ml BSA in PBS containing 0.01% Triton X-100 (PBST). Sections were then transferred to the primary antibody for incubation overnight at 4°C with appropriate dilution in PBST with 1% NGS. After washing six times in PBST, sections were incubated in the secondary antibody for 2 h at room temperature then washed again six times. 4′,6′-Diamidino-2-phenylindole (300 nm) was often applied to label nuclei, followed by a wash and postfix of the sections with PFA, and then mounting with Fluoromount-G (Electron Microscopy Sciences). Immunolabeling was visualized with a fluorescence microscope, and images were collected with a Leica DM confocal scanning microscope using 10, 20, and 63× water immersion lenses with the channels for Cy3 (red) and Alexa488 (green) used sequentially. Each image was acquired with the laser intensity adjusted to prevent over-saturation. For double-labeling with two antibodies made in different species, a mixture of the antibodies against the two proteins was applied at once. If the two antibodies were made in the same species, the primary antibodies were applied sequentially. Colocalization of antibodies that mark either presynaptic (5-HTT) or postsynaptic (MAP2) processes with an antibody for PKARIIβ was performed on 63× images using the colocalization finder function in ImageJ.
Slice electrophysiology
Acute TC slices were prepared as described previously (Lu et al., 2001). The artificial CSF (ACSF; in mm: 124 NaCl, 5 KCl, 1.25 NaH2PO4, 1.3 MgSO4, 2 CaCl2, 26 NaHCO3, and 11 glucose, pH 7.2, 290–300 mOsm) was saturated with 95% O2 and 5% CO2. The whole-cell recording solution contained the following (in mm): 99 cesium gluconate, 17.5 CsCl, 8 NaCl, 10 HEPES, 0.2 EGTA, 4 Mg-ATP, 0.3 GTP, 7 phosphocreatine, and 10 BAPTA. BAPTA was included in the pipette to prevent inadvertent potentiation of the postsynaptic neuron.
Stimuli were applied to the ventrobasal thalamus through bipolar sharpened and insulated stainless-steel microelectrodes (Frederick Haer Company, Bowdoinham, ME). Data were collected and analyzed on-line using a computer-driven acquisition system (National Instruments, Austin, TX) and software that was written under the Igor (WaveMetrics, Lake Oswego, OR) programming environment.
Input–output analysis of synaptic transmission using field potential recordings was conducted with a range of stimulus intensities (0–850 μA with 50 μA intervals). The nonspecific glutamate receptor antagonist kynurenic acid (10 mm) (Sigma) was routinely added to the perfusate at the end of the recordings to ensure the identification of fiber volley and synaptic response was correct. Fiber volley amplitudes and field EPSP slopes were averaged for 20 sweeps at each stimulus intensity.
EPSCs were measured in voltage-clamp mode using in vitro whole-cell voltage-clamp recording techniques following published protocols (Lu et al., 2001). To evaluate and monitor the health of the cell, input and series resistances were continuously monitored, with cells that had <300 MΩ input resistance or drifted >20% discarded. Only responses that exhibited short and constant latencies that did not change with increasing stimulus intensity were considered monosynaptic. Because the responses in young cells tend to drift down if the stimulation intensity or the stimulation frequency is high, half-saturating stimulation strength with a relatively low stimulation frequency (10–15 ms interval) was used to evoke stable EPSCs. Before any experimental manipulation, 10–15 min of stable baseline response was acquired. EPSC amplitudes were calculated by subtracting the mean current during a fixed 3–4 ms window before the stimulus artifact from the mean current during a similar window at the peak of the EPSC.
To measure the AMPAR/NMDAR current ratio, which is a gross measure of the relative contribution of AMPA and NMDA receptor-mediated currents across a population of synapses, we first isolated the AMPA response by voltage clamping the cell at hyperpolarized membrane potentials (−70 mV) while stimulating the thalamus. We then depolarized the cell to +40 mV to relieve the Mg2+ block of the NMDAR and added 2,3-dihydroxy-6-nitro-7-sulfonyl-benzo[f]quinoxaline (10 μm) (Tocris Cookson, Ballwin, MO) to the perfusate to block AMPA and kainate receptors, leaving a pure NMDAR response. These experiments were done at normal (half-saturating) stimulus strength as a gross measure of the relative amplitude of the AMPAR and NMDAR currents (Crair and Malenka, 1995; Lu et al., 2001, 2003) in the presence of GABAergic antagonists (0.1 mm picrotoxin, cesium ions in the whole-cell solution block GABAB responses).
For analysis of AMPA “evoked miniature” events, stable whole-cell voltage-clamp recordings were established at −70 mV holding potential, and the Ca2+ in the ACSF was exchanged for Sr2+. Sr2+-based ACSF desynchronizes release, allowing isolated evoked miniature currents to be analyzed (Xu-Friedman and Regehr, 1999). Evoked miniature events were recorded in 1 s epochs every 2 s in Igor Pro using ACSF (2 mm Sr2+) containing 100 μm picrotoxin and 50 μm d-AP-5 (Tocris Cookson) to eliminate inhibitory currents and possible NMDAR current contamination, respectively. Data was imported into Mini Analysis (Synaptosoft, Decatur, GA), amplitude thresholds were set at 2.5 times root mean square (RMS) noise, and at least 80 events were identified and used for analysis in each cell. Temporal windows were chosen to represent both the fast and slow components of miniature event decay times. A significant advantage of measuring “evoked minis” is that the population is dominated by TC synapses, as opposed to measuring spontaneous minis in TTX, which is a random sample of all synapses on the cell.
LTP was induced using a pairing protocol, in which postsynaptic depolarization is “paired” with presynaptic stimulation in whole-cell voltage-clamp mode (Lu et al., 2001). For these experiments, BAPTA was excluded from the pipette solution, and EPSCs from somatosensory cortex layer IV neurons elicited with thalamic stimulation were recorded at −70 mV until a stable baseline of 6–10 min was obtained. Pairing was then initiated by switching to a holding potential of −10 mV and stimulating at 1 Hz for 100 s. The holding potential was then switched back to −70 mV after pairing. The percentage of EPSC change was calculated as the mean EPSC amplitude of 20 sweeps at 20 min after pairing minus the mean EPSC amplitude of 20 sweeps right before pairing (baseline amplitude) divided by the baseline amplitude.
Results
PKA activity and catalytic subunit protein levels are reduced in PKARIIβ−/− mice
PKARIIβ−/− mice lack a regulatory subunit that controls PKA signaling activity. Because binding of PKA regulatory subunits to PKA catalytic subunits renders the PKA holoenzyme inactive, one would naively expect an increase in PKA activity in PKARIIβ−/− mice. However, previous studies show that PKA activity is actually reduced in PKARIIβ−/− mice (Brandon et al., 1997) because of the rapid degradation of catalytic subunits in the absence of a regulatory subunit (Amieux et al., 1997; Brandon et al., 1998).
We examined how PKA activity was affected in the developing somatosensory cortex of PKARIIβ−/− mice using tissue homogenates from P11 somatosensory cortex of PKARIIβ−/− mice (n = 3) and wild-type controls (n = 3). When compared with wild types, knock-outs had a significant reduction in stimulated PKA activity (ratio of PKA activity in the presence and absence of cAMP) (2.55 ± 0.07 vs 1.32 ± 0.03; p < 0.005; t test) (Fig. 1A). We also measured PKAC protein levels in synaptosomes prepared from P11 somatosensory cortex of PKARIIβ−/− mice and wild-type littermate controls using Western blot analysis (Fig. 1A,B). Quantification demonstrated that PKAC subunit protein levels are lower in PKARIIβ−/− somatosensory cortex compared with wild-type controls (0.49 ± 0.06; n = 5 pairs; p ≪ 0.01; t test). These data show that a decrease in the basal level of catalytic subunit protein likely causes a reduction in PKA activity in the developing somatosensory cortex of PKARIIβ−/− mice.
Figure 1.

Reduced PKA activity in somatosensory cortex of PKARIIβ−/− mice. A, The ratio of PKA activity measured in the presence (+cAMP) and absence (basal) of cAMP is significantly reduced in P11 somatosensory cortex of PKARIIβ−/− (KO) mice compared with wild-type (WT) controls (histograms on left; *p < 0.005, t test). PKA activity was measured in tissue homogenates from somatosensory cortex using a protein kinase assay kit (Promega). PKAC protein levels are also significantly reduced in PKARIIβ−/− compared with wild-type littermate control mice (histograms on right; *p ≪ 0.01, t test). B, Example Western blots of synaptosomes prepared from P11 somatosensory cortex of PKARIIβ−/− mice and their wild-type littermate controls using anti-PKARIIβ and anti-PKAC antibodies. Actin is used to normalize the protein amount. As expected, PKARIIβ−/− mice have no PKARIIβ expression. Error bars show SEM.
