

Abstract
Endogenous mechanisms underlying the remodeling of neuronal circuitry after mammalian CNS injury or disease remain primarily unknown. Here, we investigated axonal plasticity after optic nerve injury and found that macrophages recruited into the injury site and adult retinal ganglion cell (RGC) axons, which undergo injury-induced sprouting and terminal remodeling, were linked by their respective expression of a ligand and receptor pair active in axon guidance. Recruited macrophages specifically upregulated mRNA encoding the guidance molecule EphB3 and expressed EphB proteins capable of binding Ephrin B molecules in vivo and in vitro. Injured adult RGC axons in turn expressed EphrinB3, a known receptor for EphB3, and RGC axons bound recombinant EphB3 protein injected into the optic nerve. In vitro, EphB3 supported adult RGC axon outgrowth, and axons turned toward a source of this guidance molecule. In vivo, both reduction of EphB3 function in adult heterozygous animals and loss of function in homozygous animals greatly decreased RGC axon re-extension or sprouting after optic nerve injury. Comparisons of axon re-extension in EphB3 null and wild-type littermates showed that this loss of axonal plasticity was not attributable to a difference in intrinsic axon growth potential. Rather, the results indicated an essential role for local optic nerve-derived EphB3 in regulating adult RGC axon plasticity after optic nerve injury. Of note, the loss of EphB3 did not affect the ability of injured RGC axons to elaborate complex terminal branching, suggesting that additional EphB3-independent mechanisms governed adult axon branching triggered by CNS damage.
Keywords: Eph, Ephrin, regeneration, sprouting, optic nerve, trauma, guidance molecule, macrophage, visual system, retinal ganglion cell
Introduction
Neuroscientists around the turn of the 20th century made two fundamental observations on CNS response to injury. First, injured CNS axons do not regenerate to reconstitute functional pathways. Second, although long distance axon regeneration does not occur, CNS damage triggers significant remodeling of neuronal circuitry. Plasticity in the form of local axon sprouting and branching has been documented after adult CNS damage (Raisman, 1969; Cotman et al., 1973; Azmitia et al., 1978; Jones et al., 1996; Beattie et al., 1997; Raineteau and Schwab, 2001; Weidner et al., 2001; Fisher and Lewis, 2003) and in pathological conditions (Darian-Smith and Gilbert, 1994; Li et al., 1995; Fariss et al., 2000; Hirsch, 2000; Holtmaat et al., 2003). An analysis of the cellular and molecular basis of this adult nervous system, reactive plasticity is necessary for a more complete understanding of the underlying neuropathology and development of therapeutic strategies.
The optic nerve is a site of substantial axonal plasticity and remodeling after injury. As with other CNS axons, injured retinal ganglion cell (RGC) axons do not have the ability to grow over long distances to reconnect with targets. However, it has long been appreciated that injured RGC axons do, in fact, mount a significant initial attempt at regrowth (Cajal, 1928). Golgi studies showed that RGC axons first withdraw from the region of damage but then re-extend back toward the lesion site over the first 2 weeks and show axon turning, looping, and branching. Similar findings have been reported by researchers using more modern techniques (Grafstein and Ingoglia, 1982; Allcutt et al., 1984; Zeng et al., 1995; Selles-Navarro et al., 2001). Ironically, axon visualization methods favored in more recent work tend to label the entire RGC axon population and do not always reveal the trajectories and highly branched endings of individual injured RGC axons that amply document the presence of a robust sprouting and remodeling process.
The mechanisms mediating adult RGC axonal plasticity after injury have not been explored extensively. Although the sprouting response of injured RGC axons is abortive in that it does not result in successful regeneration, the mechanisms involved are of substantial interest because axon re-extension is one of the first responses of injured RGC axons and, as such, must be understood if attempts at promoting axon regeneration are to be successful. Furthermore, because adult CNS axons are generally more refractory to growth compared with their embryonic counterparts, endogenous mechanisms that can trigger significant adult axon growth are worth investigating. Finally, because neuronal plasticity after damage and disease may be a characteristic throughout the nervous system, identification of molecular mediators of RGC axon re-growth after optic nerve injury may provide useful insight into the mechanisms involved.
During normal development, axon growth and pathfinding as well as terminal branching of RGC axons are controlled by families of axon guidance proteins in the embryonic brain, major families of which include the netrins, semaphorins, Slits, Ephs/Ephrins, and members of the Ig superfamily (Flanagan and Vanderhaeghen, 1998; Stuermer and Bastmeyer, 2000; McLaughlin et al., 2003; Oster et al., 2004; van Horck et al., 2004). Ephs, and Ephrins in particular, are versatile axon guidance molecules capable of bidirectional signaling (Cowan and Henkemeyer, 2001; Lu et al., 2001) and mediate intraretinal axon pathfinding at the optic nerve head (ONH) (Birgbauer et al., 2000, 2001), the optic chiasm (Nakagawa et al., 2000; Williams et al., 2003), and the superior colliculus (Hindges et al., 2002; Knoll and Drescher, 2002; Mann et al., 2002; McLaughlin et al., 2003). Of note, the manner in which embryonic RGC axons respond to EphB proteins depends on local environmental signals and ranges from growth inhibition (Birgbauer et al., 2001), to unresponsiveness (Suh et al., 2004), to enhanced growth (Mann et al., 2002). Axon guidance molecule response switching has been described during the transition from embryonic development to early postnatal life (Cai et al., 2001). Whether and how injured adult CNS axons react to embryonic axon guidance molecules is not known.
Here, we report that EphB3, a transmembrane member of the EphB axon guidance molecule subfamily, reappears in the adult optic nerve after injury, coincident with RGC axon sprouting and remodeling. In vitro assays demonstrate that adult RGC axon growth is stimulated by EphB3 protein and RGC axons in animals with reduced or loss of EphB3 function have dramatically decreased axon sprouting after optic nerve injury. However, the sporadic RGC axons that do re-extend toward the lesion site are capable of elaborating complex branching patterns. These results indicate that axon guidance molecules, which mediate the development of neuronal connectivity in utero, come into play once again in the injured adult nervous system and govern specific aspects of axon regrowth and plasticity after CNS damage.
Materials and Methods
Animals.
Thy1-YFP transgenic mice on a C57BL/6 background [B6.Cg-Tg(Thy1-YFPH)2Jrs/J], described by Feng et al. (2000), were obtained from The Jackson Laboratory (Bar Harbor, ME) and bred at the University of California at San Francisco (UCSF). EphB3 mutant mice of CD-1 background and EphrinB3 mutant mice of mixed 129/sv × C57BL/6 background were obtained from Dr. M. Henkemeyer (University of Texas Southwestern, Dallas, TX), and breeding colonies were established at UCSF. Animals were genotyped as described previously (Orioli et al., 1996; Yokoyama et al., 2001). Thy1-YFP mice were mated with EphB3 mutant mice to produce animals in which RGC axon response to injury could be quantified in the absence of normal EphB3 function. Animals were backcrossed into the EphB3 CD-1 background for six generations. Littermate mice with wild-type EphB3 function were used as control animals. All experiments were performed in animals 6–12 months of age and were in accordance with guidelines of the UCSF Institutional Animal Care and Use Committee.
Antibodies and reagents.
The antibodies used in this study, their working concentrations, and their vendor sources were as follows: anti-green fluorescent protein (GFP) (Alexa Fluor 488 conjugated; 4 μg/ml; Molecular Probes, Eugene, OR); anti-myelin basic protein (1:200; Chemicon, Temecula, CA); anti-glial fibrillary acidic protein (GFAP) (1:600; Sigma, St. Louis, MO); anti-rat macrophage (ED1; 1:50; Chemicon); anti-mouse macrophage/monocytes (MOMA-2; 5 μg/ml; Chemicon); anti-mouse F4/80 (20 μg/ml; Serotec, Indianapolis, IN); anti-mouse EphB3 (1:100; R & D Systems, Minneapolis, MN); anti-human Ephrin-B3 (1:20; R & D Systems); anti-βIII tubulin (0.5 μg/ml; Covance Research Products, Denver, PA); anti-GAP-43 (1:200; MAB347; Chemicon); anti-L1 antiserum [1:2000 (Chung et al., 1991)]; and anti-human IgG Fcγ (1:200; Jackson ImmunoResearch, West Grove, PA). Primary antibody binding was detected using appropriate secondary antibodies conjugated to FITC, Cy2, or Cy3 (Jackson ImmunoResearch) or peroxidase (ABC kit; Vector Laboratories, Burlingame, CA). Actin was visualized using Texas Red-phalloidin (Molecular Probes). Specimens were mounted in Vectashield mounting medium and counterstained with 4',6-diamidino-2-phenylindole (DAPI) (Vector Laboratories) when required.
Histology and immunohistochemistry.
Retinas and optic nerves were fixed in 4% paraformaldehyde, infiltrated with 30% sucrose, embedded in OCT compound (TissueTek), and sectioned along the longitudinal axis of the optic nerve. Ten-micrometer-thick sections were used for in situ hybridization and immunolabeling studies, whereas 50-μm-thick sections were used for counting of yellow fluorescent protein (YFP)-labeled axons and analysis of axon terminal morphology using confocal microscopy (Zeiss, Thornwood, NY). For each of the postinjury time points examined (2, 4, 8, 12, and 18 d after lesion), tissues from at least six adult optic nerves and retinas were analyzed. The number of YFP axons identified by confocal microscopy was determined in the margin or penumbra region of the lesion, corresponding to a region extending 100 μm immediately adjacent to the lesion in the direction toward the globe. Axon trajectories, growth cone morphologies, and terminal specializations were also analyzed in this region. The location of the lesion site was confirmed by the presence of macrophage invasion and the disruption of the ensheathing nerve connective tissue.
The morphology of the ONH in wild-type and mutant mice was examined in plastic embedded sections. Tissues were fixed in 4% paraformaldehyde, 2.5% glutaraldehyde, and 0.1 m cacodylic acid, pH 7.2. Samples were postfixed in 2% OsO4 and embedded in Epon resin. One-micrometer-thick sections were cut and stained with 1% toluidine blue.
In situ hybridization.
In situ hybridization was performed using digoxigenin-labeled sense and antisense RNA probes with alkaline phosphatase detection (Roche Molecular Biochemicals, Mannheim, Germany) or by immunofluorescence (TSA-plus fluorescein system; PerkinElmer Life Sciences, Norwalk, CT). The following probes, all from American Type Culture Collection (Manassas, VA), were generated from the mouse expressed-sequence tag clones: Netrin-1 (AA 655493, IAMGE ID 1139227, ATCC 1406668); Slit2 (AA 171288, IMAGE ID 615062, ATCC 773470); and Slit3 (AA 117086, IMAGE ID 557242, ATCC 715650). The in situ probes for EphB1, EphB2, EphB3, EphrinB1, EphrinB2, EphrinB3, and Sema5A have been described previously (Birgbauer et al., 2000; Oster et al., 2003). The expression of EphB mRNA in macrophages was determined in double-labeling studies in which in situ hybridization was first performed with the standard protocol, followed by immunolabeling using appropriate antibodies.
Immunoblots.
Optic nerves or retinas were dissected from adult animals and placed in extraction buffer containing 2% Triton X-100. Tissue extracts were added to sample buffer containing SDS/β-mercaptoethanol and loaded onto 7.5% acrylamide gels for electrophoresis, with each lane containing the equivalent of protein from two optic nerves or from one retina. After transfer to nitrocellulose, blots were blocked with PBS/5% anti-donkey serum or dry milk and probed with anti-mouse EphB3 (1:1000; R & D Systems) or anti-human EphrinB3 (1:500; R & D Systems). Immunoreactivity was detected using donkey anti-goat antibodies conjugated to peroxidase (1:50,000; Jackson ImmunoResearch) and visualized using SuperSignal West Pico Chemiluminescent (Pierce, Rockford, IL). To identify protein loading levels, immunoblots were stripped using stripping buffer (Pierce) and incubated with anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH) antibody (MAB374; Chemicon) to visualize the amount of GAPDH protein in different samples.
