

Abstract
Glutamate excitotoxicity and complement attack have both been implicated separately in the generation of tissue damage in multiple sclerosis and in its animal model, experimental autoimmune encephalomyelitis. Here, we investigated whether glutamate receptor activation sensitizes oligodendrocytes to complement attack. We found that a brief incubation with glutamate followed by exposure to complement was lethal to oligodendrocytes in vitro and in freshly isolated optic nerves. Complement toxicity was induced by activation of kainate but not of AMPA receptors and was abolished by removing calcium from the medium during glutamate priming. Dose–response studies showed that sensitization to complement attack is induced by two distinct kainate receptor populations displaying high and low affinities for glutamate. Oligodendrocyte death by complement required the formation of the membrane attack complex, which in turn increased membrane conductance and induced calcium overload and mitochondrial depolarization as well as a rise in the level of reactive oxygen species. Treatment with the antioxidant Trolox and inhibition of poly(ADP-ribose) polymerase-1, but not of caspases, protected oligodendrocytes against damage induced by complement. These findings indicate that glutamate sensitization of oligodendrocytes to complement attack may contribute to white matter damage in acute and chronic neurological disorders.
Keywords: kainate receptors, glutamate, complement, calcium, oxidative stress, oligodendrocytes
Introduction
Complement is a central component of the innate immune system in protection against invading pathogens and in clearance of potentially damaging debris. Complement consists of soluble and membrane proteins that interact with one another in a regulated manner. Complement activation culminates in the formation of the membrane attack complex (MAC), also referred to as C5b-9, a protein complex that damages membranes and causes cell leakage as well as cell death (Muller-Eberhard, 1986). Sublytic levels of C5b-9 can also induce a variety of biological responses, such as the release of proinflammatory mediators (Kilgore et al., 1997), entry into the cell cycle (Rus et al., 1996), and either resistance to (Soane et al., 1999) or induction of (Nauta et al., 2002) apoptosis.
Both the lack of certain complement components and an excessive or inappropriate complement activation are causes of serious illnesses, demonstrating the requirement for a delicate balance of complement activation and regulation for health. The CNS by itself can shape an immune and inflammatory response, and brain cells, including astrocytes, microglia, neurons, and oligodendrocytes, can synthesize complement proteins to recognize and kill pathogens while preserving normal cells (Levi-Strauss and Mallat, 1987; Barnum, 1995; Scolding et al., 1998; Gasque et al., 2000; Hosokawa et al., 2003). Complement has been implicated in diverse human neurodegenerative disorders, including Alzheimer's disease (McGeer et al., 1989; Shen and Meri, 2003), Huntington's disease (Singhrao et al., 1999), and Rasmussen's syndrome (Whitney et al., 1999). A wealth of evidence has implicated the complement in models of demyelination in vitro (Appel and Bonstein, 1964), in animal models of multiple sclerosis (MS) as experimental allergic encephalomyelitis (EAE) (Linington et al., 1989; Nataf et al., 2000; Mead et al., 2002), and also in the disease in humans (Yam et al., 1980; Mollnes et al., 1987; Compston et al., 1989; Storch et al., 1998; Lucchinetti et al., 2000). Deposition of MAC was shown at the edges of active plaques, suggesting a close association with ongoing pathology (Storch et al., 1998). Recently, a study of newly forming lesion in patients with relapsing and remitting MS found that oligodendrocytes throughout the affected tissue appear apoptotic and that myelin sheaths stain positively for activated complement, including MAC deposition (Barnett and Prineas, 2004).
One mechanism of cellular injury unique to the CNS is excitotoxicity. Excitotoxic damage is produced after accumulation of excess extracellular glutamate and subsequent overstimulation of glutamate receptors (Choi, 1998). Dysregulation of glutamate homeostasis occurs in EAE, and treatment with antagonists of glutamate receptors results in substantial amelioration of both tissue damage and the neurological deficits associated with this experimental condition (Pitt et al., 2000). Notably, glutamate levels are increased in acute lesions and normal-appearing white matter in patients with MS (Srinivasan et al., 2005), a feature that correlates with altered glutamate homeostasis in lesions and with oligodendroglial and axonal damage (Werner et al., 2001).
Together, these data implicate both complement activation and glutamate excitotoxicity in the pathology of demyelination. Previous studies have shown that brief activation of ionotropic glutamate receptors sensitizes neurons to complement attack (Xiong and McNamara, 2002). Oligodendrocytes also express functional ionotropic glutamate receptors (Verkhratsky and Steinhauser, 2000) and, like neurons, are liable to be damaged by excessive glutamate signaling in vitro and in vivo (Yoshioka et al., 1996; Matute et al., 1997; McDonald et al., 1998; Sánchez-Gómez et al., 2003). In the present study, we investigated whether sublethal excitotoxic insults sensitize oligodendrocytes to complement attack. Our results indicate that glutamate, via activation of kainate receptors, primes oligodendrocytes in vitro and in situ for complement attack, a process that requires calcium (Ca2+) influx and reactive oxygen species (ROS) generation.
Materials and Methods
Materials.
DMEM for culture was from Invitrogen (Barcelona, Spain), CNTF and neurotrophin-3 were from Peprotech (London, UK), and other culture reagents were purchased from Sigma (St. Louis, MO). l-Glutamic acid, NMDA, 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX), thapsigargin (Thp), paraquat, and Trolox (Sigma), GYKI53655 [1-(4-aminophenyl)-3-methylcarbamyl-4-methyl7,8-methylenedioxy-3,4-dihydro-5H-2,3-benzodiazepine] (ABX, Radeberg, Germany), AMPA, quisqualate, DCG IV [(2S,2′R,3′R)-2-(2′,3′-dicarboxycyclopropyl)glycine], and l-AP-4 (Tocris Cookson, Bristol, UK), ZVAD-F (N-benzyloxycarbonyl-Val-Ala-Asp-fluoromethyl ketone) (Enzyme System Products, Dublin, CA), and DPQ (3,4-dihydro-5-[4-(1-piperidinyl)butoxy]-1(2H)-isoquinolinone) (Calbiochem, La Jolla, CA) were first dissolved in distilled water (l-glutamic acid, NMDA, DCG IV, CNQX, and paraquat), DMSO (GYKI53655, ZVAD-F, thapsigargin, and DPQ), or an equimolar solution of NaOH (AMPA, l-AP-4, quisqualate, and Trolox) and were added to culture medium to achieve the desired final concentration. Rabbit complement [lyophilized Low-Tox Complement (LTC); Cedarlane, Hornby, Ontario, Canada) was reconstituted in cold distilled water, diluted 1:12 in culture medium or artificial CSF (aCSF), and filtered immediately before use.
Oligodendrocyte cultures.
Primary cultures of oligodendrocytes derived from the optic nerves of 12-d-old Sprague Dawley rats were obtained as described previously (Sánchez-Gómez and Matute, 1999). Cells were seeded into 24-well plates bearing poly-d-lysine-coated glass coverslips (12 mm diameter) at a density of 10–15 × 103 cells per well and were maintained at 37°C and 5% CO2 in a chemically defined medium (Sato's medium), and fresh medium was added every day. Cells were used 2–4 d later for different experiments, and, at this time, cultures were composed of at least 98% O4/galactocerebroside (GalC)+ cells (Alberdi et al., 2002).
Toxicity assays.
After 2 d in culture, oligodendrocytes were incubated with different concentrations of glutamate receptor agonists or paraquat in culture medium for 10 min or 3 h, respectively, and then with fresh culture medium in the absence or presence of LTC or with heat-inactivated LTC (HI-LTC) (56°C, 30 min) for 24 h. For assays with glutamate receptor antagonists, the antioxidant Trolox, or inhibitors of caspases and poly(ADP-ribose) polymerase-1 (PARP-1), cells were pretreated with CNQX (30 μm) and GYKI53655 (100 μm) for 10 min and ZVAD-F (50 μm), DPQ (30 μm), and Trolox (10 μm) for 30 min before application of glutamate. In all cases, the drugs were present during incubation with glutamate (10 min). Oligodendrocyte viability was assessed by loading cells with 1 μm calcein-AM (Invitrogen) for 30 min, and fluorescence was measured using a Synergy-HT fluorimeter (Bio-Tek Instruments, Winooski, VT). Cell death was revealed with propidium iodide (20 μg/ml). All experiments were performed in duplicate, and the values provided here are the average of at least three independent experiments.
Electrophysiology.