Layer IV barrel cytoarchitecture is disturbed in PKARIIβ−/− mice
Barrels are composed of the clustered arbors of thalamocortical afferents and a surrounding ring of neurons in layer IV of the somatosensory cortex. We examined the clustering of presynaptic TCAs and the organization of postsynaptic layer IV neurons in tangential sections from flattened somatosensory cortex of P14–P21 PKARIIβ−/− mice and wild-type littermate controls (Fig. 2). CO staining, a standard method for visualizing barrels (Seligman et al., 1968; Wong-Riley and Welt, 1980; Wong-Riley et al., 1978), showed that clustering of PKARIIβ−/− TCAs (n = 6) is comparable with that of wild-type littermate controls (n = 6) (Fig. 2A,B). TCAs transiently express 5-HTT during barrel development (Lebrand et al., 1996, 1998; Hansson et al., 1998; Datwani et al., 2002). No difference in the TCA barrel pattern was observed using an antibody against 5-HTT in tangential sections through somatosensory cortex of younger animals (P5–P7) either (n = 4 for both genotypes) (Fig. 2C,D). In contrast, Nissl staining of tangential sections through barrel cortex, which reveals the pattern of postsynaptic neurons, showed that the organization of layer IV neurons into barrels is disturbed in PKARIIβ−/− mice (Fig. 2E,F). We quantified the barrel cytoarchitecture defect by measuring the relative density of neurons in barrel walls and hollows. As reported previously in wild-type mice (Pasternak and Woolsey, 1975), the cell density in barrel walls is much higher than in barrel hollows (ratio of 1.76 ± 0.05; n = 4), whereas in PKARIIβ−/− mice, there is no difference in cell density between the wall and hollow (0.93 ± 0.05; n = 4; p < 0.001 for difference between PKARIIβ−/− and wild type, t test).
Figure 2.
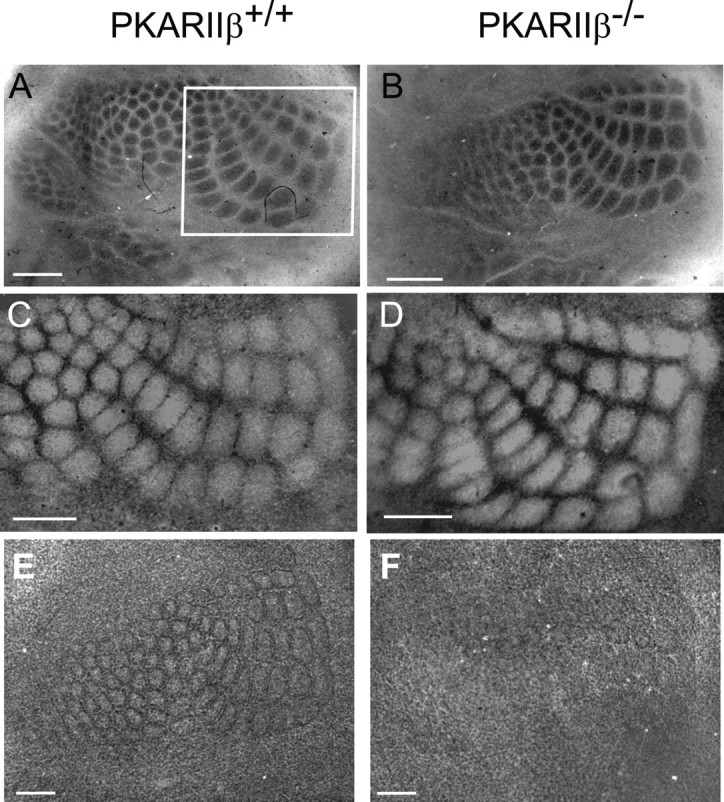
Organization of layer IV neurons but not thalamocortical axon clustering is disrupted in PKARIIβ−/− mice. A, B, Tangential sections from P16 wild-type (A) and PKARIIβ−/− (B) mice stained with CO (n = 6 for both genotypes). Posteromedial whisker barrel patterns (white box in A) are indistinguishable between genotypes. C, D, Tangential sections from P7 wild-type (C) and PKARIIβ−/− (D) mice immunostained with anti-5-HTT antibody (n = 4 for both genotypes), which selectively labels thalamocortical afferents. The barrel pattern is again indistinguishable between genotypes, showing that the gross patterning of thalamocortical afferents in PKARIIβ−/− mice is similar to that of wild types. E, F, Nissl stained tangential sections from P16 wild-type (E) and PKARIIβ−/− (F) mice. In wild-type mice (E), layer IV neurons organize into a barrel wall and hollow pattern characteristic of rodent somatosensory cortex. In PKARIIβ−/− mice (F), only a very rudimentary barrel pattern is visible in posteromedial barrel field. Quantification confirms that the density of neurons in the barrel wall relative to the barrel hollow is significantly higher in wild-type mice than in PKARIIβ−/− littermate mice (1.76 ± 0.05 in n = 4 wild-type mice; 0.93 ± 0.05 in n = 4 PKARIIβ−/− mice; p < 0.001, t test). Scale bars, 500 μm.
In addition to forming cytoarchitectonic barrel walls in somatosensory cortex, layer IV neurons also preferentially orient their dendrites toward a barrel hollow, presumably to make synapses with thalamocortical afferents relaying information from one facial whisker (Woolsey et al., 1975; Datwani et al., 2002). We examined whether dendritic asymmetry in spiny stellate cells, a layer IV cell type known to have oriented dendrites (Harris and Woolsey, 1979, 1983), was impaired by the loss of PKARIIβ function in the PKARIIβ−/− mice (Fig. 3). Dendrites of Golgi stained neurons in wild-type mice typically show substantial dendritic asymmetry (Fig. 3A,B,G), with only 16% of neurons lacking orientation bias (Fig. 3I) (arbitrarily defined as dendritic asymmetry <75%; see Materials and Methods). In contrast, 65% of layer IV neurons in PKARIIβ−/− mice lacked orientation bias (Fig. 3J), resulting in significantly less dendritic asymmetry in PKARIIβ−/− (0.71 ± 0.02; n = 20 cells from four animals) (Fig. 3G) compared with wild-type neurons (0.81 ± 0.02; n = 19 cells from four animals; p < 0.01; t test). The span of dendrites in PKARIIβ−/− mice (249.43 ± 31.77 μm; n = 20) was also significantly broader than in wild-type neurons (174.24 ± 11.96 μm; n = 19; p < 0.05; t test) (Fig. 3H), which is consistent with the barrel cytoarchitectonic deficits in these mice. Other dendritic parameters, including total dendritic length (PKARIIβ+/+ mice, 746.2 ± 77.6 μm, n = 19; PKARIIβ−/− mice, 988.7 ± 113.1 μm, n = 20) and spine density (PKARIIβ+/+ mice, 0.22 ± 0.02 per μm, n = 19; PKARIIβ−/− mice, 0.19 ± 0.02 per μm, n = 20), were normal in PKARIIβ−/− mice. In total, these data show that in PKARIIβ−/− mice, presynaptic thalamic afferents form a normal barrel pattern, but postsynaptic layer IV neurons organize into rudimentary barrel patches with dramatically disturbed neuronal morphology and cytoarchitecture.
PKARIIβ has a preferred postsynaptic expression in barrel cortex
To gain some insight into the locus of action of PKARIIβ-mediated PKA signaling in barrel development, we examined the pattern of PKARIIβ expression in barrel cortex during the time barrels are forming. Previous studies using in situ hybridization and immunohistochemistry on adult mice showed that PKARIIβ is expressed strongly in neocortex but is expressed relatively weakly in the thalamus (Cadd and McKnight, 1989; Glantz et al., 1992). In wild-type thalamocortical slices stained with an anti-PKARIIβ antibody, we found that PKARIIβ is present in layer IV at P3 (data not shown) and is enriched in a barrel pattern when the barrels emerge at around P5 (Fig. 4B,C).
Figure 4.
