

Abstract
The bed nucleus of the stria terminalis (BNST) is a key component of the CNS stress and reward circuit. Synaptic plasticity in this region could in part underlie the persistent behavioral alterations in generalized anxiety and addiction. Group I metabotropic glutamate receptors (mGluRs) have been implicated in stress, addiction, and synaptic plasticity, but their roles in the BNST are unknown. We find that activation of group I mGluRs in the dorsal BNST induces depression of excitatory synaptic transmission through two distinct mechanisms. First, a combined activation of group I mGluRs (mGluR1 and mGluR5) induces a transient depression that is cannabinoid 1 receptor dependent. Second, as with endocannabinoid-independent group I mGluR long-term depression (LTD) in the adult hippocampus, we find that activation of mGluR5 induces an extracellular signal-regulated kinase (ERK)-dependent LTD. Surprisingly, our data demonstrate that this LTD requires the ERK1 rather than ERK2 isoform, establishing a key role for this isoform in the CNS. Finally, we find that this LTD is dramatically reduced after multiple exposures but not a single exposure to cocaine, suggesting a role for this form of plasticity in the actions of psychostimulants on anxiety and reward circuitries and their emergent control of animal behavior.
Keywords: drug addiction, stress, anxiety, synaptic plasticity, extended amygdala, excitatory transmission
Introduction
Addiction is thought of as a disorder of learning-related processes involving alterations in glutamatergic signaling (Winder et al., 2002; Kalivas, 2004; Wolf et al., 2004). Substances of abuse administered both contingently and noncontingently to the animal alter synaptic plasticity in several brain regions involved in memory and reward, and it is hypothesized that long-lasting alterations at glutamatergic synapses in key regions underlie aspects of the persistence of addiction.
A large literature indicates that group I metabotropic glutamate receptors (mGluRs) play key roles in behavioral responses to psychostimulants. Group I receptors (mGluR1 and mGluR5) are Gq-coupled receptors whose activation leads to the recruitment of a variety of effectors, including phospholipase C and extracellular-signal regulated kinases (ERKs) (Peavy and Conn, 1998; Choe and Wang, 2001; Gallagher et al., 2004). Genetic disruption of either mGluR5 (Chiamulera et al., 2001) or the homer family of group I mGluR-interacting proteins (Swanson et al., 2001) produces profound alterations to responses to psychomotor stimulants, as does pharmacological inhibition of group I mGluRs (McGeehan and Olive, 2003; Herzig and Schmidt, 2004).
A major function of group I mGluR activation is to produce a persistent weakening of glutamatergic transmission at synapses in the CNS. There appears to be at least two major mechanisms through which this is accomplished. One involves the recruitment of endocannabinoid signaling and presynaptic alterations and has been observed in the dorsal and ventral striatum (Gerdeman and Lovinger, 2001; Sung et al., 2001; Robbe et al., 2002). A second major mechanism is through endocannabinoid-independent signaling mechanisms (Rouach and Nicoll, 2003) and has been extensively characterized in the hippocampus. Both presynaptic (Gereau and Conn, 1995; Watabe et al., 2002; Zakharenko et al., 2002) and postsynaptic (Huber et al., 2000, 2001; Snyder et al., 2001; Nosyreva and Huber, 2005) mechanisms have been described for endocannabinoid-independent forms of group I mGluR long-term depression (LTD).
Endocannabinoid-dependent group I mGluR-induced LTD is disrupted in the ventral striatum in animals previously exposed to cocaine, suggesting that alterations in this form of plasticity may play a role in cocaine-induced alterations in animal behavior (Swanson et al., 2001). To date, however, the role of endocannabinoid-independent group I mGluR LTD has not been explored. We addressed this question by examining effects of activation of group I mGluRs at excitatory synapses within the bed nucleus of the stria terminalis (BNST), a region critical in mediating stress–reward interactions (Delfs et al., 2000; Stewart, 2000). We find that activation of group I mGluRs produces two distinct effects. First, combined activation of mGluR1 and mGluR5 produces a transient depression of transmission that is presynaptically maintained and primarily endocannabinoid dependent. Second, mGluR5 activation induces an LTD of transmission that is postsynaptically induced, cannabinoid receptor 1 (CB1R) independent, and surprisingly ERK1 dependent. As with endocannabinoid-dependent mGluR5 LTD in the nucleus accumbens (NAc), we find that endocannabinoid-independent LTD in the BNST is ablated in animals that have been administered cocaine. However, although a single intraperitoneal injection of cocaine reduced LTD in NAc, we find that multiple exposures are required.
Materials and Methods
Animals
Male C57BL/6J mice (5–10 weeks old; The Jackson Laboratory, Bar Harbor, ME), CB1R knock-out males 5–6 weeks of age, or ERK1 (−/−, +/+) males 5–6 weeks of age, both of which were on a C57Bl/6J mixed background, were used.
Brain slice preparation
Methods are as described by Grueter and Winder (2005). Briefly, mice were decapitated under anesthesia (isoflurane). The brains were quickly removed and placed in ice-cold artificial CSF (ACSF) (in mm: 124 NaCl, 4.4 KCl, 2 CaCl2, 1.2 MgSO4, 1 NaH2PO4, 10 glucose, and 26 NaHCO3). Hemisected coronal brain slices (300 μm thick) were prepared using a vibratome (Leica, Nussloch, Germany). Rostral slices containing anterior portions of BNST are represented in Figures 1A and 6C [bregma 0.26 mm to 0.02 mm (Franklin and Paxinos, 1997)] and have been described previously (Egli and Winder, 2003; Weitlauf et al., 2004; Grueter and Winder, 2005). Slices were then transferred to an interface recording chamber (∼28°C) in which field potential recordings were performed. Slices were allowed to equilibrate for 1 h in normal ACSF before beginning experiments.
Figure 1.

Group 1 mGluR activation in the BNST results in LTD of excitatory transmission. A, Diagram of coronal mouse brain containing the BNST (shaded in gray). B, Application of DHPG-induced a persistent reduction in the synaptic response (N2), as measured by the peak amplitude of field potential responses, normalized to the average value during the basal period before drug application. Inset, Sample traces from one experiment before (dark line) and after (light line) DHPG application, average of six consecutive traces at 18–20 and 38–40 min. Error bars indicate SEM. C, Time course of DHPG-induced reduction in EPSC amplitude, normalized to the average value during the basal period before drug application. Inset, Representative EPSC traces from BNST neurons before and after DHPG application, average of 10 consecutive traces at 4–5 and 12–13 min. D, Concentration–response curve of peak DHPG-induced depression of EPSCs (n = 3–12).
Figure 6.

DHPG application results in phosphorylation of ERK. A, Representative slice illustrating size and location of BNST and striatal punches taken for biochemical analysis. B, Representative immunoblot for phosphorylated ERK1/ERK2 and pan ERK1/ERK2 from either the BNST or the striatum in the absence or presence of DHPG. DHPG (100 μm) was applied for 5 min, after which tissue punches were immediately collected. C, D, Quantitative analysis of ERK1 and ERK2 phosphorylation in the BNST (C; n = 8; *p < 0.05) and the striatum (D; n = 8; *p < 0.05). Error bars indicate SE.
Whole-cell patch-clamp recordings were performed in a submerged recording chamber (24–25°C). Slices were prepared as above, except a low-sodium/high-sucrose dissecting solution replaced the ACSF. Slices were allowed to recover in a submerged holding chamber (25°C) for a minimum recovery period of 30 min. Slices were then removed from the holding chamber and placed in the recording chamber in which they were continuously perfused with oxygenated (95% O2/5% CO2) ACSF at a rate of 2 ml/min.
