

Abstract
This work describes the first successful oligodendrocyte-based cell therapy for presymptomatic arylsulfatase A (ARSA) null neonate mice, a murine model for human metachromatic leukodystrophy (MLD). We found that oligodendrocyte progenitors (OLPs) engrafted and survived into adulthood when transplanted in the neonatal MLD brain. Transplanted cells integrated nondisruptively, did not produce tumors, and survived as proteolipid protein- and MBP-positive postmitotic myelinating oligodendrocytes (OLs) intermingled with endogenous MLD OLs within the adult MLD white matter. Transplanted MLD mice had reduced sulfatide accumulation in the CNS, increased brain ARSA activity, and full prevention of the electrophysiological and motor deficits that characterize untreated MLD mice. Our results provide direct evidence that healthy OLPs can tolerate the neurotoxic accumulation of sulfatides that evolves during the postnatal development of the MLD brain and contribute to OL cell replacement to limit the accumulation of sulfatides and the evolution of CNS defects in this lysosomal storage disease mouse model.
Keywords: myelin, oligodendrocytes, migration, transplantation, leukodystrophy, demyelination
Introduction
Patients with metachromatic leukodystrophy (MLD), a genetic disease affecting the expression of arylsulfatase A (ARSA), show accumulation of sulfatides, oligodendrocyte (OL) dysfunction, loss of myelin, and progressive neurodegeneration (Gieselmann, 2003). Currently, transplantation of hematogenous stem cells has been used to limit clinical symptoms in MLD patients (Krivit et al., 1999a,b; Koc et al., 2002). However, any central remyelination therapy for MLD will face the fundamental problem of coping with global OL degeneration/dysfunction. Direct gene therapy to hippocampal neurons (Consiglio et al., 2001; Luca et al., 2005) and cerebellar neurons (C. Croci and E. Bongarzone, unpublished observations) of MLD mice led to a substantial reduction of neuronal degeneration, in association to long-range transport of lysosomal enzymes along axons (Neufeld and Fratantoni, 1970; Passini et al., 2002; Luca et al., 2005). However, direct gene therapy to restore ARSA activity of the entire population of OLs in the CNS remains a challenge. Alternatively, transplantation of migratory oligodendrocyte progenitor cells (OLPs), capable of nondisruptive integration within the population of endogenous OLs (i.e., mosaic distribution), might serve to therapeutically limit the progression of central degeneration and contribute to OL replacement.
OLPs have intrinsic and unique properties with respect to their differentiation potential in the brain and in vivo migration, making them appealing cells for central remyelination therapies of lysosomal storage diseases. OLPs are immature neural cells committed to the oligodendroglial lineage, which respond to several mitogens with increased cell proliferation (Barres et al., 1993; Calver et al., 1998; Redwine and Armstrong, 1998; Frost et al., 2003) and have a remarkable capacity for in vivo migration. Migration of OLPs depends on combined signals from chemoattractive cues such as PDGF-A (Baron et al., 2002), Notch signaling (Wang et al., 1998; Givogri et al., 2002, 2003), and interaction with extracellular matrix signals (Frost et al., 1996; Garcion et al., 2001). OLPs can be maintained for extended periods of time as proliferative precursors without significant loss of myelinogenic potential. OLPs are graftable, particularly in the neonatal CNS of myelin mutants, where they respond remarkably well to endogenous cues with migration, integration, and long-term survival as well as the capacity to myelinate areas in which endogenous remyelination fails (Lubetzki et al., 1988; Lachapelle et al., 1990). Several elegant studies have demonstrated the therapeutic value of OLP transplantation in myelin mutants (Duncan et al., 1988; Learish et al., 1999; Yandava et al., 1999; Magy et al., 2003; Crang et al., 2004; Duncan, 2005). These studies have indicated that the outcome of cell therapy in severe mutants such as Jimpy, Shiverer, and Twitcher mice strongly relates to the extent of central and peripheral compromise and the accumulation of neurotoxic molecules such as the deposit of aberrant protein or toxic lipid metabolites.
ARSA null mice display progressive sulfatide storage throughout the CNS, mild loss of myelin, and develop behavioral deficits in adulthood, constituting a murine model for late infantile/early adult forms of human MLD (Hess et al., 1996). MLD mice have not been evaluated as recipients of OLP-based central cell therapies. In this work, we successfully treated neonate MLD mice with healthy OLPs and limited the evolution of central deficits when transplanted MLD mice entered into adulthood.
Materials and Methods
Animals.
Knock-out MLD (ARSA−/−) mice and the corresponding wild type (WT; ARSA+/+) animals were used throughout this study. MLD mice were maintained in the San Raffaele Scientific Institute animal research facility by crossing homozygous offspring from carrier ARSA−/+ mice (Hess et al., 1996). All procedures were done under the protocols approved by the Animal Care and Use Committee of our institute in accordance with Italian law.
Preparation of OLP cultures from normal and MLD nervous tissue.
OLP cells were prepared as described previously from primary glial cultures from MLD or wild-type animals (Gard and Pfeiffer, 1989; Louis at al, 1992; Bongarzone et al., 1996). Cells were grown in glial defined medium consisting of DMEM/F12 supplemented with 5 mg/L insulin, 16.1 mg/L putrescine, 50 mg/L transferrin, 0.8 μg/ml sodium selenite, and 2.2 g/l sodium bicarbonate. OLPs were maintained in proliferative conditions by the addition of 30% of medium conditioned by B104 cells (a kind gift from Dr. J. De Vellis, University of California at Los Angeles, Los Angeles, CA) (Schubert et al., 1974; Louis et al., 1992; Luo and Miller, 1997).
In vitro genetic retroviral/lentiviral labeling and fluorescent labeling of OLPs.
OLPs were grown to confluence in 75 cm2 culture flask before cells were exposed to 106 colony-forming units of either β-galactosidase-retroviral (bag cells; CRL-9560; American Type Culture Collection, Manassas, VA) or green fluorescent protein-lentiviral (GFP-LV) vector, supplemented with 8 mg/ml polybrene. Cells were left overnight in the presence of each vector. Efficiency of transduction was evaluated by 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside (X-gal) staining or GFP fluorescence and estimated to be ∼90%. Alternatively, OLPs were incubated with PKH26 red fluorescent cell linker kit (Sigma, St. Louis, MO) as indicated by the manufacturer. This fluorescent dye is transported and accumulated in lipid regions of the plasma membrane, allowing visualization of myelinating processes.
Cell transplantation.
Postnatal day 2 (P2) newborn pups (MLD and wild type) were anesthetized by brief hypothermia and received 150,000 of labeled cells in 2 μl on the left ventricle at a rate of 0.5 μl/min using sterile needles attached to a 5 μl Hamilton syringe. The number of cells injected was based on previous studies from other laboratories (Franklin et al., 1996; Meng et al., 2003) and on unpublished preliminary experiments from our laboratory. Efforts were made to avoid aggregation of cells before infusion by resuspending cells right before injection in PBS without serum, to avoid shearing and clumping. Cell viability was checked right before the injections by trypan blue vital staining (we also checked cell viability in the leftover of cell suspension after the injections) and found it to be higher than 90%. Mice were warmed and quickly returned to their mothers. Transplanted animals were not subjected to immunosuppression. In our hands, the survival rate of this procedure was >95% of recipients. In some experiments, MLD and WT pups were transplanted with OLPs previously isolated from ARSA-deficient pups and labeled to express GFP.
Bromodeoxyuridine labeling.
Mice (two or three mice per group) received three intraperitoneal injections of bromodeoxyuridine (BrdU; 50 mg/kg body weight) at 3 h intervals 6 d, 14 d, and 1 year after transplants. Mice were killed the following day and processed for BrdU immunostaining as described below.
Immunocytochemistry.
The following antibodies were used in this study: antibodies against PDGFrα (1:100; Santa Cruz Biotechnology, Santa Cruz, CA); polysialylated neural cell adhesion molecule (PSA-NCAM; 1:50; Hybridoma Bank, Manassas, VA); β-galactosidase (1:1000; F. Hoffmann-La Roche, Basel, Switzerland); proteolipid protein (PLP; 1:20; AA3; a kind gift from Dr. S. Pfeiffer, University of Connecticut, Farmington, CT); MBP (1:300; Chemicon, Temecula, CA); neuronal-specific nuclear protein (NeuN; 1:100; Chemicon); GFAP (1:500; Chemicon); monocyte chemoattractant protein-1 (MCP-1; 1:200; Santa Cruz Biotechnology); A2B5, O4, and O1 (1:50; kind gifts from Dr. T. Gard, University of South Alabama, Mobile, AL); ARSA (1:500; kind gift from Dr. V. Gieselmann and U. Matzner, Rheinische Freidrich-Wilhelms Universität, Bonn, Germany); BrdU (1:200; Chemicon); and GFP (1:250; Santa Cruz Biotechnology). Mice were killed at different time points (from 1 d to 1 year after the transplant; three to five animals per group) by transcardiac perfusion with saline followed by 4% paraformaldehyde/PBS. Brain tissue was rapidly removed and postfixed for 24 h and processed for cryosectioning. Sections were mounted on slides or alternatively used for free-floating staining as described previously (Bongarzone et al., 1999). After incubation with appropriate primary antibodies, tissue was thoroughly rinsed in PBS and processed for peroxidase-diamino bencidine staining (Vector Laboratories, Burlingame, CA) or for confocal fluorescence microscopy. In some experiments, sections were incubated with 1 μg/ml of 4′,6′-diamidino-2-phenylindole (DAPI). BrdU staining was done after pretreatment with 2N HCl for 20 min followed by 0.1 m sodium borate buffer, pH 8.6, and incubation with monoclonal anti-BrdU.
Agarose migration assay.