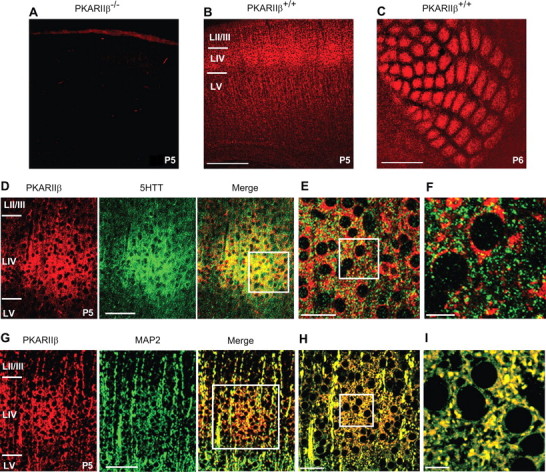
PKARIIβ is extensively colocalized with postsynaptic but not presynaptic markers. A, Control immunostaining of a P5 PKARIIβ−/− thalamocortical slice with anti-PKARIIβ antibody reveals that this antibody is specific to PKARIIβ. B, C, Cortical expression of PKARIIβ was examined by labeling wild-type thalamocortical (B) and tangential (C) slices with anti-PKARIIβ antibody. PKARIIβ is expressed in layer IV as it delaminates from layer II/III at P3 (data not shown) and forms a barrel pattern when barrels emerge at around P5 (B, C). D–I, Presynaptic and postsynaptic localization of PKARIIβ was examined by coimmunostaining P5 wild-type thalamocortical slices using anti-5-HTT (D–F) and anti-MAP2 (G–I) antibodies, respectively, together with an anti-PKARIIβ antibody. D, Low-magnification image of a single barrel stained with a 5-HTT antibody (green) to label presynaptic thalamocortical terminals and a PKARIIβ (red) antibody and their merged image. At low magnification, both show a clear barrel pattern. E, Merged image of the barrel in D (white box) at 63× magnification. F, A 4× zoom of white-boxed area in E. G, Low-magnification image of a single barrel labeled with a MAP2 antibody (green) to mark postsynaptic dendrites and a PKARIIβ antibody (red) and their merged image. H, Merged image of the barrel in G (white box) at 63× magnification. I, 4× zoom of white-boxed area in H. Colocalization analysis of high-magnification images using ImageJ shows that PKARIIβ expression colocalizes significantly more (p < 0.05; t test) with MAP2 (28.8 ± 7.5%; n = 4) than with 5-HTT (7.1 ± 2.2%; n = 4). LIV, Layer IV; LV, layer V. Scale bars: A–C, 500 μm; D, G, 100 μm; E, H, 40 μm; F, I, 10 μm.
To examine the subcellular localization of PKARIIβ, we double-labeled wild-type thalamocortical slices with antibodies for PKARIIβ and presynaptic or postsynaptic markers. Coimmunostaining of P5 wild-type thalamocortical slices with antibodies for PKARIIβ and 5-HTT, which mark presynaptic terminals on thalamocortical synapses, showed little colocalization (7.1 ± 2.2%; n = 4) (Fig. 4D–F). Similar results were obtained with antibodies to other presynaptic molecules, such as synapsin and GAP43 (data not shown). However, MAP2, a commonly used dendritic marker (Bernhardt and Matus, 1984; De Camilli et al., 1984), was extensively colocalized with PKARIIβ (28.8 ± 7.5%; n = 4; p < 0.05 for the difference between colocalization of PKARIIβ and 5-HTT and MAP2; t test) (Figs. 4G–I). These results indicate that PKARIIβ is preferentially localized in the postsynaptic terminal of layer IV neurons, which is consistent with the layer IV barrel phenotype in PKARIIβ−/− mice.
AMPAR/NMDAR current ratio does not increase with development in PKARIIβ−/− mice
Thalamocortical synaptic transmission assayed using field potential recordings in a thalamocortical brain slice was grossly normal in PKARIIβ−/− mice (Fig. 5A–C). In particular, the input–output curve, which measures synaptic response from a large population of neurons in layer IV of barrel cortex as a function of thalamocortical fiber volley amplitude caused by thalamic (VPM) stimulation, was indistinguishable in wild-type controls and PKARIIβ−/− mice at P6–P7 (n = 8 and n = 4, respectively) (Fig. 5C). This shows that there is no gross defect with functional thalamocortical synaptic innervation of layer IV in barrel cortex of PKARIIβ−/− mice.
Figure 5.
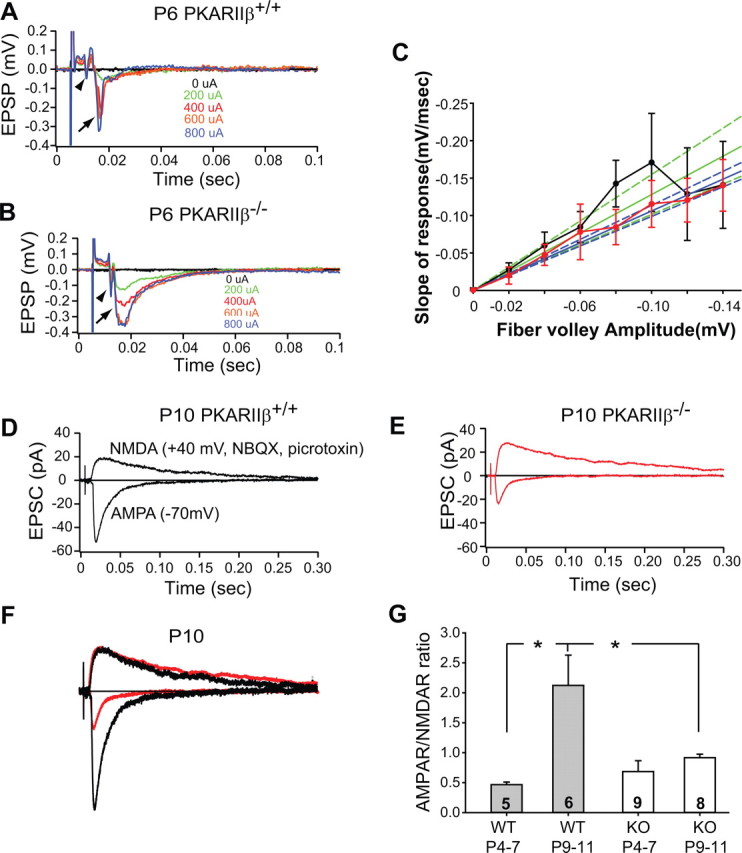
Developmental increase in AMPAR/NMDAR current ratio is absent in PKARIIβ−/− mice. A–C, Input–output analyses of extracellular field potential recordings indicate no difference in gross synaptic transmission between wild-type and PKARIIβ−/− thalamocortical synapses. A, B, Sample average responses at different stimulation strengths of P6 wild-type and PKARIIβ−/− mice. The arrowhead shows the fiber volley, and the arrow shows the postsynaptic response. C, Input–output curves in wild-type (black; n = 8) and PKARIIβ−/− (red; n = 4) mice at P6–P7 are similar. Regression analysis for wild types (green) and PKARIIβ−/− (blue) show no significant difference at 95% confidence interval (dashed lines overlap). D–F, Sample whole-cell voltage-clamp measurements of AMPAR-mediated and NMDAR-mediated thalamocortical EPSCs in a P10 wild-type littermate control (D) and P10 PKARIIβ−/− (E) mouse. F, Overlay of the responses in D and E scaled so that the NMDAR currents are the same amplitude. Note the AMPAR-mediated EPSC in PKARIIβ−/− animal is small in comparison to the littermate control. G, Summary quantification of AMPAR/NMDAR current ratios for different age groups and different genotypes. The AMPAR/NMDAR current ratio of wild-type animals increases significantly with age (*p < 0.05; t test). However, this increase is absent in PKARIIβ−/− (KO) mice (p = 0.27; t test). The AMPAR/NMDAR current ratio of P9–P11 PKARIIβ−/− mice is also significantly lower than that of P9–P11 wild-type (WT) littermate controls (*p < 0.05; t test). Error bars indicate SEM.
We used whole-cell voltage-clamp techniques to examine glutamatergic synaptic transmission at TC synapses. In wild-type mice, the ratio of AMPAR-mediated to NMDAR-mediated currents (AMPAR/NMDAR current ratio) increases during the first week after birth (0.47 ± 0.04, n = 5 for P4–P7; 2.12 ± 0.5, n = 6 for P9–P11; p < 0.05; t test) (Fig. 5G), which is typical of glutamatergic synaptic development at many central synapses (Crair and Malenka, 1995; Wu et al., 1996; Lu et al., 2001). We previously showed in brl mice that the AMPAR/NMDAR current ratio remained small during TC synapse development, but it was unclear whether this was specifically attributable to the absence of cAMP–PKA signaling in postsynaptic layer IV neurons (Lu et al., 2003). Because of the exclusive postsynaptic barrel map defect of PKARIIβ−/− mice and the preferable postsynaptic expression pattern of PKARIIβ, PKARIIβ−/− mice provided us an opportunity to analyze the effect of lowered PKA activity in layer IV neurons on TC synapse maturation. In PKARIIβ−/− mice, the normal developmental increase in AMPAR/NMDAR current ratio failed to occur (0.68 ± 0.18, n = 9 for P4–P7; 0.91 ± 0.05, n = 8 for P9–P11; p = 0.27; t test), and the AMPAR/NMDAR current ratio remained small (p < 0.05 for wild type vs PKARIIβ−/− at P9–P11; t test) (Fig. 5D–G), which is similar to what was observed in brl (Lu et al., 2003). These results indicate that the increase in AMPAR/NMDAR current ratio during TC synapse maturation is mediated by postsynaptic PKA function and interfering with this function impairs postsynaptic barrel map formation.