Recombinant proteins.
Recombinant EphB3-Fc consisted of the extracellular domain of mouse EphB3 fused to the Fc portion of human IgG (Birgbauer et al., 2001). Recombinant protein was harvested from a stably transfected cell line and purified using protein A chromatography. The purity of the protein fractions was identified by silver staining, and protein concentration was determined by Bradford assay. Human Fc control protein was produced and characterized in a similar manner (Birgbauer et al., 2001). Recombinant L1-Fc consisted of the extracellular domain of human L1 protein fused to the Fc portion of human IgG (Suh et al., 2004). Recombinant EphrinB3-Fc protein was obtained from R & D Systems.
Optic nerve injury and tissue harvesting.
Mice were anesthetized by intraperitoneal injection of 90 mg/kg ketamine HCl (Ketaset; Fort Dodge Laboratories, Overland Park, KS) and 5 mg/kg xylazine (Lloyd Laboratories, Shenandoah, IA). Before optic nerve crush, a drop of Alcaine (0.5% proparacaine HCl ophthalmic solution; Alcon Laboratories, Fort Worth, TX) was applied to the experimental eye. Bent forceps were then inserted behind the globe using a ventromedial approach, and pressure was applied for 60 s. (Nerve crushes were verified by the observation of pupillary dilation, which indicated that the ciliary parasympathetic fibers had been lesioned.) Xylocaine ointment (2%; Lidocaine HCl jelly; Astra Pharmaceuticals, Worcester, MA) was then applied over the operated eye. Animals with ischemic changes in the retina caused by damage of the ophthalmic vessels were killed and excluded from study. Animals were reanesthetized 2–32 d after surgery and perfused with 4% paraformaldehyde, and the optic nerves and retinas were harvested for study.
Optic nerve injection.
Mice were anesthetized as described above for the in vivo injection of reagents into the optic nerve. Two temporary holding sutures were placed onto the inferior eyelid and pulled to expose the inferior conjunctiva. A single cut was then made into the conjunctiva using surgical microscissors. Local blood vessels visible on the conjunctiva and the vortex vein were cauterized. The inferior rectus muscle was retracted, and the remaining conjunctiva was spread apart using a blunt dissection probe. The optic nerve was then exposed, and a glass micropipette containing a reagent of interest was inserted into the optic nerve ∼0.5 mm behind the globe under visual control through a surgical microscope. Reagent was ejected from the glass micropipette using a Picospritzer (General Valve; Parker Hannifin, Cleveland, OH). After the injection, the holding sutures were removed. Neomycin (E. Fougera & Co., Melville, NY) and 2% lidocaine ointment (Alpharma USPD, Franklin, NJ) were applied over the operated eye. Animals were reanesthetized after 8 d. The optic nerves were harvested and processed for immunocytochemistry.
Optic nerve explant culture and macrophage labeling.
C57BL/6 mice (6–12 months of age) or EphB3 null mice (6–12 months of age) that received an optic nerve crush 5–8 d before were anesthetized, and the experimental optic nerve was isolated. The nerve was dissected into 200-μm-long segments, and the segments closest to the lesion site were cultured in vitro in Neurobasal-A (Invitrogen, San Diego, CA) medium with B-27 supplement (Invitrogen) and 20 mm HEPES, pH 7.4, at 37°C under 5% CO2. Macrophages were observed to migrate out of the nerve explant cultures beginning at 24 h after culture. EphrinB3-Fc (50 μg/ml) or human Fc control protein (50 μg/ml) (B irgbauer et al., 2001) was added together with BSA (1 mg/ml) onto the nerve explant cultures for 2 h at 4°C to label cells. Experiments were also performed in which optic nerve macrophages were first incubated with Ig (from mouse serum) to bind to mouse Fc receptors before the addition of EphrinB3-Fc protein. Nerve cultures were then washed and fixed using 2% paraformaldehyde. EphrinB3-Fc and Fc binding were visualized using Cy3-conjugated anti-human Fc antibody (Jackson ImmunoResearch). Cells migrating out from nerve explants were simultaneously labeled with anti-ED1 antibody to confirm their identity as macrophages.
Macrophage quantitation.
The number of macrophages recruited after optic nerve lesion was counted in both wild-type (n = 4) and EphB3 null littermates (n = 4). Injured optic nerves were obtained from animals that had undergone a nerve crush 4 d before. Each optic nerve was cut along its long axis to obtain 12-μm-thick cryostat sections. Six cryostat sections evenly spaced ∼150 μm apart in the optic nerve were chosen for ED1-positive macrophage counting. The number of ED1-positive macrophages was counted from a 100 × 100 μm2 region in each 12-μm-thick section, and the results were used to determine an average number of macrophages per unit volume. The region of macrophage accumulation in each of the six sections was also traced using ImageJ software, and its boundaries were used to approximate the overall extent of the lesion in that section. The size of these lesion sites was then extrapolated to arrive at an estimate of the total volume of the optic nerve lesion site. In both wild-type and EphB3 null groups, the average density of macrophages was analyzed in three nerves from three different animals, and the volume of the lesion area was determined in four nerves from four different animals. The number of macrophages per unit volume was then multiplied by the total volume of the lesion to arrive at an estimated number of macrophages.
Analysis of YFP axons.
Cryostat sections (50 μm thick) were obtained along the longitudinal axis of the optic nerve including the ONH region and the margin of the lesion site. YFP-containing axons were labeled using anti-GFP antibody (Molecular Probes) and identified using confocal microscopy. The number of YFP axons was counted in each tissue section, and images for display were compiled using the z-stack mode. For analysis of axon branching after optic nerve injury, YFP axons that had one or more branches were considered as having a terminal branching morphology.
Statistical analysis.
Statistical analysis was performed using STATA 8.0 statistics software to determine whether the number of axons encountered at the ONH or the margin zones changed significantly between 2 and 18 d after injury and whether differences in axon regrowth existed between wild-type and EphB3 mutant mice. The Shapiro-Wilk test was used to determine whether each set of data from days 2, 4, 8, 12, and 18 after injury was normally distributed. The results indicated that at least some of the data in each set were not normally distributed. The data sets were then subjected to the Kruskal–Wallis test, a nonparametric test for multiple comparisons to determine whether a given data set contained significant differences among the groups. Data sets containing significant differences were also subjected to post hoc Tukey tests in which pairwise comparisons between specific groups were performed to determine whether the results were statistically different from each other. Results from the Tukey test are reported as differences of the mean between the two groups and the 95% confidence interval. Confidence intervals that did not cross the value 0 were considered significant.
In vitro retinal explants assay.
For in vitro assays, C57BL/6 mice 6–12 months of age first received an optic nerve crush 8–12 d before tissue harvesting (Meyer and Miotke, 1990). Animals were then reanesthetized, and the eye corresponding to the injured optic nerve was harvested into Ringer’s solution. The retina was isolated, and retinal explants ∼500 μm2 in size were cut and placed vitreal side down into 35 mm coverslip culture dishes (MatTek Corporation, Ashland, MA) that were coated with 1 mg/ml poly-l-lysine (Sigma), followed by either 2.5 or 10 μg/ml laminin (Invitrogen) at 37°C in 5% CO2. Explants were also cultured on dishes that were first coated with anti-human Fc antibody (5 μg/ml; Jackson ImmunoResearch), followed by 0.25–2.5 μg/ml L1-Fc recombinant protein (Suh et al., 2004). Explants were maintained in Neurobasal-A (Invitrogen) medium with B-27 supplement (Invitrogen) and 20 mm HEPES, pH 7.4, at 37°C under 5% CO2 for 5–12 d. Cultures were fixed and stained with anti-tubulin antibody and used for axon length measurements. Axons were traced and digitized, and their lengths were quantified using Photoshop 7.0 (Adobe Systems, San Jose, CA). Data for axon outgrowth on laminin and EphB3-Fc were obtained from 4–12 sets of independent experiments. In experiments using L1-Fc as a protein substratum, the axon outgrowth pattern was fasiculated and precluded a precise analysis of axon number and axon length. Instead, the number of explants with axon outgrowth was counted at each condition and expressed as a percentage of all explants tested. In an additional set of three independent experiments using the L1-Fc substratum, outgrowth length measurements were derived by tracing and digitizing the entire pattern of outgrowth from explants. Data from the three experiments were pooled to obtain an average measurement per explant in each of the conditions with different amounts of EphB3-Fc protein.
Experiments were conducted to compare the axon outgrowth from retinal explants of adult EphB3 null animals and wild-type littermates on both laminin and L1-Fc protein substrata. The protocols for tissue harvesting, substratum preparation, and analysis were as described above for C57BL/6 animals.
Growth cone turning assays.
Growth cone turning induced by EphB3 gradients was performed as described previously (Birgbauer et al., 2001; Suh et al., 2004). Glass micropipettes containing the reagent of interest were placed at an ∼45° angle to the growth trajectory of the axon being examined and at 70–100 μm away from the growing tip. The direction of growth cone extension at the beginning of the experiment was determined over a 30 min period of observation. Micropipettes contained EphB3-Fc (0.26 mg/ml) or, as a control, heat-inactivated (74°C for 20 min) EphB3-Fc (0.26 mg/ml) or Fc protein (1 mg/ml). Images were captured at 1 min intervals for 45–90 min after reagent application.
Axons >1 mm in length were used in growth cone turning assays. The turning angle was defined by the angle between the original direction of axon extension and the direction of growth cone extension after reagent exposure. Only growth cones that extended ≥20 μm over the time course of the experiment were included for analysis. The responses were categorized as growth cone collapse, turning, or no apparent response.
Results
Quantitative analysis of RGC axon re-extension in response to optic nerve injury
Although the phenomenon of RGC axon regrowth after optic nerve injury has been reported in a number of mammalian species, a quantitative analysis required for more mechanistic studies has not been performed. We began our analysis by determining the temporal characteristics of RGC axon re-extension after injury to the optic nerve in adult mice 6–12 months of age (n = 29). To obtain a quantitative measure of plasticity, studies were performed in Thy1-YFP mice (strain H), a strain of transgenic mice in which each animal has a small and fixed percentage of RGC cells and axons that express the fluorescent protein YFP. This YFP expression allows both the accurate counting of YFP-positive axon number and the detailed visualization of axon terminal morphologies of individual RGC axons. Optic nerve damage was induced by a standard retro-orbital nerve crush, and RGC axons were examined at multiple intervals between 2 and 32 d after injury. The number of YFP-labeled RGC axons was counted both at the ONH region and in a margin or “penumbra” region adjacent to the injury site. We defined the margin zone as a 100-μm-wide region of the optic nerve bordering the lesion site in which injured RGC axons actively re-extend and exhibit axon terminal remodeling. The RGC axon number at the ONH region provided an estimate of the number of axons remaining at various time intervals after nerve injury.
