

Abstract
The entorhinal cortex (EC) is a significant component of the systems that underlie certain forms of memory formation and recall. Evidence has been emerging that the dopaminergic system in the EC facilitates these and other functions of the EC. The effects of dopamine (DA) on membrane properties and excitability of EC neurons, however, are not known. We used in vitro whole-cell patch-clamp recordings from layer V pyramidal neuronal somata and dendrites of the adult rat lateral EC to investigate the effects of DA on the excitability of these neurons. We found that brief application of DA caused a reduction in the excitability of layer V EC pyramidal neurons. This effect was attributable to voltage-dependent modification of membrane properties that can best be explained by an increase in a hyperpolarization-activated conductance. Furthermore, the effects of DA were blocked by pharmacological blockade of h-channels, but not by any of a number of other ion channels. These actions were produced by a D1 receptor-mediated increase of cAMP but were independent of protein kinase A. A portion of the actions of DA can be attributed to effects in the apical dendrites. The data suggest that DA can directly influence the membrane properties of layer V EC pyramidal neurons by modulation of h-channels. These actions may underlie some of the effects of DA on memory formation.
Keywords: dopamine, entorhinal cortex, whole cell, patch clamp, rat, hyperpolarization-activated current, voltage sag
Introduction
The entorhinal cortex (EC) is part of a network that aids in the consolidation and recall of memories (Buzsaki, 1996; Murray et al., 1998; Kapur and Brooks, 1999; Lavenex and Amaral, 2000; Sybirska et al., 2000; Haist et al., 2001; Squire et al., 2004; Dolcos et al., 2005; Steffenach et al., 2005). Neuronal pathology and reduction of EC volume are potential contributive components to Alzheimer's disease (Hyman et al., 1984; Kotzbauer et al., 2001) and schizophrenia (Falkai et al., 1988; Arnold et al., 1991; Akil and Lewis, 1997; Krimer et al., 1997; Joyal et al., 2002; Prasad et al., 2004). Dopamine (DA) receptor activation within the EC enhances recall of certain forms of memory and facilitates memory consolidation (Ardenghi et al., 1997; Izquierdo et al., 1998; Barros et al., 2001). Alterations of the DA system also modulate behaviors that depend in part on the EC (Feldon and Weiner, 1991; Ruob et al., 1997; Swerdlow et al., 2003; Liu et al., 2004). Thus, it would appear that DA receptors in the EC significantly modulate several memory processes. Very little is known, however, about the actions of DA on EC neurons.
Similar to other cortical pyramidal neurons, deep layer EC neurons display a spectrum of ionic conductances, and electrophysiological features that are indicative of the presence of active conductances (Eder and Heinemann, 1996; Eder et al., 1996; Richter et al., 2000; Agrawal et al., 2001, 2003; Egorov et al., 2002a,b). For instance, many of these neurons display a voltage sag in response to hyperpolarization, and other indicators of hyperpolarization-activated currents (Ih) (Richter et al., 2000; Hamam et al., 2002). Ih potently regulates firing and synaptic integration (Mayer and Westbrook, 1983; Spain et al., 1987; Pape and McCormick, 1989; McCormick and Huguenard, 1992; Magee, 1998, 1999). Although it is known that DAergic fibers innervate the EC (Fallon et al., 1978; Descarries et al., 1987; Diop et al., 1988; Goldsmith and Joyce, 1994; Erickson et al., 2000), and DA receptors are present in the EC (Richfield et al., 1989; Weiner et al., 1991; Goldsmith and Joyce, 1994; Yokoyama et al., 1994; Tarazi et al., 1999), there has been little evidence that ion channels can be modulated by DA in the EC. In other regions, it has been demonstrated that DA alters the excitability of cortical neurons (Zhou and Hablitz, 1999; Henze et al., 2000; Gulledge and Jaffe, 2001; Lavin and Grace, 2001; West and Grace, 2002; Tseng and O'Donnell, 2004), perhaps explained by modulation of K+ channels (Malenka and Nicoll, 1986; Nicoll, 1988; Pedarzani and Storm, 1995a; Storm et al., 2000; Gorelova et al., 2002; Dong and White, 2003; Dong et al., 2004; Kroner et al., 2005) and sodium channels (Cantrell et al., 1999; Maurice et al., 2001; Cooper et al., 2003). This study used in vitro whole-cell recordings from visually identified layer V pyramidal neurons of the rat lateral EC to investigate the role of DA in modulating the excitability of these neurons. An understanding of the effects of DA in the EC may lead to insight into how DA modulates memory processes that rely on EC neuronal activity.
