

Abstract
Cannabinoids represent one of the most commonly used hallucinogenic drug classes. In addition, cannabis use is a primary risk factor for schizophrenia in susceptible individuals and can potently modulate the emotional salience of sensory stimuli. We report that systemic activation or blockade of cannabinoid CB1 receptors modulates emotional associative learning and memory formation in a subpopulation of neurons in the mammalian medial prefrontal cortex (mPFC) that receives functional input from the basolateral amygdala (BLA). Using in vivo single-unit recordings in rats, we found that a CB1 receptor agonist potentiated the response of medial prefrontal cortical neurons to olfactory cues paired previously with a footshock, whereas this associative responding was prevented by a CB1 receptor antagonist. In an olfactory fear-conditioning procedure, CB1 agonist microinfusions into the mPFC enabled behavioral responses to olfactory cues paired with normally subthreshold footshock, whereas the antagonist completely blocked emotional learning. These results are the first demonstration that cannabinoid signaling in the mPFC can modulate the magnitude of neuronal emotional learning plasticity and memory formation through functional inputs from the BLA.
Keywords: cannabinoids, prefrontal cortex, amygdala, emotional learning, electrophysiology, extracellular recordings
Introduction
Cannabinoids strongly influence emotional processing and sensory perception (Wachtel et al., 2002; Green et al., 2003). Receptors for endogenous cannabinoids are found ubiquitously throughout the CNS, with the CB1 cannabinoid receptor subtype localized to regions known to be involved in associative learning and emotional processing, such as the hippocampus, amygdala, striatum, and frontal cortical regions (Tsou et al., 1998; Marsicano and Lutz, 1999; Moldrich and Wenger, 2000; Mcdonald and Mascagni, 2001). Moreover, CB1 receptor transmission is involved in emotional learning phenomena (Marsicano et al., 2002; Bilkei-Gorzo et al., 2005; Varvel et al., 2005) and is believed to be disrupted in disorders such as schizophrenia (Leweke et al., 1999; Dean et al., 2001; Giuffrida et al., 2004; Zavitsanou et al., 2004; D’Souza et al., 2005).
Considerable evidence suggests that the cannabinoid CB1 receptor system is a crucial mediator of emotional learning. Genetic deletion of the CB1 receptor in mice blocks the extinction of conditioned fear to an auditory cue paired previously with a footshock (Marsicano et al., 2002) but not extinction of an appetitively motivated learning task, suggesting that CB1 receptor signaling may be preferentially involved in memory extinction for “aversive” emotional conditioned associations (Holter et al., 2005). Others have reported that the CB1 receptor is essential for normal emotional behaviors and stress responses because genetic deletion of the CB1 receptor in mice (Martin et al., 2002) or pharmacological modulation in rats (Onaivi et al., 1990) increases behavioral measures of aggressive and/or depressive-like behaviors. In addition, stress-induced amygdala activation is potentiated by blockade of CB1 receptors in rodents (Patel et al., 2005), further implicating the cannabinoid CB1 receptor in the processing and integration of emotionally salient information. However, no evidence presently exists concerning how cannabinoid signaling may influence emotional associative learning at the level of the single neuron specifically within the mammalian medial prefrontal cortex (mPFC) nor how inputs from the amygdala may modulate such processes. Interactions between the mPFC and the amygdala are crucial for integrating emotionally salient information (Milad and Quirk, 2002; Rosenkranz and Grace, 2002; Rosenkranz et al., 2003; Maren and Quirk, 2004; Milad et al., 2004; Laviolette et al., 2005). We have reported previously that neurons within the mPFC that receive a functional input from the basolateral amygdala (BLA) can encode and express learned associations between discrete odor stimuli and footshock (Laviolette et al., 2005). In addition, single neurons within the lateral nucleus of the amygdala encode and express emotional associative learning through a dopamine (DA)-dependent mechanism (Rosenkranz and Grace, 2002), demonstrating that neurons within both the amygdala and mPFC can encode learned associations between sensory stimuli and emotionally salient events. We therefore investigated the possible role of the CB1 receptor in the encoding, acquisition, and modulation of emotional associative learning using single-unit extracellular recordings during olfactory conditioning in chloral hydrate-anesthetized animals and olfactory fear-conditioning procedures in behaving animals. Our results demonstrate that CB1 receptors within the mPFC strongly modulate emotional learning and are essential for the acquisition and expression of this conditioned emotional learning.
Materials and Methods
Animal preparation.
All procedures were performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the University of Pittsburgh Institutional Animal Care and Use Committee. Male Sprague Dawley rats (275–350 g) were anesthetized with an intraperitoneal injection of 400 mg/kg of 8% chloral hydrate and placed in a stereotaxic device (David Kopf Instruments, Tujunga, CA). Subsequent supplements of choral hydrate were administered via a lateral tail vein catheter or intraperitoneally, as required. Body temperature was monitored with a rectal temperature probe (Precision Thermometer 4600; YSI, Yellow Springs, OH) and maintained at ∼37° using a heat control unit and heating pad (Fintronics, Orange, CT). Incisions were made in the scalp to expose the skull, burr holes were drilled, and the dura overlying the BLA or mPFC was removed. Coordinates for these areas were determined using the stereotaxic atlas of Paxinos and Watson (1997) as follows from bregma (in mm): BLA: anteroposterior (AP), −3.0; lateral (L), ±5.0; ventral (V), −8.0 from the dural surface; mPFC: AP, +3.0 rostral; L, ±0.5; V, −3.4 from the dural surface. For bilateral mPFC guide-cannula implantation, animals were anesthetized with ketamine–xylazine and placed in a stereotaxic frame. Bilateral guide cannulas were implanted in the mPFC at a 15° angle with the following coordinates from bregma (in mm): AP, +2.9; L, ±1.9; V, −3.0. Guide cannulas were secured with jeweler's screws and dental acrylic.
Single-unit recordings.
Single-barrel electrodes were constructed using a vertical microelectrode puller (Narishige, Tokyo, Japan) and filled with 2% Pontamine sky blue in 2 m NaCl (impedance measured in situ ranged between 8 and 20 MΩ measured at 1 kHz). Recording electrodes were lowered slowly into the mPFC via a hydraulic micromanipulator (Narishige). A bipolar concentric stimulating electrode (Plastics One, Roanoke, VA) was lowered into the BLA. Stimulation of the BLA was delivered using a Grass (Quincy, MA) S88 stimulator with stimulation amplitudes ranging between 100 and 800 μA. Signals from the recording electrode were amplified by a head stage connected to the preamplifier before being fed into a window discriminator/amplifier (Fintronics, Foster City, CA) and an audio monitor (AM5; Grass). Signals were filtered with a low cutoff of 200 Hz and a high cutoff of 8 kHz and displayed on an oscilloscope (Kikusui, Yokohama, Japan). Data were simultaneously collected and monitored on-line using software developed in this laboratory (Neuroscope) and stored on a computer for off-line analysis. To isolate neurons in the mPFC that responded to BLA stimulation, electrical stimulus pulses (0.5 Hz, 0.2–0.6 mA, 0.3 ms duration) were delivered during electrode penetration of the mPFC to search for single units that exhibited evoked spike activity. The latency of response to BLA input was determined as the time from the beginning of the stimulus artifact to the beginning of the evoked spike. Single-unit-evoked spikes were operationally defined as monosynaptic and orthodromic if they showed very little shift in latency with increasing stimulus intensity but some range (1–5 ms) in latency distribution (“jitter”) and if they failed to follow high-frequency stimulation (>250 Hz), thus ruling out antidromic activation. In addition, collision tests were performed on mPFC neurons responding to BLA stimulation to verify orthodromicity. The locations of the recordings were marked by ejection of Pontamine sky blue from the recording electrode using a constant −25 μA current. A nonresponsive mPFC control neuron was defined as a spontaneously active mPFC neuron that failed to show any evoked responses to at least 100 stimulations of the BLA over a range of test currents (200–800 μA, 0.5 Hz, 0.3 ms duration). Only one neuron was recorded and conditioned per animal.
Bursting analysis.
Using previously established criteria based on an analysis of baseline recordings of spontaneously active, BLA-responsive mPFC neurons, a bursting event was defined as the occurrence of two or more spikes with an interspike interval (ISI) of <45 ms (Laviolette et al., 2005). We hypothesized that the spiking of mPFC neurons could be described by two separate spiking modes, regular spiking and bursting, that produced a characteristic ISI distribution that could be modeled by an inverse Gaussian probability density (IGPD) function (Barbieri et al., 2001). We therefore fit our collective baseline ISI histogram with a sum of two IGPDs as follows:
where
 |
wi is an ISI in the distribution; μr and μb are the expected values of the regular-mode and burst-mode ISI, respectively; and μ3λ−1 describes the variance of each distribution. Fit optimization was performed in Matlab (MathWorks, Natick, MA) using a least-squares algorithm with the following four free parameters: μr, λr, μb, and λb. Analysis was performed on both the percentage of spiking events that occurred in the form of a burst and the number of individual spikes within each burst event. We compared these bursting parameters in response to conditioned stimulus-positive (CS+) and CS-negative (CS−) odor presentations after conditioning with preconditioning baseline levels. We have reported previously that mPFC neuronal bursting is not an epiphenomenon of firing frequency, because no significant correlation exists between cortical neuron firing frequency and percentage of spikes occurring within burst events (Laviolette et al., 2005).