Results showed that after optic nerve injury, the number of RGC axons at the ONH steadily declined over 18 d (Fig. 1A,C), likely reflecting the loss of RGCs that is known to occur beginning a few days after optic nerve damage (Aguayo et al., 1996). In contrast, over the same time interval, the number of axons in the margin zone showed a biphasic response. In the first phase 2–4 d after injury (Fig. 1B,D), the number of RGC axons in the margin zone was significantly reduced compared with the number of YFP axons in a similar region of uninjured optic nerves. In the second phase beginning 4–8 d after injury, an increase in the number of YFP axons was observed in the margin zone, resulting in a threefold increase in axon number in the margin zone at 18 d compared with 2 d after injury. [As expected, at any given time point, the number of axons at the margin zone was less than the number at the ONH (Fig. 1, compare C, D).] This analysis showed that the mechanisms that promote adult RGC axon plasticity and regrowth are present and operational within 4–8 d after optic nerve injury.
Figure 1.

Adult mouse RGC axon response to optic nerve injury. A, YFP-labeled RGC axons at the ONH at different intervals after nerve injury. The numbers at the bottom right represent the days after injury. UI, Uninjured. Individual images represent a composite three-dimensional stack of confocal images collected at 1 μm intervals from 50-μm-thick sections. The retina is toward the left. B, Appearance of YFP-labeled RGC axons in the uninjured optic nerve (UI) and RGC axons in the margin region at 4, 8, 12, and 18 d after injury. C, Graph showing the mean number of RGC axons per tissue section at the ONH before and at various times after injury. The numbers in parentheses represent the number of tissue sections analyzed. The data set of this graph contained a statistical difference (Kruskal–Wallis test; p = 0.0002). Tukey tests showed that the results from days 2–18 were each significantly different from the data from the uninjured nerve (differences of the mean, 11.7–17.4; 95% confidence intervals, 2.1–27.4. D, Graph showing the mean number of RGC axons per tissue section in the margin region adjacent to the injury site at various times after injury. The data set forming this graph contained a statistically significant difference (Kruskal–Wallis test; p = 0.0001). The numbers in parentheses represent the number of tissue sections analyzed. The asterisks indicate that the results from days 8, 12, and 18 were each statistically different from that from days 2 and 4 (pairwise comparisons by Tukey tests; differences of the mean, 9.8–18.7; 95% confidence intervals, 0.065–28.1). E–L, Higher-magnification views of the endings of RGC axons after optic nerve injury. The different morphologies have been arranged left to right in increasing apparent complexity. E, F, Turning axons. G, Axons with 180° loops (arrows). H, Axon with a 270° loop (arrow). I–L, Axons with complex branching morphology. Scale bars, 50 μm. Error bars indicate SEM.
RGC axons elaborate specialized endings in response to injury
RGC axons re-extending within the marginal zone 4–8 d after injury exhibited specialized endings and axon terminal remodeling. Re-extending adult RGC axons exhibited several types of trajectories and endings including turning (Fig. 1E,F), axon looping (Fig. 1G,H), and extensive axon terminal branching (Fig. 1I–L). Given that adult RGC axons in the normal optic nerve never have looping trajectories or branching, the presence of these types of specialized endings strongly indicated that the damaged CNS environment contained specific signals that can trigger adult axon terminal remodeling.
Astrocytes and oligodendrocytes after optic nerve injury
Optic nerve injury resulted in extensive rearrangements of local cell populations. In normal adult mice, oligodendrocytes were found throughout the optic nerve but were excluded from the ONH region immediately behind the globe (Fig. 2A, UI, uninjured). On the other hand, astrocytes were found within the ONH region and throughout the nerve as well. Two to 4 d after injury, both astrocytes and oligodendrocytes vacated the lesion site, leaving a region that was primarily unoccupied by either cell type (Fig. 2A). This astrocyte- and oligodendrocyte-free zone was quite well defined by day 8 and remained visible even at day 18. DAPI nuclei staining revealed this zone to be occupied by nucleated cells (Fig. 2B).
Figure 2.
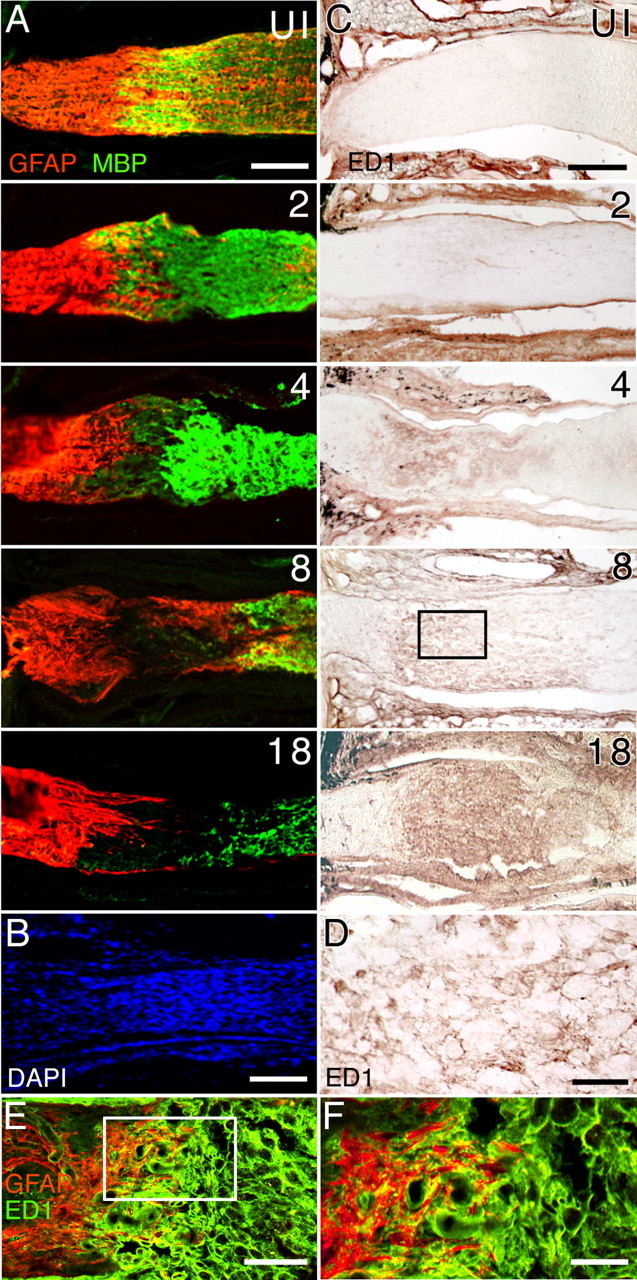
Cellular reorganization in the optic nerve after injury. A, Distribution of GFAP+ astrocytes (red) and myelin basic protein (MBP+) oligodendrocytes (green) before and after nerve injury. The numbers represent the days after injury. Retina is toward the left. By 4 d after injury, astrocytes and oligodendrocytes have begun to vacate the injured area, and a region devoid of these two cell types is observed by 8 d after injury. B, DAPI cell nuclei staining from a different optic nerve showing the presence of numerous cell bodies at the lesion site 12 d after injury. C, Accumulation of ED1+ macrophages after optic nerve injury. The boxed region at day 8 after injury is shown at a higher magnification in D. D, Higher-magnification view of ED1+ macrophages. E, Double immunostaining of an optic nerve tissue section 8 d after injury using anti-GFAP (red) and anti-ED1 (green) showing that the margin region surrounding the lesion site contains both astrocytes and macrophages. F, Higher-magnification view of the boxed region in E. Scale bars: A–C, E, 100 μm; D, F, 20 μm. UI, Uninjured nerve.
Axon response coincides with macrophage accumulation
Immunolabeling studies using antibodies against the macrophage markers F4/80, MOMA-2, and ED1 identified the cells recruited to and occupying the lesion site as macrophages. The uninjured adult optic nerve showed little or no immunoreactivity to macrophage markers (Fig. 2C, UI). Within 1–2 d after injury, faintly labeled ED1-positive cells began to appear throughout the optic nerve, beyond the immediate site of injury (Fig. 2C). Between 4 and 8 d, these cells became more strongly immunoreactive and gradually coalesced into a well organized cell population (Fig. 2C,D). (Similar findings were observed using F4/80 and MOMA-2.) A dense collection of macrophages remained at 18 d after injury. These results showed that the re-extension of injured RGC axons toward the lesion site occurred at a time when astrocytes and oligodendrocytes were actively withdrawing from the region of damage. Although injured RGC axons that are attempting to regrow back to the lesion site may also encounter astrocytes in the margin zone (Fig. 2E,F), the phenomenon of RGC axon sprouting was spatially and temporally coincident with the in-migration and accumulation of recruited macrophages.
Appearance of EphB3 in the injured optic nerve
The pattern of macrophage accumulation was mirrored by the expression of EphB3, a member of the EphB subfamily of RGC axon guidance molecules. In the uninjured adult optic nerve, EphB3 mRNA was only present faintly in cells that were arranged in a row-like manner and in some cells of the optic nerve sheath (data not shown). Two days after optic nerve injury, EphB3 mRNA was upregulated in scattered cells within the nerve (Fig. 3A). Between days 4 and 8, EphB3+ cells began to coalesce into an organized group at the lesion site. The intensity of the mRNA hybridization signal and the numbers of positive cells began to decrease by day 18, and EphB3 mRNA was not readily apparent by 32 d after nerve injury.
Figure 3.
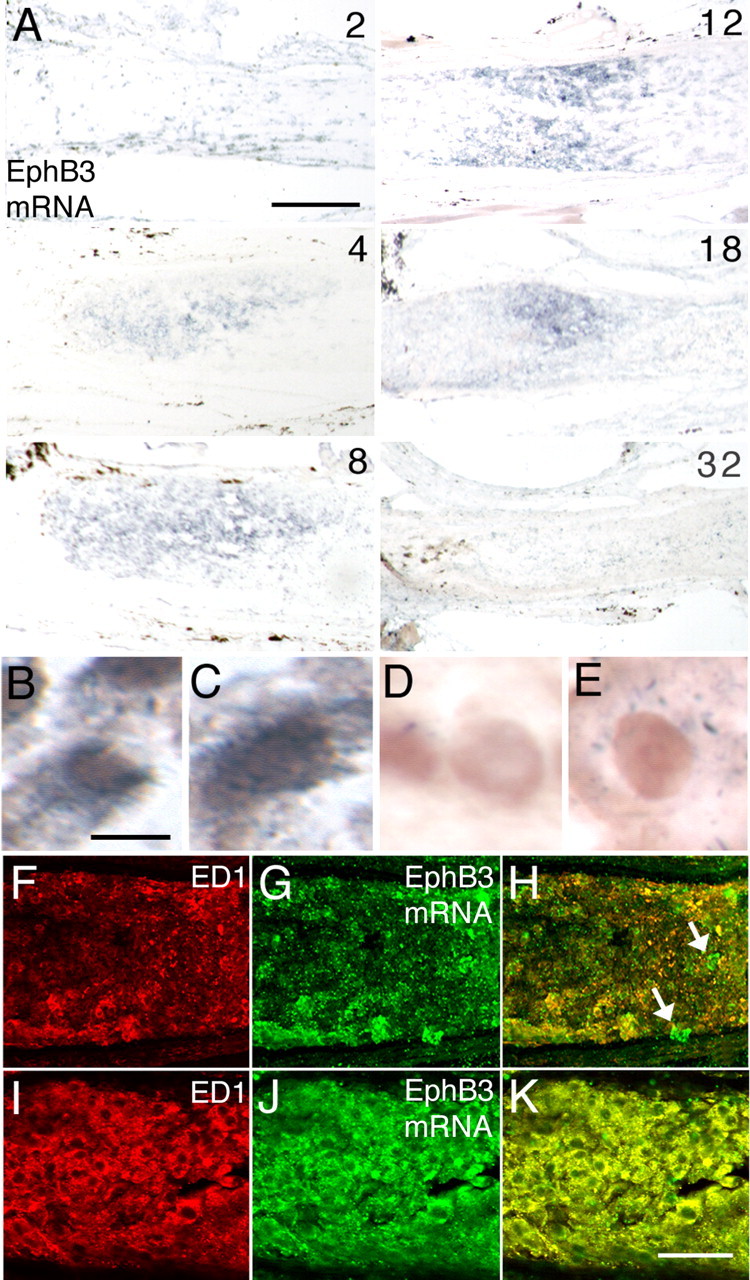
Expression of EphB3 by macrophages. A, In situ hybridization detection of EphB3 mRNA at intervals between 2 and 32 d after nerve injury. EphB3 mRNA signal can be detected as early as 2 d after injury and appears to reach a peak at ∼8–12 d. Gene expression is downregulated by 32 d. The retina is toward the left. B, C, Examples of ED1+ macrophages (brown) that simultaneously exhibit an EphB3 mRNA hybridization signal (dark blue) in the optic nerve 8 d after injury. D, ED1+ macrophages in the injured optic nerve after hybridization with a sense control strand. E, No mRNA hybridization is seen in ED1+ macrophages from EphB3 null animals with optic nerve injury. F, ED1+ macrophages (red) at the margin region 4 d after injury. G, Visualization of EphB3 mRNA fluorescence hybridization (green) in the same tissue section as in F. H, Merged image showing correspondence between ED1+ macrophages and expression of EphB3 mRNA (yellow). A few cells expressing EphB3 mRNA are not immunopositive for ED1 (arrows). I, ED1+ macrophages in the core of the injury site 12 d after injury. J, Visualization of EphB3 mRNA fluorescence hybridization in the same tissue section as in I. K, Merged image showing EphB3 mRNA expression in ED1+ macrophages. Scale bars: A, 100 μm; B–E, 5 μm; F–K, 50 μm.
