

Abstract
Oxidative damage to dopaminergic nigrostriatal (DNS) neurons plays a central role in the pathogenesis of Parkinson's disease (PD). Glucose-6-phosphate dehydrogenase (G6PD) is a key cytoprotective enzyme that provides NADPH, the major source of the reducing equivalents of a cell. Mutations of this enzyme are the most common enzymopathies worldwide. We have studied in vivo the role of G6PD overexpressed specifically in the DNS pathway and show that the increase of G6PD activity in the soma and axon terminals of DNS neurons, separately from other neurons or glial cells, protects them from parkinsonism. Analysis of DNS neurons by histological, neurochemical, and functional methods showed that even a moderate increase of G6PD activity rendered transgenic mice more resistant than control littermates to the toxic effects of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP). The neuroprotective action of G6PD was also observed in aged animals despite that they had a greater susceptibility to MPTP. Therefore, overexpression of G6PD in dopaminergic neurons or pharmacological activation of the native enzyme should be considered as potential therapeutic strategies to PD.
Keywords: dopaminergic nigrostriatal neurons, glucose-6-phosphate dehydrogenase, transgenic mice, neurodegeneration, experimental parkinsonism, gene therapy
Introduction
The main neuropathological feature of Parkinson's disease (PD) is the death of dopaminergic neurons in the substantia nigra (SN) projecting to the striatum. The mechanism of neuronal death in sporadic PD, although unknown, is likely multifactorial, involving a cascade of events among which the oxidative damage of a cell appears to have a prominent role (Dauer and Przedborski, 2003; Dawson and Dawson, 2003; Andersen, 2004; Bossy-Wetzel et al., 2004). Selective decreases in reduced glutathione (GSH) (Perry and Yong, 1986; Pearce et al., 1997), mitochondrial complex I activity (Parker et al., 1989; Schapira et al., 1990; Tieu et al., 2004) and reactive oxygen species (ROS)-destroying enzymes (Kunikowska and Jenner, 2003) or elevated concentrations of iron, which can act as a catalyst for detrimental oxidative reactions (Dexter et al., 1987; Sofic et al., 1991), have been reported within the parkinsonian SN. In addition, there is evidence of oxidative damage to lipids, proteins, and DNA in the brains and leukocytes of PD patients (Alam et al., 1997; Migliore et al., 2002). Inflammatory-related events, such as nitric oxide (NO)-derived reactive species produced by glial inducible NO synthase (iNOS), appear also to participate in SN dopaminergic degeneration (Liberatore et al., 1999; Chung et al., 2004; Yao et al., 2004). Indeed, the effects of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), a toxic agent used to produce experimental parkinsonism, appear to depend on its biotransformation to MPP+, a potent mitochondria complex I inhibitor taken up by the plasma-membrane dopamine transporter (DAT) (Langston et al., 1983; Beal, 2001).
Given the importance of ROS in the pathophysiology of PD, stimulation of antioxidant defense could be a promising therapeutic tool. Transgenic mice with general cellular overexpression of any of the H2O2 metabolizing enzymes, glutathione peroxidase (GPx), cytosolic Cu,Zn-superoxide dismutase (SOD1) and mitochondrial Mn-SOD (SOD2) are more resistant to the toxic effects of MPTP, whereas deficient animals are more sensitive (Przedborski et al., 1992; Klivenyi et al., 1998; Zhang et al., 2000; Andreassen et al., 2001). These animal models, however, do not discriminate among events occurring in glial cells and the different neuronal subtypes existing in the SN. It is also important to determine whether protective enzymes in dopaminergic nigrostriatal (DNS) neurons are transported to the striatal presynaptic terminals, probably less resistant than the soma to neurodegenerative insults. We have generated transgenic mice overexpressing glucose-6-phosphate dehydrogenase (G6PD) driven by the tyrosine hydroxylase (TH) promoter to selectively increase the reductive potential rather than the ROS metabolizing activity in dopaminergic neurons. G6PD catalyzes the first rate-limiting step in the hexose monophosphate shunt and provides NADPH, required to maintain the GSH and other sulfydryl groups of a cell and the antioxidant enzyme catalase in its active form (Kletzien et al., 1994; Salvemini et al., 1999). G6PD has widely recognized antioxidant and cytoprotective effects (Pandolfi et al., 1995; Tian et al., 1999; Felix et al., 2002; Garcia-Nogales et al., 2003) and its deficiency, normally without apparent clinical effects, is the most common human enzymopathy (Vulliamy et al., 1992). The role of this enzyme in a neurodegenerative disorder such as PD, although suggested by epidemiological studies (Abraham et al., 2005), is unknown. Herein, we show that G6PD overexpression along the DNS pathway confers resistance to MPTP-derived parkinsonism.
Materials and Methods
Generation of pTH-G6PD transgenic mice.
Animals were housed at a regulated temperature (22 ± 1°C) in a 12 h light/dark cycle. All experiments were performed in accordance with institutional guidelines approved by the ethical committee in the “Hospital Universitario Virgen del Rocío.” Animals used for studies were either young adult (3–6 months old) or aged (2–2.5 years old) mice. The 9.0 kb promoter of tyrosine hydroxylase (pTH) gene, a kind gift from J. H. Son (Cornell University Medical College, New York, NY) (Min et al., 1994), was used to direct the expression of rat G6PD into catecholaminergic brain regions and adrenal medulla. Rat G6PD coding sequence was cloned by RT-PCR using 2FG6PD as forward primer: 5′-TGGCAGCGGCAACTAAAT-3′ and 1654R as reverse primer: 5′-TAGAGGGTTAGATGGTGA-3′ and verified by sequencing. The 11.5 kb HindIII/NotI fragment, containing the pTH plus the G6PD coding sequence flanked by the β-globin intron II and a poly A signal (from pSG5; Stratagene), was cut from pcDNA3-TH/G6PD plasmid and used for injection into fertilized C57BL/6 eggs to create pTH-G6PD transgenic founder animals.
Genotyping.
Animals positive for the transgene were identified by PCR using genomic DNA as a template. The primers were designed to hybridize with the rat G6PD coding region (forward primer, F3G6PD: 5′-GAGGTCCCACAGAGGCAGATGAG-3′) and with a sequence inside the poly A signal region of pSG5 (reverse primer, RPOLA: 5′-CAGACATGATAAGATACAT-3′) to amplify specifically the transgene.
Immunocytochemistry, densitometric measurements of striatal TH immunoreactivity, and SN stereological neuronal counts.
For all of the experiments, animals were deeply anesthetized by intraperitoneal injection of chloral hydrate (490 mg/kg). After decapitation, mouse brains were removed and fixed overnight at 4°C with 4% paraformaldehyde in PBS. Slices 25–100 μm thick were cut with a vibratome [Lancer (St. Louis, MO) 1000] or with a cryostat (Leica, Wetzlar, Germany). A polyclonal anti-rat TH antibody (1:1000 dilution; Pel-Freez, Rogers, AR) or a polyclonal anti-human G6PD antibody [1:100 dilution; a kind gift from J. M. Bautista (Universidad Complutense de Madrid, Madrid, Spain) (Gomez-Gallego et al., 1996)] was used for immunohistochemical studies. A biotin-conjugated secondary antibody was used against both primary antibodies.
Densitometric measurements of TH immunoreactivity were performed for each animal from three digitized pictures of TH-immunostained striatal sections using the NIH Image software. Density of the cortex was used to subtract the general background, and, in addition, the most denervated slice was used to subtract specifically the striatal background.