Pavlovian conditioning: electrophysiological recording experiments.
A conditioning procedure was performed by pairing an odor (peppermint or almond) with a footshock. This procedure was adapted from a previously described odor-conditioning procedure used in anesthetized rats (Rosenkranz and Grace, 2002; Laviolette et al., 2005). The footshock was delivered by two 28 gauge needles inserted in the lateral side of the footpad contralateral to the neuronal recordings. During the conditioning phase, each odor (peppermint or almond) was presented a minimum of four times, for 10 s, with a 60 s delay between presentations. One odor was selected randomly to be paired with footshock. Paired odor selection was fully counterbalanced across experiments. The footshock-paired odor (CS+) was paired with the delivery of footshock (5 s, 0.8 or 0.4 mA, 20 Hz, 0.3 ms duration), which was followed by presentation of the nonpaired odor (CS−). This pairing process was performed four to six times, followed by a 2 min rest period before the test phase. During the test phase, the CS+ odor and the CS− odor were presented for 30 s, and neuronal activity was recorded for an additional 90 s period. Off-line analyses of individual mPFC neurons were then conducted that included both the use of spike discrimination and sorting software and visual analysis of each recording epoch. Neuronal responses to conditioned stimuli were analyzed both in comparison with baseline and preconditioning spontaneous activity levels, and comparisons were also performed between preconditioning responses to the odors versus postconditioning responses, as described previously (Laviolette et al., 2005).
Olfactory fear-conditioning procedure.
Rats were taken from their home cages, received sham microinfusions into the mPFC, and were habituated for 30 min in a ventilated conditioning chamber with an electric grid floor inside a sound-attenuated room. Olfactory fear conditioning took place in one of two distinct environments, counterbalanced within groups: “shock” environment A had black walls and a metallic grid shock floor, whereas shock environment B had white walls with a grid shock floor. Testing 24 h later took place in one of two alternate environments, where animals had not previously received electric shock, counterbalanced within groups: “test” environment A had black walls and a black Plexiglas floor, whereas test environment B had white walls with a black Plexiglas floor. On day 1 (habituation phase), animals were habituated to a random combination of shock environment A or B and test environment A or B in a counterbalanced order for 30 min in each environment. On day 2 (conditioning phase), animals were returned to the conditioning room. Bilateral microinfusions into the mPFC of saline vehicle, (R)-(+)-[2,3-dihydro-5-methyl-3[(4-morpholinyl)methyl] pyrrolo[1,2,3-de]-1, 4-benzoaxzinyl]-(1-naphthalenyl)methanone mesylate salt (WIN 55,212-2) (5–50 ng), or the selective and competitive CB1 receptor antagonist 1-(2,4-dichlorophenyl)-5-(4-iodophenyl)-4-methyl-N-1-piperidinyl-1H-pyrazole-3-carboxamide trifluoroacetate salt (AM-251) (5–50 ng) were performed, and the rat was placed in the previously assigned shock environment. During the conditioning phase, one of the odors (almond or peppermint) was presented to the animal for 19 s and a footshock was then delivered (0.8 or 0.4mA) through an electric grid floor (Colbourne Instruments, Lehigh Valley, PA) for 1 s. Two minutes later, the alternate odor was presented for 20 s (CS−) in the absence of footshock. This conditioning cycle was repeated five times. On the following day (test phase), rats were returned to the test room and placed in the previously assigned test environment. Before odor presentation, the rat was allowed to explore the environment for 1 min during which baseline levels of freezing and exploratory behavior were observed. Odors (CS+ or CS−) were then presented for 5 min each to the animal in a counterbalanced order, and the amount of time freezing was recorded. Freezing activity was scored by an observer blind to the experimental condition. Rat “freezing behavior” was defined as complete immobility, with the exception of respiratory-related movement. We also analyzed exploratory behavior in response to presentations of CS+ or CS− odors, as described previously (Laviolette et al., 2005). Exploratory behavior was scored as follows: 0, no locomotion; 1, ambulation across one side of the testing chamber; 2, ambulation across two sides; 3, exploration of the full perimeter of the testing chamber; 4, exploration of the center and entire perimeter of the test chamber; a score was assigned for every minute of each of the 5 min during the odor presentations.
Drug administration.
Drugs were administered either intravenously through a lateral tail vein or directly into the BLA (unilaterally) through a chematrode (Plastics One), which allows for both local drug microinfusion and electrical stimulation of the region of interest. For mPFC microinfusions, stainless steel guide cannulas (22 gauge) were implanted bilaterally into the mPFC (see Fig. 4A) and drugs were administered through a 28 gauge microinfusion injector (Plastics One). For olfactory fear-conditioning experiments in anesthetized animals, WIN 55,212-2 (Sigma, St. Louis, MO) and AM-251 (Tocris, Ellisville, MO) were dissolved in DMSO and diluted with physiological saline to obtain a pH of ∼7.4. Muscimol (Sigma) was dissolved in physiological saline. For systemic administration experiments, doses of WIN 55,212-2 (0.05–0.5 mg/kg) or AM-251 (0.1–1.0 mg/kg) were selected based on the criteria that baseline neuronal activity was not altered before olfactory conditioning and no measurable effects on body temperature, respiration, or heart rate or mPFC neuronal responses to footshock (during conditioning) were observed. Neuronal activity was recorded during the injection procedures. The effective doses of WIN 55,212-2 (0.5 mg/kg) and AM-251 (1.0 mg) were the highest doses possible that met these criteria, and thus higher concentrations were not used. After a BLA-responsive mPFC neuron was isolated, baseline activity was recorded for 2 min. The animals then received the injection over 1 min, and neuronal activity was recorded for an additional 2 min before the olfactory conditioning procedure began. For intra-BLA muscimol experiments, muscimol (500 ng/0.5 μl; Sigma) was dissolved in PBS (pH adjusted to 7.4) and microinfused unilaterally into the BLA 3 min before the start of the conditioning procedure. Neuronal activity was recorded for at least 2–3 min before and during the entire drug infusion procedure (for both intravenous and intra-BLA drug administration) to ensure that no changes in baseline activity were induced by the administered drugs.
Figure 4.
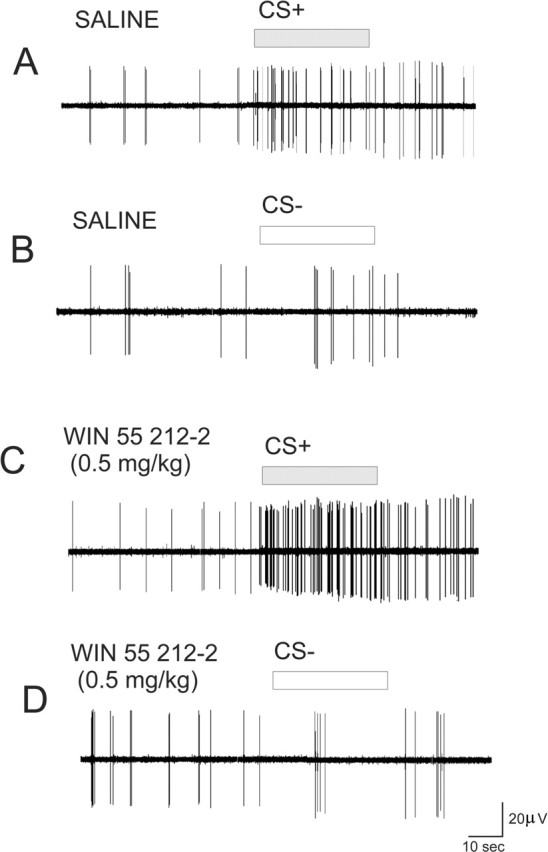
Effects of WIN 55,212-2 (0.5 mg/kg, i.v.) on neuronal activity during conditioned cue presentations. A, Recording from an mPFC neuron in a control rat showing CS+-specific responding during presentation of the footshock-paired odor. B, Recording from the same neuron during the CS− odor presentation. C, Recording from an mPFC neuron in a WIN 55,212-2 (0.5 mg/kg) pretreated rat showing CS+-specific responding during presentation of the footshock-paired odor reveals robust potentiation of the conditioned neuronal response only in the presence of the CS+ olfactory stimulus. D, Recording from the same neuron during the CS− odor presentation.
Histology.
Extracellular recording sites in the mPFC, electrical stimulation sites in the BLA, and bilateral intra-mPFC guide cannula placements were all verified histologically. At the end of the experiments, rats were deeply anesthetized and decapitated, and the brains were removed and fixed in 10% formalin solution for a minimum of 24 h. Brains were cryoprotected with 25% sucrose in 0.1 m phosphate buffer, frozen, and sectioned with a cryostat. Mounted sections were then stained with cresyl violet. Recording sites were identified with Pontamine sky blue spots. The stimulation site was determined from the ventralmost point of the stimulating electrode track identified under light microscopy. Bilateral mPFC microinfusion locations were determined by the location of the injector tip.
Statistics.
Data were analyzed with one-, two-, or three-way ANOVA or Student's t tests where appropriate. Post hoc analyses were performed with Fisher's LSD tests or Newman–Keuls tests.