Whole-cell recordings were performed at room temperature on an inverted microscope (CK-40; Olympus Optical, Tokyo, Japan) using the EPC-7 patch-clamp amplifier (HEKA Elektronik, Lambrecht, Germany). Recordings were low-pass filtered at 2 kHz, digitized at 5 kHz, and stored as data files on a computer using the pClamp 8.2 program (Molecular Devices, Palo Alto, CA) for later analysis. The extracellular bath solution contained the following (in mm): 140 NaCl, 5.4 KCl, 2 CaCl2, 1 MgCl2, and 10 HEPES, pH 7.3. Patch-clamp pipettes (3–5 MΩ) were filled with a solution containing the following (in mm): 140 KCl, 2 CaCl2, 2 MgCl2, 10 HEPES, 11 EGTA, and 2 Na-ATP, pH 7.3. Changes in membrane conductance were measured by applying 10 mV depolarizing pulses (0.1 Hz, 2 s width) at a holding membrane potential of −70 mV. Results were normalized to currents recorded at time t = 0 before LTC addition (t = 1 min).
Measurement of [Ca2+]i.
[Ca2+]i was calculated as described previously (Alberdi et al., 2002; Sánchez-Gómez et al., 2003). Cells were incubated with fura-2 AM (Invitrogen) at 5 μm in culture medium for 30–45 min at 37°C. Cells were washed of excess fura-2 AM and were pretreated with vehicle, AMPA (0.1 μm to 2 mm), glutamate (0.1 μm to 5 mm), glutamate together with 30 μm CNQX, or glutamate with 100 μm GYKI53655 for 10 min. Then, coverslips were placed in a chamber with LTC or HI-LTC with or without CaCl2 or with Thp in incubation buffer (HBSS containing 20 mm HEPES, pH 7.4, 10 mm glucose, and 2 mm CaCl2). The incubation chamber was mounted on the stage of a Zeiss (Oberkochen, Germany) inverted epifluorescence microscope (Axiovert 35) equipped with a 150 W xenon Polychrome IV lamp (T.I.L.L. Photonics, Martinsried, Germany) and a Plan Neofluar 40× oil immersion objective (Zeiss). Cells were visualized with a high-resolution digital black and white CCD camera (Orca; Hamamatsu Photonics Iberica, Spain), and image acquisition and data analysis were performed using the AquaCosmos software program (Hamamatsu Photonics). At the end of the assay, in situ calibration was performed with the successive addition of 10 μm ionomycin and 2 m Tris/50 mm EGTA, pH 8.5. The [Ca2+]i concentration was estimated by the 340/380 ratio method, using a Kd value of 224 nm. Data were analyzed with Excel (Microsoft, Seattle, WA) and Prism 4.0 software (GraphPad Software, San Diego, CA).
Measurement of mitochondrial potential.
Oligodendrocyte cultures were exposed to glutamate and subsequently incubated with culture medium or LTC, as above. Thereafter, cells were loaded with 100 nm tetramethylrhodamine ethyl ester (TMRE) (Invitrogen) and 1 μm calcein-AM for 10 min. Calcein fluorescence, a common agent used to test cell viability, was used here to quantify the number of cells within the reading field. Fluorescence was measured using a Synergy-HT fluorimeter (Bio-Tek Instruments), and data were expressed as a percentage of TMRE/calcein fluorescence with respect to controls. All experiments (n = 6) were performed at least in triplicate and plotted as mean ± SEM.
Free radical measurement.
Oligodendroglial cultures were exposed to glutamate with or without antagonists as described previously. Subsequently, cells were loaded with 40 μm 2′,7′-dichlorodihydrofluorescein diacetate (DCFDA) (Invitrogen) and 1 μm calcein red–orange fluorescence (control dye) to assay the levels of free radicals. Fluorescence was measured as above, and values were plotted as the percentage of DCFDA/calcein red–orange fluorescence with respect to controls. All experiments (n = 6) were performed at least in triplicate and plotted as mean ± SEM.
Immunocytochemistry in oligodendrocyte cultures.
Cells were exposed to glutamate for 10 min and then to LTC for 30 min. The cultures were washed three times with PBS and incubated with rabbit anti-human C5b-9 (2 μg/ml; Calbiochem) for 30 min at 4°C. Subsequently, cells were washed, fixed with 4% paraformaldehyde in 0.1 m PBS for 10 min, washed with PBS, and incubated for 1 h with a biotinylated goat anti-rabbit IgG (1:200; Vector Laboratories, Burlingame, CA). Immunodetection was performed using streptavidin, Alexa Fluor-488 conjugate (1:200; Invitrogen) for 60 min. After three washes, cultures were processed with mouse IgG3 anti-GalC (5 μg/ml; ChemiconTemecula, CA), and the antibody was visualized by red-fluorescence Alexa Fluor-594 goat anti-mouse IgG (1:200; Invitrogen) and mounted in Glycergel (DakoCytomation, Barcelona, Spain). Images were acquired with an Olympus Optical Fluoview FV500 confocal microscope using sequential acquisition to avoid overlapping of fluorescent emission spectra. In all cases, cells were counterstained with Hoechst 33258 to simultaneously evaluate nuclear condensation. Oligodendrocytes stained with antibodies to C5b-9 were counted by a blinded observer, and counts were plotted as a percentage of the total number of cells in the cultures. All experiments (n = 3) were performed in duplicates.
Isolation of optic nerves, treatment, and immunohistochemistry.
Adult male Sprague Dawley rats were anesthetized deeply with chloroform and then decapitated. Optic nerves were freed from their meninges in aCSF (in mm: 126 NaCl, 3 KCl, 2 MgSO4, 26 NaHCO3, 1.25 NaH2PO4, and 2 CaCl2.2H2O) supplemented with 10 mm glucose. Then, optic nerves were pretreated with glutamate for 10 min in the absence or presence of AMPA/kainate receptor antagonists, followed by incubation for 3 h at 37°C with oxygen-saturated aCSF alone or containing LTC. After incubation, the nerves were fixed for 2 h in 4% paraformaldehyde in 0.1 m PBS, pH 7.2, cryoprotected in 15% sucrose, and frozen. To evaluate cellular death, cryostat sections (10 μm thick) were mounted onto glass slides and stained with Hoechst 33258 to identify cells with nuclear condensation. To visualize the formation of MAC in oligodendrocytes, cryostat sections were processed for double immunofluorescence using rabbit antibody to anti-human C5b-9 (2 μg/ml; Calbiochem) and mouse monoclonal antibody against anti-adenomatous polyposis coli (APC) (Ab-7, 1 μg/ml; Oncogene Sciences, Boston, MA). Rabbit antibody was detected with biotinylated goat anti-rabbit IgG (1:200; Vector Laboratories) followed by streptavidin, Alexa Fluor-488 conjugate (1:200; Invitrogen) for 60 min, and monoclonal antibody was detected with red-fluorescence Alexa Fluor-594 goat anti-mouse IgG (1:200; Invitrogen). Cells were counterstained with Hoechst 33258 to simultaneously evaluate nuclear condensation. Stained sections were observed and photographed with an Olympus Optical Fluoview FV500 confocal microscope. Quantification of MAC immunofluorescence by densitometry was done with the Image Tool 2.03 software, and values are expressed as the percentage of background subtracted from the integrated density.
Statistical analysis.
Student's paired and non-paired t test was used to assess significance for data with two groups. Statistical p values are reported in figure legends.
Results
Glutamate sensitizes cultured oligodendrocytes to complement attack
Oligodendrocytes express functional ionotropic glutamate receptors (Verkhratsky and Steinhauser, 2000). Activation of these receptors on glial cells produces a large variety of biological responses, which suggests that they may have an active role in brain signaling. In neurons, a minimal activation of ionotropic glutamate receptors sensitizes cells to complement attack (Xiong and McNamara, 2002), but, in oligodendrocytes, this biological response has not been explored. Here, we tested whether glutamate modulates the sensitivity of oligodendrocytes in culture to complement attack. Cells were challenged with glutamate for 10 min and subsequently incubated with or without rabbit complement (LTC, 1.2 mg/ml) for 24 h. Glutamate (10 μm, 10 min) alone caused very low toxicity to oligodendrocytes (2.6 ± 0.6% of control, vehicle treated) after 24 h incubation in culture medium (Sato's medium). In contrast, pretreatment with glutamate followed by addition of LTC acted synergistically to kill oligodendrocytes (16.3 ± 2.2% of control, vehicle plus LTC treated) (Fig. 1A). Toxicity required complement activation because cell death was abolished (2.1 ± 0.7% of control, vehicle treated) by HI-LTC (56°C, 30 min) (Fig. 1A).
Figure 1.