For double-fluorescence immunocytochemistry brains were removed, fixed, and incubated in 30% sucrose before freezing. Coronal slices, 25 μm thick, were cut with a cryostat. We used primary antibodies against G6PD, TH and glial fibrillary acidic protein (GFAP; dilution 1:300; Sigma, St. Louis, MO). Secondary fluorescent antibodies were as follows: green (fluorescein; Pierce, Rockford, IL) or red (Alexa Fluor 568; Invitrogen, Eugene, OR).
SN counts of TH-immunoreactive and Nissl-positive (stained with 0.1% cresyl violet) neurons were performed in 30 μm coronal cryostat sections spaced 120 μm throughout the extent of SN pars compacta (SNpc). The number of cells were estimated by systematic random sampling using a 9260 μm2 × 20 μm optical dissector (West, 1993) excluding neurons in the superficial planes of sections. The volume of the SN was estimated according to the principle of Cavalieri (1966). Stereological measurements were done using the C.A.S.T. Grid System (Olympus, Albertslund, Denmark) with a coefficient of error ≤0.09.
In situ hybridization.
In situ hybridization against TH and G6PD mRNAs was made using digoxigenin UTP-labeled antisense riboprobes containing partial cDNA sequences of TH and G6PD, respectively (Toledo-Aral et al., 2003; Gao et al., 2004). After hybridization, the sections were incubated with an alkaline phosphatase-conjugated anti-digoxigenin antibody (1:1000; Roche Diagnostics, Mannheim, Germany).
Glucose-6-phosphate dehydrogenase activity.
Tissues were homogenized with a Dounce homogenizer in extraction buffer containing the following (in mm): 20 Tris-HCl, 1 EDTA, 3 MgCl2, 1 ε-amino-N-caproic acid, and 0.02% (w/v) β-mercaptoethanol, pH 8.0, supplemented with a protease inhibitor mixture (1:1000 dilution; Sigma). After centrifugation at 48,000 × g for 1 h, the supernatant was collected to measure G6PD activity. G6PD activity was determined by measuring the rate of NADPH production spectrophotometrically, as described previously (Bautista et al., 1992). The activity was then normalized to the amount of protein determined with the Bradford method (Bio-Rad Protein Assay; Bio-Rad, Hercules, CA).
Western blot analysis.
Striata from transgenic and control mice were homogenized in a buffer containing the following (in mm): 320 sucrose and 5 HEPES supplemented with a protease inhibitor mixture (1:1000 dilution; Sigma). Homogenates were centrifuged at 2000 × g for 5 min. Part of the supernatants was used for TH immunoblots. The rest of each supernatant fraction was subsequently centrifuged at 30,000 × g for 30 min, as described previously (Gainetdinov et al., 1998). Pellets resuspended in the same buffer supplemented with 1% of SDS were used for DAT Western blots. Twenty micrograms (for TH) or 50 μg (for DAT) of total protein from striatum of each animal were transferred to membranes and incubated with the primary antibodies: anti-TH (1:2000 dilution; Pel-Freez), anti-DAT (1:10,000 dilution; Chemicon, Temecula, CA) or anti-α-tubulin (1:10,000 dilution; Sigma), followed by horseradish peroxidase-conjugated secondary antibodies. Blots were revealed and scanned in a Storm Imaging System 840 (Amersham Biosciences, Sunnyvale, CA). Emitted fluorescent light was quantified directly with the ImageQuant (Amersham Biosciences) software.
MPTP treatment.
Young adult mice (3–6 months of age) were treated subcutaneously with a single dose of 40 mg/kg MPTP (Sigma). Aged animals (2–2.5 years of age) were treated with 30 mg/kg MPTP. For control, animals were injected with saline subcutaneously. Tissues samples were taken 1 week after the injection.
Measurement of dopamine and glutathione content and 1-methyl-4-phenylpyridinium ion (MPP+) metabolism.
Striatal dopamine content was determined by HPLC (ALEXYS 100; Antec Leyden, Zoeterwoude, The Netherlands) using a 3 μm C-18 column (ALB-215; Antec Leyden), followed by electrochemical detection with a glassy carbon electrode and an in situ ISAAC reference electrode (Antec Leyden) (Klivenyi et al., 2000b).
Total glutathione content in ventral mesencephalon was determined by both HPLC (ALEXYS 100; Antec Leyden) using a dual-electrochemical detector (Richie and Lang, 1987) and the enzymatic recycling assay (Tietze, 1969).
For the measurement of striatal MPP+ levels, mice were killed 1 h, 2 h and 4 h after subcutaneous MPTP injection (40 mg/kg) and their striata were removed and frozen. Then, the samples were sent to Dr. F. Fornai's laboratory (University of Pisa, Pisa, Italy) and were processed for MPP+ measurement using HPLC with UV detector (295 nm wavelengh) as described previously (Fornai et al., 2005).
Determination of striatal dopamine release by amperometry.
After beheading, the brains were rapidly removed and cut in coronal slices (250 μm thick) with a vibratome. Slices were incubated in Krebs'–Ringer's bicarbonate buffer [containing the following (in mm): 126 NaCl, 2.5 KCl, 25 NaHCO3, 1.5 MgSO4, 1.2 NaH2PO4, 2.5 CaCl2, and 10 glucose] bubbled with 95% O2 and 5% CO2 at room temperature for at least 60 min before recording. For amperometric measurements, slices were superfused with a control solution containing the following (in mm): 150 NaCl, 2.7 KCl, 2.5 CaCl2, 1 MgCl2, and 10 HEPES, pH 7.4, as described previously (Espejo et al., 1998). To elicit dopamine release, this solution was replaced by another solution with similar composition, except 66 mm KCl and 84 mm NaCl. Secretory events were recorded at room temperature (22–25°) with a carbon-fiber electrode connected to the input of a high-gain current to voltage converter. The electrode was polarized to a constant voltage of 650 mV and placed on top of the striatal gray matter with a micromanipulator (Ureña et al., 1994; Espejo et al., 1998). Amperometric currents were recorded with an EPC-8 patch-clamp amplifier (HEKA Elecktronik, Lambrecht/Pfalz, Germany). Data acquisition and analysis of amperometric currents were done with an ITC-16 interface (Instrutech, Great Neck, NY) and a PULSE/PULSEFIT software (version 8.11; HEKA Elecktronik). The maximum peak of dopamine release in picoampere was determined for each animal as mean obtained from three to four slices. The picoampere value for the transgenic animals was presented as percentage of one obtained from a control littermate recorded with the same electrode and experimental conditions.
Statistical analysis.
Data were presented as mean ± SEM and were analyzed with either Student's t test or one-way ANOVA followed by Student–Newman–Keuls test. p < 0.05 was considered statistically significant.
