

Abstract
Transcriptional dysregulation caused by expanded polyglutamines (polyGlns) in huntingtin (htt) may be central to cell-autonomous mechanisms for neuronal cell death in Huntington's disease (HD) pathogenesis. We hypothesized that these mechanisms may involve the dysfunction of the transcriptional regulator CA150, a putative modifier of onset age in HD, because it binds to htt and accumulates in an HD grade-dependent manner in striatal and cortical neurons. Consistently, we report herein that CA150 expression rescues striatal cell death in lentiviral overexpression (rats) and knock-in (mouse cells) conditions for mutant htt neurotoxicity. In both systems, rescue was dependent on the (Gln-Ala)38 repeat normally found in CA150. We excluded the possibility that rescue may be caused by the (Gln-Ala)38 repeat interacting with polyGlns and, by doing so, blocking mutant htt toxicity. In contrast, we found the (Gln-Ala)38 repeat is required for the nuclear restriction of exogenous CA150, suggesting that rescue requires nuclear CA150. Additionally, we found the (Gln-Ala)38 repeat was dispensable for CA150 transcriptional repression ability, suggesting further that CA150 localization is critical to rescue. Finally, rescue was associated with increased neuritic aggregation, with no reduction of nuclear inclusions, suggesting the solubilization and nuclear export of mutant htt. Together, our data indicate that mutant htt may induce CA150 dysfunction in striatal neurons and suggest that the restoration of nuclear protein cooperativity may be neuroprotective.
Keywords: Huntington, huntingtin, striatum, neuron death, transcription factor, neuroprotection
Introduction
Huntington's disease (HD) is a dominant disorder characterized by striatal neurodegeneration (Kowall et al., 1987). HD is caused by CAG expansion in exon 1 of huntingtin (htt), which results in expanded polyglutamines (polyGlns) (The Huntington's Disease Collaborative Research Group, 1993). The disease may be the combined effect of cell-autonomous (Sipione et al., 2002; Wang et al., 2005) and cell-cell interaction (Gu et al., 2005) processes involving either a loss of normal htt function or a gain of toxic properties produced by misfolded mutant N-terminal htt, or both (Landles and Bates, 2004; Li and Li, 2004). The loss-of-function may apply to the regulation of transcription (Zuccato et al., 2003), axonal transport (Gunawardena et al., 2003), and vesicle trafficking (Gauthier et al., 2004) as performed by the putative scaffolding activity of htt. The gain of toxic properties may also apply to the regulation of transcription (Steffan et al., 2000; Nucifora et al., 2001; Dunah et al., 2002), axonal transport (Szebenyi et al., 2003), and vesicle trafficking (Qin et al., 2004) through abnormal binding of mutant htt to partner proteins that may be subsequently sequestered into the aggregates formed from mutant htt seeds (for review, see Li and Li, 2004). However, the pathogenic role of aggregates is debated because they may be protective (Saudou et al., 1998; Yu et al., 2002; Arrasate et al., 2004) or coincidental (Sisodia, 1998). Although the data may be biased by a lack of sensitivity for microaggregates, the abnormal interaction of soluble mutant htt with transcriptional regulators appears to be a more consistent core mechanism for neurotoxicity (Saudou et al., 1998; Dunah et al., 2002; Jiang et al., 2003; Schaffar et al., 2004). The effects of mutant htt on transcription may be notably central to cell-autonomous mechanisms for neuronal cell death (Wang et al., 2005). Among transcriptional regulators that may bind htt and participate to these mechanisms is CA150, a protein containing a (Gln-Ala)38 repeat of unknown function, three WW domains that mediate the interaction with the transcriptional elongation factor SF1 (splicing factor 1) and splicing factors, and six FF motifs that bind the phosphorylated C-terminal domain of the largest subunit of RNA polymerase II (Sune and Garcia-Blanco, 1999; Goldstrohm et al., 2001; Carty and Greenleaf, 2002; Lin et al., 2004; Smith et al., 2004). We found previously that CA150 binds htt in vitro and increasingly accumulates in the brain of HD patients as the disease grade increases (Holbert et al., 2001). Additionally, the (Gln-Ala)38 repeat is polymorphic, with a slightly earlier than expected age at onset (AO) being detected in individuals who carried a long CA150 repeat allele (Holbert et al., 2001). This observation and similar observations described previously (Chattopadhyay et al., 2003) suggested that CA150 may modify AO in HD (Holbert et al., 2001). The CA150 protein may thus strongly participate in HD pathogenesis. We hypothesized that there may be a significant loss of CA150 function caused by mutant htt in the neuronal cell. The nuclear accumulation of CA150 in HD brains was indeed suggestive of abnormal interactions between mutant htt and the protein, as is the case for other htt partners including CREB binding protein (CBP) or Sp1 (Dunah et al., 2002; Li et al., 2002). To test for this hypothesis, we overexpressed CA150 in two complementary conditions for mutant htt neurotoxicity, including overexpression using the lentiviral-mediated delivery of N-terminal mutant htt in the rat striatum (de Almeida et al., 2002), and knock-in using striatal cells derived from HdhQ111 mice (Trettel et al., 2000). In both systems, (1) CA150 overexpression rescued mutant htt toxicity, suggesting CA150 dysfunction in HD, and (2) CA150 rescuing activity was dependent on the presence of the (Gln-Ala)38 repeat, which influenced the intracellular distribution of CA150 in rats, suggesting that the repeat may support proper CA localization and function.
Materials and Methods
Plasmid construction.
The construction of SIN-W-PGK (phosphoglycerate kinase) vectors encoding htt171-19Q and htt171-82Q was previously described (de Almeida et al., 2002). Green fluorescent protein (GFP) and LacZ constructs were previously described in (de Almeida et al., 2001; Fleury et al., 2003). The construction of DNA fragments encoding hemagglutinin (HA) tagged full-length CA150 was described previously (Holbert et al., 2001). To construct DNA fragments encoding CA150 deleted for the (Gln-Ala)38 repeat (amino acids 177–255) region (CA150ΔQA), we amplified the 5′ and 3′ portions of the cDNA by PCR and ligated the resulting products using DNA repair in yeast (Oldenburg et al., 1997). The HA-tagged CA150 and CA150ΔQA were then cloned into the BamH1/XhoI sites of the self-inactivating SIN-W-PGK transfer vector (Deglon et al., 2000) and BamH1/XhoI sites of the pCDNA3.1 vector (Invitrogen, Carlsbad, CA). To construct DNA fragments encoding the HA-tagged CA150 protein fragment that contains the (Gln-Ala)38 repeat, we amplified the corresponding cDNA region (amino acids 162–331) by PCR using the forward primer 5′-GCTATGGCTTACCCATACG-ATGTTCCAGATTACGCTAGCTTGGGTGGTCATATGATGGAGTT-AAG-3′ containing an HA tag coding sequence and the reverse primer 5′-TTATCGCTTTCGCTTCTTTCGCTTTCGCTTCTTGGTTTGCAC-AGG-3′ containing a sequence coding for the CA150 nuclear localization signal (amino acids 626–631). This PCR product was cloned into the pCRII-TOPO vector (Invitrogen) and then into the BamH1/XhoI sites of the pCDNA3.1 plasmid. All constructs were sequenced to verify integrity. The CA150 responsive reporter construct −42α4CAT and the CA150 expression construct pefBOST7CA150 have been described previously (Sune and Garcia-Blanco, 1999). PefBOST7CA150-ΔQA was constructed by replacing the Bsm1-KpnI fragment of pefBOST7CA150 with that of pcDNA3.1-CA150ΔQA (described herein). The CA150 cDNA was a gift from C. Sune and M. Garcia-Blanco (Duke University, Durham, NC).
Lentiviral vector production.
Viral particles were produced in human embryonic kidney 293T cells using a four plasmid system as described previously (de Almeida et al., 2002) and resuspended in PBS/1% bovine serum albumin (BSA). The particle content of the viral batches was determined by p24 antigen ELISA (PerkinElmer, Wellesley, MA).
Culture of primary rat striatal cells.
E16 embryos were collected from timed-pregnant Sprague Dawley rats (Charles River Laboratories, Les Oncins, France) killed by CO2 inhalation. Dissections were performed in ice-cold dissection medium (DMX) containing Ca2+- and Mg2+-free PBS, 0.6% d-glucose, 1% Pen-Strep (10,000 U/ml, 10,000 μg/ml; Invitrogen), and 10 mm HEPES (Invitrogen). Ganglionic eminences were isolated and cut into pieces. The tissue was homogenized by repeated pipetting in DMX containing BSA (Fluka). Cells were centrifuged at 4°C for 5 min at 1000 × g and resuspended in 10 ml of glutamate-free Neurobasal medium containing 1% B27 (Invitrogen), 1% Pen-Strep (10,000 U/ml, 10,000 μg/ml), 0.5 mm l-glutamine and 15 mm KCl. Cells were plated at a density of 150,000 cells/cm2 in multiwell dishes coated with 20 μg/ml poly-l-lysine (Sigma, St. Louis, MO). The cultures were maintained in a humid incubator (5% CO2, 37°C) and one-half of the medium was changed weekly. Cell cultures were infected with lentiviral vectors at a ratio of 10 ng of p24 antigen/105 cells 1 d after plating [1 d in vitro (DIV)]. Coinfections were performed using two viral vectors, each at a ratio of 10 ng p24 antigen/105 cells. At 2 DIV, one-half of the medium was replaced with fresh culture medium.