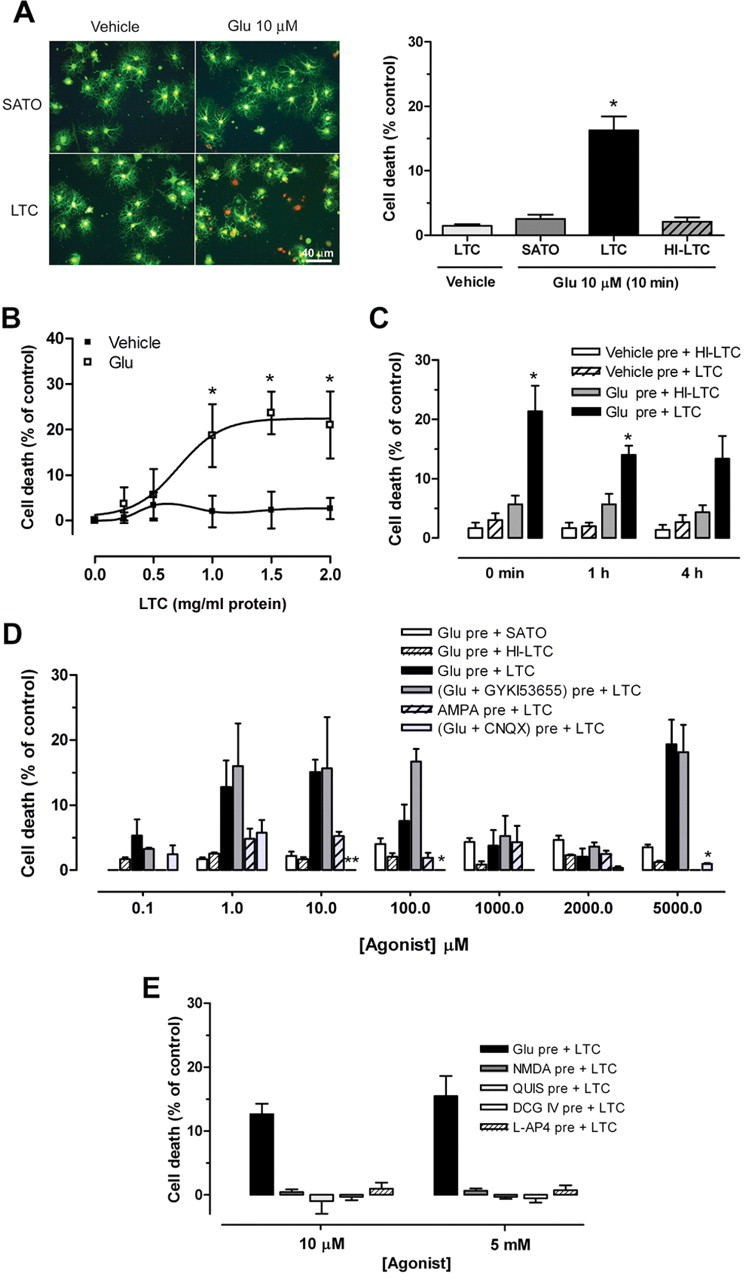
Glutamate activation of kainate receptors sensitizes cultured oligodendrocytes to complement attack. A, Left, Oligodendrocyte viability (green calcein fluorescence) and death (propidium iodide red fluorescence) 24 h after pretreatment with vehicle or glutamate (Glu; 10 μm) for 10 min, followed by exposure to culture medium (SATO) or complement (LTC). Scale bar, 40 μm. Right, Histogram illustrating oligodendrocyte death. Note that HI-LTC was ineffective. Values are the mean ± SEM of duplicates from at least three different experiments.*p < 0.01, paired Student's t test. B, Dose–response curves showing LTC toxicity (0.25–2.00 mg/ml; 24 h) after pretreatment with vehicle or glutamate (10 μm, 10 min). Values are the mean ± SEM of duplicates from at least three different experiments. *p < 0.05 compared with vehicle pretreatment plus LTC, non-paired Student's t test. C, Time course of glutamate-mediated sensitization to complement attack. Cells were pretreated with vehicle or glutamate (10 μm, 10 min) and then incubated with LTC or HI-LTC immediately (0 min), 1 h, or 4 h later. Values are the mean ± SEM of duplicates from at least three different experiments. *p < 0.05 compared with vehicle pre + LTC or Glu pre + HI-LTC, paired Student's t test. D, Dose-dependent cell death after exposure to glutamate or AMPA as above. Cell death is maximal at 10 μm and 5 mm glutamate and is prevented by the AMPA/kainate antagonist CNQX (30 μm) but not by the AMPA-selective antagonist GYKI53655 (100 μm). In contrast, AMPA pretreatment induces very low toxicity. LTC alone (1.2 mg/ml) produced no detectable cell death. *p < 0.05, **p < 0.01 compared with Glu pre + LTC, paired Student's t test. E, NMDA and agonists of group I, II, and III metabotropic glutamate receptors quisqualate (QUIS), DCG IV, and l-AP-4, respectively, all at low and high concentrations, do not sensitize oligodendrocytes to complement toxicity. Values are the mean ± SEM of duplicates from at least three experiments.
To analyze the sensitivity of oligodendrocytes to complement-induced cell death, cultures were incubated with increasing concentrations of LTC after exposure to either vehicle or glutamate (10 μm, 10 min). Treatment with LTC was toxic to oligodendrocytes in a concentration-dependent manner only after pretreatment with glutamate (Fig. 1B).
We next examined the time course of glutamate sensitization to complement attack. Oligodendrocytes were exposed to vehicle or glutamate as above and then to LTC or HI-LTC added for 24 h immediately after the agonist or at 1 and 4 h later. Oligodendrocyte cell death was only observed after pretreatment with glutamate and incubation with LTC at all of the time points examined (Fig. 1C). However, toxicity was only significant when LTC was added immediately or at 1 h after glutamate pretreatment (21.3 ± 4.3 and 14.0 ± 1.2% of control, respectively; p < 0.05).
The major types of glutamate receptors expressed by oligodendrocytes are AMPA and kainate receptors (Verkhratsky and Steinhauser, 2000). Thus, we initially studied whether or not selective activation of either receptor type sensitizes to complement attack. Oligodendrocytes were exposed to LTC after pretreatment with a wide range of AMPA or glutamate concentrations, alone or in the presence of CNQX (30 μm), an AMPA/kainate receptor antagonist, or GYKI53655 (100 μm), an AMPA receptor antagonist (Fig. 1D). We observed that the profile of glutamate sensitization to complement attack and the resulting oligodendrocyte death was dose dependent and showed two components that peaked at 10 μm glutamate (15.1 ± 1.9% of control, vehicle treated) and at 5 mm glutamate (19.4 ± 3.7% of control, vehicle treated). This effect was blocked by CNQX (30 μm) but not by GYKI53655 (100 μm). In contrast, selective activation of AMPA receptors by AMPA pretreatment did not sensitize oligodendrocytes to complement attack (Fig. 1D). Glutamate alone or followed by HI-LTC showed low toxicity in the conditions assayed (Fig. 1D).
To further characterize the pharmacology of the complement attack initiated by glutamate, we also primed oligodendrocytes with selective agonists of NMDA receptors and metabotropic glutamate receptors before exposure to LTC, as above. However, none of the agonists at the concentrations assayed (10 μm and 5 mm) induced complement attack and cell death (Fig. 1E). Collectively, these results demonstrate that a brief glutamate exposure is sufficient to sensitize oligodendrocytes to complement attack. Sensitization is mediated by two subtypes of kainate receptor with high and low affinity for glutamate but not by AMPA, NMDA, and metabotropic glutamate receptor activation.
Complement increases plasma membrane conductance and produces massive Ca2+ influx in glutamate-pretreated oligodendrocytes
Glutamate pretreatment sensitizes neurons to the membrane attack pathway of the complement cascade (Xiong and McNamara, 2002). MAC inserts into and through the membrane, creating a channel resulting in ion flux and ultimately osmotic lysis (Morgan, 1999). The earliest detectable event after MAC attack is a large influx of Ca2+ into the cell (Sala-Newby et al., 1998), which ultimately causes cell death (Kim et al., 1987). For this reason, we analyzed membrane conductance as a consequence of MAC deposition in glutamate-pretreated oligodendrocytes by whole-cell patch-clamp recordings. Cells were pretreated with glutamate (5 mm) for 10 min, and membrane currents were subsequently recorded in the presence of LTC for 40 min. After 10–12 min, LTC induced a progressive increase in membrane conductance in oligodendrocytes pretreated with glutamate, which did not occur in the absence of glutamate pretreatment (vehicle) or when the cells were exposed to HI-LTC (Fig. 2A). Glutamate exposure for 10 min did not alter membrane conductance over the time period examined (data not shown). The currents activated by LTC were dependent on the previous kainate receptor stimulation because they were blocked by CNQX but not by the AMPA antagonist GYKI53655 (Fig. 2A).
Figure 2.