Results
Selective G6PD overexpression in dopaminergic nigrostriatal neurons
Transgenic G6PD lines were generated by injection into pronuclear stage mouse embryos of a 11.5 kb DNA fragment containing the 9 kb rat tyrosine hydroxylase promoter (pTH) driving the expression of the rat G6PD gene (Fig. 1A). Integration of the pTH-G6PD construct in founder animals and identification of the transgenic lines were demonstrated by PCR using specific primers (Fig. 1B). Expression of the G6PD transgene in the brain was analyzed by in situ hybridization. Although G6PD is a housekeeping gene that to some degree is constitutively expressed in most tissues (Kletzien et al., 1994; Corcoran et al., 1996), its expression in the brain of wild-type animals was undetectable with our in situ hybridization method (Fig. 1C, left). In transgenic littermates, G6PD mRNA was highly expressed in neurons of the SN and the ventral tegmental area (VTA) (Fig. 1C, center), overlapping the areas of TH expression (Fig. 1C, right). G6PD mRNA was also abundant in the locus ceruleus and other brainstem areas, as well as in neurons scattered throughout the brain, particularly in the neocortex, hippocampus, amygdala, and septal nuclei. Outside the brain, transgenic G6PD mRNA was abundantly expressed in chromaffin cells of the adrenal medulla (AM) (Fig. 1C, center, inset).
Figure 1.

Generation of pTH-G6PD transgenic mice. A, Scheme of the pTH-G6PD construct used to create the pTH-G6PD transgenic mouse. B, Representative agarose gel of PCR products of genomic DNA from some of the offspring produced by a founder mouse. The expected size of the PCR product was 330 bp. Lanes with sample #100 and #101, wild-type littermates; lanes with sample #189 and #190, G6PD transgenic mice; lane 5, a wild-type mouse; lane 6, water. C, Analysis of the G6PD mRNA level by in situ hybridization in the ventral mesencephalon and the adrenal gland (insets) of a wild-type (left) or a transgenic mouse (center). In situ hybridization to detect TH mRNA in ventral mesencephalon and adrenal gland (right) is shown for comparison.
Overexpression of G6PD in the nigrostriatal pathway of transgenic animals was also evidenced by immunocytochemistry using an antibody against G6PD (Gomez-Gallego et al., 1996). Wild-type mice did not show any appreciable G6PD immunoreactivity either at the mesencephalon or the striatum (Fig. 2A, left, top and bottom); however, the levels of the protein were quite high in the SN, VTA (Fig. 2A, center, top), or AM (data not shown) of pTH-G6PD transgenic animals with a distribution similar to that of the dopaminergic marker enzyme TH (Fig. 2A, right, top). Noticeably, despite the high level of expression of G6PD in the soma of transgenic DNS neurons, G6PD staining in the striatum was scant and sparsely distributed (Fig. 2A, center, bottom), a pattern clearly different from that characteristic of TH-expressing dopaminergic fibers (Fig. 2A, right, bottom). This suggests that transgenic G6PD is transported along the axons of DNS neurons less efficiently than native TH. Overexpression of G6PD in the DNS system did not appear to alter significantly the synthesis of other proteins. Western blot estimates of the expression of TH and DAT in striatal dopaminergic terminals were unaffected, or even slightly increased, in transgenic with respect to wild-type mice (Fig. 2B). Moreover, brain levels of the antioxidant enzymes tested (catalase, SOD1, and GPx) were not modified in pTH-G6PD transgenics, as determined by in situ hybridization analysis (data not shown).
Figure 2.

Determination of G6PD, TH, and DAT protein levels in the nigrostriatal pathway of pTH-G6PD transgenic mice. A, Immunocytochemical study indicating the lack of G6PD signal in wild-type animals (left) either in SN (top) or in striatum (St; bottom). A strong immunostaining was observed in the substantia nigra and the ventral tegmental area of transgenic mice (center, top). At large magnification, a marked signal for G6PD protein was seen in some striatal fibers of transgenic animals. As control, immunostaining against TH was performed (right). B, Protein from striatum was immunoblotted against TH (left, top), DAT (right, top), or tubulin (for normalization). Quantification of TH and DAT density in transgenic (Tg) mice was calculated as percentage of wild-type (Wt) animals (bottom panels); n = 8 for each group used in TH and DAT quantification. Statistical analysis was done using Student's t test. Tub, α-Tubulin. Error bars represent SEM.
Selective expression of G6PD in dopaminergic neurons was confirmed by double-immunofluorescence staining of mesencephalic slices with antibodies against TH, G6PD, and the GFAP. The confocal images of Figure 3 illustrate that practically all of the G6PD+ neurons in the SN were also TH+ (A–C), whereas none of the G6PD+ cells appeared stained with the glial-specific antibody (D–F).
Figure 3.

Specific expression of G6PD in DNS neurons. Immunofluorescence staining of a slice of ventral mesencephalon using antibodies against G6PD (red, A) and TH (green, B). Superposition of the two fluorescent signals is evidenced by the yellow color in the merged image in C. The insets show a region in the substantia nigra at large magnification. A similar experiment using antibodies against G6PD (red, D) and GFAP (green, E). The lack of superposition of the two fluorescent signals (merged image in F) indicates that G6PD was not expressed in glial cells.
To check whether catecholaminergic tissues in the transgenic animals had indeed a higher amount of functionally active G6PD, we measured the enzymatic activity in several parts of the brain and the adrenal glands as well as in liver and kidney, which were used as controls (Table 1). Ventral mesencephalon, striatum, and adrenal gland of transgenic animals showed a relative increase of G6PD activity, whereas in liver and kidney it remained unchanged. The increase of G6PD activity was, however, only statistically significant in striatum (∼1.6-fold) and adrenal medulla (∼2-fold).
Table 1.
G6PD activity (UI/mg) in pTH-G6PD transgenic mice
| Wt | Tg | |
|---|---|---|
| Ventral mesencephalon | 0.038 ± 0.008 | 0.050 ± 0.010 |
| Striatum | 0.055 ± 0.008 | 0.085 ± 0.012* |
| Cortex | 0.052 ± 0.004 | 0.065 ± 0.012 |
| Adrenal gland | 0.088 ± 0.016 | 0.173 ± 0.027* |
| Liver | 0.013 ± 0.002 | 0.016 ± 0.004 |
| Kidney | 0.021 ± 0.003 | 0.019 ± 0.004 |
Data were expressed as mean ± SEM.
*p < 0.05, Student's t test; n = 9 for each group.Wt, Wild type; Tg, transgenic.
Because increased cytosolic concentration of GSH has been reported in cell lines stably overexpressing G6PD (Salvemini et al., 1999), total GSH concentration in ventral mesencephalon of transgenic and wild-type mice was determined by two different methods. A slight increase in GSH concentration was observed in transgenic mice versus control by both, HPLC (6%; n = 6 and 7 respectively, p = 0.4), and by the spectrophotometric recycling assay (16%; n = 7 and 8 respectively, p = 0.08). Although a trend for GSH increase in transgenic animals is clearly observable, it might have been underestimated because of a dilution effect produced by non-TH-expressing cells within the dissected area (see Discussion).
Transgenic pTH-G6PD mice did not seem to have any gross physiological alteration, as litter size, animal weight and life span were similar to those of wild-type littermates.
