

Abstract
The hypothalamic paraventricular nucleus (PVN) integrates preautonomic and neuroendocrine control of energy homeostasis, fluid balance, and the stress response. We recently demonstrated that glucocorticoids act via a membrane receptor to rapidly cause endocannabinoid-mediated suppression of synaptic excitation in PVN neurosecretory neurons. Leptin, a major signal of nutritional state, suppresses CB1 cannabinoid receptor-dependent hyperphagia (increased appetite) in fasting animals by reducing hypothalamic levels of endocannabinoids. Here we show that glucocorticoids stimulate endocannabinoid biosynthesis and release via a Gαs–cAMP–protein kinase A-dependent mechanism and that leptin blocks glucocorticoid-induced endocannabinoid biosynthesis and suppression of excitation in the PVN via a phosphodiesterase-3B-mediated reduction in intracellular cAMP levels. We demonstrate this rapid hormonal interaction in both PVN magnocellular and parvocellular neurosecretory cells. Leptin blockade of the glucocorticoid-induced, endocannabinoid-mediated suppression of excitation was absent in leptin receptor-deficient obese Zucker rats. Our findings reveal a novel hormonal crosstalk that rapidly modulates synaptic excitation via endocannabinoid release in the hypothalamus and that provides a nutritional state-sensitive mechanism to integrate the neuroendocrine regulation of energy homeostasis, fluid balance, and the stress response.
Keywords: leptin, glucocorticoids, endocannabinoids, neuroendocrine, stress, energy homeostasis
Introduction
We recently demonstrated that stress levels of glucocorticoids (GCs) cause fast, CB1 cannabinoid receptor-dependent retrograde inhibition of glutamatergic excitatory synaptic inputs to neurosecretory cells of the hypothalamic paraventricular nucleus (PVN) (Di et al., 2003, 2005a), which control the major endocrine systems involved in maintaining energy homeostasis and fluid balance and in the homeostatic adaptation to different kinds of stress. This effect, which we refer to here as GC-induced, endocannabinoid-mediated suppression of synaptic excitation (GSE), was demonstrated in different PVN parvocellular neuronal subpopulations that control the hypothalamic–pituitary–adrenal (HPA) axis and the hypothalamic–pituitary–thyroid (HPT) axis, as well as in the magnocellular neurons from the PVN and from the supraoptic nucleus (SON), which integrate the hypothalamic–neurohypophysial system. The HPA axis controls circulating levels of GCs, which are the main endocrine signal in the adaptation to stress and are also a major signal for the maintenance of energy homeostasis via control of metabolism and appetite (Castonguay, 1991; Sapolsky et al., 2000). Fasting is a metabolic stress that increases the circulating levels of GCs and decreases the circulating levels of leptin (Chowers et al., 1969; Ahima, 2000), the primary endocrine signal of the nutritional state. This hormonal profile sets the stage for vital behavioral and physiological adaptations that take place during caloric deficit to promote the reestablishment of energy balance. A major behavioral aspect of this homeostatic adaptation to fasting is the development of hyperphagia, which was shown to depend on a decrease in leptin levels, an increase in GC levels, and activation of central CB1 receptors (Castonguay, 1991; Ahima, 2000; Di Marzo et al., 2001). Thus, during fasting, circulating GCs and hypothalamic levels of endocannabinoids are increased and both are involved in hypothalamic stimulation of feeding (Tempel et al., 1993; Ahima, 2000; Di Marzo et al., 2001; Kirkham et al., 2002). Leptin, conversely, suppresses hyperphagia in fasting animals by reducing endocannabinoid levels in the hypothalamus, probably via blockade of endocannabinoid biosynthetic pathways (Di Marzo et al., 2001).
Together, these findings suggested to us that crosstalk between leptin and GCs may rapidly modulate hypothalamic neuronal activity via opposing regulation of endocannabinoid biosynthesis and retrograde release. Here we used liquid chromatography–tandem mass spectrometry (LC/MS/MS) and whole-cell patch-clamp recordings in acute hypothalamic slices to directly demonstrate that GCs act on a membrane receptor to rapidly induce biosynthesis and release of the endocannabinoids anandamide (AEA) and 2-arachidonoylglycerol (2-AG) in the PVN via a Gαs–cAMP–protein kinase A (PKA)-dependent mechanism. We show that leptin blocks the GC-induced endocannabinoid biosynthesis and inhibitory effect in PVN neurosecretory neurons by activating phosphodiesterase 3B (PDE-3B). This nongenomic interaction between GCs and leptin is present in both the parvocellular and magnocellular subpopulations of PVN neuroendocrine cells and represents a nutritional state-sensitive, endocannabinoid-mediated mechanism that controls the synaptic excitation of neuroendocrine cells involved in the regulation of energy homeostasis, fluid balance, and the stress response.
Materials and Methods
Animals.
Male Sprague Dawley rats (3–4 weeks) and lean Zucker (Fa/?) or obese Zucker (fa/fa) rats (5–6 weeks) (Charles River Laboratories, Wilmington, MA) were used according to a protocol approved by the Tulane University Institutional Animal Care and Use Committee and in accordance with United States Public Health Service guidelines.
Slice preparation.
Rats were decapitated under deep pentobarbital sodium anesthesia (50 mg/kg body weight). The brain was immediately transferred to ice-cold (0–1°C), oxygenated artificial CSF (aCSF), blocked, and sliced. The composition of the aCSF was as follows (in mm): 140 NaCl, 3 KCl, 1.3 MgSO4, 1.4 NaH2PO4, 2.4 CaCl2, 11 glucose, and 5 HEPES, pH adjusted to 7.2–7.3 with NaOH. Two contiguous coronal hypothalamic slices (350 μm) containing the PVN were sectioned on a vibratome, bisected along the midline (i.e., at the third ventricle), and incubated in oxygenated aCSF at room temperature for >1.5 h before being transferred to the recording chamber. Hemi-slices were transferred to the recording chamber and allowed to equilibrate at 32–34°C for at least 20 min before recordings.
Electrophysiological recording.
Whole-cell patch-clamp recordings were performed in putative magnocellular and parvocellular neurons at 32–34°C at a holding potential of −65 mV in the presence of the voltage-gated sodium channel inhibitor tetrodotoxin (TTX) (1 μm) and the GABAA receptor blocker bicuculline (10 μm). Patch pipettes were pulled from borosilicate glass (1.65 mm outer diameter, 1.2 mm inner diameter; KG33; Garner Glass, Claremont, CA) with a Flaming/Brown P-97 micropipette puller (Sutter Instruments, Novato, CA) to a resistance of 4–6 MΩ. The pipette solution contained the following (in mm): 120 K-gluconate, 10 KCl, 1 NaCl, 1 MgCl2, 1 EGTA, 2 Mg-ATP, 0.3 Na-GTP, and 10 HEPES, pH adjusted to 7.3 with KOH. The osmolarity of the solution was adjusted to 300 mOsm with 20 mm d-sorbitol. Putative magnocellular and parvocellular cells were distinguished on the basis of their morphology, positions within the PVN, as well as the transient outward rectification seen in magnocellular and not in parvocellular neurons (Luther et al., 2000), and the parvocellular neuroendocrine cells were distinguished from the parvocellular preautonomic neurons by location and by their lack of low-threshold calcium spike, as described by Stern (2001) and Luther et al. (2002) (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). After the whole-cell recording configuration was obtained, all cells were observed for 5–10 min for stability before a 3 min baseline control recording of miniature EPSCs (mEPSCs) was recorded. Drugs were applied immediately after baseline recordings.
Drugs applied in the perfusion bath.