Extracellular field recordings
Experiments were performed as reported previously (Egli and Winder, 2003; Weitlauf et al., 2004; Egli et al., 2005; Grueter and Winder, 2005). Low-resistance (1–3 MΩ) extracellular electrodes were filled with ACSF. Stimulating electrodes were placed on the dorsal anterolateral BNST border of the internal capsule ∼200–500 μm dorsal to the anterior commissure as illustrated in Figure 1A. Field potential responses were evoked at a frequency of 0.05 Hz using a stimulus range of 5–15 V at a duration of 100–150 μs. Picrotoxin (25 μm) was added to block GABAA receptor-mediated currents. The synaptic component of the field potential, the N2, is an AMPA receptor-mediated potential because it is sensitive to the AMPA receptor antagonist CNQX (data not shown) (Weitlauf et al., 2004; Egli et al., 2005; Grueter and Winder, 2005). Data points are represented as averages of the peak amplitude at 1 min intervals.
Whole-cell voltage-clamp recordings
Whole-cell recordings were performed as reported by Grueter and Winder (2005). Briefly, electrodes of 3.0–6.0 MΩ were filled with the following (in mm): 135 K Gluconate, 5 NaCl, 10 HEPES, 0.6 EGTA, 4 Na2+ATP, 0.4 Na2+GTP, and 0.1% biocytin. As in the interface chamber studies, picrotoxin (25 μm) was included in the ACSF. Evoked EPSCs of 100–400 pA were recorded while voltage clamped at −70 mV. After whole-cell configuration was achieved, cells were allowed to equilibrate a minimum of 10 min before baseline recordings were started. Postsynaptic parameters were all monitored continuously throughout the duration of the experiments. Events were recorded at a frequency of 0.17 Hz. Data is represented as a 30 s average of the peak amplitudes. In experiments in which behavioral manipulations were made, data acquisition and analysis were performed blinded to in vivo treatment. A paired-pulse ratio (PPR) was acquired by applying a second stimulus of equal intensity 50 ms after the first stimulus as described in detail by Grueter and Winder (2005).
Biochemistry
Tissue incubation.
Hemisected slices (300 μm) were made as described above and transferred to multiwell submerged chambers containing oxygenated ACSF at 25–27°C. After a 90 min incubation period, 25 μm picrotoxin was added. Two hours after slicing, 100 μm (RS)-3,5-dihydroxyphenylglycine (DHPG) was added for 5 min. The slices were removed and immediately frozen in dry ice. Tissue punches of the BNST and dorsal striatum were taken using a 0.41 mm punch from VWR Scientific (West Chester, PA). Additionally, hemisected slices (400 μm) were taken from mice that received repeated intraperitoneal injections to measure total protein levels. These slices were treated with 25 μm picrotoxin 90 min after slicing, and punches were taken 30 min later. The punches were stored at −80°C until homogenization.
Western blotting.
Briefly, tissue punches were homogenized in ice-cold homogenization buffer (2% SDS, 2 mm sodium orthovanadate, 2 mm sodium fluoride, 0.1 mm benzanimide, 10 μg/ml aprotinin, and 10 μg/ml leupeptin) and centrifuged for 20 min at 1000 × g, and protein levels were obtained using a BCA protein assay kit from Pierce (Rockford, IL). Homogenate protein levels were normalized across samples and then processed according to Vanhoose et al. (2002), with one notable alteration. The SDS-PAGE procedure was done using a reducing sample buffer containing 5% SDS and 40 mm DTT. Primary antibodies were used in the following concentrations: phosphorylated ERK (Cell Signaling Technology, Beverly, MA), 1:5000; ERK (Cell Signaling Technology), 1:1000; and mGluR5 (Upstate Biotechnology, Lake Placid, NY), 1:800.
Cocaine self-administration
All procedures were approved by the Institutional Animal Care and Use Committee of Vanderbilt University. After at least 1 week acclimation in the animal care facility, male C57BL/6J mice at 5–6 weeks of age were weighed and food restricted. Food and water were taken away between 3:00 P.M. and 5:00 P.M. and then returned between 9:00 A.M. and 11:00 A.M. the following day. They were allowed to eat and drink for 6 h per day with this regimen for 4 d. This procedure did not cause significant weight loss in the mice.
Food training.
Food-restricted mice were trained to lever press in Med Associates (E. Fairfield, VT) operant chambers on an fixed ratio 1 (FR-1) schedule of reinforcement with 25% vanilla Ensure as the reinforcer, with one noncontingent reinforcer available at the start of the session. Presses on the inactive lever were recorded but had no programmed consequences. Sessions were 1 h each and were initiated between 9:00 A.M. and 12:00 P.M. Illumination of the house light indicated the start of the session, and cue lights indicated availability of the liquid reinforcer. Mice were trained in this way until they received 50 reinforcers for two consecutive sessions; otherwise, they were excluded from the rest of the study.
Self-administration training
Mice successfully completing food training were catheterized in the right jugular vein with a SILASTIC catheter as described previously (Schramm-Sapyta et al., 2005). After 2–5 d of recovery with food and water available ad libitum, cocaine/saline self-administration was commenced. During self-administration, the mice were food restricted: one pellet was available per day, with water available ad libitum. This method maintained the animals at ∼90% of their free-feeding weight. Self-administration was performed on an FR-1 schedule for 3 h sessions, in which pressing of the active lever resulted in delivery of cocaine (0.3 mg/kg per infusion in 0.040 ml). Infusions were 2.5 s, during which time the cue lights were illuminated. Active lever presses during the infusion and presses on the inactive lever were recorded but had no programmed consequences. Self-administration sessions were terminated if the mouse received 100 infusions. Self-administration sessions were conducted for 11 total days: 10 d with cocaine (n = 6) or saline (n = 6), followed by a 1 d saline self-administration. Immediately after the 11th session, catheter patency was verified by infusion of ketamine through the catheter. Only animals that had patent catheters were included in the electrophysiological analysis.
Intraperitoneal cocaine administration
C57BL/6J mice at 5–6 weeks of age were weighed and given intraperitoneal (intraperitoneal) injections of either saline or cocaine (20 mg/kg). For the chronically treated experiments, injections were repeated for 10 consecutive days. For the experiments in which a single exposure to cocaine was examined, mice received saline injection for 5 d before the cocaine injection. At 24 h after the last injection, slices from these animals were prepared as stated above.
Drugs
All experiments were performed in the presence of picrotoxin (25 μm). When indicated in the text, the following drugs were bath applied: DHPG (10–100 μm), dl-2-amino-5-phosphonopentanoic acid (dl-AP-5) (100 μm), 2-methy-6-(phenylethynyl)pyridine hydrochloride (MPEP) (10 μm), (S)-(+)-α-amino-4-carboxy-2-methybenzeneacetic acid (LY367385) (100 μm), guanosine-5′-O-(2-thiodiphosphate) (GDPβS) (1 mm), and 1,4-diamino-2,3-dicyano-1,4-bis[2-aminophenylthio]butadiene (U0126) (20 μm) were purchased from Tocris Cookson (Ballwin, MO). N-Piperidino-5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methylpyrazole-3-carboxamide (SR141716) (100 μm) was acquired from National Institutes of Health. Cocaine HCl and picrotoxin were purchased from Sigma (St. Louis, MO). Dimethylsulfoxide was the carrier for picrotoxin, MPEP, and U0126 (0.02% v/v).
Results
Group I mGluRs depress excitatory transmission in the BNST
To study the effects of group I mGluR activation on excitatory synaptic transmission in the BNST, we used local afferent stimulation and both extracellular and whole-cell patch-clamp recording of postsynaptic responses as described previously (Fig. 1A) (Weitlauf et al., 2004; Egli et al., 2005; Grueter and Winder, 2005). To activate group I mGluRs, the specific agonist DHPG was briefly bath applied. Under extracellular recording conditions, after 15 min superfusion of DHPG (100 μm), the amplitude of the N2 (synaptic, glutamatergic component) decreased to 79 ± 4% of baseline. The N2 amplitude 40 min after DHPG remained depressed at 86 ± 2% of baseline (p < 0.05) (Fig. 1B). These effects were independent of changes in the N1, the nonsynaptic, predominantly axonally mediated response, indicating that depression of the synaptically evoked response occurs independent of gross alteration of the number of axons recruited by the local afferent stimulation.