OLPs were resuspended at 1 × 104 per 3 μl of medium containing 0.3% of low melting point agarose and maintained at 37°C (Milner et al., 1996). Three microliters of the cell suspension were plated at the bottom of wells in a 24 well plate percolated with poly-d-lysine (10 μg/ml) and left at 4°C until agarose hardened. Coronal slices (0.5 mm thick), sectioned from bregma 0 to bregma −5 of P2 MLD or wild-type brains, were prepared separately and maintained in HBSS medium on ice. Brain slices were sectioned in halves at the midline, and each tissue slice was positioned on the top of each well maintaining a distance of 5 mm from the OLP–agarose drops. Tissue easily attached to the poly-lysine-coated plastic for up to 3–4 d, time after which sections would start lifting. Three groups in triplicate (six wells each group) consisting of wild-type or MLD OLPs were maintained in the agarose drops with or without MLD tissue or wild-type tissue for 24 h before fixation of cells with 0.5% glutaraldehyde for 10 min and β-gal staining.
X-gal staining.
Tissue was fixed in 2% paraformaldehyde for 1 h and cryosectioned at 20 μm. Free-floating sections were preincubated in 0.01% sodium deoxycholate, 0.1 m NaPO4, pH 7.3, 2 mm MgCl2, and 0.02% IGPAL CA-630. Staining was performed at 37°C overnight with fresh X-gal stain solution [1 mg X-gal, 0.01% sodium deoxycholate, 0.1 m NaPO4, pH7.3, 5 mm K3Fe(CN)6, 5 mm K4Fe(CN)63H2O, 2 mm MgCl2, and 0.02% IGPAL CA-630, a nonionic detergent (Sigma)]. Sections were counterstained with neutral red, postfixed with 0.6% of paraformaldehyde for 15 min, dehydrated, and mounted.
β-Galactosidase activity.
β-Gal activity was determined using the β-Gal reporter assay kit (F. Hoffmann-La Roche) in brain homogenates (50 μg total protein) as recommended by the manufacturer. Incubating samples at 60°C for 1 h inactivated endogenous β-Gal activity. Proteins were measured using Bradford assay.
Flow cytometry.
MLD mice transplanted with GFP+ OLPs and untreated controls were killed 7 d after grafting and perfused with saline (three mice per group). Brains were dissected and mechanically dissociated. Cell suspension was filtered through a 40 μm cell strainer and cells centrifuged at 1500 rmp. Cell pellets were resuspended in fluorescence-activated cell sorting (FACS) solution (PBS without calcium and magnesium containing 2% FBS), and viable cells were counted using trypan blue. GFP+ cells were detected from an input of 106 cells (from whole-brain cell suspension) using FACScan (Becton Dickinson, Mountain View, CA). Results were expressed as percentage of GFP+ cells from the total number of cells transplanted.
Counting of exogenous myelinating OLs.
To calculate the percentage of exogenous LacZ-positive OLs from the total population of interfascicular OLs in the corpus callosum, 15 μm coronal sections from 1-year-old treated MLD brains (covering bregma 0 to bregma 1) were stained with anti-β-galactosidase antibodies as described above and examined with light microscopy. One of every four sections was used for toluidine blue histological staining for morphological identification of the entire population of interfascicular callosal OLs. Four random digital pictures were taken per section covering an area of 230 × 290 mm per picture, and the number of interfascicular OLs that expressed β-galactosidase contained in that area was counted. The average number of β-galactosidase-positive OLs was expressed as the percentage from the total number of interfascicular callosal OLs. Three treated MLD mice were used for this experiment. To calculate the number of MBP-positive myelin sheaths that coexpressed GFP (i.e., myelin produced by exogenous grafted OLs), brain sections from 1-year-old treated MLD mice were double-stained for GFP and MBP and observed by confocal microscopy. Four random digital pictures per each section (three or four serial sections from three treated MLD mice) were taken in the cortex and the striatum. Results are expressed as the percentage of double GFP/MBP-positive myelin sheath profiles from the total MBP positive myelin profiles.
ARSA enzyme assay and sulfatide deposit determination.
ARSA activity was determined as described previously (Luca et al., 2005). Brain coronal sections (20 μm) of 1-year-old transplanted and nontransplanted MLD were stained with 1% Alcian blue in 1N HCl for 30 min, dried with filter paper, dehydrated, and mounted. The number of sulfatide deposits that were Alcian blue positive were counted in different areas and expressed as percentage of deposits in treated respect nontreated MLD mice, representing the results from five animals per group. Counting was done on sections at the same anatomical level [correlated with the Paxinos and Franklin (1997) anatomical atlas for adult mouse brain].
Motor-evoked potential recording.
One-year-old untreated and treated MLD and wild-type mice (7–10 per group) were anesthetized with tribromoethanol (0.02 ml/g of body weight) and placed under a heating lamp to avoid hypothermia. Motor-evoked potentials (MEPs), muscle responses evoked by transcranial electrical stimulation of the motor cortex, were recorded as described by Dolcetta et al. (2005).
Rotarod test.
One-year-old untreated and treated MLD and wild-type mice (7–10 per group) were tested on an accelerating Rotarod apparatus (4–40 rpm in 5 min). Latencies in falling off the rod were recorded over 3 d with three consecutive trials per day. The average of each day performance was used for statistical comparison by two-way ANOVA using Scheffe's test as post hoc analysis.
Results
In vitro properties of the neural oligodendroglial progenitors
Healthy OLPs (i.e., with normal ARSA activity) were maintained at low density in proliferative conditions to favor the oligodendroglial phenotype, characterized by the expression of PSA-NCAM, PDGFrα, and immunolabeling with the monoclonal antibody A2B5 (Table 1). More than 90% of the cells differentiated into mature cellular profiles associated with elaborated and delicate processes after incubation in the presence of 1% FCS and T3 (Fig. 1A,B, Table 1). Differentiated cells acquired the expression of mature cell markers for OLs such as those recognized with antibodies for PLP, MBP, and galactocerebrosides (O1) (Table 1). If cells were incubated in 10% FCS, most of them rapidly acquired the morphology of astrocytes expressing GFAP (Table 1). Proliferating OLPs were easily transduced with a β-gal-retroviral vector (Fig. 1B) or a GFP-lentiviral vector (Fig. 1C) or alternatively, loaded with PKH26 dye, which specifically labels plasma membrane (Fig. 1D). None of these labeling methods affected the capacity of OLPs to differentiate in vitro (evidenced by MBP and PLP gene expression analysis; data not-shown) nor their in vitro survival and importantly allowed us to identify transplanted cells up to 1 year after the surgery, the longest time tested in this study.
Table 1.
In vitro phenotypic characterization of graftable progenitors
| Cell marker | Proliferating in B104 CM | Differentiated in T3/1% FCS | Differentiated in 10% FCS |
|---|---|---|---|
| PSA-NCAM | + | −/+ | − |
| PDGFrα | + | −/+ | − |
| A2B5 | + | − | −/+ |
| O4 | + | + | − |
| O1 | −/+ | + | − |
| MBP | − | + | − |
| PLP | − | + | − |
| GFAP | − | − | + |
| Tuj1 | − | − | − |
Progenitors were incubated with B104 conditioned medium (CM) to promote their proliferation or in low (1%) or high (10%) FCS to stimulate their differentiation. Low serum culture medium contained T3 hormone. Cells were then immunostained with cell-lineage-specific antibodies to study the acquired phenotype in the different incubation conditions. Positive and negative indicates presence or absence of immunolabeling, respectively. Results were replicated in three independent experiments. Tuj1, Class III β-tubulin.
Figure 1.
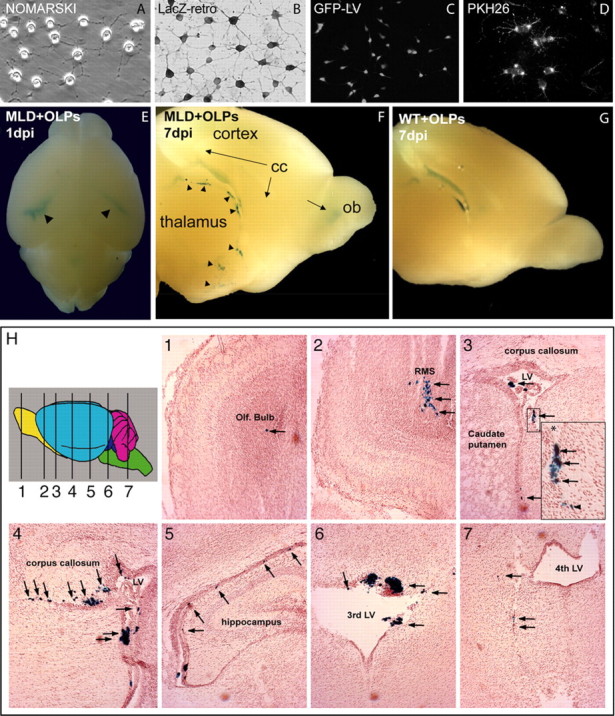
In vitro labeling of oligodendrocyte progenitors and detection of LacZ ARSA OLPs in different regions of the neonate MLD brain after transplantation. OLPs were grown on coated coverslips in differentiating conditions for 3 d. A, OLPs displayed multiple processes. OLPs were transduced with β-gal retroviruses (LacZ-retro; B), GFP lentiviruses (C), or loaded with the cell membrane dye PKH26 (D). Neonate (P2) MLD and healthy pups received a single injection of β-gal OLPs in their left ventricle. One day postinjection (dpi), OLPs moved within the ventricular system (E, arrowheads), migrating latter (7 dpi) toward the cortex, thalamus, and olfactory bulb (F, arrowheads and arrows). At 7 dpi, OLPs grafted in healthy controls remained primarily within the ventricular system (G) with few cells seen in the parenchyma. H1–H7, Serial coronal sections stained for β-gal and neutral red from an MLD mouse 1 d after surgery. Transplanted OLPs were detected along the rostrocaudal axis and within the olfactory bulb (H1), RMS (H2), lateral ventricle (LV), choroids plexus, SVZ (H3 and its higher-magnification inset, H4, H5), third ventricle (H6), and fourth ventricle (H7). The arrows depict LacZ OLPs that migrated within the brain parenchyma. n = 3 mice per group per time point. cc, Corpus callosum; ob or Olf. Bulb, olfactory bulb.
