

Abstract
The transcription factor cAMP response element-binding protein (CREB) is implicated in mediating the actions of chronic morphine in the locus ceruleus (LC), but direct evidence to support such a role is limited. Here, we investigated the influence of CREB on LC neuronal activity and opiate withdrawal behaviors by selectively manipulating CREB activity in the LC using viral vectors encoding genes for CREBGFP (wild-type CREB tagged with green fluorescent protein), caCREBGFP (a constitutively active CREB mutant), dnCREBGFP (a dominant-negative CREB mutant), or GFP alone as a control. Our results show that in vivo overexpression of CREBGFP in the LC significantly aggravated particular morphine withdrawal behaviors, whereas dnCREBGFP expression attenuated these behaviors. At the cellular level, CREBGFP expression in the LC in vivo and in vitro had no significant effect on neuronal firing at baseline but enhanced the excitatory effect of forskolin (an activator of adenylyl cyclase) on these neurons, which suggests that the cAMP signaling pathway in these neurons was sensitized after CREB expression. Moreover, in vitro studies showed that caCREBGFP-expressing LC neurons fired significantly faster and had a more depolarized resting membrane potential compared with GFP-expressing control cells. Conversely, LC neuronal activity was decreased by dnCREBGFP, and the neurons were hyperpolarized by this treatment. Together, these data provide direct evidence that CREB plays an important role in controlling the electrical excitability of LC neurons and that morphine-induced increases in CREB activity contribute to the behavioral and neural adaptations associated with opiate dependence and withdrawal.
Keywords: cAMP pathway, morphine, dependence, addiction, viral gene transfer, drug abuse
Introduction
The locus ceruleus (LC), the largest cluster of noradrenergic neurons in the brain, has served as a useful model of opiate action (Nestler and Aghajanian, 1997). Acutely, opiates decrease LC neuronal activity and inhibit activity of the cAMP signaling pathway in LC neurons. Chronically, these neurons show tolerance to opiates, as demonstrated by the return of their firing rates and cAMP signaling activity toward pretreatment levels during continuous opiate exposure, as well as dependence on opiates, as demonstrated by activation of LC firing rates and cAMP signaling levels after opiate withdrawal (Rasmussen et al., 1990; Kogan et al., 1992; Nestler, 1992; Ivanov and Aston-Jones, 2001). Several lines of evidence indicate that these opiate-induced adaptations in the LC contribute to opiate physical dependence and withdrawal. Local infusion of an opioid receptor antagonist into the LC induces opiate withdrawal (Koob et al., 1992). Moreover, blockade of LC neuronal activity or of the cAMP pathway specifically within the LC attenuates opiate physical dependence and withdrawal (Punch et al., 1997; Taylor et al., 1998). The LC is thought to exert these effects via its widespread projections to numerous CNS areas (Aston-Jones and Harris, 2004). Although the precise role of the LC in opiate dependence and withdrawal continues to be debated (Caille et al., 1999; Williams et al., 2001), studies of opiate effects in the LC have taught us a great deal about the molecular basis of opiate action in the nervous system, with similar changes in neural excitability and the cAMP pathway now documented in numerous brain regions (Nestler, 2004).
Indeed, studies of the LC were the first to implicate the cAMP-regulated transcription factor cAMP response element-binding protein (CREB) in opiate-induced neural plasticity. Opiates acutely decrease CREB phosphorylation (required for CREB activity) in the LC, CREB phosphorylation reverts to normal during continuous opiate exposure, and CREB phosphorylation and transcriptional activity increase above control levels after precipitation of opiate withdrawal (Guitart et al., 1992; Shaw-Lutchman et al., 2002). This phenomenon is partly mediated by chronic opiate induction of CREB expression in the LC (Widnell et al., 1994). Consistent with these biochemical data, Maldonado et al. (1996) showed that mice deficient in CREB exhibit markedly attenuated signs of physical opiate withdrawal, and we showed that local knockdown of CREB activity in the LC specifically, using CREB antisense oligonucleotides, caused a similar attenuation of certain withdrawal behaviors (Lane-Ladd et al., 1997). This antisense oligonucleotide treatment also prevented the opiate withdrawal-induced increase in LC neuronal activity (Lane-Ladd et al., 1997). However, these findings are limited by legitimate concerns about the potential toxicity and lack of specificity of antisense oligonucleotide treatments in vivo and by the lack of knowledge of what effect induction of CREB, as seen in response to chronic opiates, would have on LC neuronal activity. In the present study, we used viral-mediated gene transfer to more definitively investigate the effect of CREB in the LC on opiate physical dependence and withdrawal and on LC neuronal activity by selectively increasing or decreasing CREB function in the LC.
Materials and Methods
Animals.
Adult male Sprague Dawley rats (Charles River, Kingston, NY) were used for in vivo viral injections and drug treatments (initial weight, 200–250 g), and younger rats were used for slice cultures (25–30 d old).
Surgical procedures.