Decrease of MPTP toxicity in pTH-G6PD transgenic mice
To assess the possible neuroprotective role of G6PD overexpressed in DNS neurons, transgenic and control littermates were rendered parkinsonian by systemic administration of a single dose of MPTP (40 mg/kg) and killed 1 week later. This pharmacological treatment reproduced the major hallmarks of PD, including substantial dopaminergic cell loss in the SN and marked striatal denervation, with <50% animal mortality. Higher MPTP doses resulted in death of most of the animals. The representative example in Figure 4A shows that both striatal denervation (top) and dopaminergic mesencephalic neuronal loss (bottom) induced by MPTP were partially prevented in transgenic animals overexpressing G6PD. The percentage of striatal dopaminergic innervation that remained after MPTP treatment in wild-type mice (32.9 ± 4.8% with respect to untreated animals; n = 29) increased significantly in MPTP-treated pTH-G6PD transgenics (48.3 ± 6.3%; n = 30; p < 0.05) (Fig. 4B). In parallel, we also performed stereological estimates of TH- and Nissl-positive cells in the SNpc of MPTP-treated transgenic and wild-type animals compared with untreated littermates (Fig. 4C,D). The number of SN TH-positive neurons that remained after MPTP treatment decreased to 52% in wild-type mice (3946 ± 563; n = 10) with respect to untreated animals (7587 ± 491; n = 8) but only to 72% in transgenic mice (5436 ± 408; n = 11) (p < 0.05) (Fig. 4C). Interestingly, the decrease in the number of Nissl-positive cells in the SNpc after MPTP treatment was clearly larger in wild-type than in transgenic animals, thus indicating that MPTP-induced cell death was prevented by G6PD overexpression (Fig. 4D).
Figure 4.
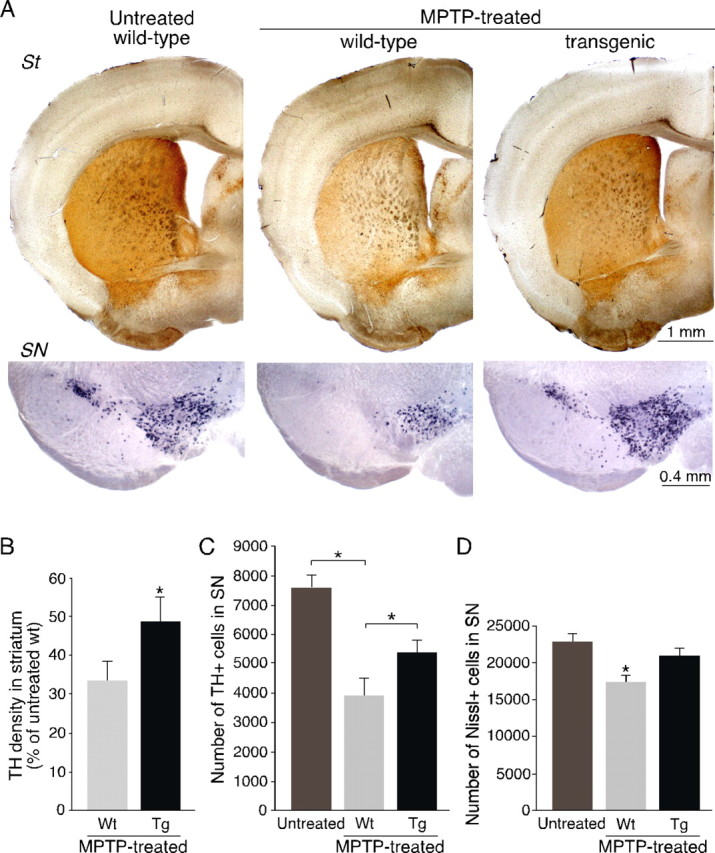
Protection against MPTP-induced toxicity by G6PD overexpression in young animals. A, Analysis of TH expression by immunohistochemistry (top panels) and in situ hybridization (bottom panels) in striatum (St) and SN of saline-treated (MPTP untreated; left) or MPTP-treated (center and right) animals. B, Densitometric measurements of TH immunoreactivity in striata of MPTP-treated mice. In each experiment, all of the data from MPTP-treated animals were normalized to saline-treated wild-type (Wt) animal levels; n = 9 for saline-treated mice, n = 29 for wild-type MPTP-treated group and n = 30 for transgenic (Tg) MPTP-treated group. C, D, Stereological estimations of TH-positive neurons (C) and Nissl-positive cells (D) in the SNpc of untreated and MPTP-treated transgenic and nontransgenic animals 7 d after acute MPTP. The estimated Nissl-stained cell number was as follows: 22915 ± 972 in untreated wild-type; 17388 ± 698 in MPTP-treated wild-type; and 20960 ± 815 in MPTP-treated transgenic animals). The asterisk indicates statistically significantly different with respect to the other two groups. C, D, The number of experiments are as follows: 8 saline-injected wild-type, 10 MPTP-treated wild-type, and 11 MPTP-treated transgenic animals. Statistical analysis was done using one-way ANOVA. Error bars represent SEM.
Functional evaluation of the protection of pTH-G6PD transgenics to MPTP toxicity was initially performed by HPLC. Dopamine content (expressed as nanograms of dopamine per milligram of protein) measured in striatal homogenates of control animals (231.5 ± 5.5; n = 24), was reduced to ∼20% in MPTP-treated wild type mice (42.9 ± 4.5; n = 60). Dopamine content in MPTP-treated pTH-G6PD transgenic littermates increased to 56.0 ± 12.6, (n = 29; p = 0.19 compared with the value of wild-type MPTP-treated animals). Although with low statistical significance, there was a clear trend for attenuation of the deleterious effect of MPTP in pTH-G6PD transgenics compared with control animals. This was further confirmed by the amperometric detection of depolarization-evoked catecholamine release (Ureña et al., 1994) from presynaptic terminals in striatal slices. This method provides a good quantitative correlate of the functional status of the nigrostriatal pathway in animals subjected to the various experimental conditions (Espejo et al., 1998). Typical striatal catecholamine secretory responses to high K+ are shown in Figure 5A. In slices from untreated wild-type and transgenic animals, high K+-elicited secretion was similar (Fig. 5A,B). MPTP treatment induced in wild-type mice a marked decrease of striatal catecholamine release; however, the average amount of high K+-induced catecholamine secretion was ∼2.3-fold higher in transgenic mice (Fig. 5B). Therefore, the direct estimation of catecholamine release from striatal terminals near the recording electrode indicates a more pronounced protection by G6PD than the measurement of overall striatal catecholamine content reported above. These differences could be explained, at least in part, if after MPTP treatment, the pool of rapid releasable catecholamines (detected by amperometry) recovers faster that the overall catecholamine content of the cells (see Discussion).
Figure 5.

Dopamine released from striatum in MPTP-treated young adult mice. A, Amperometric signals attributable to oxidation of dopamine released from striatal slices of a wild-type (Wt) animal (left, top) or a transgenic (Tg) littermate (right, top) after exposure to an external solution with 66 mm K+. Dopamine released from wild-type (left, bottom) or transgenic (right, bottom) striatal slices after MPTP treatment. B, Quantification of dopamine released from striatum after MPTP treatment. In each group (untreated or MPTP-treated) the data from transgenic animals were normalized to the respective wild-type values. n = 8 pairs for the untreated group, and n = 17 pairs for MPTP-treated group. Statistical analysis was done using Student's t test. ∗p < 0.05. Error bars represent SEM.
MPTP pharmacokinetics did not significantly change in transgenic animals compared with wild-type littermates. Striatal MPP+ levels (in micrograms of MPP+ per milligram of protein) measured at 1, 2, and 4 h after MPTP administration were, respectively, the following: 0.708 ± 0.081 versus 0.562 ± 0.081; 1.019 ± 0.087 versus 0.915 ± 0.223; and 0.416 ± 0.037 versus 0.626 ± 0.116 (n = 3–4). Altogether, these results indicate that G6PD overexpression protects DNS neurons against MPTP-derived neurotoxicity.