DHPG also reduced EPSCs in a concentration-dependent manner (Fig. 1C,D). DHPG at 100 μm reduced EPSCs to 52 ± 5% of baseline at the peak effect that remained depressed (62 ± 6% of baseline) 30 min after DHPG application (Fig. 1C). The DHPG-induced LTD of EPSCs was independent of NMDA receptor function because robust LTD was elicited in slices pretreated with the NMDA receptor antagonist dl-AP-5 (100 μm) (peak effect, 46 ± 11% of baseline; 30 min after DHPG, 65 ± 8% of baseline) (Fig. 2A).
Figure 2.
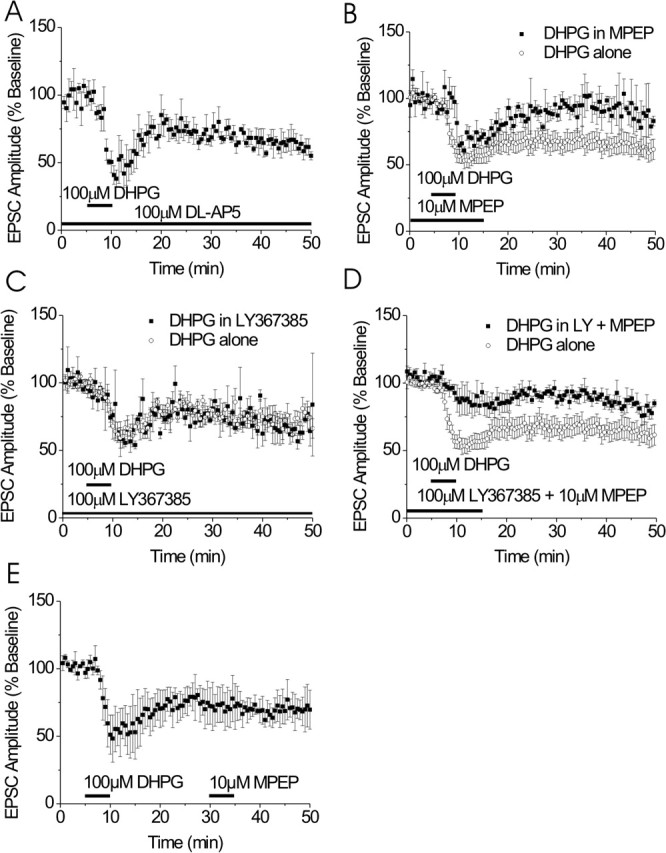
DHPG induces a dual-component persistent depression in the BNST. A, The NMDA receptor antagonist, dl-AP-5 (100 μm) does not alter DHPG-induced depression of EPSCs (n = 3). B, MPEP blocks the late but not the peak effect of DHPG on EPSCs (n = 3). C, DHPG effects on BNST EPSCs are unchanged in the presence of LY367385 (n = 3). D, Both the mGluR1 antagonist LY367385 and MPEP are required to attenuate the peak DHPG effect on EPSCs (n = 5). E, Late MPEP application does not reverse DHPG-induced LTD (n = 3). Error bars indicate SE.
To begin to identify the group I mGluR subtypes, mGluR1 and/or mGluR5, involved in the DHPG-induced depression of EPSCs, we applied DHPG in the presence of selective mGluR1- and mGluR5-specific antagonists. In the presence of the mGluR5-specific antagonist MPEP (10 μm), the initial component of the depression was not significantly altered (69 ± 8 vs 52 ± 5% of baseline); however, 30 min after DHPG, the depression was significantly attenuated (94 ± 3 vs 62 ± 6% of baseline; p < 0.05) (Fig. 2B). To test for a role of mGluR1 in the DHPG-induced depression of synaptic transmission, 100 μm DHPG was applied in the presence of the mGluR1-specific antagonist LY367385 (100 μm). Neither the initial DHPG-mediated depression (60 ± 6 vs 65 ± 5% of baseline; p > 0.05) nor the late component (77 ± 6 vs 73 ± 5% of baseline; p > 0.05) was significantly altered in the presence of LY367385 compared with DHPG alone (Fig. 2C). When DHPG was then applied in the presence of both of these antagonists, both the initial DHPG-induced depression (74 ± 8 vs 52 ± 6% of baseline; p < 0.05) (Fig. 2D) and the persistent depression (89 ± 7 vs 62 ± 6% of baseline; p < 0.05) were significantly attenuated compared with DHPG alone. Thus, these data suggest that DHPG-induced persistent depression of excitatory transmission in the BNST requires mGluR5 activation and that combined mGluR5 and mGluR1 activity is required for the early component.
To verify that DHPG-induced persistent depression was not simply the result of poor washout of DHPG or perhaps of induction of constitutive activity of mGluR5, we again applied MPEP, this time 20 min after washout of DHPG (Fig. 2E). At this time point, MPEP failed to alter EPSC amplitude, suggesting that transient activation of mGluR5 induces LTD.
Postsynaptic induction of mGluR5-dependent LTD in BNST
In the hippocampus, mGluR5-dependent LTD is thought to be induced postsynaptically (Watabe et al., 2002). To begin to determine the site of induction of mGluR5-dependent LTD in the BNST, we replaced GTP with 1 mm GDPβS in our standard potassium gluconate-based electrode-filling solution to inhibit activation of postsynaptic G-proteins. Dialyzing GDPβS into the postsynaptic cell blocked DHPG-induced LTD (114 ± 20% of baseline) in the BNST but had no effect on the early component (66 ± 8% of baseline) (Fig. 3A). GDPβS did not alter basal transmission in time-matched recordings (data not shown). These results indicate that a postsynaptic G-protein is necessary for induction of LTD, although it does not rule out the possibility of presynaptic maintenance of this LTD.
Figure 3.

Characterization of DHPG-induced LTD. A, Blockade of postsynaptic G-protein signaling with GDPβS prevents DHPG-induced depression of EPSCs (filled squares; n = 5) but not control cells containing GTP (open circles). B, Time course of normalized PPR illustrating transient increase in release probability in response to DHPG (left axis). As illustrated, DHPG causes a transient increase in PPR during the peak effect on EPSC amplitude but returns to basal levels during the LTD (filled squares, PPR; open circles, EPSC amplitude). Error bars indicate SE.
Synaptic locus of group I mGluR-mediated effects
To begin to assess the synaptic mechanisms involved in DHPG-induced depression of transmission, we examined the effects of DHPG application on the ratio of EPSC amplitudes in response to closely spaced (50 ms) paired stimuli (PPR) to query whether DHPG produces alterations in presynaptic release probability. During the initial peak depression of the EPSC produced by DHPG, we found a significant increase in the PPR (120 ± 6%; p < 0.05) (Figs. 3B, 4C). However, there was no significant change in PPR during DHPG LTD (104 ± 5%; p = 0.37) (Figs. 3C, 4C). In comparison, activation of either group 2 or group 3 mGluRs, which commonly function presynaptically as autoreceptors, caused a persistent increase in PPR in the BNST (Grueter and Winder, 2005). These data suggest that, although the mGluR1- and mGluR5-dependent early depression involves an alteration in release probability, the mGluR5-dependent LTD does not.
Figure 4.