Stereotaxic surgery and intra-LC injections were performed as described previously (Lane-Ladd et al., 1997). Rats were used 24 h after viral injections. The injected viral vectors were as follows: herpes simplex virus (HSV)–green fluorescent protein (GFP) or HSV-LacZGFP as controls; and HSV-CREBGFP (CREB tagged with GFP), HSV-dnCREBGFP (a dominant-negative mutant of CREB that lacks Ser 133; also known as mCREB), and HSV-caCREBGFP (a constitutively active form of CREB that represents a CREB-VP16 fusion protein). These vectors have been used successfully in previous studies to produce the expected changes in CREB-mediated transcription (Barrot et al., 2002; Chao et al., 2002; Olson et al., 2005).
Drug treatments.
After bilateral injection of HSV vectors, morphine-dependent rats were produced as described by Rasmussen et al. (1990). Briefly, rats were given daily subcutaneous implantations of sham or morphine pellets for 3 d. On day 4, they were given injections of naltrexone (100 mg/kg in 0.9% saline, s.c.) and housed individually for assessing withdrawal behaviors exactly as described previously (Rasmussen et al., 1990; Lane-Ladd et al., 1997).
Slice cultures.
Slice cultures were prepared according to Stoppini et al. (1991). Acute brain slices (250 μm) containing the LC were obtained in sucrose–artificial CSF (ACSF) (see below, Electrophysiological recordings). After a 1 h incubation, HSV vectors were pipetted onto the LC area of the slice surface. Slices were maintained overnight at 34°C. The culture medium used in this study was MEM (Invitrogen, Gaithersburg, MD) containing 30 mm HEPES, 20 mm d-glucose, 5% B27, 5.0 mm l-glutamine, and 25 U/ml streptomycin/penicillin. We tested several culture media used previously by other investigators (Stoppini et al., 1991). However, the tonic firing of LC neurons was lost in those media. Our culture medium successfully maintained stable firing of these neurons.
Electrophysiological recordings.
Recordings were obtained from LC neurons in slice cultures and in acute brain slices from HSV vector-injected rats. Acute brain slices at the level of the LC were prepared as described previously (Lane-Ladd et al., 1997). The ACSF contained the following (in mm): 128 NaCl, 3 KCl, 1.25 NaH2PO4, 10 d-glucose, 24 NaHCO3, 2 CaCl2, and 2 MgSO4, oxygenated with 95% O2 and 5% CO2 (pH 7.35, 295–305 mOsm). Sucrose ACSF was derived by fully replacing NaCl with 254 mm sucrose in ACSF. Patch pipettes (3–5 MΩ) for whole-cell and partial access recordings (see below, Validation of partial access recording) were filled with an internal solution containing the following (in mm): 115 potassium gluconate, 20 KCl, 10 HEPES, 1.0 EGTA, 4 ATP-Mg, and 0.3 GTP (pH 7.2, 280–290 mOsm). The firing rate and resting membrane potential (RMP) of LC neurons were recorded in bridge mode of amplifier AxoClamp 2B (Molecular Devices, Union City, CA), and data acquisition was realized with pClamp 8.2 (Molecular Devices). All data in this study are presented as means ± SEM. Except for forskolin (Calbiochem, La Jolla, CA), all reagents were purchased from Sigma (St. Louis, MO).
Validation of partial access recording.
In this study, the partial access recording mode was used to measure the firing rate and membrane potential (MP) of LC neurons, because it was associated with less disturbance of the neuronal milieu. This recording mode was obtained as described by Alreja and Aghajanian (1995). After making a giga seal, the partial access recording mode was established by maintaining the giga seal for a short period (3–5 min). Regular spontaneous spikes of LC neurons could be seen clearly after the mode establishment. The partial access mode was characterized with a high access resistance (∼40 MΩ) and small spike amplitude (20–50 mV) compared with that of whole-cell recordings in LC neurons (Alreja and Aghajanian, 1991). The internal solution in the recording pipette had full access to cytoplasm in whole-cell recordings (access resistance, 4–6 MΩ; spike amplitude, 80–100 mV). Most LC neurons tested in our study became silent with whole-cell recordings, because of a time-dependent dialysis of cytoplasm by the pipette solution, as observed in previous studies (Alreja and Aghajanian, 1991, 1995). Therefore, the whole-cell recording mode was not suitable for measuring the effect of CREB on the frequency of pacemaker activity in LC neurons. In contrast, the partial access mode provided only partial access to cytoplasm. The tested neurons were less disturbed by the pipette solution, and the highly stable basal firing of these neurons could be recorded using this mode (Fig. 1). Moreover, we found that the MP of LC neurons could be reliably measured after the partial access was stable (after 10–15 min) and remained constant throughout the remainder of the recording period (Fig. 1B,C). The MP measured with the partial access mode was not significantly different from the RMP measured by whole-cell recordings (55.9 ± 0.9 mV, n = 16 vs 55.5 ± 1.5 mV, n = 11; p > 0.5) (Fig. 1D). These results suggest that the MP obtained with partial access was a close measurement of true whole-cell RMP in LC neurons. It should be pointed out that the MP recorded in partial access was not derived during the resting state of the neurons. This is because tetrodotoxin (TTX) induced obvious depolarization of LC neurons as seen by other investigators (Ishimatsu et al., 2002; Jedema and Grace, 2004). Moreover, there were calcium spikes in LC neurons in the presence of TTX (Ivanov and Aston-Jones, 2001; Han and Nestler, 2005). The MP and firing rates measured here were in the normal range for LC neurons (Alreja and Aghajanian, 1993; Van den Pol et al., 2002). Finally, Figure 1 shows that the amplitude of spikes exhibited by LC neurons varied as a function of recording time during partial access, with spike amplitudes increasing over time presumably as a consequence of gradually decreasing access resistance. However, Figure 1C also clearly demonstrates that both the firing rate and MP of the neurons remained extremely stable despite the changing spike amplitude during the course of the experiment. Indeed, the opposite effect of caCREBGFP and of dnCREBGFP on the LC firing rate and MP were apparent throughout the entire recording period (data not shown).