Results
Activation of cannabinoid CB1 receptors potentiates emotional learning in the mPFC through functional amygdala inputs
To examine the role of cannabinoid CB1 receptor signaling in the acquisition and encoding of learned emotional associations in this BLA–mPFC neural circuit, we recorded extracellularly from single units in the rat mPFC (Fig. 1A) in chloral-hydrate-anesthetized rats during a pavlovian odor fear-conditioning procedure (Rosenkranz and Grace, 2002; Rosenkranz et al., 2003; Laviolette et al., 2005). Recordings were limited to neurons that responded to orthodromic, presumably monosynaptic, input from the BLA (Fig. 1B,C). The average ± SEM response latency for all tested neurons receiving BLA input (n = 72) was 20.8 ± 1.6 ms, and they exhibited an average firing frequency (recorded over a 2 min baseline) of 1.32 ± 0.37 Hz. Established criteria for mPFC neuronal subtypes classifies mPFC neurons into two classes: regular spiking neurons with firing rates <5 Hz that are believed to correspond to pyramidal neurons versus fast-spiking neurons with firing rates >10 Hz and shorter duration waveforms that are believed to correspond to interneurons (Degenetais et al., 2002; Jackson et al., 2004). In the present study, neurons with firing rates >5 Hz (presumed interneurons) did not show reliable responses to BLA stimulation. Thus, our olfactory conditioning procedures were restricted to pyramidal mPFC neurons, consistent with our previous report (Laviolette et al., 2005).
Figure 1.

Olfactory associative learning in single mPFC neurons responding to BLA excitatory input. A, Left, Schematic presentation of neuronal recording sites in the mPFC. For clarity, only nonoverlapping sites are shown representing the anatomical distribution of recording in the mPFC. Black circles, 0.5 mg/kg WIN 55,212-2; open circles, 0.05 mg/kg WIN 55,212-2; open squares, 1.0 mg/kg AM-251; gray circles, 0.1 mg/kg AM-251; open polygons, saline controls. Right, Photomicrograph of a coronal section of the mPFC showing a representative recording site, located within the white circle. B, Left, Representative stimulation sites in the BLA corresponding to the mPFC recording sites shown in A. Symbols are the same as in A. Right, Photomicrograph of a coronal section of the BLA showing a representative stimulation site, indicated by black arrowheads. CeA, Central nucleus of amygdala; LaN, lateral nucleus of amygdala. C, Left, Orthodromic spike evoked by stimulation of the BLA. Right, The evoked response latency for the same neuron over 100 stimulations of the BLA at 600 μA.
Emotional associative learning in BLA-responsive mPFC neurons is expressed by significant increases in the number of spontaneous spikes fired, relative to baseline, in response to postconditioning presentations of odors paired previously with footshock (CS+) but not to odors that were paired with the absence of footshock (CS−) (Fig. 2A,B). BLA-responsive mPFC neurons demonstrate a stimulus-locked response to the CS+ odor during the test phase (Fig. 2B), and this response is absent during CS− presentation (Fig. 2A).
Figure 2.

BLA-responsive mPFC neurons exhibited a stimulus-locked discharge in response to CS+ presentations. A, Rate histogram showing the firing frequency during baseline, CS+, and CS− olfactory stimulus presentations. Activity increases specifically in response to CS+ presentation and not during CS− presentation. B, The firing activity of the same neuron, immediately before CS+ presentation onset and during the course of the CS+ presentation, again illustrating CS+ stimulus-locked responding.
To examine the potential role of CB1 receptors in the neuronal encoding of emotional associative learning, we examined the effects of either a selective CB1 receptor agonist (WIN 55,212-2; 0.05–0.5 mg/kg, i.v.) or antagonist (AM-251; 0.1–1.0 mg/kg, i.v.) administered before the olfactory conditioning procedure (see Materials and Methods). In rats pretreated with the CB1 agonist WIN 55,212-2, we observed a significant increase in neuronal activity in response to CS+ presentations relative to baseline (F(12,116) = 123.4; p < 0.0001), with all groups demonstrating significance (p < 0.01) (Fig. 3A). However, relative to saline control (n = 13) and animals pretreated with 0.05 mg/kg WIN-55,212-2 (n = 10), animals pretreated with 0.5 mg/kg WIN-55,212-2 displayed dramatically increased associative neuronal activity in response to the CS+ presentation (p < 0.01) (Fig. 3A). There were no significant differences in baseline activity levels (all p > 0.05) or in neuronal responding to the CS− presentation across groups (all p > 0.05). Thus, activation of CB1 receptors induces a potentiation in neuronal associative plasticity in BLA-responsive mPFC neurons specifically in response to CS+ odor presentations.
Figure 3.

Activation or blockade of CB1 receptors with WIN 55,212-2 (0.05–0.5 mg/kg, i.v.) or AM-251 (0.1–1.0 mg/kg, i.v.) modulates emotional learning in BLA-responsive neurons of the mPFC. A, After a subthreshold dose of WIN 55,212-2 (0.05 mg/kg), mPFC neurons display associative responding to the CS+ at a similar magnitude to saline controls. A higher dose (0.5 mg/kg) causes a potentiation in the neuronal associative response to the CS+ presentation. Pharmacological inactivation of the BLA with muscimol (500 ng) before olfactory conditioning but after WIN 55,212-2 (0.5 mg/kg) administration prevents neuronal associative responding at testing. However, if the BLA is inactivated after conditioning in animals given WIN 55,212-2 (0.5 mg/kg) before conditioning, BLA-responsive mPFC neurons still display robust potentiation of associative responding to the CS+ presentation relative to saline control animals. Pre-Cond, Preconditioning; Post-Cond, postconditioning. B, Comparing preconditioning and postconditioning 10 s odor presentations demonstrates that CB1 receptor activation with WIN 55,212-2 (0.5 mg/kg) potentiates associative neuronal responding specifically to CS+ presentations relative to saline control or animals pretreated with a lower dose of WIN 55,212-2 (0.05 mg/kg). C, Blockade of CB1 receptors with a competitive CB1 receptor antagonist, AM-251, blocked neuronal associative learning in BLA-responsive neurons of the mPFC at an intravenous dose of 1.0 mg/kg. A lower dose (0.1 mg/kg, i.v.) had no effect on associative neuronal responding relative to saline control animals. Pretreatment with AM-251 (1.0 mg/kg, i.v.) blocks the associative learning potentiation induced by WIN 55,212-2 (0.5 mg/kg, i.v.). D, Comparing preconditioning and postconditioning 10 s odor presentations after AM-251 pretreatment (0.1 or 1.0 mg/kg, i.v.) demonstrates that CB1 receptor blockade at the higher dose (1.0 mg/kg) prevents neuronal associative responding in response to CS+ presentations. E, Neither administration of the CB1 antagonist AM-251 (0.1 and 1.0 mg/kg, i.v.) nor the CB1 receptor agonist WIN 55,212-2 (0.05–0.5 mg/kg, i.v.) caused any significant alterations in spontaneous neuronal activity after injection. Pre, Preconditioning; Post, postconditioning. Error bars indicate mean ± SEM.
To determine whether active input from the BLA is required for the acquisition and/or expression of neuronal associative plasticity in the mPFC and the learning potentiation effect of WIN 55,212-2, we pharmacologically inactivated the BLA by local administration of GABAA receptor agonist muscimol (500 ng/0.5 μl) after WIN-55,212-2 administration (0.5 mg/kg, i.v.) either before the olfactory associative conditioning (preconditioning BLA muscimol; n = 8) or after conditioning (postconditioning BLA muscimol) and immediately before the testing phase (n = 9). Inactivation of the BLA before conditioning prevented the expression of neuronal associative learning in BLA-responsive mPFC neurons (F(1,65) = 18.9; p < 0.0001; p > 0.05) (Fig. 3A). In contrast, BLA inactivation after conditioning (but before testing) had no effect on the ability of WIN-55,212-2 (0.5 mg/kg) to potentiate associative learning, because these neurons displayed potentiated associative learning responses above control levels (p < 0.01) (Fig. 3A). Thus, BLA input is required during the conditioning phase for the encoding of emotional learning in BLA-responsive mPFC neurons, as reported previously (Laviolette et al., 2005). In addition, the potentiation of mPFC neuronal associative learning by CB1 receptor activation seems only to require BLA input during the conditioning phase but not after encoding has taken place, because the expression of potentiated associative plasticity in mPFC neurons is not affected by BLA inactivation (Fig. 3A).
We compared mPFC neuronal activity in response to CS+ or CS− odor presentations during the 10 s time periods of odor presentation before or after conditioning. In all groups, there was a significant increase in neuronal activity in response to CS+ presentations before versus after conditioning (F(2,100) = 14.4; p < 0.001; p < 0.05) (Fig. 3B). However, relative to saline control (n = 13) and animals pretreated with 0.05 mg/kg WIN-55,212-2 (n = 10), the animals pretreated with 0.5 mg/kg WIN-55,212-2 (n = 11) displayed significantly greater associative neuronal activity in response to the CS+ odor presentation after conditioning (p < 0.01) (Fig. 3B).