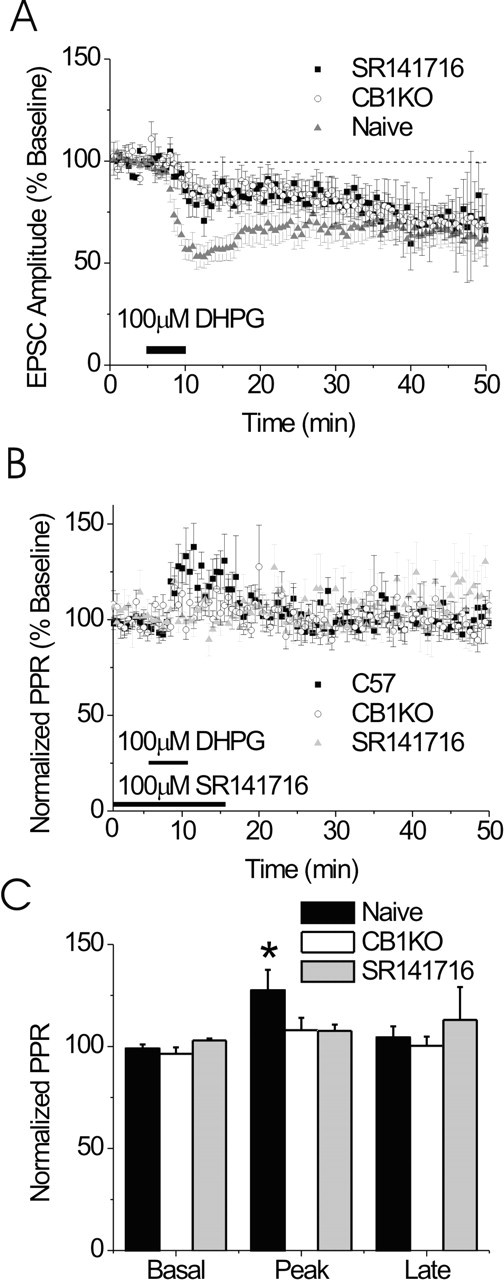
The early component of DHPG-induced depression of the EPSC is CB1R dependent but the LTD is CB1R independent. A, Summary of the effects of DHPG (100 μm) on EPSCs in BNST from CB1R knock-out (KO) mice (white circles; n = 9) in the presence of SR141716 (black squares; n = 3) and control mice (gray triangles; n = 12). B, The PPR is unchanged in the CB1R knock-out or in the presence of SR141716 after DHPG application. C, Bar graph representing the basal, peak, and late effects of DHPG on PPR in control CB1R knock-out mice and in the presence of SR141716 (n = 9 and 12, respectively; *p < 0.05 compared with basal). Error bars indicate SE.
mGluR1- and mGluR5-dependent early depression, but not mGluR5 LTD, is CB1R dependent
Group I mGluR-induced LTD has been demonstrated to be presynaptically maintained and dependent on CB1R signaling in the nucleus accumbens and striatum (Maejima et al., 2001; Robbe et al., 2003) but independent of cannabinoid signaling in the hippocampus (Rouach and Nicoll, 2003). To test whether the CB1R is involved in effects of DHPG on EPSCs in BNST, we examined the effects of DHPG (100 μm) on EPSCs recorded from BNST neurons either in the presence of the CB1R antagonist SR141716 (100 μm) or in slices obtained from CB1R knock-out mice (Zimmer et al., 1999). Interestingly, the initial depression of the EPSC by DHPG was significantly blunted both in neurons from CB1R knock-out mice (84 ± 5% of baseline; p < 0.05) and in the presence of SR141716 (84 ± 7% of baseline; p > 0.05) compared with control animals at the same time point (Fig. 4A). Consistent with these data, we found that, in contrast to results obtained in wild-type mice, PPR after DHPG application was unaltered in these experiments (108 ± 6 and 108 ± 2% of baseline for CB1R knock-out and in the presence of SR141716 respectively; p > 0.05) (Fig. 4B,C). The CB1R knock-out mice did not have a generalized deficit in receptor regulation of presynaptic function, however, because activation of group II mGluRs in the BNST of CB1R knock-out mice resulted in an increase in PPR (data not shown) as described previously in wild-type mice (Grueter and Winder, 2005).
Whereas the acute effect of DHPG on EPSCs was blunted in the CB1R knock-out mice and in the presence of SR141716, DHPG-induced LTD was still robustly elicited in these animals (73 ± 7 and 65 ± 14 vs 62 ± 6% of baseline 30 min after DHPG administration) (Fig. 4A). These data suggest that the initial component of the DHPG-induced depression of the EPSC, as in the hippocampus, is mediated through the CB1R, whereas the mGluR5-mediated LTD in the BNST is independent of CB1R signaling.
DHPG-induced LTD of synaptic transmission in the BNST is ERK1 dependent
CB1R-independent group I mGluR-dependent LTD in the hippocampus has been reported to require ERK activation (Gallagher et al., 2004). Furthermore, group I mGluRs recruit ERK activity in a variety of contexts (Mao and Wang, 2003; Mao et al., 2005). To examine the role of ERK in DHPG-induced depression of EPSCs in the BNST, we used two converging approaches. First, we applied DHPG (100 μm) to BNST slices in the presence of an mitogen-activated protein kinase (MAPK) kinase (MEK) inhibitor (U0126, 20 μm) while recording EPSCs. The presence of the MEK inhibitor significantly attenuated, but did not ablate, the early component of the DHPG-mediated depression of transmission to 69 ± 5 versus 52 ± 5% of baseline (Fig. 5A). However, the DHPG-induced LTD was abolished in the presence of the MEK inhibitor (92 ± 10% of baseline, 30 min after DHPG) (Fig. 5A).
Figure 5.
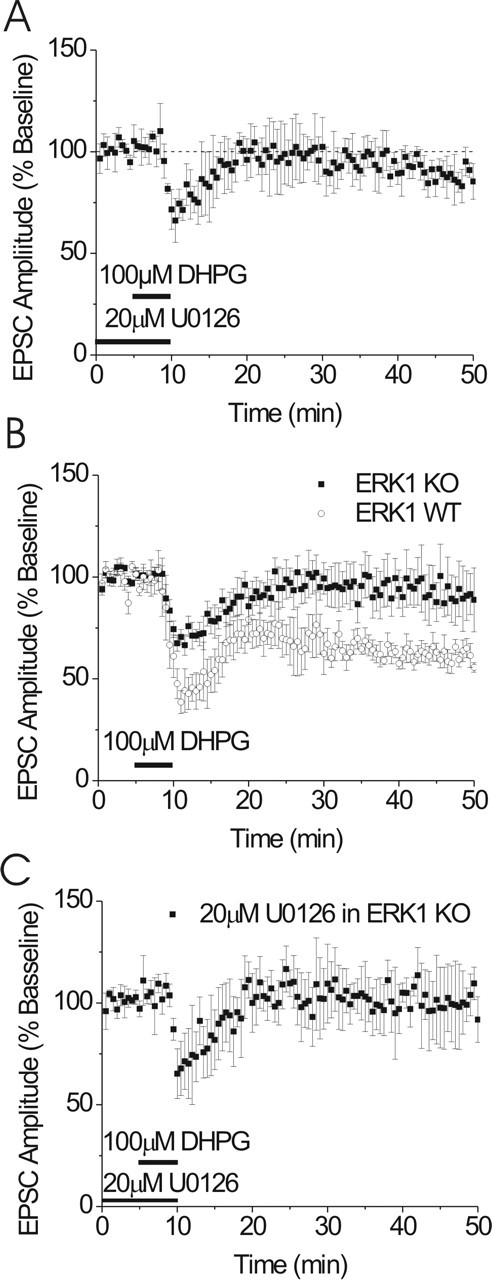
mGluR5-dependent LTD in the BNST is ERK dependent. A, DHPG-induced LTD of EPSCs is abolished by the presence of the MEK inhibitor U0126 (20 μm; n = 6). U0126 was applied for a minimum of 30 min before DHPG application. B, EPSCs from BNST neurons from ERK1 knock-out (KO) mice do not elicit LTD after DHPG application (n = 6). WT, Wild type. Inset represents presence/absence of ERK1 protein in tissue punches containing BNST in control and ERK1 knock-out mice. Error bars indicate SE.