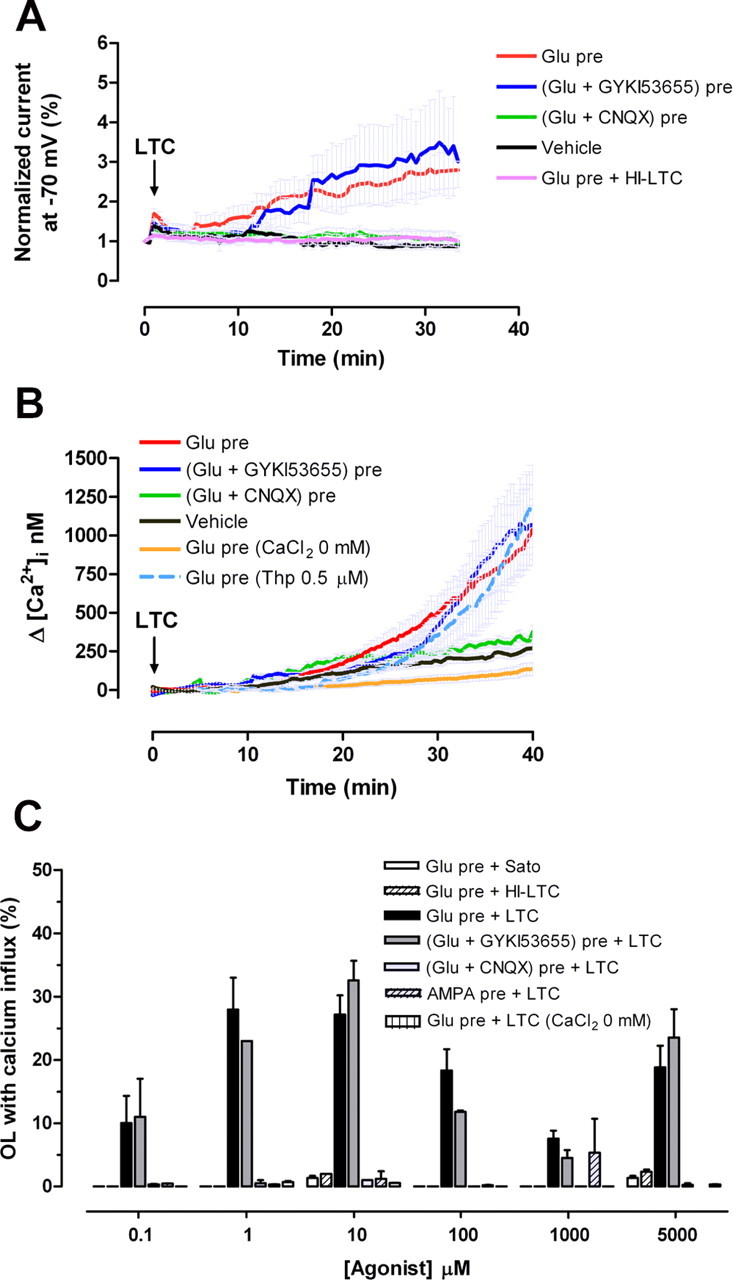
Complement increases plasma membrane conductance and [Ca2+]i in glutamate-sensitized oligodendrocytes. A, Effect of complement (LTC) on oligodendroglial membrane conductance after pretreatment with glutamate (Glu; 5 mm for 10 min; n = 15). Current induced by LTC after glutamate pretreatment was blocked in the presence of CNQX (30 μm; n = 6) but not in the presence of GYKI53655 (100 μm; n = 8). Note that the membrane conductance of oligodendrocytes exposed exclusively to LTC (vehicle; n = 7) or to HI-LTC (n = 6) was not altered. B, Increase in [Ca2+]i in oligodendrocytes exposed to glutamate (Glu; 10 μm) and then to LTC is blocked by CNQX (30 μm) and by removal of Ca2+ from the medium but not by GYKI53655 (100 μm) or by Thp (0.5 μm). The curves illustrate average ± SEM responses of 15–52 cells from at least five different experiments. C, Percentage of oligodendrocytes showing massive Ca2+ influx during LTC incubation subsequent to exposure to different concentrations of glutamate or AMPA. Values represent average ± SEM of at least five experiments (n = 750 cells).
We next monitored by microfluorimetry whether or not the increase in membrane conductance induced by complement attack raised [Ca2+]i. Indeed, we observed that [Ca2+]i increased irreversibly at 15 min of LTC incubation in oligodendrocytes pretreated with 10 μm glutamate alone or together with 100 μm GYKI53655 (Fig. 2A,B). In contrast, [Ca2+]i did not increase substantially during LTC incubation of oligodendrocytes pretreated with glutamate applied together with 30 μm CNQX (Fig. 2B,C). In addition, inhibition of endoplasmic reticulum Ca2+-ATPase with thapsigargin did not alter the [Ca2+]i increase in glutamate-pretreated oligodendrocytes during LTC incubation (Fig. 2B). However, removal of Ca2+ from the medium during LTC incubation abolished the [Ca2+]i increase in glutamate-primed cells (Fig. 2B,C), indicating that the [Ca2+]i increase was caused by Ca2+ influx and not by its release from the intracellular stores.
To further characterize Ca2+ influx induced by glutamate and LTC, we performed dose–response experiments as illustrated above for the toxicity assays and then quantified the number of responding oligodendrocytes (Fig. 2C). The percentage of oligodendrocytes showing Ca2+ influx were maximal at 1–10 μm glutamate (27.9 ± 5 and 27.1 ± 3% of cells, respectively) and at 5 mm glutamate (18.8 ± 3.5% of cells). All responses were blocked by pretreatment of glutamate with CNQX, but not with GYKI53655, and by removing Ca2+ from the medium. AMPA and glutamate alone or followed by HI-LTC were ineffective at all the concentrations assayed.
Together, these data indicate that a brief activation of high- and low-affinity kainate receptors marks oligodendrocytes for complement attack and that this results in an increase in membrane conductance and a massive Ca2+ influx. These features point to MAC as the effector mechanism mediating complement attack.
Glutamate pretreatment induces MAC formation in oligodendrocytes in culture
The previous results provide evidence that glutamate pretreatment induces MAC formation in the oligodendrocyte membrane. Next, we investigated the presence of rabbit complement activation products resulting in MAC formation on the surface of oligodendrocytes by double immunofluorescence. Cells were treated with either glutamate (10 μm; 10 min) or vehicle, incubated with LTC for 30 min, and subsequently processed for double immunofluorescence with antibodies to the C5b-9 complex and the sulfatide ester of GalC, an oligodendrocyte marker. Pretreatment with glutamate induced extensive C5b-9 immunoreactivity on the membrane of oligodendrocyte cell bodies and processes in a large population of cells (43.2 ± 1.6% of total cells). In contrast, C5b-9 staining after vehicle pretreatment was present in much fewer oligodendrocytes (24.7 ± 1.7% of total cells) (Fig. 3A,B).
Figure 3.