Materials and Methods
Slice preparation.
Male Sprague Dawley rats (4–6 weeks of age) were anesthetized with an intraperitoneal injection of a combination of ketamine (42 mg/ml), xylazine (8 mg/ml), and, in some experiments, acepromazine (1.4 mg/ml). No differences were observed in the results from animals in which acepromazine was included in the anesthetic mixture. Once anesthetized, rats were perfused intracardially with an ice-cold solution containing the following (in mm): 110 choline chloride, 2.5 KCl, 1.25 NaH2PO4, 25 NaHCO3, 0.5 CaCl2, 7 MgCl2, 7 dextrose, 1.3 ascorbic acid, 3 pyruvic acid (all drugs were from Sigma, St. Louis, MO, unless otherwise specified). The solution was saturated with a 95% O2, 5% CO2 gas mixture. After perfusion, the rat was quickly decapitated, and the brain was removed and sliced in 350 μm sections in the same solution (Vibratome Series 1000; Vibratome, St. Louis, MO). Brain slices were then incubated at 37°C for 10 min in a solution containing the following (in mm): 125 NaCl, 2.5 KCl, 1.25 NaH2PO4, 25 NaHCO3, 2 CaCl2, 2 MgCl2, 10 dextrose, 1.3 ascorbic acid, 3 pyruvic acid. After incubation, brain slices stabilized at room temperature for at least 50 min before transfer to the recording chamber.
Whole-cell recordings.
Brain slices were transferred to the recording chamber with a solution containing the following (in mm): 125 NaCl, 2.5 KCl, 1.25 NaH2PO4, 25 NaHCO3, 2 CaCl2, 2 MgCl2, 10 dextrose, 0.075 sodium metabisulfite (an antioxidant that prevents degradation of DA). Layer V of the lateral EC was visually identified using infrared differential interference contrast microscopy. Pyramidal neurons were selected based on the protrusion of a long apical dendrite and a nonstellate appearance. Patch electrodes were constructed from borosilicate glass with an outer diameter of 2.0 mm and an inner diameter of 1.16 mm (Sutter Instruments, Novato, CA). Electrodes were pulled using a Flaming/Brown micropipette puller (model P-97; Sutter Instruments). Electrodes were filled with the following (in mm): 120 K+ methylsulfate, 20 KCl, 0.2 EGTA, 10 HEPES, 2 MgCl2, with a pH of 7.3. On the day of recordings, 4 mm Na2-ATP, 0.3 mm Tris-GTP, 7 mm phosphocreatine, and 0.1% Neurobiotin were added to the internal recording solution (yielding an osmolarity of ∼290 mOsm). The electrode junction potential was corrected in the bath, but the diffusion potential was not compensated after seal formation and membrane rupture. Based on relative ionic mobilities and charge, this potential was calculated to be 7–8 mV. Therefore, the actual membrane potentials could be up to 8 mV more hyperpolarized than the measured voltage. The liquid junction potential between the intracellular and extracellular solutions was also measured to be 3–4 mV using the method described by Neher (1992), suggesting that this difference between measured and actual membrane potentials may be even less. In some experiments, Sylgard was applied to the electrode tips, or the tips were wrapped with Parafilm, to minimize electrode capacitance. After seal formation and membrane rupture for whole-cell configuration, the neuron was given at least 5 min to stabilize before data were collected. Most recordings were performed at 30–31°C, except when noted.
Synaptic stimulation.