We next examined the effects of DHPG on EPSCs in the BNST of ERK1 knock-out mice. Similar to the U0126 experiments, application of DHPG in the BNST of ERK1 knock-out mice produced an initial depression of the EPSC that was significantly attenuated, although still present (74 ± 4% of baseline). Surprisingly, however, LTD was completely ablated (102 ± 7% of baseline) (Fig. 5B). The effect of DHPG on EPSCs in the BNST from wild-type littermates was not different from C57BL/6J mice (Fig. 5B). To test for potential compensatory actions of ERK2 in the ERK1 knock-out, we repeated the MEK inhibitor experiments in the ERK1 knock-out animals. Pretreatment with U0126 (20 μm) did not alter the phenotype observed in the ERK1 knock-out animals (104 ± 8% of baseline) (Fig. 5C).
DHPG activates ERK in BNST
The above results suggest that ERK1 activity is required for DHPG-induced LTD. Because group I mGluR activation has been shown to activate ERK1/2 in other brain regions (Choe and Wang, 2001; Adwanikar et al., 2004; Gallagher et al., 2004), we tested whether DHPG application activated ERK1 and ERK2 in the dorsal BNST. Frozen tissue punches from the BNST and striatum were obtained after a 5 min exposure to DHPG (100 μm) and analyzed by Western blot to determine ERK activation (Fig. 6C,D).
First, as has been reported previously, we found that DHPG produced robust activation of ERK in striatum (Choe and Wang, 2001) (Fig. 6B,D). Striatal ERK2 phosphorylation was increased almost twofold in response to DHPG (185 ± 35% of basal; p < 0.05) (Fig. 6B,D), whereas ERK1 phosphorylation was not significantly altered (75 ± 3% of basal; p > 0.05) (Fig. 6B,D). Thus, as is the case in many brain regions, ERK2 appears to be the predominant isoform activated in the striatum. Interestingly, in the BNST, the relative ratio of ERK1 to ERK2 expression was higher than in striatum (Fig. 6B). Moreover, we observed a relatively robust increase in ERK1 phosphorylation in the BNST in the presence of 100 μm DHPG (137 ± 9% of basal; p < 0.05) (Fig. 6B,C). DHPG treatment caused only a very small increase in ERK2 activation (111 ± 4% of basal; p < 0.05) (Fig. 6B,C). These data suggest that activation of group I mGluRs results in ERK1 activation in the BNST.
mGluR5-dependent LTD in the BNST is greatly attenuated by contingent in vivo cocaine administration
In the ventral lateral BNST, Dumont et al. (2005) report that contingent cocaine administration induces a change in AMPA/NMDA ratios. Thus, we examined the effects of self-administration of cocaine on group I mGluR-dependent LTD in the BNST. After food training, mice were allowed 3 h daily sessions of access to either cocaine or saline on an FR-1 schedule. Animals in the cocaine group lever pressed significantly more than saline controls (Fig. 7A). Twenty-four hours after the last day of self-administration (10 d total), the mice were killed, and brain slices were prepared for electrophysiological analysis. Analysis of EPSC recordings from these mice demonstrate that the initial component of the DHPG-induced depression did not differ between slices prepared from cocaine self-administering (51 ± 6% of baseline) (Fig. 7B) and saline administering (61 ± 4% of baseline) (Fig. 7B) mice. However, the persistent depression of EPSCs by DHPG was significantly attenuated in slices prepared from cocaine self-administering mice (83 ± 9% of baseline; when compared with saline control animals, 63 ± 5% of baseline; p < 0.05). Operant training did not alter DHPG-induced LTD because there is no difference between animals self-administering saline and naive animals (61 ± 4 vs 62 ± 6%, respectively). PPR from cocaine-treated and saline-treated groups were not different than naive animals (data not shown).
Figure 7.

mGluR5-dependent LTD is attenuated by contingent and noncontingent in vivo cocaine. A, Summary of active lever presses for cocaine pressing versus saline pressing mice (*p < 0.05). B, Summary of the effects of cocaine self-administration on mGluR5-induced LTD in the BNST (cocaine, n = 9; saline, n = 13). C, Summary of the effects of intraperitoneal administration of cocaine on mGluR-induced LTD in the BNST (repeated intraperitoneal cocaine, gray triangles, n = 8; single intraperitoneal cocaine, black squares, n = 6; saline, white circles, n = 8). D, Quantification of total levels of mGluR5, ERK1, and ERK2 protein from BNST punches obtained from chronically treated (intraperitoneally) mice (saline, n = 14; cocaine, n = 12). Error bars indicate SE.
Repeated but not single noncontingent in vivo cocaine exposure attenuates mGluR5-dependent LTD in the BNST
A single intraperitoneal cocaine administration disrupts endocannabinoid-dependent, group I mGluR-induced LTD in the nucleus accumbens (Swanson et al., 2001; Fourgeaud et al., 2004) and activates ERK in the BNST (Valjent et al., 2004). As mentioned above, in the neighboring ventral lateral BNST, changes in synaptic signaling after cocaine administration occur after self-administration but are unaltered after a single intraperitoneal injection of cocaine. As shown in Figure 7C, a single exposure to cocaine is not sufficient to alter mGluR5 LTD in the dorsal BNST (75 ± 2 vs 69 ± 6% of baseline; p > 0.05) (Fig. 7C). However, intraperitoneal injection of cocaine for 10 consecutive days abolished mGluR5 LTD (98 ± 5 vs 69 ± 6% of baseline; p < 0.05), demonstrating that contingency in the dorsal BNST is not strictly required. To begin to determine a mechanism of cocaine-mediated changes in group I mGluR LTD, we assessed ERK1/2 and mGluR5 levels. No change in total ERK1 (109 ± 4 vs 103 ± 4% saline vs cocaine; p > 0.05) (Fig. 7D), total ERK2 (105 ± 5 vs 107 ± 5% saline vs cocaine; p > 0.05), or mGluR5 (100 ± 4 vs 99 ± 4%, saline vs cocaine; p > 0.05) (Fig. 7D) protein levels were observed 1 d after cocaine administration.
Discussion
Group I mGluRs are necessary for many of the behavioral alterations produced by cocaine administration (Chiamulera et al., 2001; Herzig and Schmidt, 2004), and signaling by mGluR5 in the nucleus accumbens is altered by cocaine administration (Swanson and Kalivas, 2000; Swanson et al., 2001). The BNST is uniquely positioned to modulate the integration of motivational and higher-order processive innervations, receiving excitatory input from limbic regions, such as the amygdala and hippocampus, and projecting to areas involved in the reward/motivation and stress circuitry, the ventral tegmental area (VTA), NAc and paraventricular nucleus (Herman and Cullinan, 1997; McDonald et al., 1999; Georges and Aston-Jones, 2002). Thus, we examined group I mGluR regulation of excitatory transmission in the BNST.
Group I mGluR activation depresses excitatory transmission in the BNST via temporally distinct phenomena
Activation of group I mGluRs in the BNST produces two temporally and mechanistically distinct effects on excitatory synaptic transmission. DHPG-induced acute depression differs from the persistent depression in a number of respects. Using mGluR-subtype specific antagonists, we find that the persistent depression is induced by mGluR5 activation, whereas mGluR1 activation is additionally required for the acute depression resulting from DHPG application. This differentiation between the role of mGluR1 and mGluR5 in the acute depression and mGluR5 in the LTD is consistent with the distinct action of these group I mGluR subtypes in the hippocampus on LTD and excitability of hippocampal neurons (Mannaioni et al., 2001; Faas et al., 2002). We used pharmacological, genetic, and biophysical tools to further differentiate these processes.