Water-soluble dexamethasone (Dex) (1 or 2.5 μm) and corticosterone (Cort) (1 μm) (Sigma, St. Louis, MO) were dissolved directly in aCSF to their final concentrations. Dexamethasone hemisuccinate–bovine serum albumin (Dex-BSA) (Steraloids, Newport, RI) was stored as a 2 mm solution in water and was freshly washed in ether to remove any residual free dexamethasone. AM251 [N-(piperidin-1-yl)-5-(4-iodophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxamide] (Tocris Cookson, Ballwin, MO) was stored as a 10 mm solution in DMSO. 2-AG (Tocris Cookson) was stored at 5 mg/ml in anhydrous ethanol. Murine leptin (Sigma) was reconstituted and stored as a 78 μm solution in 15 mm HCl plus 7.5 mm NaOH. Cilostamide (Cil) (Tocris Cookson) was stored as a 100 μm solution in DMSO. H-89 (N-[2-(p-bromo-cinnamylamino)-ethyl]-5-isoquinoline-sulfon-amide 2HCl) (Calbiochem, La Jolla, CA) was stored as a 100 μm solution in DMSO. All bath-applied drugs were stored at −20°C and dissolved to their final concentrations in aCSF just before bath application.
Drugs applied via the patch pipette.
(R)-Adenosine, cyclic 3′,5′-(hydrogenphosphorothioate) triethylammonium (cAMPS-Rp), triethylammonium salt (Tocris Cookson) was stored as a 40 mm solution in water and diluted to its final concentration with the recording pipette solution. Rabbit antisera against rat Gαs and Gαq/11 (Chemicon, Temecula, CA) were directly diluted in the recording pipette solution to a final dilution of 1:2000. Pipette tips were filled with regular pipette solution, and the pipettes were backfilled with the solution containing the test drug.
Quantitative analysis of endocannabinoids in acute slices.
Two contiguous 350 μm coronal hypothalamic slices containing the PVN were sectioned on a vibratome, and the slices were bisected along the midline and trimmed around the PVN by cutting laterally at the medial edge of the fornix, dorsally at the vertex of the third ventricle, and ventrally at ∼1.5 mm from the base of the brain. Horizontal cerebellar slices (350 μm) were used as controls. Trimmed hemi-slices were stored for 2–3 h in aCSF at room temperature and then preincubated for 20 min in aCSF at 34°C. The two contiguous hemi-slices from each side were pooled together into control and test groups. Test slices were transferred to modified aCSF (maCSF) (aCSF containing 1 μm TTX, 10 μm bicuculline, and 0.025% BSA) with the specific drug treatment, whereas the contralateral control slices were transferred to maCSF without treatment for the same incubation time at 34°C. Test groups were treated as follows: Dex, 10 min of 1 μm dexamethasone in maCSF; Dex + Lep, preincubation in maCSF with leptin (10 nm) for 5 min before transfer to maCSF with leptin (10 nm) plus Dex (1 μm); Dex + H-89, preincubation in maCSF with H-89 (1 μm) for 5 min before transfer to maCSF with H-89 (1 μm) plus Dex (1 μm). Immediately after treatment, slices were collected and homogenized in 1 ml of ice-cold methanol in preparation for LC/MS/MS analysis. One milliliter samples of incubating buffer also were collected from each assay to control for changes in the levels of endocannabinoids in solution. LC/MS/MS analysis of the endocannabinoids AEA and 2-AG was performed on chloroform methanol (2:1) lipid extracts, which were loaded with deuterated standard mixture (AEA-d8, 2-AG-d8) and purified by solid-phase extraction on C18 columns (Varian, Walnut Creek, CA). Samples were eluted with 10 ml of 1% methanol in ethyl acetate (Electron Microscopy Sciences, Fort Washington, PA) and concentrated on an N2 stream evaporator before LC/MS/MS analysis. Samples were loaded on a Biobasic-AX column (100 × 2.1 mm, 5 μm particle size; Thermo-Hipersyl-Keystone, Bellefonte, PA). The column was run with a 45 min gradient protocol starting with solvent solution A (40:60:0.01 methanol/water/acetic acid, pH 4.5) at a flow rate of 300 μl/min, reached 100% of solvent B (99.99:0.01 methanol/acetic acid) in 30 min, and run isocratic for 5 min, after which the system returned to 100% solvent solution A in 10 min. LC effluents were diverted to an electrospray-ionization probe (Elkins-Sinn, Cherry Hill, NJ) on a TSQ Quantum triple quadrupole mass spectrometer (Thermo-Finnigan, San Jose, CA) running on negative ion detection mode. Electrospray voltage was 3 kV; sheath gas was argon at 1.5 mTorr. The instrument was run on full scan mode to detect MS2 spectra and selected reaction mode for quantitative analysis to detect parent/daughter ion pairs simultaneously. The selected parent/daughter ion pairs were 346.3/259.3, 377.2/285.2, 353.2/266.3, and 385.4/310.2 mass/charge for AEA, 2-AG, AEA-d8, and 2-AG-d8, respectively.
Tissue punch reverse transcription-PCR.
The PVN and SON were microdissected from 400-μm-thick acute coronal hypothalamic slices, rapidly homogenized in ice-cold guanidinium thiocyanate containing lysis buffer, and immediately frozen in an ethanol dry-ice slurry. Total RNA was extracted using the RNeasy total RNA isolation kit (Qiagen, Valencia, CA) and treated with 10 U of RNase-free DNase I (Invitrogen, Carlsbad, CA) for 30 min at 37°C. Reverse transcription (RT) was performed using the Superscript III first-strand synthesis system (Invitrogen) according to the protocol of the manufacturer. As a control for genomic DNA carryover, a similar procedure was performed for each PVN and SON sample without including the Superscript III enzyme. Amplification of cDNAs via PCR was performed using primers designed to amplify the long (Ob-Rb) isoform of the leptin receptor (Levin et al., 2003).
Statistical analysis.
For the statistical analysis of mEPSC frequency and amplitude, the Student’s paired t test was used for within-cell statistical comparisons of mean values recorded during 3 min intervals before and during (7–10 min) drug treatment as specified in the text. For biochemical analyses, one-way ANOVA was used for comparison between the mean endocannabinoid concentration in control slices and the mean endocannabinoid concentration in treated groups, and between treated groups. For this analysis, the values of control hemi-slices used in all experiments were pooled together. Probability values of <0.05 were considered significant.
Results
Leptin blocks glucocorticoid-induced biosynthesis of 2-AG and AEA in the PVN
Acute hypothalamic slices trimmed around the PVN were homogenized immediately after a 10 min treatment with the synthetic GC Dex (1 μm) and analyzed by LC/MS/MS. Dex caused a significant increase in the levels of the endocannabinoids AEA (control, 12.5 ± 2.1 pmol/mg protein; Dex, 40.5 ± 10.5 pmol/mg protein; n = 6; p < 0.0005) and 2-AG (control, 302.5 ± 38.9 pmol/mg protein; Dex, 685.5 ± 125.0 pmol/mg protein; n = 6; p < 0.001) (Fig. 1). The same treatment in cerebellum slices had no effect on levels of AEA (control, 4.1 ± 0.5 pmol/mg protein; Dex, 5.1 ± 0.8 pmol/mg protein; n = 6; p = 0.36) or 2-AG (control, 33.8 ± 10.7 pmol/mg protein; Dex, 52.3 ± 14.8 pmol/mg protein; n = 6; p = 0.34) (Fig. 1). A 5 min pretreatment with leptin (10 nm) blocked the Dex-induced increase in endocannabinoid levels and caused a rather significant reduction in 2-AG levels (155.4 ± 15.3 pmol/mg protein; n = 6; p < 0.05) and a nonsignificant reduction in AEA levels (8.6 ± 2.4 pmol/mg protein; n = 6; p = 0.29) (Fig. 1). To control for possible elution of endocannabinoids into the incubating buffer, we also analyzed buffer samples, which revealed only negligible amounts of endocannabinoids in solution in both control and treated groups (data not shown). These results demonstrate a fast, GC-induced biosynthesis of AEA and 2-AG that is rapidly and completely blocked by leptin, thus revealing a nongenomic GC–leptin interaction that regulates endocannabinoid levels in the PVN.
