

Abstract
Glial cell line-derived neurotrophic factor (GDNF) is an important neurotrophic factor that has therapeutic implications for neurodegenerative disorders. We previously showed that leucine-isoleucine (Leu-Ile), an analog of a dipeptide-like structure of FK506 (tacrolimus), induces GDNF expression both in vivo and in vitro. In this investigation, we sought to clarify the cellular mechanisms underlying the GDNF-inducing effect of this dipeptide. Leu-Ile transport was investigated using fluorescein isothiocyanate-Leu-Ile in cultured neurons, and the results showed the transmembrane mobility of this dipeptide. By liquid chromatography-mass spectrometry and quartz crystal microbalance assay, we identified heat shock cognate protein 70 as a protein binding specifically to Leu-Ile, and molecular modeling showed that the ATPase domain is the predicted binding site. Leu-Ile stimulated Akt phosphorylation, which was attenuated significantly by heat shock protein 90 (Hsp90) inhibitor geldanamycin (GA). Moreover, enhanced interaction between phosphorylated Akt and Hsp90 was detected by immunoprecipitation. Leu-Ile elicited an increase in cAMP response element binding protein (CREB) phosphorylation, which was inhibited by GA, indicating that CREB is a downstream target of Hsp90/Akt signaling. Leu-Ile elevated the levels of GDNF mRNA and protein expression, whereas inhibition of CREB blocked such effects. Leu-Ile promoted the binding activity of phosphorylated CREB with cAMP response element. These findings show that CREB plays a key role in transcriptional regulation of GDNF expression induced by Leu-Ile. In conclusion, Leu-Ile activates Hsp90/Akt/CREB signaling, which contributes to the upregulation of GDNF expression. It may represent a novel lead compound for the treatment of dopaminergic neurons or motoneuron diseases.
Keywords: GDNF, dipeptide, FK506, Hsp90, Hsc70, CREB
Introduction
Glial cell line-derived neurotrophic factor (GDNF) is an important neurotrophic factor that regulates the development, migration, and survival of neurons, and has therapeutic implications for neurodegenerative disorders (Airaksinen and Saarma, 2002). We have demonstrated that leucine-isoleucine (Leu-Ile), an analog of a dipeptide-like structure of FK506, shows nonimmunosuppressive activity and promotes neuronal survival through induction of GDNF in both in vivo and in vitro studies (Nitta et al., 2004), but the mechanism is unclear.
Studies have indicated the complex regulatory mechanisms of GDNF expression. It can be induced by diverse extracellular stimuli (Verity et al., 1999; Castro et al., 2005), and multiple transcription factor binding sites have been identified in the promoter sequence of the GDNF gene (Woodbury et al., 1998), such as cAMP response element (CRE) binding protein (CREB) (Matsushita et al., 1997; Baecker et al., 1999). Previous studies showed that CREB activation is associated with GDNF expression (Young et al., 1999; Lenhard et al., 2002), implying that CREB may participate in regulating GDNF expression as a transcriptional factor. Phosphorylation of CREB at serine-133 (Ser133) within the kinase-inducible domain is critical for its function as a stimulus-dependent transcriptional activator, and multiple kinases have been implicated as activators of CREB in neurons, including protein kinase C (PKC) (Roberson et al.,1999), calmodulin kinase II (CaMKII) (Lee et al., 2004), extracellular signal-regulated kinase 1/2 (ERK1/2) (Schinelli et al., 2001), and serine/threonine kinase Akt (Brunet et al., 2001). Different extracellular stimuli may activate distinct signalings, which contribute to CREB phosphorylation and cellular responses. In particular, CREB is considered to be a regulatory target for Akt, and Akt can promote cell survival by stimulating the expression of cellular genes via the CREB-dependent pathway (Du and Montminy, 1998; Pugazhenthi et al., 2000).
Although phosphoinositide 3-kinase (PI3-k) is an important activator for Akt, increasing evidence has indicated that Akt can be regulated in PI3-k independent manners in neurons, such as ERK1/2 and CaMK cascades (Yano et al., 1998; Brami-Cherrier et al., 2002). Moreover, Akt is a well characterized heat shock protein 90 (Hsp90)-dependent kinase (Basso et al., 2002; Xu et al., 2003), and chaperones Hsp90 and heat shock cognate protein 70 (Hsc70) have been demonstrated to play a role in Akt regulation through distinct mechanisms. For instance, Hsp90–Akt interaction increases Akt activity by protecting it from dephosphorylation by protein phosphatase 2A (PP2A) (Sato et al., 2000; Yun and Matts, 2005); the binding of client molecule to Hsc70 maintains Akt phosphorylation and downstream cascade activation by inhibiting its proteasomal degradation based on Hsc70/Hsp90 machinery (Doong et al., 2003). Therefore, modulation of Hsp90 resulting from a variety of physiological or pharmacological factors may alter Akt signaling, contributing to the regulation of cellular function and response (Pratt and Toft, 2003). It is known that Hsp90 ATPase activity is highly regulated by the binding of cochaperone or client proteins, such as FK506 binding protein (FKBP) and Hsc70 (McLaughlin et al., 2002).
We defined a signaling cascade in which Leu-Ile promotes CREB phosphorylation via Hsp90/Akt signaling, which plays an important role in the transcriptional regulation of the GDNF gene. Moreover, Hsc70 is likely to cooperate with Hsp90 as a cochaperone to modulate Akt activity.
Materials and Methods
Materials.
Leu-Ile, proline-leucine (Pro-Leu), and isoleucine-proline (Ile-Pro) were synthesized by Kokusan Chemical (Tokyo, Japan). Leu-Ile labeled with fluorescein isothiocyanate (FITC) was prepared by Thermo Electron Corporation (Ulm, Germany). Geldanamycin (GA) and 2-(4-morpholinyl)-8-phenyl-1(4H)-benzopyran-4-one (LY294002) were purchased from Sigma (St. Louis, MO). FK506 was gifted from Fujisawa Pharmaceutical (Osaka, Japan). Anti-Hsp70, anti-Hsp90, anti-phosphorylated Akt (pAkt; Thr308), anti-Akt, anti-pCREB (Ser133), anti-CREB, anti-ERK1/2, anti-pERK1/2 (Thr202/Tyr204), anti-pCaMKIIα,β, anti-phosphorylated p38 mitogen-activated protein kinase (pP38MAPK) (Thr180/Tyr182), anti-phosphorylated stress-activated protein kinase/Jun-terminal kinase (pSAPK/JNK) (Thr183/Tyr185), and anti-microtubule-associated protein 2 (MAP2) antibodies were purchased from Cell Signaling Technology (Beverly, MA). Anti-PKC, anti-c-Src, and anti-actin antibodies were from Santa Cruz Biotechnology (Santa Cruz, CA). Anti-Hsc70 antibody and recombinant Hsc70 were from Stressgen Biotechnologies (Victoria, Canada). Anti-GDNF antibody was from R&D Systems (Minneapolis, MN). Anti-glial fibrillary acidic protein (GFAP) antibody was from Chemicon International (Temecula, CA).
Primary hippocampal neuron cultures.
Primary hippocampal neuronal cultures were prepared from day 17 embryos of rats. Briefly, hippocampi were dissected and digested with 0.25% trypsin at 37°C for 30 min. Hippocampal cells were plated in polyornithine-coated plates in DMEM/F12 medium containing 10% fetal bovine serum. The medium was replaced with DMEM/F12 medium containing 1% N2 supplement (Invitrogen, San Diego, CA) 24 h later. MAP2-positive cells accounted for over 95% of the total in cultures.
Transmembrane transport of Leu-Ile.
Cultured neurons were incubated with FITC-Leu-Ile or FITC at various concentrations for the indicated periods at 37°C. Cells were washed three times with PBS and collected in 300 μl PBS using a rubber scratcher. After samples were sonicated and centrifuged at 10,000 × g for 30 min at 4°C, the supernants (200 μl) were collected for fluorescent density measurement at an excitation wavelength of 485 nm and an emission wavelength of 518 nm by Fluoroskan Ascent (Thermo Labsystems, Waltham, MA). The intracellular amount of FITC-Leu-Ile or FITC was calculated according to the standard curve.
Identification of binding protein for Leu-Ile in mouse brain.
Brains were removed from 7-week-old male Institute of Cancer Research mice (Nippon SLC, Shizuoka, Japan), and homogenized in radioimmunoprecipitation assay (RIPA) buffer (20 mm Tris-HCl, pH7.4, 0.25 m NaCl, 5 mm EDTA, 1% Triton X-100, 1 mm PMSF, and 1 μg/ml each of leupeptin, aprotinin, and pepstatin A). After centrifugation at 10,000 × g for 60 min at 4°C, the supernants were collected and reacted with FITC-Leu-Ile for 60 min at 37°C. Samples from the reaction complex were subjected to gel electrophoresis, and the gels were analyzed directly by FluorImager595 (Molecular Dynamics, Sunnyvale, CA).
Preparation of Sepharose Affigel-10 (Amersham Biosciences, Arlington Heights, IL) coupled with Leu-Ile was performed according to manufacturer’s instructions. The column with Affigel-10 coupled with Leu-Ile was loaded with brain homogenates and equilibrated with 10 mm Tris-HCl buffer, pH 7.4, at 4°C overnight. The column was washed extensively with 0.1 m PBS, pH 7.4, followed by elution with 0.17 m glycine-HCl buffer, pH 3.0. The eluants were separated by gel electrophoresis. To identify the binding protein by liquid chromatography-mass spectrometry (LC-MS), the specific protein band was excised from the gel after silver staining and digested in-gel with trypsin. The digested peptide fragments were directly sprayed into a Q-Tof hybrid mass spectrometer equipped with an electrospray source (Q-Tof 2; Micromass, Manchester, UK). The analysis was conducted using Mascot Search (Matrix Science, Boston, MA) with reference to the protein sequence database at the National Center for Biotechnology Information (NCBI) (http://www.ncbi.nlm.nih.gov/Entrez). To validate the binding protein, samples from the reaction complex of brain homogenates with Leu-Ile were subjected to gel electrophoresis and then immunoblotted with anti-Hsc70 antibody. In addition, recombinant Hsc70 protein was reacted with Leu-Ile at 37°C for 60 min, and the reaction complex was separated by gel electrophoresis followed by Coomassie brilliant blue (CBB) staining.