AMPAR-evoked miniature currents are small at PKARIIβ−/− TC synapses
Theoretically, the small AMPAR/NMDAR current ratio in PKARIIβ−/− mice could be attributable to small AMPAR-mediated currents or large NMDAR-mediated currents or both. We examined miniature AMPAR-mediated synaptic currents (“AMPA minis”) in P9–P11 mice to determine whether AMPAR currents are specifically affected by the loss of PKARIIβ function. Substitution of Ca2+ with Sr2+ in the extracellular ACSF leads to a reduction in synchronous evoked response and the appearance of delayed miniature responses as a result of the persistence of asynchronous quantal release (Xu-Friedman and Regehr, 1999) (Fig. 6A,B). This makes Sr2+ a useful tool to analyze evoked miniature EPSCs (evoked mini-EPSCs). Evoked mini-EPSCs in PKARIIβ−/− neurons (n = 6) were on average smaller than comparable wild-type neurons (n = 7; 7.3 ± 0.59 vs 10.6 ± 1.3 pA; p < 0.05; t test) (Fig. 6C, inset). Comparison of the evoked mini-EPSC amplitude histograms shows a shift in the peak of the distribution, with the histogram in PKARIIβ−/− mice peaking at a smaller amplitude than wild-type littermate controls (Fig. 6C). In addition, wild-type controls often had responses with very large amplitudes (Fig. 6A,C). The mean cumulative probability distributions also revealed a shift in evoked mini EPSCs in PKARIIβ−/− mice toward smaller amplitudes compared with wild-type littermate controls (Fig. 6D). The difference between genotypes was not caused by an intrinsic difference in RMS noise levels (1.98 ± 0.06 vs 2.06 ± 0.09 pA; p = 0.45; t test). These results indicate that in the absence of PKARIIβ-mediated PKA signaling, the number of functional AMPARs at TC synapses is lower on average, leading to smaller AMPAR mediated currents.
Figure 6.

Small AMPAR mini-EPSCs at PKARIIβ−/− thalamocortical synapses. A, B, Sample traces of evoked AMPAR-mediated current responses in Ca2+–ACSF (gray) and Sr2+–ACSF (black) from recordings in P9–P11 PKARIIβ−/− (B) and wild-type littermate control (A) neurons. In the presence of Sr2+–ACSF, responses to evoked synchronous release is lower and quantal events (evoked mini-EPSCs) in response to asynchronous release appear. The amplitudes of these quantal events are smaller on average in PKARIIβ−/− mice. C, Average frequency histogram of evoked mini-EPSCs. The PKARIIβ−/− histogram (white bars) peaks at a smaller amplitude than wild-type littermate controls (gray bars), which have a longer large amplitude tail. Inset, Mean evoked mini-EPSC amplitude is significantly lower (*p < 0.05; t test) for PKARIIβ−/− mice (white) with respect to wild-type littermate controls (gray). D, The cumulative probability distribution in PKARIIβ−/− (KO) animals (black) also shows a shift toward smaller amplitudes compared with wild-type (WT) littermate controls (gray). Comparison of root mean square noise shows no difference (p = 0.45; t test) between genotypes (data not shown). Error bars indicate SEM.
LTP is impaired at developing thalamocortical synapses in PKARIIβ−/− mice
The lack of a developmental increase in AMPAR currents could be attributable to deficits in thalamocortical LTP at developing thalamocortical synapses in PKARIIβ−/− mice (Lu et al., 2003). In this scenario, the cellular mechanisms responsible for LTP mediate the increase in AMPAR currents during thalamocortical synapse development via PKA-mediated phosphorylation of AMPA receptor subunits (Ehlers, 2000; Lee et al., 2000, 2003; Esteban et al., 2003). We examined this possibility using a standard LTP pairing protocol in thalamocortical slices from P4–P6 PKARIIβ−/− and wild-type littermate control mice (Fig. 7). In wild-type mice, pairing induced a robust potentiation of thalamocortical response (Fig. 7A,C,D) (122.96 ± 41.04%; n = 7). In contrast, neurons in littermate PKARIIβ−/− mice showed significantly less potentiation (1.59 ± 6.92%; n = 5; p < 0.05; t test) (Fig. 7B–D). This suggests that an activity-dependent process that is mediated by PKARIIβ is responsible for the normal functional development of thalamocortical synapses.
Figure 7.

LTP deficits in PKARIIβ−/− thalamocortical synapses. A, B, Example whole-cell voltage-clamp recordings from P6 wild-type control (A) and PKARIIβ−/− (B) neurons show that thalamocortical response in wild-type but not PKARIIβ−/− layer IV neurons can be potentiated using an LTP pairing protocol. Traces on the right (average of 20 sweeps) from before (1) and 30 min after pairing (2) show significant potentiation in wild-type but not PKARIIβ−/− neurons. C, Summary graph of pairing experiments showing the EPSC percentage change for PKARIIβ−/− mice (black circles) and their wild-type littermate controls (gray circles). PKARIIβ−/− thalamocortical synapses show no sign of potentiation. D, Summary histogram of EPSC percentage change from the average of 20 sweeps starting at 20 min after pairing for all neurons. PKARIIβ−/− (KO) thalamocortical synapses show almost no potentiation (1.59 ± 6.92%; n = 5), and their EPSC percentage change is significantly lower (*p < 0.05; t test) than that of wild-type (WT) littermate controls (122.96 ± 41.04%; n = 7). Error bars indicate SEM.
Phosphorylation of postsynaptic but not presynaptic PKA targets is reduced in PKARIIβ−/− mice
There are a number of PKA targets in the presynaptic and postsynaptic terminal of thalamocortical synapses that are potentially affected by decreased PKA activity in PKARIIβ−/− mice. We examined the phosphorylation levels of several of the most prominent targets, some of which have demonstrably reduced phosphorylation in brl mice (Lu et al., 2003, 2006). Western blot analysis on synaptosomes prepared from P11 somatosensory cortex of major presynaptic PKA targets, including phospho-synapsin (at Ser 9) and phospho-Rim, shows no difference between PKARIIβ−/− mice and wild-type littermate controls (0.91 ± 1.66, p = 0.26 and 1.03 ± 0.07, p = 0.56, respectively; t test; n = 5 for both). In contrast, phosphorylation of two postsynaptic PKA targets, GluR1 (at Ser 845) and NR1 (at Ser 897), had a significant reduction in PKARIIβ−/− mice compared with wild-type littermate controls (0.45 ± 0.1 and 0.61 ± 0.08, respectively; n = 4; p < 0.005 for both; t test) (Fig. 8A,B). PKA also regulates MAPK signaling in neurons (Morozov et al., 2003; Waltereit and Weller, 2003). However, analysis of phosphorylation levels with an anti-phospho MAPK [extracellular signal-regulated kinase (ERK)] antibody that recognizes both p42ERK and p44ERK showed no significant difference between genotypes (1.23 ± 0.34; n = 5; p = 0.51; t test) (Fig. 8A). These data are consistent with the small AMPAR-mediated currents we observe physiologically, because PKA phosphorylation of GluR1 through an LTP-like process is known to regulate AMPAR trafficking at the synapse (Ehlers, 2000; Lee et al., 2000, 2003; Esteban et al., 2003). The reduced PKA phosphorylation of postsynaptic but not presynaptic targets is also consistent with the postsynaptic localization of PKARIIβ and the postsynaptic barrel phenotype observed in PKARIIβ−/− mice, reinforcing a postsynaptic function for PKARIIβ during barrel map formation.
Figure 8.