Macrophage expression of EphB3 axon guidance molecule
Because the spatiotemporal pattern of EphB3 mRNA expression closely paralleled the pattern of macrophage accumulation, double-labeling experiments were performed to determine whether macrophages recruited in response to optic nerve injury were the source of EphB3. The EphB3 mRNA hybridization signal was evident in ED1-immunopositive cells, confirming the presence of EphB3 mRNA in macrophages (Fig. 3B,C,F–K). No signal was detected using the sense control stand for hybridization (Fig. 3D). Furthermore, no hybridization signal was detected in macrophages in the injured optic nerve of EphB3 null animals (Fig. 3E), supporting the specificity of the hybridization probe. Analysis of the frequency of concurrent ED1 immunoreactivity and EphB3 mRNA expression in cells revealed that >95% of cells that expressed EphB3 mRNA were immunopositive for ED1 and were thus macrophages (Fig. 3F–K). Of note, macrophages that were scattered around the margin zone migrating into the core of the lesion site clearly expressed EphB3 mRNA (Fig. 3F–H). Studies were also performed to examine whether additional axon guidance molecules were present in the adult optic nerve after injury. Analysis using mRNA probes against Netrin-1, Slit2, Slit3, EphB1, EphB2, EphrinB1, EphrinB2, and Semaphorin5A failed to demonstrate positive hybridization signal in the 2 week period after injury (data not shown).
Consistent with the presence of EphB3 mRNA, immunoblots using optic nerve tissue extracts from normal optic nerves (UI) and nerves 8 d after injury (8), revealed a protein band at ∼110 kDa, consistent with the molecular weight of EphB3 protein (Fig. 4A). This band was not detected in immunoblots using optic nerve tissue extracts from EphB3 null animals (KO), further supporting the identity of the detected band as EphB3. [The faint immunoreactive band in the uninjured nerve may reflect the presence of EphB3 mRNA in the normal optic nerve sheath connective tissue (X. Liu and D. W. Sretavan, unpublished observation).] Protein loading controls were performed using GAPDH as a marker. The amount of EphB3 protein detected at 8 d after injury was apparently upregulated compared with that in the uninjured nerve. The expression of EphB protein was detected in macrophages migrating out of cultured adult optic nerve segments subjected to injury 5 d before explantation. Double-labeling studies showed that EphrinB3-Fc protein applied to cultured optic nerve segments specifically bound to the surface of ED1-positive macrophages (Fig. 4B,C) in a punctate–aggregate manner. Application of the Fc portion alone (human origin) to mouse macrophages did not result in detectable Fc binding, indicating that the binding of EphrinB3-Fc on mouse macrophages was primarily accomplished through the EphrinB3 domain (data not shown). Furthermore, EphrinB3-Fc binding was not detected on optic nerve macrophages derived from EphB3 null animals (Fig. 4D,E), indicating that EphrinB3-Fc can reliably detect EphB protein. EphrinB3-Fc protein was also injected into the optic nerves of mice in vivo to determine whether macrophages in the injured optic nerve expressed EphB protein. The results showed that EphrinB3-Fc protein injection followed by visualization of the Fc tag resulted in the labeling of macrophages (Fig. 4F–H), indicating that the expression of EphB protein on macrophages is indeed part of a normal response to nerve injury.
Figure 4.

Expression of EphB3 protein in the injured adult optic nerve and macrophages. A, Immunoblots of protein preparations from uninjured (UI) and injured adult optic nerves 8 d after injury (8) probed using an anti-EphB3 antibody. A signal at ∼110 kDa corresponding to the expected size for EphB3 protein was detected in both samples. This protein band was not present in optic nerve tissue obtained from EphB3 null animals (KO). GAPDH-immunopositive bands used as protein loading controls are shown below. B, ED1-positive macrophages isolated from segments of injured adult optic nerve in culture. C, The macrophages shown in B bound exogenously applied EphrinB3-Fc protein resulting in a punctate–aggregate pattern of cell-surface labeling. D, ED1-positive macrophages from segments of injured adult optic nerve in culture derived from an EphB3 homozygous null animal. E, Exogenously applied EphrinB3-Fc protein failed to bind to the EphB3 null macrophages shown in D. F, ED1-positive macrophages in an adult optic nerve 5 d after injury (green). G, EphrinB3-Fc binding in the same region shown in D (red). H, Colocalization of ED1 immunoreactivity with EphrinB3-Fc binding. Scale bars: (in B) B–E, 20 μm; (in F) F–H, 10 μm.
Retinal expression of EphrinB3 and EphB3 binding on RGC axons
During development, EphB binds EphrinB proteins on cells to activate intracellular signaling cascades and influence cell or growth cone behavior (Holland et al., 1998; Cowan and Henkemeyer, 2001; Lu et al., 2001). To examine whether EphB3 expressed by macrophages could affect adult RGC axons, we investigated and confirmed the presence of EphrinB expression in RGC layer of the retina and on RGC axons using four different approaches. First, EphrinB3 mRNA was detected in the RGC layer of the uninjured adult retina (Fig. 5A). After injury, EphrinB3 mRNA was maintained in the RGC layer at 4 and 12 d (Fig. 5B,C) and was present even at 18 d (data not shown). (EphrinB1 and EphrinB2 mRNA were not detected.) EphrinB3 mRNA was also detected in the uninjured nerve (Fig. 5E), and after injury, EphrinB3 was found in cells that accumulated at the lesion site and in the surrounding connective tissue (Fig. 5F,G). Second, a 65 kDa band consistent with the molecular weight of EphrinB3 protein was detected in immunoblots using adult retinal (data not shown) and optic nerve tissues after nerve injury (Fig. 5H). Third, anti-EphrinB3 immunoreactivity was detectable at low levels in the uninjured optic nerve (Fig. 5I), and after injury, EphrinB3 immunoreactivity appeared upregulated and was present on RGC axons within the optic nerve, in the surrounding connective tissues, and in cells accumulating at the lesion site (Fig. 5J). Finally, recombinant EphB3-Fc protein (EphB3 extracellular domain fused to Fc protein) injected in vivo into the optic nerve of adult mice resulted in the specific labeling of long thin profiles reminiscent of RGC axons (Fig. 5K). Injection of control Fc protein alone resulted only in diffuse punctate staining (Fig. 5L). These findings indicated that axonal processes within the adult optic nerve expressed a receptor protein, likely EphrinB3, capable of binding EphB3.
Figure 5.

EphrinB3 mRNA and protein expression in the adult retina and on RGC axons. A, EphrinB3 mRNA expression in the ganglion cell layer and the inner nuclear layer of the retina from a normal adult mouse. [See D for location of ganglion cell layer (GCL) and inner nuclear layer (INL).] B, EphrinB3 mRNA expression in the ganglion cell layer and the inner nuclear layer 4 d after optic nerve injury. C, EphrinB3 mRNA expression in the ganglion cell layer and the inner nuclear layer 12 d after optic nerve injury. D, Retinal section from a normal adult mouse exposed to the EphrinB3 sense control probe. E, EphrinB3 mRNA expression in the optic nerve of a normal adult mouse. A faint hybridization signal is detected in cells distributed in a row-like manner. F, EphrinB3 mRNA expression in the optic nerve of an adult mouse 4 d after optic nerve injury. G, EphrinB3 mRNA expression in the optic nerve of an adult mouse 12 d after optic nerve injury. H, Anti-EphrinB3 immunoblot. A faint signal is detected in the protein sample from uninjured optic nerves (UI). A band at ∼65 kDa, the expected size of EphrinB3, is present in the optic nerve samples 8 d after injury (8). I, Uninjured optic nerve section after staining using anti-EphrinB3 antibody. There is diffuse staining particularly at the ONH region. J, Optic nerve (ON) tissue section 12 d after injury stained with anti-EphrinB3 antibody. A strong signal was detected in the optic nerve proximal to the injury site. CT, Connective tissue; L, lesion site. K, Section from the same optic nerve shown in J after staining with a secondary antibody alone. L, Recombinant EphB3-Fc protein injected into the adult optic nerve binds to RGC axons. EphB3-Fc was detected using an anti-Fc antibody. M, Fc control protein injected into the adult optic nerve is distributed as diffuse punctate aggregates and does not result in apparent RGC axon binding. Scale bars: A–G, I–K, 100 μm; L, M, 20 μm.
Collectively, these data suggested the following. First, the presence of EphrinB3 in the normal adult retina suggested that this molecule may be involved in some aspect of adult retinal function. Second, the expression of EphrinB3 by cells at the lesion site, together with the fact that EphB3-positive macrophages were located in the same region, suggested a role for EphB–EphrinB interactions in local cellular function after injury. Third, the preservation of EphrinB3 protein expression on injured adult RGC axons suggested that these axons were in a position to respond to EphB proteins found on macrophages in damaged optic nerves.
EphB3 supports adult RGC axon growth in vitro
To examine whether EphB3 can influence the growth of adult RGC axons, recombinant EphB3 protein was used as a protein substratum in an in vitro RGC axon outgrowth assay modified from Meyer and colleagues (Meyer and Miotke, 1990; Bates and Meyer, 1997). In this assay, retinal explants from adult animals 6–12 months of age were grown on a laminin substratum and allowed to extend axons up to several millimeters long. These axons had small club-like growth cones (Fig. 6C,D) and expressed the axon marker GAP-43 (Fig. 6G). Although these axons have a well formed microtubule cytoskeleton (Fig. 6E), their growth cones were noticeably sparse in actin cytoskeleton (Fig. 6F), unlike typical embryonic retinal growth cones. To determine whether EphB3 can influence adult RGC axon outgrowth, the amount of laminin used for substratum coating was first decreased to a level at which adult RGC axon growth was barely supported (Fig. 6A). Recombinant EphB3 was then added to the substratum in increasing amounts, and the effects on adult RGC axon growth were quantified. The addition of EphB3 promoted the outgrowth of RGC axons (Fig. 6B). A dose–response relationship was evident as the amount of EphB3 was increased, whereas laminin levels were unchanged (Fig. 6I,J). Both the number of axons issued by adult retinal tissue explants as well as the mean total length of the RGC axon increased with larger doses of EphB3.