Leu-Ile- or FK506-conjugated Affigel-10 was incubated with brain homogenates at 4°C overnight. The columns were washed extensively followed by elution with 0.17 m glycine-HCl buffer, pH 3.0. The eluants were separated by SDS-PAGE and immunoblotted with anti-Hsc70 antibody.
Interaction between Leu-Ile and Hsc70.
The binding of Leu-Ile or FK506 for Hsc70 was examined by quartz crystal microbalance (QCM), which is useful for studying mass-measuring and molecular interaction in aqueous solutions (Motomiya et al., 2003). Briefly, 100 μl of each dipeptide (10 μg/ml in PBS) or FK506 [10 μg/ml in chloroform or chloroform/ethanol (1:1)] was immobilized into a QCM plate for 1 h at room temperature, and then removed. After washing three times with PBS, the plates were soaked in PBS at 25°C. Hsc70 or heat-denatured Hsc70 was applied to the equilibrated solution, and the change in resonance frequency was recorded using AFFINIX Q User Analysis software (AQUA; Initium, Tokyo, Japan). The binding affinity was indicated by frequency changes of QCM, and disassociation constant (Kd) was calculated with the AQUA software.
Modeling of functional domains of Hsc70 and the predicted binding site for Leu-Ile.
To further understand the interaction between Hsc70 and Leu-Ile, molecular models of the ATPase domain, substrate-binding domain, and C-terminal domain of Hsc70 were generated using the three-dimensional structural data from the Protein Data Bank (http://pdbbeta.rcsb.org/pdb/Welcome.do) in the Research Collaboratory for Structural Bioinformatics (Flaherty et al., 1990; Morshauser et al., 1999; Chou et al., 2003). Interaction of each domain with Leu-Ile was analyzed using Molecular Operating Environment (MOE) software (Chemical Computing Group, Montreal, Quebec, Canada). All calculations used an MMFF94x force field and a cutoff distance of 9.5 Å for nonbinding interactions. The Alpha Site Finder of the MOE program was used for docking stimulation.
Western blotting.
Cultured neurons were lysed in RIPA buffer (20 mm Tris-HCl, pH 7.6, 150 mm NaCl, 1 mm sodium orthovanadate, 2 mm EDTA, 50 mm NaF, 1% Nonidet P-40, 1 mm PMSF, and 2 μg/ml each of aprotinin, leupeptin, and pepstatin). Lysates were sonicated and centrifuged at 9000 × g for 15 min. Protein concentrations were determined by protein assay reagents (Bio-Rad, Hercules, CA). The samples were subjected to SDS-PAGE and then electrotransferred to polyvinylidene difluoride membranes. The membranes were immunoblotted and developed using chemiluminescence detection reagents. To calculate the amount of phosphorylated form versus total protein, the same membranes were stripped, incubated with the primary antibody for total protein, and examined as described above. The relative amount of immunoreactive protein in each band was assayed by scanning densitometric analysis using the Atto Densitograph 4.1 system (Atto, Tokyo, Japan).
Immunoprecipitation.
After centrifugation, the supernatants of cell lysates were normalized for protein concentration. A fraction (500 μg) of the total protein was incubated by gently rocking at 4°C overnight in the presence of anti-Hsp90 antibody. The immunocomplexes were captured by protein A Sepharose (Amersham Biosciences), washed out by lysis buffer, and then subjected to SDS-PAGE and immunoblotting.
Immunostaining.
Cultured neurons attached to glass coverslips were fixed with 4% paraformaldehyde in PBS for 20 min, and then blocked in 3% normal serum and 0.1% Triton X-100 for 1 h. The coverslips were incubated with the primary antibodies at 4°C overnight, washed with PBS, and then incubated with appropriate secondary antibodies (Invitrogen) for 2 h. After being washed and mounted, stained neurons were observed under a fluorescent microscope (Axioscop 2 plus; Zeiss, Thornwood, NY).
Real-time RT-PCR.
The level of GDNF mRNA was determined by real-time reverse transcription (RT)-PCR using an iCycler system (Bio-Rad). Briefly, isolation of total RNA was performed using RNeasy mini kit (Qiagen, Hilden, Germany). For reverse transcription, 1 μg RNA was converted into a cDNA by a standard 20 μl reverse transcriptase reaction using oligo-dT primers and Superscript II RT (Invitrogen). Total cDNA (1 μl) was amplified in a 25 μl reaction mixture using 0.1 μm each of forward and reverse primers and Platinum Quantitative PCR SuperMix-UDG (Invitrogen). Ribosomal mRNA was used and determined as the control for RNA integrity with TaqMan Ribosomal RNA control reagents (Applied Biosystems, Foster City, CA). The primer and dye probes were designed by Nippon Gene (Tokyo, Japan) using Primer Express software. The GDNF forward was 5′-AGCTGCCAGCCCAGAGAATT-3′ (base pair 288–307), with the reverse being 5′-GCACCCCCGATTTTTGC-3′ (base pair 354–370) and the dye probe being 5′-CAGAGGGAAAGGTCGCAGAGGCC-3′ (base pair 309–331).
Antisense oligonucleotide of CREB.
CREB expression was inhibited by an oligodeoxynucleotide (ODN) targeting the initiation codon of CREB mRNA as previously reported (Johnson et al., 2000; Saini et al., 2004). The phosphorothioate ODNs were synthesized by Nisshinbo Industries (Tokyo, Japan). The sequence of antisense ODN was 5′-GCTCCAGAGTCCATGGTCAT-3′, with a sense ODN with the sequence 5′-ATGACCATGGACTCTGGAGC-3′ as a control. Transfections were performed using Lipofectamine reagent (Invitrogen), and oligonucleotide was added to culture medium at a final concentration of 4 μm. The inhibition of CREB expression after transfection was assessed by Western blotting. To investigate the role of CREB in transcriptional regulation of GDNF expression, cultures were incubated with CREB ODN before Leu-Ile treatment.
pCREB-CRE binding activity.
Cultured neurons were collected and nuclear extracts were prepared by using BD TransFactor extraction kits (BD Biosciences, Franklin Lakes, NJ) according to the manufacturer’s protocol. The CRE-pCREB binding activity was determined using TransAM pCREB/CREB transcription factor assay kits (Active Motif Carlsbad, CA). Briefly, nuclear extract was applied to each well immobilized with oligonucleotide containing CRE 5′-TGACGTCA-3′, and incubated for 3 h. After washing, the wells were incubated with anti-pCREB antibody, followed by HRP-conjugated secondary antibody. After development with tetramethylbenzidine, the absorbance was measured with a microplate reader at 450 nm with reference at 655 nm. The specificity of the binding of pCREB to CRE was confirmed by conducting competitive experiments with 20 pmol of wild-type oligonucleotide probe or mutant probe containing the consensus CRE.
Statistical analysis.
All data were expressed as means ± SEM. Statistical significance was determined by a one-way ANOVA, followed by the Student–Newman–Keuls test for multigroup comparisons. Differences were considered significant when p < 0.05.
Results
Transmembrane transport
As shown in Fig. 1A, uptake of FITC-Leu-Ile by neurons was increased with the elevation of extracellular concentration. Time-course studies showed that uptake of FITC-Leu-Ile by neurons was a quick process and appeared to be saturated after incubation for 60 min (Fig. 1B). Although transmembrane transport of FITC was observed in dose- and time-course studies, its penetration amount was much lower than that of FITC-Leu-Ile (Fig. 1A,B). Because specific inhibitor for neuronal peptide transporters is not available, competitive transport was investigated using high concentrations of Leu-Ile. Simultaneous incubation with various concentrations of Leu-Ile for 30 min significantly inhibited FITC-Leu-Ile transport in a concentration-dependent manner (Fig. 1C), but failed to inhibit FITC transport (Fig. 1D), suggesting that FITC-Leu-Ile and Leu-Ile are transported by the same pathway. Together, these results strongly imply that FITC-Leu-Ile transport is mainly caused by the transmembrane activity of Leu-Ile rather than FITC and that the kinetics of FITC-Leu-Ile transport, at least in some degree, reflects that of Leu-Ile.
Figure 1.