Synaptic inputs to the recorded neuron were activated by discrete electrical stimulation using theta glass pipettes with a tip size between 2–4 μm [filled with the following (in mm): 125 NaCl, 2.5 KCl, 1.25 NaH2PO4, 10 HEPES, 2 CaCl2, 2 MgCl2, 25 dextrose]. The electrodes were positioned within 20 μm of the apical dendrite at a distance of 150–200 μm from the soma. Stimulations were performed as single pulses (0.2 ms duration; 0.005–0.05 mA) or as trains (10 stimuli; 20 Hz).
Drug application.
Chemicals were bath applied in the recording solution, and included DA (1–10 μm; all drugs were from Sigma unless otherwise specified), (±)-6-chloro-7,8-dihydroxy-1-phenyl-2,3,4,5-tetrahydro-1H-3-benzazepine hydrobromide (SKF81297) (10 μm), quinpirole (10 μm), R(+)-7-chloro-8-hydroxy-3-methyl-1-phenyl-2,3,4,5-tetrahydro-1H-3-benzazepine hydrochloride (SCH23390) (5 μm), sulpiride (5 μm), TTX (1–2 μm), 4-AP (3 mm), CsCl (10 mm), CdCl2 (200 μm), BaCl2 (250 μm), tetraethylammonium (TEA) (1 mm), NiCl2 (50 μm), nimodipine (10 μm), 6,10-diaza-3(1,3)8,(1,4)-dibenzena-1,5(1,4)-diquinolinacy clodecaphane (UCL1684) (100 nm), forskolin (10–50 μm), 8-Br-cAMP (100–250 μm), and 4-ethylphenylamino-1,2-dimethyl-6-methylamino-pyrimidinium chloride (ZD7288) (20 μm). In a subset of experiments, as indicated, BAPTA tetrapotassium (20 mm), 9-(tetrahydro-2′-furyl)adenine (SQ22536) (1 mm; adenylyl cyclase blocker), (9R,10S,12S)-2,3,9,10,11,12-hexahydro-10-hydroxy-9-methyl-1-oxo-9,12-epoxy-1H-diindolo[1,2,3-fg-3′,2′,1′-K1]pyrrolo[3,4–1][1,6]benzodiazocine-10-carboxylic acid (KT5720) (1.2 μm; protein kinase A blocker), or ZD7288 (20 μm) were included in the recording pipette. When mentioned, synaptic transmission was blocked by the inclusion of CNQX (10 μm), dl-AP-5 (50 μm), bicuculline (10 μm; in DMSO), and (2S)-3-[[(1S)-1-(3,4-dichlorophenyl)ethyl]amino-2-hydroxypropyl](phenylmethyl)phosphinic acid (CGP55845) (2 μm; in DMSO) in the extracellular recording solution. The total concentration of DMSO in the bath remained <0.15%. On completion of the experiment, brain slices were transferred to 4% paraformaldehyde in 10% sucrose solution, in which they remained for at least 24 h. Slices were stained for Neurobiotin using the ABC Vectastain kit (Vector Laboratories, Burlingame, CA).
Data collection and analysis.
Voltage signals were amplified and filtered at 2 kHz (SEC-05L; NPI, Tamm, Germany) and collected via an ITC18 interface board (Instrutech, Port Washington, NY), transmitting to an Apple computer (PowerPC G5: Apple, Cupertino, CA) running Igor Pro software (WaveMetrics, Lake Oswego, OR). Before formation of a seal, the electrode tip potential was negated, and the open-tip resistance was measured (typically 4–6 MΩ). After successful transition to whole-cell configuration, series resistance was monitored and compensated throughout the experiment using built-in bridge circuitry. Only recordings with a stable series resistance <15 MΩ were retained for additional analysis. Membrane potential was tracked throughout the experiment in the presence and absence of a holding current. Only neurons that had a resting membrane potential of at least −60 mV and action potentials (APs) that exceed 0 mV were examined. To hold the membrane at various potentials, direct current was applied through the recording electrode.