Figure 1.

Endocannabinoid levels in the PVN are regulated by glucocorticoids and leptin. A 10 min bath application of Dex (1 μm) caused a significant increase in the levels of both AEA and 2-AG in PVN slices (n = 6) but not in cerebellum slices (n = 6) compared with their respective controls (PVN, n = 16; cerebellum, n = 6). Previous application of Lep (10 nm) for 5 min blocked the dexamethasone-induced release of both AEA and 2-AG in PVN slices (n = 6). Means ± SE. *p < 0.05 versus control group; **p < 0.001 versus control group; ***p < 0.0005 versus control group; ##p < 0.005 versus PVN (Dex) group.
Leptin blocks the glucocorticoid-induced, endocannabinoid-mediated suppression of excitation in PVN magnocellular and parvocellular neuroendocrine cells
Our LC/MS/MS analysis of PVN slices containing both parvocellular and magnocellular subdivisions showed a complete blockade of GC-induced endocannabinoid release by leptin, suggesting that leptin suppresses endocannabinoid release from each of the magnocellular and parvocellular subpopulations of the PVN; we showed previously that multiple cell types from both the magnocellular and parvocellular neuroendocrine cell groups undergo GSE (Di et al., 2003, 2005a). To determine the neurophysiologic significance and the intracellular mechanisms of the GC–leptin interactions that regulate endocannabinoid biosynthesis and release, whole-cell patch-clamp recordings were performed in putative PVN magnocellular and parvocellular neuroendocrine cells to test for leptin blockade of GSE. In the magnocellular neurons, a 10 min bath application of Cort (1 μm) or Dex (2.5 μm) caused a rapid (∼1 min onset) reduction in the frequency of mEPSCs (Cort, −33.2 ± 9.4%, n = 5, p < 0.05; Dex, −39.2 ± 10.5%, n = 9, p < 0.05) without affecting mEPSC amplitude (Fig. 2A,D), indicating a presynaptic suppression of glutamate release consistent with our previous findings (Di et al., 2005a). Confirming our previous work demonstrating that GSE is mediated by CB1 receptor activation, the GC inhibitory effect was mimicked by bath application of 2-AG (3 μm) (−31.2 ± 6.1%; n = 7; p < 0.05) and was blocked by the CB1 antagonist AM251 (1 μm) (+30.1 ± 18.0%; n = 7; p = 0.17) (Fig. 2B,D). A 5 min preapplication of leptin (10 nm), although without effect alone on mEPSCs, completely abolished GSE in all of the PVN magnocellular neurons tested (Lep + 2.5 μm Dex, +13.3 ± 7.0%, n = 6, p = 0.08; Lep + 1 μm Cort, +9.9 ± 23.1%, n = 6, p = 0.32) (Fig. 2C,D).
Figure 2.
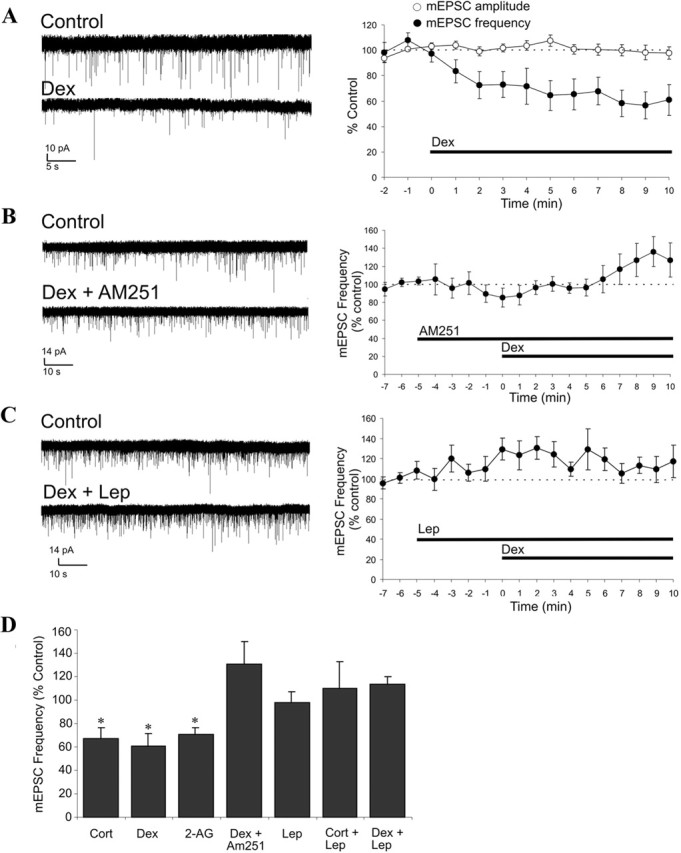
Leptin blockade of GSE in PVN magnocellular neurons. A–C, Left, Representative recordings before (control) and after ∼9 min of drug application; right, time course of normalized mEPSC amplitude and/or frequency. A, Dex (2.5 μm) caused a reduction of mEPSC frequency but not mEPSC amplitude (n = 9). B, The CB1 antagonist AM251 (1 μm) blocked the dexamethasone-induced decrease in mEPSC frequency (n = 8). C, Preapplication of Lep for 5 min blocked the dexamethasone-induced decrease in mEPSC frequency (n = 6). D, Summary histogram showing the mean normalized mEPSC frequency recorded over the last 3 min of a 10 min application of Cort (n = 5), Dex (n = 9), 2-AG (n = 8), Dex + AM251 (n = 8), Lep (n = 7), Cort + Lep (n = 5), and Dex + Lep (n = 6). Means ± SE. *p < 0.05.
In putative parvocellular neuroendocrine cells, as reported previously (Di et al., 2003), a 10 min bath application of Dex (2.5 μm) also caused a rapid reduction (∼1 min onset) in the frequency of mEPSCs (Dex, −36.6 ± 3%; n = 6; p < 0.01) without affecting mEPSC amplitude (Fig. 3A,C). Like in the magnocellular neurons, a 5 min preapplication of leptin (10 nm), although without effect alone on mEPSCs, also completely abolished GSE in all of the parvocellular neurons tested (Lep + 2.5 μm Dex, 9.6 ± 4%; n = 5; p = 0.28) (Fig. 3B,C). This indicated that leptin blockade of GSE is a common feature among the different PVN neurosecretory subpopulations.
Figure 3.