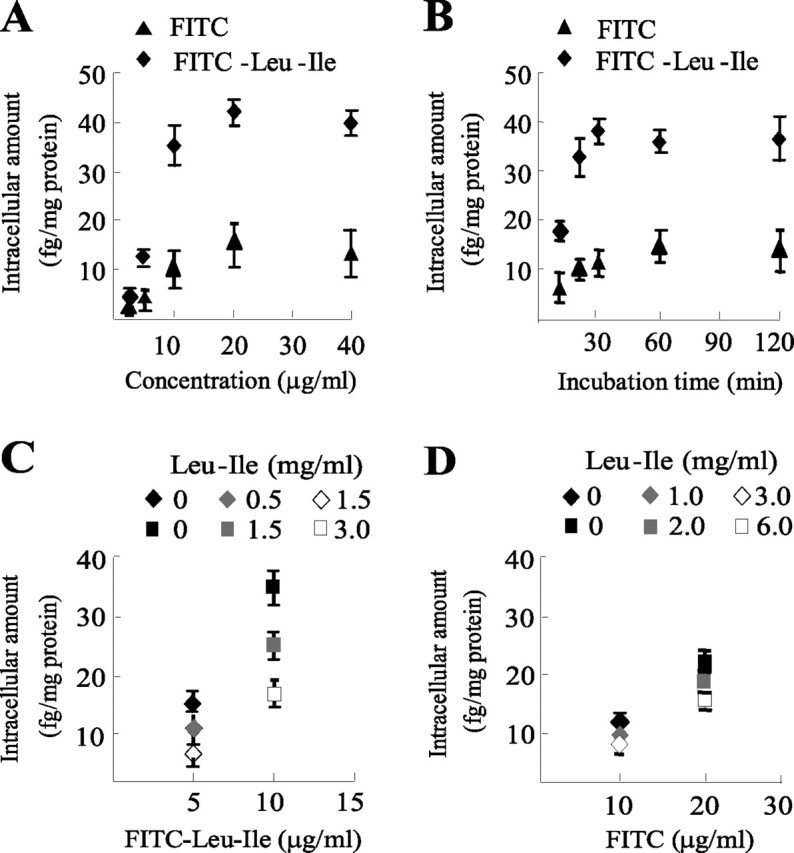
Transmembrane transport of Leu-Ile. A, Cultured neurons were exposed to FITC-Leu-Ile or FITC at various concentrations for 30 min, and uptake was analyzed according to intracellular fluorescent densities (n = 3). B, Neurons were incubated with 10 μg/ml FITC-Leu-Ile or FITC at 37°C for the indicated time periods. Time course uptake was analyzed (n = 3). C, Neurons were exposed to FITC-Leu-Ile for 30 min in the presence of various concentrations of Leu-Ile, which were indicated by different symbols. Penetration of FITC-Leu-Ile was significantly inhibited by competitive Leu-Ile. D, High concentrations of Leu-Ile could not inhibit FITC transport.
Identification of target protein for Leu-Ile in mouse brain
FITC-Leu-Ile was incubated with mouse brain homogenate, and the reaction complexes were subjected to electrophoresis. By fluorescent scanning, one specific fluorescent protein band with a molecular weight of ∼70 kDa was detected, suggesting that this protein has the specific affinity for Leu-Ile (Fig. 2A, arrow). To further identify the protein binding to Leu-Ile, brain homogenate was applied to Leu-Ile-conjugated Affigel-10, and the eluants were subjected to electrophoresis and detected by silver staining. We found a specific protein band of ∼70 kDa with stronger density in the gel (Fig. 2B, arrow), which was similar to that detected by fluorescent scanning. It has been known that the family of heat shock protein 70 represents an important cellular mechanism in neuroprotection (Rubio et al., 2002; Zhang et al., 2004); moreover, Leu-Ile derives from FK506, which exerts neuroprotective action through Hsp70 (Gold et al., 2004). Therefore, the band of ∼70 kDa was selected and processed to generate tryptic peptides, which was analyzed by direct nanoflow LC-MS. All of the digested peptide fragments could be assigned to Hsc70 with 100% homology by Mascot Search, and the matching score was 496 (Fig. 2C,D). To confirm the mass spectrometric-based identification of the Leu-Ile-binding protein, reaction complexes were subjected to SDS-PAGE and immunoblotted with Hsc70 antibody. We detected Hsc70 (open arrow) as well as an Hsc70-Leu-Ile complex (closed arrow), which showed a slight retardation of electrophoretic mobility because of the increased molecular weight compared with that of Hsc70 (Fig. 2E). Moreover, Hsc70-Leu-Ile (closed arrow) and Hsc70 (open arrow) were also identified by CBB staining in the reaction complex of Leu-Ile with recombinant Hsc70 (Fig. 2F). These results confirm that Hsc70 is a specific binding protein for Leu-Ile.
Figure 2.
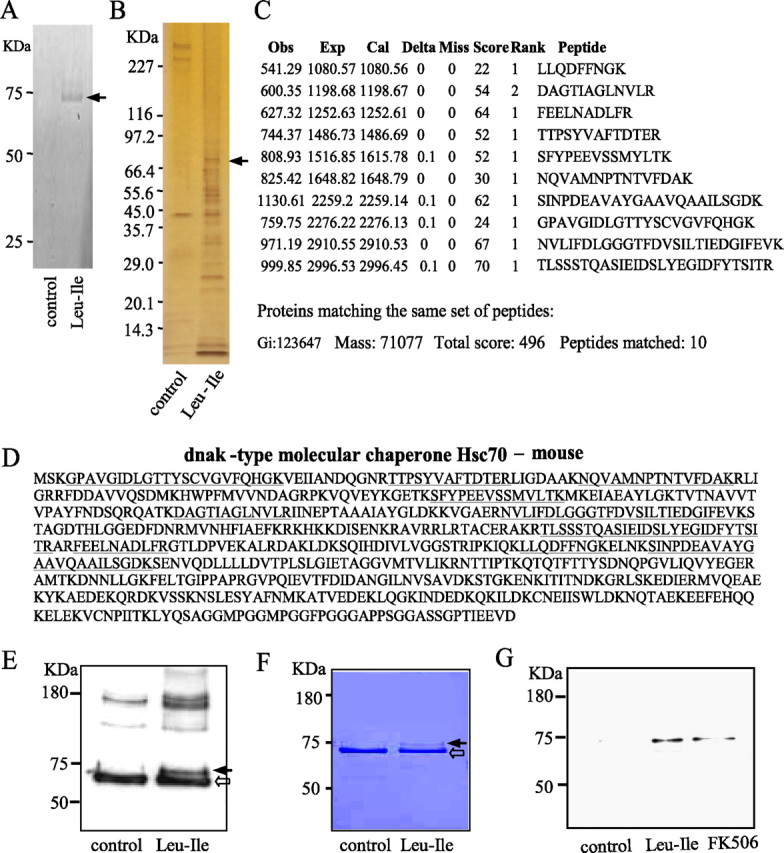
Identification of the specific protein binding to Leu-Ile. A, The reaction complexes of brain homogenate and FITC-Leu-Ile were subjected to gel electrophoresis, followed by fluorescent scanning. FITC-Leu-Ile alone was used as a control. The protein binding with FITC-Leu-Ile is marked by an arrow. B, Brain homogenate was incubated with Leu-Ile Affigel-10 followed by washing and elution. The eluates were separated by electrophoresis, followed by silver staining. The protein band (arrow) was analyzed by mass spectrometry. C, The figure incorporates the observed mass (Obs), expected nominal mass (Exp), and calculated mass (Cal), together with the Miss, Score, Rank from Mascot Search, and Peptide sequence. D, The picture shows the amino acid sequence assigned to each peptide (underlined) and their position in the Hsc70 sequence (NCBI, Gi:123647). E, Brain homogenate was or was not (control) reacted with Leu-Ile, and the reaction complex was subjected to SDS-PAGE followed by immunoblotting with anti-Hsc70 antibody. Leu-Ile-Hsc70 (closed arrow) and Hsc70 (open arrow) are shown. F, Recombinant Hsc70 was or was not (control) reacted with Leu-Ile, and the reaction complexes were subjected to SDS-PAGE followed by CBB staining. Leu-Ile-Hsc70 (closed arrow) and Hsc70 (open arrow) are shown. G, Brain homogenate was incubated with Leu-Ile- or FK506-Affigel, followed by washing and elution. The eluates were separated by electrophoresis and probed with anti-Hsc70 antibody.
To study whether FK506 binds to Hsc70, Leu-Ile or FK506 Affigel-10 were incubated with brain homogenate at 4°C overnight. The eluants were subjected to SDS-PAGE and probed with anti-Hsc70 antibody. Interestingly, Hsc70 was detected in the eluants from both Leu-Ile and FK506 Affigel-10, suggesting that Hsc70 may bind to FK506 directly or indirectly (Fig. 2G).
Interaction between Leu-Ile and Hsc70
QCM was applied to investigate more directly the interaction between Leu-Ile and Hsc70. The resonance frequency change (−δ F) of QCM responding to Leu-Ile decreased over time, indicating that Hsc70 had a significant affinity for Leu-Ile in a time-dependent manner (Fig. 3A). However, Pro-Leu and Ile-Pro had no effect on the resonance frequency change. The resonance frequency was decreased dose-dependently by Hsc70, showing the affinity of Hsc70 for Leu-Ile with Kd equal to 1.83 × 10−8 m (Fig. 3B). Leu-Ile had no influence on resonance frequency change when it was incubated with heat-denatured Hsc70 (data not shown). These results indicate the binding specificity of these two molecules and the requirement of the three-dimensional conformations of Hsc70 and Leu-Ile. However, resonance frequency change was not observed when Hsc70 was added to the QCM plate immobilized with FK506, suggesting that FK506 may not bind directly to Hsc70 (Fig. 3C).
Figure 3.

Affinity of Leu-Ile and Hsc70 was assayed by QCM. A, Time course of frequency change (−δ F) of dipeptide-immobilized QCM is shown, responding to the addition of Hsc70 to the aqueous solution. B, Binding behavior of Leu-Ile to Hsc70 is dependent on Leu-Ile concentration. C, Frequency change of FK506-immobilized QCM was not observed with Hsc70.