Neuronal emotional learning in neurons of the mPFC requires signaling through cannabinoid CB1 receptors
Administration of the selective CB1 receptor antagonist AM-251 (0.1–1.0 mg/kg, i.v.) before conditioning blocked neuronal associative learning in BLA-responsive mPFC neurons at a dose of 1.0 mg/kg but not at 0.1 mg/kg (Fig. 3C,D), without affecting baseline neuronal activity before (Fig. 2C) (F(12,116) = 31.4; p < 0.0001). The saline control group (n = 13) and animals pretreated with a lower dose of AM-251 (0.1 mg/kg, n = 8) demonstrated significantly increased neuronal activity in response to CS+ presentations relative to baseline (p < 0.01). However, relative to saline control and animals pretreated with 0.1 mg/kg AM-251, animals pretreated with 1.0 mg/kg AM-251 displayed no associative neuronal activity in response to the CS+ presentation (difference between CS+ and CS− responses during testing (p > 0.05) (Fig. 3C,D). In addition, across groups, no significant differences in neuronal responding to the CS− presentation (all p > 0.05) or between baseline activity levels (all p > 0.05) were observed. Moreover, pretreatment with the CB1 receptor antagonist AM-251 (1.0 mg/kg, i.v.) completely blocked associative neuronal potentiation induced by WIN 55,212-2 (0.5 mg/kg, i.v.) before treatment (n = 7; F(6,20) = 0.17; p > 0.05), demonstrating that the effects of the CB1 agonist were dependent on CB1 receptor signaling (Fig. 3C). This blockade of associative conditioning was evident during the 10 s CS+ or CS− odor presentation epochs measured before and after conditioning (Fig. 3D) (F(2,88) = 11.9; p < 0.001). PFC neuron activity increased specifically in response to CS+ presentations recorded after conditioning in saline controls (n = 13; p < 0.01) and in animals pretreated with a lower dose of AM-251 (0.1 mg/mg; p < 0.01). However, a higher dose of AM-251 (1.0 mg/kg) completely blocked associative neuronal plasticity with no significant difference in the response to CS+ and CS− presentations during the test phase (Fig. 3D). No significant differences in responses to the CS− presentation relative to baseline activity levels were observed (all p > 0.05). Furthermore, the selected doses of WIN 55,212-2 (0.05–0.5 mg/ml, i.v.) or AM-251 (0.1–1.0 mg/ml, i.v.) had no effect on the spontaneous firing activity of BLA-responsive mPFC neurons recorded for 5 min after drug injection (Fig. 3E) (F(4,45) = 0.82; p > 0.05; between groups, all p > 0.05). The potentiation in the response to the CS+ is illustrated in a representative neuronal trace from a saline control neuron (Fig. 4A,B) versus a neuron pretreated with WIN 55,212-2 (0.5 mg/kg) (Fig. 4C,D). Thus, WIN 55,212-2 produces a robust potentiation specifically in response to CS+ stimulus presentation compared with the saline control neuron (Fig. 4). When analyzed in terms of changes in absolute firing frequencies between saline controls and effective doses of WIN 55,212-2 (0.5 mg/kg) or AM-251 (1.0 mg/kg), a significant difference between CS+ and CS− was consistently observed (F(2,77) = 21.6; p > 0.001). However, only the increased firing in response to CS+ presentations for the saline control and WIN 55,212-2 groups were significant (p < 0.05). No significant differences were observed in absolute firing frequency relative to baseline for any of the CS− presentations relative to baseline across groups (p > 0.05).
Cannabinoid receptor signaling modulates associative neuronal bursting in the mPFC
Administration of the CB1 agonist increased the percentage of spikes occurring in bursts specifically in response to the CS+ presentation (Fig. 5A) (F(2,83) = 14.7; p < 0.0001). Although all groups showed significantly greater percentages of bursting in response to CS+ presentations (p < 0.05), in animals pretreated with WIN 55,212-2 (0.5 mg/kg; n = 11), BLA responsive mPFC neurons showed significantly greater bursting in response to the CS+ (n = 13; p < 0.01) compared with the saline control group or the group receiving the lower dose of WIN-55,212-2 (n = 8; 0.05 mg/kg; p < 0.05) (Fig. 5A). In contrast, pretreatment with AM-251 (1.0 mg/kg, i.v.), blocked associative bursting in response to CS+ presentations (Fig. 5B) (F(11,107) = 3.5; p < 0.05), because the percentage of spikes occurring in bursts was not significantly different from that of saline control animals or in animals pretreated with a lower dose of AM-251 (0.1 mg/kg, i.v.; p > 0.05). Furthermore, pretreatment with AM-251 (1.0 mg/kg, i.v.) blocked the ability of WIN-55,212-2 (0.5 mg/kg, i.v.) to potentiate the percentage of burst-related spikes in response to the CS+ (n = 7) relative to baseline (F(6,20) = 0.13; p > 0.05) (Fig. 5B). Thus, CB1 receptor activation potentiates associative bursting in response to an emotionally salient CS+, whereas blockade of the CB1 receptor prevents this associative bursting.
Figure 5.

Cannabinoid CB1 receptors modulate associative neuronal bursting in BLA-responsive mPFC neurons. A, WIN 55,212-2 potentiates associative neuronal bursting in BLA-responsive mPFC neurons at the intravenous dose of 0.5 mg/kg compared with a lower dose of WIN 55,212-2 (0.05 mg/kg, i.v.) and saline controls. B, In contrast, AM-251 blocked neuronal associative bursting reflected in the percentage of spontaneous spikes occurring in burst events at a dose of 1.0 mg/kg but not a lower intravenous dose of 0.1 mg/kg. Pretreatment with the effective dose of AM-251 (1.0 mg/kg) competitively blocked the ability of WIN 55,212-2 (0.5 mg/kg) to potentiate neuronal associative bursting in BLA-responsive mPFC neurons. C, Activation of CB1 receptors potentiated the number of spikes per burst event specifically in response to CS+ presentations at a dose of 0.5 mg/kg but not at a lower dose of 0.05 mg/kg, both given intravenously. D, In contrast, AM-251 prevented this associative increase in the number of spikes per burst at the dose of 1.0 mg/kg but not at the lower dose of 0.1 mg/kg. Pretreatment with the effective dose of AM-251 (1.0 mg/kg) blocked the ability of WIN 55,212-2 (0.5 mg/kg) to potentiate the number of spikes per burst event in response to the CS+ presentation. E, Left, In a saline-pretreated control animal, the percentage of bursting during baseline spontaneous activity of mPFC neuronal responding compared with that during CS+ and CS− presentation is shown. Control neurons increase the percentage of bursting during baseline specifically in response to the CS+ presentation. Right, Activity trace for this neuron during the first second of CS+ odor presentation. In control neurons, bursts typically take place in doublets or triplets. F, Left, A BLA-responsive mPFC neuron from a WIN 55,212-2 (0.5 mg/kg, i.v.) pretreated animal showing the percentage of bursting activity over baseline, CS+, and CS− presentations. This neuron displays a potentiation in the percentage of bursting in response to the CS+ presentation. Right, Activity trace for this same neuron during the first second of CS+ presentation. G, Left, In a BLA-responsive mPFC neuron from an AM-251 (1.0 mg/kg) pretreated animal, there is a blockade of associative neuronal bursting in response to CS+ and CS− presentations. Right, The neuronal activity trace from this same neuron during the first second of CS+ presentation; no bursting activity is present. H, I, There is no significant correlation between firing frequency and percentage of spike events occurring in bursts. The difference between baseline firing frequency and CS+ firing frequency is plotted as a function of the difference between baseline percentage of bursting and CS+ percentage of bursting in the same neurons for either saline (H) or WIN 55,212-2 (I; 0.5 mg/kg) pretreated neurons. *p < 0.01. Error bars indicate mean ± SEM.