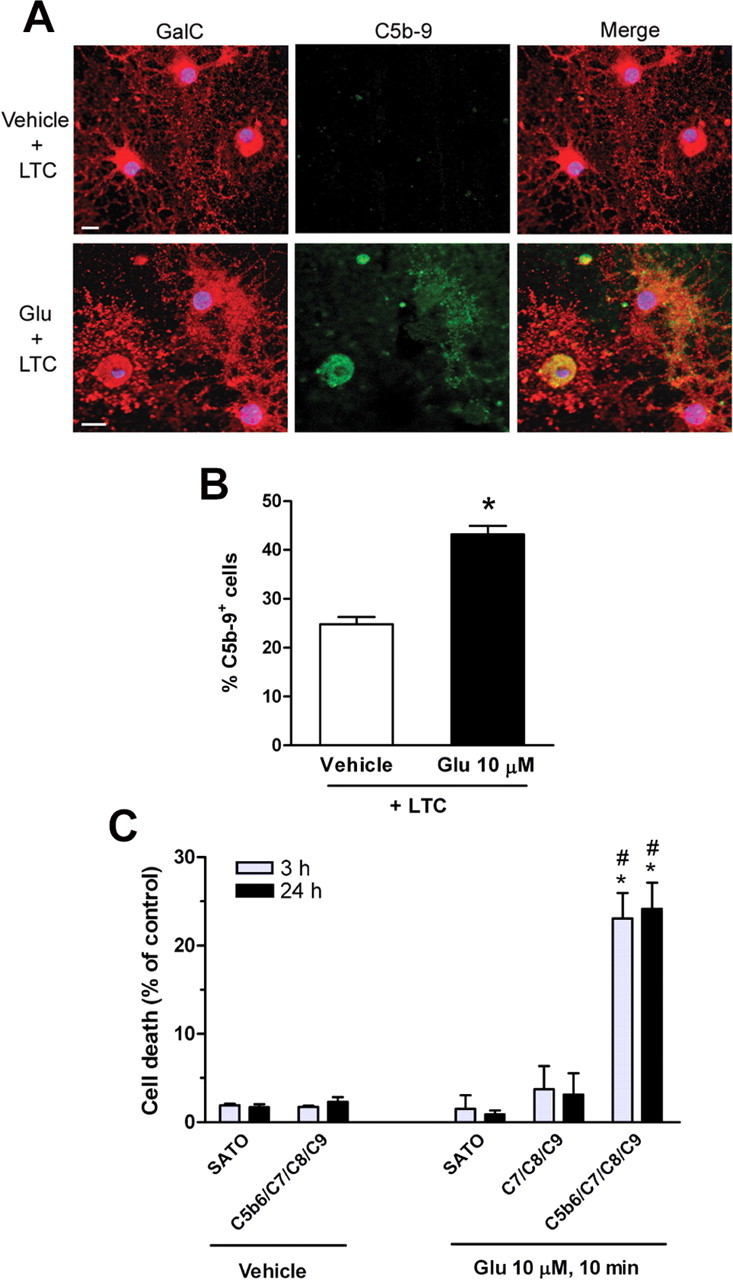
Glutamate pretreatment induces MAC formation in oligodendrocytes in culture. A, Cells were pretreated with vehicle (top) or glutamate (Glu; bottom; 10 μm, 10 min) and subsequently incubated with rabbit complement (LTC; 30 min). Cells are stained with antibodies to oligodendrocyte-type marker GalC (red; left) and to C5b-9 (green; middle) immunoreactivity. Colocalization of GalC and C5b-9 immunofluorescence is shown in merged images (yellow; right). Pretreatment with glutamate specifically induces MAC formation in oligodendrocyte. Blue fluorescence denotes oligodendrocyte nuclei as revealed by Hoechst 33258. Scale bar, 10 μm. B, Histogram represents the average ± SEM of double-labeled cells from duplicates of three different experiments. *p < 0.0001 compared with vehicle pretreatment plus LTC, non-paired Student's t test. C, Cell death at 3 and 24 h after pretreatment with vehicle or glutamate (10 μm, 10 min) followed by addition of MAC proteins C5b-6, C7, C8, and C9 (0.5 μg/ml each). Values represent average ± SEM. *p < 0.005 compared with vehicle pretreatment followed by C5b-6 plus C7/C8/C9; #p < 0.01 compared with glutamate pretreatment followed by C7/C8/C9.
To further assess the activation of the membrane attack pathway in glutamate-sensitized oligodendrocytes, we added purified proteins contributing to MAC formation after pretreatment with either glutamate or vehicle and examined cell death at 3 and 24 h of incubation. Thus, sequential addition of C5b-6, C7, C8, and C9 showed cytotoxic effects in glutamate (23.1 ± 2.9 and 24.2 ± 3% of control at 3 and 24 h after incubation, respectively) but not in vehicle-pretreated oligodendrocytes. Moreover, MAC formation was observed in cultures treated with glutamate followed by addition of C5b-6, C7, C8, and C9 and was absent after omitting incubation with C5b-6, the first component required to initiate the membrane attack pathway (Fig. 3C) (supplemental Fig. 1, available at www.jneurosci.org as supplemental material).
Ca2+ influx and ROS generation mediate glutamate sensitization to complement attack
A previous study has described that excitotoxic sensitization in neurons to complement attack requires extracellular Ca2+ and generation of ROS (Xiong and McNamara, 2002). In oligodendrocytes, glutamate receptor agonists AMPA and kainate evoke a rise in the level of intracellular Ca2+ and ROS specifically triggered by receptor activation (Sánchez-Gómez et al., 2003). First, we determined whether Ca2+ influx was necessary to induce glutamate sensitization of oligodendrocytes to complement attack. Cells were prestimulated with glutamate (10 μm and 5 mm) in normal or Ca2+-free media for 10 min and subsequently incubated with LTC for 24 h. Oligodendrocyte death induced by complement was totally blocked by removing Ca2+ from the incubation buffer, indicating that sensitization requires the presence of Ca2+ in the extracellular media (Fig. 4A). Next, we determined whether ROS generation during glutamate sensitization determines complement toxicity. Indeed, treatment with the antioxidant Trolox (10 μm) for 30 min before and during treatment with glutamate (both at 10 μm and 5 mm) abolished cell death caused by LTC (Fig. 4A).
Figure 4.

Glutamate sensitization to complement attack in oligodendrocytes requires Ca2+ influx and the generation of ROS. A, Toxicity induced by complement (LTC) is prevented if glutamate (Glu) is added to the medium in the absence of Ca2+ or in the presence of the antioxidant Trolox (10 μm). Values represent the mean ± SEM of duplicates from three different experiments. #p < 0.005 compared with Glu pre + SATO; *p < 0.05, **p < 0.01 compared with Glu pre + LTC; paired Student's t test. B, Paraquat-sensitized oligodendrocytes to complement attack. Cells were exposed to vehicle or 100 μm paraquat for 3 h and then exposed to HI-LTC or LTC for an additional 24 h. *p < 0.005 compared with vehicle pretreatment followed by LTC, paired Student's t test.
To determine whether ROS are sufficient to sensitize oligodendrocytes to complement attack, cells were pretreated with paraquat (100 μm, 3 h), an agent capable of generating ROS intracellularly (Patel et al., 1996) and subsequently incubated with LTC or HI-LTC for 24 h. Paraquat-pretreated cells followed by incubation with HI-LTC produced a modest oligodendroglial cell death (4.9 ± 2.3% of control, paraquat plus Sato's medium) (Fig. 4B). However, pretreatment of paraquat caused a remarkable sensitization to the toxic effects of LTC (28.54 ± 2.30% of control, paraquat plus Sato's medium) (Fig. 4B). Together, these results demonstrate that Ca2+ influx and ROS generation are specific mediators in glutamate sensitization to complement attack in oligodendrocytes and that intracellular ROS formation is sufficient to sensitize the cells to complement attack.
Complement produces mitochondrial dysfunction, oxidative stress, and PARP-1 activation in glutamate-pretreated oligodendrocytes
The mitochondrion is a major regulator of [Ca2+]i, and alterations in Ca2+ homeostasis in this organelle triggers cell demise. In the present study, we detected a large Ca2+ influx into glutamate-pretreated oligodendrocytes after complement attack. To determine whether a mitochondrial dysfunction provoked by complement would be involved in oligodendrocyte death, we monitored by fluorimetry mitochondrial potential and intracellular ROS accumulation in oligodendrocytes, using TMRE and DCFDA, respectively. Cells were pretreated with glutamate at 10 μm or 5 mm for 10 min and then incubated with culture medium (Sato's medium) or LTC. Reductions in TMRE fluorescence (for each concentration, respectively, 85.9 ± 2.4 and 89.2 ± 2.7% of control, vehicle-treated cells) were observed after 45–60 min of LTC incubation (Fig. 5A). Also, intracellular ROS accumulation (for each concentration, respectively, 120 ± 8.4 and 113 ± 4% of control, vehicle-treated cells) was detected after 30 min of LTC incubation (Fig. 5B). In contrast, no changes in TMRE and DCFDA fluorescence at these time points were detected in glutamate-pretreated oligodendrocytes that were left in Sato's medium with no LTC (Fig. 5A,B). In addition, ROS generation was significantly prevented by glutamate coincubation with CNQX (30 μm) but not with GYKI53655 (100 μm) (Fig. 5C).
Figure 5.

Complement causes mitochondrial depolarization (A), oxidative stress (B, C), and PARP-1-mediated apoptosis in glutamate (Glu)-sensitized oligodendrocytes (D). Each culture measurement was normalized to calcein fluorescence (A) or calcein red–orange fluorescence (B, C), indicators of cell viability, and 100% represents control values in the absence of agonist. D, PARP-1 inhibition with DPQ (30 μm) reduces cell death, whereas the pan-caspase inhibitor ZVAD-F (50 μm) was ineffective. In turn, the antioxidant Trolox (10 μm) fully protected oligodendrocytes from complement (LTC) toxicity. Values represent mean ± SEM of triplicates from six different experiments. A, B, *p < 0.05 compared with Glu pre + SATO. C, *p < 0.05 compared with vehicle + LTC; #p < 0.05 compared with Glu pre + LTC. D, *p < 0.05, **p < 0.01 compared with Glu pre + LTC. All by paired Student's t test.