In contrast to the persistent depression described in detail below, the acute depression is associated with a robust increase in PPR of evoked EPSCs, suggesting that it involves a decrease in release probability. At many CNS synapses, presynaptic actions of group I mGluRs are mediated by the endocannabinoid system (Maejima et al., 2001). Similar to findings in the hippocampus (Rouach and Nicoll, 2003) and NAc (Robbe et al., 2002), we found that the initial phase of the DHPG-mediated depression is ablated in CB1R knock-out mice and in the presence of the CB1R antagonist. These data suggest that the acute depression is mediated by mGluR1/5-dependent production of endocannabinoids, which then act on presynaptic CB1Rs to depress transmission. It is interesting that the acute depression was not blocked by inclusion of GDPβS in the pipette. A likely possibility is that endocannabinoids generated by neighboring cells produce this depression in a heterosynaptic process, as described by Wilson and Nicoll (2001).
Group I mGluR activation also leads to a mechanistically distinct persistent depression. As mentioned above, pharmacological evidence suggests the DHPG-induced LTD is mGluR5 dependent and CB1R independent. The induction of this form of LTD has a postsynaptic component, because dialysis of the postsynaptic cell with GDPβS blocked DHPG-induced LTD. The LTD was not associated with alterations in PPR, consistent with the lack of CB1R dependence, suggesting that the maintenance of this LTD is not through an alteration of release probability.
A novel role for ERK1 in mGluR5 LTD
The MAPK/ERK signaling cascade plays a crucial role in a variety of cell regulatory events. Much emphasis has been placed on the role of this signaling cascade in learning and memory and in glutamatergic synaptic plasticity at a variety of synapses (Adams and Sweatt, 2002; Sweatt, 2004). In particular, MEK inhibitors block the induction of several forms of NMDA receptor-dependent long-term potentiation (LTP) and LTD throughout synapses of the hippocampus and the amygdala (for review, see Sweatt, 2004). A more recent, growing literature indicates that the ERK signaling cascade plays a critical role in behavioral alterations produced by psychostimulants. Inhibitors of the ERK signaling cascade administered in vivo reduce psychostimulant-mediated behavioral sensitization (Valjent et al., 2005). It has also been suggested that ERK activation is involved in the incubation of cocaine craving in a region-specific manner (Lu et al., 2005). Furthermore, in vivo psychostimulant administration produces activation of ERK in the reward circuitry, including strong activation within the BNST (Valjent et al., 2004).
To date, evidence has suggested that the primary ERK isoform involved in these processes is ERK2 (Sweatt, 2004). Here, however, we provide evidence that ERK1 rather than ERK2 plays a critical role in mGluR5-induced LTD in the BNST. By using an inhibitor (U0126) targeting the MEKs that activate ERK1/ERK2, we show that ERK is necessary for mGluR5-induced LTD in the BNST. MEK inhibitors such as U0126 do not allow for distinction of the relative roles of ERK1 and ERK2. Because ERK1 expression relative to ERK2 is stronger in the BNST than many other regions, we took advantage of the availability of mice with a targeted deletion of ERK1 and found that DHPG-induced LTD is absent in ERK1 knock-out mice. We demonstrate that the disruption of LTD in the ERK1 knock-out is not through altered function of ERK2 because the effects of U0126 were occluded in the ERK1 knock-out. Furthermore, consistent with a role specifically for ERK1 in DHPG-induced LTD in the BNST, we find that DHPG application preferentially activates ERK1 in the BNST, in contrast to the striatum, in which ERK2 is the predominant isoform activated. It is important to note that, at present, it is unclear where in the BNST the ERK1 required for this LTD is located.
At present, very little is known about the distinctive roles of the ERK1 and ERK2 isoforms, because they are highly homologous (Chen et al., 2001). However, several studies, primarily in non-neuronal systems, have identified proteins and/or drugs that produce or facilitate selective activation of ERK1 versus ERK2 (Schaeffer et al., 1998). Although little information exists for substrates differentially phosphorylated by ERK1 and ERK2, data from knock-out mice point to differences in function. For example, ERK1 knock-out mice are viable and fertile (Selcher et al., 2001; Pages and Pouyssegur, 2004), whereas targeted disruption of ERK2 produces a lethal mutation (Adams and Sweatt, 2002). Furthermore, although evidence suggests that ERK1/2 plays an important role in hippocampal synaptic plasticity and hippocampal-based learning and memory, these phenotypes are either unimpaired (Selcher et al., 2001) or modulated in complex ways in ERK1 null mice (Mazzucchelli et al., 2002).
Group I mGluRs and drug addiction
Our results indicate that mGluR5 is responsible for the DHPG-induced LTD in the BNST. Multiple studies indicate that mGluR5 participates in behavioral responses to cocaine (Chiamulera et al., 2001; Herzig and Schmidt, 2004; McGeehan et al., 2004; Lee et al., 2005). Although the specific site of action of mGluR5 function in cocaine-mediated behavioral and rewarding properties are still unknown, we show that, like in the NAc (Swanson et al., 2001; Fourgeaud et al., 2004), in vivo cocaine administration attenuates DHPG-induced LTD. However, unlike the NAc, a single exposure to cocaine is not sufficient to alter mGluR5-induced LTD. This suggests that the threshold for cocaine administration required for modification of these different forms of group I mGluR LTD may differ across brain regions, which may have important implications for differential neural circuit recruitment during repeated cocaine administration.
At present, the mechanisms by which repeated cocaine administration produces an ablation of LTD in the BNST are unknown. We find that mGluR5, ERK1, and ERK2 protein levels are not altered in the BNST after in vivo cocaine administration, suggesting that either alteration of the levels of another key signaling protein or posttranslation modifications likely play key roles.
Excitatory transmission and drug addiction
Plasticity of glutamatergic synaptic transmission in specific brain regions has been suggested to play an important role in the neuroadaptations that occur after substance abuse (Winder et al., 2002; Kalivas, 2004; Wolf et al., 2004). Recent work suggests both psychostimulants and stress induce LTP in the VTA, and psychostimulants alter NAc synaptic plasticity (Swanson et al., 2001; Thomas et al., 2001). Because the BNST projects to the VTA, it is possible that the BNST plays a role in the integration of information leading to adaptive changes in synaptic responses after administration of drugs of abuse. Indeed, this brain region plays a critical role in stress-induced reinstatement of drug seeking (Erb et al., 1996; Shaham et al., 2000), as well as morphine withdrawal-induced conditioned place aversion (Aston-Jones et al., 1999). Moreover, evidence suggests that this region, and in particular glutamatergic transmission within this region, plays a critical role in anxiety-like behavior in rodents (Walker et al., 2003). Thus, persistent regulation of glutamatergic transmission in this region by group I mGluRs may play an important role in regulating generalized anxiety states.
It is interesting to note that cocaine self-administration has been reported recently to enhance AMPA/NMDA response ratios in the ventral BNST (Dumont et al., 2005), and intraperitoneal administration of cocaine enhances this ratio on dopaminergic neurons within the VTA (Borgland et al., 2004), a major efferent site from the BNST. Our data provide a potential mechanism contributing to these changes, because a reduction in the inhibitory constraint provided by group I mGluRs, as we have demonstrated, would likely facilitate the induction of NMDA receptor-dependent synaptic plasticity in these regions. In conclusion, we establish a role for ERK1 in the CNS, participating in cocaine-sensitive mGluR5-dependent LTD in the BNST.
Footnotes
This work was supported by grants from the National Alliance for Research on Schizophrenia and Depression, National Institute on Drug Abuse, and National Institute on Alcohol Abuse and Alcoholism. We thank Drs. S. K. Dey and Haibin Wang for providing the CB1 null mice generated by A. Zimmer.