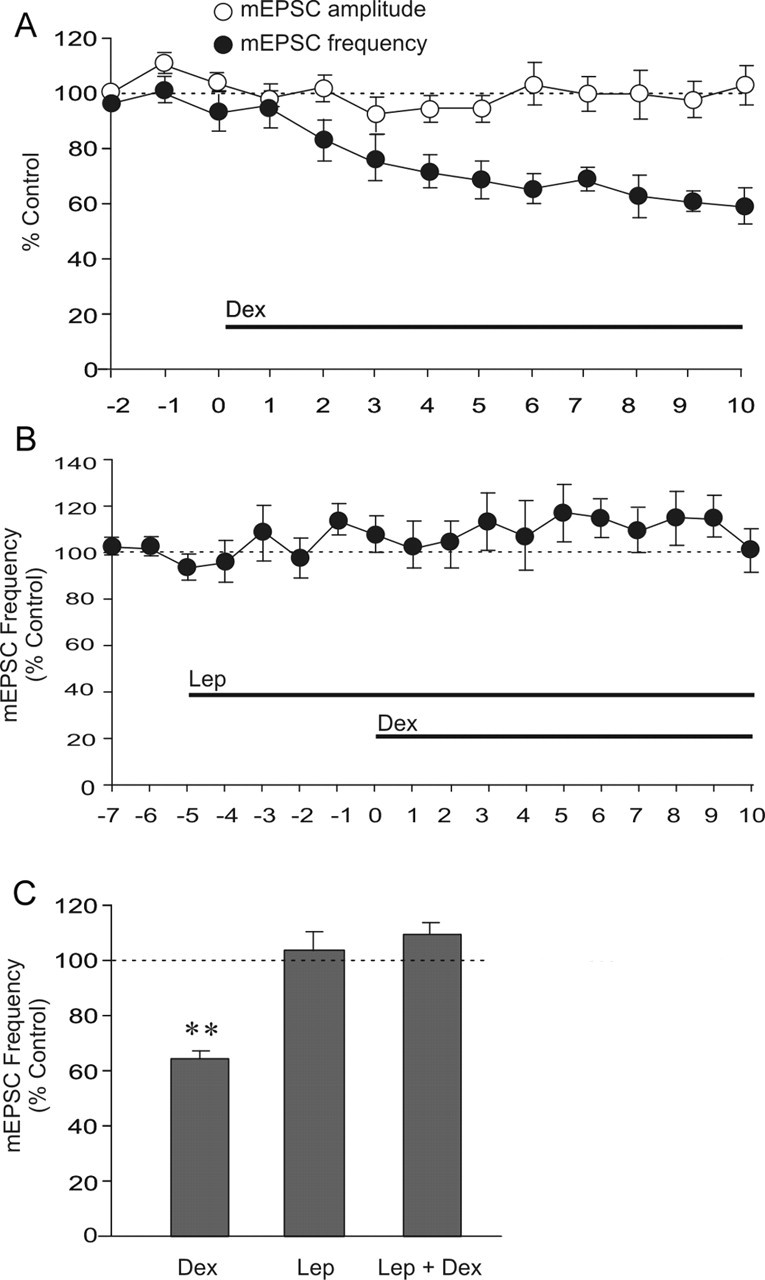
Leptin blockade of GSE in PVN parvocellular neurons. A, Time course of normalized mEPSC amplitude and frequency showing that Dex (2.5 μm) caused a reduction of mEPSC frequency but not mEPSC amplitude (n = 6). B, Preapplication of Lep for 5 min blocked the dexamethasone-induced decrease in mEPSC frequency (n = 5). C, Summary histogram showing the mean normalized mEPSC frequency recorded over the last 3 min of 10 min application of Dex (n = 6) and Dex + Lep (n = 6) or over the last 3 min of 5 min application of Lep (n = 5). Means ± SE. **p < 0.01.
Leptin blockade of GSE is mediated by Ob-Rb-dependent activation of PDE-3B
Genetically obese Zucker rats (fa/fa) are homozygous for a defective leptin receptor gene, which prevents leptin receptor signaling in these animals, whereas lean Zucker rats (FA/?) express at least one normal allele for the leptin receptor and present a normal response to leptin. Obese Zucker rats chronically show a physiological, hormonal, and behavioral profile similar to those of fasted rats, which includes hyperphagia, elevated circulating levels of GCs, and increased hypothalamic levels of endocannabinoids (Freedman et al., 1985; Ahima, 2000; Di Marzo et al., 2001). Leptin failed to block GSE in PVN magnocellular neurons in slices from obese Zucker rats (−32.9 ± 10.1%; n = 7; p < 0.01) but had an inhibitory effect on GSE in slices from lean Zucker rats (+3.0 ± 16.8%; n = 6; p = 0.39) (Fig. 4A–C) similar to that seen in slices from normal Sprague Dawley rats.
Figure 4.

Leptin fails to suppress GSE in PVN neurons from leptin receptor-deficient obese Zucker rats. A, Representative recording of mEPSCs in a PVN magnocellular neuron from an obese Zucker rat before (control) and after ∼9 min application of Dex (2.5 μm) in the presence of 10 nm leptin (Dex + Lep). B, Time course of mean normalized mEPSC frequency recorded in PVN magnocellular neurons from lean (n = 6) and obese (n = 7) Zucker rats showing that 5 min leptin (10 nm) preapplication prevents GSE in lean but not in genetically obese Zucker rats. C, Summary histogram showing the mean normalized mEPSC frequency recorded over the last 3 min of a 10 min application of Dex or Lep + Dex in PVN magnocellular neurons from lean (n = 7 and 6, respectively) and obese (n = 5 and 7) Zucker rats. Means ± SE. *p < 0.05; **p < 0.01.
Because the anorexigenic effects of central leptin injection are mediated by leptin receptor-dependent activation of PDE-3B (Zhao et al., 2002), we tested whether the leptin blockade of GSE is PDE-3B dependent in magnocellular and parvocellular neuroendocrine cells from normal Sprague Dawley rats. Bath application of the PDE-3B inhibitor cilostamide (1 μm) prevented the leptin-mediated blockade of GSE in both putative magnocellular (−29.9 ± 5.9%; n = 6; p < 0.05) and parvocellular neuroendocrine (−31.4 ± 2%; n = 5; p < 0.05) cells (Fig. 5A–C). These results together indicate that the leptin blockade of GSE is mediated by a receptor encoded for by the Ob gene. RT-PCR analysis of PVN and supraoptic nucleus tissue punches confirmed the presence of mRNA encoding for Ob-Rb, the long splice variant of the leptin receptor encoded for by the Ob gene, in these hypothalamic nuclei (Fig. 6).
Figure 5.
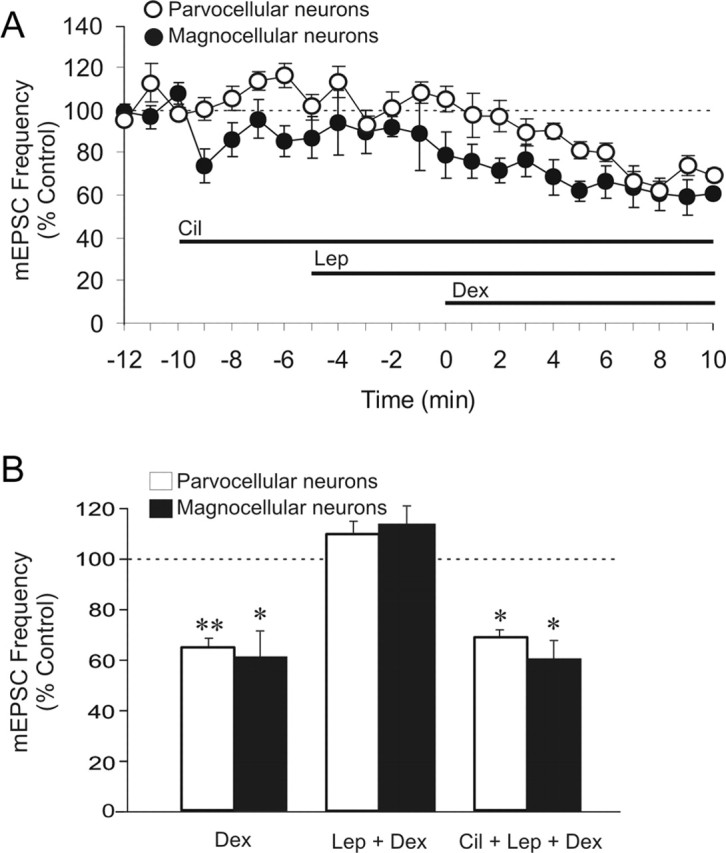
Leptin blocks GSE in both magnocellular and parvocellular PVN neurons by activating PDE-3B. A, Time course of normalized mean mEPSC frequency recorded from PVN magnocellular and parvocellular neurons showing that, in the presence of cilostamide (1 μm), 5 min preapplication of leptin (10 nm) failed to prevent GSE during 10 min application of 2.5 μm Dex. B, Summary histogram showing mean normalized mEPSC frequency recorded over the last 3 min of a 10 min application of Dex (n = 9), Lep + Dex (n = 6), or Cil + Lep + Dex (n = 6) in magnocellular neurons and of Dex (n = 6), Lep + Dex (n = 5), or Cil + Lep + Dex (n = 5) in parvocellular neurons. Means ± SE. *p < 0.05; **p < 0.01. In the summary histogram (B), the data from the groups Dex and Lep + Dex for both magnocellular and parvocellular neurons are repeated here for comparison.