CB1 receptor signaling was also found to modulate the number of spikes occurring in each burst that occurred during the CS+ presentation. We compared the mean number of spikes occurring within an identified burst for each experimental group in response to saline, CS+, or CS− presentations. Note that although a burst cannot contain less than two spikes, some mPFC neurons do not display spontaneous burst activity during baseline or CS− presentations but then display strong bursting activity specifically during the CS+ presentations. Therefore, the group “mean” number of spikes in bursts can appear as less than two during the group analysis, although our burst-sorting procedure requires a minimum of two spikes to be counted as a burst. All groups displayed significantly greater numbers of burst-related spikes in response to CS+ presentations versus CS− and baseline levels (F(2,86) = 5.5; p < 0.001; all p <.05) (Fig. 5C); however, pretreatment with WIN 55,212-2 (0.5 mg/kg) significantly increased the mean number of spikes per burst in response to the CS+ presentation relative to the saline control group (p < 0.01) (Fig. 5C). In contrast, pretreatment with AM-251 blocked the associative increase in the mean number of spikes per burst (F(10,98) = 7.82; p < 0.001) (Fig. 5D). The saline control group (n = 13) and animals pretreated with the lower dose of AM-251 (0.1 mg/kg; n = 8) displayed significantly greater numbers of spikes per burst in response to CS+ presentations (p < 0.05); however, this effect was blocked in animals receiving the higher dose of AM-251 (n = 10; 1.0 mg/kg) (Fig. 5D). Furthermore, pretreatment with AM-251 (1.0 mg/kg, i.v.) blocked the ability of WIN 55,212-2 (0.5 mg/kg, i.v.) to potentiate the number of burst-related spikes in response to the CS+ (n = 8), because these neurons displayed no differences in spikes per burst in response to the CS+ or CS− relative to baseline (F(6,20) = 1.55; p > 0.05) (Fig. 5D). Thus, CB1 receptors also modulate emotional associative learning in mPFC neurons receiving input from the BLA by potentiating (CB1 receptor activation) or preventing (CB1 receptor blockade) the encoding and expression of associative bursting responses to CS+ presentations, both in terms of the frequency of bursts and in the number of spikes occurring in each burst. Figure 5E–G illustrates examples of the increase in the percentage of bursting in animals receiving the CB1 receptor agonist and the increased number of spikes within each burst that occurred during the first second of the CS+ presentation. As reported previously, increases in mPFC neuronal bursting occur specifically in response to presentations of a CS+ odor and are not simply reflective of a gross increase in firing frequency (Laviolette et al., 2005). To examine whether the increased bursting produced during CB1 receptor activation was independent of increased firing frequency in the current study, we performed a linear regression analysis comparing the percentage of neuronal bursting as a function of firing frequency. There was no significant correlation between firing frequency and percentage of bursting either during baseline levels or during the presentation of the CS+ odor for either saline control neurons (r2 = 1.7; p > 0.05) or in neurons pretreated with the effective dose of WIN 55,212-2 (0.5 mg/kg; r2 = 3.1; p > 0.05) (Fig. 5H,I). Our ISI criterion for an mPFC neuronal burst is ≤45 ms (see Materials and Methods) (Laviolette et al., 2005). Before off-line spike-sorting burst analysis for the experimental groups, we rigorously examined the individual ISI distributions for each single mPFC neuron for baseline, CS+, and CS− recording epochs to determine how CB1 receptor modulation altered the bursting parameter activity of mPFC neurons. Our burst percentage analyses based on the ISI distributions across groups revealed that CB1 receptor activation increases the percentage of spikes occurring in bursts (Fig. 5); this is illustrated as a shift in neuronal ISI distribution toward the “burst” portion of the ISI specifically in response to CS+ presentations (Fig. 6A,B). Although the saline control neuron displays a moderate shift and increase in ISIs taking place below the 45 ms burst criterion (Fig. 6A), a much more robust shift toward the burst ISI criterion is evident in the mPFC neuron recorded in the animal pretreated with WIN 55,212-2 (0.5 mg/mg) (Fig. 6B).
Figure 6.

CB1 receptor activation potentiates the frequency of burst events specifically in response to CS+ odor presentations. A, ISI histogram from a single BLA-responsive mPFC saline control rat showing the distribution of ISIs over the 2 min baseline, CS−, and CS+ recording epochs. A moderate shift in ISI frequency occurring below the 45 ms burst criterion (see Materials and Methods) takes place in response to the CS+ presentation relative to baseline. B, An ISI histogram from a single BLA-responsive mPFC neuron pretreated with the effective dose of WIN 55,212-2 (0.5 mg/kg) shows a strong shift toward ISI frequencies occurring below the 45 ms burst ISI criterion relative to baseline and CS− recording epochs, demonstrating that CB1 receptor activation can strongly potentiate associative bursting in response to emotionally salient conditioned stimuli. For all panels, the black arrows indicate the 45 ms burst ISI threshold cutoff point (see Materials and Methods).
Local activation of medial prefrontal cortical CB1 receptors potentiates emotional associative learning in behaving animals
Cannabinoid CB1 receptor activation potentiates the encoding and expression of neuronal associative plasticity in BLA-responsive neurons of the mPFC as reflected in firing frequency and levels of bursting activity. This modulatory action was also found to be expressed at the behavioral level. In an olfactory fear-conditioning procedure, animals received bilateral microinfusions of either WIN 55,212-2 (5–50 ng/0.5 μl) or AM-251 (5–50 ng/0.5 μl) into the mPFC (see Materials and Methods) before olfactory conditioning. Bilateral microinfusions of the CB1 receptor antagonist AM-251 blocked olfactory fear conditioning in animals tested 24 h after conditioning, using a suprathreshold footshock intensity (0.8 mA) that normally produces robust freezing in response to the footshock-paired odor (Rosenkranz and Grace, 2002, 2003; Laviolette et al., 2005) (Fig. 7). Only the higher dose of intra-mPFC AM-251 (50 ng) significantly attenuated fear conditioning relative to saline controls (p < 0.01; F(2,39) = 45.6; p < 0.0001) (Fig. 7A). To determine whether intra-mPFC AM-251-induced blockade of fear conditioning was attributable to state-dependent effects, animals were also tested under the presence of the effective dose of intra-mPFC AM-251 (50 ng). Under these conditions, the expression of olfactory fear conditioning was still blocked relative to saline controls (t(6) = 0.13; p > 0.05), demonstrating that the observed block of fear conditioning was not attributable to nonspecific, state-dependency effects (Fig. 7A). In contrast, intra-mPFC microinfusions of the CB1 agonist WIN 55,212-2 (50 ng) had no effect on olfactory fear conditioning at this suprathreshold level of footshock intensity relative to saline controls (p < 0.05) (Fig. 7A). A similar response was observed with exploratory behavior (F(3,80) = 22.7; p < 0.0001), a behavior known to be significantly attenuated by fear-producing stimuli (Rosenkranz and Grace, 2003). The higher dose of AM-251 (50 ng) blocked exploratory behavior suppression in response to CS+ presentations relative to saline controls (p < 0.01), whereas neither the lower dose of AM-251 (5 ng) nor WIN 55,212-2 (50 ng) had any effect on exploratory behavior in response to CS+ presentations (p <.05) (Fig. 7B). In addition, no significant differences were observed between baseline, CS− or CS+ presentations across these groups (p > 0.05).
Figure 7.
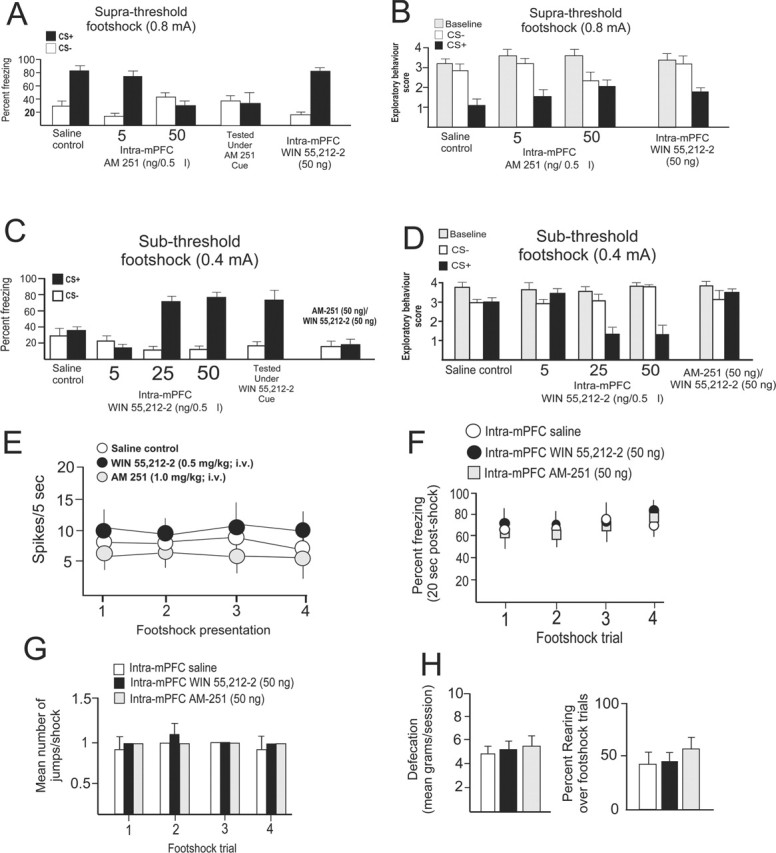
CB1 receptor activation or blockade in the mPFC modulates the behavioral expression of associative olfactory fear conditioning; the effects of intra-mPFC CB1 receptor modulation on neuronal and behavioral sensitivity to footshock presentation are shown. A, Rats show conditioned freezing behavior to an olfactory cue paired with a suprathreshold level of footshock (0.8 mA) 24 h after conditioning. Relative to saline controls, intra-mPFC microinfusions of AM-251 (50 ng/0.5 μl) before conditioning blocks olfactory fear conditioning relative to saline controls, whereas WIN 55,212-2 (50 ng/0.5 μl) has no effect on olfactory fear conditioning. Rats tested in the presence of intra-mPFC AM-251 (50 ng) still demonstrated a block in olfactory fear-conditioning expression, thus ruling out state-dependency effects. B, Similarly, AM-251 dose-dependently attenuates spontaneous exploratory behavior measures (see Materials and Methods) in response to CS+ odor presentations, whereas WIN 55,212-2 (50 ng) has no effect at this level of footshock (0.8 mA). C, A subthreshold level of footshock (0.4 mA) produces no fear conditioning in saline-pretreated control animals. However, intra-mPFC infusions of WIN 55,212-2 (25 or 50 ng/0.5 μl) potentiated the effects of this subthreshold level of footshock by enabling freezing to CS+ presentations relative to saline controls and a subthreshold dose of intra-mPFC WIN 55,212-2 (5 ng). This effect was blocked by simultaneous administration of the CB1 antagonist AM-251 (50 ng) with the highest effective dose of intra-mPFC WIN 55,212-2 (50 ng). Animals tested in the presence of intra-mPFC WIN 55,212-2 (50 ng) demonstrated strong olfactory fear-conditioning expression to subthreshold footshock, thus ruling out state-dependency effects. D, Subthreshold footshock failed to induce conditioned attenuation in exploratory behavior in response to postconditioning presentations of the CS+. However, in animals receiving intra-mPFC WIN 55,212-2 (25 or 50 ng), strong conditioned attenuation of exploratory behavior was observed during presentation of the CS+. This effect was blocked by coadministration of the CB1 antagonist AM-251 (50 ng) with the highest effective dose of intra-mPFC WIN 55,212-2 (50 ng). E, There were no significant differences in neuronal responsiveness in animals pretreated with effective systemic doses of either WIN 55,212-2 (0.5 mg/kg) or AM-251 (1.0 mg/kg). F, Bilateral intra-mPFC microinfusions of either WIN 55,212-2 (50 ng) or AM-251 (50 ng) produce no change in the percentage of freezing in response to suprathreshold footshock (0.8 mA) presentations during conditioning. G, H, Similarly, no differences were observed between intra-mPFC WIN 55,212-2 (50 ng) or AM-251 (50 ng) in terms of the number of jumps in response to footshock presentations (G) nor in the amount of defecation or in the percentage of animals displaying rearing behavior in response to footshock (H). Error bars indicate mean ± SEM.