Input resistance was determined from the linear fit of the neuronal voltage response to small (+40 to −40 pA; 500–700 ms) square-step direct-current injections from −70 mV. The initial membrane time constant of these small voltage responses was quantified by fitting with a least-squares fit to two exponentials. The voltage sag ratio in response to hyperpolarizing direct current steps of 700 ms was calculated as the peak voltage deflection divided by the amplitude of the steady-state voltage deflection. A hyperpolarizing direct current step that caused a peak 5–15 mV deflection was used for the purpose of this measurement. The normalized rebound voltage overshoot in response to hyperpolarizing direct current step was quantified as the peak rebound voltage deflection divided by the steady-state voltage deflection during the current step.
The action potential rise time was quantified as the peak of the first derivative (dV/dT) of the action potential waveform. The action potential threshold was defined as the peak of the third derivative of the action potential waveform. The action potential amplitude was defined as the difference between the action potential peak voltage and the initial membrane potential. The rheobase current was defined as the amplitude of the current injection to evoke a single action potential. Excitability was quantified as the number of action potentials in response to a fixed amplitude square step current injection. A slow afterhyperpolarizing potential (sAHP) was evoked by 5–10 suprathreshold direct current steps of 2–5 ms duration at 50 Hz, at a membrane potential of approximately −55 mV. The peak amplitude of the sAHP was measured relative to the baseline membrane potential. The fast afterhyperpolarizing potential (fAHP) evoked by a single action potential was measured as the peak hyperpolarizing deflection from action potential threshold. PSP amplitude was measured as the peak voltage deflection in response to synaptic stimulation. PSP summation was quantified as the ratio of the last PSP to the first PSP. Paired-pulse facilitation was quantified as the amplitude of the second PSP divided by the amplitude of the first PSP. In some experiments, a current shaped similar to a synaptic current was injected into the neuron (αPSP, current shape described by A(t/α)e−αt, where A is the amplitude of the injected current, t is time, and time to peak equals 1/α). The αPSPs evoked by this current injection were quantified and analyzed in the same manner as synaptic PSPs.
All planned comparisons were made with paired t tests, with an α level of 0.05 considered significant. Additional post hoc comparisons were made with Student's t tests. Bonferroni corrections of α values were used for multiple comparisons.
Results
A total of 241 neurons was recorded from layer V of lateral EC that displayed electrophysiological characteristics consistent with previously identified EC layer V pyramidal neurons (Hamam et al., 2002). Thus, they displayed a baseline action potential amplitude (104.9 ± 0.8 mV), half-width (1.16 ± 0.02 ms), Vrest (−69.3 ± 0.9 mV), and input resistance (89.3 ± 3.7 MΩ) consistent with previous reports, and regular spiking or adapting firing patterns. Neurons that were stained for biocytin (n = 67) displayed morphological characteristics consistent with EC layer V pyramidal neurons, such as a prominent apical dendrite and pyramidal or oblong-shaped soma (Fig. 1A).
Figure 1.
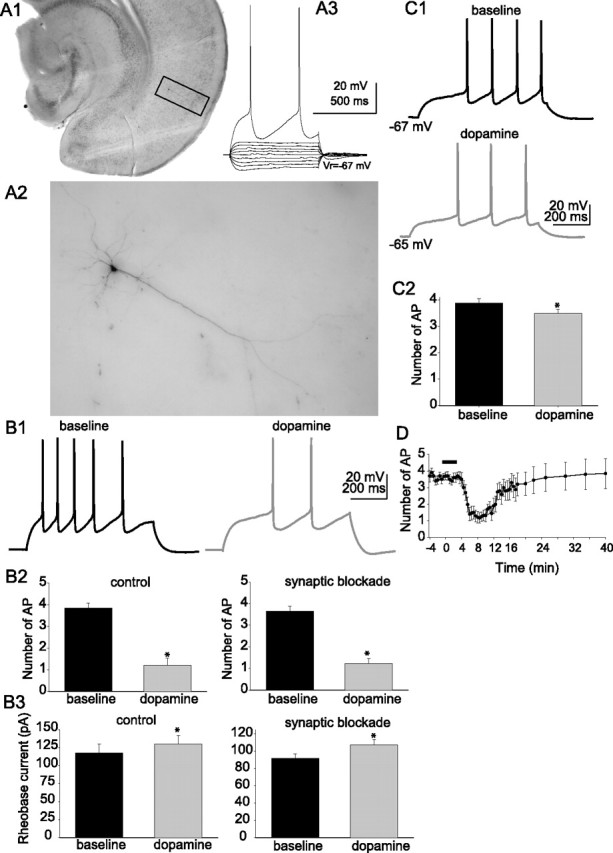