PKARIIβ−/− mice have a significant reduction in phosphorylation of postsynaptic but not presynaptic PKA targets. A, Western blot examples of synaptosomes prepared from P11 somatosensory cortex of PKARIIβ−/− mice and their littermate controls using antibodies against PKA phosphorylation targets. B, Quantification of normalized ratio of anti-phospho GluR1 (PGluR1, Ser845) to total GluR1 and anti-phospho NR1 (PNR1, Ser 897) to total NR1 reveals a significant difference between PKARIIβ−/− (KO) and wild-type (WT) littermate control mice (*p < 0.005 for both; t test). None of the presynaptic PKA phosphorylation targets such as phospho-synapsin (p = 0.26; t test) and phospho-Rim (p = 0.56; t test) reveal a difference between PKARIIβ−/− mice and their wild-type littermate controls (data not shown). Quantification of the band labeled with an anti-phospho-MAPK (PMAPK) antibody, which recognizes both p44 (top band in A) and p42 (bottom band in A) phosphorylation of MAPK (or ERK), also shows no difference (data not shown; p = 0.51; t test). Error bars indicate SEM.
PKARIIβ−/− mice have normal TCA map plasticity
Another important feature of cortical maps is their developmental plasticity, in which the size of cortex devoted to a particular peripheral sense organ changes if the pattern of sensory input changes. For example, if one row of whiskers is removed during the first week after birth, the area of cortex devoted to this row will shrink and the neighboring rows will expand at the expense of the deprived row. In mammals, cortical maps have a critical period for this type of plasticity. In mouse barrel cortex, the critical period (P0–P5) coincides with barrel map formation. Therefore, it is intriguing to investigate whether barrel map development and plasticity are mediated by common molecular pathways.
Recently, map plasticity in visual cortex (ocular dominance plasticity) was shown to be defective in PKARIIβ−/− mice (Fischer et al., 2004). Because PKARIIβ−/− mice have poorly formed barrel (Nissl) maps, we were unable to quantitatively examine cytoarchitectural plasticity in PKARIIβ−/− mice. However, TCA clustering is grossly normal in PKARIIβ−/− mice, so we examined map plasticity of TCA clustering using cytochrome oxidase stain in PKARIIβ−/− mice by removing a row of whiskers at P1 (Fig. 9A–D,H) or P5 (Fig. 9E,F,H). We found that PKARIIβ−/− mice have a similar degree of thalamocortical map plasticity (MPI) as wild-type littermate control mice at P1 (MPI for wild type, 0.22 ± 0.04, n = 5; MPI for PKARIIβ−/− mice, 0.23 ± 0.02, n = 7; p = 0.93; t test) (Fig. 9H). Thalamocortical map plasticity is significantly reduced in both PKARIIβ−/− (1.00 ± 0.03; n = 4) and their wild-type littermate control (1.03 ± 0.03; n = 4; p = 0.57 for difference between PKARIIβ−/− and PKARIIβ+/+; t test) mice by P5. This suggests that loss of PKARIIβ does not affect the developmental plasticity of thalamocortical afferents.
Figure 9.
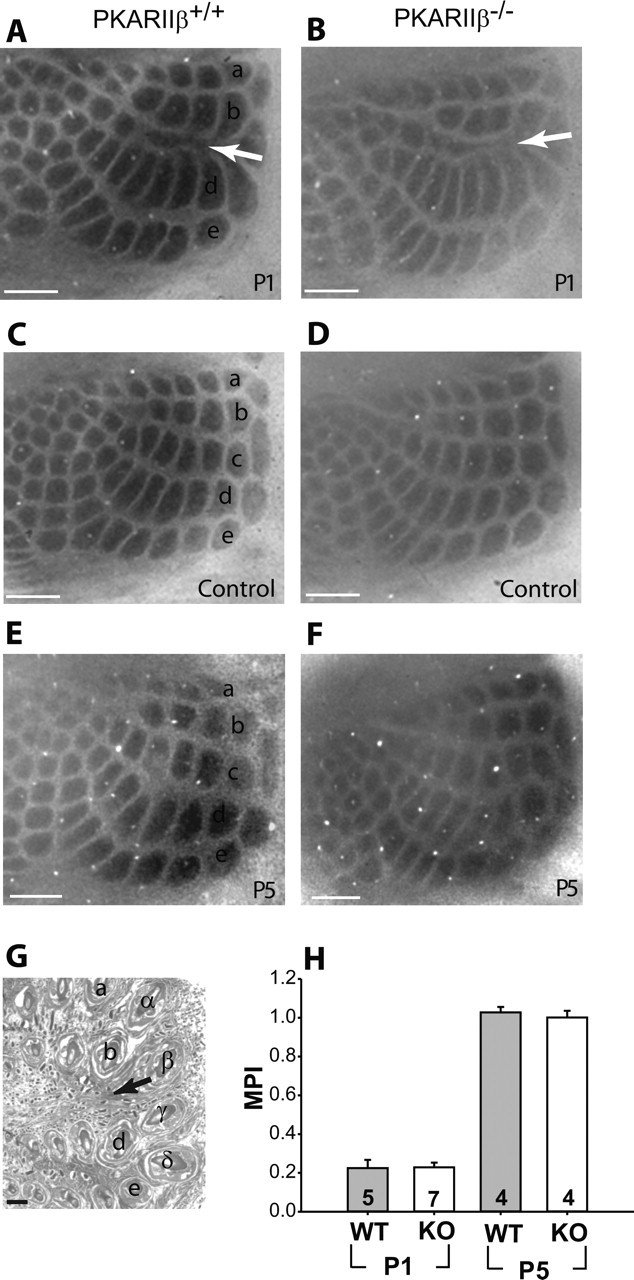
TCA barrel map plasticity is normal in PKARIIβ−/− mice. A, B, CO staining of tangential sections from barrel cortex shows that C-row whisker cauterization at P1 induced robust map plasticity in TCAs of both PKARIIβ−/− mice (B) and their littermate controls (A). C, D, Barrel cortex of control hemispheres where corresponding whiskers were left intact show a normal barrel pattern. E, F, Map plasticity critical period ends by P5 for both PKARIIβ−/− (F) and wild-type control mice (E). G, H&E staining of the cauterized whisker pad confirms that the cauterizations were limited to the C row of whiskers. H, Summary quantification of barrel map plasticity measured with an MPI (see Materials and Methods). CO barrel map plasticity is significant for wild-type littermate control and PKARIIβ−/− (KO) mice at P1 (p ≪ 0.001; t test) compared with control hemispheres (data not shown). However, comparison of MPIs for PKARIIβ−/− mice and wild-type (WT) littermate controls show no significant difference neither at P1 (p = 0.93; t test) nor at P5 (p = 0.57; t test). Scale bars, 500 μm. Error bars indicate SEM.
Discussion
We examined the cellular mechanisms mediating neural circuit formation, focusing on the role of PKA signaling in barrel map development. We showed that PKARIIβ−/− mice have decreased PKA activity in somatosensory cortex and associated barrel map deficits in the morphology and cytoarchitectural organization of layer IV cortical neurons but not thalamocortical afferents. Using immunohistochemical and biochemical techniques, we localized PKARIIβ expression to postsynaptic terminals in barrel cortex and showed that postsynaptic, but not presynaptic, PKA targets have decreased phosphorylation in PKARIIβ−/− synapses. These findings are consistent with previous reports in adult mice that analyzed different brain regions using immunohistochemical and electron microscopy techniques to show that PKARIIβ is localized to dendrites, perikaya, and postsynaptic densities but is rarely present in axon terminals (Ludvig et al., 1990; Glantz et al., 1992). In addition, we examined thalamic barrelloids using CO staining at P5 and found that organization of barrelloids is intact in PKARIIβ−/− mice (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Moreover, we did not detect any difference in anatomical plasticity of TCAs in PKARIIβ−/− and wild-type mice. These findings suggest that the PKARIIβ−/− barrel phenotype is specific to the cortex, and PKARIIβ-mediated PKA signaling is not necessary for the gross development and plasticity of TCA clusters, although it is required for postsynaptic barrel map development.