References
- Adams JP, Sweatt JD (2002). Molecular psychology: roles for the ERK MAP kinase cascade in memory. Annu Rev Pharmacol Toxicol 42:135–163. [DOI] [PubMed] [Google Scholar]
- Adwanikar H, Karim F, Gereau RW IV (2004). Inflammation persistently enhances nocifensive behaviors mediated by spinal group I mGluRs through sustained ERK activation. Pain 111:125–135. [DOI] [PubMed] [Google Scholar]
- Aston-Jones G, Delfs JM, Druhan J, Zhu Y (1999). The bed nucleus of the stria terminalis. A target site for noradrenergic actions in opiate withdrawal. Ann NY Acad Sci 877:486–498. [DOI] [PubMed] [Google Scholar]
- Borgland SL, Malenka RC, Bonci A (2004). Acute and chronic cocaine-induced potentiation of synaptic strength in the ventral tegmental area: electrophysiological and behavioral correlates in individual rats. J Neurosci 24:7482–7490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Gibson TB, Robinson F, Silvestro L, Pearson G, Xu B, Wright A, Vanderbilt C, Cobb MH (2001). MAP kinases. Chem Rev 101:2449–2476. [DOI] [PubMed] [Google Scholar]
- Chiamulera C, Epping-Jordan MP, Zocchi A, Marcon C, Cottiny C, Tacconi S, Corsi M, Orzi F, Conquet F (2001). Reinforcing and locomotor stimulant effects of cocaine are absent in mGluR5 null mutant mice. Nat Neurosci 4:873–874. [DOI] [PubMed] [Google Scholar]
- Choe ES, Wang JQ (2001). Group I metabotropic glutamate receptor activation increases phosphorylation of cAMP response element-binding protein, Elk-1, and extracellular signal-regulated kinases in rat dorsal striatum. Brain Res Mol Brain Res 94:75–84. [DOI] [PubMed] [Google Scholar]
- Delfs JM, Zhu Y, Druhan JP, Aston-Jones G (2000). Noradrenaline in the ventral forebrain is critical for opiate withdrawal-induced aversion. Nature 403:430–434. [DOI] [PubMed] [Google Scholar]
- Dumont EC, Mark GP, Mader S, Williams JT (2005). Self-administration enhances excitatory synaptic transmission in the bed nucleus of the stria terminalis. Nat Neurosci 8:413–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egli RE, Winder DG (2003). Dorsal and ventral distribution of excitable and synaptic properties of neurons of the bed nucleus of the stria terminalis. J Neurophysiol 90:405–414. [DOI] [PubMed] [Google Scholar]
- Egli RE, Kash TL, Choo K, Savchenko V, Matthews RT, Blakely RD, Winder DG (2005). Norepinephrine modulates glutamatergic transmission in the bed nucleus of the stria terminalis. Neuropsychopharmacology 30:657–668. [DOI] [PubMed] [Google Scholar]
- Erb S, Shaham Y, Stewart J (1996). Stress reinstates cocaine-seeking behavior after prolonged extinction and a drug-free period. Psychopharmacology (Berl) 128:408–412. [DOI] [PubMed] [Google Scholar]
- Faas GC, Adwanikar H, Gereau RW IV, Saggau P (2002). Modulation of presynaptic calcium transients by metabotropic glutamate receptor activation: a differential role in acute depression of synaptic transmission and long-term depression. J Neurosci 22:6885–6890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fourgeaud L, Mato S, Bouchet D, Hemar A, Worley PF, Manzoni OJ (2004). A single in vivo exposure to cocaine abolishes endocannabinoid-mediated long-term depression in the nucleus accumbens. J Neurosci 24:6939–6945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franklin K, Paxinos G (1997). In: The mouse brain in stereotaxic coordinates New York: Academic.
- Gallagher SM, Daly CA, Bear MF, Huber KM (2004). Extracellular signal-regulated protein kinase activation is required for metabotropic glutamate receptor-dependent long-term depression in hippocampal area CA1. J Neurosci 24:4859–4864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georges F, Aston-Jones G (2002). Activation of ventral tegmental area cells by the bed nucleus of the stria terminalis: a novel excitatory amino acid input to midbrain dopamine neurons. J Neurosci 22:5173–5187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerdeman G, Lovinger DM (2001). CB1 cannabinoid receptor inhibits synaptic release of glutamate in rat dorsolateral striatum. J Neurophysiol 85:468–471. [DOI] [PubMed] [Google Scholar]
- Gereau RW IV, Conn PJ (1995). Roles of specific metabotropic glutamate receptor subtypes in regulation of hippocampal CA1 pyramidal cell excitability. J Neurophysiol 74:122–129. [DOI] [PubMed] [Google Scholar]
- Grueter BA, Winder DG (2005). Group II and III metabotropic glutamate receptors suppress excitatory synaptic transmission in the dorsolateral bed nucleus of the stria terminalis. Neuropsychopharmacology 30:1302–1311. [DOI] [PubMed] [Google Scholar]
- Herman JP, Cullinan WE (1997). Neurocircuitry of stress: central control of the hypothalamo-pituitary-adrenocortical axis. Trends Neurosci 20:78–84. [DOI] [PubMed] [Google Scholar]
- Herzig V, Schmidt WJ (2004). Effects of MPEP on locomotion, sensitization and conditioned reward induced by cocaine or morphine. Neuropharmacology 47:973–984. [DOI] [PubMed] [Google Scholar]
- Huber KM, Kayser MS, Bear MF (2000). Role for rapid dendritic protein synthesis in hippocampal mGluR-dependent long-term depression. Science 288:1254–1257. [DOI] [PubMed] [Google Scholar]
- Huber KM, Roder JC, Bear MF (2001). Chemical induction of mGluR5- and protein synthesis-dependent long-term depression in hippocampal area CA1. J Neurophysiol 86:321–325. [DOI] [PubMed] [Google Scholar]
- Kalivas PW (2004). Glutamate systems in cocaine addiction. Curr Opin Pharmacol 4:23–29. [DOI] [PubMed] [Google Scholar]
- Lee B, Platt DM, Rowlett JK, Adewale AS, Spealman RD (2005). Attenuation of behavioral effects of cocaine by the metabotropic glutamate receptor 5 antagonist 2-methyl-6-(phenylethynyl)-pyridine in squirrel monkeys: comparison with dizocilpine. J Pharmacol Exp Ther 312:1232–1240. [DOI] [PubMed] [Google Scholar]
- Lu L, Hope BT, Dempsey J, Liu SY, Bossert JM, Shaham Y (2005). Central amygdala ERK signaling pathway is critical to incubation of cocaine craving. Nat Neurosci 8:212–219. [DOI] [PubMed] [Google Scholar]
- Maejima T, Hashimoto K, Yoshida T, Aiba A, Kano M (2001). Presynaptic inhibition caused by retrograde signal from metabotropic glutamate to cannabinoid receptors. Neuron 31:463–475. [DOI] [PubMed] [Google Scholar]
- Mannaioni G, Marino MJ, Valenti O, Traynelis SF, Conn PJ (2001). Metabotropic glutamate receptors 1 and 5 differentially regulate CA1 pyramidal cell function. J Neurosci 21:5925–5934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao L, Wang JQ (2003). Group I metabotropic glutamate receptor-mediated calcium signalling and immediate early gene expression in cultured rat striatal neurons. Eur J Neurosci 17:741–750. [DOI] [PubMed] [Google Scholar]