In the anesthetized preparation, single mPFC neurons demonstrate robust associative learning in terms of electrophysiological activation in response to odor cues paired previously with 0.8 mA of footshock (Figs. 3A,B, 4). However, in awake, behaving animals, this amplitude of current represents a suprathreshold level of footshock (0.8 mA) in which behavioral responding reaches a ceiling level (Rosenkranz and Grace, 2003; Laviolette et al., 2005). Using this suprathreshold level of footshock (0.8 mA), we observed that although olfactory fear conditioning is blocked by the CB1 antagonist, the CB1 agonist has no observable effect because saline control animals already display strong fear conditioning, thereby obscuring any possible CB1 receptor activation-mediated potentiation in emotional associative learning (Fig. 7A,B). To determine whether mPFC CB1 receptor activation could potentiate emotional learning in terms of behavioral output, we performed a series of olfactory fear-conditioning experiments using a behaviorally subthreshold footshock intensity.
In pilot studies, we determined that a footshock intensity of 0.4 mA produced no measurable fear conditioning in this olfactory fear-conditioning assay. However, if WIN 55,212-2 (5, 25, and 50 ng) was microinfused into the mPFC bilaterally before olfactory fear conditioning, this subthreshold footshock intensity (0.4 mA) now elicited conditioned responses to the CS+ (Fig. 7C,D) (F(3,53) = 64.3; p < 0.0001). Intra-mPFC WIN 55,212-2 (25 or 50 ng) microinfusions potentiated olfactory fear conditioning relative to saline controls (25 ng, n = 7; 50 ng, n = 7; p < 0.01) (Fig. 7C), whereas a lower dose did not (5 ng, n = 6; p > 0.05) (Fig. 7C). Similarly, intra-mPFC WIN 55,212-2 pretreatment (25 or 50 ng) potentiated exploratory behavior suppression in response to CS+ presentations after subthreshold footshock conditioning (p < 0.05; F(3,80) = 28.8; p < 0.0001) relative to saline controls or animals pretreated with a lower dose of WIN 55,212-2 (n = 6; 5 ng; p > 0.05), which displayed no suppression of exploratory behavior (Fig. 7D). These potentiating effects of intra-mPFC WIN 55,212-2 (50 ng) were completely blocked in a separate group of animals (n = 7) by coinfusion of AM-251 (50 ng) bilaterally into the mPFC before olfactory conditioning [i.e., no differences were observed in terms of freezing behavior (t(6) = 0.44; p > 0.05) or exploratory behavior measures (F(6,13) = 1.0; p > 0.05)] (Fig. 7D). To determine whether the intra-mPFC WIN 55,212-2-induced potentiation of fear conditioning was attributable to state-dependent effects, animals were also tested under the presence of the effective dose of intra-mPFC WIN 55,212-2 (50 ng). Under these conditions, the expression of olfactory fear conditioning was still potentiated relative to controls (t(6) = 4.78; p < 0.05), demonstrating that the observed block of fear conditioning was not as a result of nonspecific, state-dependency effects (Fig. 7C). Thus, similar to the potentiating effects on emotional associative learning and encoding observed at the level of the single mPFC neuron, we report that CB1 receptor activation directly in the mPFC amplifies the emotional salience and subsequent conditioned association between an olfactory CS and a subthreshold footshock unconditioned stimulus.
In addition to their psychotropic effects, cannabinoids have profound effects on nociceptive processing. Indeed, CB1 receptor activation may produce anti-nociceptive effects (Gingold and Bergasa, 2003; Papanastassiou et al., 2004; Hohmann et al., 2005). To determine whether the administration of the effective doses of WIN 55,212-2 (0.5 mg/kg) or AM-251 (1.0 mg/kg) produced any effects on neuronal footshock sensitivity, we analyzed mPFC neuronal responses to footshocks during the conditioning procedure. We observed no significant differences in mPFC neuronal responses to footshock presentations during the conditioning procedure between saline control, WIN 55,212-2, or AM-251 pretreated groups (F(2,111) = 0.22; p > 0.05) during any of the footshock presentations (Fig. 7E). To determine whether the effective doses of intra-mPFC WIN 55,212-2 (n = 6) or AM-251 (n = 6) produced any alterations in nociceptive sensitivity to footshock in our olfactory fear-conditioning experiment relative to intra-mPFC saline (n = 5), we performed a separate control experiment examining four dimensions of footshock sensitivity: (1) percentage of freezing immediately after shock; (2) number of jumps in response to the shock; (3) amount of defecation in response to footshock; and, finally, (4) percentage of animals that displayed rearing in response to footshock. These behavioral parameters are well established indices of footshock sensitivity in rats (Antoniadis and McDonald, 1999). No group differences were observed for the percentage of time spent freezing in response to footshock (Fig. 7F) (F(2,63) = 1.24; p > 0.05), in the mean number of jumps in response to footshock presentations (Fig. 7G) (F(2,63) = 1.26; p > 0.05), for the amount of defecation in response to footshocks (Fig. 7H) (F(4,14) = 0.13; p > 0.05), or in the percentage of animals rearing in response to footshock (Fig. 7H) (F(2,63) = 0.75; p > 0.05). Thus, neither systemic nor intra-mPFC administration of WIN 55,212-2 or AM-251 produced any measurable alterations in sensitivity to footshock. This is consistent with the finding that mPFC CB1 receptors are not involved in nociceptive processing (Martin et al., 1999). Thus, the observed neuronal and behavioral potentiation of emotional learning after systemic or intra-mPFC CB1 agonist administration is likely attributable to a specific effect on the encoding and acquisition of emotional associative learning within the BLA–mPFC circuit rather than any nonspecific alterations in pain sensitivity.
Discussion
In human users, cannabinoids are frequently reported to profoundly alter sensory perception and the emotional salience of sensory stimuli (Wachtel et al., 2002; Green et al., 2003). However, the neurobiological circuitry underlying these effects is not well understood. The present results demonstrate that CB1 receptors within the amygdala–prefrontal cortical circuit can potently modulate emotional associative learning processes during both the acquisition and expression of learned, emotionally salient conditioned associations. Cannabinoid CB1 receptor activation potentiated the encoding of emotional associative learning at the level of the single mPFC neuron, and a similar potentiation was demonstrated in a behavioral fear-conditioning assay via direct infusions of a CB1 receptor agonist into the mPFC. To our knowledge, this is the first report that cannabinoid receptor activation can potentiate emotional associative learning at the single cortical neuron level in vivo and in an olfactory fear-conditioning assay in behaving animals. It is important to note that the observed modulation in neuronal associative learning was induced via systemic administration of CB1 receptor drugs in the electrophysiological conditioning experiments. As noted previously, CB1 receptors are widely distributed within the mammalian brain and the BLA in particular contains high concentrations of CB1 receptors. Moreover, we have demonstrated that active input from the BLA is required for the WIN 55,212-2-mediated neuronal learning potentiation. Nevertheless, the observed behavioral effects of intra-mPFC microinfusions of the same CB1 receptor compounds on emotional learning modulation indicates that CB1 receptors localized within the mPFC are sufficient for the potentiation or blockade of emotional learning processes in behaving animals. Therefore, although we presently cannot rule out the possibility that BLA CB1 receptors play a role in the systemic actions of CB1 drugs on our electrophysiological results in mPFC neurons, the correspondence between changes in neuronal firing and the behavioral actions of CB1 drugs administered directly into the mPFC supports the role of the mPFC as a common site of action of these drugs in emotional learning processes.
Interestingly, considerable evidence suggests that activation of CB1 receptors can induce learning and memory impairments (Sullivan, 2000; Robinson et al., 2003; O’Shea et al., 2004; Bilkei-Gorzo et al., 2005; Varvel et al., 2005). Nevertheless, CB1 receptors are essential for the extinction of conditioned fear associations (Marsicano et al., 2002), indicating an important role for this receptor in neuronal emotional learning and memory. Although exogenous cannabinoids have been shown to disrupt associative learning and attenuate the induction of long-term potentiation (LTP) (Sullivan, 2000), endocannabinoids have been shown to strongly facilitate the induction of LTP in the hippocampus (Carlson et al., 2002), suggesting that under certain conditions, cannabinoid receptor activation may facilitate neuronal learning processes, consistent with the present findings.