OLPs migrated extensively within the MLD neonatal brain
We evaluated the in vivo capacity of β-gal+ OLPs to survive and migrate within the brain of newborn MLD pups and their healthy controls. One day after the graft, we observed that β-gal+ cells distributed throughout the brain ventricular system, including the choroids plexus. However, β-gal+ cells migrated rapidly within the MLD brain parenchyma and less within the wild-type tissue (Fig. 1E, arrowheads). Many β-gal+ cells were seen exiting the ventricles 3 d after transplantation in the MLD brain, and by 7 d after grafting, LacZ+ cells were detected in different areas such as subcortical white matter, caudate–putamen, and olfactory bulbs (Fig. 1F, arrows and arrowheads). At this time point, wild-type recipients showed β-gal cells mostly associated to the ventricular system (Fig. 1G, but fewer β-gal+ cells were detected within the wild-type brain parenchyma. Coronal sections 1 d after the transplant allowed a more detailed localization of migratory β-gal+ cells within the MLD brain. We observed a large number of these cells lining on the walls of the ventricular system (Fig. 1H3,H4,H6). At this time point, many cells were found within the choroids plexus, whereas others migrated away from the ventricles and were seen within the brain parenchyma (Fig. 1H2,H4,H5,H7, arrows). OLPs were also detected in the subventricular zone (SVZ) (Fig. 1H3 and its inset), rostral migratory stream (RMS) (Fig. 1H2), and olfactory bulb (Fig. 1H1). In 1 year long-term experiments, grafted OLPs were detected in the corpus callosum (see Fig. 4P,S), cortex (see Fig. 4P,Q,T), striatum (see Fig. 4U), and cerebellum (see Fig. 4R).
Figure 4.

Transplanted ARSA+/+ OLPs differentiated into mature OLs within white matter territories. Transplanted MLD mice were analyzed by immunostaining for β-galactosidase (A–I, P–S) or β-gal histochemistry (J–M) and for the expression of myelin genes. A–C, Double immunohistochemistry performed at 7 d postinjection (dpi) against β-galactosidase (in green) and PDGFrα (in red) identified numerous double-positive cells (seen in yellow) in the SVZ (A), RMS (B), and olfactory bulb (C). D–E show background staining with secondary antibodies labeled with FITC and Texas Red (TR). F–I, OLPs loaded with the membrane dye PKH26 (PHK; in red) were abundant 30 d after transplant in the external capsule (F) with O4 immunostaining (G). H, The merged image on the three channels, PKH26, O4, and nuclear staining with DAPI. β-gal-positive OLPs were detected at corpus callosum (J–K) and cortex (L) 60 d after grafting. β-gal OLs were seen colocalizing with mature myelin specific gene expression domains such as MBP (K, arrows) and PLP (J). Most of the cells appeared connected to cell processes in a parallel disposition, typical of interfascicular OLs (I). Transplanted cells (filled arrow) did not show coexpression with the neuronal nuclear marker NeuN (L, open arrow). Few LacZ cells were found in close proximity to blood vessels and expressed the astrocytic marker GFAP (M, arrow). N–U, Images of WT (N), untreated MLD (O), and transplanted MLD mice (P–U) 1 year after treatment [year postinjection (ypi)]. β-Galactosidase-positive cells were detected in cortex (Q, T), cerebellar white matter (R), corpus callosum (S), and striatum (U). U, The number of MBP-positive myelin sheaths that coexpressed GFP fluorescence was counted per section in the cortex and the striatum. Results are expressed as percentage of the total MBP-positive myelin profiles in four random fields from three or four serial sections from three treated MLD mice. Scale bar: (in I) B–E, 25 μm; I–K, 100 μm.
Survival of OLPs is accompanied by early in vivo proliferation in the MLD brain
We conducted two transplantation experiments using either LacZ or GFP labeled OLPs to evaluate the efficiency of the engraftment (i.e., the percentage of surviving grafted cells over time at 1, 7, 15, and 30 d after transplant). In the first experiment, we measured the activity of the reporter gene β-gal and correlated it with the β-gal activity of the input of cells at the moment of the graft as an indirect parameter of grafted cells remaining within the host tissue. Figure 2A shows that ∼15% of the total input of β-gal activity remained in MLD brains 1 d after the grafting. However, whereas β-gal activity decreased to almost background levels in transplanted aged healthy controls, ∼8% of LacZ activity was detected in the brain of transplanted MLD mice at 7, 15, and 30 d after surgery. In the second experiment, we analyzed the absolute number of GFP-positive cells at 7 d after transplantation by FACS of total brain cell suspension. Figure 2B shows an example of a MLD brain 7 d after the transplant displaying ∼9% of GFP-positive cells, in agreement with the results obtained from β-gal activity.
Figure 2.
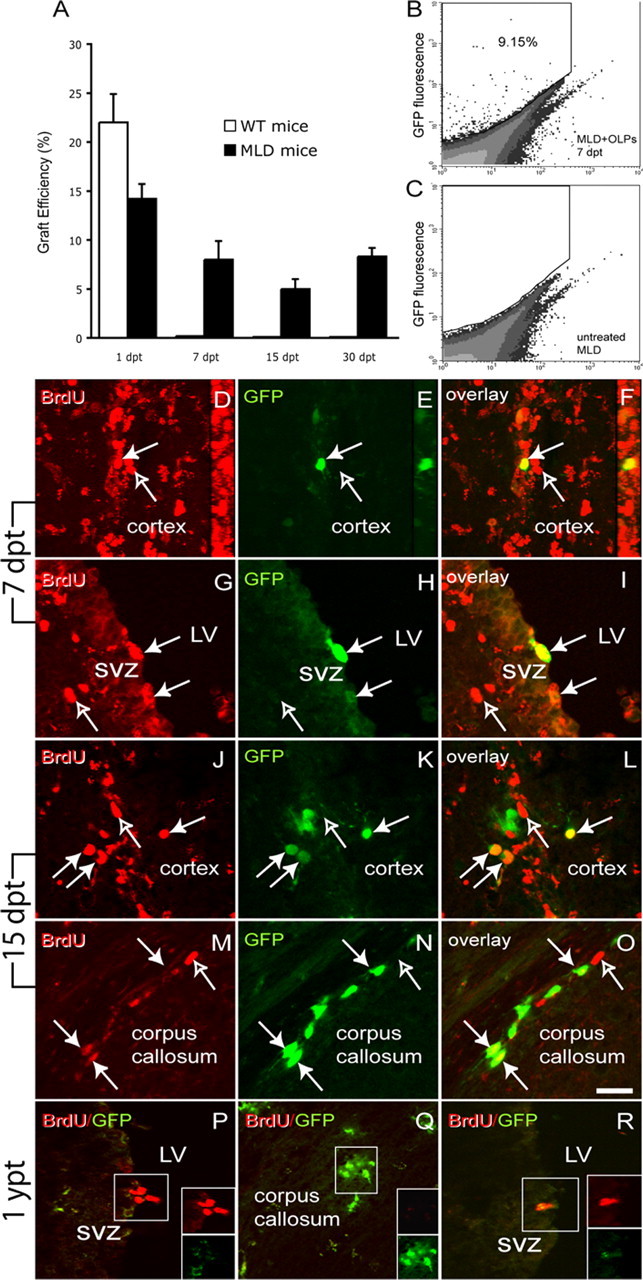
MLD brain promoted long-term survival and proliferation of OLPs. A, To estimate the efficiency of the engraftment, β-galactosidase activity was measured in total brain homogenates from transplanted MLD and healthy mice and correlated to the total enzyme activity of the input of cells at the moment of the graft. The plot shows that after an initial phase of cell death, cells transplanted in MLD pups survived better than in healthy mice, and ∼9% of the original amount of enzyme activity remained 1 month after surgery. B, C, We validated our findings on LacZ enzyme activity by FACS analysis of surviving GFP-positive cells in mice that received GFP+ OLPs. B, An example of the cytometric analysis for the presence of GFP+ cells from 7 d postinjection transplanted MLD mice, whereas C shows the FACS analysis of an untreated control. A total of 106 cells from total brain cell suspensions were acquired. To evaluate whether grafted OLPs continued to divide once transplanted in the neonate brain, we performed pulses of BrdU to detect DNA synthesis. D–R, Coronal sections of transplanted mice displaying GFP+ OLPs, counterstained with anti-BrdU antibodies (in red) and analyzed by confocal microscopy. Seven days after transplantation (D–I), GFP+ OLPs transplanted in the MLD brain were detected dividing within the cortex (D–F and the 90o rotation confocal reconstructions) and the SVZ (G–I). Fifteen days after the graft (J–O), most of the double BrdU/GFP-positive cells (filled arrows) were primarily detected in the cortex (J–L) and the corpus callosum (M–O). One year after the grafts (P–Q), BrdU+ cells were in the SVZ (P) and the hippocampal granule layer (data not shown). Transplanted GFP+ cells were found in white matter areas such as the corpus callosum (Q) but did not immunolabel with anti-BrdU, indicating their withdrawal from the cell cycle (Q). R, BrdU+ cells in the SVZ of an untreated 1-year-old MLD mouse. Scale bar: (in O) 25 μm. n = 3 mice per group per time point. dpt, Days post-transplantation; ypt, year post-transplantation; LV, lateral ventricle. Results are expressed as mean value ± SEM.