Figure 1.

Measurement of LC neuronal firing rates and MP using the partial access recording mode. A, Top, Sample trace of partial access recording. The MP became stable with the development of partial access compared with the beginning of recording (within 10–15 min). Bottom, Sample trace of on-line firing rate analysis with pClamp (analysis on at ∼4 min after making giga-seal). The firing rate remained stable during the measurement (>60 min), although the amplitude of spikes changed. B, Sample traces in a 20 s window at 15, 40, and 60 min time points from A. MPs were −58.1, −57.6, and −58.3 mV. Note that the firing rate and MP were extremely stable, although the amplitude of spikes progressively increased over time. C, Despite the shifting spike amplitude, the firing rate and MP remained constant throughout the experiment. D, MP measured with the partial access mode is not different from the RMP recorded using the whole-cell mode (n = 16 and 11, respectively; p > 0.5).
Results
In vivo manipulations of CREB activity in the LC regulate morphine withdrawal
In a previous study, intra-LC infusion of antisense oligonucleotides to CREB was shown to attenuate certain behaviors of opiate withdrawal (Lane-Ladd et al., 1997). However, it has remained unknown whether morphine withdrawal can be influenced by upregulation of CREB activity in the LC. This is an important question, because chronic opiate exposure and induction of withdrawal robustly increases CREB function in the LC (see Introduction). To systematically investigate the role of CREB in the regulation of opiate withdrawal, we used HSV vectors expressing GFP, CREBGFP, or dnCREBGFP to manipulate CREB activity in the LC. The activity of these vectors on CREB-mediated transcription has been verified both in vivo and in vitro (see Materials and Methods). Here, we further verified these vectors by showing that HSV-CREBGFP increased (134 ± 19% of HSV-GFP) whereas HSV-dnCREBGFP decreased (60 ± 25% of HSV-GFP) expression of tyrosine hydroxylase, a well established CREB target, in the LC (n = 4; p < 0.05).
Rats received morphine pellet implantations after bilateral injection of vehicle, HSV-GFP, HSV-CREBGFP, or HSV-dnCREB into the LC. This method allows the highly selective targeting of HSV vectors to the LC (Fig. 2A–D). Opiate withdrawal was then induced by administration of naltrexone, an opioid receptor antagonist. We found that certain signs of withdrawal (e.g., wet dog shakes and ptosis) were aggravated by CREBGFP expression compared with vehicle and GFP control, whereas these withdrawal behaviors showed significant attenuation after dnCREBGFP expression (Fig. 2E). Teeth chatter, another sign of withdrawal, showed similar regulation: worsening by CREBGFP (2.5 ± 0.2; p < 0.05) and attenuation by dnCREBGFP (1.2 ± 0.2; p < 0.05) compared with vehicle (2.1 ± 0.2) and GFP (2.0 ± 0.4) controls. Other signs, including lacrimation (Fig. 2E) and irritability, piloerection, and diarrhea (data not shown), were not affected by these treatments. The attenuated opiate withdrawal caused by dnCREBGFP is consistent with the previous findings using CREB antisense oligonucleotide infused into the LC (Lane-Ladd et al., 1997), whereas the exacerbation of withdrawal caused by CREBGFP demonstrated that upregulation of CREB in the LC is capable of worsening opiate physical dependence and withdrawal.
Figure 2.

Effect of in vivo manipulation of CREB activity in the LC of morphine-dependent rats on opiate withdrawal. A, Vehicle or viral injection target sites are shown in orange. B, GFP expression in the injected site of a representative animal. C, Map matching the injected brain region in B. 4V, Fourth ventricle. D, GFP-expressing neurons in an injected LC. E, Bar graphs showing morphine withdrawal scores as function of CREB manipulations. n = 12 animals in each group. ∗p < 0.05.