Culture of striatal cell lines.
7Q/7Q and 109Q/109Q cells established from HdhQ111 knock-in mice (Trettel et al., 2000) were cultured in DMEM (Sigma) supplemented with 10% fetal bovine serum, Pen-Strep (10,000 U/ml, 10,000 μg/ml), and 1 mm l-glutamine and grown at 33°C with 5% CO2. transfections were performed using using lipofectamine 2000 (Invitrogen) following manufacturer's indications, with the PCDNA3.1 plasmid encoding β-galactosidase and either the empty PCDNA3.1 plasmid or PCDNA3.1 plasmids encoding CA150 species using a CA150:β-galactosidase DNA ratio of 4:1. Cell death assays were performed as described previously (Gauthier et al., 2004).
Immunoblot analysis of cell extracts.
Cells were rinsed in ice-cold PBS and collected in PBS with complete mini protease inhibitor cocktail (Roche Diagnostics, Indianapolis, IN), sonicated on ice for 25 s and centrifugated at 16,000 × g for 10 min. Protein concentrations of supernatants were determined using the Coomassie blue protein assay reagent (Pierce, Rockford, IL). Protein samples (30 μg) in loading buffer (625 mm Tris, pH 6.8, 0.001% bromophenol blue, 10% glycerol, 2% SDS) were separated by SDS/PAGE and analyzed by Western blotting with the Myc antibody 4A6 (diluted 1:1000; Upstate Biotechnology, Lake Placid, NY), the GFP (diluted 1:1000; Invitrogen) or actin (diluted 1:2000; Invitrogen) antibody for the assays in primary cultures of rat striatal cells, and the htt (4C8; diluted 1:800; Chemicon, Temecula, CA) or actin (diluted 1:2000; Invitrogen) antibody for the assays in HdhQ111 mouse cells.
Injection of lentiviruses.
Female adult Wistar rats received stereotaxical injections in the striatum using a Hamilton syringe with a 34 gauge blunt-tip needle (Hamilton, Reno, NV). Viral particles were concentrated at 125,000 ng of p24 per milliliter. Viral suspensions (4 μl) were injected in each striatum at 0.2 μl/min. One group of experiments was stopped at 8 weeks after infection, and another group was stopped at 12 weeks after infection. All tests were based on seven to eight animals per experiment.
Tissue preparation for immunohistochemical analysis.
After anesthesia with a sodium pentobarbital overdose (150 mg/kg), the animals were transcardially perfused, first with phosphate buffer and Liquemin (5000 U/L), and then with phosphate buffer containing 4% paraformaldehyde and 10% picric acid. Brains were fixed in 4% paraformaldehyde for 24 h, cryoprotected in 25% sucrose and 0.1 m phosphate buffer for 48–72 h, and then frozen on dry ice. Coronal sections (thickness, 20 μm) of the striatum were prepared in a −20°C cold cryostat (Cryocut 1800; Leica Microsystems, Bannockburn, IL). Free-floating slices were then collected in PBS containing 0.12 μm sodium azide.
Immunohistochemical procedure.
Brain sections were first treated at 37°C in a PBS solution containing 0.1% diphenylhydrazine, then incubated in 0.1 m PBS containing 10% neonatal goat serum (NGS) (Dako, High Wycombe, UK), followed by incubation with the following antibodies: htt antibody 2B4 (diluted 1:200), htt antibody EM8 (MAB5374; diluted 1:2000; Chemicon), neuronal-specific nuclear protein (Neu-N) antibody (diluted 1:500; Chemicon), ubiquitin antibody (diluted 1:1000; Dako), dopamine and cAMP-regulated phosphoprotein (DARPP-32) antibody (AB 1656; diluted 1:7500; Chemicon), and HA-tag antibody (MMS 101R; diluted 1:1000; Covance) for detection of CA150, diluted in PBS containing 5% NGS solution. For immunostaining with the htt antibody EM48, the brain sections were treated with 1% cyanoborohydride and washed with PBS 0.4% Triton X-100, and no serum was used at any step of the immunostaining procedure. After three washings, the brain sections were incubated with the secondary antibodies coupled to either a fluorophore (Jackson ImmunoResearch, West Grove, PA) or biotin (Vector Laboratories, Peterborough, UK). Nuclear staining was performed using acridine orange (1 μg/ml) or 4′,6′-diamidino-2-phenylindole dihydrochloride (2.5 μg/ml). The brain sections incubated with biotin-coupled secondary antibodies were further processed using the Vectastain Elite ABC detection kit (Vector Laboratories) with 3,3′-diaminobenzidine tetrahydrochloride (Pierce) as a substrate. For Nissl staining, slides were mounted and stained with 0.15% cresyl violet.
Lesion measurements.
The DARPP-32 and Neu-N expression levels were used to determine the severity of the lesions. The volume of the lesions was assessed based on the loss of nuclear DARPP-32 and Neu-N expression after immunostaining. For each animal, the images of six to eight serial sections were acquired at 4× magnification and analyzed using an image analysis system (Morphostar 5.12; IMSTAR, Paris, France). For each brain section, the shape of the lesioned area was manually delineated on the digitized images. The volume of each lesion was determined using the formula V = ΣS * k, where ΣS is the sum of the lesion surface (S) in each of the section, and k is the interval between the serial sections (0.6 mm). Striatal shrinkage was assessed based on the percentage of area occupied by the bundles of fibers around the injection point. In coronal sections stained with cresyl violet, fibers appear as large pale circles. Cresyl-violet-stained sections with a clearly visible injection point were acquired at 10× magnification. Using intensity thresholding, the areas of the bundles of fibers were measured and compared with the total area of the field of view (0.15 mm2). Image analysis was performed using Metaview (Roper Scientific, Tucson, AZ).
Analysis of 2B4-positive signals.
Coronal sections showing the striatum, e.g., rostrocaudal levels plus 1 mm (Paxinos et al., 1985), were scanned with a 50× objective using a Zeiss (Oberkochen, Germany) Axioplan2 imaging microscope motorized for X, Y, and Z displacements using the image acquisition and analysis system (Morphostar 5.12; IMSTAR). Analyzed areas in the striatum encompassed the entire region showing mutant htt aggregates as revealed using the htt antibody 2B4 (∼2–4 mm2, depending on the specimens acquired). This represented, on average, 300–500 contiguous digitized images per animal, corresponding to contiguous 112 × 91 μm fields of view. Image pixels were 0.12 × 0.12 μm in size. Section lighting was similar for all of the image acquisitions performed, and the homogeneity of lighting was automatically corrected using blank images. Pixel intensity histograms showed absence of saturation and no dark pixels at gray level 0. To avoid underestimation of the small 2B4-positive signals (small aggregates), each field of view was acquired at 12 equidistant different focal planes over 5 μm along the z-axis within the section. The set of stacked images was projected over the z-axis to show the large as well as all of the very small particles in the volume analyzed. Projection (stacked) images were segmented for quantification of dark objects (aggregates). The segmentation of objects was obtained using light-intensity thresholding followed by the size of the object and shape filtering. Using this procedure, 2B4-positive objects with an apparent cross section area >0.5 μm2 were reliably detected. For each image, the objects touching one of the two X or Y borders of the fields of view were eliminated. Depending on the animal considered, ∼1500 objects/section were detected and analyzed. The area of each object was attributed to one of the 40 classes of 1 μm2 bin of the distribution histogram. In addition to this automated procedure, for all animals, we performed manual cell counts to estimate the number of nuclear inclusions (NIs) and small neuritic-like aggregates using five fields of view (112 × 91 μm).
Additionaly, we performed confocal analysis of 2B4 signals in brain sections counterstained with a nuclear marker. Images of 2B4-stained sections counterstained with the nuclear marker acridine orange were acquired at 100× magnification on a Leica confocal microscope and analyzed using the Metaview image analysis software (Roper Scientific). Each field of view was acquired at 10 equidistant different focal planes over 15 μm along the z-axis within the section in the red (2B4 staining) and green (acridine orange staining) channels. The set of stacked images was projected over the z-axis to analyze the intracellular distribution of htt aggregates (2B4 staining) compared with nuclear staining (acridine orange staining) using five fields of view (150 × 150 μm) per animal.
Confocal analysis of HA-positive signals.
Images of HA-stained sections counterstained with acridine orange were acquired using the 100× objective of a Leica confocal microscope and morphometric analysis was performed using Metaview (Roper Scientific). Analyses were performed for three rats using six fields of view (150 × 150 μm) per animal. For each of the focal planes, HA signals were quantified from the nucleus (acridine orange staining). To assess cytoplasmic expression, nuclear HA signals were blackened, and the remaining signals were quantified. Cytoplasmic analysis was performed only if nuclear HA signals were detected in the same confocal plane and cell.
Aggregation assays.