Next, MLD mice were transplanted with GFP+ OLPs and BrdU-pulsed on day 6, day 14, and 1 year after transplant to evaluate whether surviving OLPs continued to proliferate. We found numerous GFP+ OLPs to have incorporated BrdU 7 d after transplantation in the MLD brain (Fig. 2D–I, filled arrows; open arrows denote endogenous proliferating cells). There was no apparent anatomical preference for the grafted cells to proliferate, because we detected GFP/BrdU double-positive cells within the cortex (Fig. 2D–F), ventricular walls (Fig. 2G–I), and corpus callosum (data not shown). Fifteen days after the graft, proliferating BrdU/GFP double-positive OLPs were less frequent and primarily located within the cortex (Fig. 2J–L) and corpus callosum (Fig. 2M–O, filled arrows). One year after transplantation, we detected numerous GFP+ OLs within the cortex (data not shown), striatum (data not shown), corpus callosum (Fig. 2Q), and SVZ (Fig. 2P) but none of GFP+ cells incorporated BrdU. As positive controls, BrdU uptake was detected in endogenous dividing precursors within the SVZ of treated and untreated MLD mice (Fig. 2P,R). These results indicate that grafted cells withdrew from the cell cycle in long-term transplants.
MLD neural tissue is permissive to the migration of ARSA+/+ OLPs in vitro
Our results indicated that MLD brain supported long-term migration and survival of OLPs, whereas healthy brain was significantly less permissive to transplanted cells. We used the agarose drop assay to test the capacity of brain-derived tissue to influence the migration of OLPs in vitro to examine whether MLD and healthy brain might influence differently OLP migration in an in vitro system. Exposure of OLPs to MLD brain slices showed reproducible migration out of the agarose drop in a 1 d assay (Fig. 3A,B). In contrast, migration of OLPs out of the agarose drop was reduced when cells were coincubated with healthy neonatal brain samples (Fig. 3A,C), in support of the findings of neonatal transplantation experiments.
Figure 3.

The MLD brain promotes migration of OLPs and absence of ARSA is not a limiting factor for their migration. OLPs were plated on agarose drops and incubated in the presence of brain slices prepared from MLD or wild-type pups for 24 h before X-gal staining. A, The number of cells per area surrounding the agarose drop was counted. MLD tissue promoted a migration of wild-type or MLD OLPs out of the agarose drop, similar to the OLP migration seen in B104 conditioned medium. In contrast, healthy tissue promoted migration much less efficiently. B, C, Examples of β-gal-stained wild-type OLPs that migrated out the agarose drop after 24 h of culture with MLD (B) and wild-type (C) brain slices. The arrows demarcate the limit of the agarose drop. D–H, Transplanted MLD and wild-type mice were grafted with ARSA−/− OLPs and killed at 7 and 15 d after transplantation. Grafted cells migrated extensively to the corpus callosum (cc) and cortex (F) and SVZ (D, E). In contrast, ARSA−/− OLPs migrated poorly in wild-type brain, even 15 d after transplant (G–H). cp, Caudate–putamen; LV, lateral ventricle; dpi, days postinjection. Results are expressed as mean value ± SEM.
Absence of ARSA activity does not influence the migration properties of MLD OLPs
To determine whether impairment in the migration of MLD OLPs contributes to the pathogenesis of MLD, we measured the migration capacity of ARSA−/− OLPs after transplantation in wild-type and MLD neonatal brains. In agarose migration assays, we found that MLD OLPs behaved similarly to healthy OLPs (Fig. 3A). For the transplantation experiments, MLD OLPs were isolated from newborn knock-out mice and transduced with GFP-LV before grafting. Recipient mice were killed 7 and 15 d after transplantation to evaluate their migratory patterns. We found that ARSA−/− OLPs were able to migrate extensively in the MLD brain, with similar migration patterns to those of healthy OLPs (Fig. 3D–F). Grafted ARSA−/− OLPs migrated to the SVZ (Fig. 3D,E) and corpus callosum (Fig. 3F) between 7 and 15 d after the grafting. However, ARSA−/− OLPs migrated poorly and remained nearby the area of the graft in wild-type brains (Fig. 3G,H) without integration in white matter areas. ARSA−/− OLPs were detected 15 d after the graft (the longest time point in this experiment), adopting extensive cell processes and expressing MBP (data not shown), indicating their capacity to differentiate into mature OLs.
Grafted progenitors differentiated into mature myelinating OLs, expressed myelin genes, and integrated in myelinated areas of the MLD brain
Many LacZ+ OLPs moved into the SVZ–RMS–olfactory bulb system where most of them expressed PDGFrα, an early marker for the oligodendroglial lineage 7 d after transplantation (Fig. 4A–C). Double-positive β-gal/PDGFrα precursors and β-gal/O4 OLs were also detected in other areas such as the hippocampal dentate gyrus, the wall of the third ventricle, corpus callosum, and external capsule of MLD mice (data not shown). At later time points (30 d after transplant), most of OLPs were found in white matter areas such as the external capsule (Fig. 4F–H) coexpressing myelin sphingolipids recognized by the monoclonal antibody O4. Figure 4I shows PKH26-loaded OLs connected to processes adopting a parallel orientation to axons within the corpus callosum, characteristic of interfascicular OLs. Sixty days after transplant, numerous β-gal+ cells residing within the corpus callosum (Fig. 4J,K) coexpressed mature OL markers such as PLP (Fig. 4J) and MBP (Fig. 4K). Grafted cells (or their progeny) were detected 1 year after the transplant (Fig. 4P–S) using anti-β-Gal antibodies. β-Gal-positive cells showed delicate and elaborated processes, characteristic of myelinating OLs (Fig. 4Q–S). Untreated wild-type (Fig. 4N) or untreated MLD brains (Fig. 4O) showed no specific anti-β-Gal staining.
To study whether transplanted OLs were indeed forming myelin sheaths in the MLD brain, we used anti-MBP antibodies as a specific marker to visualize myelin sheaths and counted the number of MBP+/GFP+ myelin sheaths 1 year after transplants by double confocal microscopy. Figure 4, T and U, shows the presence of many double MBP+/GFP+ myelin sheaths in the cortex (Fig. 4T and its insets) and the striatum (Fig. 4U and its insets) amid endogenous myelin sheaths that are MBP positive but GFP negative. We estimated the abundance of myelin sheaths originated from transplanted GFP+ OLPs by counting the number of MBP+/GFP+ sheaths per area. Figure 4V shows that ∼27 and 15% of the MBP+ myelin sheaths in the cortex and striatum were derived from GFP+ OLs. Because of the high density of myelin sheaths in heavily myelinated structures such as the corpus callosum, we were unable to accurately count GFP+ myelin sheaths in this area. On the other hand, counting of myelinating cell bodies showed, for example, that ∼5% of the population of OLs in the corpus callosum and the cortex derived from grafted OLPs.
We did not find transplanted β-gal+ cells expressing NeuN, indicative that grafted OLPs did not acquire a neuronal phenotype in vivo (Fig. 4L, compare cell pointed to by filled arrow to cell pointed to by open arrow). We rarely observed grafted cells expressing GFAP, and only in close association to blood vessels (Fig. 4M, arrow).
Reduction of astroglial and microglial reaction in the brain of transplanted MLD mice
MLD mice develop a mild astrogliosis over the evolution of the disease. This can be visualized by immunohistochemical analysis with anti-GFAP antibodies. Figure 5B shows that GFAP+ cellular profiles, characteristic of reactive astroglia, are more abundantly distributed within the corpus callosum as well as the cortex of 1-year-old untreated MLD brain compared with healthy brains (Fig. 5A). In contrast, transplanted MLD mice showed a remarkable reduction of reactive astrocytes within the cortex (Fig. 5C).
Figure 5.
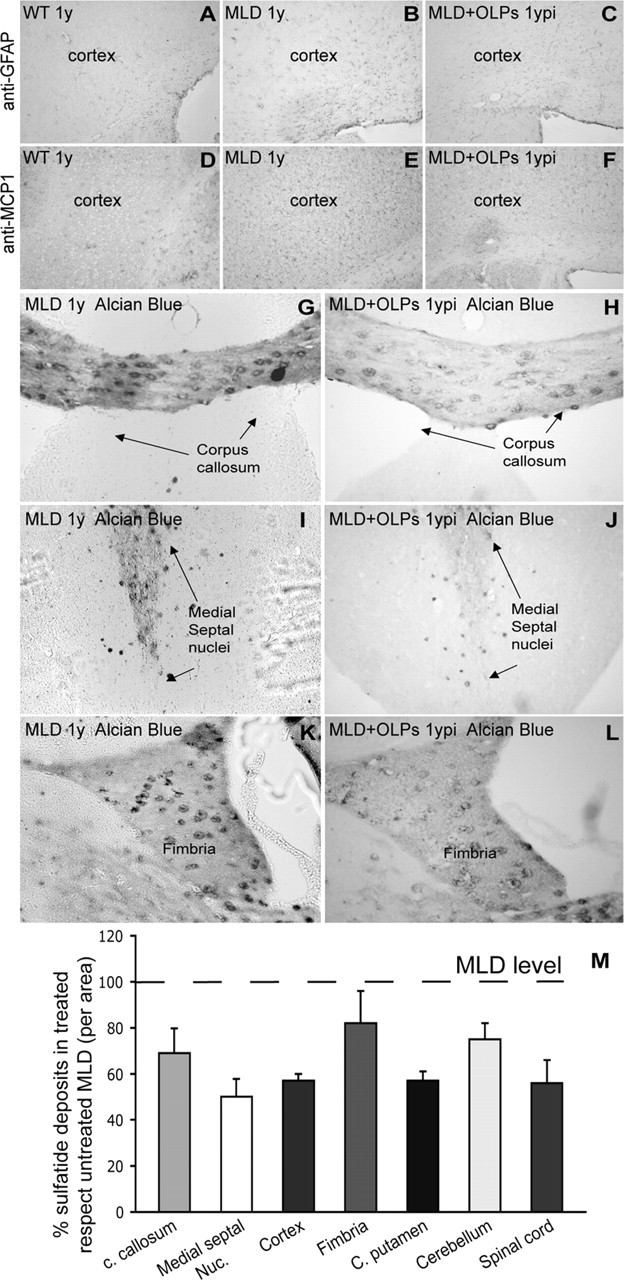
Sulfatide deposits and gliosis were reduced in MLD mice grafted with OLPs 1 year after surgery. Decreased level of astrogliosis (GFAP) and microgliosis (MCP-1) was observed in treated animals (C, F) in comparison to 1-year-old untreated MLD (B and E, respectively). A, D, Endogenous expression of GFAP and MCP-1 in healthy wild-type brain. Sections of transplanted (H–L) and nontransplanted (G–K) MLD brains were stained with Alcian blue to detect sulfatide deposits. Corpus callosum (H), medial septal nuclei (J), and the fimbria (L) of transplanted MLD mice showed reduction in the number and size of sulfatide deposits in comparison to the untreated MLD brain (G, I, K). M, The graph shows the abundance of sulfatide deposits in different areas of the brain expressed as a percentage of deposits in the same areas from untreated mice. n = 5 mice per group. ypi, Year postinjection; c. callosum, corpus callosum; Medial septal Nuc., medial septal nuclei; C. putamen, caudate–putamen. Results are expressed as mean value ± SEM.