Damage to mitochondria can result in both caspase-dependent and caspase-independent cell death. To characterize whether caspases can mediate oligodendrocyte cell death caused by complement, we used a pan-caspase inhibitor, ZVAD-F (50 μm), that blocks caspase proteolytic activity. Maximal activation of high- and low-affinity kainate receptors with glutamate followed by LTC caused cell death that was not significantly diminished by ZVAD-F (Fig. 5D). However, inhibition of PARP-1 with DPQ (30 μm) greatly attenuated toxicity (5.1 ± 3.2 and 2.0 ± 2.0% cell death after sensitization with 10 μm and 5 mm glutamate, respectively) (Fig. 5D). Notably, the antioxidant Trolox (10 μm) protected oligodendrocytes from complement damage, indicating that ROS generation is relevant to toxicity (Fig. 5D).
These results indicate that complement, in glutamate-pretreated oligodendrocytes, produces mitochondrial depolarization, oxidative stress, and PARP-1-dependent cell death.
Glutamate sensitizes oligodendrocytes in situ to MAC formation
Next, we studied whether optic nerve oligodendrocytes in situ are a target of complement attack after glutamate sensitization. Isolated optic nerves were preincubated with oxygen-saturated aCSF containing vehicle or 0.1–10 μm glutamate for 10 min at 37°C and were then incubated in aCSF alone or containing LTC for 3 h. The number of cells with changes in nuclear integrity, such as reduction in nuclear volume together with condensed chromatin, was quantified using Hoechst 33258 staining. Brief exposure of glutamate (10 min) did not cause cell death in optic nerves incubated in the absence of LTC (Fig. 6A, −LTC). In contrast, pretreatment with glutamate (0.1–10 μm) increased complement-dependent cell death (Fig. 6A, +LTC). The highest number of cells with condensed chromatin was observed in nerves pretreated with 1 μm glutamate (Fig. 6A). Pretreatment with CNQX, but not with GYKI53655, prevented chromatin condensation, indicating that the LTC-associated cell death was specifically triggered by kainate receptor activation (Fig. 6B,C).
Figure 6.
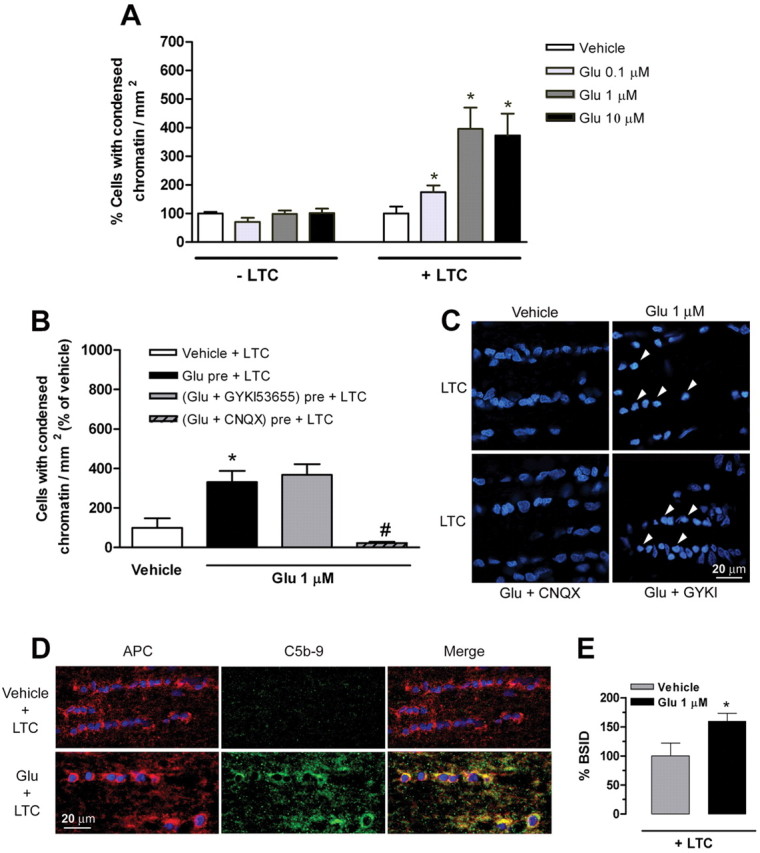
Glutamate sensitizes oligodendrocytes in situ to complement attack. A, Maximal sensitization to complement (LTC) attack is observed after pretreatment of isolated optic nerves with 1 μm glutamate (Glu). Values (average ± SEM) were obtained from at least three optic nerves. *p < 0.01 by non-paired Student's t test. B, Glutamate with CNQX, but not with GYKI536555, significantly reduces the number of cells with condensed chromatin in optic nerves incubated with LTC. Values (average ± SEM) were obtained from at least three optic nerves. *p < 0.05 compared with vehicle + LTC; #p < 0.001 compared with Glu pre + LTC. C, Representative images showing nuclei of optic nerve cells labeled with Hoechst 33258 after exposure to artificial CSF (vehicle) and glutamate alone or in conjunction with CNQX and GYKI53655, followed by LTC. Notice that cells with condensed chromatin (arrows) are only evident in conditions in which kainate receptors are activated by glutamate. D, Formation of MAC (green) around the oligodendrocytes (red) is absent in optic nerves treated with vehicle (top row) but occurs in nerves incubated with glutamate (bottom row), as visualized with anti-C5b-9 (green) and APC antibodies (red). Colocalization of APC and C5b-9 immunofluorescence is shown in merged images (yellow; right). Nuclei are labeled with Hoechst 33258. E, Quantification of MAC immunofluorescence in nerves treated with vehicle or glutamate followed by LTC. Values (average ± SEM) were obtained from four optic nerves and at least 20 fields per nerve were evaluated.*p < 0.05 by paired Student's t test. Scale bars, 20 μm.
We then assessed by immunocytochemistry the presence of MAC in optic nerves treated with either vehicle or 1 μm glutamate for 10 min and subsequent LTC incubation for 3 h. Double immunofluorescence showed that the majority of cells affected by MAC formation (green; C5b-9) were mature interfascicular oligodendrocytes (red; APC+) (Fig. 6D), in optic nerves pretreated with 1 μm glutamate. In contrast, low staining was present in nerves treated with vehicle and LTC (Fig. 6D,E). Together, these results indicate that glutamate, via activation of kainate receptors, sensitizes oligodendrocytes in situ to complement attack.
Discussion
The results reported here show that a brief and nontoxic activation of kainate receptors by glutamate sensitizes oligodendrocytes in vitro and in situ to complement attack. The mechanism by which glutamate induces sensitization requires Ca2+ influx and the generation of ROS. Complement-mediated toxicity is initiated by MAC, which induces an increase of the plasma membrane conductance, causing Ca2+ overload and loss of mitochondrial membrane potential, as well as the acute generation of ROS. Finally, we observed that complement induces apoptosis in sensitized oligodendrocytes by a caspase-independent, PARP-1-dependent pathway.
Sublethal activation of kainate receptor sensitizes oligodendrocytes to complement attack
Oligodendrocytes express primarily AMPA and kainate receptors (Verkhratsky and Steinhauser, 2000), and excessive activation of these receptors causes oligodendrocyte death (Yoshioka et al., 1996; Matute et al., 1997; McDonald et al., 1998; Sánchez-Gómez et al., 2003). In the current study, we found that a brief exposure to nontoxic concentrations of glutamate rendered oligodendrocytes sensitive to complement attack, an effect that is blocked by CNQX, an antagonist of AMPA and kainate receptors, and not triggered by agonists of NMDA and metabotropic glutamate receptors. Notably, sensitization by glutamate occurs even if AMPA receptors are selectively blocked with GYKI53655, indicating that kainate receptor activation is required to initiate complement attack. In line with these findings, another study found that glutamate pretreatment transforms neurons from a complement-resistant to a complement-sensitive cell type via activation of NMDA and non-NMDA ionotropic glutamate receptors (Xiong and McNamara, 2002); however, the pharmacology of the latter receptors was not defined in that study.
Our previous immunocytochemical observations showed that kainate receptors in oligodendrocytes derived from the optic nerve are made up of glutamate receptors GluR6 and GluR7 and kainate receptors KA1 and KA2 (Sánchez-Gómez and Matute, 1999). In addition, excitotoxic insults mediated by kainate receptors indicated that two distinct populations of receptors with high and low affinity trigger oligodendrocyte death (Sánchez-Gómez and Matute, 1999). Consistent with these findings, the dose–response profile of sensitization by glutamate in oligodendrocytes revealed that activation of either high- or low-affinity kainate receptors can induce complement attack.
Sensitization depended on the influx of Ca2+ during kainate receptor activation because removing this cation from the culture medium prevented sensitization. This feature is consistent with the high permeability of kainate receptors to Ca2+ (Alberdi et al., 2002). Thus, sustained activation of these receptors causes Ca2+ overload, which depolarizes mitochondria and generates oxidative stress and, ultimately, oligodendrocyte demise (Sánchez-Gómez et al., 2003). In addition, sensitization to complement attack was induced by paraquat, an agent that produces ROS, and prevented by the antioxidant Trolox, suggesting that it is mediated by oxidative stress. Likewise, sensitization of neurons on glutamate receptor activation depends on the generation of ROS because a superoxide dismutase mimetic abolishes the induction of complement attack (Xiong and McNamara, 2002). However, the mechanisms that link oxidative stress to complement attack are still unknown.