Figure 6.
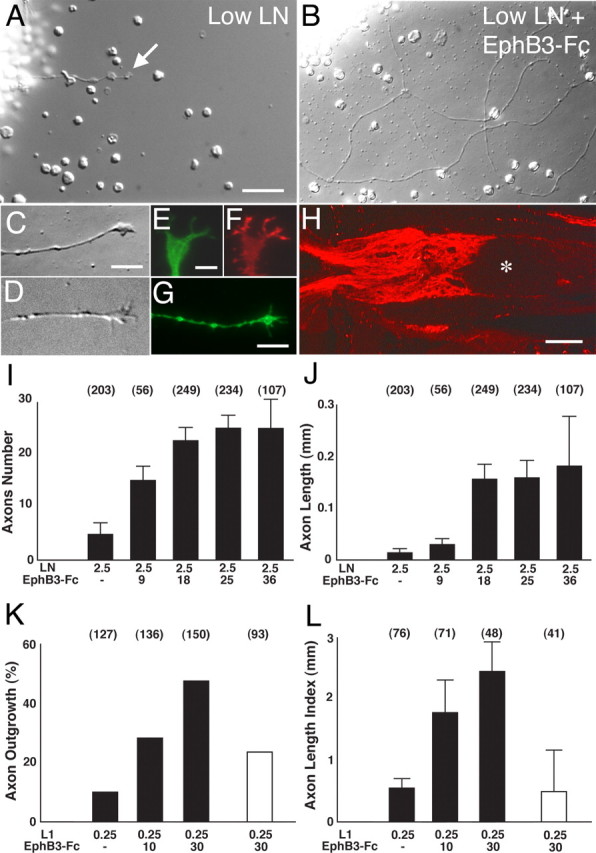
EphB3-Fc protein supports adult RGC axon outgrowth. A, Adult retinal explant grown on a substratum coated with low levels of laminin (2.5 μg/ml). This level of laminin coating supports only minimal axon outgrowth (arrow points to an axon). B, Adult retinal explant grown for 3 d on a substratum coated with low laminin (2.5 μg/ml) and EphB3-Fc (9 μg/ml). The presence of EphB3-Fc increased the amount of axon outgrowth from adult retinal explants. C, Growth cone extending on low laminin (2.5 μg/ml) and EphB3-Fc (9 μg/ml). D, Growth cone extending on L1 (0.25 μg/ml) and EphB3-Fc (10 μg/ml). E, Anti-tubulin staining in a growth cone similar to C. F, Phalloidin staining in the same growth cone as in E. G, Anti-GAP-43 immunostaining in a retinal axon grown on a laminin substratum (10 μg/ml). H, Immunostaining showing L1 protein expression on RGC axons within the optic nerve 12 d after injury. The asterisk indicates the injury site. I, Graph showing the dose–response relationship between the mean axon number with increasing concentrations of EphB3-Fc protein used for coating onto a laminin substratum. The numbers at the top of each column indicate the number of explants examined for the given experimental condition. J, Graph showing the relationship between total axon length (mean ± SEM) with increasing concentrations of EphB3-Fc protein used for substratum coating. The numbers at the top of each column indicate the number of explants examined for the given experimental condition. K, Graph showing the dose–response relationship between the percentage of retinal explants with axon outgrowth and increasing amounts of EphB3-Fc protein used for coating onto an L1 substratum. The numbers in parentheses represent the numbers of explants used in each condition. Data from five independent experiments (mean ± SEM) are shown. The percentage of retinal explants from EphrinB3 null animals with axon outgrowth on an L1/EphB3-Fc substratum is indicated by the open bar. Data from three independent experiments (mean ± SEM) are shown. L, Graph showing the dose–response relationship between the amount of outgrowth with increasing amounts of EphB3-Fc protein used for coating onto a, L1 substratum. The numbers in parentheses represent the total number of explants used in each condition. Data from three independent experiments (mean ± SEM) are shown. The amount of outgrowth from retinal explants of EphrinB3 null animals on an L1/EphB3-Fc substratum is indicated by the open bar. Data from three independent experiments (mean ± SEM) are shown. Scale bars: A, B, 50 μm; C, D, G, 5 μm; E, F, 2 μm; H, 100 μm. LN, Laminin.
The growth-promoting effects of EphB3 were not limited to adult RGC axons cultured on laminin. EphB3 was also effective in promoting adult RGC axon outgrowth on explants cultured on a substratum of L1 protein (Fig. 6K,L). L1 is a member of the Ig superfamily of cell adhesion molecules that is expressed by specific populations of axons and can mediate multiple developmental functions including axon fasciculation (Walsh and Doherty, 1997; Panicker et al., 2003). It is of note that L1 is expressed by adult RGC axons after optic nerve injury (Fig. 6H), and thus the exposure of RGC axons to L1 and EphB3 in vitro mimics the in vivo situation in which injured RGC axons are likely simultaneously exposed to both axonal L1 and macrophage EphB3. The growth-promoting effects of EphB3 were absent in retinal axons from adult EphrinB3 null mice (Fig. 6K,L, open bar), suggesting that this particular member of the Ephrin B family is involved in EphB3 signal transduction for this axon population.
RGC axons turn toward an EphB3 source
Figure 7.
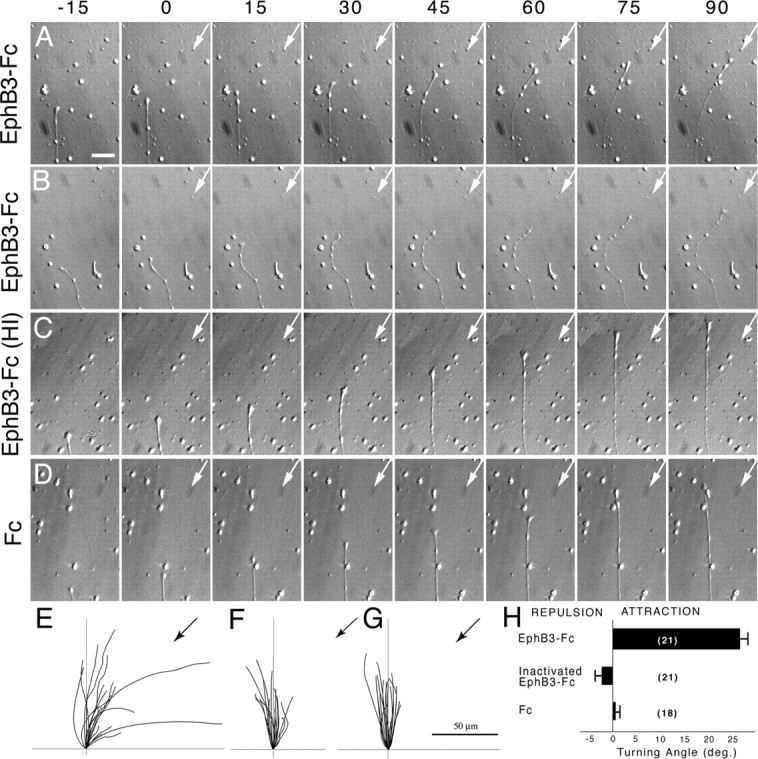
Time-lapse images of adult retinal axon responses to protein reagents applied in axon turning assays. The numbers at the top of each panel indicate the minutes elapsed. The pipette was loaded with test protein, and protein was expelled beginning at time 0. Arrows indicate the 45° orientation of the pipette with respect to the direction of axon growth at the start of protein application. A, Retinal axon turning toward the source of EphB3-Fc protein delivered from a glass micropipette. B, Another retinal axon turning toward the source of EphB3-Fc protein delivered from the glass micropipette. C, Example of a retinal axon that did not respond to a heat-inactivated EphB3-Fc protein source. D, Example of a retinal axon that did not respond to an Fc protein source. E, Graph showing the trajectories of retinal axons exposed to an EphB3-Fc protein source over a period of 1 h. The black arrow indicates the 45° orientation of the pipette with respect to the direction of axon growth at the start of protein application. F, Graph showing the trajectories of retinal axons exposed to a source of heat-inactivated EphB3-Fc protein over a period of 1 h. G, Graph showing the trajectories of retinal axons exposed to an Fc protein source over a period of 1 h. H, Graph illustrating the mean turning angle of retinal axons exposed to different protein sources. EphB3-Fc, n = 21; heat-inactivated EphB3-Fc, n = 21; Fc, n = 18. A positive turning angle indicates axon turning toward the protein source. A mean turning angle close to zero indicates that retinal axons did not respond to the applied test protein. Error bars indicate SEM. The turning angle induced by EphB3-Fc was significantly different from the other conditions (Student’s t test; p < 0.001). Scale bars: (in A) A–D, 5 μm; E–G, 50 μm.
Regrowth of injured RGC axons in EphB3 mutant animals
The potential functional involvement of macrophage EphB3 in RGC axon response to optic nerve injury was examined in EphB3 mutant animals. During development, the mouse retina expresses multiple members of the EphB protein family including EphB1, EphB2, and EphB3 (Birgbauer et al., 2000; Hindges et al., 2002). These EphB molecules appear to have overlapping functions during development, because the loss of a single EphB molecule such as EphB3 does not have an apparent effect on retinal or optic nerve formation (Birgbauer et al., 2000). EphB3 null mice are viable and fertile, and the histological appearance of the retina and the optic nerves from adult EphB3 null animals was not noticeably different from that of wild-type littermates (Fig. 8B,C). EphB3 mRNA was not detected in the RGC layer of the adult mouse retina, nor was it induced after optic nerve injury, consistent with previous work documenting little or no EphB protein on injured or uninjured adult optic axons (Knoll et al., 2001).
Figure 8.

Analysis of the optic nerve, macrophage accumulation, and in vitro retinal axon growth in EphB3 mutant animals. A, Plastic embedded section through a portion of the retina and the optic nerve of a wild-type adult mouse of the mixed EphB3/Thy1-YFP background. B, Plastic embedded section through a portion of the retina and the optic nerve of a homozygous EphB3 null adult mouse from the same litter as in A. C, EphB3 mRNA expression in the optic nerve of a wild-type animal of the mixed EphB3/Thy1-YFP background 8 d after injury. D, No EphB3 mRNA expression in the optic nerve of an EphB3 homozygous null littermate 8 d after injury. E, ED1+ macrophages in the optic nerve of a wild-type animal of the mixed EphB3/Thy1-YFP background 8 d after injury. F, ED1+ macrophages in the optic nerve of an EphB3 homozygous null littermate 8 d after injury. G, Graph showing the average number of macrophages per unit volume in an optic nerve lesion site. The results from three nerves in each group are shown. H, Graph showing the average total volume of the optic nerve lesion site. The results from four nerves in each group are shown. I, Graph showing the percentage of retinal explants from EphB3 null and wild-type littermate animals with axon outgrowth on a laminin substratum. The numbers in parentheses represent the number of explants in each group. No statistically significant difference was found between the genotypes (Student’s t test; p = 0.53). Data pooled from three independent experiments are shown. J, Graph showing the mean axon length per retinal explant from EphB3 null or wild-type littermate animals on a laminin substratum. No statistically significant difference was observed between the comparison groups (Student’s t test; p = 0.4). Data from three independent experiments are shown. Error bars indicate SEM. Scale bars: A, B, 100 μm; C–F, 50 μm. WT, Wild type; KO, EphB3 null.