Figure 6.

RT-PCR analysis of the Ob-Rb leptin receptor isoform expression in rat PVN and SON. Using Ob-Rb-specific primers that yielded a 92 bp amplimer, the expression of Ob-Rb was detected in punches of PVN and SON from three individual rats (1, 2, 3) but not in rat liver. Although the same primers could amplify rat brain genomic DNA, no PCR product was detected in any of the PVN or SON samples in which reverse transcriptase was omitted (−RT), indicating no genomic DNA contamination. M, Marker; L, liver; G, genomic DNA.
Glucocorticoid-induced synthesis of endocannabinoids is mediated by a Gαs–cAMP–PKA pathway
Because PDE-3B activation causes a reduction of cellular cAMP levels by converting it to 5′-AMP (Zhao et al., 2002; Sahu, 2004), our data indicated that the crosstalk between leptin and GC signaling pathways regulates endocannabinoid biosynthesis in PVN neuroendocrine cells by controlling cellular cAMP levels. Therefore, we tested in putative magnocellular neurons whether GCs are coupled to endocannabinoid biosynthesis via G-protein-coupled receptor activation of a cAMP-dependent pathway. Intracellular application (via the patch pipette) of an antiserum against the Gαs-protein subunit completely blocked GSE (−2.4 ± 8.2%; n = 6; p = 0.41), whereas a Gαq/11 antiserum had no effect on GSE (Fig. 7A,C). Intracellular application of the cAMP/PKA antagonist cAMPS-Rp (50 μm) also blocked GSE (Fig. 7B,C). These findings suggest that GSE depends on the postsynaptic activation of a glucocorticoid receptor coupled to a stimulatory G-protein and cAMP. Additional LC/MS/MS provided direct evidence that PKA activation is necessary for the GC-induced endocannabinoid biosynthesis and release in the PVN. Thus, bath application of the PKA inhibitor H-89 (1 μm) blocked the Dex-induced increase in endocannabinoid levels in trimmed PVN slices, causing instead a significant decrease in 2-AG levels (AEA, 12.7 ± 4.9 pmol/mg protein, p = 0.99; 2AG, 105.4 ± 48.0 pmol/mg protein, n = 5, p < 0.05) (Fig. 7D). Consistent with GC actions at a putative membrane GC receptor, as shown in our previous studies (Di et al., 2003, 2005a), GSE in PVN magnocellular neurons was also seen with the membrane-impermeant Dex-BSA conjugate (2.5 μm) (−42.6 ± 5.2%; n = 9; p < 0.01) (Fig. 7C).
Figure 7.

Signaling mechanism mediating glucocorticoid-induced retrograde endocannabinoid release. A–C, Left, Representative recordings before (control) and after ∼9 min of drug application; right, time course of mean normalized mEPSC frequency. A, The dexamethasone effect was inhibited by intracellular application of a Gαs antiserum (n = 6) but not a Gαq/11 antiserum (n = 4). B, Intracellular application of a PKA antagonist, cAMPS-Rp (50 μm), inhibited the dexamethasone effect (n = 8). C, Summary histogram showing mean normalized mEPSC frequency recorded over the last 3 min of a 10 min application of membrane-impermeant Dex-BSA (n = 9) and of dexamethasone in the presence of the Gαs antiserum (n = 6), the Gαq/11 antiserum (n = 4), and cAMPS-Rp (n = 8); means ± SE. *p < 0.05; **p < 0.01. D, LC/MS/MS analysis showing that the PKA antagonist H-89 (1 μm) (n = 5) blocked the Dex-induced rise in PVN levels of AEA and 2-AG; means ± SE. *p < 0.05 versus control group; **p < 0.001 versus control group; ***p < 0.0005 versus control group; #p < 0.05; ##p < 0.005 versus PVN (Dex) group. The PVN (control) (n = 16) and PVN (Dex) (n = 5) groups are the same presented in the Figure 1, which were used here for statistical comparison and for clarity.
Discussion
Here we demonstrate a crosstalk between GC- and leptin-activated signaling pathways that rapidly controls endocannabinoid biosynthesis and release, thereby modulating CB1-dependent inhibition of excitatory synaptic inputs to PVN neurosecretory cells involved in the regulation of energy homeostasis, fluid balance, and the adaptation to stress. The rapid onset of GSE (∼1 min) and of the leptin suppression of GSE (∼5 min) indicates a nongenomic interaction between these hormones. Our data show that GCs stimulate endocannabinoid synthesis by activation of a membrane receptor coupled to a Gαs–cAMP–PKA pathway, which is consistent with studies showing that GCs activate a nongenomic, PKA-dependent signaling pathway in mouse neuroblastoma cells (Han et al., 2005) and that cAMP and PKA are required for GC-induced apoptosis in leukemic cells (Tiwari et al., 2005). Furthermore, Cadas et al. (1996) demonstrated that agents that stimulate cAMP formation cause PKA-mediated potentiation of the Ca2+-dependent biosynthesis of the AEA precursor N-arachidonyl-phosphatidyl-ethanolamine (NAPE). NAPE is thought to be synthesized by the Ca2+-dependent transacylation between phosphatidylcholine and phosphatidilethanolamine, a reaction that also generates sn-1-lyso-2-arachidonoyl-phosphatidylcholine, which has been proposed to serve as a 2-AG precursor in neurons (Di Marzo and Deutsch, 1998). Therefore, our results suggest that GCs bind to a putative Gαs-coupled membrane GC receptor to promote cAMP synthesis, leading downstream to PKA-dependent stimulation of a biosynthetic pathway common to 2-AG and AEA. Endocannabinoids synthesized from the membrane then cause CB1-dependent inhibition of presynaptic glutamate release (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). Additional investigation will be necessary to demonstrate directly that GCs activate adenylyl cyclase and to confirm the potential concomitant involvement of intracellular Ca2+ as well as phospholipase D and phospholipase C, which are required for the synthesis of AEA and 2-AG, respectively (Di Marzo and Deutsch, 1998).
Glucocorticoid-induced biosynthesis of endocannabinoids and the subsequent CB1-dependent suppression of excitatory inputs to PVN neurons were blocked completely by the PDE-3B-dependent actions of leptin, indicating that leptin prevents PKA-dependent endocannabinoid biosynthesis by reducing cAMP levels. The dependence on PDE-3B and the absence of effect in genetically obese Zucker rats indicate that leptin blockade of GSE is likely to be mediated by Ob-Rb, which is thought to be the only leptin receptor capable of activating PDE-3B in the brain (Sahu, 2004). Our RT-PCR data and previous immunohistochemical studies (Hakansson et al., 1998; Yarnell et al., 1998; Matsuda et al. 1999) indicate that Ob-Rb is expressed in the PVN. Because leptin suppresses activity-dependent release of endocannabinoids in the lateral hypothalamus (Jo et al., 2005), it will be interesting to test whether leptin can also block the oxytocin (OT)-, α-melanocyte-stimulating hormone- and activity-dependent release of endocannabinoids that has been demonstrated in different hypothalamic nuclei (Hirasawa et al., 2004; Di et al., 2005b; Jo et al., 2005; Sabatier and Leng, 2006).