In vivo dnCREBGFP expression decreases the firing rate of LC neurons
To examine the cellular mechanisms underlying the regulation of opiate withdrawal behaviors by CREB, electrophysiological recordings were performed using brain slices obtained from drug-naive rats 24 h after bilateral viral injection into the LC. Here, we focused on LC neuronal firing rates, which have been shown to reflect aspects of opiate tolerance, dependence, and withdrawal (see Introduction). Our results showed that HSV-mediated expression of a LacZGFP fusion protein in LC neurons had no effect on their firing rate compared with uninfected (GFP−) neighboring neurons in the same slices, or to neurons in slices from uninjected rats (Fig. 3). Neurons expressing dnCREBGFP fired at dramatically lower rates compared with dnCREBGFP− neighboring neurons from the same brain slices or the other controls mentioned above (Fig. 3). This finding is consistent with previous observations using CREB antisense oligonucleotides (Lane-Ladd et al., 1997). In contrast, CREBGFP expression in LC neurons had no effect on their firing rate compared with controls (Fig. 3).
Figure 3.

Effect of in vivo expression of CREBGFP or dnCREBGFP on firing rates of LC neurons in brain slices. A, Sample recordings from LC neurons of control rats (untreated and LacZGFP injection) and rats treated with CREBGFP or dnCREBGFP. The basal firing rate of GFP+ neurons from dnCREB-injected rats was decreased. B, Bar graphs show that dnCREBGFP+ neurons fired significantly lower compared with GFP− neighboring neurons in the same slices, to neurons transfected with a control virus (LacZGFP), and to neurons from untreated slices (n = 42 for untreated control; n = 7–12 in each other group). ∗∗∗p < 0.001. C, Cumulative probability plot of LC firing rates from dnCREBGFP-, LacZGFP-, and CREBGFP-expressing neurons (n = 10–12 per group).
The excitatory effect of forskolin on LC neurons is enhanced by in vivo expression of CREBGFP
Several groups have shown that activation of the cAMP signaling pathway in LC neurons increases neuronal firing (North et al., 1987; Alreja and Aghajanian, 1995; Ivanov and Aston-Jones, 2001). Accordingly, evidence suggests that upregulation of the cAMP pathway in the LC caused by chronic opiate exposure contributes to the increased firing of the neurons seen under these conditions (Kogan et al., 1992; Ivanov and Aston-Jones, 2001). Because previous work implicated CREB in controlling the activity of the cAMP pathway in the LC (Lane-Ladd et al., 1997), we examined the sensitivity of the cAMP pathway in LC neurons after in vivo overexpression of CREBGFP. Forskolin (an activator of adenylyl cyclase) increased the basal firing rate of LC neurons significantly more in CREBGFP+ cells than in CREBGFP− neurons (Fig. 4A,B). In contrast, forskolin increased basal LC firing to an identical extent in LacZGFP-expressing or nonexpressing neurons (Fig. 4C). Similar results were observed in vitro (using a slice culture system described below): responses of LC neurons to forskolin were sensitized by CREBGFP expression over a dose range of the drug (Fig. 4D). These results demonstrate that, although CREB overexpression does not alter basal activity of LC neurons, it is sufficient for enhancing the excitatory effect of forskolin on these cells.
Figure 4.
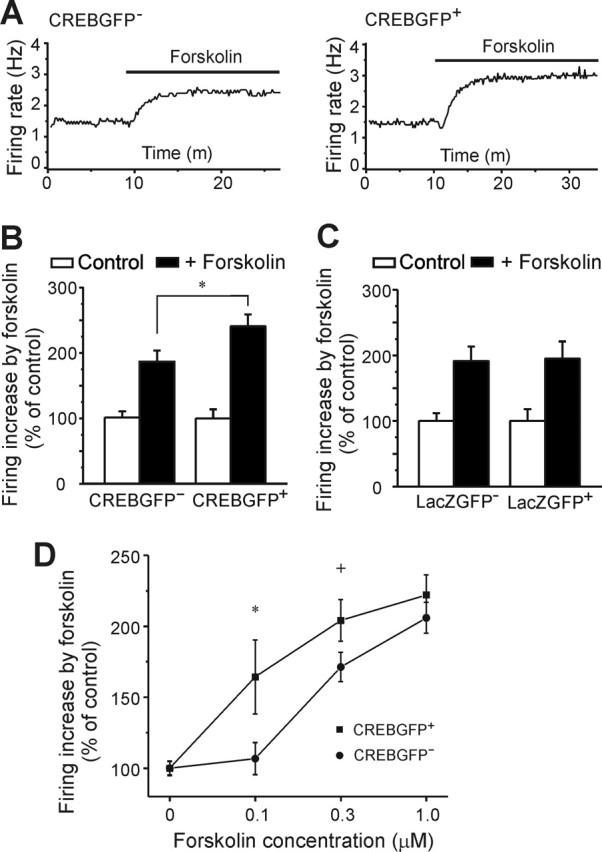
Effect of CREBGFP expression in vivo (A–C) or in vitro (D) on forskolin activation of LC neurons. A, Sample traces show the firing rate increase in CREBGFP− and CREBGFP+ neurons in response to 0.3 μm forskolin in acute slices. B, Bar graph shows that forskolin induces a greater increase in the firing rate in CREBGFP+ neurons than in CREBGFP− neurons (n = 15). ∗p < 0.05. C, In vivo expression of LacZGFP has no effect, with similar forskolin-induced increases in the LC neuronal firing rate in LacZGFP− and LacZGFP+ neurons (n = 10–15; p > 0.1). D, In vitro expression of CREBGFP increases the response of LC neurons in slice cultures to forskolin over a dose range of the drug (∗p < 0.05; +p = 0.06; n = 33–65).