Although wild-type littermates responded to optic nerve injury by the upregulation of EphB3 expression (Fig. 8D), EphB3 null mice, as expected, showed no detectable EphB3 mRNA expression after nerve damage (Fig. 8E). The absence of EphB3 protein however, did not appear to alter the recruitment and accumulation of macrophages after optic nerve injury (Fig. 8F,G). A quantitative estimate of macrophage numbers after optic nerve injury in wild-type littermates and EphB3 null animals showed approximately equal numbers of these cells recruited after optic nerve injury (Fig. 8H,I) [mean: wild-type, 25,140 (n = 3); EphB3 null, 24,170 (n = 3)]. Adult RGC axons from retinal explants of EphB3 null animals and wild-type littermates did not differ in their ability to grow in culture when tested on laminin (Fig. 8J,K) or L1 protein (data not shown). The percentage of retinal explants with axon growth as well as the mean total amount of axon growth per explant did not differ between these two genotypes. These data indicated that adult RGC axons derived from EphB null animals, if provided with a favorable growth substratum, could readily assemble cytoskeleton and form a new axon.
Reduced RGC axon regrowth in EphB3 mutant mice
To assess whether the loss of EphB3 function affected RGC axon re-extension after optic nerve injury, EphB3 mutant mice were crossed with Thy1-YFP animals. Adult RGC axon re-extension after injury was then determined by the analysis of YFP axons in wild-type (n = 26), heterozygous (n = 31), and homozygous null (n = 41) littermates of the mixed EphB3/Thy1-YFP strain background. Adult animals of the EphB3/Thy1-YFP mixed-strain background have a lower total number of RGC axons in their optic nerves compared with animals of the Thy1-YFP background (data not shown). Nevertheless, wild-type mixed-strain animals clearly exhibited an axon re-extension phenomenon similar to that observed in Thy1-YFP animals (Fig. 9A,F). RGC axon re-extension in wild-type mixed-strain animals reached a peak between 8 and 12 d after injury.
Figure 9.
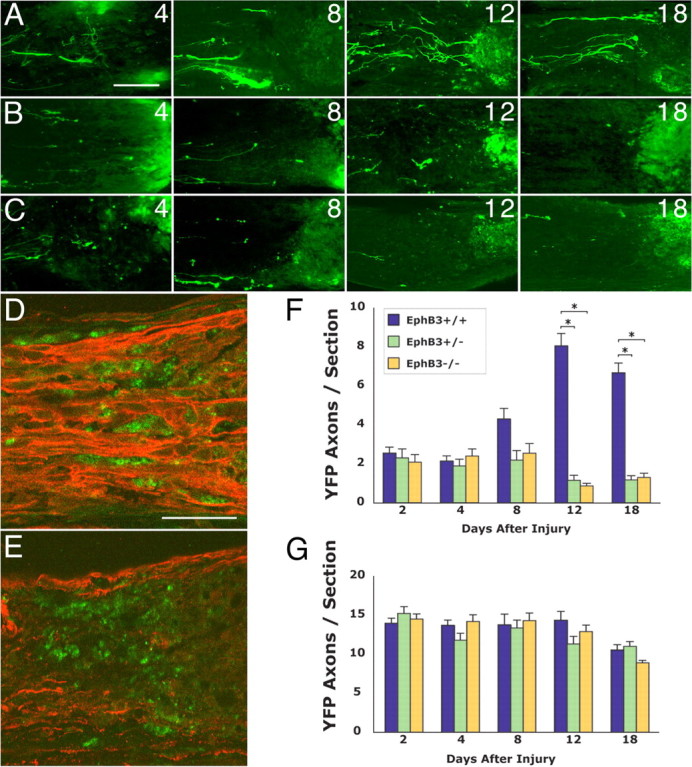
Effects of reduced EphB3 function on RGC axon plasticity and remodeling in vivo. A, YFP RGC axons in the optic nerve of wild-type littermates showing RGC regrowth at 4–18 d after injury. B, YFP RGC axons in the optic nerve of adult EphB3 heterozygous littermates at 4–18 d after injury. C, YFP RGC axons in the optic nerve of adult EphB3 homozygous null littermates at 4–18 d after injury. D, The spatial relationship between regrowing retinal axons (red, anti-βIII tubulin staining) and macrophages (green, ED1 staining) 8 d after optic nerve injury in an EphB3 wild-type animal. Retinal axons course between and maintain a close spatial relationship with individual macrophages. E, The spatial relationship between retinal axons (red, anti-βIII tubulin staining) and macrophages (green, ED1 staining) 8 d after optic nerve injury in an EphB3 homozygous null animal. F, Graph showing RGC axon regrowth (quantified as axons per section) within the margin zone adjacent to the optic nerve injury site between 2 and 18 d after injury in wild-type, heterozygous, and homozygous littermates. The data from wild-type, EphB3 heterozygote, and EphB3 homozygous null mice showed a statistical difference at days 12 and 18 by the Kruskal–Wallis test (p = 0.0001). The results from EphB3 heterozygous and homozygous null animals (asterisks) were each statistically different from those from wild-type animals at 12 and 18 d as determined by pairwise comparisons using the Tukey test (differences of the mean, 5.4–7.2; 95% confidence intervals, 4.3–8.3). G, Graph showing the number of RGC axons in the ONH region between 2 and 18 d after injury in wild-type, heterozygous, and homozygous littermates. No statistically significant difference was found in the data from wild-type, heterozygous, and homozygous groups at all time points by the Kruskal–Wallis test (p = 0.12 to p = 0.96). Error bars indicate SEM. Scale bars: A–C, 50 μm; D, E, 50 μm.
RGC axon re-extension was perturbed both in adult EphB3 heterozygous (Fig. 9B,F) and homozygous null mutant (Fig. 9C,F) littermates. In contrast to wild-type littermates, which showed significant RGC axon re-extension by 12–18 d after optic nerve injury, RGC axons in heterozygous and homozygous littermates did not exhibit a significant re-extension phenomenon, and fewer RGC axons were detected in the margin zone 12–18 d after injury than at 8 d [Kruskal–Wallis test (p = 0.0001), post hoc Tukey pairwise comparison with differences of the mean 5.4–7.2; 95% confidence intervals, 4.3–8.3].
Altered relationship between macrophages and RGC axons in EphB3 mutant animals
The relationship between macrophages and adult RGC axons re-extending toward the lesion site was altered in EphB3 null animals. In wild-type animals 8 d after optic nerve injury, re-extending RGC axons frequently coursed in between individual macrophages, and many instances of RGC axons winding around and encircling single macrophages were apparent (Fig. 9D). In contrast, RGC axons were noticeably lacking in regions populated by macrophages in EphB3 null animals (Fig. 9E). Because in these experiments the entire population of RGC axons was visualized using an anti-βIII tubulin antibody, the results demonstrated that the lack of significant regrowth toward the injury site, as demonstrated by quantitative analysis in YFP-labeled axons (Fig. 9A–C,F), applied to the general RGC axon population.
Similar RGC axon survival in wild-type, heterozygous, and homozygous animals
There was no significant difference in the number of YFP-labeled RGC axons at the ONH region of wild-type, heterozygous, and homozygous animals at each of the time points examined (2–18 d) after optic nerve injury (Fig. 9G) (Kruskal–Wallis test; p = 0.12 to p = 0.96). Animals of all three genotypes showed a decline in the number of RGC axons at the ONH by day 18, consistent with the gradual loss of RGCs after nerve damage. The finding that the number of RGC axons at the ONH of EphB3 heterozygous and homozygous null animals was comparable to that in wild-type littermates indicated that there was no accelerated loss of RGCs in EphB3 mutant animals that might account for the low numbers of YFP axons. Instead, it appeared that normal numbers of adult RGC axons were present in the ONH of EphB3 mutant animals, but in the absence of full EphB3 protein expression in the optic nerve, RGC axonal plasticity was compromised, and these axons did not re-extend toward the lesion site.
Axon terminal branching occurs in EphB3 mutant animals
Although few RGC axons re-extended into the margin zone of EphB3 mutant animals, sufficient numbers of these axons could be counted when pooled together from multiple animals to permit an analysis of their axon terminal morphologies at 8 and 12 d after nerve injury. RGC axons in EphB3 heterozygous and homozygous mutant animals showed axon terminals that appeared similar to those in wild-type littermates (Fig. 10). Axons in EphB3 mutants exhibited simple branching (Fig. 10A) as well as extensive branching patterns (Fig. 10G) that were equivalent in complexity to the most complex axons in wild-type animals. Eight days after injury, ∼20% of axons in wild-type, EphB3 heterozygous, and homozygous mutant animals had at least more than one axon terminal branch (Table 1). By 12 d after injury, 17% of wild-type RGC axons showed a branching morphology, whereas up to 40% of axons from heterozygous (χ2 test; p = 0.03) and homozygous null animals (χ2 test; p = 0.03) exhibited branching. These results suggested that the ability of adult RGC axons to elaborate complex terminal branches remained intact and perhaps even showed an increase after a reduction or loss of EphB3 function. Thus, the molecular mechanisms that mediated adult RGC axon re-extension toward the site of injury were different from those that triggered the elaboration of axon terminal branching by the same axons.
Figure 10.

RGC axon branching in EphB3 mutant animals after optic nerve injury. Axons arranged in increasing level of complexity of their terminal morphology are shown. A, Adult EphB3 heterozygous mutant animal. B–G, Adult EphB3 null animals. Scale bar, 50 μm.
Table 1.
Percentage of axons with terminal branching
| Genotype | 8 d after injury |
12 d after injury |
||
|---|---|---|---|---|
| Number of axons | % Branching | Number of axons | % Branching | |
| EphB3+/+ | 86 | 21 | 161 | 17 |
| EphB3+/− | 49 | 25 | 30 | 40* |
| EphB3−/− | 73 | 21 | 36 | 39* |
*p<0.05, statistical significance compared with EphB3+/+ (χ2 test).
Discussion
RGC axon re-extension and remodeling after optic nerve injury occurred in close spatiotemporal relationship to macrophage recruitment into the lesion site. Recruited macrophages expressed the axon guidance molecule EphB3, whereas injured RGC axons expressed EphrinB3, a known receptor for EphB3. Adult RGC axons in vivo bound exogenously applied EphB3 protein, and adult RGC axon outgrowth in vitro was promoted by an EphB3 protein substratum and by soluble EphB3 protein. Both EphB3 heterozygous and homozygous null adult animals showed a significant reduction in axon re-extension in the injured nerve that was not attributable to a reduced intrinsic ability for axon extension or an accelerated axon loss after injury. Rather, EphB3 seemed to act as a local promoter of adult RGC axon re-extension after injury. Furthermore, in EphB3 mutant animals, the ability of damaged RGC axons to elaborate terminal branching was unperturbed suggesting that axon terminal remodeling in the damaged CNS was governed by additional EphB3-independent mechanisms.
Embryonic versus adult RGC axon response to EphB3
In mouse embryos, both in vitro and in vivo studies indicate that EphB proteins serve as inhibitory RGC axon guidance molecules during intraretinal axon pathfinding toward the ONH (Birgbauer et al., 2000, 2001). Embryonic and adult RGC axon responses to EphB protein do, however, differ in one significant manner. Although embryonic RGC growth cones are inhibited by these guidance molecules, the current study showed that adult RGC axons in the optic nerve responded to EphB3 as a growth-promoting molecule. The activity of EphB3 as an “attractive” axon guidance molecule is reminiscent of its role during development of the Xenopus retinotectal projection (Mann et al., 2002). A difference in response characteristic is also consistent with reports that axon response to a particular guidance molecule can switch from inhibition to growth promotion during the progression from embryonic development to postnatal life and may be related to levels of growth cone cyclic nucleotides (Cai et al., 2001). If so, our results extend this previous observation and indicate that the switch that occurs perinatally may be stable even at 6–12 months of age in mice.