We also examined the physiological development of thalamocortical synapses in PKARIIβ−/− mice and found that thalamocortical LTP and AMPAR function were significantly impaired. The smaller AMPAR currents we observed in older animals are consistent with the absence of LTP and the decreased PKA phosphorylation of GluR1 detected biochemically in PKARIIβ−/− mice. In total, these experiments support an activity-dependent model for barrel map formation in which the selective addition of thalamocortical synapses based on Hebbian mechanisms for synapse formation act through a PKA-dependent pathway that strengthens connections by increasing the number of functional AMPARs at the synapse.
cAMP–PKA signaling in barrel development
Brl (Adcy1−/−) mice, which have been studied in great detail, have a barrel map defect that is more severe than PKARIIβ−/− mice, because they lack organization of both presynaptic and postsynaptic elements of the barrel map (Welker et al., 1996; Abdel-Majid et al., 1998), whereas TCA clustering is intact in PKARIIβ−/− mice. It is possible that the less severe barrel phenotype in PKARIIβ−/− mice stems from the remaining 50% of PKA activity in the somatosensory cortex of these mutants. The phenotypic difference between PKARIIβ−/− and brl mice also suggests that different signaling components downstream of Adcy1 play a role in the formation of presynaptic and postsynaptic barrel patterns. Data presented here and in our previous report (Lu et al., 2003) suggest that Adcy1 acts via PKARIIβ to regulate postsynaptic barrel map development. However, in presynaptic TCA terminals, Adcy1 may act through signaling molecules other than PKA or through a PKAR subunit distinct from PKARIIβ. Unlike in brl mice, the phosphorylation of presynaptic PKA targets is not reduced in PKARIIβ−/− mice, strengthening the suggestion that a different PKAR subunit is responsible for TCA clustering in barrel cortex (Lu et al., 2003, 2006).
Our previous studies with brl mice provided indirect evidence for the role of PKA activity in TC synapse maturation. Similar to the results we report here, we found that the AMPAR/NMDAR current ratio, which normally increases during TC synapse maturation, remained unchanged in brl mice (Lu et al., 2003). The smaller AMPAR/NMDAR current ratio was attributable, at least in part, to smaller AMPAR currents (Lu et al., 2003). As in brl mice, the reduced number of functional AMPARs at TC synapses of PKARIIβ−/− mice may be because of a change in AMPAR properties and/or because of an impairment in AMPAR insertion into the synapse, both of which are regulated by PKA phosphorylation of the GluR1 subunit through an LTP-like process (Banke et al., 2000; Ehlers, 2000; Lee et al., 2000, 2003; Esteban et al., 2003). Consistent with the LTP deficit we observe in PKARIIβ−/− mice, we previously showed that LTP was a PKA-dependent process that is impaired in brl mice (Lu et al., 2003). Our results here provide direct evidence for the requirement of postsynaptic PKA activity in the regulation of AMPAR function via synaptic plasticity mechanisms, such as LTP, and the activity-dependent maturation of TC synapses during barrel map development.
What triggers cAMP–PKA signaling in barrel development?
Compelling evidence exists that NMDAR and AMPAR activity mediates the development of dendritic arbors in a variety of brain regions (Rajan and Cline, 1998; Rajan et al., 1999; Zhou et al., 2004). At early stages of development, NMDARs mediate the formation and extension of dendritic branches. Later, AMPAR function contributes to dendritic morphogenesis as the synapses mature (Wu et al., 1996), and both NMDAR and AMPAR activity are required for the maintenance and stabilization of dendritic arbors (Rajan and Cline, 1998; Wu et al., 1999). NMDARs may play a similar role during barrel cortex development, because dendritic morphogenesis of layer IV neurons is impaired in transgenic mice that specifically lack NMDA receptor (NR1) function only in the cortex (Ctx-NR1−/− mice) (Iwasato et al., 1997, 2000; Datwani et al., 2002). Ctx-NR1−/− mice have larger dendritic arbors and dendritic span, increased branching, and no orientation bias toward barrel hollows, interfering with barrel wall formation. We also found similar dendritic deficits in PKARIIβ−/− mice, with larger dendritic spans and fewer neurons with asymmetric dendrites, despite normal total dendritic length. The similarity between the postsynaptic barrel map phenotypes in Ctx-NR1−/− and PKARIIβ−/− mice, and the absence of a developmental increase in AMPAR function as well as NMDAR-dependent thalamocortical LTP during TC synapse development in PKARIIβ−/− mice, support the hypothesis that NMDAR-mediated PKA function mediates the morphological and cytoarchitectural development of layer IV neurons in somatosensory cortex.
On the other hand, Ctx-NR1−/− mice have a somewhat more severe barrel phenotype than PKARIIβ−/− mice. Not only do cortical layer IV neurons fail to form a barrel pattern, but TCA clustering in barrel cortex is also modestly impaired, with the TCAs representing the large whiskers organized into patches that are smaller and less distinct than wild-type TCA clusters (Iwasato et al., 2000), and single TCAs projecting aberrantly to inappropriate barrels (Lee et al., 2005). This suggests that signaling through NMDARs may act via molecules other than PKARIIβ for patterning of TCAs, whereas PKARIIβ-mediated PKA signaling plays a role downstream of NMDARs for postsynaptic neuronal organization during barrel map development.
Other possible upstream regulators of cAMP signaling include G-protein-coupled receptors, such as metabotropic glutamate receptors (mGluRs) and serotonin receptors. 5-HT signaling has been implicated in barrel map development, because monoamine oxidase (MAOA) and 5-HTT knock-out mice have barrel map deficits as a result of elevated levels of extracellular 5-HT (Cases et al., 1996; Salichon et al., 2001). Knock-outs of two components of the mGluR signaling pathway, mGluR5 and phospholipase C-β1, have stronger postsynaptic than presynaptic barrel map defects, like we observe in PKARIIβ−/− mice (Erzurumlu and Kind, 2001; Hannan et al., 2001). Stimulation of mGluRs increases cAMP levels in the hippocampus and the visual cortex (Winder and Conn, 1993; Reid et al., 1996), but it is not clear how mGluRs regulate cAMP levels during barrel cortex development. Although our results indicate that regulation of AMPAR function by PKA is required for proper maturation of TC synapses and barrel map development, we do not know whether cAMP–PKA signaling is regulated exclusively by NMDARs, or whether mGluR5 receptors, 5-HT receptors, or some other trigger is also involved.
Barrel map plasticity in PKARIIβ−/− mice
Ctx-NR1−/− mice lack cortical barrel cytoarchitecture and have some defects in the development of TCA clusters representing barrels. However, lesion induced plasticity of TCAs is intact (Iwasato et al., 2000). Another recent study using MAOA knock-out mice also suggests that there is a dissociation between TCA barrel pattern development and plasticity (Rebsam et al., 2005). Both of these reports indicate that signaling pathways mediating barrel map plasticity and the initial development of TCAs are distinct, or at a minimum a defect in one pathway that disrupts pattern development is insufficient to disrupt map plasticity. Because the barrel map phenotype of PKARIIβ−/− mice is exclusively postsynaptic, we were able to analyze TCA map plasticity. We saw no difference in TCA map plasticity in PKARIIβ−/− mice and wild-type controls, indicating that reducing PKA activity in layer IV neurons is not sufficient to disrupt map plasticity in TCAs.
Role of PKARIIβ in PKA function
cAMP and PKA mediate a variety of signaling cascades, even within the same cell (Colledge and Scott, 1999; Carnegie and Scott, 2003; Bauman et al., 2004). Specificity of action for each signaling pathway is achieved by localizing PKA to distinct subcellular compartments via interaction of PKA regulatory subunits with A kinase anchoring proteins (AKAPs) (Coghlan et al., 1995; Westphal et al., 1999; Colledge et al., 2000). AKAPs that interact with type II PKAR, such as AKAP79/150, bind to scaffolding proteins that associate with glutamate receptors at the postsynaptic density and regulate AMPAR currents (Rosenmund et al., 1994; Colledge et al., 2000). This interaction is specifically required for the phosphorylation of GluR1, so the anchoring of PKA in close proximity to its substrates allows for tight regulation and signal integration in response to neuronal activity (Colledge et al., 2000; Tavalin et al., 2002). Endogenous PKARIIβ is likely compartmentalized near AMPARs at thalamocortical synapses and may be responsible for their activity-dependent trafficking during barrel formation. Therefore, the functional and anatomical deficits observed in barrel cortex of PKARIIβ−/− mice may not only be caused by lower overall PKA activity, but may be specifically caused by the improper localization of PKA and the consequent misregulation of local PKA activity in postsynaptic layer IV neurons.
Footnotes
This work was supported by National Institutes of Health Grant R01 MH62639, the Mental Retardation and Developmental Disabilities Research Center at Baylor College of Medicine, and a National Alliance for Research on Schizophrenia and Depression Sidney R. Baer Jr Foundation Investigator award to M.C.C. We thank Dr. G. Lonart for phospho-specific antibodies and Bobby Antalffy for help with histology.