In vitro expression of caCREBGFP and dnCREBGFP has opposite effects on LC neuronal excitability
To further investigate the relationship between CREB and LC neuronal activity, we developed a slice culture system in which LC neurons in brain slices in vitro could be infected with HSV vectors. Acute brain slices could not be used for this purpose, because HSV vectors require 12–24 h to express their encoded transgenes and LC neurons do not remain healthy under such conditions. Conditions used for slice cultures of other brain regions were not suitable for the LC, because LC neurons completely lost their pacemaker activity after a few hours in culture. However, by altering the culturing conditions (see Materials and Methods), we succeeded in preparing slice cultures of LC neurons in which the cells showed a stable level of regular tonic firing for up to 48 h (Fig. 5A–D). As a control experiment, the firing rate of LC neurons in untreated slice cultures was compared with that in LC neurons expressing GFP. There was no difference between these two groups of neurons (Fig. 5D,E). Expression of dnCREBGFP in LC neurons dramatically decreased their firing rate compared with controls, which is consistent with the effect seen with dnCREBGFP in vivo (Fig. 5D,E,G). Conversely, LC neuronal firing rates were greatly increased by overexpression of caCREBGFP (Fig. 5D,E,G). The RMP of the neurons was also measured in each group. Expression of caCREB depolarized the cells compared with controls, whereas dnCREB hyperpolarized the LC neurons (Fig. 5D,F,H).
Figure 5.
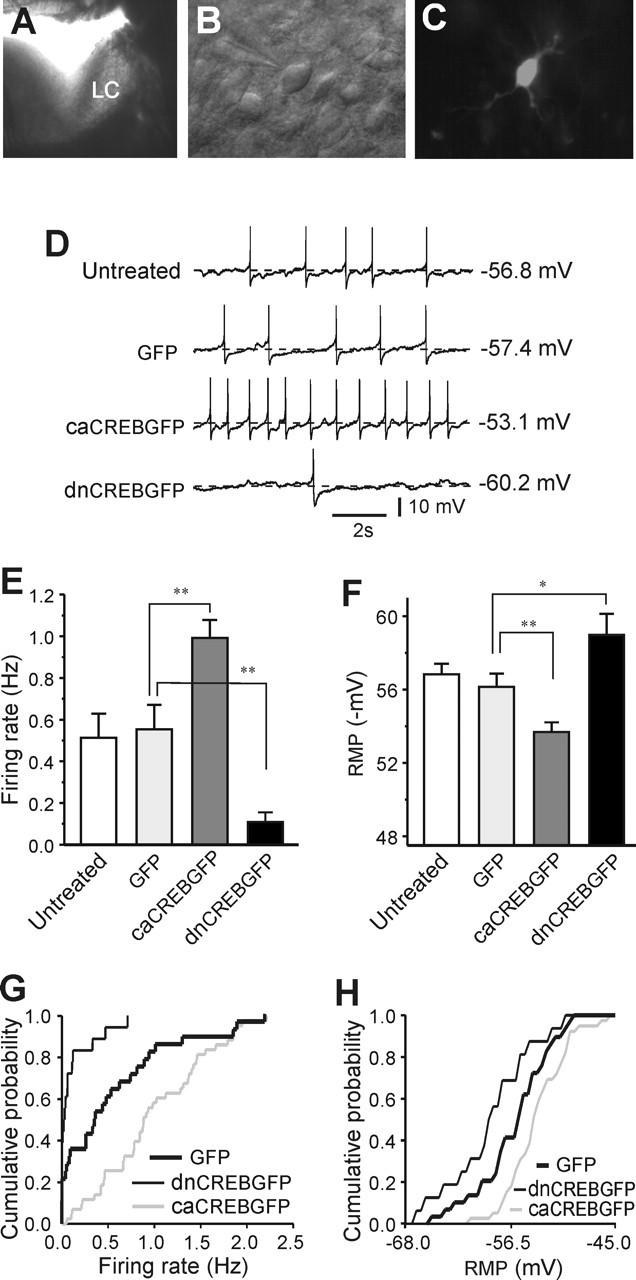
Opposite effects of in vitro expression of caCREBGFP and dnCREBGFP on LC neuronal excitability. A, Photograph under dissection microscope showing an overnight-cultured slice with the LC, on which an HSV vector expressing GFP was pipetted. B, LC neurons in a slice culture. C, A caCREBGFP+ neuron in a slice culture. D, Sample recordings were from untreated, GFP-treated, caCREBGFP-treated, or dnCREBGFP-treated slice cultures. E, There was no difference in the firing rate between untreated cells and neurons expressing GFP (n = 20–28; p > 0.1). The firing rate of caCREBGFP-expressing neurons was dramatically increased compared with GFP control neurons (n = 43 in caCREBGFP group), whereas dnCREBGFP expression decreased the LC firing rate (n = 18 in dnCREBGFP group). F, The RMP was also reciprocally regulated by caCREBGFP and dnCREBGFP (n = 16–32 neurons per group). G, Cumulative probability plot of the firing rate from dnCREBGFP-, GFP-, and caCREBGFP-expressing LC neurons (n = 18–43). H, Cumulative probability plot of the RMP from dnCREBGFP-, GFP-, and caCREBGFP-expressing LC neurons (n = 16–32). ∗p < 0.05; ∗∗p < 0.01.