- Mao L, Yang L, Tang Q, Samdani S, Zhang G, Wang JQ (2005). The scaffold protein Homer1b/c links metabotropic glutamate receptor 5 to extracellular signal-regulated protein kinase cascades in neurons. J Neurosci 25:2741–2752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzucchelli C, Vantaggiato C, Ciamei A, Fasano S, Pakhotin P, Krezel W, Welzl H, Wolfer DP, Pages G, Valverde O, Marowsky A, Porrazzo A, Orban PC, Maldonado R, Ehrengruber MU, Cestari V, Lipp HP, Chapman PF, Pouyssegur J, Brambilla R (2002). Knockout of ERK1 MAP kinase enhances synaptic plasticity in the striatum and facilitates striatal-mediated learning and memory. Neuron 34:807–820. [DOI] [PubMed] [Google Scholar]
- McDonald AJ, Shammah-Lagnado SJ, Shi C, Davis M (1999). Cortical afferents to the extended amygdala. Ann NY Acad Sci 877:309–338. [DOI] [PubMed] [Google Scholar]
- McGeehan AJ, Olive MF (2003). The mGluR5 antagonist MPEP reduces the conditioned rewarding effects of cocaine but not other drugs of abuse. Synapse 47:240–242. [DOI] [PubMed] [Google Scholar]
- McGeehan AJ, Janak PH, Olive MF (2004). Effect of the mGluR5 antagonist 6-methyl-2-(phenylethynyl)pyridine (MPEP) on the acute locomotor stimulant properties of cocaine, d-amphetamine, and the dopamine reuptake inhibitor GBR12909 in mice. Psychopharmacology (Berl) 174:266–273. [DOI] [PubMed] [Google Scholar]
- Nosyreva ED, Huber KM (2005). Developmental switch in synaptic mechanisms of hippocampal metabotropic glutamate receptor-dependent long-term depression. J Neurosci 25:2992–3001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pages G, Pouyssegur J (2004). Study of MAPK signaling using knockout mice. Methods Mol Biol 250:155–166. [DOI] [PubMed] [Google Scholar]
- Peavy RD, Conn PJ (1998). Phosphorylation of mitogen-activated protein kinase in cultured rat cortical glia by stimulation of metabotropic glutamate receptors. J Neurochem 71:603–612. [DOI] [PubMed] [Google Scholar]
- Robbe D, Kopf M, Remaury A, Bockaert J, Manzoni OJ (2002). Endogenous cannabinoids mediate long-term synaptic depression in the nucleus accumbens. Proc Natl Acad Sci USA 99:8384–8388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbe D, Alonso G, Manzoni OJ (2003). Exogenous and endogenous cannabinoids control synaptic transmission in mice nucleus accumbens. Ann NY Acad Sci 1003:212–225. [DOI] [PubMed] [Google Scholar]
- Rouach N, Nicoll RA (2003). Endocannabinoids contribute to short-term but not long-term mGluR-induced depression in the hippocampus. Eur J Neurosci 18:1017–1020. [DOI] [PubMed] [Google Scholar]
- Schaeffer HJ, Catling AD, Eblen ST, Collier LS, Krauss A, Weber MJ (1998). MP1: a MEK binding partner that enhances enzymatic activation of the MAP kinase cascade. Science 281:1668–1671. [DOI] [PubMed] [Google Scholar]
- Schramm-Sapyta NL, Olsen CM, Winder DG (2005). Cocaine self-administration reduces excitatory responses in the mouse nucleus accumbens shell. Neuropsychopharmacology in press. [DOI] [PubMed]
- Selcher JC, Nekrasova T, Paylor R, Landreth GE, Sweatt JD (2001). Mice lacking the ERK1 isoform of MAP kinase are unimpaired in emotional learning. Learn Mem 8:11–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaham Y, Erb S, Stewart J (2000). Stress-induced relapse to heroin and cocaine seeking in rats: a review. Brain Res Brain Res Rev 33:13–33. [DOI] [PubMed] [Google Scholar]
- Snyder EM, Philpot BD, Huber KM, Dong X, Fallon JR, Bear MF (2001). Internalization of ionotropic glutamate receptors in response to mGluR activation. Nat Neurosci 4:1079–1085. [DOI] [PubMed] [Google Scholar]
- Stewart J (2000). Pathways to relapse: the neurobiology of drug- and stress-induced relapse to drug-taking. J Psychiatry Neurosci 25:125–136. [PMC free article] [PubMed] [Google Scholar]
- Sung KW, Choi S, Lovinger DM (2001). Activation of group I mGluRs is necessary for induction of long-term depression at striatal synapses. J Neurophysiol 86:2405–2412. [DOI] [PubMed] [Google Scholar]
- Swanson CJ, Kalivas PW (2000). Regulation of locomotor activity by metabotropic glutamate receptors in the nucleus accumbens and ventral tegmental area. J Pharmacol Exp Ther 292:406–414. [PubMed] [Google Scholar]
- Swanson CJ, Baker DA, Carson D, Worley PF, Kalivas PW (2001). Repeated cocaine administration attenuates group I metabotropic glutamate receptor-mediated glutamate release and behavioral activation: a potential role for Homer. J Neurosci 21:9043–9052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweatt JD (2004). Mitogen-activated protein kinases in synaptic plasticity and memory. Curr Opin Neurobiol 14:311–317. [DOI] [PubMed] [Google Scholar]
- Thomas MJ, Beurrier C, Bonci A, Malenka RC (2001). Long-term depression in the nucleus accumbens: a neural correlate of behavioral sensitization to cocaine. Nat Neurosci 4:1217–1223. [DOI] [PubMed] [Google Scholar]
- Valjent E, Pages C, Herve D, Girault JA, Caboche J (2004). Addictive and non-addictive drugs induce distinct and specific patterns of ERK activation in mouse brain. Eur J Neurosci 19:1826–1836. [DOI] [PubMed] [Google Scholar]
- Valjent E, Pascoli V, Svenningsson P, Paul S, Enslen H, Corvol JC, Stipanovich A, Caboche J, Lombroso PJ, Nairn AC, Greengard P, Herve D, Girault JA (2005). Regulation of a protein phosphatase cascade allows convergent dopamine and glutamate signals to activate ERK in the striatum. Proc Natl Acad Sci USA 102:491–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanhoose AM, Emery M, Jimenez L, Winder DG (2002). ERK activation by G-protein-coupled receptors in mouse brain is receptor identity-specific. J Biol Chem 277:9049–9053. [DOI] [PubMed] [Google Scholar]
- Walker DL, Toufexis DJ, Davis M (2003). Role of the bed nucleus of the stria terminalis versus the amygdala in fear, stress, and anxiety. Eur J Pharmacol 463:199–216. [DOI] [PubMed] [Google Scholar]
- Watabe AM, Carlisle HJ, O'Dell TJ (2002). Postsynaptic induction and presynaptic expression of group 1 mGluR-dependent LTD in the hippocampal CA1 region. J Neurophysiol 87:1395–1403. [DOI] [PubMed] [Google Scholar]
- Weitlauf C, Egli RE, Grueter BA, Winder DG (2004). High-frequency stimulation induces ethanol-sensitive long-term potentiation at glutamatergic synapses in the dorsolateral bed nucleus of the stria terminalis. J Neurosci 24:5741–5747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson RI, Nicoll RA (2001). Endogenous cannabinoids mediate retrograde signalling at hippocampal synapses. Nature 410:588–592. [DOI] [PubMed] [Google Scholar]
- Winder DG, Egli RE, Schramm NL, Matthews RT (2002). Synaptic plasticity in drug reward circuitry. Curr Mol Med 2:667–676. [DOI] [PubMed] [Google Scholar]
- Wolf ME, Sun X, Mangiavacchi S, Chao SZ (2004). Psychomotor stimulants and neuronal plasticity. Neuropharmacology 47:[Suppl 1], 61–79. [DOI] [PubMed] [Google Scholar]
- Zakharenko SS, Zablow L, Siegelbaum SA (2002). Altered presynaptic vesicle release and cycling during mGluR-dependent LTD. Neuron 35:1099–1110. [DOI] [PubMed] [Google Scholar]
- Zimmer A, Zimmer AM, Hohmann AG, Herkenham M, Bonner TI (1999). Increased mortality, hypoactivity, and hypoalgesia in cannabinoid CB1 receptor knockout mice. Proc Natl Acad Sci USA 96:5780–5785. [DOI] [PMC free article] [PubMed] [Google Scholar]