Dopamine decreases the excitability of layer V EC pyramidal neurons. A1–A3, Example of a Neurobiotin-filled pyramidal neuron from layer V of lateral EC. Also shown is the electrophysiological response of that filled neuron to a set of subthreshold step current injections from −100 to +100 pA in 25 pA steps, and a step current injection of 200 pA that evokes action potentials. The filled neuron displays morphological and electrophysiological similarities to previously described layer V EC pyramidal neurons. B1–B3, Application of DA (10 μm) decreased the excitability of layer V EC pyramidal neurons, measured as the number of action potentials evoked by a set current step (B1) from the same membrane potential (−70 mV; n = 11). This effect of DA persisted in the presence or absence of blockers of synaptic transmission (B2) (10 μm CNQX, 10 μm bicuculline, 2 μm CGP55845, 50 μm APV; n = 27) and increased the rheobase current (B3) (n = 27). C1, C2, DA decreased the excitability of layer V EC pyramidal neurons to a much lesser extent when the membrane was permitted to depolarize as a result of DA application (n = 6). D, After a 2–3 min application of DA (applied during black bar in plot), the excitability was reduced, and partially recovered after a prolonged washout. This plot was constructed from the data from 12 neurons whose excitability was continuously monitored. The last six data points are from eight of those neurons, whose excitability was monitored for a prolonged period of time. *Significant p value of at least 0.05. Error bars indicate SE.
DA modulation of excitability
Excitability was measured as the number of action potentials evoked by a fixed amplitude of current injection (700 ms duration) from a membrane potential of −70 mV. A current amplitude was chosen that evoked three to five action potentials. After short (2–3 min) bath application of 10 μm DA, there was a significant decrease in the number of evoked action potentials (Fig. 1B) (baseline, 3.86 ± 0.23 action potentials; post-DA, 1.13 ± 0.35 action potentials; n = 11; t = 8.59; p < 0.001; df = 11; paired t test). This was accompanied by an increase in the rheobase current (baseline, 117.3 ± 12.4 pA; post-DA, 129.6 ± 12.4 pA; n = 11; t = −4.50; p = 0.0011; df = 10; paired t test). In most instances, a small, ∼2 mV depolarization from Vrest was observed, which was offset with direct current injection to hold the membrane potential close to −70 mV.
In a separate set of experiments, excitability was examined in the absence of a holding current. This allowed the membrane potential to depolarize after DA application (n = 6; baseline, −68.8 ± 0.9 mV; post-DA, −65.8 ± 0.9 mV; t = −5.81; p = 0.0021; df = 5; paired t test). Nevertheless, there was a significant, albeit smaller, decrease in excitability (Fig. 1C) (baseline, 3.9 ± 0.16 action potentials; post-DA, 3.5 ± 0.15 action potentials; t = 2.88; p = 0.034; df = 5; paired t test).
In some preliminary observations, it was noted that application of DA was accompanied by changes in the occurrence of postsynaptic potentials. Because alterations of synaptic conductances can conceivably induce changes of excitability, these and all subsequent experiments were performed in the presence of blockers for glutamatergic and GABAergic inputs, unless otherwise noted (see Materials and Methods). Application of 10 μm DA still resulted in a decrease of excitability (baseline, 3.65 ± 0.23 action potentials; post-DA, 1.22 ± 0.21 action potentials; n = 27; t = 7.39; p < 0.001; df = 26; paired t test), an increase in rheobase current (baseline, 91.9 ± 4.9 pA; post-DA, 107.0 ± 6.1 pA; n = 27; t = −6.27; p < 0.0001; df = 26; paired t test) (Fig. 1B), and a 2–3 mV depolarization. The persistence of these effects of DA when synaptic inputs were blocked indicated that they were likely attributable to direct postsynaptic actions. The onset of the effect on excitability could be observed within 3–4 min after the start of the DA application and persisted after the 2–3 min DA application (Fig. 1D). The effects of DA on excitability reversed to near-baseline values after washout periods close to 15 min.