Discussion
The involvement of CREB in the chronic actions of opiates was hypothesized originally based on the observation that prolonged exposure to opiates upregulates the cAMP signaling pathway in the LC (Guitart et al., 1992; Nestler, 1992). Because CREB is known to mediate many of the effects of cAMP on gene expression, it was hypothesized that CREB may be an important mediator of opiate-induced changes in gene expression in the LC that contribute to a state of opiate dependence (Nestler and Aghajanian, 1997; Nestler, 2004). In the present study, using viral-mediated gene transfer, we provide several important new lines of evidence to support this scheme. We demonstrate that upregulating CREB activity in the LC aggravates opiate dependence and withdrawal, enhances the excitatory effect of forskolin on LC neurons, increases the LC neuronal firing rate, and depolarizes these neurons. In contrast, downregulating CREB activity in the LC opposes opiate dependence and withdrawal, decreases LC firing rate, and hyperpolarizes the neurons. These observations support a scheme whereby the activity of CREB is a key control point for the excitability of LC neurons and raise the possibility that chronic opiates, by altering CREB function, induce changes in LC neurons that are part of the mechanisms underlying opiate dependence and withdrawal.
The observation that CREB overexpression increases forskolin-induced activation of LC neurons suggests that among the many target genes for CREB in these cells are components of the cAMP pathway. Indeed, we and others have shown previously that chronic opiate administration upregulates the cAMP pathway in the LC, including increased expression of two isoforms of adenylyl cyclase, ACI and ACVIII, and of the catalytic and regulatory subunits of protein kinase A (Lane-Ladd et al., 1997; Chao et al., 2002). Of these various cAMP signaling proteins, only ACVIII appears to be a direct target for CREB (Lane-Ladd et al., 1997). Consistent with these observations, CREB, or caCREB to an even greater extent, induces ACVIII promoter activity, whereas dnCREB inhibits promoter activity, both in vitro and within the brain in vivo (Chao et al., 2002). Because activation of the cAMP pathway excites LC neurons (Wang and Aghajanian, 1987; Alreja and Aghajanian, 1995; Ivanov and Aston-Jones, 2001), these observations support the hypothesis that chronic opiate exposure, through the induction of CREB and the consequent induction of ACVIII, increases LC neuronal excitability. Additional work is needed to directly investigate this possibility.
It is likely that many additional genes are involved in the effects of CREB on LC neuronal excitability. A recent microarray study identified numerous genes that are upregulated or downregulated in the rat and mouse LC in response to chronic opiate administration (McClung et al., 2005). Interestingly, ∼20% of these opiate-regulated genes are known to be regulated by cAMP or CREB. Several of these genes (e.g., the α1 subunit of Na+/K+ ATPase and the GluR1 AMPA glutamate receptor subunit) would be expected, like ACVIII, to alter LC excitability. Additional work is needed to study the role of these and other genes as downstream targets of CREB that mediate the excitatory effect of CREB on the LC. The ionic mechanisms underlying the tonic firing activity of these neurons, and their activation after stimulation of the cAMP pathway, remain unclear (North et al., 1987; Wang and Aghajanian, 1990; Ivanov and Aston-Jones, 2001; Alreja and Aghajanian, 1993). We were unable to identify the ionic mechanisms by which CREB regulates LC neuronal activity, because the effects of caCREB and dnCREB on the RMP were lost in the whole-cell configuration. This is not surprising, because rundown of the cAMP pathway occurs under these conditions (Alreja and Aghajanian, 1995). This important question, therefore, must await future investigations.