ATPase domain of Hsc70 is the predicted binding site of Leu-Ile
To get further insights for Leu-Ile-Hsc70 interaction, three-dimensional structural models of ATPase domain, substrate-binding domain, or C-terminal domain of Hsc70 were produced, and the potential interaction of each domain with Leu-Ile was analyzed by MOE software. The ATPase domain showed the strongest interaction potential with Leu-Ile among these three domains. The predicted binding site of Leu-Ile in this domain appears to be a pocket structure, which is near the ADP docking site (supplemental Fig. 1A, available at www.jneurosci.org as supplemental material). It has been demonstrated that substrates, binding at this domain, affect the ATP cycle and cause conformational regulation of Hsc70 and its cochaperones (Hernández et al., 2002). The substrate-binding domain showed no stable docking site for Leu-Ile (supplemental Fig. 1B, available at www.jneurosci.org as supplemental material).
Leu-Ile stimulates Akt phosphorylation
Because Hsc70/Hsp90 cochaperones modulate the activities of a restricted number of tyrosine and serine/threonine kinases (Nollen and Morimoto, 2002), we first investigated whether c-Src, P38MAPK, SAPK/JNK, and Akt were affected by Leu-Ile. The pAkt level was elevated by Leu-Ile (10 μg/ml) treatment for 20 or 30 min (Fig. 4A), whereas no changes in levels of other kinases were observed. In addition, expressions of Hsp90, Hsp70, and Hsc70 were not affected by Leu-Ile treatment for the indicated time points, showing that Leu-Ile is not a heat shock response inducer (Fig. 4B). Another two peptides, Pro-Leu and Ile-Pro, could not promote Akt phosphorylation (Fig. 4C).
Figure 4.
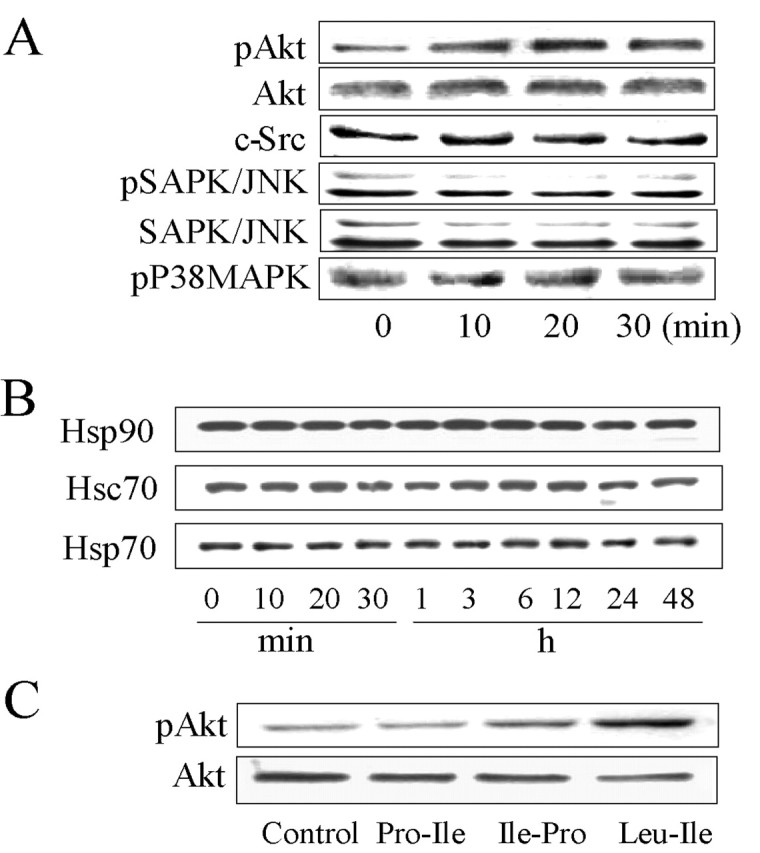
Leu-Ile stimulates Akt phosphorylation. A, Neurons were exposed to Leu-Ile (10 μg/ml) for the indicated times. Cell lysates were subjected to SDS-PAGE and probed with various antibodies. The representative immunoblots are shown. B, Neurons were exposed to Leu-Ile (10 μg/ml) for the indicated times. Immunoblots were probed with antibodies against Hsp90, Hsp70, and Hsc70. C, Neurons were stimulated with Leu-Ile, Pro-Leu, and Ile-Pro (10 μg/ml) for 30 min. Cell lysates were subjected to SDS-PAGE and probed with antibodies against pAkt and Akt.
Leu-Ile-induced Akt phosphorylation is mediated by Hsp90
As shown in Figure 5A, Akt phosphorylation was stimulated after Leu-Ile treatment for 20 or 30 min [F(5,18) = 11.30; p < 0.01 and p < 0.01 respectively, compared with the control (0 min)]. Because Hsp90 is a modulator for Akt, we thus investigated whether Akt activation by this dipeptide was mediated through Hsp90. The cultures were exposed to an Hsp90 inhibitor GA (10 μm) for 3 h, followed by Leu-Ile stimulation for 30 min. We found that the increase in pAkt level induced by Leu-Ile was obviously abolished by GA pretreatment (p < 0.01, compared with Leu-Ile treatment for 30 min). GA did not cause toxicity to neurons in our cultures (data not shown). To evaluate the involvement of PI3-k, an upstream activator for Akt, the cultures were stimulated by Leu-Ile after pretreatment with a PI3-k inhibitor LY294002 (15 μm) for 2 h. Although the pAkt level induced by Leu-Ile was inhibited by LY294002 to some degree, it was still much higher than that of LY294002 alone (F(5,18) = 11.05; p < 0.01) (Fig. 5B), suggesting that Leu-Ile-activated Akt may not be mediated by PI3-k. Furthermore, immunoblotting of immunoprecipitates with anti-Hsp90 antibody was performed using anti-Akt or anti-pAkt antibody. The level of immunoprecipitated pAkt was elevated significantly by Leu-Ile stimulation for 20 or 30 min [F(3,12) = 7.75; p < 0.01 and p < 0.01 respectively, versus control (0 min)] (Fig. 5C, top), indicating the enhanced interaction between Hsp90 and pAkt. Although immunoprecipitated Akt did not differ significantly, the increase tendency was obvious (Fig. 5C, middle). Direct interaction between Hsc70 and Akt was not observed in our immunoprecipitation assays (data not shown). Together, Leu-Ile is considered to activate Akt in an Hsp90-dependent manner.
Figure 5.
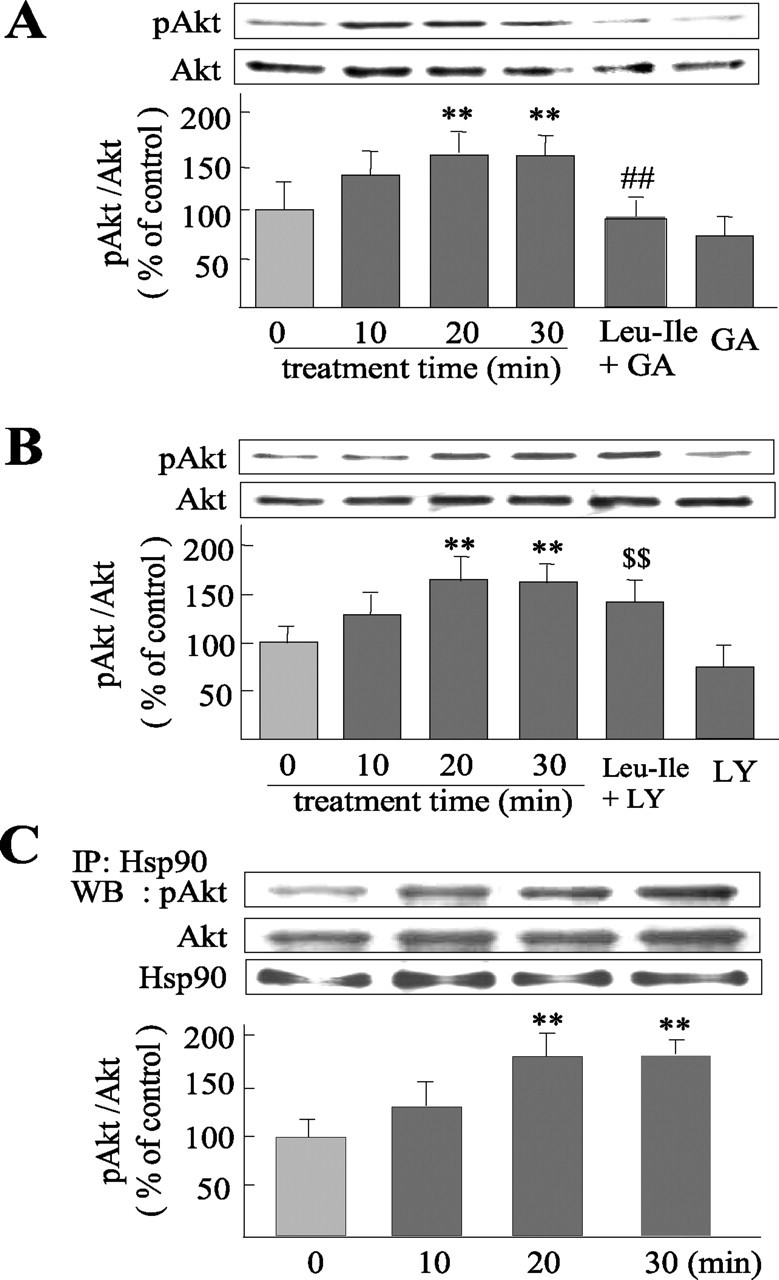
Akt activation induced by Leu-Ile is mediated by Hsp90. A, B, Neurons were stimulated with Leu-Ile (10 μg/ml) alone for 0, 10, 20, and 30 min, or pretreated with GA (10 μm) for 3 h (A) or LY294002 (15 μm) for 2 h (B), followed by Leu-Ile (10 μg/ml) treatment for 30 min. Cell lysates were immunoblotted with antibodies against pAkt and Akt. Each column represents the mean ± SEM (n = 4). Leu-Ile + GA neurons were pretreated with GA followed by Leu-Ile; GA neurons were pretreated with GA alone; Leu-Ile + LY neurons were pretreated with LY294002 followed by Leu-Ile treatment; LY neurons were pretreated with LY294002. **p < 0.01 versus control (0 min); ##p < 0.01 versus Leu-Ile (30 min); $$p < 0.01 versus LY294002. C, Cultures were exposed to 10 μg/ml of Leu-Ile for the periods indicated. Cell extracts were immunoprecipitated (IP) with anti-Hsp90 antibody or control rabbit IgG, followed by immunoblotting (WB). Densitometric data are presented as the mean ± SEM (n = 4). **p < 0.01 versus control (0 min).