KKQ40KK and KK(QA)20KK peptides were chemically synthesized by the Keck Biotechnology Center (Yale University, New Haven, CT) and supplied as crude reaction products. The peptides were purified to homogeneity by reverse-phase HPLC and characterized by electrospray mass spectrometry. For spontaneous aggregation kinetics, the peptides were disaggregated and solubilized in pH 3 water and then adjusted to the desired concentration in PBS (Chen and Wetzel, 2001). The concentrations of the peptides were checked on analytical reverse-phase HPLC by converting the peptide peak area (A215) to micrograms of peptide using a standard curve generated from a polyGln solution of known concentration (amino acid analysis) (Wetzel, 2004). Aggregates used in seeding reactions were grown at 37°C in PBS and collected by centrifuging at 14,000 rpm for 30 min (Chen et al., 2002). The concentrations of the aggregates were determined by disaggregating an aliquot of aggregate, solubilizing in pH 3 water, and were then quantified as above by analytical HPLC. Known weight concentrations of aggregates were sonicated with five 30 s pulses on ice with a probe sonicator to prepare the seeds. Monomer elongation reactions were seeded with 10% aggregates by weight. Both spontaneous and seeded reactions were performed at 37°C and kinetics of aggregation monitored by thioflavin T (ThT) fluorescence (Chen et al., 2002) as follows. A 10 μl aliquot of a 2.5 mm ThT solution was added to a cuvette containing 300 μl of the peptide suspension and fluorescence quantified on a PerkinElmer LS50B luminescence spectrophotometer at the following settings: excitation wavelength, 450 nm (slit width, 5 nm); emission wavelength, 489 nm (slit width, 10 nm).
α4-integrin transcriptional repression assay.
Human 293T cells were grown in DMEM–low glucose (Invitrogen) supplemented with 10% fetal calf serum, penicillin, and streptomycin. Twenty-four hours before transfection, 2.5 × 105 cells per well were seeded in six-well dishes. CA150 expression plasmid (62.5–250 ng) or 250 ng of empty vector, and 166 ng of −42a4CAT reporter, was transfected per well using calcium phosphate. Transfections were performed in triplicate, and cell lysate was prepared at 48 h after transfection by 3× freeze/thaw in 100 mm Tris, pH 8.0. CAT assays were performed using the diffusion method of Neumann (Neumann et al., 1987). The average slope of CAT activity versus time for triplicate samples was determined and designated CAT activity. Levels of CA150 and CA150ΔQA expression were determined by Western blot using antibody to the T7 epitope tag (Novagen, Madison, WI). Polypyrimidine-tract binding protein was used as a loading control.
Statistical analyses.
ANOVA tests were used for statistical analysis. ANOVA was followed by Tukey's multiple-comparison tests for the analysis of cell culture data.
Results
CA150 overexpression rescues mutant htt toxicity in primary striatal rat cultures
To test for the effect of CA150 overexpression on striatal cells expressing mutant htt, we used rat primary striatal cultures subjected to lentiviral infection. In this model, htt171-82Q expression under the control of the PGK promoter produces a 50% reduction in Neu-N expression compared with htt171-19Q, an effect observed at 2 months and accompanied by the formation of htt-positive neuritic aggregates (NAs) and nuclear aggregates (Zala et al., 2005). At 8 weeks, CA150 coexpression with htt171-82Q restored Neu-N expression to htt171-19Q levels, an effect dependent on the presence of the (Gln-Ala)38 repeat normally found in the protein (Fig. 1A). To rule out a reduction of mutant htt expression levels by CA150, as a transcriptional regulator (Sune and Garcia-Blanco, 1999; Lin et al., 2004; Smith et al., 2004), we tested for the effect of CA150 and CA150ΔQA on the expression level of GFP driven by the PGK promoter at 6 weeks, using Western blot. Cells coexpressing GFP and CA150 species showed no change in the level of GFP expression compared with cells coexpressing GFP and LACZ (Fig. 1B). Additionally, we tested for the effect of CA150 species on the expression levels of normal and mutant htt species at 3 weeks (e.g., before the appearance of aggregates). As shown by Western blot of htt migrating as a single band, CA150 overexpression did not reduce the expression level of htt (Fig. 1C). Thus, CA150 appeared to rescue mutant htt toxicity with no effect on PGK-mediated transcription.
Figure 1.

CA150 overexpression protects primary striatal cells from mutant htt toxicity. A, Eight weeks after infection, neuronal dysfunction was measured by Neu-N immunohistochemistry. Neu-N expression was restored after htt171-82Q and CA150 infection compared with cells infected with htt171-82Q alone (∗∗p < 0.005; average rescue is 83.3%). Data are mean ± SD (n = 6) expressed as percentages of control, namely htt171-19Q (27.2 ± 9 Neu-N-positive cells/field of view). The mean rescue of htt171-82Q-infected cells by CA150 was 83.3%. B, CA150, and CA150 deleted for the (Gln-Ala)38 repeat (CA150ΔQA), did not alter PGK-driven expression as assessed by GFP immunoblot analysis 6 weeks after infection. C, No noticeable difference of htt transgene expression by CA150 or CA150ΔQA overexpression was observed 3 weeks after infection, as tested by immunoblot for the myc tag of htt.
CA150 overexpression rescues striatal pathology in rats
Having observed that CA150 overexpression may be protective against polyGlns toxicity in a cellular model of mutant htt striatal neurotoxicity, we sought to examine whether CA150 expression could be protective in vivo using a lentiviral-based model of mutant htt neurotoxicity in rats (de Almeida et al., 2001). In this model, N-terminal mutant htt (first 171 amino acids) under the control of the PGK promoter produces strong striatal dysfunction 8 weeks after intracerebral injection, as revealed by DARPP-32 immunostaining (de Almeida et al., 2001). The introduction of a stereotactic needle and sham striatal injections did not produce striatal lesions as detected using DARPP-32 immunostaining (data not shown). First, we tested for the toxicity of CA150 overexpression using DARPP-32 and Neu-N as an early and late marker of striatal dysfunction, respectively (de Almeida et al., 2001). The expression of CA150 was monitored using HA epitope staining. No striatal dysfunction across serial sections was observed for CA150 or CA150ΔQA compared with htt171-19Q (Fig. 2A). We then tested for CA150 toxicity at a later stage (12 weeks) and under conditions of coexpression of htt171-19Q with either CA150 or CA150ΔQA. No striatal degeneration (as inferred from DARPP-32 and Neu-N immunostaining) was observed in these conditions (Fig. 2B). Thus, expressing CA150 either alone or in combination with htt171-19Q was not toxic to the rat striatum at 8–12 weeks after injection, a stage at which mutant htt produces striatal degeneration (de Almeida et al., 2001).
Figure 2.
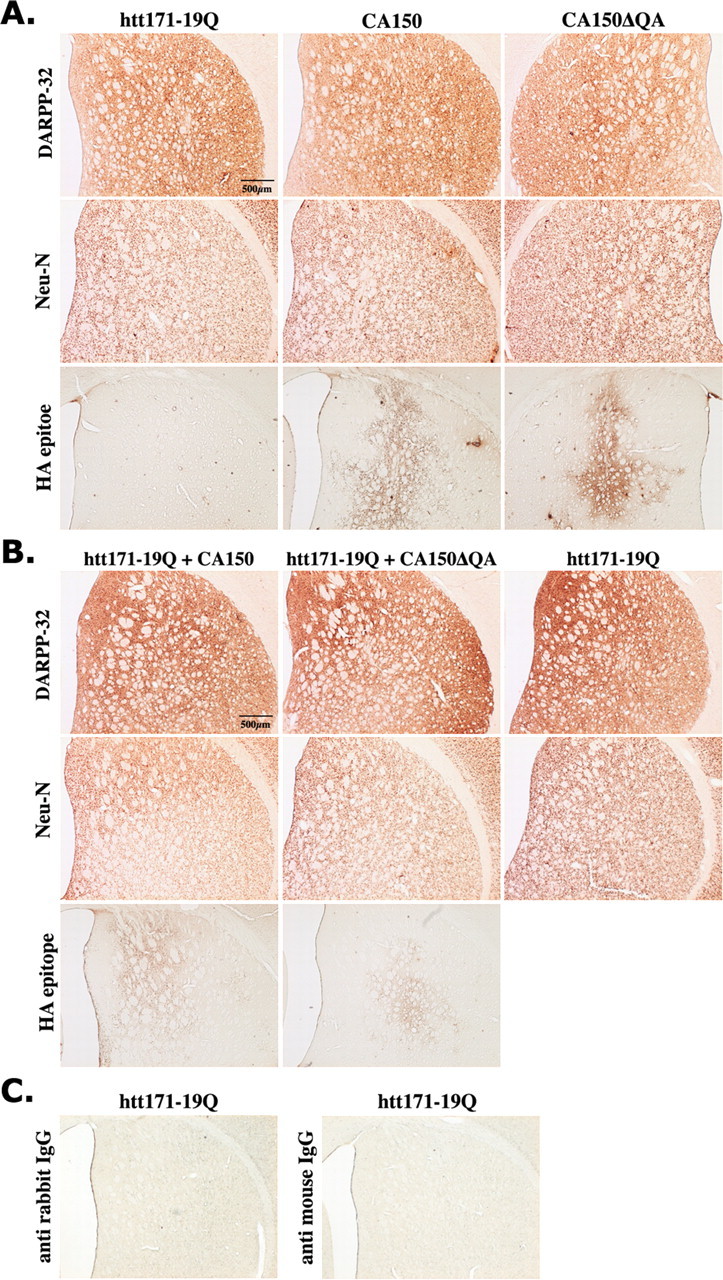
The overexpression of CA150 or CA150ΔQA does not produce a striatal pathology in rats. Striatal toxicity was assessed using DARPP-32 and Neu-N immunostaining. Immunostaining for extragenous CA150 is shown (HA epitope). No lesion was observed at 8 or 12 weeks compared with animals injected with htt171-19Q lentiviruses. A, Simple infections analyzed at 8 weeks. B, Double infections analyzed at 12 weeks. C, Striatal sections incubated only with the secondary antibodies (left panel, no primary DARPP-32 antiserum; right panel, no Neu-N antibody) show no immunostaining.