MPTP toxicity and G6PD neuroprotection in aged mice
Because the incidence rate of PD and most other sporadic neurodegenerative disorders increases with age, we sought to test whether neuroprotection is preserved in old (>24 months) pTH-G6PD transgenic mice. Striatal TH levels (an indication of dopaminergic innervation) were of the same magnitude in young (3–6 months) and aged mice; nevertheless, MPTP toxicity, assessed by Western blots of TH in striatal homogenates, was markedly increased in both old wild-type (Fig. 6A,B) and transgenic (data not shown) animals. The decrease of depolarization-evoked striatal catecholamine release induced by MPTP treatment was also more drastic in aged (reduction to 8.8 ± 3.5% of control, n = 6) than in young (reduction to 28.8 ± 5.8% of control, n = 4) animals, although the former were subjected to a milder MPTP treatment (30 mg/kg instead of 40 mg/kg) to decrease animal mortality. However, G6PD overexpression conferred on old mice a protection against MPTP of the same magnitude observed in young animals. An analysis similar to that in Figure 5B indicated that catecholamine secretion increased a nonsignificant 1.26 ± 0.19-fold in untreated transgenics (n = 4) versus untreated wild-type (n = 4) animals but 2.33 ± 0.58-fold in MPTP-treated transgenics (n = 9) with respect to the MPTP-treated wild-type (n = 9) mice (p < 0.05). The box diagrams in Figure 6C illustrate that although, on average, G6PD protection was similar in the two animal groups, the distribution of the amount of depolarization-evoked striatal dopamine release (normalized to the wild-type littermates) was rather scattered in young transgenics, suggesting individual variability in the effect of systemically administered MPTP. In contrast, the same distribution was more compact and clearly displaced toward higher values in aged transgenic animals. Thus, although MPTP toxicity on DNS neurons increases in old animals, its attenuation by genetic overexpression of G6PD is maintained or even favored by aging.
Figure 6.
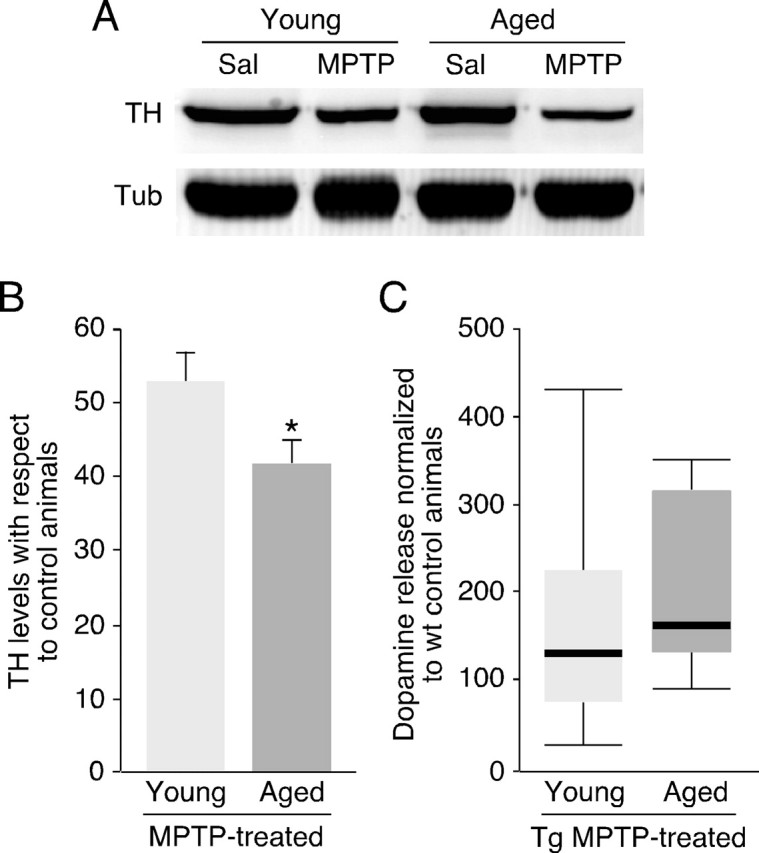
Attenuation of MPTP-induced dopaminergic striatal denervation by G6PD overexpression in aged mice. A, Representative Western blot showing the decrease of TH expression in striatum after MPTP treatment in young or aged wild-type mice. Sal, Saline; Tub, α-tubulin. B, Quantification of decrease in TH levels in wild-type MPTP-treated animals with respect to age-matched untreated mice by Western blot analysis. Young mice, n = 10 for untreated, and n = 15 for MPTP-treated animals. Aged mice, n = 12 for untreated mice, and n = 16 for the group treated with MPTP. Statistical analysis was done using Student's t test. Error bars represent SEM. C, Box diagram comparing distribution of values of dopamine released from striata of young and old transgenic animals after normalization with their respective MPTP-treated wild-type littermates.
Discussion
Although sporadic PD has probably a multifactorial etiology, dysfunction of the cellular redox regulatory mechanisms, affecting the highly susceptible DNS neurons, is thought to be among the major causes that predispose to the disease (Adams and Odunze, 1991; Olanow, 1993; Jenner, 2003; Andersen, 2004; Greenamyre and Hastings, 2004). In this paper, we describe a novel genetic animal model that overexpresses the enzyme G6PD specifically in DNS neurons. G6PD is a key enzyme that provides NADPH, the major source of the reducing equivalents of a cell and has a broadly recognized role as antioxidant and cytoprotective (Pandolfi et al., 1995; Salvemini et al., 1999; Tian et al., 1999; Nicol et al., 2000; Garcia-Nogales et al., 2003). We showed that increase of G6PD activity in DNS neurons protects them against MPTP-induced neurotoxicity. This form of experimental parkinsonism does not perfectly model human PD; nevertheless, it reproduces most of the symptoms of the disease and therefore is used as a tool for testing new therapeutics for PD (Beal, 2001; Sedelis et al., 2001). We also observed that susceptibility to MPTP toxicity increases with mouse age, as does the incidence rate of PD in the human population. However, neuroprotection attributable to G6PD overexpression is preserved, or even accentuated, in old animals.
There is a plethora of clinical and experimental data indicating that oxidative cell damage attributable to either ROS or NO-derived reactive species is central to PD and other neurodegenerative diseases (Perry and Yong, 1986; Dexter et al., 1987; Sofic et al., 1991; Alam et al., 1997; Pearce et al., 1997; Knott et al., 2000; Migliore et al., 2002; Kunikowska and Jenner, 2003). In recent years, several genetically engineered mouse lines have been examined for their ability to model the pathology of PD. Mice that are null for GPx, SOD1, or SOD2 are more sensitive to the toxic effects of MPTP, whereas transgenics overexpressing SOD1 or SOD2 are more resistant (Przedborski et al., 1992; Klivenyi et al., 1998, 2000b; Zhang et al., 2000; Andreassen et al., 2001). S-nitrosylation of parkin, and possibly other proteins, favors neurodegeneration (Chung et al., 2004; Yao et al., 2004), and both iNOS null animals and mice treated with inhibitors against the neuronal form of NOS (nNOS) are also more resistant to MPTP (Dehmer et al., 2000; Klivenyi et al., 2000a). Nevertheless, the role of NO on this process appears to be quite complex. For example, NO-derived peroxynitrite has been reported to exert a protective effect on NO-mediated apoptosis attributable to activation of G6PD-dependent penthose pathway (Garcia-Nogales et al., 2003). Overexpression of ferritin, an iron-binding protein that chelates intracellular iron, in dopaminergic neurons also reduces their sensitivity to MPTP (Kaur et al., 2003).