CREB is a downstream target of Hsp90/Akt signaling activated by Leu-Ile
CREB was chosen to study intensively because it is a regulatory target of Akt and closely associated with GDNF expression. The amount of pCREB at Ser133 was increased after Leu-Ile stimulation for 20 or 30 min (Fig. 6A), whereas both Pro-Leu and Ile-Pro could not promote CREB phosphorylation (Fig. 6B). Moreover, increased pCREB immunoreactivity and nucleus translocation induced by Leu-Ile were observed in MAP2-positive cells (Fig. 6C, middle), which is thought to be an early step for gene transcriptional regulation of CREB. To study the possibility of Leu-Ile acting on glial cells, both GFAP and pCREB were stained, despite that the culture contained few glial cells. We found that enhanced pCREB immunoreactivity and nucleus translocation induced by Leu-Ile were not located in GFAP-positive cells, which showed lower pCREB immunoreactivity (Fig. 6D). To assess the involvement of Hsp90/Akt signaling in CREB phosphorylation induced by Leu-Ile, we investigated the change of pCREB after inhibition of this pathway. The cultures were stimulated with Leu-Ile alone for 10, 20, or 30 min, or pretreated with GA (10 μm) for 3 h, followed by Leu-Ile treatment for 30 min. We found that the pCREB level was elevated after Leu-Ile exposure for 20 or 30 min [F(5,18) = 11.32; p < 0.01 and p < 0.01 respectively, compared with control (0 min)], whereas the increase was inhibited by GA pretreatment (p < 0.01 versus Leu-Ile for 30 min) (Fig. 6E). Double-staining supported these results, showing the loss of pCREB immunoreactivity and nucleus translocation in GA-treated neurons (Fig. 6C, bottom). The increase in pCREB induced by Leu-Ile could not be inhibited by LY294002, and showed a higher level compared with LY294002 group (F(5,18) = 10.36; p < 0.01) (Fig. 6F). Because a wide range of neuromodulators can converge on CREB via various cascades in neurons, we examined pERK1/2, PKC, and pCaMKIIα/β. However, no changes of these kinases were observed responding to Leu-Ile (Fig. 6G). Collectively, these results show that CREB is a downstream target of Hsp90/Akt signaling activated by Leu-Ile.
Figure 6.
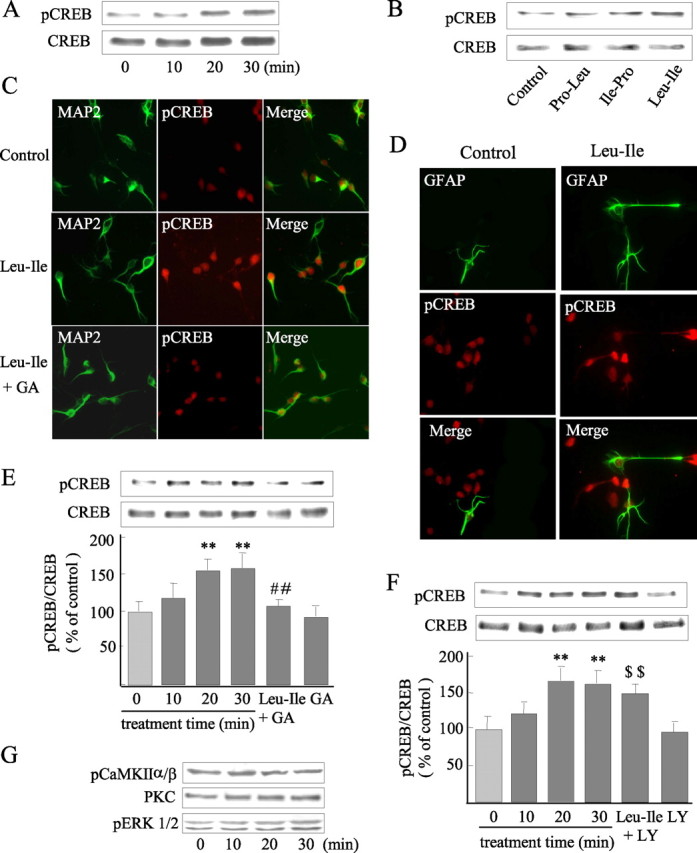
CREB is a downstream target of Hsp90/Akt signaling activated by Leu-Ile. A, Cultured neurons were exposed to 10 μg/ml of Leu-Ile for 0, 10, 20, and 30 min, and pCREB was measured by immunoblotting. B, Western blotting with anti-pCREB antibody reveals CREB activation induced by Leu-Ile but not Pro-Leu and Ile-Pro. C, Visualization of CREB phosphorylation (red) in MAP2-positive neurons (green) induced by Leu-Ile. D, Phosphorylated CREB (red) induced by Leu-Ile is not located in GFAP-positive cells (green). E, F, Neurons were treated with Leu-Ile (10 μg/ml) for 0 (control), 10, 20, and 30 min respectively, or pretreated with GA (10 μm) for 3 h (E) or LY294002 (15 μm) for 2 h (F), followed by Leu-Ile (10 μg/ml) treatment for 30 min. Each column represents the mean ± SEM (n = 4). Leu-Ile + GA neurons were pretreated with GA followed by Leu-Ile; GA neurons were pretreated with GA; Leu-Ile + LY neurons were pretreated with LY294002 followed by Leu-Ile; LY neurons were pretreated with LY294002. **p < 0.01 versus control (0 min); ##p < 0.01 versus Leu-Ile (30 min); $$p < 0.01 versus LY294002. G, PKC, pERK1/2, and pCaMKIIα/β were measured after Leu-Ile (10 μg/ml) treatment by immunoblotting.
Leu-Ile increases GDNF expression in a CREB-dependent manner
After the cultures were exposed to 10 μg/ml Leu-Ile, Pro-Leu, or Ile-Pro for 24 h respectively, the levels of GDNF expression were measured. We found that Leu-Ile, but not Pro-Leu and Ile-Pro, significantly promoted GDNF production (Fig. 7A). Moreover, the levels of GDNF mRNA were obviously elevated when neurons were incubated with Leu-Ile (10 μg/ml) for 12 or 18 h, as evidenced by real-time RT-PCR measurement (Fig. 7B). These results were well consistent with our previous report (Nitta et al., 2004). To investigate the role of CREB in transcriptional regulation of the GDNF gene induced by Leu-Ile, CREB antisense ODN was used to downregulate CREB expression. CREB expression was inhibited when neurons were transfected with CREB antisense ODN for 24 h, whereas sense ODN showed no effect (Fig. 7C). GDNF mRNA levels were measured after Leu-Ile treatment for 18 h in the presence of CREB antisense ODN or sense ODN, and we found that GDNF mRNA induced by Leu-Ile was inhibited significantly by CREB antisense ODN (Fig. 7D). Furthermore, cellular GDNF expression was analyzed after Leu-Ile treatment in the presence of CREB antisense ODN. The GDNF level was dramatically elevated to ∼188% after neurons were incubated with Leu-Ile for 24 h (F(5,18) = 25.74; p < 0.001 versus control). However, the induction of GDNF expression by this dipeptide was significantly attenuated by CREB antisense ODN (p < 0.01 versus Leu-Ile or Leu-Ile plus CREB sense ODN). CREB antisense ODN did not significantly influence the basal expression of GDNF (Fig. 7E). Similarly, immunostaining revealed both stronger GDNF and nuclear pCREB immunoreactivities in Leu-Ile-treated neurons (Fig. 7F, middle), whereas such actions were blocked by CREB inhibition resulted from antisense ODN (Fig. 7F, right). These observations indicated that GDNF expression was in parallel with CREB phosphorylation. Additionally, pCREB-CRE binding activity was obviously promoted after Leu-Ile treatment for 30 min (F(3,12) = 51.28; p < 0.01 compared with control) (Fig. 7G, left two columns). Competitive experiments showed the specificity of pCREB-CRE binding, because the increased pCREB-CRE binding activity was almost totally blocked when competitive wild-type ODN probe was added (p < 0.001 versus mutant ODN treatment), but not the mutant one (Fig. 7G, right two columns). Collectively, these results showed that CREB plays a key role in transcriptional regulation of the GDNF gene induced by Leu-Ile.
Figure 7.