Despite the large number of studies that have implicated diverse roles for CREB in the regulation of neural and behavioral plasticity (Carlezon et al., 2005), there have been surprisingly few studies that have directly examined the influence of CREB on neuronal activity per se. A recent study by Marie et al. (2005), using viral vector techniques similar to those used here, demonstrated that CREB promotes glutamatergic transmission mediated via NMDA glutamate receptors at excitatory synapses in the hippocampus. A preliminary study of nucleus accumbens neurons indicates that CREB also promotes the excitability of these cells, although via distinct mechanisms possibly involving regulation of voltage-gated Na+, K+, and Ca2+ channels (Dong et al., 2006). Our findings show that CREB exerts a similar, excitatory effect on LC neurons, although apparently via still distinct mechanisms. These studies are important, because they show that CREB tends to exert the same net functional effect (enhancing excitability) in numerous neuronal cell types, although perhaps via distinct mechanisms in each cell type. As we identify these ionic mechanisms, we will gain an increasingly complete understanding, at the detailed molecular level, by which chronic perturbations such as opiates induce long-term changes in the brain. These studies are a critical step in our efforts to identify the molecular and cellular basis of neural and behavioral plasticity.
Footnotes
This work was supported by grants from the National Institute on Drug Abuse. We thank Sarah B. Lane-Ladd for technical assistance.
References
- Alreja M, Aghajanian GK (1991). Pacemaker activity of locus coeruleus neurons: whole-cell recordings in brain slices show dependence on cAMP and protein kinase A. Brain Res 556:339–343. [DOI] [PubMed] [Google Scholar]
- Alreja M, Aghajanian GK (1993). Opiates suppress a resting sodium-dependent inward current and activate an outward potassium current in locus coeruleus neurons. J Neurosci 13:3525–3532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alreja M, Aghajanian GK (1995). Use of the whole-cell patch-clamp method in studies on the role of cAMP in regulating the spontaneous firing of locus coeruleus neurons. J Neurosci Methods 59:67–75. [DOI] [PubMed] [Google Scholar]
- Aston-Jones G, Harris GC (2004). Brain substrates for increased drug seeking during protracted withdrawal. Neuropharmacology 47:[Suppl 1], 167–179. [DOI] [PubMed] [Google Scholar]
- Barrot M, Olivier JD, Perrotti LI, DiLeone RJ, Berton O, Eisch AJ, Impey S, Storm DR, Neve RL, Yin JC, Zachariou V, Nestler EJ (2002). CREB activity in the nucleus accumbens shell controls gating of behavioral responses to emotional stimuli. Proc Natl Acad Sci USA 99:11435–11440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caille S, Espejo EF, Reneric JP, Cador M, Koob GF, Stinus L (1999). Total neurochemical lesion of noradrenergic neurons of the locus ceruleus does not alter either naloxone-precipitated or spontaneous opiate withdrawal nor does it influence ability of clonidine to reverse opiate withdrawal. J Pharmacol Exp Ther 290:881–892. [PubMed] [Google Scholar]
- Carlezon WA Jr, Duman RS, Nestler EJ (2005). The many faces of CREB. Trends Neurosci 28:436–445. [DOI] [PubMed] [Google Scholar]
- Chao JR, Ni YG, Bolanos CA, Rahman Z, DiLeone RJ, Nestler EJ (2002). Characterization of the mouse adenylyl cyclase type VIII gene promoter: regulation by cAMP and CREB. Eur J Neurosci 16:1284–1294. [DOI] [PubMed] [Google Scholar]
- Dong Y, Green T, Saal D, Marie H, Neve R, Nestler EJ, Malenka RC (2006). CREB modulates excitability of nucleus accumbens neurons. Nat Neurosci 9:475–477. [DOI] [PubMed] [Google Scholar]
- Guitart X, Thompson MA, Mirante CK, Greenberg ME, Nestler EJ (1992). Regulation of cyclic AMP response element-binding protein (CREB) phosphorylation by acute and chronic morphine in the rat locus coeruleus. J Neurochem 58:1168–1171. [DOI] [PubMed] [Google Scholar]
- Han MH, Nestler EJ (2005). Persistent sodium and calcium-activated chloride currents in rat locus ceruleus: roles in pacemaker activity. Soc Neurosci Abstr 31:971–15. [Google Scholar]
- Ishimatsu M, Kidani Y, Tsuda A, Akasu T (2002). Effects of methylphenidate on the membrane potential and current in neurons of the rat locus coeruleus. J Neurophysiol 87:1206–1212. [DOI] [PubMed] [Google Scholar]
- Ivanov A, Aston-Jones G (2001). Local opiate withdrawal in locus coeruleus neurons in vitro. J Neurophysiol 85:2388–2397. [DOI] [PubMed] [Google Scholar]
- Jedema HP, Grace AA (2004). Corticotropin-releasing hormone directly activates noradrenergic neurons of the locus ceruleus recorded in vitro. J Neurosci 24:9703–9713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kogan JH, Nestler EJ, Aghajanian GK (1992). Elevated basal firing rates and enhanced responses to 8-Br-cAMP in locus coeruleus neurons in brain slices from opiate-dependent rats. Eur J Pharmacol 211:47–53. [DOI] [PubMed] [Google Scholar]