Next, we tested for the rescue of htt171-82Q toxicity by CA150 overexpression using serial sections (Fig. 3). At 8 weeks, the DARPP-32 staining indicated that CA150 rescued up to 77% of the degeneration produced by Htt171-82Q, whereas CA150ΔQA rescued toxicity to a lesser extent (up to 37%) (Fig. 3A,B). At 12 weeks, rescue was observed neither with CA150 nor CA150ΔQA, suggesting that CA150 species do not block but delay the striatal degeneration induced by N-terminal mutant htt, a progressive phenomenon that reaches a plateau at 12 weeks in this model (de Almeida et al., 2001). A similar profile of activity was observed using Neu-N staining, with a partial rescue (70% reduction of Neu-N loss) by CA150 at 8 weeks (Fig. 3A,C). At 12 weeks, and likewise with DARPP-32 immunostaining, Neu-N staining showed no rescue by CA150.
Figure 3.
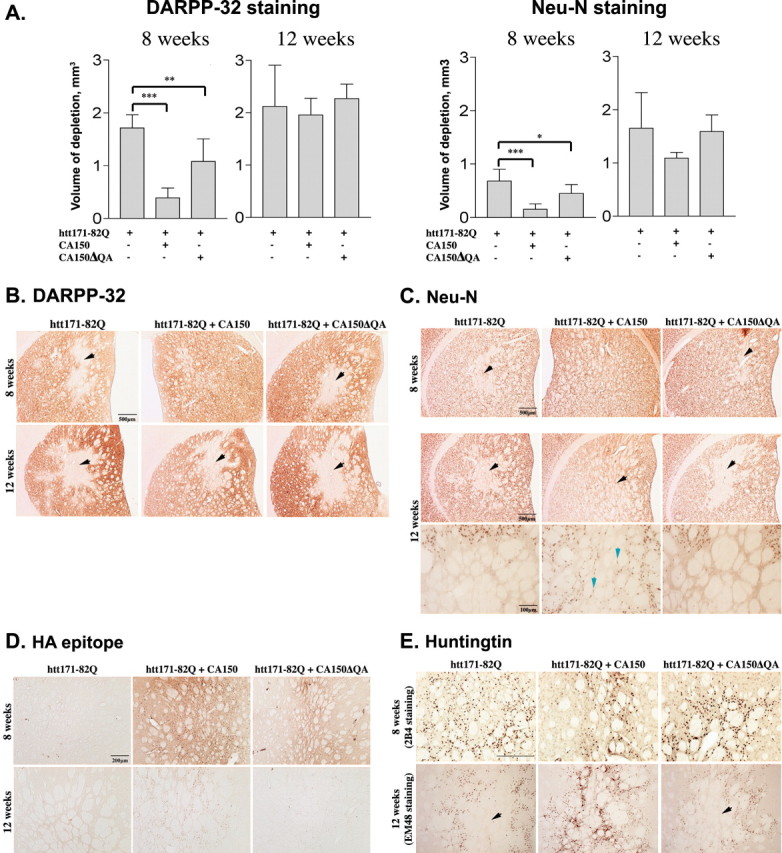
CA150 attenuates the striatal neuropathology induced by mutant htt overexpression in a (Gln-ala)38-dependent manner. Rat striata injected with the indicated lentiviruses were examined for Neu-N and DARPP-32 expression 8 or 12 weeks after infection. A, Quantification of striatal lesions at 8 and 12 weeks. At 8 weeks, CA150 rescued 77.3% of the DARPP-32 downregulation and 70% of the Neu-N downregulation, whereas CA150ΔQA rescued 37.16% of the DARPP-32 downregulation and 34.1% of the Neu-N downregulation. No effect was observed at 12 weeks. Data are mean ± SD from seven to eight animals (∗∗∗p < 0.0001, ∗∗p < 0.001, and ∗p < 0.05 compared with rats injected with htt171-82Q alone). B, Examples of DARPP-32 immunostaining. The arrowheads indicate lesions. C, Examples of Neu-N immunostaining. The black arrowheads indicate lesions at 8 or 12 weeks. Also shown are high-magnification views (bottom panels) of the lesions at 12 weeks. The blue arrowheads indicate Neu-N-positive cells that survived in the lesion of rats coinjected with htt171-82Q and CA150. D, Examples of immunostaining (HA epitope) revealing the expression of extragenous CA150 species. Shown here are the lesioned areas. The most intense staining was observed in the presence of CA150 rescuing activity. E, Examples of human mutant htt expression revealed by 2B4 (8 weeks) and EM48 (12 weeks) immunostaining. The black arrowheads show the lesions produced by mutant htt.
Finally, we tested for the effects of mutant htt and CA150 on striatal morphology at 8 and 12 weeks (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Compared with wild-type htt expression, mutant htt expression produced striatal shrinkage as detected by the increase in the percentage area occupied by the bundles of fibers that cross the striatum. Striatal shrinkage, here likely to result from the loss of medium size spiny neurons as inferred from DARPP-32 and cresyl violet staining, was fully rescued at 8 weeks by CA150 expression and partially rescued by CA150ΔQA expression. At 12 weeks, CA150 showed partial rescuing activity, and CA150ΔQA showed no rescuing activity.
Together, these data suggested that CA150 overexpression was able to delay the progressive neuropathology induced by mutant htt, an effect strongly dependent on the presence of the (Gln-Ala)38 repeat in the protein.
CA150 overexpression increases neuritic aggregates in rats expressing mutant htt
We noticed that CA150 overexpression may be associated with a redistribution of htt aggregates and further analyzed this phenomenon using the ratio for NAs versus NIs. To test whether the rescue by CA150 may be accompanied by a change in aggregate distribution, we quantified the number and size of 2B4 signals at 8 weeks. Compared with htt171-82Q expression, the coexpression of htt171-82Q and CA150 strongly increased the number of small (2–6 μm2) aggregates and slightly decreased the number of large (16–23 μm2) aggregates, an effect not observed for the coexpression of htt171-82Q and CA150ΔQA (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). To test whether this effect reflected changes in the frequency of NAs versus NIs, we assessed the aggregation patterns using visual inspection, and confocal analysis in the presence of nuclear staining. Compared with htt171-82Q expression, the coexpression of htt171-82Q and CA150 strongly increased the NA/NI ratio, an effect not observed for the coexpression of htt171-82Q and CA150ΔQA (Fig. 4A,B). Similar results were obtained at 12 weeks after infection (data not shown). Altogether, these observations indicated that the rescue of mutant htt toxicity was accompanied by an increased proportion of NAs. To further characterize the nature of these aggregates, we sought to examine whether they may contain the exogenous CA150 protein. The coimmunostaining with HA epitope (tag for CA150) and ubiquitin antibodies indicated that NIs were positive for exogenous CA150 (Fig. 4C,D). A round and diffuse expression of exogenous CA150 was also observed around the ubiquitin-positive NIs, which is suggestive of nuclear expression (Fig. 4C,D). In contrast, the small and aligned ubiquitin-positive aggregates were negative for HA, suggesting that NAs did not contain exogenous CA150 (Fig. 4C,D). These observations suggested that the increased proportion of NAs caused by CA150 overexpression was not attributable to a direct interaction of mutant htt with CA150. Rather, the occurrence of CA150-negative NAs was suggestive of an indirect effect of nuclear CA150 on the nucleo-cytoplasmic distribution and accumulation of mutant htt.
Figure 4.

CA150 overexpression increases NA formation in the rat striatum. At 8 weeks, htt-containing aggregates were revealed by immunohistochemistry using the antibody 2B4 and nuclei stained using acridine orange. A, Number of NAs, NIs, and NA/NI ratio. The coexpression of htt171-82Q and CA150 led to a 5.65-fold increase in the number of NAs compared with rats injected with htt171-82Q alone (∗∗p < 0.001) without decreasing NIs, which corresponds to a 7.04-fold increase in the NA/NI ratio (∗∗∗p < 0.0001). The coexpression of htt171-82Q and CA150ΔQA did not modify the number of NAs or NIs compared with expression of htt171-82Q alone. Aggregate counts were performed on double-stained sections (2B4, acridine orange) for five fields of view/section at high magnification. Data are mean ± SD from six to eight animals. B, Examples of 2B4 immunostaining at 100× magnification. Top panels show 2B4 staining with gray arrowheads for NIs and blue arrowheads for NAs. Bottom panels show confocal images with 2B4 (red) and nuclear (green) staining. The NIs (white arrowheads) and NAs (yellow arrowheads) are also shown. C, Area and number of objects positive for either exogenous CA150 (HA epitope) or ubiquitin, or both, eight weeks after coinjection of htt171-82Q and CA150. D, Example of HA and ubiquitin coimmunostaining. The white arrowheads show ubiquitinated, HA-positive large objects likely to represent NIs. The blue arrowheads show ubiquitin-positive and HA-negative small objects likely to represent NAs.