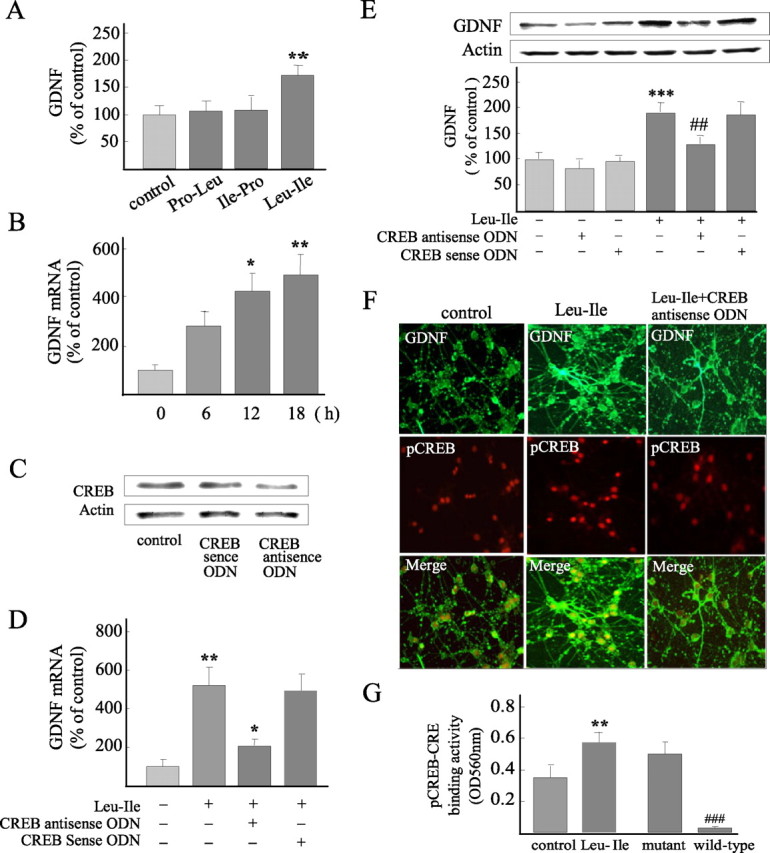
Leu-Ile increases GDNF expression in a CREB-dependent manner. A, Leu-Ile significantly increased GDNF expression, whereas Pro-Leu and Ile-Pro showed no GDNF-inducing activities. **p < 0.01 versus control (n = 4). B, GDNF mRNA levels induced by Leu-Ile for various periods were studied by real-time RT-PCR. *p < 0.05 and ** p < 0.01 versus control (0 h). C, CREB expression was blocked by CREB antisense ODN, as revealed by Western blotting. D, Neurons were incubated with Leu-Ile (10 μg/ml) for 24 h in the presence of CREB antisense ODN or sense ODN. Data are expressed as a percentage of the control (mean ± SEM; n = 4). **p < 0.01 versus control; *p < 0.05 versus Leu-Ile or Leu-Ile plus CREB sense ODN. E, Neurons were incubated with Leu-Ile (10 μg/ml) for 24 h in the presence of CREB antisense ODN or sense ODN. Data are expressed as a percentage of control (mean ± SEM; n = 4). ***p < 0.001 versus control; ##p < 0.01 versus Leu-Ile or Leu-Ile plus CREB sense ODN. F, Neurons were labeled with anti-GDNF (green) and anti-pCREB antibodies (red). G, CRE-pCREB binding activities were quantified after Leu-Ile treatment for 30 min. **p < 0.01 versus control; ###p < 0.001 verse mutant ODN (n = 4).
Discussion
Using the principles of structure-based drug design, we synthesized three dipeptide analogs which resemble the dipeptide-like binding site of FK506 for immunophilin. Among these dipeptides, hydrophobic Leu-Ile was demonstrated to promote GDNF expression. Transport studies revealed the transmembrane mobility of Leu-Ile, although it is not clear which pathway is responsible for this process. Peptide transporter PTH1 is considered to transport oligopeptides, especially dipeptides, into neurons (Yamashita et al.,1997), and Leu-Ile is possibly transported by this transporter.
A series of experiments indicated that Leu-Ile binds specially to Hsc70, a member of the heat shock protein 70 family, which represents an important cellular mechanism in chaperone-mediated neuroprotection (Muchowski, 2002). The binding of Hsc70 to Leu-Ile was time and dose dependent, as suggested by QCM measurement. Moreover, such binding depended on the dimensional structure of both Hsc70 and Leu-Ile, because heat-denatured Hsc70 failed to bind this dipeptide, and another two similar dipeptides, Pro-Leu and Ile-Pro, showed no affinity for Hsc70. These findings indicate that Leu-Ile-Hsc70 interaction may be not a transient association but a specific binding dependent on their dimensional structure. By molecule modeling and docking stimulation, the ATPase domain of Hsc70 rather than the substrate-binding domain is shown to be the predicted binding site for Leu-Ile. It is known that Hsc70 interacts with cochaperones through the ATPase domain and that binding of exposed stretches of hydrophobic residues in proteins or peptides is regulated by ATP-hydrolysis-induced conformational changes in the ATPase domain (Nollen et al., 2001). Therefore, the interaction between Leu-Ile and Hsc70 may result in conformational and functional regulation of Hsc70.
Hsc70/Hsp90 chaperones are specially considered to be an integrated cochaperone machinery. They often work together as essential components of a process that alters the conformations of a certain number of signaling transducers to states that respond in signal transduction, such as glucocorticoid receptors, Akt, and Src kinases (Rajapandi et al., 2000; Pearl and Prodromou, 2001). Moreover, activities of Hsc70/Hsp90 machinery are affected by a wide range of cofactor proteins that interact directly and specifically with either Hsc70 or Hsp90, and modulation of Hsc70 ATPase may affect the functions of Hsp90 toward its client proteins. We thus proposed that Leu-Ile, after binding to Hsc70, influenced Hsc70/Hsp90 chaperoning function toward client signaling proteins, resulting in mobilization of downstream signaling. To explore this hypothesis, we first studied some tyrosine and serine/threonine kinases, including mitogen-activated protein kinases, Akt, and Src, which are closely associated with Hsp90 and neuron survival (Richter and Buchner, 2001). Akt phosphorylation was elevated apparently by Leu-Ile, whereas other kinases showed no change, implying the functional modulation of Hsc70 by Leu-Ile and involvement of Hsc70/Hsp90 cochaperone in the regulation of Akt phosphorylation. Thulasiraman et al. (2002) reported a similar finding that a small hydrophobic peptide, binding to the ATPase domain of Hsc70, affects ATPase activity and Hsp90/Hsc70-dependent transformation of eukaryotic initiation factor 2α kinase into an active form.
Heat shock response has been implicated in mediating the neuroprotective effect of FK506 (Klettner and Herdegen, 2003; Gold et al., 2004) and in activating Akt by conformational regulation of this molecule (Konishi et al., 1999; Matsuzaki et al., 2004). However, Leu-Ile did not affect the expression of Hsc70, Hsp70, or Hsp90, indicating that it unlikely exerts neuroprotective action by a mechanism of heat shock response. Given the possibility that the binding of FK506 to Hsp90/steroid receptor complexes might dissociate Hsp90 from heat shock factor, thus inducing heat shock response (Gold et al., 1999, 2004; Klettner and Herdegen, 2003), Leu-Ile unlikely affects the association between Hsp90 and steroid receptor, which may underlie its incapability in inducing heat shock response.
GA is known to bind the ATP-binding pocket of Hsp90 and to inhibit ATP binding and hydrolysis, thereby disrupting its function (Basso et al., 2002). GA significantly blocked the increased pAkt levels induced by Leu-Ile, whereas PI3-k inhibitor LY294002 failed. On the basis of these findings, Leu-Ile is considered to activate Akt through Hsp90. It is clear that Akt interacts with Hsp90 via its catalytic domain and that Hsp90 promotes Akt activity by reducing PP2A-mediated pAkt dephosphorylation at the threonine 308 residue (Sato et al., 2000). Therefore, Leu-Ile, after binding to Hsc70, may facilitate Hsp90-Akt interaction through conformational regulation, resulting in an increase in pAkt through protecting it from dephosphorylation. Immunoprecipitation assays demonstrated such hypothesis because it revealed a significant increase in pAkt-Hsp90 interaction. We also observed an elevation in total Akt immunoprecipitated by Hsp90 despite that the difference was not significant. Considering that GA causes the ubiquitin-mediated degradation of client Akt (Prodromou et al., 1997), Leu-Ile may inhibit proteasomal degradation of Akt mediated by Hsc70/Hsp90, resulting, accordingly, in pAkt elevation. This notion is supported by previous studies, which show that CAIR-1, after binding to the Hsc70 ATPase domain, increases Akt phosphorylation by inhibiting its shift from Hsp90 to Hsc70, where Akt is ubiquitinated and degraded (Doong et al., 2003). Our data cannot distinguish between these two mechanisms. Anyway, Hsc70, after binding to Leu-Ile, appears to be crucial for the transmitting of neurotrophic signals of this dipeptide, which brings about Hsp90/Akt signaling. Nakagomi et al. (2003) reported that Hsp27 promotes survival in PC12 cells and ganglion neurons by promoting Akt activity, which is independent of upstream activators. A chaperone-like protein, β-synuclein, exerts a neuroprotective effect by directly stabilizing Akt activity rather than by acting on PI3-k (Hashimoto et al., 2004). Therefore, these findings, together with ours, further support a notion that chaperones like Hsp90 may participate in neuroprotection by conformational or chaperoning modulation of Akt rather than by acting on upstream effectors of the pathways. Additionally, Akt activation is required for increased expression of astroglial GDNF induced by melatonin (Lee et al., 2006).