- Koob GF, Maldonado R, Stinus L (1992). Neural substrates of opiate withdrawal. Trends Neurosci 15:186–191. [DOI] [PubMed] [Google Scholar]
- Lane-Ladd SB, Pineda J, Boundy VA, Pfeuffer T, Krupinski J, Aghajanian GK, Nestler EJ (1997). CREB (cAMP response element-binding protein) in the locus coeruleus: biochemical, physiological, and behavioral evidence for a role in opiate dependence. J Neurosci 17:7890–7901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maldonado R, Blendy JA, Tzavara E, Gass P, Roques BP, Hanoune J, Schutz G (1996). Reduction of morphine abstinence in mice with a mutation in the gene encoding CREB. Science 273:657–659. [DOI] [PubMed] [Google Scholar]
- Marie H, Morishita W, Yu X, Calakos N, Malenka RC (2005). Generation of silent synapses by acute in vivo expression of CaMKIV and CREB. Neuron 45:741–752. [DOI] [PubMed] [Google Scholar]
- McClung CA, Nestler EJ, Zachariou V (2005). Regulation of gene expression by chronic morphine and morphine withdrawal in the locus ceruleus and ventral tegmental area. J Neurosci 25:6005–6015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nestler EJ (1992). Molecular mechanisms of drug addiction. J Neurosci 12:2439–2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nestler EJ (2004). Historical review: molecular and cellular mechanisms of opiate and cocaine addiction. Trends Pharmacol Sci 25:210–218. [DOI] [PubMed] [Google Scholar]
- Nestler EJ, Aghajanian GK (1997). Molecular and cellular basis of addiction. Science 278:58–63. [DOI] [PubMed] [Google Scholar]
- North RA, Williams JT, Surprenant A, Christie MJ (1987). Mu and delta receptors belong to a family of receptors that are coupled to potassium channels. Proc Natl Acad Sci USA 84:5487–5491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson VG, Zabetian CP, Bolanos CA, Edwards S, Barrot M, Eisch AJ, Hughes T, Self DW, Neve RL, Nestler EJ (2005). Regulation of drug reward by cAMP response element-binding protein: evidence for two functionally distinct subregions of the ventral tegmental area. J Neurosci 25:5553–5562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Punch LJ, Self DW, Nestler EJ, Taylor JR (1997). Opposite modulation of opiate withdrawal behaviors on microinfusion of a protein kinase A inhibitor versus activator into the locus coeruleus or periaqueductal gray. J Neurosci 17:8520–8527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen K, Beitner-Johnson DB, Krystal JH, Aghajanian GK, Nestler EJ (1990). Opiate withdrawal and the rat locus coeruleus: behavioral, electrophysiological, and biochemical correlates. J Neurosci 10:2308–2317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw-Lutchman TZ, Barrot M, Wallace T, Gilden L, Zacharou V, Impey S, Duman RS, Storm D, Nestler EJ (2002). Regional and cellular mapping of cAMP response element-mediated transcription during naltrexone-precipitated morphine withdrawal. J Neurosci 22:3663–3672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoppini L, Buchs PA, Muller D (1991). A simple method for organotypic cultures of nervous tissue. J Neurosci Methods 37:173–182. [DOI] [PubMed] [Google Scholar]
- Taylor JR, Punch LJ, Elsworth JD (1998). A comparison of the effects of clonidine and CNQX infusion into the locus coeruleus and the amygdala on naloxone-precipitated opiate withdrawal in the rat. Psychopharmacology 138:133–142. [DOI] [PubMed] [Google Scholar]
- Van den Pol AN, Ghosh PK, Liu RJ, Li Y, Aghajanian GK, Gao XB (2002). Hypocretin (orexin) enhances neuron activity and cell synchrony in developing mouse GFP-expressing locus coeruleus. J Physiol (Lond) 541:169–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YY, Aghajanian GK (1987). Excitation of locus coeruleus neurons by an adenosine 3′,5′-cyclic monophosphate-activated inward current: extracellular and intracellular studies in rat brain slices. Synapse 1:481–487. [DOI] [PubMed] [Google Scholar]
- Wang YY, Aghajanian GK (1990). Excitation of locus coeruleus neurons by vasoactive intestinal peptide: role of cAMP and protein kinase A. J Neurosci 10:3335–3343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Widnell KL, Russel DS, Nestler EJ (1994). Regulation of expression of cAMP response-binding protein in the locus coeruleus in vivo and in a locus coeruleus-like cell line in vitro. Proc Natl Acad Sci USA 91:10947–10951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams JT, Christie MJ, Manzoni O (2001). Cellular and synaptic adaptations mediating opioid dependence. Physiol Rev 81:299–343. [DOI] [PubMed] [Google Scholar]