References
- Abdel-Majid RM, Leong WL, Schalkwyk LC, Smallman DS, Wong ST, Storm DR, Fine A, Dobson MJ, Guernsey DL, Neumann PE (1998). Loss of adenylyl cyclase I activity disrupts patterning of mouse somatosensory cortex. Nat Genet 19:289–291. [DOI] [PubMed] [Google Scholar]
- Amieux PS, Cummings DE, Motamed K, Brandon EP, Wailes LA, Le K, Idzerda RL, McKnight GS (1997). Compensatory regulation of RIalpha protein levels in protein kinase A mutant mice. J Biol Chem 272:3993–3998. [DOI] [PubMed] [Google Scholar]
- Banke TG, Bowie D, Lee H, Huganir RL, Schousboe A, Traynelis SF (2000). Control of GluR1 AMPA receptor function by cAMP-dependent protein kinase. J Neurosci 20:89–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauman AL, Goehring AS, Scott JD (2004). Orchestration of synaptic plasticity through AKAP signaling complexes. Neuropharmacology 46:299–310. [DOI] [PubMed] [Google Scholar]
- Beaver CJ, Ji Q, Fischer QS, Daw NW (2001). Cyclic AMP-dependent protein kinase mediates ocular dominance shifts in cat visual cortex. Nat Neurosci 4:159–163. [DOI] [PubMed] [Google Scholar]
- Beaver CJ, Fischer QS, Ji Q, Daw NW (2002). Orientation selectivity is reduced by monocular deprivation in combination with PKA inhibitors. J Neurophysiol 88:1933–1940. [DOI] [PubMed] [Google Scholar]
- Bernhardt R, Matus A (1984). Light and electron microscopic studies of the distribution of microtubule-associated protein 2 in rat brain: a difference between dendritic and axonal cytoskeletons. J Comp Neurol 226:203–221. [DOI] [PubMed] [Google Scholar]
- Brandon EP, Idzerda RL, McKnight GS (1997). PKA isoforms, neural pathways, and behaviour: making the connection. Curr Opin Neurobiol 7:397–403. [DOI] [PubMed] [Google Scholar]
- Brandon EP, Logue SF, Adams MR, Qi M, Sullivan SP, Matsumoto AM, Dorsa DM, Wehner JM, McKnight GS, Idzerda RL (1998). Defective motor behavior and neural gene expression in RIIβ-protein kinase A mutant mice. J Neurosci 18:3639–3649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadd G, McKnight GS (1989). Distinct patterns of cAMP-dependent protein kinase gene expression in mouse brain. Neuron 3:71–79. [DOI] [PubMed] [Google Scholar]
- Carnegie GK, Scott JD (2003). A-kinase anchoring proteins and neuronal signaling mechanisms. Genes Dev 17:1557–1568. [DOI] [PubMed] [Google Scholar]
- Cases O, Vitalis T, Seif I, De Maeyer E, Sotelo C, Gaspar P (1996). Lack of barrels in the somatosensory cortex of monoamine oxidase A-deficient mice: role of a serotonin excess during the critical period. Neuron 16:297–307. [DOI] [PubMed] [Google Scholar]
- Cline H (2003). Sperry and Hebb: oil and vinegar? Trends Neurosci 26:655–661. [DOI] [PubMed] [Google Scholar]
- Coghlan VM, Perrino BA, Howard M, Langeberg LK, Hicks JB, Gallatin WM, Scott JD (1995). Association of protein kinase A and protein phosphatase 2B with a common anchoring protein. Science 267:108–111. [DOI] [PubMed] [Google Scholar]
- Colledge M, Scott JD (1999). AKAPs: from structure to function. Trends Cell Biol 9:216–221. [DOI] [PubMed] [Google Scholar]
- Colledge M, Dean RA, Scott GK, Langeberg LK, Huganir RL, Scott JD (2000). Targeting of PKA to glutamate receptors through a MAGUK-AKAP complex. Neuron 27:107–119. [DOI] [PubMed] [Google Scholar]
- Crair MC, Malenka RC (1995). A critical period for long-term potentiation at thalamocortical synapses. Nature 375:325–328. [DOI] [PubMed] [Google Scholar]
- Datwani A, Iwasato T, Itohara S, Erzurumlu RS (2002). NMDA receptor-dependent pattern transfer from afferents to postsynaptic cells and dendritic differentiation in the barrel cortex. Mol Cell Neurosci 21:477–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Camilli P, Miller PE, Navone F, Theurkauf WE, Vallee RB (1984). Distribution of microtubule-associated protein 2 in the nervous system of the rat studied by immunofluorescence. Neuroscience 11:817–846. [PubMed] [Google Scholar]
- Ehlers MD (2000). Reinsertion or degradation of AMPA receptors determined by activity-dependent endocytic sorting. Neuron 28:511–525. [DOI] [PubMed] [Google Scholar]
- Erzurumlu RS, Kind PC (2001). Neural activity: sculptor of ‘barrels’ in the neocortex. Trends Neurosci 24:589–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esteban JA, Shi SH, Wilson C, Nuriya M, Huganir RL, Malinow R (2003). PKA phosphorylation of AMPA receptor subunits controls synaptic trafficking underlying plasticity. Nat Neurosci 6:136–143. [DOI] [PubMed] [Google Scholar]
- Feldman DE, Knudsen EI (1998). Experience-dependent plasticity and the maturation of glutamatergic synapses. Neuron 20:1067–1071. [DOI] [PubMed] [Google Scholar]
- Fischer QS, Beaver CJ, Yang Y, Rao Y, Jakobsdottir KB, Storm DR, McKnight GS, Daw NW (2004). Requirement for the RIIβ isoform of PKA, but not calcium-stimulated adenylyl cyclase, in visual cortical plasticity. J Neurosci 24:9049–9058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glantz SB, Amat JA, Rubin CS (1992). cAMP signaling in neurons: patterns of neuronal expression and intracellular localization for a novel protein, AKAP 150, that anchors the regulatory subunit of cAMP-dependent protein kinase II beta. Mol Biol Cell 3:1215–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannan AJ, Blakemore C, Katsnelson A, Vitalis T, Huber KM, Bear M, Roder J, Kim D, Shin HS, Kind PC (2001). PLC-beta1, activated via mGluRs, mediates activity-dependent differentiation in cerebral cortex. Nat Neurosci 4:282–288. [DOI] [PubMed] [Google Scholar]
- Hansson SR, Mezey E, Hoffman BJ (1998). Serotonin transporter messenger RNA in the developing rat brain: early expression in serotonergic neurons and transient expression in non-serotonergic neurons. Neuroscience 83:1185–1201. [DOI] [PubMed] [Google Scholar]
- Harris RM, Woolsey TA (1979). Morphology of Golgi-impregnated neurons in mouse cortical barrels forming vibrissae damage at different post-natal ages. Brain Res 161:143–149. [DOI] [PubMed] [Google Scholar]
- Harris RM, Woolsey TA (1983). Computer assisted analyses of barrel neuron axons and their putative synaptic contacts. J Comp Neurol 220:63–79. [DOI] [PubMed] [Google Scholar]
- Howe DG, Wiley JC, McKnight GS (2002). Molecular and behavioral effects of a null mutation in all PKA C beta isoforms. Mol Cell Neurosci 20:515–524. [DOI] [PubMed] [Google Scholar]
- Huang YY, Kandel ER, Varshavsky L, Brandon EP, Qi M, Idzerda RL, McKnight GS, Bourtchouladze R (1995). A genetic test of the effects of mutations in PKA on mossy fiber LTP and its relation to spatial and contextual learning. Cell 83:1211–1222. [DOI] [PubMed] [Google Scholar]
- Iwasato T, Erzurumlu RS, Huerta PT, Chen DF, Sasaoka T, Ulupinar E, Tonegawa S (1997). NMDA receptor-dependent refinement of somatotopic maps. Neuron 19:1201–1210. [DOI] [PubMed] [Google Scholar]
- Iwasato T, Datwani A, Wolf AM, Nishiyama H, Taguchi Y, Tonegawa S, Knopfel T, Erzurumlu RS, Itohara S (2000). Cortex-restricted disruption of NMDAR1 impairs neuronal patterns in the barrel cortex. Nature 406:726–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kind PC, Neumann PE (2001). Plasticity: downstream of glutamate. Trends Neurosci 24:553–555. [DOI] [PubMed] [Google Scholar]
- Lebrand C, Cases O, Adelbrecht C, Doye A, Alvarez C, El Mestikawy S, Seif I, Gaspar P (1996). Transient uptake and storage of serotonin in developing thalamic neurons. Neuron 17:823–835. [DOI] [PubMed] [Google Scholar]
- Lebrand C, Cases O, Wehrle R, Blakely RD, Edwards RH, Gaspar P (1998). Transient developmental expression of monoamine transporters in the rodent forebrain. J Comp Neurol 401:506–524. [PubMed] [Google Scholar]
- Lee HK, Barbarosie M, Kameyama K, Bear MF, Huganir RL (2000). Regulation of distinct AMPA receptor phosphorylation sites during bidirectional synaptic plasticity. Nature 405:955–959. [DOI] [PubMed] [Google Scholar]