MAC deposition and mitochondrial dysfunction cause oligodendrocyte death
Complement can exert cytotoxic effects by diverse mechanisms. In oligodendrocytes sensitized with glutamate, complement toxicity is induced by MAC deposition into the plasma membrane only if all of the components of MAC are available. MAC deposition occurs rapidly after sensitization and causes within minutes an increase in the plasma membrane conductance accompanied by a rise in [Ca2+]i. This effect was blocked by removal of Ca2+ from the culture media and indicates that MAC forms a functional pore in the membrane of sensitized oligodendrocytes. These results are in agreement with previous studies in non-neural cells that found an immediate rise in [Ca2+]i during the earliest phase of C5b-9 complex formation (Sala-Newby et al., 1998).
Lytic cell death, the classical role of complement and MAC, is associated with necrosis, a proinflammatory form of cell death in tissues in which the cell content is released to propagate inflammation (Bohana-Kashtan et al., 2004). MAC, among its many nonlytic effects, also plays a role in modulating apoptosis (Nauta et al., 2002). Apoptotic cell death does not involve the release of cell content and is considered to be non-inflammatory in nature. Active compounds generated during complement activation may exert either proapoptotic or anti-apoptotic effects. Both apoptotic and anti-apoptotic effects are likely to occur under sublytic conditions of complement activation (Bohana-Kashtan et al., 2004). In our study, exposure to complement of sensitized oligodendrocytes caused apoptosis by a caspase-independent mechanism that involves PARP-1 activation subsequent to Ca2+ overload, mitochondrial depolarization, and the production of ROS. In neurons, increased levels of free radicals and Ca2+ overload in mitochondria lead to the release into the cytoplasm of proapoptotic factors (Nicholls and Budd, 2000). The apoptosis-inducing factor, a caspase-independent death factor, is released from mitochondria and translocates to the nucleus, in which it induces lysis of chromatin and cell death by PARP-1 activation (Yu et al., 2002). The fact that PARP-1 inhibition rescues oligodendrocytes from complement toxicity strongly suggests that MAC, at sublytic conditions, kills oligodendrocytes by apoptosis involving a caspase-independent pathway.
Glutamate sensitization to complement attack and white matter damage
Excitotoxicity, an exclusive mechanism of the CNS, is implicated in neuronal death after insults such as ischemia and seizures (Choi, 1998). Likewise, white matter is vulnerable to enhanced glutamate signaling via AMPA/kainate receptor activation, as observed in perinatal ischemia (Follett et al., 2000), stroke (McCracken et al., 2002), traumatic injury (Xu et al., 2004), experimental allergic encephalomyelitis (Pitt et al., 2000; Smith et al., 2000), and MS (Pitt et al., 2003). The findings reported here extend the known deleterious effects of glutamate excitotoxicity to oligodendrocytes, under conditions in which nontoxic glutamate stimuli occur concomitantly with a derailment of the complement system within the CNS or after blood–brain barrier opening.
Cells of the human brain are capable of synthesizing most, if not all, of the complement proteins (Barnum, 1995). Oligodendrocytes and neurons are vulnerable to complement attack, a feature that is particularly observed in MS (Compston et al., 1989; Prineas et al., 2001; Schwab and McGeer, 2002) and in neurodegenerative conditions, including Alzheimer's, Pick's, Huntington's, and Parkinson's diseases (Yamada and McGeer, 1990). This vulnerability may be related to a deficiency of the ability to express complement regulatory proteins (Scolding et al., 1998). As a consequence, the simultaneous occurrence of excitotoxic and complement insults in vivo could act synergistically to destroy neurons (Xiong and McNamara, 2002). Furthermore, subtle, nontoxic increases in glutamate levels may be sufficient to initiate or aggravate complement attack in oligodendrocytes, as shown in the current study.
The present results may be relevant to lesion formation in MS, a disease in which many components of the complement machinery are present in CSF and in CNS tissue (Yam et al., 1980; Mollnes et al., 1987; Compston et al., 1989; Storch et al., 1998; Lucchinetti et al., 2000). Moreover, magnetic resonance spectroscopy has shown that glutamate concentration is elevated in acute lesions and in normal-appearing white matter (Srinivasan et al., 2005), probably as a consequence of impaired glutamate uptake (Werner et al., 2001). Importantly, MAC deposition occurs at the edges of active plaques (Storch et al., 1998) and in myelin sheaths within freshly forming lesions displaying numerous apoptotic oligodendrocytes (Barnett and Prineas, 2004), suggesting that a concomitant increase of glutamate levels and the presence of complement may indeed initiate lesion formation and/or contribute to damage in MS.
In summary, our results show that activation of kainate receptors sensitizes oligodendrocytes to complement toxicity and suggest that this mechanism is deleterious to white matter in neurological disorders with primary or secondary altered glutamate homeostasis and complement derailment.
Footnotes
This work was supported by grants from Ministerio de Educación y Ciencia, Ministerio de Sanidad y Consumo, Gobierno Vasco, and the University of País Vasco. E.A. is a Ramón y Cajal research fellow. We thank Dr. M. Mallat for his suggestions and comments on this manuscript.
References
- Alberdi E, Sánchez-Gómez MV, Marino A, Matute C (2002). Ca2+ influx through AMPA or kainate receptors alone is sufficient to initiate excitotoxicity in cultured oligodendrocytes. Neurobiol Dis 9:234–243. [DOI] [PubMed] [Google Scholar]
- Appel SH, Bonstein MB (1964). The application of tissue culture to the study of experimental allergic encephalomyelitis. II. Serum factors responsible for demyelination. J Exp Med 119:303–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnett MH, Prineas JW (2004). Relapsing and remitting multiple sclerosis: pathology of the newly forming lesion. Ann Neurol 55:458–468. [DOI] [PubMed] [Google Scholar]
- Barnum SR (1995). Complement biosynthesis in the central nervous system. Crit Rev Oral Biol Med 6:132–146. [DOI] [PubMed] [Google Scholar]
- Bohana-Kashtan O, Ziporen L, Donin N, Kraus S, Fishelson Z (2004). Cell signals transduced by complement. Mol Immunol 41:583–597. [DOI] [PubMed] [Google Scholar]
- Choi DW (1998). Glutamate neurotoxicity and diseases of the nervous system. Neuron 1:623–634. [DOI] [PubMed] [Google Scholar]
- Compston DA, Morgan BP, Campbell AK, Wilkins P, Cole G, Thomas ND, Jasani B (1989). Immunocytochemical localization of the terminal complement complex in multiple sclerosis. Neuropathol Appl Neurobiol 15:307–316. [DOI] [PubMed] [Google Scholar]
- Follett PL, Rosenberg PA, Volpe JJ, Jensen FE (2000). NBQX attenuates excitotoxic injury in developing white matter. J Neurosci 20:9235–9241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasque P, Dean YD, McGreal EP, VanBeek J, Morgan BP (2000). Complement components of the innate immune system in health and disease in the CNS. Immunopharmacology 49:171–186. [DOI] [PubMed] [Google Scholar]
- Hosokawa M, Klegeris A, Maguire J, McGeer PL (2003). Expression of complement messenger RNAs and proteins by human oligodendrocytes. Glia 42:417–423. [DOI] [PubMed] [Google Scholar]
- Kilgore KS, Schmid E, Shanley TP, Flory CM, Maheswari V, Tramontini NL, Cohen H, Ward PA, Friedl HP, Warren JS (1997). Sublytic concentrations of the membrane attack complex of complement induce endothelial interleukin-8 and monocyte chemoattractant protein-1 through nuclear factor-kappa B activation. Am J Pathol 150:2019–2031. [PMC free article] [PubMed] [Google Scholar]