The animal model generated in this work was designed to enhance selectively antioxidant defense in DNS neurons by increasing their reductive power rather than ROS metabolizing activity. Thus, we could test in an in vivo model the neuroprotective effects of G6PD overexpression in the DNS pathway separately from events occurring in other SN neurons or glial cells. This transgenic animal model is unique because it permits the minimizing of oxidation-dependent alterations intrinsic to DNS neurons, which probably precedes the circle of events (neuroinflammation, production of ROS and NO-derived species, aberrant protein interactions and activation of apoptosis, among other) leading to PD (Andersen, 2004). Indeed, depletion of GSH within dopaminergic SN neurons is the earliest known biochemical marker of nigral degeneration. This could result from dysfunction of dopamine metabolism, as dopamine quinones (DAQs), products resulting from dopamine oxidation that polymerize into the dark pigment neuromelanin characteristic of SN neurons, can conjugate GSH and reduce its concentration. Moreover, high levels of 5cysDAQ, a derivative of the GSH-conjugated DAQs that inhibits mitochondrial complex I as MPTP, have been found in the SN of individuals with PD (Andersen, 2004).
An interesting observation in our study is that a nonphenotypic marker (Nissl staining) showed a clear SN cell death protection in transgenic animals. In addition, G6PD overexpression also rescued TH-positive cells damaged by the neurotoxin. In this respect, our TH-positive cell counts (and striatal densitometry measurements) might have underestimated the ability of G6PD overexpression to recover the dopaminergic phenotype that was probably still in progress at the time of animals being killed. This could also explain why at this point the striatal dopamine content, which requires de novo transmitter synthesis, was only mildly improved.
The oxidative stress in the DNS pathway inherent to dopamine metabolism is probably more challenging for striatal dopaminergic terminals than for the cell bodies in the SN, because the former are exposed to high local concentrations of dopamine and are likely less resistant to neurodegenerative insults. We have shown in our animal model that histological and functional protection of the striatal dopaminergic terminals is paralleled by higher striatal G6PD immunoreactivity and enzymatic activity. Although the increase of striatal G6PD activity in transgenic animals was only moderate (∼1.6-fold), they exhibited a higher (∼2.3-fold) resistance to MPTP. In accord with this observation, it has been shown in cell lines that small increases in G6PD are sufficient to accelerate the penthose pathway (Stanton et al., 1991). Thus, stimulation of G6PD activity in striatal terminals appears to be a highly efficient mechanism to fight against MPTP toxicity. The marked protective effect of G6PD on DNS fibers occurred, although in transgenic animals only a small fraction of the G6PD protein seemed to be transported from the somas to the striatal terminals. In fact, the increase of striatal G6PD activity in transgenic animals (∼1.6-fold) is relatively modest compared with the values (threefold to sixfold) obtained in permanently transfected HeLa cells (Salvemini et al., 1999) or transgenic animals expressing G6PD in other tissues (Tang et al., 1993; Corcoran et al., 1996). G6PD activity (or GSH level) measured in the whole mesencephalon or the striatum of pTH-transgenic animals is surely an underestimate of the actual enzymatic activity in dopaminergic neurons because of dilution by the nondopaminergic tissue. However, our observations could also indicate that part of the transgenic G6PD is functionally inactive and unable to be transported along the DNS axons. G6PD is a highly regulated enzyme (Kletzien et al., 1994; Corcoran et al., 1996; Salvemini et al., 1999; Gao et al., 2004) that can be rapidly activated by hormones and growth factors, which induce its translocation from a bound, nonactive pool to a soluble active form (Stanton et al., 1991; Tian et al., 1994, 1999). Therefore, a fraction of the G6PD overexpressed in transgenic dopaminergic neurons could form nonactive aggregasomes, bound to particulate elements unable to be transported along the DNS pathway.
There are several hundred G6PD variants, many of which are genetically polymorphic, and G6PD deficiency is the most common enzymopathy, affecting some 400 millions people worldwide (Vulliamy et al., 1992). Although the majority of affected individuals have minor or moderate G6PD deficiency and are asymptomatic, in some extreme cases they develop hemolytic anemia as a result of decreased oxidative defense in red blood cells (Vulliamy et al., 1992). Given the marked protective role of G6PD in DNS neurons described here, it can be speculated that G6PD deficiency could predispose to sporadic PD. The gene that encodes G6PD is located on the long arm of the X chromosome, and, indeed, the higher incidence rate of PD in men than in women has been attributed to X linkage of genetic risk factors (Wooten et al., 2004).
In conclusion, we show that selective enhancement of G6PD activity in the dopaminergic nigrostriatal system has a marked neuroprotective effect. Thus, selective overexpression of G6PD in dopaminergic neurons, pharmacological activation of the native enzyme, and facilitation of its axonal transport are potential therapeutic strategies to PD that should be explored in future work. Epidemiological studies should also be designed to investigate the possible association between G6PD deficiency and sporadic PD.
Footnotes
This work was supported by the “Ayuda a la investigación 2000” of the Juan March Foundation and by grants from the Lilly Foundation, Spanish Ministry of Education and Science (Genómica and Proteómica and SAF programs), Spanish Ministry of Health (Redes Centrode Investigaciones de Enfermedades Neurodegenerativas and Terapia Celular), and the Andalusian Government. R.M. and J.V. were supported by predoctoral fellowships of the Spanish Ministry of Education and Fondo de Investigación Sanitaria. We gratefully acknowledge the technical assistance of José A. Rodríguez-Gómez, José M. Martín, and Paco Fernández Matute. Striatal MPP+ determinations were generously done at the laboratory of Dr. Francesco Fornai (University of Pisa, Pisa, Italy).
References
- Abraham S, Soundararajan CC, Vivekanandhan S, Behari M (2005). Erythrocyte antioxidant enzymes in Parkinson's disease. Indian J Med Res 121:111–115. [PubMed] [Google Scholar]
- Adams JD Jr, Odunze IN (1991). Oxygen free radicals and Parkinson's disease. Free Radic Biol Med 10:161–169. [DOI] [PubMed] [Google Scholar]
- Alam ZI, Daniel SE, Lees AJ, Marsden DC, Jenner P, Halliwell B, Jenner A, Cairns N, Marsden CD (1997). A generalised increase in protein carbonyls in the brain in Parkinson's but not incidental Lewy body disease. J Neurochem 69:1326–1329. [DOI] [PubMed] [Google Scholar]
- Andersen JK (2004). Oxidative stress in neurodegeneration: cause or consequence? Nat Med 10:S18–25. [DOI] [PubMed] [Google Scholar]
- Andreassen OA, Ferrante RJ, Dedeoglu A, Albers DW, Klivenyi P, Carlson EJ, Epstein CJ, Beal MF (2001). Mice with a partial deficiency of manganese superoxide dismutase show increased vulnerability to the mitochondrial toxins malonate, 3-nitropropionic acid, and MPTP. Exp Neurol 167:189–195. [DOI] [PubMed] [Google Scholar]
- Bautista JM, Mason PJ, Luzzatto L (1992). Purification and properties of human glucose-6-phosphate dehydrogenase made in E. coli. Biochim Biophys Acta 1119:74–80. [DOI] [PubMed] [Google Scholar]
- Beal MF (2001). Experimental models of Parkinson's disease. Nat Rev Neurosci 2:325–334. [DOI] [PubMed] [Google Scholar]
- Bossy-Wetzel E, Schwarzenbacher R, Lipton SA (2004). Molecular pathways to neurodegeneration. Nat Med 10:S2–9. [DOI] [PubMed] [Google Scholar]
- Cavalieri B (1966). In: Geometria degli indivisibili Torino, Italy: Unione Tipografica Editrice.