GA blocked CREB activation induced by Leu-Ile, indicating that CREB activation proceeds via Hsp90/Akt signaling. These findings are supported by previous studies that show that modulation of Akt based on Hsc70/Hsp90 cochaperones results in the maintenance of downstream CREB activation (Doong et al., 2003). Although CRE exists in the promoter sequence of the GDNF gene, there is no direct evidence showing the role of CREB in GDNF transcriptional regulation. Moreover, Akt promotes phosphorylation of CREB, stimulates recruitment of CREB to promoters, and activates gene expression (Pugazhenthi et al., 2000; Leinninger et al., 2004). We thus intensively investigated the role of Leu-Ile-activated pCREB in GDNF expression. We found that Leu-Ile-induced GDNF mRNA production and protein expression were attenuated when CREB was inhibited. Furthermore, CREB activation was accompanied by an increased capacity to activate transcription of target genes, because pCREB-CRE-binding activity was promoted. These results demonstrated that CREB-dependent transcriptional regulation is responsible for the GDNF-inducing properties of Leu-Ile. Although GDNF expression likely involves combinatorial interactions with multiple transcription factors including CREB, nuclear factor κB (NF-κB), and AP-2 (Woodbury et al., 1998), Leu-Ile-induced CREB activation is sufficient for inducing GDNF expression, indicating that CREB functions as an important transcriptional factor for the GDNF gene. Similarly, FK960 induces GDNF expression in CREB-dependent mechanisms (Koyama et al., 2004). The extent to which a transcription factor is required for GDNF transcriptional regulation is likely to depend on the character, strength, or duration of extracellular stimuli. For example, NF-κB seems to play a role in GNDF induction in response to cytokines (Tanaka et al., 2000), whereas CREB likely participates in GNDF induction by growth factors like basic fibroblast growth factor (Lenhard et al., 2002). The defined Hsp90/Akt/CREB pathway may provide a novel significant signaling that regulates GDNF expression.
Several cascades have been implicated in underlying neurotrophic activity of FK506. For example, FK506 potentiates NGF-induced neurite outgrowth via the Ras/Raf/MAPK pathway and involves PI3-k signaling (Price et al., 2003, 2005). Gold et al. (1999) reported that GA blocked neurotrophic action of FK506, suggesting FK506 interaction with Hsp90 via binding to FKBP52 is important for its neuroregenerative properties. However, FKBP-12 is not necessary for its neurotrophic effects (Gold et al., 1999, 2005). As suggested from QCM findings, FK506 may not interact with Hsc70 directly. Although the role of Hsc70 in mediating neuroprotective action of FK506 is unclear at present, it is tempting to speculate that FK506 might regulate the chaperoning function of Hsp90/Hsc70 through FKBP and, thus, modulate certain signaling kinases. It remains to be investigated intensively.
GDNF is a promising therapeutic agent for the treatment of neurodegenerative diseases. However, the delivery of GDNF to the CNS provides an interesting challenge, because GDNF is unable to cross the blood–brain barrier (Kirik et al., 2004), and use of low-molecular-weight drugs is an interesting alternative. FK506 exerts neuroprotective action, which is thought to depend on its GDNF-promoting effect (Tanaka et al., 2003). However, it cannot be used in therapy for neurological disorders because of its immunosuppressive effects. Leu-Ile, a small hydrophobic molecule, can penetrate neurons and promote GDNF expression, although it shows no immunosuppressive activity. Thus, it may represent a novel lead compound for treatment of dopaminergic neuron or motoneuron diseases such as Parkinson disease.
In conclusion, Leu-Ile targets the Hsc70/Hsp90 cochaperone and, thus, triggers Akt/CREB signaling, resulting in upregulation of GDNF expression. This defined cascade may provide a deep insight into the cellular mechanism of GDNF expression regulation.
Footnotes
*A.N. and X.C. contributed equally to this work.
This work was supported in part by a grant-in-aid for Science Research and Special Coordination Funds for Promoting Science and Technology, Target-Oriented Brain Science Research Program KAKENHI, and the 21st Century Center of Excellence Program “Integrated Molecular Medicine for Neuronal and Neoplastic Disorders” from the Ministry of Education, Culture, Sports, Science and Technology of Japan; by a grant-in-aid for Health Science Research on Regulatory Science of Pharmaceuticals and Medical Devices, and Dementia and Fracture from the Ministry of Health, Labor and Welfare of Japan; and by a Smoking Research Foundation grant for Biomedical Research.
References
- Airaksinen MS, Saarma M (2002). The GDNF family: signalling, biological functions and therapeutic value. Nat Rev Neurosci 3:383–394. [DOI] [PubMed] [Google Scholar]
- Baecker PA, Lee WH, Verity AN, Eglen RM, Johnson RM (1999). Characterization of a promoter for the human glial cell line-derived neurotrophic factor gene. Mol Brain Res 69:209–222. [DOI] [PubMed] [Google Scholar]
- Basso AD, Solit DB, Chiosis G, Giri B, Tsichlis P, Rosen N (2002). Akt forms an intracellular complex with heat shock protein 90 (Hsp90) and Cdc37 and is destabilized by inhibitors of Hsp90 function. J Biol Chem 277:39858–39866. [DOI] [PubMed] [Google Scholar]
- Brami-Cherrier K, Valjent E, Garcia M, Pagès C, Hipskind RA, Caboche J (2002). Dopamine induces a PI3-kinase-independent activation of Akt in striatal neurons: a new route to cAMP response element-binding protein phosphorylation. J Neurosci 22:8911–8921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunet A, Datta SR, Greenberg ME (2001). Transcription-dependent and -independent control of neuronal survival by the PI3K-Akt signaling pathway. Curr Opin Neurobiol 11:297–305. [DOI] [PubMed] [Google Scholar]
- Castro LM, Gallant M, Niles LP (2005). Novel targets for valproic acid: up-regulation of melatonin receptors and neurotrophic factors in C6 glioma cells. J Neurochem 95:1227–1236. [DOI] [PubMed] [Google Scholar]
- Chou CC, Forouhar F, Yeh YH, Shr HL, Wang C, Hsiao CD (2003). Crystal structure of the C-terminal 10-kDa subdomain of Hsc70. J Biol Chem 278:30311–30316. [DOI] [PubMed] [Google Scholar]
- Doong H, Rizzo K, Fang S, Kulpa V, Weissman AM, Kohn EC (2003). CAIR-1/BAG-3 abrogates heat shock protein-70 chaperone complex-mediated protein degradation. J Biol Chem 278:28490–28500. [DOI] [PubMed] [Google Scholar]
- Du K, Montminy M (1998). CREB is a regulatory target for the protein kinase Akt/PKB. J Biol Chem 273:2377–32379. [DOI] [PubMed] [Google Scholar]
- Flaherty KM, DeLuca-Flaherty C, McKay DB (1990). Three-dimensional structure of the ATPase fragment of a 70K heat-shock cognate protein. Nature 346:623–628. [DOI] [PubMed] [Google Scholar]
- Gold BG, Densmore V, Shou W, Matzuk MM, Gordon HS (1999). Immunophilin FK506-binding protein 52 (not FK506-binding protein 12) mediates the neurotrophic action of FK506. J Pharmacol Exp Ther 289:1202–1210. [PubMed] [Google Scholar]
- Gold BG, Voda J, Yu X, Gordon H (2004). The immunosuppressant FK506 elicits a neuronal heat shock response and protects against acrylamide neuropathy. Exp Neurol 187:160–170. [DOI] [PubMed] [Google Scholar]
- Gold BG, Armistead DM, Wang MS (2005). Non-FK506-binding protein-12 neuroimmunophilin ligands increase neurite elongation and accelerate nerve regeneration. J Neurosci Res 80:56–65. [DOI] [PubMed] [Google Scholar]
- Hashimoto M, Bar-On P, Ho G, Takenouchi T, Rockenstein E, Crews L, Masliah E (2004). β-Synuclein regulates Akt activity in neuronal cells. A possible mechanism for neuroprotection in Parkinson’s disease. J Biol Chem 279:23622–23629. [DOI] [PubMed] [Google Scholar]
- Hernández MP, Sullivan WP, Toft DO (2002). The assembly and intermolecular properties of the hsp70-Hop-hsp90 molecular chaperone complex. J Biol Chem 277:38294–38304. [DOI] [PubMed] [Google Scholar]
- Johnson JR, Chu AK, Sato-Bigbee C (2000). Possible role of CREB in the stimulation of oligodendrocyte precursor cell proliferation by neurotrophin-3. J Neurochem 74:1409–1417. [DOI] [PubMed] [Google Scholar]
- Kirik D, Georgievska B, Bjorklund A (2004). Localized striatal delivery of GDNF as a treatment for Parkinson disease. Nat Neurosci 7:105–110. [DOI] [PubMed] [Google Scholar]
- Klettner A, Herdegen T (2003). The immunophilin-ligands FK506 and V-10,367 mediate neuroprotection by the heat shock response. Br J Pharmacol 138:1004–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konishi H, Fujiyoshi T, Fukui Y, Matsuzaki H, Yamamoto T, Ono Y, Andjelkovic M, Hemmings BA, Kikkawa U (1999). Activation of protein kinase B induced by H2O2 and heat shock through distinct mechanisms dependent and independent of phosphatidylinositol 3-kinase. J Biochem 126:1136–1143. [DOI] [PubMed] [Google Scholar]