The (Gln-Ala)38 repeat in CA150 influences the localization of the protein
Having observed that the (Gln-Ala)38 repeat may be important to CA150 rescuing activity and that CA150 rescuing activity may originate from nuclear forms of the protein, we investigated the influence of the (Gln-Ala)38 repeat on the intracellular distribution of the protein in nontoxic conditions, namely the expression of CA150 species alone or in combination with htt171-19Q. When full-length CA150 was expressed alone or in combination with the nonpathogenic htt171-19Q protein, its expression was restricted to the nucleus in confocal planes (Fig. 5A,B). In contrast, CA150ΔQA localization was decreased in the nucleus, and cytoplasmic expression was detected (Fig. 5A,B). These observations suggested that CA150 rescuing activity was partially dependent on the (Gln-Ala)38 repeat because this repeat may participate to the retention of the protein in the nucleus.
Figure 5.
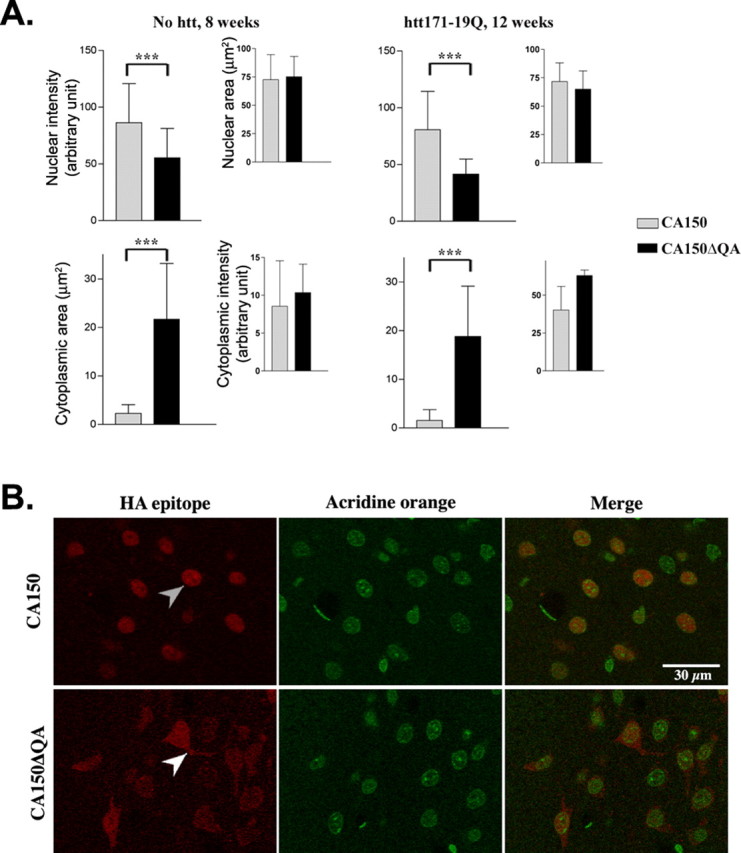
The (Gln-Ala)38 repeat of CA150 influences the nuclear localization of the protein. Eight or 12 weeks after infection of rat striata, the localization of exogenous CA150 and CA150ΔQA was revealed using HA epitope staining. Nuclei were stained using acridine orange. A, Quantification of cytoplasmic and nuclear signals from confocal planes for CA150 compared with CA150ΔQA. Shown are nuclear intensity of HA signals and cytoplasmic area of HA staining. Rats injected with CA150ΔQA alone showed increased cytoplasmic signals and weaker nuclear signals compared with rats injected with CA150 alone, an effect not influenced by htt171-19Q or the time after infection (∗∗∗p < 0.0001). Data are mean ± SD from six fields of view. B, Confocal images of HA epitope staining (red) and nuclear counterstaining (green). The gray arrowheads show nuclear CA150 and white arrowheads show nucleo-cytoplasmic CA150ΔQA.
CA150 rescues 109Q/109Q striatal cell death in a (Gln-Ala)38-dependent manner
Having shown that CA150 rescued cell loss in overexpression conditions for mutant htt neurotoxicity, we sought to examine the effect of CA150 overexpression on striatal toxicity in a model that reflects a closer situation to the genetic conditions HD patients, namely striatal cells derived from HdhQ111 knock-in mice (Trettel et al., 2000). In this system, β-galactosidase overexpression and serum deprivation produces polyGln-dependent cell death (Gauthier et al., 2004). The overexpression of CA150 fully rescued the cell death induced in 109Q/109Q cells back to 7Q/7Q levels, an effect lost for CA150ΔQA (Fig. 6A). The CA150 rescuing activity was not caused by a reduction in the expression level of full-length mutant htt as shown by Western blot (Fig. 6B). Thus, CA150 overexpression was able to rescue mutant htt neurotoxicity in knock-in conditions.
Figure 6.

Effect of overexpression of CA150 species in 109Q/109Q striatal lines. A, CA150 overexpression reduces cell death induced in 109Q/109Q striatal lines compared with cells transfected with empty vector (∗∗∗p < 0.0001). No difference in cell death was observed in cells overexpressing CA150ΔQA compared with cells transfected with the empty vector. Overexpression of a CA150 fragment containing the QA repeat and 16 aa N-terminal and 76 aa C-terminal to the repeat (CA150162–331) was toxic for 7Q/7Q cells compared with cells transfected with empty vector (p < 0.0001). Data are mean ± SD of six independent experiments (100–150 cells counted per experiment). B, No noticeable difference of htt expression by CA150 species or (QA)38 overexpression was observed as tested by Western blotting.
CA150 rescuing activity is unrelated to a blockage of polyGlns in mutant htt by the (Gln-Ala)38 repeat
The biological profile for CA150 rescuing activity in the overexpression system pointed to a role of the (Gln-Ala)38 repeat in restricting CA150 localization to the nucleus and to an indirect modification of mutant htt nucleo-cytoplasmic distribution/trafficking that did not involve a reduction of NI formation. The rescue in the knock-in conditions, a system that does not show NIs, further supported the notion that CA150 rescuing activity was unlinked to a decrease in potentially toxic NIs. To explore further the mode of action of CA150, we sought to examine the contribution of the (Gln-Ala)38 repeat to the rescue of mutant htt toxicity. Broken sequences of polyGln are capable of interacting with pure polyGln sequences, for example in the ability to seed aggregate growth (Thakur and Wetzel, 2002), raising the possibility that the Gln-Ala repeat in CA150 may be able to interact with polyGlns in mutant htt, thereby blocking their toxicity. To test for this possibility, we used two assays. First, we tested a 168 amino acid CA150 fragment (amino acids 162–331) containing the (Gln-Ala)38 repeat for rescue of cell death in the striatal cells derived from knock-in mice. The CA150 fragment was found to be toxic in 7Q/7Q cells (Fig. 6A), an effect unrelated to an increase in htt expression (Fig. 6B). This suggested that, should Gln-Ala repeats be capable of binding to the polyGln tract in htt, the poly(Gln-Ala) is less likely to be protective than to enhance toxicity, possibly through enhancing the aggregation of polyGln proteins. Second, we compared the spontaneous and seeded aggregation of two peptides containing either a Q40 sequence or a (QA)20 repeat sequence. As a repeat sequence above the repeat length threshold for HD, Q40 is known to rigorously aggregate in vitro (Scherzinger et al., 1999; Chen et al., 2001). Interestingly, the (QA)20 peptide spontaneously aggregated significantly more aggressively than Q40 (Fig. 7). Additionally, (QA)20 aggregates were very efficient seeds for Q40 elongation (Fig. 7), which is surprising, given the very low seeding efficiencies observed in efforts to promote elongation by one amyloidogenic peptide using amyloid fibril seeds from other peptides (O'Nuallain et al., 2004). Because they were observed using repeat sequences in simple buffers at nonphysiological concentrations, these effects may not fully recapitulate the activity in the context of the carrier proteins. However, using repeat sequences has previously allowed the biological consequences of aggregation to be best understood (RICK), and our data suggest that poly(Gln-Ala) sequences in vivo might be able to promote polyGln aggregation by rapidly aggregating to form seeds capable of very efficient cross-seeding of polyGln elongation. Together, the two assays used above strongly suggested that, should Gln-Ala repeats be able to interact with polyGlns in the context of proteins, this does not translate in reduced toxicity of mutant htt.
Figure 7.

Formation of amyloid-like aggregates from glutamine containing repeat peptides. Monomeric KKQ40KK (▪, □) and KK(QA)20KK (•, ○) were incubated in the 10–20 μm range at 37°C in PBS and amyloid formation monitored by the ThT reaction. Monomer alone (○, □), monomeric Q40 peptide plus 10% by weight of (QA)20 aggregate seeds (•), and monomeric (QA)20 peptide plus 10% by weight of Q40 aggregate seeds (▪) are shown.