- Chung KK, Thomas B, Li X, Pletnikova O, Troncoso JC, Marsh L, Dawson VL, Dawson TM (2004). S-nitrosylation of parkin regulates ubiquitination and compromises parkin's protective function. Science 304:1328–1331. [DOI] [PubMed] [Google Scholar]
- Corcoran CM, Fraser P, Martini G, Luzzatto L, Mason PJ (1996). High-level regulated expression of the human G6PD gene in transgenic mice. Gene 173:241–246. [DOI] [PubMed] [Google Scholar]
- Dauer W, Przedborski S (2003). Parkinson's disease: mechanisms and models. Neuron 39:889–909. [DOI] [PubMed] [Google Scholar]
- Dawson TM, Dawson VL (2003). Molecular pathways of neurodegeneration in Parkinson's disease. Science 302:819–822. [DOI] [PubMed] [Google Scholar]
- Dehmer T, Lindenau J, Haid S, Dichgans J, Schulz JB (2000). Deficiency of inducible nitric oxide synthase protects against MPTP toxicity in vivo. J Neurochem 74:2213–2216. [DOI] [PubMed] [Google Scholar]
- Dexter DT, Wells FR, Agid F, Agid Y, Lees AJ, Jenner P, Marsden CD (1987). Increased nigral iron content in postmortem parkinsonian brain. Lancet 2:1219–1220. [DOI] [PubMed] [Google Scholar]
- Espejo EF, Montoro RJ, Armengol JA, Lopez-Barneo J (1998). Cellular and functional recovery of Parkinsonian rats after intrastriatal transplantation of carotid body cell aggregates. Neuron 20:197–206. [DOI] [PubMed] [Google Scholar]
- Felix K, Rockwood LD, Pretsch W, Nair J, Bartsch H, Bornkamm GW, Janz S (2002). Moderate G6PD deficiency increases mutation rates in the brain of mice. Free Radic Biol Med 32:663–673. [DOI] [PubMed] [Google Scholar]
- Fornai F, Schluter OM, Lenzi P, Gesi M, Ruffoli R, Ferrucci M, Lazzeri G, Busceti CL, Pontarelli F, Battaglia G, Pellegrini A, Nicoletti F, Ruggieri S, Paparelli A, Sudhof TC (2005). Parkinson-like syndrome induced by continuous MPTP infusion: convergent roles of the ubiquitin-proteasome system and alpha-synuclein. Proc Natl Acad Sci USA 102:3413–3418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gainetdinov RR, Fumagalli F, Wang YM, Jones SR, Levey AI, Miller GW, Caron MG (1998). Increased MPTP neurotoxicity in vesicular monoamine transporter 2 heterozygote knockout mice. J Neurochem 70:1973–1978. [DOI] [PubMed] [Google Scholar]
- Gao L, Mejias R, Echevarria M, Lopez-Barneo J (2004). Induction of the glucose-6-phosphate dehydrogenase gene expression by chronic hypoxia in PC12 cells. FEBS Lett 569:256–260. [DOI] [PubMed] [Google Scholar]
- Garcia-Nogales P, Almeida A, Bolanos JP (2003). Peroxynitrite protects neurons against nitric oxide-mediated apoptosis. A key role for glucose-6-phosphate dehydrogenase activity in neuroprotection. J Biol Chem 278:864–874. [DOI] [PubMed] [Google Scholar]
- Gomez-Gallego F, Garrido-Pertierra A, Mason PJ, Bautista JM (1996). Unproductive folding of the human G6PD-deficient variant A. FASEB J 10:153–158. [DOI] [PubMed] [Google Scholar]
- Greenamyre JT, Hastings TG (2004). Biomedicine. Parkinson's—divergent causes, convergent mechanisms. Science 304:1120–1122. [DOI] [PubMed] [Google Scholar]
- Jenner P (2003). Oxidative stress in Parkinson's disease. Ann Neurol 53:S26–S36. discussion S36–S38. [DOI] [PubMed] [Google Scholar]
- Kaur D, Yantiri F, Rajagopalan S, Kumar J, Mo JQ, Boonplueang R, Viswanath V, Jacobs R, Yang L, Beal MF, DiMonte D, Volitaskis I, Ellerby L, Cherny RA, Bush AI, Andersen JK (2003). Genetic or pharmacological iron chelation prevents MPTP-induced neurotoxicity in vivo: a novel therapy for Parkinson's disease. Neuron 37:899–909. [DOI] [PubMed] [Google Scholar]
- Kletzien RF, Harris PK, Foellmi LA (1994). Glucose-6-phosphate dehydrogenase: a “housekeeping” enzyme subject to tissue-specific regulation by hormones, nutrients, and oxidant stress. FASEB J 8:174–181. [DOI] [PubMed] [Google Scholar]
- Klivenyi P, St Clair D, Wermer M, Yen HC, Oberley T, Yang L, Flint Beal M (1998). Manganese superoxide dismutase overexpression attenuates MPTP toxicity. Neurobiol Dis 5:253–258. [DOI] [PubMed] [Google Scholar]
- Klivenyi P, Andreassen OA, Ferrante RJ, Lancelot E, Reif D, Beal MF (2000a). Inhibition of neuronal nitric oxide synthase protects against MPTP toxicity. NeuroReport 11:1265–1268. [DOI] [PubMed] [Google Scholar]
- Klivenyi P, Andreassen OA, Ferrante RJ, Dedeoglu A, Mueller G, Lancelot E, Bogdanov M, Andersen JK, Jiang D, Beal MF (2000b). Mice deficient in cellular glutathione peroxidase show increased vulnerability to malonate, 3-nitropropionic acid, and 1-methyl-4-phenyl-1,2,5,6-tetrahydropyridine. J Neurosci 20:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knott C, Stern G, Wilkin GP (2000). Inflammatory regulators in Parkinson's disease: iNOS, lipocortin-1, and cyclooxygenases-1 and -2. Mol Cell Neurosci 16:724–739. [DOI] [PubMed] [Google Scholar]
- Kunikowska G, Jenner P (2003). Alterations in m-RNA expression for Cu,Zn-superoxide dismutase and glutathione peroxidase in the basal ganglia of MPTP-treated marmosets and patients with Parkinson's disease. Brain Res 968:206–218. [DOI] [PubMed] [Google Scholar]
- Langston JW, Ballard P, Tetrud JW, Irwin I (1983). Chronic Parkinsonism in humans due to a product of meperidine-analog synthesis. Science 219:979–980. [DOI] [PubMed] [Google Scholar]
- Liberatore GT, Jackson-Lewis V, Vukosavic S, Mandir AS, Vila M, McAuliffe WG, Dawson VL, Dawson TM, Przedborski S (1999). Inducible nitric oxide synthase stimulates dopaminergic neurodegeneration in the MPTP model of Parkinson disease. Nat Med 5:1403–1409. [DOI] [PubMed] [Google Scholar]
- Migliore L, Petrozzi L, Lucetti C, Gambaccini G, Bernardini S, Scarpato R, Trippi F, Barale R, Frenzilli G, Rodilla V, Bonuccelli U (2002). Oxidative damage and cytogenetic analysis in leukocytes of Parkinson's disease patients. Neurology 58:1809–1815. [DOI] [PubMed] [Google Scholar]
- Min N, Joh TH, Kim KS, Peng C, Son JH (1994). 5′ upstream DNA sequence of the rat tyrosine hydroxylase gene directs high-level and tissue-specific expression to catecholaminergic neurons in the central nervous system of transgenic mice. Brain Res Mol Brain Res 27:281–289. [DOI] [PubMed] [Google Scholar]