- Lee HK, Takamiya K, Han JS, Man H, Kim CH, Rumbaugh G, Yu S, Ding L, He C, Petralia RS, Wenthold RJ, Gallagher M, Huganir RL (2003). Phosphorylation of the AMPA receptor GluR1 subunit is required for synaptic plasticity and retention of spatial memory. Cell 112:631–643. [DOI] [PubMed] [Google Scholar]
- Lee LJ, Iwasato T, Itohara S, Erzurumlu RS (2005). Exuberant thalamocortical axon arborization in cortex-specific NMDAR1 knockout mice. J Comp Neurol 485:280–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Bendito G, Molnar Z (2003). Thalamocortical development: how are we going to get there? Nat Rev Neurosci 4:276–289. [DOI] [PubMed] [Google Scholar]
- Lu HC, Gonzalez E, Crair MC (2001). Barrel cortex critical period plasticity is independent of changes in NMDA receptor subunit composition. Neuron 32:619–634. [DOI] [PubMed] [Google Scholar]
- Lu HC, She WC, Plas DT, Neumann PE, Janz R, Crair MC (2003). Adenylyl cyclase I regulates AMPA receptor trafficking during mouse cortical “barrel” map development. Nat Neurosci 6:939–947. [DOI] [PubMed] [Google Scholar]
- Lu HC, Butts BA, Kaeser PS, She WC, Janz R, Crair MC (2006). Barrel map development relies on efficient neurotransmitter release. J Neurosci 26:2692–2703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludvig N, Ribak CE, Scott JD, Rubin CS (1990). Immunocytochemical localization of the neural-specific regulatory subunit of the type II cyclic AMP-dependent protein kinase to postsynaptic structures in the rat brain. Brain Res 520:90–102. [DOI] [PubMed] [Google Scholar]
- Morozov A, Muzzio IA, Bourtchouladze R, Van-Strien N, Lapidus K, Yin D, Winder DG, Adams JP, Sweatt JD, Kandel ER (2003). Rap1 couples cAMP signaling to a distinct pool of p42/44MAPK regulating excitability, synaptic plasticity, learning, and memory. Neuron 39:309–325. [DOI] [PubMed] [Google Scholar]
- Pasternak JR, Woolsey TA (1975). The number, size and spatial distribution of neurons in lamina IV of the mouse SmI neocortex. J Comp Neurol 160:291–306. [DOI] [PubMed] [Google Scholar]
- Qi M, Zhuo M, Skalhegg BS, Brandon EP, Kandel ER, McKnight GS, Idzerda RL (1996). Impaired hippocampal plasticity in mice lacking the Cbeta1 catalytic subunit of cAMP-dependent protein kinase. Proc Natl Acad Sci USA 93:1571–1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajan I, Cline HT (1998). Glutamate receptor activity is required for normal development of tectal cell dendrites in vivo. J Neurosci 18:7836–7846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajan I, Witte S, Cline HT (1999). NMDA receptor activity stabilizes presynaptic retinotectal axons and postsynaptic optic tectal cell dendrites in vivo. J Neurobiol 38:357–368. [DOI] [PubMed] [Google Scholar]
- Rao Y, Fischer QS, Yang Y, McKnight GS, LaRue A, Daw NW (2004). Reduced ocular dominance plasticity and long-term potentiation in the developing visual cortex of protein kinase A RII alpha mutant mice. Eur J Neurosci 20:837–842. [DOI] [PubMed] [Google Scholar]
- Rebsam A, Seif I, Gaspar P (2005). Dissociating barrel development and lesion-induced plasticity in the mouse somatosensory cortex. J Neurosci 25:706–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid SN, Daw NW, Gregory DS, Flavin H (1996). cAMP levels increased by activation of metabotropic glutamate receptors correlate with visual plasticity. J Neurosci 16:7619–7626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenmund C, Carr DW, Bergeson SE, Nilaver G, Scott JD, Westbrook GL (1994). Anchoring of protein kinase A is required for modulation of AMPA/kainate receptors on hippocampal neurons. Nature 368:853–856. [DOI] [PubMed] [Google Scholar]
- Salichon N, Gaspar P, Upton AL, Picaud S, Hanoun N, Hamon M, De Maeyer E, Murphy DL, Mossner DL, Lesch KP, Hen R, Seif I (2001). Excessive activation of serotonin (5-HT) 1B receptors disrupts the formation of sensory maps in monoamine oxidase A and 5-HT transporter knock-out mice. J Neurosci 21:884–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seligman AM, Karnovsky MJ, Wasserkrug HL, Hanker JS (1968). Nondroplet ultrastructural demonstration of cytochrome oxidase activity with a polymerizing osmiophilic reagent, diaminobenzidine (DAB). J Cell Biol 38:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strominger RN, Woolsey TA (1987). Templates for locating the whisker area in fresh flattened mouse and rat cortex. J Neurosci Methods 22:113–118. [DOI] [PubMed] [Google Scholar]
- Sweatt JD (2001). Memory mechanisms: the yin and yang of protein phosphorylation. Curr Biol 11:R391–R394. [DOI] [PubMed] [Google Scholar]
- Tavalin SJ, Colledge M, Hell JW, Langeberg LK, Huganir RL, Scott JD (2002). Regulation of GluR1 by the A-kinase anchoring protein 79 (AKAP79) signaling complex shares properties with long-term depression. J Neurosci 22:3044–3051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiele TE, Willis B, Stadler J, Reynolds JG, Bernstein IL, McKnight GS (2000). High ethanol consumption and low sensitivity to ethanol-induced sedation in protein kinase A-mutant mice. In: J Neurosci 20: pp. RC75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waltereit R, Weller M (2003). Signaling from cAMP/PKA to MAPK and synaptic plasticity. Mol Neurobiol 27:99–106. [DOI] [PubMed] [Google Scholar]
- Wang H, Storm DR (2003). Calmodulin-regulated adenylyl cyclases: cross-talk and plasticity in the central nervous system. Mol Pharmacol 63:463–468. [DOI] [PubMed] [Google Scholar]
- Welker E, Armstrong-James M, Bronchti G, Ourednik W, Gheorghita-Baechler F, Dubois R, Guernsey DL, Van der Loos H, Neumann PE (1996). Altered sensory processing in the somatosensory cortex of the mouse mutant barrelless. Science 271:1864–1867. [DOI] [PubMed] [Google Scholar]
- Westphal RS, Tavalin SJ, Lin JW, Alto NM, Fraser ID, Langeberg LK, Sheng M, Scott JD (1999). Regulation of NMDA receptors by an associated phosphatase-kinase signaling complex. Science 285:93–96. [DOI] [PubMed] [Google Scholar]
- Winder DG, Conn PJ (1993). Activation of metabotropic glutamate receptors increases cAMP accumulation in hippocampus by potentiating responses to endogenous adenosine. J Neurosci 13:38–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong-Riley MT, Welt C (1980). Histochemical changes in cytochrome oxidase of cortical barrels after vibrissal removal in neonatal and adult mice. Proc Natl Acad Sci USA 77:2333–2337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong-Riley MT, Merzenich MM, Leake PA (1978). Changes in endogenous enzymatic reactivity to DAB induced by neuronal inactivity. Brain Res 141:185–192. [DOI] [PubMed] [Google Scholar]
- Woolsey TA, Dierker ML, Wann DF (1975). Mouse SmI cortex: qualitative and quantitative classification of Golgi-impregnated barrel neurons. Proc Natl Acad Sci USA 72:2165–2169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu G, Malinow R, Cline HT (1996). Maturation of a central glutamatergic synapse. Science 274:972–976. [DOI] [PubMed] [Google Scholar]
- Wu GY, Zou DJ, Rajan I, Cline H (1999). Dendritic dynamics in vivo change during neuronal maturation. J Neurosci 19:4472–4483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu-Friedman MA, Regehr WG (1999). Presynaptic strontium dynamics and synaptic transmission. Biophys J 76:2029–2042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuda H, Barth AL, Stellwagen D, Malenka RC (2003). A developmental switch in the signaling cascades for LTP induction. Nat Neurosci 6:15–16. [DOI] [PubMed] [Google Scholar]
- Zhou Q, Homma KJ, Poo MM (2004). Shrinkage of dendritic spines associated with long-term depression of hippocampal synapses. Neuron 44:749–757. [DOI] [PubMed] [Google Scholar]