Our LC/MS/MS analysis of PVN slices along with the data from our electrophysiological experiments indicates that leptin-mediated blockade of GSE is a common feature among PVN parvocellular and magnocellular neurosecretory cells. Such a broad regulatory influence over PVN neuroendocrine output is consistent with the well established nutritional state-dependent regulation by leptin, GCs, and endocannabinoids of different fundamental aspects of homeostasis. For instance, in fasting animals, circulating levels of leptin decrease whereas both circulating levels of GCs and hypothalamic levels of endocannabinoids increase, and a hyperphagia that is GC and CB1 dependent follows (Castonguay, 1991; Ahima, 2000; Di Marzo et al., 2001; Kirkham et al., 2002). Given that the inhibition of PVN neural activity stimulates appetite and weight gain (Grandison and Guidotti, 1977) and the fact that the PVN is the only hypothalamic site in which local GC application triggers hyperphagia and obesity (Tempel et al., 1993), our results strongly suggest that GCs may cause hyperphagia in the PVN by triggering endocannabinoid biosynthesis and inhibition of excitatory inputs to different PVN cell populations. Conversely, the leptin blockade of GSE is consistent with the reduction in hypothalamic levels of endocannabinoids caused by exogenous leptin application in fasting rats, which results in the suppression of CB1-dependent hyperphagia (Di Marzo et al., 2001). In additional agreement with our data, both fasting Sprague Dawley rats and obese Zucker rats fed ad libitum have their characteristic hyperphagia suppressed by CB1 blockade and GC depletion by adrenalectomy (Freedman et al., 1985; Castonguay, 1991; Di Marzo et al., 2001; Jeong et al., 2004), indicating that endocannabinoid- and GC-dependent hyperphagia develops whenever leptin signaling is reduced or impaired.
It is noteworthy that the fasting-induced drop in leptin levels leads to the suppression of both the norepinephrine-dependent satiety response to cholecystokinin (CCK) and CCK-induced c-fos expression in both PVN parvocellular and magnocellular neurons (McMinn et al., 2000). Conversely, feeding, gastric distension, and systemic administration of CCK activate both parvocellular and magnocellular OT-secreting neurons of the PVN, causing a transient rise in circulating OT and central OT receptor-mediated anorexia (Verbalis et al., 1986; Renaud et al., 1987; Olson et al., 1991). Preautonomic parvocellular OT neurons are thought to mediate CCK-induced short-term satiety (Olson et al., 1991, 1992; Ueta et al., 1993; Blevins et al., 2003), whereas peripheral OT secreted by magnocellular neurons has been shown to promote insulin-like anabolic adaptations appropriate to the sated state (Pittman et al., 1961). In adipose tissue, for instance, OT stimulates in vitro synthesis of protein and glycogen and facilitates lipogenesis but suppresses lipolysis (Mirsky and Perisutti, 1961; Krahl, 1964; Muchmore et al., 1981). Furthermore, peripheral OT may favor satiety by stimulating CCK release (Wu et al., 2002) and by potentiating glucose-induced insulin secretion in vitro and in vivo (Altszuler and Hampshire, 1981; Knudtzon, 1983; Gao et al., 1991; VanderWeele, 1994). It is likely, therefore, that the combination of high GC levels with concomitant low levels of leptin during fasting favors the release of endocannabinoids in the PVN, reducing the excitation of both parvocellular and magnocellular OT neurons in response to inputs triggered by satiety factors, such as CCK. This would contribute to hyperphagia by causing a delay in satiation until leptin and GCs return to basal levels after nutritional recovery. This hypothesis is consistent with the findings of Blevins et al. (2004) showing that leptin potentiates the CCK-induced activation of PVN parvocellular OT neurons, which in turn activate brainstem neurons that control gastric motility and cause meal termination. However, it remains to be determined whether the GSE–leptin crosstalk also applies to these OT-secreting preautonomic neurons of the PVN.
Another important aspect of the adaptation to fasting that has been shown to be under modulation by GCs, leptin, and endocannabinoids is the decline in metabolic rate and energy expenditure resulting from the downregulation of the PVN TRH-secreting neurons and reduced HPT axis outflow (Harris et al., 1978). Whereas GCs (Brabant et al., 1987) and exogenous cannabinoids (Hillard et al., 1984) were shown to suppress the HPT axis, leptin prevents the fasting-induced suppression of the HPT axis (Ahima, 2000). We previously demonstrated GSE in PVN TRH neurons (Di et al., 2003), and our current results suggest that the drop in leptin levels in combination with the increase in GC levels during fasting is likely to favor GSE in PVN TRH-releasing neurons, contributing to the fasting-induced inhibition of the HPT axis.
Our findings are also consistent with abnormal HPA axis tone giving rise to increased appetite, obesity, and metabolic dysfunction (Freedman et al., 1985; Castonguay et al., 1986; Dallman and Stern, 1986; Argiles, 1989). For instance, in obese Zucker rats, which show characteristic eating and metabolic disorders, an increased sensitivity to the central inhibitory GC feedback causes abnormally low basal secretion of both corticotrophin releasing hormone and vasopressin by parvocellular PVN neurons that control HPA axis at the posterior pituitary level (Plotsky et al., 1992). Our findings, along with the increased basal levels of hypothalamic endocannabinoids observed in obese Zucker rats (Di Marzo et al., 2001), suggest that the fast negative GC feedback is tonically enhanced in these animals because of the failure of leptin to suppress GSE in the PVN. Our data also suggest that a similar failure of leptin-mediated disinhibition may be expected in obese Zucker rats for the other parvocellular and magnocellular neurosecretory subpopulations involved in metabolic control and fluid balance (Knudtzon, 1983; Verbalis et al., 1986; Langhans et al., 1991; Veltmar et al., 1992; Bealer and Abell, 1995; McCann et al., 2002) and could contribute to the metabolic and cardiovascular alterations observed in obese individuals with leptin resistance (Zimmet et al., 1999; Sahu, 2003). Furthermore, deregulation of the GC–leptin crosstalk could also contribute to the metabolic syndrome associated with chronic stress (Dallman et al., 1995; Chrousos, 2000).
In conclusion, the leptin blockade of GSE in PVN neurosecretory neurons demonstrated here provides the cellular mechanism for a central leptin-mediated regulation of major neuroendocrine axis and therefore represents an endocannabinoid-mediated, nutritional state-sensitive mechanism to coordinate the hormonal control of energy homeostasis, fluid balance, cardiovascular regulation, and the stress response. Sustained elevated circulating levels of GCs (e.g., by chronic stress, depression, or in Cushing’s syndrome) or leptin (e.g., in obesity) or GC deficiency (e.g., in Addison’s disease) could result in deregulation of this mechanism, leading to the eating, metabolic, and cardiovascular disorders associated with these pathological conditions.
Footnotes
This work was supported by National Institutes of Health Grants MH066958 (J.G.T.), NS23002 (N.G.B.), and P20RR016816 (N.G.B., J.G.T.). We thank Bret Smith, Ivan de Araujo, and Sidarta Ribeiro for providing critical feedback on a draft of this manuscript, Johnny Porter for providing us with Zucker rats, Katalin Halmos for technical assistance, and Claudine Ferrão for assistance with graphics.