Application of DA also resulted in a small decrease of the measured input resistance (Fig. 2A) (RN baseline, 105.3 ± 6.7 MΩ; post-DA, 95.6 ± 4.9 MΩ; n = 32; t = 2.63; p = 0.013; df = 31; paired t test), measured from −70 mV. The membrane time constant, measured at the beginning of 3–5 mV hyperpolarizing steps from the same membrane potential, also decreased after application of DA (Fig. 2B) (baseline, 41.7 ± 4.1 ms; post-DA, 33.0 ± 2.4 ms; n = 32; t = 3.99; p = 0.003; df = 31; most waveforms were well fit with the first, primary, exponential, which is the value reported here). The faster membrane time constant and decreased RN are consistent with enhancement of a conductance that is active near Vrest. These changes of excitability and input resistance were not attributable to washout of intracellular components, or other time-dependent factors, because no substantial change of excitability or input resistance was observed in a series of control experiments that were performed in the same manner, but with the exclusion of DA application (baseline excitability, 3.8 ± 0.1 action potentials; post-pseudo-DA application, 3.9 ± 0.2 action potentials; n = 6; t = 1.35; p = 0.235; paired t test; baseline RN, 101. 2 ± 4.4 MΩ; post-pseudo-DA application, 103.0 ± 4.3 MΩ; n = 6; t = 1.86; p = 0.122; paired t test).
Figure 2.
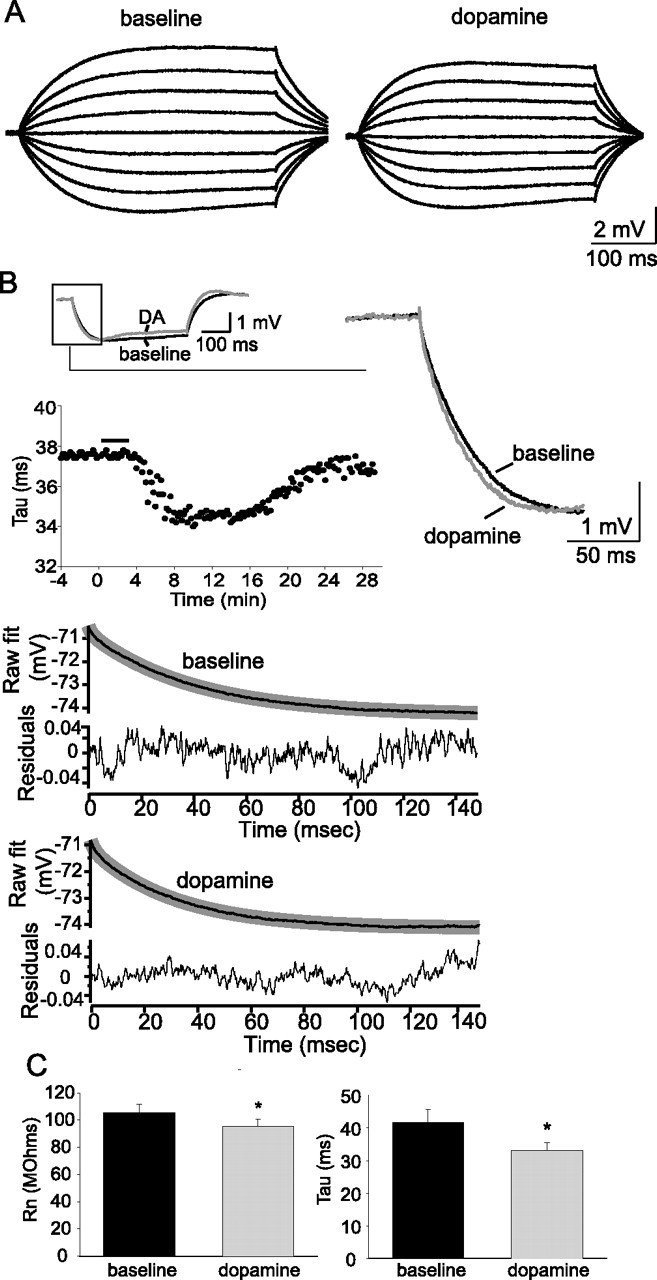
Dopamine modulates membrane properties of layer V EC pyramidal neurons. A, Application of DA (10 μm) reduced the input resistance, measured from −70 mV as the response to small current steps (−40 to +40 pA). B, Application of DA also sped the membrane time constant (Tau), measured by least-squares fitting of two exponentials to the beginning of the voltage response to a small current step (traces displayed are an average of 10 consecutive sweeps). The time course of the change of membrane time constant is plotted below the trace. The bottom plots demonstrate the fitted exponential (thin black line) for the averaged voltage response displayed in B and the residuals. C, Group data for the effects of DA on RN (n = 32) and Tau (n = 32). *Significant p value of at least 0.05. Error bars indicate SE.