- Nicol CJ, Zielenski J, Tsui LC, Wells PG (2000). An embryoprotective role for glucose-6-phosphate dehydrogenase in developmental oxidative stress and chemical teratogenesis. FASEB J 14:111–127. [DOI] [PubMed] [Google Scholar]
- Olanow CW (1993). A radical hypothesis for neurodegeneration. Trends Neurosci 16:439–444. [DOI] [PubMed] [Google Scholar]
- Pandolfi PP, Sonati F, Rivi R, Mason P, Grosveld F, Luzzatto L (1995). Targeted disruption of the housekeeping gene encoding glucose 6-phosphate dehydrogenase (G6PD): G6PD is dispensable for pentose synthesis but essential for defense against oxidative stress. EMBO J 14:5209–5215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker WD Jr, Boyson SJ, Parks JK (1989). Abnormalities of the electron transport chain in idiopathic Parkinson's disease. Ann Neurol 26:719–723. [DOI] [PubMed] [Google Scholar]
- Pearce RK, Owen A, Daniel S, Jenner P, Marsden CD (1997). Alterations in the distribution of glutathione in the substantia nigra in Parkinson's disease. J Neural Transm 104:661–677. [DOI] [PubMed] [Google Scholar]
- Perry TL, Yong VW (1986). Idiopathic Parkinson's disease, progressive supranuclear palsy and glutathione metabolism in the substantia nigra of patients. Neurosci Lett 67:269–274. [DOI] [PubMed] [Google Scholar]
- Przedborski S, Kostic V, Jackson-Lewis V, Naini AB, Simonetti S, Fahn S, Carlson E, Epstein CJ, Cadet JL (1992). Transgenic mice with increased Cu/Zn-superoxide dismutase activity are resistant to N-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced neurotoxicity. J Neurosci 12:1658–1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richie JP Jr, Lang CA (1987). The determination of glutathione, cyst(e)ine, and other thiols and disulfides in biological samples using high-performance liquid chromatography with dual electrochemical detection. Anal Biochem 163:9–15. [DOI] [PubMed] [Google Scholar]
- Salvemini F, Franze A, Iervolino A, Filosa S, Salzano S, Ursini MV (1999). Enhanced glutathione levels and oxidoresistance mediated by increased glucose-6-phosphate dehydrogenase expression. J Biol Chem 274:2750–2757. [DOI] [PubMed] [Google Scholar]
- Schapira AH, Cooper JM, Dexter D, Clark JB, Jenner P, Marsden CD (1990). Mitochondrial complex I deficiency in Parkinson's disease. J Neurochem 54:823–827. [DOI] [PubMed] [Google Scholar]
- Sedelis M, Schwarting RK, Huston JP (2001). Behavioral phenotyping of the MPTP mouse model of Parkinson's disease. Behav Brain Res 125:109–125. [DOI] [PubMed] [Google Scholar]
- Sofic E, Paulus W, Jellinger K, Riederer P, Youdim MB (1991). Selective increase of iron in substantia nigra zona compacta of parkinsonian brains. J Neurochem 56:978–982. [DOI] [PubMed] [Google Scholar]
- Stanton RC, Seifter JL, Boxer DC, Zimmerman E, Cantley LC (1991). Rapid release of bound glucose-6-phosphate dehydrogenase by growth factors. Correlation with increased enzymatic activity. J Biol Chem 266:12442–12448. [PubMed] [Google Scholar]
- Tang TK, Tam KB, Huang SC (1993). High-level and erythroid-specific expression of human glucose-6-phosphate dehydrogenase in transgenic mice. J Biol Chem 268:9522–9525. [PubMed] [Google Scholar]
- Tian WN, Pignatare JN, Stanton RC (1994). Signal transduction proteins that associate with the platelet-derived growth factor (PDGF) receptor mediate the PDGF-induced release of glucose-6-phosphate dehydrogenase from permeabilized cells. J Biol Chem 269:14798–14805. [PubMed] [Google Scholar]
- Tian WN, Braunstein LD, Apse K, Pang J, Rose M, Tian X, Stanton RC (1999). Importance of glucose-6-phosphate dehydrogenase activity in cell death. Am J Physiol 276:C1121–C1131. [DOI] [PubMed] [Google Scholar]
- Tietze F (1969). Enzymic method for quantitative determination of nanogram amounts of total and oxidized glutathione: applications to mammalian blood and other tissues. Anal Biochem 27:502–522. [DOI] [PubMed] [Google Scholar]
- Tieu K, Perier C, Vila M, Caspersen C, Zhang HP, Teismann P, Jackson-Lewis V, Stern DM, Yan SD, Przedborski S (2004). l-3-Hydroxyacyl-CoA dehydrogenase II protects in a model of Parkinson's disease. Ann Neurol 56:51–60. [DOI] [PubMed] [Google Scholar]
- Toledo-Aral JJ, Mendez-Ferrer S, Pardal R, Echevarria M, Lopez-Barneo J (2003). Trophic restoration of the nigrostriatal dopaminergic pathway in long-term carotid body-grafted parkinsonian rats. J Neurosci 23:141–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ureña J, Fernandez-Chacon R, Benot AR, Alvarez de Toledo GA, Lopez-Barneo J (1994). Hypoxia induces voltage-dependent Ca2+ entry and quantal dopamine secretion in carotid body glomus cells. Proc Natl Acad Sci USA 91:10208–10211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vulliamy T, Mason P, Luzzatto L (1992). The molecular basis of glucose-6-phosphate dehydrogenase deficiency. Trends Genet 8:138–143. [DOI] [PubMed] [Google Scholar]
- West MJ (1993). New stereological methods for counting neurons. Neurobiol Aging 14:275–285. [DOI] [PubMed] [Google Scholar]
- Wooten GF, Currie LJ, Bovbjerg VE, Lee JK, Patrie J (2004). Are men at greater risk for Parkinson's disease than women? J Neurol Neurosurg Psychiatry 75:637–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao D, Gu Z, Nakamura T, Shi ZQ, Ma Y, Gaston B, Palmer LA, Rockenstein EM, Zhang Z, Masliah E, Uehara T, Lipton SA (2004). Nitrosative stress linked to sporadic Parkinson's disease: S-nitrosylation of parkin regulates its E3 ubiquitin ligase activity. Proc Natl Acad Sci USA 101:10810–10814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Graham DG, Montine TJ, Ho YS (2000). Enhanced N-methyl-4-phenyl-1,2,3,6-tetrahydropyridine toxicity in mice deficient in CuZn-superoxide dismutase or glutathione peroxidase. J Neuropathol Exp Neurol 59:53–61. [DOI] [PubMed] [Google Scholar]