MLD mice also develop signs of microglia activation and infiltration of macrophages, which can be studied by the level of immunoreactivity for MCP-1 (Fig. 5E, compare with background levels in healthy controls in D). Treatment with OLPs reduced the number of MCP-1-positive cells within the brain (Fig. 5F), indicating a less severe recruitment of macrophages in the CNS of treated MLD mice.
Diffusion of ARSA and in vivo cross-correction to endogenous neural cells reduced sulfatide deposits in transplanted MLD mice
Sulfatides accumulate progressively in MLD mice and are clearly detectable by Alcian blue staining after the first year of life. Microscopic examination of brain sections of untreated MLD mice revealed the presence of sulfatide deposits distributed abundantly throughout the brain, particularly in white matter areas. Figure 5 shows example pictures of Alcian blue-stained sulfatide deposits in diverse areas of the MLD brain such as corpus callosum (Fig. 5G), medial septal nuclei (Fig. 5I), and fimbria (Fig. 5K). In contrast, MLD mice that received OLP transplants showed a lower density of sulfatide deposits in these areas (Fig. 5H,J,L) as well as in other areas such as cortex, caudate–putamen, cerebellum, and spinal cord (Fig. 5M). Quantification of the density of Alcian blue spots per area showed that OLP transplants led to a reduction between 20 and 50% of sulfatide deposits in the MLD CNS (Fig. 5M). The reduction of sulfatide deposits was accompanied by an increase in ARSA activity. Enzyme activity in brain homogenates from 1-year-old treated MLD mice was 0.94 ± 0.28 nmol P-nitrocatechol sulfate (PNC) per milligram per hour compared with 3.04 ± 0.9 nmol PNC per milligram per hour in healthy brains (n = 5). These results indicate that neonatal transplants of OLPs in the brain of MLD mice led to ∼31% reconstitution of the normal ARSA activity.
Immunohistochemical analysis using anti-ARSA antibodies allowed us to confirm that ARSA enzyme diffused to many areas of the brain in transplanted MLD mice (Fig. 6). ARSA was detected primarily as punctuated pattern in the cytoplasm of cells throughout the wild-type cortex (Fig. 6A, arrow), corpus callosum (Fig. 6D, arrow), striatum (Fig. 6G, arrow), basal ganglia (data not shown), and cerebellum (data not shown) and was absent in the brain of untreated MLD mice (Fig. 6B,E,H). In contrast, numerous ARSA+ cells were seen in the cortex (Fig. 6C, arrow), corpus callosum (Fig. 6F, arrow), striatum (Fig. 6I, arrow), fimbria (Fig. 6J,K, arrows), the vicinity of the ventricular areas (data not shown), and cerebellum (data not shown). Interestingly, we found that not all neural cells in the brain of transplanted MLD brains contained ARSA (Fig. 6, compare distribution of ARSA in the cortex of treated MLD mice in C with that of the corresponding area in the wild-type tissue in A). Figure 6K shows an example of cells in the fimbria clearly containing immunodetectable ARSA (Fig. 6, arrows) and cells that are not containing significant levels of ARSA (Fig. 6, arrowheads). Secretion, diffusion and uptake of ARSA by deficient cells (cross-correction) are thought to play a central role in correction of the deficit of lysosomal enzymes in gene- and cell-based therapies. Our results confirmed that this process occurred in MLD mice transplanted with OLPs. Figure 6L–N shows an example field from 1-year-treated MLD cortex, in which a transplanted OLP (Fig. 6L, arrow), coimmunostained with anti-β-galactosidase and anti-ARSA antibodies, is in close proximity to an endogenous cell that took up the enzyme (anti-β-galactosidase negative; anti-ARSA positive).
Figure 6.
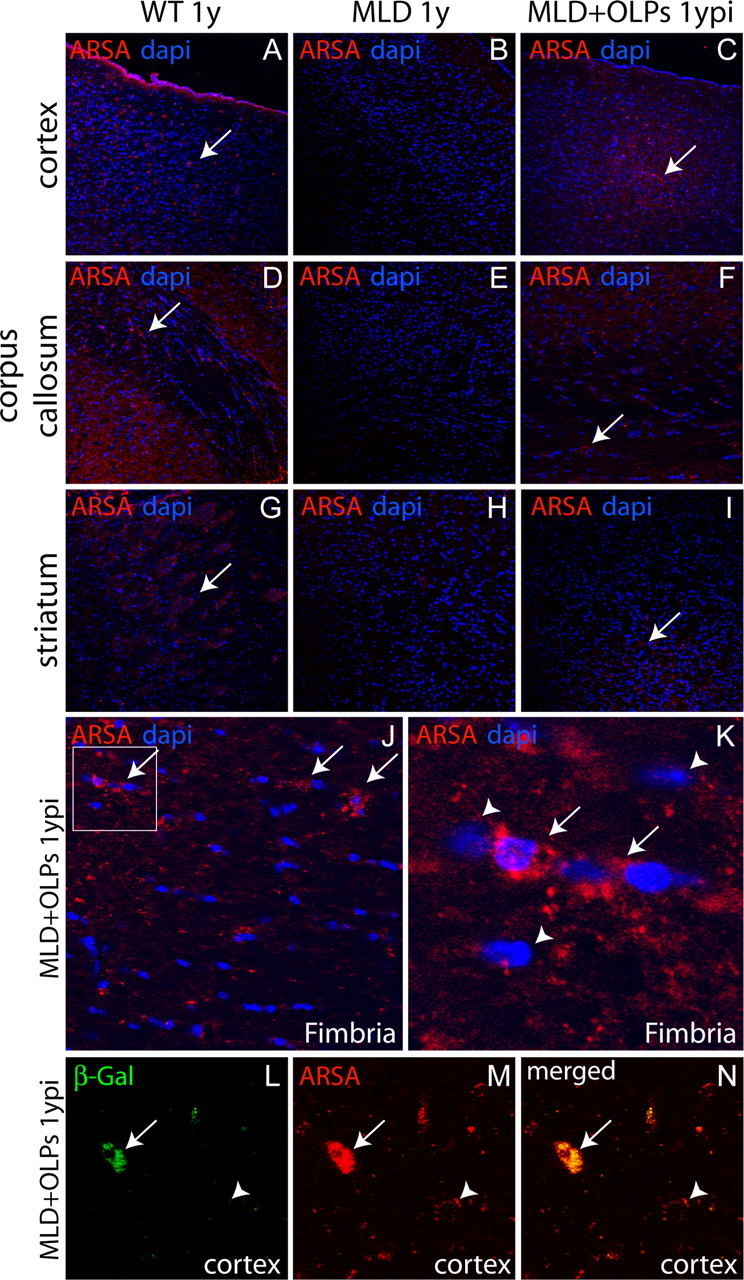
Diffusion of ARSA and in vivo cross-correction in endogenous neural cells. ARSA enzyme was immunodetected in several areas including cortex (A), corpus callosum (D), and striatum (G) in 1-year-old WT brains. Untreated MLD mice showed absence of immunodetectable ARSA, as expected (B, E, H). Transplanted MLD brain showed accumulation of ARSA in cortex (C), corpus callosum (F), striatum (I), and fimbria (J). The arrows in J and K point to cells with oligodendroglial morphology, which contain significant levels of immunodetectable ARSA. The arrowheads in K point to cells that did not show significant immunolabeling with anti-ARSA antibodies. L–N, An example field of 1-year-old transplanted MLD brain in which a transplant-derived β-galactosidase-positive OL (L, green, arrow) shows colocalization of immunolabeling using anti-ARSA antibodies (M, red, arrow). The same field shows an endogenous (β-galactosidase negative) cell, which showed significant immunolabeling with anti-ARSA antibodies, confirming in vivo cross-correction in the brain of MLD transplanted mice (N, arrowhead). ypi, Year postinjection.
Prevention of central electrophysiological and motor learning impairments in transplanted MLD mice
We then examined whether the engraftment of OLPs had a beneficial impact on central electrophysiology in the MLD mice by measuring MEPs. Central conduction time (CCT) and cortical MEP showed prolonged latencies in MLD mice after 1 year of age (Fig. 7A, black bars). Treatment with OLPs led to a complete normalization in the latency values for both CCT and cortical MEPs on MLD mice (Fig. 7A, gray bars).
Figure 7.