In a separate group of neurons, the effects of DA were examined with the membrane potential held at −70 mV with continuous direct current, and also at a more depolarized potential that was still subthreshold to spontaneous action potential firing (close to −50 mV, smaller direct current steps were used to evoke action potentials). The effect of DA on excitability was eliminated at the depolarized membrane potential (Fig. 3A,B) (n = 5; −70 mV baseline, 3.7 ± 0.2; post-DA, 0.63 ± 0.3; t = 8.34; p = 0.001; df = 4; paired t test; close to threshold baseline, 3.8 ± 0.2; post-DA 4.3 ± 0.3; t = 2.6; p = 0.06; df = 7; paired t test). To examine this more systematically, the membrane potential was shifted through a range of voltages (−85 to −50 mV). A current intensity was used that evoked approximately four action potentials from a baseline membrane potential of −70 mV. To exclude the possibility that the effects of DA were specific to a small range of current intensities, two different amplitudes of current steps were also used, while shifting the membrane potential through this range of voltages. The two other current intensities evoked one action potential from a baseline membrane potential of −70 mV or approximately eight action potentials from −70 mV. As above, application of DA reduced excitability measured from −70 mV at these three frequencies. However, the reduction of excitability observed after application of DA was reduced at depolarized potentials, often reversing at more depolarized potentials (Fig. 3C) (n = 7). This may be consistent with two possibilities: (1) DA may modulate an ionic conductance that is deactivated by prolonged depolarization, or (2) the effect is simply dependent on the amplitude of the direct current step used to evoke action potentials, and the effects will not be observed with a wider range of steps. To determine whether this alone, independent of the membrane potential, could explain the results, a series of direct current step amplitudes was examined, from a potential close to −70 mV. The effects of DA remained throughout a range of stimulus amplitudes (Fig. 3B2) (n = 12). However, the effects were larger with greater amplitude direct current steps. One possibility is that DA modulated a current that was both dependent on the absolute “resting” potential as well as the relative change of potential during a direct current step. These features are consistent with a current that is voltage dependent, in that the effects of DA were greatest when greater changes of voltage were used, and the current was deactivated by prolonged depolarization, as the effects were minimized at continuous depolarized potentials.
Figure 3.

The effects of dopamine depend on the membrane voltage. A1, A2, Application of DA reduced the excitability of layer V EC neurons, when action potentials were evoked from −70 mV, but not from a more depolarized membrane potential close to action potential threshold (−55 to −50 mV in this plot; n = 8). B1, B2, The effects of DA on excitability persisted over a range of stimulation intensities (n = 12; the x-axis represents multiplicatives of the threshold stimulation intensity). The action potentials were evoked by current injection at a baseline membrane potential of −70 mV. C, The voltage dependence of the effects of DA on excitability were maintained over a series of firing frequencies when the neuron was shifted through a range of membrane potentials (n = 7) (for more details, see Results). In this panel, three different amplitudes of step current injection were alternatingly used while the membrane potential was shifted through a range of voltages: stimulation intensity 1, a current injection that evokes one AP from a baseline membrane potential of −70 mV; stimulation intensity 2, a current injection that evokes approximately four APs from a baseline membrane potential of −70 mV; and stimulation intensity 3, a current injection that evokes approximately eight APs from a baseline membrane potential of −70 mV. *Significant p value of at least 0.05. Error bars indicate SE.