The adult injured visual system compared with the embryonic visual system seems to be more sensitive to the loss of EphB3 function. The developing retina expresses multiple EphB molecules in overlapping patterns; the lack of a single EphB, such as EphB2 or EphB3, has little apparent consequence on RGC axon pathfinding (Birgbauer et al., 2000; Hindges et al., 2002); and EphB molecules appear to functionally compensate for each other. However, in the adult optic nerve, only EphB3 was detected after nerve injury, and EphB1 and EphB2 were not present. In this case, the lack of a single copy of the EphB3 gene in heterozygous animals was sufficient to alter the phenomenon of RGC axon re-extension.
Models of EphB3 function
Several lines of evidence support a model in which adult RGC axonal plasticity after injury is regulated by EphB protein present in the injured optic nerve. First, EphB3 mRNA is specifically upregulated, and EphB3 protein can be detected in the injured optic nerve. Second, EphB protein binding sites/receptors are present on adult RGC axons in vivo, and RGC axons respond to EphB recombinant proteins in in vitro outgrowth and growth cone guidance assays. Third, retinal explants from adult EphB3 null mice and wild-type littermates exhibit similar axon outgrowth in vitro, indicating that the loss of EphB3 function does not appear to affect the basic ability of RGC axons to re-extend after axonal damage. Fourth, quantitative analysis shows that approximately equivalent numbers of RGC axons remain at the ONH of both adult EphB3 null mice and wild-type littermates after optic nerve injury, indicating that the loss of EphB3 function does not accelerate loss of injured RGC axons. Finally, EphB3 null animals show a significant reduction in RGC axon number that re-extend toward the lesion site compared with wild-type littermates. Based on these data, we tentatively conclude that the regrowth of injured adult RGC axons is governed by local EphB3 protein expression in the optic nerve.
One way in which EphB3 may play a role in RGC axon regrowth is by serving as a local guidance molecule in the injured nerve. Our in vitro findings demonstrating the ability of recombinant EphB3 protein to support RGC axon outgrowth is consistent with this possibility, and we currently favor this explanation. However, findings from the present study do not yet rule out a more complex mechanism in which EphB3 is essential, but by itself is not sufficient, for the sprouting of retinal axons in response to injury in vivo and that additional factors such as astrocyte withdrawal and the removal of myelin debris may be involved. One scenario that seems unlikely is that the loss of EphB3 function affects RGC axon re-extension by interfering with macrophage migration, which in turn affects RGC axon re-growth.
Mediators of macrophage function after nerve injury
The presence of macrophages in regions of neuronal injury, including in the optic nerve, is well described (Perry et al., 1987; David et al., 1990; Blaugrund et al., 1992; Frank and Wolburg, 1996; Selles-Navarro et al., 2001). Although macrophage accumulation has been proposed to support axon regeneration (Lazarov-Spiegler et al., 1996), the mechanisms involved are not fully understood. After adult lens injury, macrophages secrete factors into the vitreous that can act intraocularly on RGC cell bodies and stimulate axon regeneration (Fischer et al., 2000; Yin et al., 2003). Results from the present study suggest that macrophages in the injured optic nerve exert local effects on injured RGC axons via a mechanism involving EphB3, a transmembrane axon guidance molecule that mediates RGC axon pathfinding during embryonic development. The expression of EphB3 by macrophages is consistent with the fact that guidance molecules such as L1, semaphorins, and Slits have been demonstrated on leukocyte populations (Kadmon et al., 1998; Kumanogoh and Kikutani, 2001; Wong et al., 2002). The findings that macrophages in the injured optic nerve express EphB3 and that EphB3 is an essential component for adult RGC axon re-extension may provide some mechanistic insight into how macrophages perform their supportive functions after CNS injury.
Separate control of axon terminal branching
Results from this study show that CNS axon re-extension and axon terminal branching after injury can be dissociated. Although axon re-extension was greatly reduced in the absence of EphB3 function, the ability of injured axons to elaborate complex axon terminal branching in EphB3 mutants was unaffected or even increased. The mechanisms governing RGC axon terminal branching remain to be identified. One possibility is that the adult RGC axon growth intrinsically gives rise to a branching morphology. We have not typically observed significant branching by adult RGC axons in vitro, and the occasional branching does not approach the complexity observed in the injured optic nerve (Liu and Sretavan, unpublished observations). During development, axonal and dendritic branching is actively controlled by local signals (Heffner et al., 1990; Roskies and O’Leary, 1994; Sato et al., 1994; Kalil et al., 2000) such as FGF2 (Szebenyi et al., 2001), Slit2 (Wang et al., 1999), Semaphorin3A, and Netrin-1 (Dent et al., 2004). For these reasons, we speculate that the injured CNS environment contains other axon guidance molecules in addition to EphB3 that regulate axonal branching. Given that, in addition to macrophages, astrocytes are also present around the optic nerve lesion, both cell populations are potential sources of such factors.
Axon guidance molecules and CNS injury
In recent years, axon guidance molecules of the Eph and semaphorin families have been reported in deafferented CNS targets (Knoll et al., 2001) and around or within lesion sites at late stages after CNS injury (Miranda et al., 1999; Pasterkamp et al., 1999; De Winter et al., 2002; Willson et al., 2002; Bundesen et al., 2003; Goldshmit et al., 2004). These studies have primarily hypothesized that axon guidance molecules negatively affect adult axon growth and may thus underlie the failure of CNS regeneration. The results from the current study show that, rather than a late event, axon guidance molecules can, in fact, appear within days after injury. Furthermore, guidance molecules that inhibit axon growth during development may not necessarily also inhibit the same axon population in the adult nervous system. The expression of axon guidance molecules at early, intermediate, and late times after CNS injury suggests that cell–cell signaling mediated by guidance molecule/receptor interactions may play a host of functions at multiple time points. Given the lessons learned from the molecular control of axon pathfinding during development, significant opportunity may exist to use the biology of axon guidance molecules to enhance functional recovery in neurological trauma and disease.
Footnotes
This work was supported by the National Institutes of Health, the Research to Prevent Blindness, and That Man May See. We thank Dr. Mark Henkemeyer for EphB3 and EphrinB3 mutant animals, Dr. Jacqueline Leung for assistance with statistical analysis, and members of the D. Sretavan and M. LaVail laboratories for technical assistance and comments.
References
- Aguayo AJ, Clarke DB, Jelsma TN, Kittlerova P, Friedman HC, Bray GM (1996). Effects of neurotrophins on the survival and regrowth of injured retinal neurons. Ciba Found Symp 196:135–144. [DOI] [PubMed] [Google Scholar]
- Allcutt D, Berry M, Sievers J (1984). A qualitative comparison of the reactions of retinal ganglion cell axons to optic nerve crush in neonatal and adult mice. Brain Res 318:231–240. [DOI] [PubMed] [Google Scholar]
- Azmitia EC, Buchan AM, Williams JH (1978). Structural and functional restoration by collateral sprouting of hippocampal 5-HT axons. Nature 274:374–376. [DOI] [PubMed] [Google Scholar]
- Bates CA, Meyer RL (1997). The neurite-promoting effect of laminin is mediated by different mechanisms in embryonic and adult regenerating mouse optic axons in vitro. Dev Biol 181:91–101. [DOI] [PubMed] [Google Scholar]
- Beattie MS, Bresnahan JC, Komon J, Tovar CA, Van Meter M, Anderson DK, Faden AI, Hsu CY, Noble LJ, Salzman S, Young W (1997). Endogenous repair after spinal cord contusion injuries in the rat. Exp Neurol 148:453–463. [DOI] [PubMed] [Google Scholar]
- Birgbauer E, Cowan CA, Sretavan DW, Henkemeyer M (2000). Kinase independent function of EphB receptors in retinal axon pathfinding to the optic disc from dorsal but not ventral retina. Development 127:1231–1241. [DOI] [PubMed] [Google Scholar]
- Birgbauer E, Oster SF, Severin CG, Sretavan DW (2001). Retinal axon growth cones respond to EphB extracellular domains as inhibitory axon guidance cues. Development 128:3041–3048. [DOI] [PubMed] [Google Scholar]
- Blaugrund E, Duvdevani R, Lavie V, Solomon A, Schwartz M (1992). Disappearance of astrocytes and invasion of macrophages following crush injury of adult rodent optic nerves: implications for regeneration. Exp Neurol 118:105–115. [DOI] [PubMed] [Google Scholar]
- Bundesen LQ, Scheel TA, Bregman BS, Kromer LF (2003). Ephrin-B2 and EphB2 regulation of astrocyte-meningeal fibroblast interactions in response to spinal cord lesions in adult rats. J Neurosci 23:7789–7800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai D, Qiu J, Cao Z, McAtee M, Bregman BS, Filbin MT (2001). Neuronal cyclic AMP controls the developmental loss in ability of axons to regenerate. J Neurosci 21:4731–4739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cajal RS (1928). In: Degeneration and regeneration of the nervous system London: Oxford UP.