References
- Ahima RS (2000). Leptin and the neuroendocrinology of fasting. Front Horm Res 26:42–56. [DOI] [PubMed] [Google Scholar]
- Altszuler N, Hampshire J (1981). Oxytocin infusion increases plasma insulin and glucagon levels and glucose production and uptake in the normal dog. Diabetes 30:112–114. [DOI] [PubMed] [Google Scholar]
- Argiles JM (1989). The obese Zucker rat: a choice for fat metabolism 1968–1988: twenty years of research on the insights of the Zucker mutation. Prog Lipid Res 28:53–66. [DOI] [PubMed] [Google Scholar]
- Bealer SL, Abell SO (1995). Paraventricular nucleus histamine increases blood pressure by adrenoreceptor stimulation of vasopressin release. Am J Physiol 269:H80–H85. [DOI] [PubMed] [Google Scholar]
- Blevins JE, Eakin TJ, Murphy JA, Schwartz MW, Baskin DG (2003). Oxytocin innervation of caudal brainstem nuclei activated by cholecystokinin. Brain Res 993:30–41. [DOI] [PubMed] [Google Scholar]
- Blevins JE, Schwartz MW, Baskin DG (2004). Evidence that paraventricular nucleus oxytocin neurons link hypothalamic leptin action to caudal brain stem nuclei controlling meal size. Am J Physiol Regul Integr Comp Physiol 287:R87–R96. [DOI] [PubMed] [Google Scholar]
- Brabant G, Brabant A, Ranft U, Ocran K, Kohrle J, Hesch RD, von zur Muhlen A (1987). Circadian and pulsatile thyrotropin secretion in euthyroid man under the influence of thyroid hormone and glucocorticoid administration. J Clin Endocrinol Metab 65:83–88. [DOI] [PubMed] [Google Scholar]
- Cadas H, Gaillet S, Beltramo M, Venance L, Piomelli D (1996). Biosynthesis of an endogenous cannabinoid precursor in neurons and its control by calcium and cAMP. J Neurosci 16:3934–3942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castonguay TW (1991). Glucocorticoids as modulators in the control of feeding. Brain Res Bull 27:423–428. [DOI] [PubMed] [Google Scholar]
- Castonguay TW, Dallman MF, Stern JS (1986). Some metabolic and behavioral effects of adrenalectomy on obese Zucker rats. Am J Physiol 251:R923–R933. [DOI] [PubMed] [Google Scholar]
- Chowers I, Einat R, Feldman S (1969). Effects of starvation on levels of corticotrophin releasing factor, corticotrophin and plasma corticosterone in rats. Acta Endocrinol (Copenh) 61:687–694. [DOI] [PubMed] [Google Scholar]
- Chrousos GP (2000). The role of stress and the hypothalamic-pituitary-adrenal axis in the pathogenesis of the metabolic syndrome: neuro-endocrine and target tissue-related causes. Int J Obes Relat Metab Disord 24:[Suppl 2], S50–S55. [DOI] [PubMed] [Google Scholar]
- Dallman MF, Akana SF, Strack AM, Hanson ES, Sebastian RJ (1995). The neural network that regulates energy balance is responsive to glucocorticoids and insulin and also regulates HPA axis responsivity at a site proximal to CRF neurons. Ann NY Acad Sci 771:730–742. [DOI] [PubMed] [Google Scholar]
- Di Marzo V, Deutsch DG (1998). Biochemistry of the endogenous ligands of cannabinoid receptors. Neurobiol Dis 5:386–404. [DOI] [PubMed] [Google Scholar]
- Di Marzo V, Goparaju SK, Wang L, Liu J, Batkai S, Jarai Z, Fezza F, Miura GI, Palmiter RD, Sugiura T, Kunos G (2001). Leptin-regulated endocannabinoids are involved in maintaining food intake. Nature 410:822–825. [DOI] [PubMed] [Google Scholar]
- Di S, Malcher-Lopes R, Halmos KC, Tasker JG (2003). Nongenomic glucocorticoid inhibition via endocannabinoid release in the hypothalamus: a fast feedback mechanism. J Neurosci 23:4850–4857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di S, Malcher-Lopes R, Marcheselli VL, Bazan NG, Tasker JG (2005a). Rapid glucocorticoid-mediated endocannabinoid release and opposing regulation of glutamate and gamma-aminobutyric acid inputs to hypothalamic magnocellular neurons. Endocrinology 146:4292–4301. [DOI] [PubMed] [Google Scholar]
- Di S, Boudaba C, Popescu IR, Weng FJ, Harris C, Marcheselli VL, Bazan NG, Tasker JG (2005b). Activity-dependent release and actions of endocannabinoids in the rat hypothalamic supraoptic nucleus. J Physiol (Lond) 569:751–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freedman MR, Castonguay TW, Stern JS (1985). Effect of adrenalectomy and corticosterone replacement on meal patterns of Zucker rats. Am J Physiol 249:R584–R594. [DOI] [PubMed] [Google Scholar]
- Gao ZY, Drews G, Henquin JC (1991). Mechanisms of the stimulation of insulin release by oxytocin in normal mouse islets. Biochem J 276:169–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grandison L, Guidotti A (1977). Stimulation of food intake by muscimol and beta endorphin. Neuropharmacology 16:533–536. [DOI] [PubMed] [Google Scholar]
- Hakansson ML, Brown H, Ghilardi N, Skoda RC, Meister B (1998). Leptin receptor immunoreactivity in chemically defined target neurons of the hypothalamus. J Neurosci 18:559–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han JZ, Lin W, Chen YZ (2005). Inhibition of ATP-induced calcium influx in HT4 cells by glucocorticoids: involvement of protein kinase A. Acta Pharmacol Sin 26:199–204. [DOI] [PubMed] [Google Scholar]
- Harris AR, Fang SL, Azizi F, Lipworth L, Vagenakis AG, Barverman LE (1978). Effect of starvation on hypothalamic-pituitary-thyroid function in the rat. Metabolism 27:1074–1083. [DOI] [PubMed] [Google Scholar]
- Hillard CJ, Farber NE, Hagen TC, Bloom AS (1984). The effects of delta 9-tetrahydrocannabinol on serum thyrotropin levels in the rat. Pharmacol Biochem Behav 20:547–550. [DOI] [PubMed] [Google Scholar]
- Hirasawa M, Schwab Y, Natah S, Hillard CJ, Mackie K, Sharkey KA, Pittman QJ (2004). Dendritically released transmitters cooperate via autocrine and retrograde actions to inhibit afferent excitation in rat brain. J Physiol (Lond) 559:611–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong KH, Sakihara S, Widmaier EP, Majzoub JA (2004). Impaired leptin expression and abnormal response to fasting in corticotropin-releasing hormone-deficient mice. Endocrinology 145:3174–3181. [DOI] [PubMed] [Google Scholar]
- Jo YH, Chen YJ, Chua SC Jr, Talmage DA, Role LW (2005). Integration of endocannabinoid and leptin signaling in an appetite-related neural circuit. Neuron 48:1055–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkham TC, Williams CM, Fezza F, Di Marzo V (2002). Endocannabinoid levels in rat limbic forebrain and hypothalamus in relation to fasting, feeding and satiation: stimulation of eating by 2-arachidonoyl glycerol. Br J Pharmacol 136:550–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knudtzon J (1983). Acute effects of oxytocin and vasopressin on plasma levels of glucagon, insulin and glucose in rabbits. Horm Metab Res 15:103–104. [DOI] [PubMed] [Google Scholar]