- Kim SH, Carney DF, Hammer CH, Shin ML (1987). Nucleated cell killing by complement: effects of C5b-9 channel size and extracellular Ca2+ on the lytic process. J Immunol 138:1530–1536. [PubMed] [Google Scholar]
- Levi-Strauss M, Mallat M (1987). Primary cultures of murine astrocytes produce C3 and factor B, two components of the alternative pathway of complement activation. J Immunol 139:2361–2366. [PubMed] [Google Scholar]
- Linington C, Morgan BP, Scolding NJ, Wilkins P, Piddlesden S, Compston DA (1989). The role of complement in the pathogenesis of experimental allergic encephalomyelitis. Brain 112:895–911. [DOI] [PubMed] [Google Scholar]
- Lucchinetti C, Bruck W, Parisi J, Scheithauer B, Rodriguez M, Lassmann H (2000). Heterogeneity of multiple sclerosis lesions, implications for the pathogenesis of demyelination. Ann Neurol 47:707–717. [DOI] [PubMed] [Google Scholar]
- Matute C, Sánchez-Gómez MV, Martínez-Millán L, Miledi R (1997). Glutamate receptor-mediated toxicity in optic nerve oligodendrocytes. Proc Natl Acad Sci USA 94:8830–8835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCracken E, Fowler JH, Dewar D, Morrison S, McCulloch J (2002). Grey matter and white matter ischemic damage is reduced by the competitive AMPA receptor antagonist, SPD 502. J Cereb Blood Flow Metab 22:1090–1097. [DOI] [PubMed] [Google Scholar]
- McDonald JW, Althomsons SP, Hyrc KL, Choi DW, Goldberg MP (1998). Oligodendrocytes from forebrain are highly vulnerable to AMPA/kainate receptor-mediated excitotoxicity. Nat Med 4:291–297. [DOI] [PubMed] [Google Scholar]
- McGeer PL, Akiyama H, Itagaki S, McGeer EG (1989). Activation of the classical complement pathway in brain tissue of Alzheimer patients. Neurosci Lett 107:341–346. [DOI] [PubMed] [Google Scholar]
- Mead RJ, Singhrao SK, Neal JW, Lassmann H, Morgan BP (2002). The membrane attack complex of complement causes severe demyelination associated with acute axonal injury. J Immunol 168:458–465. [DOI] [PubMed] [Google Scholar]
- Mollnes TE, Vandvik B, Lea T, Vartdal F (1987). Intrathecal complement activation in neurological diseases evaluated by analysis of the terminal complement complex. J Neurol Sci 78:17–28. [DOI] [PubMed] [Google Scholar]
- Morgan BP (1999). Regulation of the complement membrane attack pathway. Crit Rev Immunol 19:173–198. [PubMed] [Google Scholar]
- Muller-Eberhard HJ (1986). The membrane attack complex of complement. Annu Rev Immunol 4:503–528. [DOI] [PubMed] [Google Scholar]
- Nataf S, Carroll SL, Wetsel RA, Szalai AJ, Barnum SR (2000). Attenuation of experimental autoimmune demyelination in complement-deficient mice. J Immunol 165:5867–5873. [DOI] [PubMed] [Google Scholar]
- Nauta AJ, Daha MR, Tijsma O, van de Water B, Tedesco F, Roos A (2002). The membrane attack complex of complement induces caspase activation and apoptosis. Eur J Immunol 32:783–792. [DOI] [PubMed] [Google Scholar]
- Nicholls DG, Budd SL (2000). Mitochondria and neuronal survival. Physiol Rev 80:315–360. [DOI] [PubMed] [Google Scholar]
- Patel M, Day BJ, Crapo JD, Fridovich I, McNamara JO (1996). Requirement for superoxide in excitotoxic cell death. Neuron 16:345–355. [DOI] [PubMed] [Google Scholar]
- Pitt D, Werner P, Raine CS (2000). Glutamate excitotoxicity in a model of multiple sclerosis. Nat Med 6:67–70. [DOI] [PubMed] [Google Scholar]
- Pitt D, Nagelmeier IE, Wilson HC, Raine CS (2003). Glutamate uptake by oligodendrocytes: implications for excitotoxicity in multiple sclerosis. Neurology 61:1113–1120. [DOI] [PubMed] [Google Scholar]
- Prineas JW, Kwon EE, Cho ES, Sharer LR, Barnett MH, Oleszak EL, Hoffman B, Morgan BP (2001). Immunopathology of secondary-progressive multiple sclerosis. Ann Neurol 50:646–657. [DOI] [PubMed] [Google Scholar]
- Rus HG, Niculescu F, Shin ML (1996). Sublytic complement attack induces cell cycle in oligodendrocytes. J Immunol 15:4892–4900. [PubMed] [Google Scholar]
- Sala-Newby GB, Taylor KM, Badminton MN, Rembold CM, Campbell AK (1998). Imaging bioluminescent indicators shows Ca2+ and ATP permeability thresholds in live cells attacked by complement. Immunology 93:601–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sánchez-Gómez MV, Matute C (1999). AMPA and kainate receptors each mediate excitotoxicity in oligodendroglial cultures. Neurobiol Dis 6:475–485. [DOI] [PubMed] [Google Scholar]
- Sánchez-Gómez MV, Alberdi E, Ibarretxe G, Torre I, Matute C (2003). Caspase-dependent and caspase-independent oligodendrocyte death mediated by AMPA and kainate receptors. J Neurosci 23:9519–9528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwab C, McGeer PL (2002). Complement activated C4d immunoreactive oligodendrocytes delineate small cortical plaques in multiple sclerosis. Exp Neurol 174:81–88. [DOI] [PubMed] [Google Scholar]
- Scolding NJ, Morgan BP, Compston DA (1998). The expression of complement regulatory proteins by adult human oligodendrocytes. J Neuroimmunol 1:69–75. [DOI] [PubMed] [Google Scholar]
- Shen Y, Meri S (2003). Yin and Yang, complement activation and regulation in Alzheimer's disease. Prog Neurobiol 70:463–472. [DOI] [PubMed] [Google Scholar]
- Singhrao SK, Neal JW, Morgan BP, Gasque P (1999). Increased complement biosynthesis by microglia and complement activation on neurons in Huntington's disease. Exp Neurol 159:362–376. [DOI] [PubMed] [Google Scholar]
- Smith T, Groom A, Zhu B, Turski L (2000). Autoimmune encephalomyelitis ameliorated by AMPA antagonists. Nat Med 6:62–66. [DOI] [PubMed] [Google Scholar]
- Soane L, Rus H, Niculescu F, Shin ML (1999). Inhibition of oligodendrocyte apoptosis by sublytic C5b-9 is associated with enhanced synthesis of bcl-2 and mediated by inhibition of caspase-3 activation. J Immunol 163:6132–6138. [PubMed] [Google Scholar]
- Srinivasan R, Sailasuta N, Hurd R, Nelson S, Pelletier D (2005). Evidence of elevated glutamate in multiple sclerosis using magnetic resonance spectroscopy at 3 T. Brain 128:1016–1025. [DOI] [PubMed] [Google Scholar]
- Storch MK, Piddlesden S, Haltia M, Iivanainen M, Morgan P, Lassmann H (1998). Multiple sclerosis: in situ evidence for antibody- and complement-mediated demyelination. Ann Neurol 43:465–471. [DOI] [PubMed] [Google Scholar]
- Verkhratsky A, Steinhauser C (2000). Ion channels in glial cells. Brain Res Brain Res Rev 32:380–412. [DOI] [PubMed] [Google Scholar]
- Werner P, Pitt D, Raine CS (2001). Multiple sclerosis: altered glutamate homeostasis in lesions correlates with oligodendrocyte and axonal damage. Ann Neurol 50:169–180. [DOI] [PubMed] [Google Scholar]
- Whitney KD, Andrews PI, McNamara JO (1999). Immunoglobulin G and complement immunoreactivity in the cerebral cortex of patients with Rasmussen's encephalitis. Neurology 53:699–708. [DOI] [PubMed] [Google Scholar]
- Xiong Z, McNamara JO (2002). Fleeting activation of ionotropic glutamate receptors sensitizes cortical neurons to complement attack. Neuron 36:363–374. [DOI] [PubMed] [Google Scholar]
- Xu GY, Hughes MG, Ye Z, Hulsebosch CE, McAdoo DJ (2004). Concentrations of glutamate released following spinal cord injury kill oligodendrocytes in the spinal cord. Exp Neurol 187:329–336. [DOI] [PubMed] [Google Scholar]
- Yam P, Petz LD, Tourtellotte WW, Ma BI (1980). Measurement of complement components in cerebral spinal fluid by radioimmunoassay in patients with multiple sclerosis. Clin Immunol Immunopathol 17:492–505. [DOI] [PubMed] [Google Scholar]
- Yamada T, McGeer PL (1990). Oligodendroglial microtubular masses: an abnormality observed in some human neurodegenerative diseases. Neurosci Lett 120:163–166. [DOI] [PubMed] [Google Scholar]
- Yoshioka A, Bacskai B, Pleasure D (1996). Pathophysiology of oligodendroglial excitotoxicity. J Neurosci Res 46:427–437. [DOI] [PubMed] [Google Scholar]
- Yu SW, Wang H, Poitras MF, Coombs C, Bowers WJ, Federoff HJ, Poirier GG, Dawson TM, Dawson VL (2002). Mediation of poly(ADP-ribose) polymerase-1-dependent cell death by apoptosis-inducing factor. Science 297:259–263. [DOI] [PubMed] [Google Scholar]