- Koyama Y, Egawa H, Osakada M, Baba A, Matsuda T (2004). Increase by FK960, a novel cognitive enhancer, in glial cell line-derived neurotrophic factor production in cultured rat astrocytes. Biochem Pharmacol 68:275–282. [DOI] [PubMed] [Google Scholar]
- Lee SH, Chun W, Kong PJ, Han JA, Cho BP, Kwon OY, Lee HJ, Kim SS (2006). Sustained activation of Akt by melatonin contributes to the protection against kainic acid-induced neuronal death in hippocampus. J Pineal Res 40:79–85. [DOI] [PubMed] [Google Scholar]
- Lee SJ, Campomanes CR, Sikat PT, Greenfield AT, Allen PB, McEwen BS (2004). Estrogen induces phosphorylation of cyclic AMP response element binding (pCREB) in primary hippocampal cells in a time-dependent manner. Neuroscience 124:549–560. [DOI] [PubMed] [Google Scholar]
- Leinninger GM, Backus C, Uhler MD, Lentz SI, Feldman EL (2004). Phosphatidylinositol 3-kinase and Akt effectors mediate insulin-like growth factor-I neuroprotection in dorsal root ganglia neurons. FASEB J 18:1544–1556. [DOI] [PubMed] [Google Scholar]
- Lenhard T, Schober A, Suter-Crazzolara C, Unsicker K (2002). Fibroblast growth factor-2 requires glial-cell-line-derived neurotrophic factor for exerting its neuroprotective actions on glutamate-lesioned hippocampal neurons. Mol Cell Neurosci 20:181–197. [DOI] [PubMed] [Google Scholar]
- Matsuzaki H, Yamamoto T, Kikkawa U (2004). Distinct activation mechanisms of protein kinase B by growth-factor stimulation and heat-shock treatment. Biochemistry 43:4284–4293. [DOI] [PubMed] [Google Scholar]
- Matsushita N, Fujita Y, Tanaka M, Nagatsu T, Kiuch T (1997). Cloning and structural organization of the gene encoding the mouse glial cell line-derived neurotrophic factor, GDNF. Gene 203:149–157. [DOI] [PubMed] [Google Scholar]
- McLaughlin SH, Smith HW, Jackson SE (2002). Stimulation of the weak ATPase activity of human Hsp90 by a client protein. J Mol Biol 315:787–798. [DOI] [PubMed] [Google Scholar]
- Morshauser RC, Hu W, Wang H, Pang Y, Flynn GC, Zuiderweg ER (1999). High-resolution solution structure of the 18 kDa substrate-binding domain of the mammalian chaperone protein Hsc70. J Mol Biol 289:1387–1403. [DOI] [PubMed] [Google Scholar]
- Motomiya Y, Ando Y, Haraoka K, Sun X, Iwamoto H, Uchimura T, Maruyama I (2003). Circulating level of α2-macroglobulin-β2-microglobulin complex in hemodialysis patients. Kidney Int 64:2244–2252. [DOI] [PubMed] [Google Scholar]
- Muchowski PJ (2002). Protein misfolding, amyloid formation, and neurodegeneration: a critical role for molecular chaperones? Neuron 35:9–12. [DOI] [PubMed] [Google Scholar]
- Nakagomi S, Suzuki Y, Namikawa K, Kiryu-Seo S, Kiyama H (2003). Expression of the activating transcription factor 3 prevents c-Jun N-terminal kinase-induced neuronal death by promoting heat shock protein 27 expression and Akt activation. J Neurosci 23:5187–5196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nitta A, Nishioka H, Fukumitsu H, Furukawa Y, Sugiura H, Shen L, Furukawa S (2004). Hydrophobic dipeptide Leu-Ile protects against neuronal death by inducing brain-derived neurotrophic factor and glial cell line-derived neurotrophic factor synthesis. J Neurosci Res 78:250–258. [DOI] [PubMed] [Google Scholar]
- Nollen EA, Morimoto RI (2002). Chaperoning signaling pathways: molecular chaperones as stress-sensing “heat shock” proteins. J Cell Sci 115:2809–2816. [DOI] [PubMed] [Google Scholar]
- Nollen EA, Kabakov AE, Brunsting JF, Kanon B, Hohfeld J, Kampinga HH (2001). Modulation of in vivo HSP70 chaperone activity by Hip and Bag-1. J Biol Chem 276:4677–4682. [DOI] [PubMed] [Google Scholar]
- Pearl LH, Prodromou C (2001). Structure, function and mechanism of the Hsp90 molecular chaperone. Adv Protein Chem 59:157–186. [DOI] [PubMed] [Google Scholar]
- Pratt WB, Toft DO (2003). Regulation of signaling protein function and trafficking by the hsp90/hsp70-based chaperone machinery. Exp Biol Med 228:111–133. [DOI] [PubMed] [Google Scholar]
- Price RD, Yamaji T, Matsuoka N (2003). FK506 potentiates NGF-induced neurite outgrowth via the Ras/Raf/MAP kinase pathway. Br J Pharmacol 140:825–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price RD, Yamaji T, Yamamoto H, Higashi Y, Hanaoka K, Yamazaki S, Ishiye M, Aramori I, Matsuoka N, Mutoh S, Yanagihara T, Gold BG (2005). FK1706, a novel non-immunosuppressive immunophilin: neurotrophic activity and mechanism of action. Eur J Pharmacol 509:11–19. [DOI] [PubMed] [Google Scholar]
- Prodromou C, Roe SM, O’Brien R, Ladbury JE, Piper PW, Pearl LH (1997). Identification and structural characterization of the ATP/ADP-binding site in the Hsp90 molecular chaperone. Cell 90:65–75. [DOI] [PubMed] [Google Scholar]
- Pugazhenthi S, Nesterova A, Sable C, Heidenreich KA, Boxer LM, Heasley LE, Reusch JE (2000). Akt/protein kinase B up-regulates Bcl-2 expression through cAMP-response element-binding protein. J Biol Chem 275:10761–10766. [DOI] [PubMed] [Google Scholar]
- Rajapandi T, Greene LE, Eisenberg E (2000). The molecular chaperones Hsp90 and Hsc70 are both necessary and sufficient to activate hormone binding by glucocorticoid receptor. J Biol Chem 275:22597–22604. [DOI] [PubMed] [Google Scholar]
- Richter K, Buchner J (2001). Hsp90: chaperoning signal transduction. J Cell Physiol 188:281–290. [DOI] [PubMed] [Google Scholar]
- Roberson ED, English JD, Adams JP, Selcher JC, Kondratick C, Sweatt JD (1999). The mitogen-activated protein kinase cascade couples PKA and PKC to cAMP response element binding protein phosphorylation in area CA1 of hippocampus. J Neurosci 19:4337–4348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubio E, Valenciano AI, Segundo C, Sanchez N, de Pablo F, de la Rosa EJ (2002). Programmed cell death in the neurulating embryo is prevented by the chaperone heat shock cognate 70. Eur J Neurosci 15:1646–1654. [DOI] [PubMed] [Google Scholar]
- Saini HS, Gorse KM, Boxer LM, Sato-Bigbee C (2004). Neurotrophin-3 and a CREB-mediated signaling pathway regulate Bcl-2 expression in oligodendrocyte progenitor cells. J Neurochem 89:951–961. [DOI] [PubMed] [Google Scholar]
- Sato S, Fujita N, Tsuruo T (2000). Modulation of Akt kinase activity by binding to Hsp90. Proc Natl Acad Sci USA 97:10832–10837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schinelli S, Zanassi P, Paolillo M, Wang H, Feliciello A, Gallo V (2001). Stimulation of endothelin B receptors in astrocytes induces cAMP response element-binding protein phosphorylation and c-fos expression via multiple mitogen-activated protein kinase signaling pathways. J Neurosci 21:8842–8853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka K, Fujita N, Ogawa N (2003). Immunosuppressive (FK506) and non-immunosuppressive (GPI1046) immunophilin ligands activate neurotrophic factors in the mouse brain. Brain Res 970:250–253. [DOI] [PubMed] [Google Scholar]
- Tanaka M, Ito S, Kiuchi K (2000). Novel alternative promoters of mouse glial cell line-derived neurotrophic factor gene. Biochim Biophys Acta 1494:63–74. [DOI] [PubMed] [Google Scholar]
- Thulasiraman V, Yun BG, Uma S, Gu Y, Scroggins BT, Matts RL (2002). Differential inhibition of Hsc70 activities by two Hsc70-binding peptides. Biochemistry 41:3742–3753. [DOI] [PubMed] [Google Scholar]
- Verity AN, Wyatt TL, Lee W, Hajos B, Baecker PA, Eglen RM, Johnson RM (1999). Differential regulation of glial cell line-derived neurotrophic factor (GDNF) expression in human neuroblastoma and glioblastoma cell lines. J Neurosci Res 55:187–197. [DOI] [PubMed] [Google Scholar]
- Woodbury D, Schaar DG, Ramakrishnan L, Black IB (1998). Novel structure of the human GDNF gene. Brain Res 803:95–104. [DOI] [PubMed] [Google Scholar]
- Xu W, Yuan X, Jung YJ, Yang Y, Basso A, Rosen N, Chung EJ, Trepel J, Neckers L (2003). The heat shock protein 90 inhibitor geldanamycin and the ErbB inhibitor ZD1839 promote rapid PP1 phosphatase-dependent inactivation of AKT in ErbB2 overexpressing breast cancer cells. Cancer Res 63:7777–7784. [PubMed] [Google Scholar]
- Yamashita T, Shimada S, Guo W, Sato K, Kohmura E, Hayakawa T, Takagi T, Tohyama M (1997). Cloning and functional expression of a brain peptide/histidine transporter. J Biol Chem 272:10205–10211. [DOI] [PubMed] [Google Scholar]
- Yano S, Tokumitsu H, Soderling TR (1998). Calcium promotes cell survival through CaM-K kinase activation of the protein-kinase-B pathway. Nature 396:584–587. [DOI] [PubMed] [Google Scholar]
- Young D, Lawlor PA, Leone P, Dragunow M, During MJ (1999). Environmental enrichment inhibits spontaneous apoptosis, prevents seizures and is neuroprotective. Nat Med 5:448–453. [DOI] [PubMed] [Google Scholar]
- Yun BG, Matts RL (2005). Hsp90 functions to balance the phosphorylation state of Akt during C2C12 myoblast differentiation. Cell Signal 17:1477–1485. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Champagne N, Beitel LK, Goodyer CG, Trifiro M, LeBlanc A (2004). Estrogen and androgen protection of human neurons against intracellular amyloid beta1–42 toxicity through heat shock protein 70. J Neurosci 24:5315–5321. [DOI] [PMC free article] [PubMed] [Google Scholar]