Cannabinoids produce a variety of cognitive effects. Cannabis users report sedative and euphoric subjective effects, but alterations in sensory perception are commonly reported (Berke and Hernton, 1974; Wachtel et al., 2002; Green et al., 2003). Sensory stimuli frequently become more salient, and their emotional valence may be amplified (Berke and Hernton, 1974; Wachtel et al., 2002; Green et al., 2003). Indeed, there are similarities in the cognitive impairments observed in psychosis and during cannabis use, and CB1 receptor activation has been shown to transiently exacerbate core psychotic and cognitive deficits in schizophrenia (Giuffrida et al., 2004; D’Souza et al., 2005). Clinical studies with at least one CB1 receptor antagonist have not proven effective in treating schizophrenic psychopathology (Meltzer et al., 2004). Nonetheless, such a result would be consistent with our results, because one might predict that complete blockade of emotional learning with a CB1 antagonist, as with abnormal potentiation of typically nonsalient stimuli with a CB1 agonist, would be profoundly disruptive to emotional learning processes. Indeed, cannabis use has been identified as a major risk factor in the development of schizophrenia (Arsenault et al., 2004; Semple et al., 2005). This evidence, combined with the reported abnormalities in CB1 receptor expression and in the levels of endogenous cannabinoids in schizophrenia subjects (Dean et al., 2001; Giuffrida et al., 2004; Zavitsanou et al., 2004), further implicates this system as a potential mediator of the aberrant emotional processing observed in this disorder.
Previous reports have implicated medial prefrontal cortical neurons as important mediators of the extinction of conditioned fear associations (Garcia et al., 1999), and specifically within the infralimbic cortex (Milad and Quirk, 2002; Milad et al., 2004). However, medial prefrontal cortical neurons display conditioned activity during the expression and acquisition of pavlovian fear associations (Baeg et al., 2001; Laviolette et al., 2005), whereas other cortical regions, such as the anterior cingulate, are critical for long-term emotional memory encoding (Frankland et al., 2004). The present results demonstrate that systemic or intra-mPFC microinfusions of a CB1 antagonist block the acquisition of a conditioned fear association. Initially, this result seems contradictory to previous findings with CB1 receptor knock-out mice (Marsicano et al., 2002). However, several important differences exist between these studies. First, we used an olfactory versus auditory cue in our associative conditioning assay. One possibility is that olfactory associative encoding is more sensitive to CB1 receptor blockade compared with other sensory modalities such as audition. Our behavioral effects were localized to the mPFC, whereas the above noted study consistently found that auditory tone presentation during extinction trials resulted in elevated levels of endocannabinoids specifically in the BLA complex, which may suggest preferential involvement of a population of BLA CB1 receptors in the extinction of auditory fear conditioning. Given that extinction requires “new learning,” one possibility is that CB1 receptors localized within the mPFc are required for neuronal encoding of this new association.
Our results demonstrate that functional input from the BLA to the mPFC is critical for emotional learning within neurons of the mPFC. Local CB1 receptor signaling within the mPFC or systemic activation of CB1 receptors is sufficient to potentiate emotional associative learning in behaving animals and in single neurons, whereas blockade of CB1 receptors in the mPFC is sufficient to prevent this associative learning. Interestingly, the potentiation of neuronal learning plasticity induced by CB1 receptor activation requires BLA input during the learning acquisition process, but once the associative learning has been encoded in the mPFC neuron, the plasticity effect no longer requires BLA input for its expression. This suggests that the association is transferred from the BLA to the mPFC for behavioral expression of the learned association.
We have reported previously that neuronal associative learning takes place within the BLA and that this learning can be modulated by descending input from the cortex (Rosenkranz and Grace, 2002; Rosenkranz et al., 2003). In this context, inputs from the cortex activate inhibitory BLA interneurons that in turn decrease the activity of the BLA principal neurons, blocking the neuronal encoding of associative learning (Rosenkranz and Grace, 2001). The ability of cannabinoids to modulate emotional learning within the mPFC may similarly depend on network interactions between pyramidal neurons and inhibitory interneurons. For example, at the cellular level, endocannabinoids can regulate local neuronal circuit interactions through depolarization-induced suppression of inhibition in which neuronal depolarization induces the local release of endogenous cannabinoids (Piomelli, 2003). Within the hippocampus, neocortex, and striatum, retrograde endocannabinoid signaling can activate inhibitory CB1 receptors on GABAergic interneurons, removing inhibitory input and increasing the excitability of these neurons (Piomelli, 2003; Trettel et al., 2004; Kofalvi et al., 2005). Thus, activation of CB1 receptors located on inhibitory cortical GABAergic interneurons may serve to remove tonic inhibition on principal neurons, increasing the excitability of this system and/or allowing a potentiation of inputs (e.g., from the BLA or mesolimbic DA system) onto a prefrontal cortical neuronal network that may be responsible for the encoding and integration of emotionally salient information. Indeed, recent anatomical evidence reports a localization of CB1 receptors on presynaptic GABAergic terminals within the mPFC forming symmetric synapses with cortical pyramidal neurons (Bodor et al., 2005). We have demonstrated recently that signaling through DA D4 receptors is also essential for emotional associative learning in neurons of the mPFC receiving functional BLA input (Laviolette et al., 2005). Combined with the present evidence that blockade of CB1 receptors also disrupts this learning, this suggests that mPFC CB1 and DA receptors may share a functional commonality within the mPFC in terms of the transmission of emotionally salient conditioned associations. We are presently exploring the possible functional interactions between DA D4 and CB1 receptor substrates in the mPFC in the modulation of emotional associative learning.
In summary, we demonstrate a critical role for cannabinoid signaling in the encoding, acquisition, and expression of behavioral and neuronal emotional associative learning. The identification of a CB1 receptor-mediated mechanism within the BLA–mPFC circuit that can modulate the amplitude of emotional associative learning may lead to improved understanding of how abnormalities in this system may be related to the sensory processing and emotional learning deficits observed in disorders such as schizophrenia and addiction. In addition, the present results provide the first neurobiological explanation that may account for the heightened emotional processing and sensory perception abnormalities reported in cannabis users and implicate the BLA–mPFC circuit as a critical mediator of the psychotropic effects of cannabis.
Footnotes
This work was supported by the Canadian Institutes of Health Research and a National Alliance for Research on Schizophrenia and Depression Young Investigator Award to S.R.L. and by United States Public Health Service Grants DA15408 and MH45156.
References
- Antoniadis EA, McDonald RJ (1999). Discriminative fear conditioning to context expressed by multiple measures of fear in the rat. Behav Brain Res 101:1–13. [DOI] [PubMed] [Google Scholar]
- Arsenault L, Cannon M, Witton J, Murray RM (2004). Causal association between cannabis and psychosis: examination of the evidence. Br J Psychiatry 184:110–117. [DOI] [PubMed] [Google Scholar]
- Baeg EH, Kim YB, Jang J, Kim HT, Mook-Jung I, Jung MW (2001). Fast spiking and regular spiking neural correlates of fear conditioning in the medial prefrontal cortex of the rat. Cereb Cortex 11:441–451. [DOI] [PubMed] [Google Scholar]
- Barbieri R, Quirk MC, Frank LM, Wilson MA, Brown EN (2001). Construction and analysis of non-Poisson stimulus-response models of neural spiking activity. J Neurosci Methods 105:25–37. [DOI] [PubMed] [Google Scholar]
- Berke J, Hernton C (1974). In: The cannabis experience: an interpretive study of the effects of marijuana and hashish London: Peter Owen.