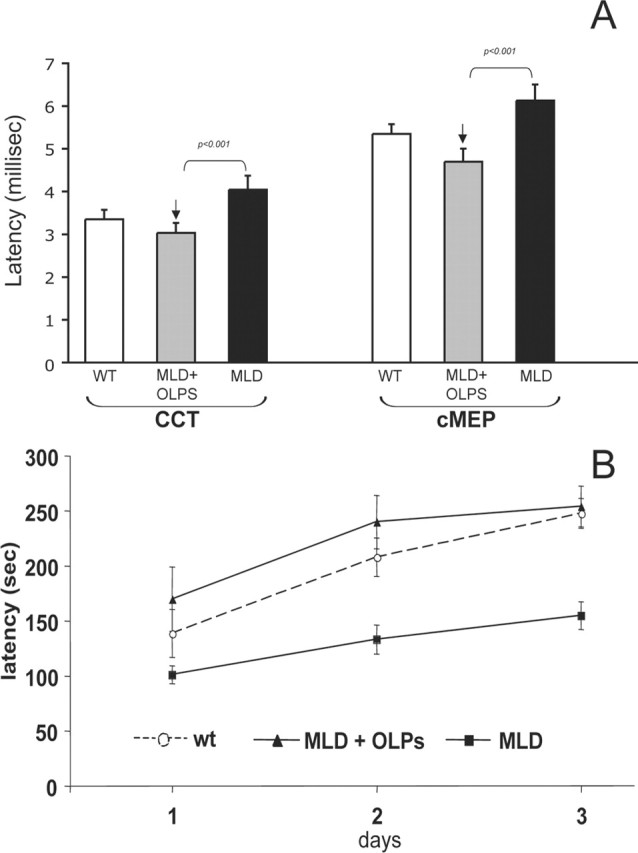
MLD mice treated with OLPs showed normalization of central electrophysiological parameters and of motor deficits. A, Electrophysiological measurement of central motor-evoked potentials (CMEPs) in transplanted mice showed significant protection of CCT and cMEPs 1 year after OLP transplantation. n = 7–10 mice per group. B, One-year-old MLD mice treated with OLPs (n = 7) or untreated (n = 11) were compared with wild-type animals (n = 10) on an accelerating Rotarod. Latencies were recorded over 3 d (3 trials each day). Means ± SEM of each day performances are indicated. Statistical analysis revealed motor recovery of the MLD transplanted animals compared with untreated MLD mice.
Motor learning was evaluated in 1-year-old mice on an accelerating Rotarod apparatus. MLD mice were found severely impaired, whereas OLP-transplanted MLD mice showed a full prevention of their motor learning impairment. In fact, two-way ANOVA indicated a significant interaction between genotype and treatment (F(2,25) = 4.97; p < 0.05). (Fig. 7B). In particular, whereas post hoc comparison revealed that MLD mice manifested a significant difference versus both WT and MLD-treated animals (p < 0.05 and p < 0.01, respectively), no statistical difference was seen between WT and treated mice (p = 0.61).
Discussion
We have examined the hypothesis that healthy OLPs transplanted in the brain of MLD pups at a time when myelination is not completed can migrate, integrate in white matter, and contribute to myelination, limiting the progression of central neurological deficits in this mutant. In vitro characterization confirmed the capacity of OLPs to generate mature MBP+ PLP+ OLs in T3/low serum (or astrocytes only in high serum). When OLPs were transplanted in the unmyelinated brain of MLD newborns, almost all of surviving cells became MBP+ PLP+ OLs. These results are consistent with other reports using similar preparations of OLPs in models of myelin disorders (Tontsch et al., 1994; Franklin and Blakemore, 1995; Learish et al. 1999; Espinosa de los Monteros et al. 2001; Crang et al., 2004).
Migration of grafted OLPs was significantly higher and targeted to white matter tracts when OLPs were intraventricularly grafted in neonate MLD pups. In contrast, a more reduced distribution of OLPs was observed when transplantation was performed in healthy pups and even when grafted in MLD mice 2 months after birth (M. Givogri and E. Bongarzone, unpublished observations). OLPs can migrate and survive better after transplantation in the brain of myelin mutants than in healthy animals (Gaillard et al., 1998; Bentlage et al., 1999). We found that ∼10% of OLPs survived during the first month and migrated to prospective areas of myelination in the MLD brain. Furthermore, these cells appeared to contribute to the pool of adult OLs present after 1 year of treatment. In fact, ∼4.5% of the total number of glial cells in the corpus callosum was derived from the transplant. Surviving OLs produced a substantial amount of myelin ranging from 15 to 27% of myelin sheaths. This indicates that surviving OLPs were able to myelinate several internodes each, responding to myelinogenic cues from the developing MLD brain.
Numerous factors might have contributed to the remarkable plasticity of the MLD brain to OLPs. First, healthy OLPs were grafted at a developmental time when the brain undergoes robust gliogenesis and there is intense migration of glial progenitors in response to migratory cues (Levison et al., 1993, Luskin and McDermott, 1994; Zerlin et al., 1995). Recent data indicate that OLPs originate from three different sites in the developing brain; Emx1-expressing progenitors in the cortex is the source for postnatal OLPs (Kessaris et al., 2006). Our experiments transplanted OLPs when this wave of Emx1 precursors are believed to be actively generating endogenous OLPs; thus, it is feasible that grafted OLPs have responded to the endogenous cues governing oligodendrogenesis during the first postnatal weeks. Several types of signals are expressed in the developing brain, which might have been preferentially recognized by grafted OLPs, stimulating their migration in the MLD brain. OLPs express members of the integrin family such as α6β1 and αvβ1 integrins, which bind extracellular matrix molecules such as fibronectin, laminins, and vitronectin (Frost et al., 1996; Garcion et al., 2001). Integrins play a fundamental role in cell–cell interactions during homotypic migration of OLPs (Milner et al., 1996; Jacques et al., 1998; Decker et al., 2000; Vitry et al., 2001). MLD pups showed slightly higher expression of laminin β2 in the brain than controls, without significant differences in other extracellular matrix component members such as fibronectin and collagen, which might have facilitated OLPs migration (Givogri and Bongarzone, unpublished observations). Interestingly, this migratory property was observed also in vitro, where brain slices from MLD mice promoted a more efficient migration of OLPs than brain slices from healthy mice. Furthermore, transplantation of MLD OLPs showed that ARSA deficiency did not influence the migratory capacity of the mutant OLPs. This result shows that the pathogenesis of MLD mice is not caused by deficient colonization of white matter territories by endogenous OLPs (Yaghootfam et al., 2005) nor caused by a toxic effect from the MLD brain environment. Instead, it suggests that pathogenesis is caused by a cell-autonomous defect of ARSA deficiency after cell migration. Additional studies will investigate whether this cell autonomous defect is affecting both OLPs and neurons in the MLD brain.
Second, transplants were done at a postnatal time when MLD pups had a delayed myelination (Yaghootfam et al., 2005). This condition likely facilitated transplanted OLPs, carrying the selective advantage of a normal sphingolipid catabolism, to migrate and myelinate the MLD brain. Accumulation of sulfatides might be considered as a potential neurotoxic agent for neural cells, in particular leading to a progressive depletion of functional OLs and the consequent loss of myelin. However, the direct link between the accumulation of sulfatides and the loss of OLs and myelin in MLD individuals has not been unequivocally established yet. Furthermore, it is unclear whether the accumulation of sulfatides in perinatal OLPs in the MLD mouse model, which might still be at a low concentration at the time of the transplantation, has an impact on their survival and/or differentiation. Some recent work elegantly illustrates the role of sphingolipids during oligodendroglial differentiation (Bansal et al., 1999; Hirahara et al., 2004; Yaghootfam et al., 2005). For example, absence of sulfatides stimulates a premature differentiation of OLPs. Interestingly, Yaghootfam and coworkers showed that MLD mice have a significant delay in early postnatal myelination without a substantial compromise of ulterior myelination. Thus, the progressive accumulation of sulfatides might induce MLD OLPs to persist for longer time as progenitors and delaying, but not blocking, their capacity to terminally differentiate and produce myelin. Under this scenario, the potential delay of endogenous MLD OLPs to differentiate would give space to grafted OLPs, the differentiation program of which is unaffected as a result of a normal sulfatide catabolism, to terminally differentiate and myelinate MLD axons, as we have seen in this study.
OLPs also migrated within the MLD SVZ–RMS. Although neurogenesis and gliogenesis are primarily embryonic events, the postnatal mammalian brain retains areas such as the SVZ where a continuum production of new neuronal and glial progenitors is maintained throughout life (Doetsch et al., 1997). Cells born in the SVZ migrate along the RMS, a restricted area connecting the SVZ with the OB, where they replace dying neurons (Lois and Alvarez-Buylla, 1994). The adult SVZ is also a source of astrocytes and OLs, which migrate radially toward the cortex, particularly in response to brain insults (Goings et al., 2004; Gotts and Chesselet, 2005). Whether the MLD SVZ reacts to the progressive neuropathology by increasing the production of neuronal and glial progenitors for neurorepair is presently uncertain. BrdU proliferation experiments indicated that grafted OLPs could home in the SVZ where they proliferated during the first weeks after transplantation but not in aged MLD mice. Together, these findings indicate that exogenous OLPs can be attracted to gliogenic sites in the brain, such as the SVZ, where mitogenic signals might stimulate their proliferation.
Sulfatide accumulation in MLD brain was reduced after transplantation of OLPs. Untreated mutants progressively accumulate sulfatides in the brain, which is accompanied by the appearance of neurological deficits in adulthood (Hess et al., 1996; Pernber et al., 2002; Luca et al., 2005). Sulfatides and other sphingolipids are important components of lipid rafts and have a fundamental role for the targeting and assembly of numerous proteins in myelin and neuronal plasma membranes (for review see Lee, 2001). Abnormal sulfatide metabolism might change rafts composition, influencing on the biogenesis of plasma membranes, contributing to MLD neuropathogenesis. Then, reducing the endogenous levels of sulfatides after infusing cells with a normal expression of ARSA, capable of long distance migration and myelination of axons in the MLD brain, might have limited the evolution of neurological impairments in treated mice. We observed in fact a good correlation between the increase in ARSA activity in the brain, the reduction of sulfatide storage lesions, and the improved central electrophysiology and motor coordination. Previous studies demonstrated that gene transfer of ARSA to neural cells directly contribute to the reduction of sulfatides and to the prevention of central neurological deficits in MLD mice (Consiglio et al., 2001; Luca et al., 2005; Sevin et al., 2006). Extracellular secretion and receptor-mediated reuptake is a general property of lysosomal enzymes such as ARSA, contributing to cross-correction of enzyme deficiency in neighboring cells. In fact, we found that ARSA appeared to diffuse and accumulate in different areas of the treated brain in direct and indirect contact to the sites of initial transplantation. Furthermore, our results showed that endogenous cells can take up ARSA, constituting in vivo evidence of the process of cross-correction and that transplanted OLPs can serve as a source of enzyme. Our study and that of others contribute direct evidence to the notion of using neural progenitors as migratory vehicles to cross-correct endogenous cells in models of lysosomal storage disorders (Snyder et al., 1995; Matzner et al., 2000; Kondo et al., 2005). Therefore, the therapeutic benefits seen in this study may arise from both improved myelination by engrafted OLs and the cross-correction of ARSA deficiency in endogenous MLD neurons and glial cells.