- Chung W-W, Lagenaur CF, Yan Y, Lund JS (1991). Developmental expression of neural cell adhesion molecules in the mouse neocortex and olfactory bulb. J Comp Neurol 314:290–305. [DOI] [PubMed] [Google Scholar]
- Cotman CW, Matthews DA, Taylor D, Lynch G (1973). Synaptic rearrangement in the dentate gyrus: histochemical evidence of adjustments after lesions in immature and adult rats. Proc Natl Acad Sci USA 70:3473–3477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowan CA, Henkemeyer M (2001). The SH2/SH3 adaptor Grb4 transduces B-ephrin reverse signals. Nature 413:174–179. [DOI] [PubMed] [Google Scholar]
- Darian-Smith C, Gilbert CD (1994). Axonal sprouting accompanies functional reorganization in adult cat striate cortex. Nature 368:737–740. [DOI] [PubMed] [Google Scholar]
- David S, Bouchard C, Tsatas O, Giftochristos N (1990). Macrophages can modify the nonpermissive nature of the adult mammalian central nervous system. Neuron 5:463–469. [DOI] [PubMed] [Google Scholar]
- Dent EW, Barnes AM, Tang F, Kalil K (2004). Netrin-1 and semaphorin 3A promote or inhibit cortical axon branching, respectively, by reorganization of the cytoskeleton. J Neurosci 24:3002–3012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Winter F, Oudega M, Lankhorst AJ, Hamers FP, Blits B, Ruitenberg MJ, Pasterkamp RJ, Gispen WH, Verhaagen J (2002). Injury-induced class 3 semaphorin expression in the rat spinal cord. Exp Neurol 175:61–75. [DOI] [PubMed] [Google Scholar]
- Fariss RN, Li ZY, Milam AH (2000). Abnormalities in rod photoreceptors, amacrine cells, and horizontal cells in human retinas with retinitis pigmentosa. Am J Ophthalmol 129:215–223. [DOI] [PubMed] [Google Scholar]
- Feng G, Mellor RH, Bernstein M, Keller-Peck C, Nguyen QT, Wallace M, Nerbonne JM, Lichtman JW, Sanes JR (2000). Imaging neuronal subsets in transgenic mice expressing multiple spectral variants of GFP. Neuron 28:41–51. [DOI] [PubMed] [Google Scholar]
- Fischer D, Pavlidis M, Thanos S (2000). Cataractogenic lens injury prevents traumatic ganglion cell death and promotes axonal regeneration both in vivo and in culture. Invest Ophthalmol Vis Sci 41:3943–3954. [PubMed] [Google Scholar]
- Fisher SK, Lewis GP (2003). Muller cell and neuronal remodeling in retinal detachment and reattachment and their potential consequences for visual recovery: a review and reconsideration of recent data. Vision Res 43:887–897. [DOI] [PubMed] [Google Scholar]
- Flanagan JG, Vanderhaeghen P (1998). The ephrins and Eph receptors in neural development. Annu Rev Neurosci 21:309–345. [DOI] [PubMed] [Google Scholar]
- Frank M, Wolburg H (1996). Cellular reactions at the lesion site after crushing of the rat optic nerve. Glia 16:227–240. [DOI] [PubMed] [Google Scholar]
- Goldshmit Y, Galea MP, Wise G, Bartlett PF, Turnley AM (2004). Axonal regeneration and lack of astrocytic gliosis in EphA4-deficient mice. J Neurosci 24:10064–10073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grafstein B, Ingoglia NA (1982). Intracranial transection of the optic nerve in adult mice: preliminary observations. Exp Neurol 76:318–330. [DOI] [PubMed] [Google Scholar]
- Heffner CD, Lumsden AG, O’Leary DD (1990). Target control of collateral extension and directional axon growth in the mammalian brain. Science 247:217–220. [DOI] [PubMed] [Google Scholar]
- Hindges R, McLaughlin T, Genoud N, Henkemeyer M, O’Leary DD (2002). EphB forward signaling controls directional branch extension and arborization required for dorsal-ventral retinotopic mapping. Neuron 35:475–487. [DOI] [PubMed] [Google Scholar]
- Hirsch EC (2000). Nigrostriatal system plasticity in Parkinson’s disease: effect of dopaminergic denervation and treatment. Ann Neurol 47:S120–S111. discussion. [PubMed] [Google Scholar]
- Holland SJ, Peles E, Pawson T, Schlessinger J (1998). Cell-contact-dependent signalling in axon growth and guidance: Eph receptor tyrosine kinases and receptor protein tyrosine phosphatase beta. Curr Opin Neurobiol 8:117–127. [DOI] [PubMed] [Google Scholar]
- Holtmaat AJ, Gorter JA, De Wit J, Tolner EA, Spijker S, Giger RJ, Lopes da Silva FH, Verhaagen J (2003). Transient downregulation of Sema3A mRNA in a rat model for temporal lobe epilepsy. A novel molecular event potentially contributing to mossy fiber sprouting. Exp Neurol 182:142–150. [DOI] [PubMed] [Google Scholar]
- Jones TA, Kleim JA, Greenough WT (1996). Synaptogenesis and dendritic growth in the cortex opposite unilateral sensorimotor cortex damage in adult rats: a quantitative electron microscopic examination. Brain Res 733:142–148. [DOI] [PubMed] [Google Scholar]
- Kadmon G, Montgomery AM, Altevogt P (1998). L1 makes immunological progress by expanding its relations. Dev Immunol 6:205–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalil K, Szebenyi G, Dent EW (2000). Common mechanisms underlying growth cone guidance and axon branching. J Neurobiol 44:145–158. [PubMed] [Google Scholar]
- Knoll B, Drescher U (2002). Ephrin-As as receptors in topographic projections. Trends Neurosci 25:145–149. [DOI] [PubMed] [Google Scholar]
- Knoll B, Isenmann S, Kilic E, Walkenhorst J, Engel S, Wehinger J, Bahr M, Drescher U (2001). Graded expression patterns of ephrin-As in the superior colliculus after lesion of the adult mouse optic nerve. Mech Dev 106:119–127. [DOI] [PubMed] [Google Scholar]
- Kumanogoh A, Kikutani H (2001). The CD100-CD72 interaction: a novel mechanism of immune regulation. Trends Immunol 22:670–676. [DOI] [PubMed] [Google Scholar]
- Lazarov-Spiegler O, Solomon AS, Zeev-Brann AB, Hirschberg DL, Lavie V, Schwartz M (1996). Transplantation of activated macrophages overcomes central nervous system regrowth failure. FASEB J 10:1296–1302. [DOI] [PubMed] [Google Scholar]
- Li ZY, Kljavin IJ, Milam AH (1995). Rod photoreceptor neurite sprouting in retinitis pigmentosa. J Neurosci 15:5429–5438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Q, Sun EE, Klein RS, Flanagan JG (2001). Ephrin-B reverse signaling is mediated by a novel PDZ-RGS protein and selectively inhibits G protein-coupled chemoattraction. Cell 105:69–79. [DOI] [PubMed] [Google Scholar]
- Mann F, Ray S, Harris W, Holt C (2002). Topographic mapping in dorsoventral axis of the Xenopus retinotectal system depends on signaling through Ephrin-B ligands. Neuron 35:461–473. [DOI] [PubMed] [Google Scholar]
- McLaughlin T, Hindges R, O’Leary DD (2003). Regulation of axial patterning of the retina and its topographic mapping in the brain. Curr Opin Neurobiol 13:57–69. [DOI] [PubMed] [Google Scholar]
- Meyer RL, Miotke J (1990). Rapid initiation of neurite outgrowth onto laminin from explants of adult mouse retina induced by optic nerve crush. Exp Neurol 107:214–221. [DOI] [PubMed] [Google Scholar]
- Miranda JD, White LA, Marcillo AE, Willson CA, Jagid J, Whittemore SR (1999). Induction of Eph B3 after spinal cord injury. Exp Neurol 156:218–222. [DOI] [PubMed] [Google Scholar]
- Nakagawa S, Brennan C, Johnson KG, Shewan D, Harris WA, Holt CE (2000). Ephrin-B regulates the Ipsilateral routing of retinal axons at the optic chiasm. Neuron 25:599–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orioli D, Henkemeyer M, Lemke G, Klein R, Pawson T (1996). Sek4 and Nuk receptors cooperate in guidance of commissural axons and in palate formation. EMBO J 15:6035–6049. [PMC free article] [PubMed] [Google Scholar]
- Oster SF, Bodeker MO, He F, Sretavan DW (2003). Invariant Sema5A inhibition serves an ensheathing function during optic nerve development. Development 130:775–784. [DOI] [PubMed] [Google Scholar]
- Oster SF, Deiner M, Birgbauer E, Sretavan DW (2004). Ganglion cell axon pathfinding in the retina and optic nerve. Semin Cell Dev Biol 15:125–136. [DOI] [PubMed] [Google Scholar]
- Panicker AK, Buhusi M, Thelen K, Maness PF (2003). Cellular signalling mechanisms of neural cell adhesion molecules. Front Biosci 8:900–911. [DOI] [PubMed] [Google Scholar]
- Pasterkamp RJ, Giger RJ, Ruitenberg MJ, Holtmaat AJ, De Wit J, De Winter F, Verhaagen J (1999). Expression of the gene encoding the chemorepellent semaphorin III is induced in the fibroblast component of neural scar tissue formed following injuries of adult but not neonatal CNS. Mol Cell Neurosci 13:143–166. [DOI] [PubMed] [Google Scholar]
- Perry VH, Brown MC, Gordon S (1987). The macrophage response to central and peripheral nerve injury. A possible role for macrophages in regeneration. J Exp Med 165:1218–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raineteau O, Schwab ME (2001). Plasticity of motor systems after incomplete spinal cord injury. Nat Rev Neurosci 2:263–273. [DOI] [PubMed] [Google Scholar]
- Raisman G (1969). Neuronal plasticity in the septal nuclei of the adult rat. Brain Res 14:25–48. [DOI] [PubMed] [Google Scholar]
- Roskies AL, O’Leary DD (1994). Control of topographic retinal axon branching by inhibitory membrane-bound molecules. Science 265:799–803. [DOI] [PubMed] [Google Scholar]
- Sato M, Lopez-Mascaraque L, Heffner CD, O’Leary DD (1994). Action of a diffusible target-derived chemoattractant on cortical axon branch induction and directed growth. Neuron 13:791–803. [DOI] [PubMed] [Google Scholar]
- Selles-Navarro I, Ellezam B, Fajardo R, Latour M, McKerracher L (2001). Retinal ganglion cell and nonneuronal cell responses to a microcrush lesion of adult rat optic nerve. Exp Neurol 167:282–289. [DOI] [PubMed] [Google Scholar]
- Stuermer CA, Bastmeyer M (2000). The retinal axon’s pathfinding to the optic disk. Prog Neurobiol 62:197–214. [DOI] [PubMed] [Google Scholar]
- Suh LH, Oster SF, Soehrman SS, Grenningloh G, Sretavan DW (2004). L1/laminin modulation of growth cone response to EphB triggers growth pauses and regulates the microtubule destabilizing protein SCG10. J Neurosci 24:1976–1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szebenyi G, Dent EW, Callaway JL, Seys C, Lueth H, Kalil K (2001). Fibroblast growth factor-2 promotes axon branching of cortical neurons by influencing morphology and behavior of the primary growth cone. J Neurosci 21:3932–3941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Horck FP, Weinl C, Holt CE (2004). Retinal axon guidance: novel mechanisms for steering. Curr Opin Neurobiol 14:61–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh FS, Doherty P (1997). Neural cell adhesion molecules of the immunoglobulin superfamily: role in axon growth and guidance. Annu Rev Cell Dev Biol 13:425–456. [DOI] [PubMed] [Google Scholar]
- Wang KH, Brose K, Arnott D, Kidd T, Goodman CS, Henzel W, Tessier-Lavigne M (1999). Biochemical purification of a mammalian slit protein as a positive regulator of sensory axon elongation and branching. Cell 96:771–784. [DOI] [PubMed] [Google Scholar]
- Weidner N, Ner A, Salimi N, Tuszynski MH (2001). Spontaneous corticospinal axonal plasticity and functional recovery after adult central nervous system injury. Proc Natl Acad Sci USA 98:3513–3518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams SE, Mann F, Erskine L, Sakurai T, Wei S, Rossi DJ, Gale NW, Holt CE, Mason CA, Henkemeyer M (2003). Ephrin-B2 and EphB1 mediate retinal axon divergence at the optic chiasm. Neuron 39:919–935. [DOI] [PubMed] [Google Scholar]
- Willson CA, Irizarry-Ramirez M, Gaskins HE, Cruz-Orengo L, Figueroa JD, Whittemore SR, Miranda JD (2002). Upregulation of EphA receptor expression in the injured adult rat spinal cord. Cell Transplant 11:229–239. [PubMed] [Google Scholar]
- Wong K, Park HT, Wu JY, Rao Y (2002). Slit proteins: molecular guidance cues for cells ranging from neurons to leukocytes. Curr Opin Genet Dev 12:583–591. [DOI] [PubMed] [Google Scholar]
- Yin Y, Cui Q, Li Y, Irwin N, Fischer D, Harvey AR, Benowitz LI (2003). Macrophage-derived factors stimulate optic nerve regeneration. J Neurosci 23:2284–2293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoyama N, Romero MI, Cowan CA, Galvan P, Helmbacher F, Charnay P, Parada LF, Henkemeyer M (2001). Forward signaling mediated by ephrin-B3 prevents contralateral corticospinal axons from recrossing the spinal cord midline. Neuron 29:85–97. [DOI] [PubMed] [Google Scholar]
- Zeng BY, Anderson PN, Campbell G, Lieberman AR (1995). Regenerative and other responses to injury in the retinal stump of the optic nerve in adult albino rats: transection of the intracranial optic nerve. J Anat 186:495–508. [PMC free article] [PubMed] [Google Scholar]
- Zheng JQ, Felder M, Connor JA, Poo MM (1994). Turning of nerve growth cones induced by neurotransmitters. Nature 368:140–144. [DOI] [PubMed] [Google Scholar]