The CA150 protein deleted for the (Gln-Ala)38 repeat is able to repress transcription
Having observed that CA150 rescuing activity may not involve a direct blockage of the polyGln tract in htt by the (Gln-Ala) repeat normally found in the protein, we sought to examine whether the decreased ability of CA150ΔQA to rescue mutant htt toxicity may be explained by a decreased ability of the deleted protein to regulate transcription using an α4-integrin repression assay in HEK293T cells (Goldstrohm et al., 2001). We observed that CA150 and CA150ΔQA both localize to HEK293T nuclei (supplemental Fig. 3A, available at www.jneurosci.org as supplemental material), thus allowing the effect of the two protein species on transcriptional repression to be readily compared in these cells. Compared with full-length CA150, CA150ΔQA showed no appreciable difference in repressing the activity of the α4-integrin promoter as tested in dose–response assays (supplemental Fig. 3B, available at www.jneurosci.org as supplemental material), suggesting that the (Gln-Ala)38 repeat is dispensable for transcriptional repression by CA150. Thus, the reduced ability of CA150ΔQA to rescue mutant htt neurotoxicity was unlikely to directly result from a loss of transcriptional repression properties of the deleted protein.
Discussion
Several studies support the notion that the abnormal regulation of transcription is central to neuronal cell death in HD, a mechanism that may be notably pertinent to cell-autonomous processes (Steffan et al., 2001; Dunah et al., 2002; Jiang et al., 2003; Marcora et al., 2003; Sugars and Rubinsztein, 2003; Zuccato et al., 2003; Gu et al., 2005). In addition to the loss of normal htt function in retaining transcriptional repressors in the cytoplasm (Cattaneo et al., 2001; Marcora et al., 2003), another mechanism may be the abnormal interactions of misfolded mutant htt species with transcription factors in the nucleus as is for example the case for Sp1 (Dunah et al., 2002). Although normal htt is primarily a cytoplasmic protein associated with the cytoskeleton (Legendre-Guillemin et al., 2002; Gauthier et al., 2004), it may also be present in the nucleus (Kegel et al., 2002), and N-terminal mutant htt species may be prone to enter and stay in the nucleus, producing NIs and cytotoxicity (Cornett et al., 2005). While NI formation is strongly associated with polyglutamine expansion in HD (Cummings and Zoghbi, 2000), the pathogenic role of NIs in HD and other polyglutamine diseases is controversial. Several model-system studies suggest that NIs may be protective (Saudou et al., 1998; Arrasate et al., 2004; Bowman et al., 2005), or coincidental (Sisodia, 1998), but it is also possible that toxicity may derive from difficult-to-detect microaggregates. Altered regulation of transcription may thus originate from either the abnormal interaction of soluble mutant htt with transcription factors (Yu et al., 2002), the sequestration of transcription factors into forming NIs (Nucifora et al., 2001), or both. Among the transcriptional regulators that accumulate and may be strongly altered in HD is CA150 (Holbert et al., 2001). In contrast to well characterized htt partners that belong to the transcriptional machinery such as CBP (Nucifora et al., 2001) or Sp1 (Li et al., 2002), the mechanisms involving CA150 in HD pathogenesis are poorly understood. It is important to understand these mechanisms for two reasons. First, CA150 may not be a general regulator of transcription but may regulate the expression of specific TATA-box-containing promoters (Sune and Garcia-Blanco, 1999), and recent findings suggest that CA150 can activate nuclear factor κB-dependent transcription, an effect that may be influenced by htt (J. Pearson and M. Garcia-Blanco, unpublished observations). Second, CA150 may be a modifier gene, the large (Gln-Ala) repeats in CA150 explaining a small but significant portion of the AO variability in HD (Holbert et al., 2001; Chattopadhyay et al., 2003). The strong CA150 rescuing activity reported herein suggests that CA150 function may be greatly altered by mutant htt in HD. Additionally, our study provides clues about the potential mechanism(s) for rescue by CA150.
The rescue by CA150 was dependent on the (Gln-Ala)38 repeat, suggesting a role for the repeat domain in the normal function of the protein. Interestingly, CA150ΔQA expression was no longer restricted to the nucleus in rat brains compared with CA150 expression alone, some level of expression being observed in the cytoplasm, thus linking the nuclear restriction of exogenous CA150 to its ability to rescue mutant htt neurotoxicity. This is in contrast to the rescuing activity of the transcriptional regulator and htt partner CBP. The CBP rescuing activity was indeed reported not to be dependent on the presence of the polyGln domain normally present in the protein, suggesting that the polyGln domain may not be essential to the CBP function(s) involved in the rescue (Nucifora et al., 2001). Regarding CA150, because the (Gln-Ala)38 repeat does not appear to be required for the transcriptional repression ability of the protein, other mechanisms may be considered to explain CA150 rescuing activity, such as, for example, the restoration of transcriptional activation (Sune et al., 1997).
The rescue by CA150 was unrelated to a beneficial interaction of the (Gln-Ala)38 repeat in CA150 with the polyGln domain in mutant htt as the nuclear expression of a short CA150 fragment that contains the repeat did not rescue but enhanced the vulnerability of striatal cells to cell death. Additionally, a (Gln-Ala)20 peptide potentiated polyGln elongation in vitro, consistent with the finding that the CA150 N-terminal fragment may be toxic to striatal cells through enhancing htt aggregation.
Finally, the rescue by CA150 was unlikely to involve a reduction of NIs as (1) rescue was observed in striatal cells derived from HdhQ111 knock-in mice that do not show htt accumulation, and (2) the increase in the NA/NI ratio in rat brains was the result of NA increase, with no appreciable NI decrease detected. This suggested that the rescue by CA150 may involve soluble proteins. One may speculate about two possibilities that are not mutually exclusive. First, the striking increase in NAs negative for CA150 was suggestive of an indirect effect of exogenous CA150 on mutant htt nucleo-cytoplasmic distribution. This may be caused by an increased amount of soluble mutant htt in the nucleus, allowing its export from the nucleus (Cornett et al., 2005), and a higher cytoplasmic concentration, which may result in more NAs. This is also consistent with the notion that impaired regulation of N-terminal htt nucleo-cytoplasmic trafficking may cause the protein to be toxic in the nucleus (Schilling et al., 2004; Cornett et al., 2005). Second, rescue could involve a restoration of normal levels of nuclear protein cooperativity for transcriptional signaling. The splicing machinery may not be primarily involved because, although CA150 associates with the spliceosome, it may be dispensable for splicing (Lin et al., 2004). The (Gln-Ala)38 repeat is dispensable for transcriptional repression by CA150 in human 293T cells, suggesting that the decreased ability of CA150ΔQA to rescue mutant htt neurotoxicity is unlikely to directly result from a loss of transcriptional repression properties of the deleted protein. However, one cannot exclude the possibility that the abnormal presence of CA150ΔQA in the cytoplasm of rat striatal cells may compromise its ability to repress transcription. Thus, cellular transcriptional processes may be involved in CA150 rescuing activity. Additional studies will be needed to identify rescue processes that may involve either CA150 transcriptional repression ability or CA150 transcriptional activation ability (Sune et al., 1997) or both. Pathways that modulate transcription and contain partner proteins common to htt and CA150 (Goehler et al., 2004) are of particular interest because they may be used by exogenous CA150 to tip the cell toward survival. Of note is PIASy, a repressor of the transforming growth factor-β/Smad and nuclear factor-κB pathways with putative SUMO E3 ligase activity (Sachdev et al., 2001; Goehler et al., 2004; Zhang et al., 2004; Zhu et al., 2004) that binds to htt and CA150 (Goehler et al., 2004). Additionally, the molecular interaction networks containing htt and CA150 (Goehler et al., 2004) may suggest genetic markers to best explain the variability of the AO in HD and biomarkers to monitor HD progression.
Finally, together with our previous finding that enlarged alleles of CA150 may influence the AO in HD (Holbert et al., 2001), the data reported herein suggest that, like CA150 lacking the (Gln-Ala)38 repeat, CA150 species containing an enlarged repeat allele may be hypomorphic. Long alleles of CA150 may indeed themselves cause CA150 dysfunction because the influence of CA150 on the AO in HD was independent from the polyGln length in mutant htt (Holbert et al., 2001).
In summary, our data indicate that CA150 dysfunction may be an important component of cell-autonomous mechanisms for striatal cell death in HD pathogenesis. Additional studies will be needed to determine whether CA150 overexpression may improve behavior in animal models of HD (Bates et al., 1998; Lin et al., 2001; Wheeler et al., 2002; Van Raamsdonk et al., 2005), and to identify the mechanisms for CA150 to modulate mutant htt neurotoxicity. Our data indeed suggest that restoring normal protein cooperativity and transport around CA150 and htt may be neuroprotective in HD, which calls for the characterization of CA150 partner proteins essential to neuronal cell survival, and the search for compounds that may act as surrogates of CA150 normal activity toward these targets.
Footnotes
*N.D. and C.N. contributed equally to this work.
This work was supported by Institut National de la Santé et de la Recherche Médicale (INSERM), the Région Ile-de-France and the Fondation pour la Recherche Médicale (M.A.), INSERM Avenir (C.N.), the Swiss National Science Foundation (Switzerland) (N.D.), National Institute of Health Grants R01 AG19322 (R.W., A.T.) and R01 GM071037 (M. A. Garcia Blanco), and the Huntington's Disease Society of America (J.P.). We thank Xiao-Jang Li for critical reading, E. Cattaneo for providing striatal cells derived from HdhQ111 knock-in mice, Y. Trottier for providing the htt antibody 2B4, M. A. Garcia-Blanco for the α4-integrin repression assays performed in his laboratory, E. Duchesnay for help in the statistical analysis of 2B4-positive aggregates distributions, and C. Escartin (Universities Research Association, Atomic Energy Commission/Centre National de la Recherche Scientifique 2210) for testing sham striatal injections.