Brain insults increase microgliosis and astrogliosis to remove cell debris and to contain secondary damage (for review, see Fawcett and Asher, 1999). Adult MLD mice show mild demyelination (Hess et al., 1996), likely consequent to OL death, and as expected, they show both microglial/astroglial responses. Introduction of healthy OLPs in developing MLD brains both reduced the presence of microglial cells, which contribute to cell death of neurons and myelinating cells (Wada et al., 2000; Ohmi et al., 2003), as well as astrogliosis. These results likely represent an amelioration of OLs turnover in treated MLD mice, consequent to the establishment of a mosaic situation of grafted OLs intermingled with endogenous MLD OLs.
In summary, our results indicate that OLPs have an intrinsic ability to populate the MLD brain, with appropriate migration, nontumorigenic integration in white matter, and differentiation into myelinating OLs. Our findings, analyzed within the context of other reports, present evidence for a potential mechanism of myelin pathogenesis in MLD CNS, in which sulfatides seem to interfere with terminal differentiation of OLs.
Footnotes
This work was supported by a Program Project Grant from the Telethon Foundation to E.B. We are grateful to Drs. Volkmar Gieselmann and Uli Metzner (Bonn, Germany) for providing the initial MLD founder ARSA null mice and anti-ARSA antibodies and Giuliana Ferrari and Francesco Lotti (Milano, Italy) for generously sharing GFP lentiviral vectors. We thank the anonymous reviewers for their constructive comments.
References
- Bansal R, Winkler S, Bheddah S (1999). Negative regulation of oligodendrocyte differentiation by galactosphingolipids. J Neurosci 19:7913–7924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baron W, Shattil SJ, ffrench-Constant C (2002). The oligodendrocyte precursor mitogen PDGF stimulates proliferation by activation of alpha(v)beta3 integrins. EMBO J 21:1957–1966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barres BA, Schmid R, Sendnter M, Raff MC (1993). Multiple extracellular signals are required for long-term oligodendrocyte survival. Development 118:283–295. [DOI] [PubMed] [Google Scholar]
- Bentlage C, Nikkhah G, Cunningham MG, Bjorklund A (1999). Reformation of the nigrostriatal pathway by fetal dopaminergic micrografts into the substantia nigra is critically dependent on the age of the host. Exp Neurol 159:177–190. [DOI] [PubMed] [Google Scholar]
- Bongarzone ER, Foster LM, Byravan S, Verity AN, Landry CF, Schonmann V, Amur-Umarjee S, Campagnoni AT (1996). Conditionally immortalized neural cell lines: potential models for the study of neural cell function. Methods 10:489–500. [DOI] [PubMed] [Google Scholar]
- Bongarzone ER, Campagnoni CW, Kampf K, Jacobs EC, Handley VW, Schonmann V, Campagnoni AT (1999). Identification of a new exon in the myelin proteolipid protein gene encoding novel protein isoforms that are restricted to the somata of oligodendrocytes and neurons. J Neurosci 19:8349–8357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calver AR, Hall AC, Yu WP, Walsh FS, Heath JK, Betsholtz C, Richardson WD (1998). Oligodendrocyte population dynamics and the role of PDGF in vivo. Neuron 20:869–882. [DOI] [PubMed] [Google Scholar]
- Consiglio A, Quattrini A, Martino S, Bensadoun JC, Dolcetta D, Trojani A, Benaglia G, Marchesini S, Cestari V, Oliverio A, Bordignon C, Naldini L (2001). In vivo gene therapy of metachromatic leukodystrophy by lentiviral vectors: correction of neuropathology and protection against learning impairments in affected mice. Nat Med 7:310–316. [DOI] [PubMed] [Google Scholar]
- Crang AJ, Gilson JM, Li WW, Blakemore WF (2004). The remyelinating potential and in vitro differentiation of MOG-expressing oligodendrocyte precursors isolated from adult rat CNS. Eur J Neurosci 20:1445–1460. [DOI] [PubMed] [Google Scholar]
- Decker L, Avellana-Adalid V, Nait-Oumesmar B, Durbec P, Baron-Van Evercooren A (2000). Oligodendrocyte precursor migration and differentiation: combined effects of PSA residues, growth factors, and substrates. Mol Cell Neurosci 16:422–439. [DOI] [PubMed] [Google Scholar]
- Doetsch F, Garcia-Verdugo JM, Alvarez-Buylla A (1997). Cellular composition and three-dimensional organization of the subventricular germinal zone in the adult mammalian brain. J Neurosci 17:5046–5061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolcetta D, Amadio S, Guerrini U, Givogri MI, Perani L, Galbiati F, Sironi L, Del Carro U, Roncarolo MG, Bongarzone E (2005). Myelin deterioration in Twitcher mice: motor evoked potentials and magnetic resonance imaging as in vivo monitoring tools. J Neurosci Res 81:597–604. [DOI] [PubMed] [Google Scholar]
- Duncan ID (2005). Oligodendrocytes and stem cell transplantation: their potential in the treatment of leukoencephalopathies. J Inherit Metab Dis 28:357–368. [DOI] [PubMed] [Google Scholar]
- Duncan ID, Hammang JP, Jackson KF, Wood PM, Bunge RP, Langford L (1988). Transplantation of oligodendrocytes and Schwann cells into the spinal cord of the myelin-deficient rat. J Neurocytol 17:351–360. [DOI] [PubMed] [Google Scholar]
- Espinosa de los Monteros A, Baba H, Zhao PM, Pan T, Chang R, de Vellis J, Ikenaka K (2001). Remyelination of the adult demyelinated mouse brain by grafted oligodendrocyte progenitors and the effect of B-104 cografts. Neurochem Res 26:673–682. [DOI] [PubMed] [Google Scholar]
- Fawcett JW, Asher RA (1999). The glial scar and central nervous system repair. Brain Res Bull 49:377–391. [DOI] [PubMed] [Google Scholar]
- Franklin RJ, Blakemore WF (1995). Glial-cell transplantation and plasticity in the O-2A lineage implications for CNS repair. Trends Neurosci 18:151–156. [DOI] [PubMed] [Google Scholar]
- Franklin RJ, Bayley S, Blakemore W (1996). Transplanted CG4 cells (an oligodendrocyte progenitor cell line) survive, migrate, and contribute to repair of areas of demyelination in ex-irradiated and damaged spinal cord but not in normal spinal cord. Exp Neurol 137:263–276. [DOI] [PubMed] [Google Scholar]
- Frost E, Kiernan BW, Faissner A, ffrench-Constant C (1996). Regulation of oligodendrocyte precursor migration by extracellular matrix: evidence for substrate-specific inhibition of migration by tenascin-C. Dev Neurosci 18:266–273. [DOI] [PubMed] [Google Scholar]
- Frost EE, Nielsen JA, Le TQ, Armstrong RC (2003). PDGF and FGF2 regulate oligodendrocyte progenitor responses to demyelination. J Neurobiol 54:457–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaillard A, Gaillard F, Roger M (1998). Neocortical grafting to newborn and adult rats: developmental, anatomical and functional aspects. Adv Anat Embryol Cell Biol 148:1–86. [DOI] [PubMed] [Google Scholar]
- Garcion E, Faissner A, ffrench-Constant C (2001). Knockout mice reveal a contribution of the extracellular matrix molecule tenascin-C to neural precursor proliferation and migration. Development 128:2485–2496. [DOI] [PubMed] [Google Scholar]
- Gard AL, Pfeiffer SE (1989). Oligodendrocyte progenitors isolated directly from developing telencephalon at a specific phenotypic stage: myelinogenic potential in a defined environment. Development 106:119–132. [DOI] [PubMed] [Google Scholar]
- Gieselmann V (2003). Metachromatic leukodystrophy: recent research developments. J Child Neurol 18:591–595. [DOI] [PubMed] [Google Scholar]
- Givogri MI, Costa RM, Schonmann V, Silva AJ, Campagnoni AT, Bongarzone ER (2002). Central nervous system myelination in mice with deficient expression of Notch1 receptor. J Neurosci Res 67:309–320. [DOI] [PubMed] [Google Scholar]
- Givogri MI, Schonmann V, Cole R, De Vellis J, Bongarzone ER (2003). Notch1 and Numb genes are inversely expressed as oligodendrocytes differentiate. Dev Neurosci 25:50–64. [DOI] [PubMed] [Google Scholar]
- Goings GE, Sahni V, Szele FG (2004). Migration patterns of subventricular zone cells in adult mice change after cerebral cortex injury. Brain Res 996:213–226. [DOI] [PubMed] [Google Scholar]
- Gotts JE, Chesselet MF (2005). Migration and fate of newly born cells after focal cortical ischemia in adult rats. J Neurosci Res 80:160–171. [DOI] [PubMed] [Google Scholar]
- Hess B, Saftig P, Hartmann D, Coenen R, Lullmann-Rauch R, Goebel HH, Evers M, von Figura K, D’Hooge R, Nagels G, De Deyn P, Peters C, Gieselmann V (1996). Phenotype of arylsulfatase A-deficient mice: relationship to human metachromatic leukodystrophy. Proc Natl Acad Sci USA 93:14821–14826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirahara Y, Bansal R, Honke K, Ikenaka K, Wada Y (2004). Sulfatide is a negative regulator of oligodendrocyte differentiation: development in sulfatide-null mice. Glia 45:269–277. [DOI] [PubMed] [Google Scholar]