- Krahl ME (1964). Specificity of insulin or oxytocin stimulation of protein synthesis in adipose tissue. Am J Physiol 207:1169–1172. [DOI] [PubMed] [Google Scholar]
- Langhans W, Delprete E, Scharrer E (1991). Mechanisms of vasopressin’s anorectic effect. Physiol Behav 49:169–176. [DOI] [PubMed] [Google Scholar]
- Levin BE, Dunn-Meynell AA, Banks WA (2003). Obesity-prone rats have normal blood-brain barrier transport but defective central leptin signaling prior to obesity onset. Am J Physiol Regul Integr Comp Physiol 286:R143–R150. [DOI] [PubMed] [Google Scholar]
- Luther JA, Halmos KC, Tasker JG (2000). A slow transient potassium current expressed in a subset of neurosecretory neurons of the hypothalamic paraventricular nucleus. J Neurophysiol 84:1814–1825. [DOI] [PubMed] [Google Scholar]
- Luther JA, Daftary SS, Boudaba C, Gould GC, Halmos KC, Tasker JG (2002). Neurosecretory and non-neurosecretory parvocellular neurones of the hypothalamic paraventricular nucleus express distinct electrophysiological properties. J Neuroendocrinol 14:929–932. [DOI] [PubMed] [Google Scholar]
- Matsuda J, Yokota I, Tsuruo Y, Murakami T, Ishimura K, Shima K, Kuroda Y (1999). Development changes in long-form leptin receptor expression and localization in rat brain. Endocrinology 140:5233–5238. [DOI] [PubMed] [Google Scholar]
- McCann SM, Antunes-Rodrigues J, Jankowski M, Gutkowska J (2002). Oxytocin, vasopressin and atrial natriuretic peptide control body fluid homeostasis by action on their receptors in brain, cardiovascular system and kidney. Prog Brain Res 139:309–328. [DOI] [PubMed] [Google Scholar]
- McMinn JE, Sindelar DK, Havel PJ, Schwartz MW (2000). Leptin deficiency induced by fasting impairs the satiety response to cholecystokinin. Endocrinology 141:4442–4448. [DOI] [PubMed] [Google Scholar]
- Mirsky IA, Perisutti G (1961). The insulin-like action of oxytocin on adipose tissue. Biochim Biophys Acta 50:603–604. [DOI] [PubMed] [Google Scholar]
- Muchmore DB, Little SA, de Haen CJ (1981). A dual mechanism of action of ocytocin in rat epididymal fat cells. Biol Chem 256:365–372. [PubMed] [Google Scholar]
- Olson BR, Drutarosky MD, Stricker EM, Verbalis JG (1991). Brain oxytocin receptor antagonism blunts the effects of anorexigenic treatments in rats: evidence for central oxytocin inhibition of food intake. Endocrinology 129:785–791. [DOI] [PubMed] [Google Scholar]
- Olson BR, Hoffman GE, Sved AF, Stricker EM, Verbalis JG (1992). Cholecystokinin induces c-fos expression in hypothalamic oxytocinergic neurons projecting to the dorsal vagal complex. Brain Res 569:238–248. [DOI] [PubMed] [Google Scholar]
- Pittman JA, Boshell BR, Williams BH, Hamner D, Hill P (1961). Insulin-like activity of vasopressin and oxytocin. Biochem Biophys Res Commun 6:29–32. [DOI] [PubMed] [Google Scholar]
- Plotsky PM, Thrivikraman KV, Watts AG, Hauger RL (1992). Hypothalamic-pituitary-adrenal axis function in the Zucker obese rat. Endocrinology 130:1931–1941. [DOI] [PubMed] [Google Scholar]
- Renaud LP, Tang M, McCann MJ, Stricker EM, Verbalis JG (1987). Cholecystokinin and gastric distension activate oxytocinergic cells in rat hypothalamus. Am J Physiol 253:R661–R665. [DOI] [PubMed] [Google Scholar]
- Sabatier N, Leng G (2006). Presynaptic actions of endocannabinoids mediate alpha-MSH-induced inhibition of oxytocin cells. Am J Physiol Regul Integr Comp Physiol 290:R577–R584. [DOI] [PubMed] [Google Scholar]
- Sahu A (2003). Leptin signaling in the hypothalamus: emphasis on energy homeostasis and leptin resistance. Front Neuroendocrinol 24:225–253. [DOI] [PubMed] [Google Scholar]
- Sahu A (2004). Minireview: a hypothalamic role in energy balance with special emphasis on leptin. Endocrinology 145:2613–2620. [DOI] [PubMed] [Google Scholar]
- Sapolsky RM, Romero LM, Munck AU (2000). How do glucocorticoids influence stress responses? Integrating permissive, suppressive, stimulatory, and preparative actions. Endocr Rev 21:55–89. [DOI] [PubMed] [Google Scholar]
- Stern JE (2001). Electrophysiological and morphological properties of pre-autonomic neurones in the rat hypothalamic paraventricular nucleus. J Physiol (Lond) 537:161–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tempel DL, Kim T, Leibowitz SF (1993). The paraventricular nucleus is uniquely responsive to the feeding stimulatory effects of steroid hormones. Brain Res 614:197–204. [DOI] [PubMed] [Google Scholar]
- Tiwari S, Dong H, Kim EJ, Weintraub L, Epstein PM, Lerner A (2005). Type 4 cAMP phosphodiesterase (PDE4) inhibitors augment glucocorticoid-mediated apoptosis in B cell chronic lymphocytic leukemia (B-CLL) in the absence of exogenous adenylyl cyclase stimulation. Biochem Pharmacol 69:473–483. [DOI] [PubMed] [Google Scholar]
- Ueta Y, Kannan H, Higuchi T, Negoro H, Yamashita H (1993). CCK-8 excites oxytocin-secreting neurons in the paraventricular nucleus in rats—possible involvement of noradrenergic pathway. Brain Res Bull 32:453–459. [DOI] [PubMed] [Google Scholar]
- VanderWeele DA (1994). Insulin is a prandial satiety hormone. Physiol Behav 56:619–622. [DOI] [PubMed] [Google Scholar]
- Veltmar A, Culman J, Qadri F, Rascher W, Unger T (1992). Involvement of adrenergic and angiotensinergic receptors in the paraventricular nucleus in the angiotensin II-induced vasopressin release. J Pharmacol Exp Ther 263:1253–1260. [PubMed] [Google Scholar]
- Verbalis JG, McCann MJ, McHale CM, Stricker EM (1986). Oxytocin secretion in response to cholecystokinin and food: differentiation of nausea from satiety. Science 232:1417–1419. [DOI] [PubMed] [Google Scholar]
- Wu CL, Hung CR, Chang FY, Pau KY, Wang JL, Wang PS (2002). Involvement of cholecystokinin receptor in the inhibition of gastric emptying by oxytocin in male rats. Pflügers Arch 445:187–193. [DOI] [PubMed] [Google Scholar]
- Yarnell DO, Knight DS, Hamilton K, Tulp O, Tso P (1998). Localization of leptin receptor immunoreactivity in the lean and obese Zucker rat brain. Brain Res 785:80–89. [DOI] [PubMed] [Google Scholar]
- Zhao AZ, Huan JN, Gupta S, Pal R, Sahu A (2002). A phosphatidylinositol 3-kinase phosphodiesterase 3B-cyclic AMP pathway in hypothalamic action of leptin on feeding. Nat Neurosci 5:727–728. [DOI] [PubMed] [Google Scholar]
- Zimmet P, Boyko EJ, Collier GR, de Courten M (1999). Etiology of the metabolic syndrome: potential role of insulin resistance, leptin resistance, and other players. Ann NY Acad Sci 892:25–44. [DOI] [PubMed] [Google Scholar]