References
- Arrasate M, Mitra S, Schweitzer ES, Segal MR, Finkbeiner S (2004). Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature 431:805–810. [DOI] [PubMed] [Google Scholar]
- Bates GP, Mangiarini L, Davies SW (1998). Transgenic mice in the study of polyglutamine repeat expansion diseases. Brain Pathol 8:699–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowman AB, Yoo SY, Dantuma NP, Zoghbi HY (2005). Neuronal dysfunction in a polyglutamine disease model occurs in the absence of ubiquitin-proteasome system impairment and inversely correlates with the degree of nuclear inclusion formation. Hum Mol Genet 14:679–691. [DOI] [PubMed] [Google Scholar]
- Carty SM, Greenleaf AL (2002). Hyperphosphorylated C-terminal repeat domain-associating proteins in the nuclear proteome link transcription to DNA/chromatin modification and RNA processing. Mol Cell Proteomics 1:598–610. [DOI] [PubMed] [Google Scholar]
- Cattaneo E, Rigamonti D, Goffredo D, Zuccato C, Squitieri F, Sipione S (2001). Loss of normal huntingtin function: new developments in Huntington's disease research. Trends Neurosci 24:182–188. [DOI] [PubMed] [Google Scholar]
- Chattopadhyay B, Ghosh S, Gangopadhyay PK, Das SK, Roy T, Sinha KK, Jha DK, Mukherjee SC, Chakraborty A, Singhal BS, Bhattacharya AK, Bhattacharyya NP (2003). Modulation of age at onset in Huntington's disease and spinocerebellar ataxia type 2 patients originated from eastern India. Neurosci Lett 345:93–96. [DOI] [PubMed] [Google Scholar]
- Chen S, Wetzel R (2001). Solubilization and disaggregation of polyglutamine peptides. Protein Sci 10:887–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Berthelier V, Yang W, Wetzel R (2001). Polyglutamine aggregation behavior in vitro supports a recruitment mechanism of cytotoxicity. J Mol Biol 311:173–182. [DOI] [PubMed] [Google Scholar]
- Chen S, Berthelier V, Hamilton JB, O'Nuallain B, Wetzel R (2002). Amyloid-like features of polyglutamine aggregates and their assembly kinetics. Biochemistry 41:7391–7399. [DOI] [PubMed] [Google Scholar]
- Cornett J, Cao F, Wang CE, Ross CA, Bates GP, Li SH, Li XJ (2005). Polyglutamine expansion of huntingtin impairs its nuclear export. Nat Genet 37:198–204. [DOI] [PubMed] [Google Scholar]
- Cummings CJ, Zoghbi HY (2000). Trinucleotide repeats: mechanisms and pathophysiology. Annu Rev Genomics Hum Genet 1:281–328. [DOI] [PubMed] [Google Scholar]
- de Almeida LP, Zala D, Aebischer P, Deglon N (2001). Neuroprotective effect of a CNTF-expressing lentiviral vector in the quinolinic acid rat model of Huntington's disease. Neurobiol Dis 8:433–446. [DOI] [PubMed] [Google Scholar]
- de Almeida LP, Ross CA, Zala D, Aebischer P, Deglon N (2002). Lentiviral-mediated delivery of mutant huntingtin in the striatum of rats induces a selective neuropathology modulated by polyglutamine repeat size, huntingtin expression levels, and protein length. J Neurosci 22:3473–3483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deglon N, Tseng JL, Bensadoun JC, Zurn AD, Arsenijevic Y, Pereira de Almeida L, Zufferey R, Trono D, Aebischer P (2000). Self-inactivating lentiviral vectors with enhanced transgene expression as potential gene transfer system in Parkinson's disease. Hum Gene Ther 11:179–190. [DOI] [PubMed] [Google Scholar]
- Dunah AW, Jeong H, Griffin A, Kim YM, Standaert DG, Hersch SM, Mouradian MM, Young AB, Tanese N, Krainc D (2002). Sp1 and TAFII130 transcriptional activity disrupted in early Huntington's disease. Science 296:2238–2243. [DOI] [PubMed] [Google Scholar]
- Fleury S, Simeoni E, Zuppinger C, Deglon N, von Segesser LK, Kappenberger L, Vassalli G (2003). Multiply attenuated, self-inactivating lentiviral vectors efficiently deliver and express genes for extended periods of time in adult rat cardiomyocytes in vivo. Circulation 107:2375–2382. [DOI] [PubMed] [Google Scholar]
- Gauthier LR, Charrin BC, Borrell-Pages M, Dompierre JP, Rangone H, Cordelieres FP, De Mey J, MacDonald ME, Lessmann V, Humbert S, Saudou F (2004). Huntingtin controls neurotrophic support and survival of neurons by enhancing BDNF vesicular transport along microtubules. Cell 118:127–138. [DOI] [PubMed] [Google Scholar]
- Goehler H, Lalowski M, Stelzl U, Waelter S, Stroedicke M, Worm U, Droege A, Lindenberg KS, Knoblich M, Haenig C, Herbst M, Suopanki J, Scherzinger E, Abraham C, Bauer B, Hasenbank R, Fritzsche A, Ludewig AH, Buessow K, Coleman SH (2004). A protein interaction network links GIT1, an enhancer of huntingtin aggregation, to Huntington's disease. Mol Cell 15:853–865. [DOI] [PubMed] [Google Scholar]
- Goldstrohm AC, Albrecht TR, Sune C, Bedford MT, Garcia-Blanco MA (2001). The transcription elongation factor CA150 interacts with RNA polymerase II and the pre-mRNA splicing factor SF1. Mol Cell Biol 21:7617–7628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu X, Li C, Wei W, Lo V, Gong S, Li SH, Iwasato T, Itohara S, Li XJ, Mody I, Heintz N, Yang XW (2005). Pathological cell-cell interactions elicited by a neuropathogenic form of mutant Huntingtin contribute to cortical pathogenesis in HD mice. Neuron 46:433–444. [DOI] [PubMed] [Google Scholar]
- Gunawardena S, Her LS, Brusch RG, Laymon RA, Niesman IR, Gordesky-Gold B, Sintasath L, Bonini NM, Goldstein LS (2003). Disruption of axonal transport by loss of huntingtin or expression of pathogenic polyQ proteins in Drosophila. Neuron 40:25–40. [DOI] [PubMed] [Google Scholar]
- Holbert S, Denghien I, Kiechle T, Rosenblatt A, Wellington C, Hayden MR, Margolis RL, Ross CA, Dausset J, Ferrante RJ, Neri C (2001). The Gln-Ala repeat transcriptional activator CA150 interacts with huntingtin: Neuropathologic and genetic evidence for a role in Huntington's disease pathogenesis. Proc Natl Acad Sci USA 98:1811–1816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Huntington's Disease Collaborative Research Group. (1993). A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. Cell 72:971–983. [DOI] [PubMed] [Google Scholar]
- Jiang H, Nucifora FC Jr, Ross CA, DeFranco DB (2003). Cell death triggered by polyglutamine-expanded huntingtin in a neuronal cell line is associated with degradation of CREB-binding protein. Hum Mol Genet 12:1–12. [DOI] [PubMed] [Google Scholar]
- Kegel KB, Meloni AR, Yi Y, Kim YJ, Doyle E, Cuiffo BG, Sapp E, Wang Y, Qin ZH, Chen JD, Nevins JR, Aronin N, DiFiglia M (2002). Huntingtin is present in the nucleus, interacts with the transcriptional corepressor C-terminal binding protein, and represses transcription. J Biol Chem 277:7466–7476. [DOI] [PubMed] [Google Scholar]
- Kowall NW, Ferrante RJ, Beal MF, Richardson EP Jr, Sofroniew MV, Cuello AC, Martin JB (1987). Neuropeptide Y, somatostatin, and reduced nicotinamide adenine dinucleotide phosphate diaphorase in the human striatum: a combined immunocytochemical and enzyme histochemical study. Neuroscience 20:817–828. [DOI] [PubMed] [Google Scholar]
- Landles C, Bates GP (2004). Huntingtin and the molecular pathogenesis of Huntington's disease. Fourth in molecular medicine review series. EMBO Rep 5:958–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legendre-Guillemin V, Metzler M, Charbonneau M, Gan L, Chopra V, Philie J, Hayden MR, McPherson PS (2002). HIP1 and HIP12 display differential binding to F-actin, AP2, and clathrin: Identification of a novel interaction with clathrin light chain. J Biol Chem 277:19897–19904. [DOI] [PubMed] [Google Scholar]
- Li SH, Li XJ (2004). Huntingtin and its role in neuronal degeneration. Neuroscientist 10:467–475. [DOI] [PubMed] [Google Scholar]
- Li SH, Cheng AL, Zhou H, Lam S, Rao M, Li H, Li XJ (2002). Interaction of huntington disease protein with transcriptional activator sp1. Mol Cell Biol 22:1277–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin CH, Tallaksen-Greene S, Chien WM, Cearley JA, Jackson WS, Crouse AB, Ren S, Li XJ, Albin RL, Detloff PJ (2001). Neurological abnormalities in a knock-in mouse model of Huntington's disease. Hum Mol Genet 10:137–144. [DOI] [PubMed] [Google Scholar]