- Jacques TS, Relvas JB, Nishimura S, Pytela R, Edwards GM, Streuli CH, ffrench-Constant C (1998). Neural precursor cell chain migration and division are regulated through different beta1 integrins. Development 125:3167–3177. [DOI] [PubMed] [Google Scholar]
- Kessaris N, Fogarty M, Iannarelli P, Grist M, Wegner M, Richardson WD (2006). Competing waves of oligodendrocytes in the forebrain and postnatal elimination of an embryonic lineage. Nat Neurosci 9:173–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koc ON, Day J, Nieder M, Gerson SL, Lazarus HM, Krivit W (2002). Allogeneic mesenchymal stem cell infusion for treatment of metachromatic leukodystrophy (MLD) and Hurler syndrome (MPS-IH). Bone Marrow Transplant 30:215–222. [DOI] [PubMed] [Google Scholar]
- Kondo Y, Wenger DA, Gallo V, Duncan ID (2005). Galactocerebrosidase-deficient oligodendrocytes maintain stable central myelin by exogenous replacement of the missing enzyme in mice. Proc Natl Acad Sci USA 102:18670–18675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krivit W, Peters C, Shapiro EG (1999a). Bone marrow transplantation as effective treatment of central nervous system disease in globoid cell leukodystrophy, metachromatic leukodystrophy, adrenoleukodystrophy, mannosidosis, fucosidosis, aspartylglucosaminuria, Hurler, Maroteaux-Lamy, and Sly syndromes, and Gaucher disease type III. Curr Opin Neurol 12:167–176. [DOI] [PubMed] [Google Scholar]
- Krivit W, Aubourg P, Shapiro E, Peters C (1999b). Bone marrow transplantation for globoid cell leukodystrophy, adrenoleukodystrophy, metachromatic leukodystrophy, and Hurler syndrome. Curr Opin Hematol 6:377–382. [DOI] [PubMed] [Google Scholar]
- Lachapelle F, Lapie P, Nussbaum JL, Gumpel M (1990). Immunohistochemical studies on cross-transplantations between Jimpy, Shiverer, and normal newborn mice. J Neurosci Res 27:324–331. [DOI] [PubMed] [Google Scholar]
- Learish RD, Brustle O, Zhang SC, Duncan SC (1999). Intraventricular transplantation of oligodendrocyte progenitors into fetal myelin mutant results in widespread formation of myelin. Ann Neurol 46:716–722. [PubMed] [Google Scholar]
- Lee AG (2001). Myelin: delivery by raft. Curr Biol 11:R60–R62. [DOI] [PubMed] [Google Scholar]
- Levison SW, Chuang C, Abramson BJ, Goldman JE (1993). The migrational patterns and developmental fates of glial precursors in the rat subventricular zone are temporally regulated. Development 119:611–622. [DOI] [PubMed] [Google Scholar]
- Lois C, Alvarez-Buylla A (1994). Long-distance neuronal migration in the adult mammalian brain. Science 264:1145–1148. [DOI] [PubMed] [Google Scholar]
- Louis JC, Magal E, Muir D, Manthorpe M, Varon S (1992). CG-4, a new bipotential glial cell line from rat brain, is capable of differentiating in vitro into either mature oligodendrocytes or type-2 astrocytes. J Neurosci Res 31:193–204. [DOI] [PubMed] [Google Scholar]
- Lubetzki C, Gansmuller A, Lachapelle F, Lombrail P, Gumpel M (1988). Myelination by oligodendrocytes isolated from 4–6-week-old rat central nervous system and transplanted into newborn Shiverer brain. J Neurol Sci 88:161–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luca T, Givogri MI, Perani L, Galbiati F, Follenzi A, Naldini L, Bongarzone ER (2005). Axons mediate the distribution of arylsulfatase A within the mouse hippocampus upon gene delivery. Mol Ther 12:669–679. [DOI] [PubMed] [Google Scholar]
- Luo J, Miller MW (1997). Basic fibroblast growth factor- and platelet-derived growth factor-mediated cell proliferation in B104 neuroblastoma cells: effect of ethanol on cell cycle kinetics. Brain Res 770:139–150. [DOI] [PubMed] [Google Scholar]
- Luskin MB, McDermott K (1994). Divergent lineages for oligodendrocytes and astrocytes originating in the neonatal forebrain subventricular zone. Glia 11:211–226. [DOI] [PubMed] [Google Scholar]
- Magy L, Mertens C, Avellana-Adalid V, Keita M, Lachapelle F, Nait-Oumesmar B, Fontaine B, Baron-Van Evercooren A (2003). Inducible expression of FGF2 by a rat oligodendrocyte precursor cell line promotes CNS myelination in vitro. Exp Neurol 184:912–922. [DOI] [PubMed] [Google Scholar]
- Matzner U, Harzer K, Learish RD, Barranger JA, Gieselmann V (2000). Long-term expression and transfer of arylsulfatase A into brain of arylsulfatase A-deficient mice transplanted with bone marrow expressing the arylsulfatase A cDNA from a retroviral vector. Gene Ther 7:1250–1257. [DOI] [PubMed] [Google Scholar]
- Meng X, Shen J, Ohashi T, Maeda H, Kim S, Eto Y (2003). Brain transplantation of genetically engineered human neural stem cells globally corrects brain lesions in the mucopolysaccharidosys type VII mouse. J Neurosci Res 74:266–277. [DOI] [PubMed] [Google Scholar]
- Milner R, Edwards G, Streuli C, ffrench-Constant C (1996). A role in migration for the αvβ1 integrin expressed on oligodendrocyte precursors. J Neurosci 16:7240–7252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neufeld EF, Fratantoni JC (1970). Inborn errors of mucopolysaccharide metabolism. Science 169:141–146. [DOI] [PubMed] [Google Scholar]
- Ohmi K, Greenberg DS, Rajavel KS, Ryazantsev S, Li HH, Neufeld EF (2003). Activated microglia in cortex of mouse models of mucopolysaccharidoses I and IIIB. Proc Natl Acad Sci USA 100:1902–1907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passini MA, Lee EB, Heuer GG, Wolfe JH (2002). Distribution of a lysosomal enzyme in the adult brain by axonal transport and by cells of the rostral migratory stream. J Neurosci 22:6437–6446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G, Franklin KBJ (1997). In: The mouse brain in stereotaxic coordinates New York: Academic.
- Pernber Z, Molander-Melin M, Berthold CH, Hansson E, Fredman P (2002). Expression of the myelin and oligodendrocyte progenitor marker sulfatide in neurons and astrocytes of adult rat brain. J Neurosci Res 69:86–93. [DOI] [PubMed] [Google Scholar]
- Redwine JM, Armstrong RC (1998). In vivo proliferation of oligodendrocyte progenitors expressing PDGFalphaR during early remyelination. J Neurobiol 37:413–428. [DOI] [PubMed] [Google Scholar]
- Schubert D, Heinemann S, Carlisle W, Tarikas H, Kimes B, Patrick J, Steinbach JH, Culp W, Brandt BL (1974). Clonal cell lines from the rat central nervous system. Nature 249:224–227. [DOI] [PubMed] [Google Scholar]
- Sevin C, Benraiss A, Van Dam D, Binnin D, Nagels G, Verot L, Laurendeau I, Vidaud M, Gieselmann V, Venier M, De Deyn P, Aubourg P, Cartier N (2006). Intracerebral adeno-associated virus-mediated gene transfer in rapidly progressive forms of metachromatic leukodystrophy. Hum Mol Genet 15:53–64. [DOI] [PubMed] [Google Scholar]
- Snyder EY, Taylor RM, Wolfe JH (1995). Neural progenitor cell engraftment corrects lysosomal storage throughout the MPS VII mouse brain. Nature 374:367–370. [DOI] [PubMed] [Google Scholar]
- Tontsch U, Archer DR, Dubois-Dalcq M, Duncan ID (1994). Transplantation of an oligodendrocyte cell line leading to extensive myelination. Proc Natl Acad Sci USA 91:11616–11620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitry S, Avellana-Adalid V, Lachapelle F, Evercooren AB (2001). Migration and multipotentiality of PSA-NCAM+ neural precursors transplanted in the developing brain. Mol Cell Neurosci 17:983–1000. [DOI] [PubMed] [Google Scholar]
- Wada R, Tifft CJ, Proia RL (2000). Microglial activation precedes acute neurodegeneration in Sandhoff disease and is suppressed by bone marrow transplantation. Proc Natl Acad Sci USA 97:10954–10959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Sdrulla AD, diSibio G, Bush G, Nofziger D, Hicks C, Weinmaster G, Barres BA (1998). Notch receptor activation inhibits oligodendrocyte differentiation. Neuron 21:63–75. [DOI] [PubMed] [Google Scholar]
- Yaghootfam A, Gieselmann V, Eckhardt M (2005). Delay of myelin formation in arylsulphatase A-deficient mice. Eur J Neurosci 21:711–720. [DOI] [PubMed] [Google Scholar]
- Yandava BD, Billinghurst LL, Snyder EY (1999). “Global” cell replacement is feasible via neural stem cell transplantation: evidence from the dysmyelinated Shiverer mouse brain. Proc Natl Acad Sci USA 96:7029–7034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zerlin M, Levison SW, Goldman JE (1995). Early patterns of migration, morphogenesis, and intermediate filament expression of subventricular zone cells in the postnatal rat forebrain. J Neurosci 15:7238–7249. [DOI] [PMC free article] [PubMed] [Google Scholar]