- Bilkei-Gorzo A, Racz I, Valverde O, Otto M, Michel K, Sastre M, Zimm A (2005). Early age-related cognitive impairments in mice lacking cannabinoid CB1 receptors. Proc Natl Acad Sci USA 102:15670–15675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodor AL, Katona I, Nyiri G, Mackie K, Ledent C, Hajos N, Freund TF (2005). Endocannabinoid signaling in rat somatosensory cortex: laminar differences and involvement of specific interneuron types. J Neurosci 25:6845–6856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson G, Wang Y, Alger BE (2002). Endocannabinoids facilitate the induction of LTP in the hippocampus. Nat Neurosci 5:8–9. [DOI] [PubMed] [Google Scholar]
- Dean B, Sundram S, Bradbury R, Scarr E, Copolov D (2001). Studies on [3H] CP-55940 binding in the human central nervous system: regional specific changes in density of cannabinoid 1 receptors associated with schizophrenia and cannabis use. Neuroscience 103:9–15. [DOI] [PubMed] [Google Scholar]
- Degenetais E, Thierry AM, Glowinski J, Gioanni Y (2002). Electrophysiological properties of pyramidal neurons during surgery with propofol anesthesia. Br J Anaesth 92:171–177. [Google Scholar]
- D’Souza DC, Abi-Saab WM, Madonick S, Forselius-Bielen K, Doersch B, Brayley G, Gueorguiva R, Cooper TB, Krystal JH (2005). Delta-9-tetrahydrocannabinol effects in schizophrenia: implications for cognition, psychosis and addiction. Biol Psychiatry 57:594–608. [DOI] [PubMed] [Google Scholar]
- Frankland PW, Bontempi B, Talton LE, Kaczmarek L, Silva A (2004). The involvement of the anterior cingulated cortex in contextual fear memory. Science 304:829–830. [DOI] [PubMed] [Google Scholar]
- Garcia R, Vouimba RM, Baudry M, Thompson RF (1999). The amygdala modulates prefrontal cortex activity relative to conditioned fear. Nature 402:294–296. [DOI] [PubMed] [Google Scholar]
- Gingold AR, Bergasa NV (2003). The cannabinoid agonist WIN 55,212-2 increases nociception threshold in cholestatic rats: implications for the treatment of the pruritis of cholestasis. Life Sci 10:2741–2747. [DOI] [PubMed] [Google Scholar]
- Giuffrida A, Leweke FM, Gerth CW, Schreiber D, Koethe D, Faulhaber J, Klosterkotter J, Piomelli D (2004). Cerebrospinal anandamide levels are elevated in acute schizophrenia and are inversely correlated with psychotic symptoms. Neuropsychopharmacology 29:2108–2114. [DOI] [PubMed] [Google Scholar]
- Green B, Kavanagh D, Young R (2003). Being stoned: a review of self-reported cannabis effects. Drug Alcohol Rev 22:453–460. [DOI] [PubMed] [Google Scholar]
- Hohmann AG, Suplita RL, Bolton NM, Neely MH, Fegley D, Mangieri R, Krev JF, Walker JM, Holmes PV, Crystal JD, Duranti A, Tontini A, Mor M, Tarzia G, Piomelli D (2005). An endocannabinoid mechanism for stress-induced analgesia. Nature 435:1108–1112. [DOI] [PubMed] [Google Scholar]
- Holter SM, Kallnik M, Wurst W, Marsicano G, Lutz B, Wotjak CT (2005). Cannabinoid CB1 receptor is dispensable for memory extinction in an appetitively motivated learning task. Eur J Pharmacol 510:69–74. [DOI] [PubMed] [Google Scholar]
- Jackson ME, Homayoun H, Moghaddam B (2004). NMDA receptor hypofunction produces concomitant firing rate potentiation and burst activity reduction in the prefrontal cortex. Proc Natl Acad Sci USA 101:8467–8472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kofalvi A, Rodrigues RJ, Ledent C, Mackie K, Vizi ES, Cunha RA, Sperlagh B (2005). Involvement of cannabinoid receptors in the regulation of neurotransmitter release in the rodent striatum: a combined immunochemical and pharmacological analysis. J Neurosci 25:2874–2884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laviolette SR, Lipski WJ, Grace AA (2005). A subpopulation of neurons in the medial prefrontal cortex encodes emotional learning through burst and frequency codes through a dopamine D4 receptor-dependent basolateral amygdala input. J Neurosci 25:6066–6075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leweke FM, Giuffrida A, Wurster U, Emrich HM, Piomelli D (1999). Elevated endogenous cannabinoids in schizophrenia. NeuroReport 10:1665–1669. [DOI] [PubMed] [Google Scholar]
- Maren S, Quirk GJ (2004). Neuronal signaling of fear memory. Nat Rev Neurosci 5:844–852. [DOI] [PubMed] [Google Scholar]
- Marsicano G, Lutz B (1999). Expression of the cannabinoid receptor CB1 in distinct neuronal subpopulations in the adult mouse forebrain. Eur J Neurosci 11:4213–4225. [DOI] [PubMed] [Google Scholar]
- Marsicano G, Wotjak CT, Azad SC, Bisogno T, Rammes G, Cascio MG, Hermann H, Tang J, Hofmann G, Zieglansberger W, Di Marzo V, Lutz B (2002). The endogenous cannabinoid system controls extinction of aversive memories. Nature 418:530–534. [DOI] [PubMed] [Google Scholar]
- Martin M, Ledent C, Parmentier M, Maldonado R, Valverde R (2002). Involvement of CB1 receptors in emotional behaviour. Psychopharmacology 159:379–387. [DOI] [PubMed] [Google Scholar]
- Martin WJ, Coffin PO, Attias E, Balinsky M, Tsou K, Walker JM (1999). Anatomical basis for cannabinoid-induced antinociception as revealed by intracerebral microinjections. Brain Res 822:237–242. [DOI] [PubMed] [Google Scholar]
- Mcdonald AJ, Mascagni F (2001). Localization of the CB1 type cannabinoid receptor in the rat basolateral amygdala: high concentrations in a subpopulation of cholecystokinin-containing interneurons. Neuroscience 107:641–652. [DOI] [PubMed] [Google Scholar]
- Meltzer HY, Arvanitis L, Bauer D, Rein W (2004). Placebo-controlled evaluation of four novel compounds for the treatment of schizophrenia and schizoaffective disorder. Am J Psychiatry 161:975–984. [DOI] [PubMed] [Google Scholar]
- Milad MR, Quirk GJ (2002). Neurons in the medial prefrontal cortex signal memory for fear extinction. Nature 420:70–74. [DOI] [PubMed] [Google Scholar]
- Milad MR, Vidal-Gonzalez I, Quirk GJ (2004). Electrical stimulation of the medial prefrontal cortex reduces conditioned fear in a temporally specific manner. Behav Neurosci 118:389–394. [DOI] [PubMed] [Google Scholar]
- Moldrich G, Wenger T (2000). Localization of the CB1 cannabinoid receptor in the rat brain. An immunohistochemical study. Peptides 21:1735–1742. [DOI] [PubMed] [Google Scholar]
- Onaivi ES, Green MR, Martin BR (1990). Pharmacological characterization of cannabinoids in the elevated plus maze. J Pharmacol Exp Ther 253:1002–1009. [PubMed] [Google Scholar]
- O’Shea M, Singh ME, McGregor IS, Mallet PE (2004). Chronic cannabinoid exposure produces lasting memory impairment and increased anxiety in adolescent but not adult rats. J. Psychopharmacol 18:502–508. [DOI] [PubMed] [Google Scholar]
- Papanastassiou AM, Fields HL, Meng ID (2004). Local application of the cannabinoid receptor agonist, WIN 55,212-2, to spinal trigeminal nucleus caudalis differentially affects nociceptive and non-nociceptive neurons. Pain 107:267–275. [DOI] [PubMed] [Google Scholar]
- Patel S, Cravatt BF, Hillard CJ (2005). Synergistic interactions between cannabinoids and environmental stress in the activation of the central amygdala. Neuropsychopharmacology 30:497–507. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C (1997). In: The rat brain in stereotaxic coordinates Compact 3rd edition San Diego: Academic.
- Piomelli D (2003). The molecular logic of endocannabinoid signaling. Nat Rev Neurosci 4:873–884. [DOI] [PubMed] [Google Scholar]
- Robinson L, Hinder L, Pertwee RG, Riedel G (2003). Effects of delta-9-THC and WIN 55,212-2 on place preference in the water maze in rats. Psychopharmacology 166:40–50. [DOI] [PubMed] [Google Scholar]
- Rosenkranz JA, Grace AA (2001). Dopamine attenuates prefrontal cortical suppression of sensory inputs to the basolateral amygdala of rats. J Neurosci 21:4090–4103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenkranz JA, Grace AA (2002). Dopamine-mediated modulation of odor-evoked amygdala potentials during pavlovian conditioning. Nature 417:82–287. [DOI] [PubMed] [Google Scholar]
- Rosenkranz JA, Moore HM, Grace AA (2003). The prefrontal cortex regulates lateral amygdala neuronal plasticity and responses to previously conditioned stimuli. J Neurosci 23:11054–11064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semple DM, McIntosh AM, Lawrie SM (2005). Cannabis use as a risk factor for psychosis: systematic review. J Psychopharmacol 19:187–194. [DOI] [PubMed] [Google Scholar]
- Sullivan JM (2000). Cellular and molecular mechanisms underlying learning and memory impairments produced by cannabinoids. Learn Mem 7:132–139. [DOI] [PubMed] [Google Scholar]
- Trettel J, Fortin DA, Levine ES (2004). Endocannabinoid signaling selectively targets perisomatic inhibitory inputs to pyramidal neurons in juvenile mouse neocortex. J Physiol (Lond) 1:95–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsou K, Brown S, Sanudo-Pena MC, Mackie K, Walker JM (1998). Immunohistochemical distribution of cannabinoid CB1 receptors in the rat central nervous system. Neuroscience 83:393–411. [DOI] [PubMed] [Google Scholar]
- Varvel SA, Anum EA, Lichtman AH (2005). Disruption of CB1 receptor signaling impairs extinction of spatial memory in mice. Psychopharmacology 179:863–872. [DOI] [PubMed] [Google Scholar]
- Wachtel SR, Elsohly MA, Ross SA, Ambre J, de Wit H (2002). Comparison of the subjective effects of delta-9-tetrahydrocannabinol and marijuana use in humans. Psychopharmacology 161:331–339. [DOI] [PubMed] [Google Scholar]
- Zavitsanou K, Garrick T, Huang XF (2004). Selective antagonist SR141716A binding to cannabinoid CB1 receptors is increased in the anterior cingulated cortex in schizophrenia. Prog Neuropsychopharmacol Biol Psychiatry 28:355–360. [DOI] [PubMed] [Google Scholar]