- Lin KT, Lu RM, Tarn WY (2004). The WW domain-containing proteins interact with the early spliceosome and participate in pre-mRNA splicing in vivo. Mol Cell Biol 24:9176–9185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcora E, Gowan K, Lee JE (2003). Stimulation of NeuroD activity by huntingtin and huntingtin-associated proteins HAP1 and MLK2. Proc Natl Acad Sci USA 100:9578–9583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann JR, Morency CA, Russian KO (1987). A novel rapid assay for chloramphenicol acetyltransferase gene expression. Biotechniques 5:444–448. [Google Scholar]
- Nucifora FC Jr, Sasaki M, Peters MF, Huang H, Cooper JK, Yamada M, Takahashi H, Tsuji S, Troncoso J, Dawson VL, Dawson TM, Ross CA (2001). Interference by huntingtin and atrophin-1 with cbp-mediated transcription leading to cellular toxicity. Science 291:2423–2428. [DOI] [PubMed] [Google Scholar]
- Oldenburg KR, Vo KT, Michaelis S, Paddon C (1997). Recombination-mediated PCR-directed plasmid construction in vivo in yeast. Nucleic Acids Res 25:451–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Nuallain B, Williams AD, Westermark P, Wetzel R (2004). Seeding specificity in amyloid growth induced by heterologous fibrils. J Biol Chem 279:17490–17499. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C, Pennisi M, Topple A (1985). Bregma, lambda and the interaural midpoint in stereotaxic surgery with rats of different sex, strain and weight. J Neurosci Methods 13:139–143. [DOI] [PubMed] [Google Scholar]
- Qin ZH, Wang Y, Sapp E, Cuiffo B, Wanker E, Hayden MR, Kegel KB, Aronin N, DiFiglia M (2004). Huntingtin bodies sequester vesicle-associated proteins by a polyproline-dependent interaction. J Neurosci 24:269–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sachdev S, Bruhn L, Sieber H, Pichler A, Melchior F, Grosschedl R (2001). PIASy, a nuclear matrix-associated SUMO E3 ligase, represses LEF1 activity by sequestration into nuclear bodies. Genes Dev 15:3088–3103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saudou F, Finkbeiner S, Devys D, Greenberg ME (1998). Huntingtin acts in the nucleus to induce apoptosis but death does not correlate with the formation of intranuclear inclusions. Cell 95:55–66. [DOI] [PubMed] [Google Scholar]
- Schaffar G, Breuer P, Boteva R, Behrends C, Tzvetkov N, Strippel N, Sakahira H, Siegers K, Hayer-Hartl M, Hartl FU (2004). Cellular toxicity of polyglutamine expansion proteins: mechanism of transcription factor deactivation. Mol Cell 15:95–105. [DOI] [PubMed] [Google Scholar]
- Scherzinger E, Sittler A, Schweiger K, Heiser V, Lurz R, Hasenbank R, Bates GP, Lehrach H, Wanker EE (1999). Self-assembly of polyglutamine-containing huntingtin fragments into amyloid-like fibrils: implications for Huntington's disease pathology. Proc Natl Acad Sci USA 96:4604–4609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schilling G, Savonenko AV, Klevytska A, Morton JL, Tucker SM, Poirier M, Gale A, Chan N, Gonzales V, Slunt HH, Coonfield ML, Jenkins NA, Copeland NG, Ross CA, Borchelt DR (2004). Nuclear-targeting of mutant huntingtin fragments produces Huntington's disease-like phenotypes in transgenic mice. Hum Mol Genet 13:1599–1610. [DOI] [PubMed] [Google Scholar]
- Sipione S, Rigamonti D, Valenza M, Zuccato C, Conti L, Pritchard J, Kooperberg C, Olson JM, Cattaneo E (2002). Early transcriptional profiles in huntingtin-inducible striatal cells by microarray analyses. Hum Mol Genet 11:1953–1965. [DOI] [PubMed] [Google Scholar]
- Sisodia SS (1998). Nuclear inclusions in glutamine repeat disorders: are they pernicious, coincidental, or beneficial? Cell 95:1–4. [DOI] [PubMed] [Google Scholar]
- Smith MJ, Kulkarni S, Pawson T (2004). FF domains of CA150 bind transcription and splicing factors through multiple weak interactions. Mol Cell Biol 24:9274–9285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steffan JS, Kazantsev A, Spasic-Boskovic O, Greenwald M, Zhu YZ, Gohler H, Wanker EE, Bates GP, Housman DE, Thompson LM (2000). The Huntington's disease protein interacts with p53 and CREB-binding protein and represses transcription. Proc Natl Acad Sci USA 97:6763–6768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steffan JS, Bodai L, Pallos J, Poelman M, McCampbell A, Apostol BL, Kazantsev A, Schmidt E, Zhu YZ, Greenwald M, Kurokawa R, Housman DE, Jackson GR, Marsh JL, Thompson LM (2001). Histone deacetylase inhibitors arrest polyglutamine-dependent neurodegeneration in Drosophila. Nature 413:739–743. [DOI] [PubMed] [Google Scholar]
- Sugars KL, Rubinsztein DC (2003). Transcriptional abnormalities in Huntington disease. Trends Genet 19:233–238. [DOI] [PubMed] [Google Scholar]
- Sune C, Garcia-Blanco MA (1999). Transcriptional cofactor CA150 regulates RNA polymerase II elongation in a TATA-box-dependent manner. Mol Cell Biol 19:4719–4728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sune C, Hayashi T, Liu Y, Lane WS, Young RA, Garcia-Blanco MA (1997). CA150, a nuclear protein associated with the RNA polymerase II holoenzyme, is involved in Tat-activated human immunodeficiency virus type 1 transcription. Mol Cell Biol 17:6029–6039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szebenyi G, Morfini GA, Babcock A, Gould M, Selkoe K, Stenoien DL, Young M, Faber PW, MacDonald ME, McPhaul MJ, Brady ST (2003). Neuropathogenic forms of huntingtin and androgen receptor inhibit fast axonal transport. Neuron 40:41–52. [DOI] [PubMed] [Google Scholar]
- Thakur AK, Wetzel R (2002). Mutational analysis of the structural organization of polyglutamine aggregates. Proc Natl Acad Sci USA 99:17014–17019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trettel F, Rigamonti D, Hilditch-Maguire P, Wheeler VC, Sharp AH, Persichetti F, Cattaneo E, MacDonald ME (2000). Dominant phenotypes produced by the HD mutation in STHdh(Q111) striatal cells. Hum Mol Genet 9:2799–2809. [DOI] [PubMed] [Google Scholar]
- Van Raamsdonk JM, Murphy Z, Slow EJ, Leavitt BR, Hayden MR (2005). Selective degeneration and nuclear localization of mutant huntingtin in the YAC128 mouse model of Huntington disease. Hum Mol Genet 14:3823–3835. [DOI] [PubMed] [Google Scholar]
- Wang W, Duan W, Igarashi S, Morita H, Nakamura M, Ross CA (2005). Compounds blocking mutant huntingtin toxicity identified using a Huntington's disease neuronal cell model. Neurobiol Dis 20:500–508. [DOI] [PubMed] [Google Scholar]
- Wetzel R (2004). Protein folding and aggregation in the expanded polyglutamine repeat diseases. In: The protein folding handbook, Part II (Wells RD, Ashizawa T, eds) pp. 1170–1214. Weinheim, Germany: Wiley-VCH.
- Wheeler VC, Gutekunst CA, Vrbanac V, Lebel LA, Schilling G, Hersch S, Friedlander RM, Gusella JF, Vonsattel JP, Borchelt DR, MacDonald ME (2002). Early phenotypes that presage late-onset neurodegenerative disease allow testing of modifiers in Hdh CAG knock-in mice. Hum Mol Genet 11:633–640. [DOI] [PubMed] [Google Scholar]
- Yu ZX, Li SH, Nguyen HP, Li XJ (2002). Huntingtin inclusions do not deplete polyglutamine-containing transcription factors in HD mice. Hum Mol Genet 11:905–914. [DOI] [PubMed] [Google Scholar]
- Zala D, Benchoua A, Brouillet E, Perrin V, Gaillard MC, Zurn A, Aebischer P, Déglon N (2005). Progressive and selective striatal degeneration in primary neuronal cultures using lentiviral vector coding for a mutant huntingtin fragment. Neurobiol Dis 20:785–798. [DOI] [PubMed] [Google Scholar]
- Zhang J, Xu LG, Han KJ, Wei X, Shu HB (2004). PIASy represses TRIF-induced ISRE and NF-kappaB activation but not apoptosis. FEBS Lett 570:97–101. [DOI] [PubMed] [Google Scholar]
- Zhu Y, Culmsee C, Klumpp S, Krieglstein J (2004). Neuroprotection by transforming growth factor-beta1 involves activation of nuclear factor-kappaB through phosphatidylinositol-3-OH kinase/Akt and mitogen-activated protein kinase-extracellular-signal regulated kinase1,2 signaling pathways. Neuroscience 123:897–906. [DOI] [PubMed] [Google Scholar]
- Zuccato C, Tartari M, Crotti A, Goffredo D, Valenza M, Conti L, Cataudella T, Leavitt BR, Hayden MR, Timmusk T, Rigamonti D, Cattaneo E (2003). Huntingtin interacts with REST/NRSF to modulate the transcription of NRSE-controlled neuronal genes. Nat Genet 35:76–83. [DOI] [PubMed] [Google Scholar]