Excitability: ion channels
Layer V EC neurons are likely to express an array of voltage-dependent and -independent conductances. The results above suggest that a voltage-dependent conductance may be involved in the effects of DA. To further understand what ion channels may be modulated by DA, and thereby alter excitability, the contributions of voltage-dependent and -independent ion channels were pharmacologically probed. Application of 4-AP (3 mm), a blocker of the voltage-dependent delayed K+ current (ID) (Hermann and Gorman, 1981; Segal and Barker, 1984; Zagotta et al., 1988), and partial blocker of the voltage-dependent transient K+ current (IA) (Thompson, 1982; Hoffman et al., 1997) enhanced the excitability of layer V EC neurons (Table 1). Application of TEA (1 mm), a blocker of sustained K+ currents (Zagotta et al., 1988; Hoffman et al., 1997) increased the number of action potentials in response to depolarizing direct current step injection (Table 1). Application of Ba2+ (250 μm), a blocker of inwardly rectifying K+ currents (IRK) and M-currents, did not significantly increase the excitability of layer V EC neurons (Table 1). Application of UCL1684 (100 nm), a blocker of BK channels, had little effect on excitability (Table 1). Intracellular application of BAPTA (20 mm), a buffer of intracellular calcium, had no significant effect on excitability, compared with control neurons (Table 1), although BAPTA and UCL1684 caused bursting in some neurons, a feature otherwise absent. Extracellular application of Ni2+ (50 μm)/nimodipine (10 μm) or Cd2+ (200 μm), all of which are expected to block certain types of Ca2+ channels, had no significant effect on excitability (Table 1). This is additional evidence that calcium-activated K+ channels have minimal influence on the excitability of these neurons as measured in these experiments. In conjunction with the previous experiments, this indicates that L- or T-type calcium channels are unlikely to influence excitability under these conditions, as quantified here. Application of Cs+ (10 mm) greatly increased the excitability of these neurons, indicating a potential role of IRK or hyperpolarization-activated currents (Ih) in regulating the excitability of these neurons (Table 1). Furthermore, application of ZD7288 (20 μm) enhanced the excitability of layer V EC neurons (Table 1), further suggesting that h-channels modulate the excitability. Application of TTX (1–2 μm) blocked action potentials in these neurons. From these data, it appears that several K+ currents and Ih strongly influence the excitability of layer V EC pyramidal neurons under basal conditions, and may be the most likely targets for the observed actions of DA on excitability.
Table 1.
Effects of ion channel blockers on excitability
| Drug | Baseline | After drug |
|---|---|---|
| K+-channel blocker | ||
| TEA (1 mm; n = 8) | 3.76 ± 0.31 | 4.71 ± 0.44* |
| 4-AP (3 mm; n = 9) | 3.54 ± 0.28 | 6.10 ± 0.29* |
| Ba2+ (250 μm; n = 6) | 3.81 ± 0.25 | 4.13 ± 0.26† |
| Cs+ (10 mm; n = 6) | 3.72 ± 0.20 | 6.83 ± 0.23* |
| Ca2+-channel blocker | ||
| Cd2+ (200 μm; n = 5) | 3.69 ± 0.24 | 3.73 ± 0.23† |
| Nimodipine/Ni2+ (10/50 μm; n = 6) | 3.85 ± 0.22 | 3.92 ± 0.22† |
| Ca2+-activated K+-channel blocker | ||
| UCL1684 (100 nm; n = 7) | 3.81 ± 0.23 | 3.94 ± 0.27† |
| h-channel blocker | ||
| ZD7288 (20 μm; n = 11) | 3.61 ± 0.26 | 6.47 ± 0.25* |
* p < 0.05, paired t test.
†Not significant.
Although these data demonstrate that numerous ion channels may be involved in regulating the excitability of layer V EC neurons, the effects of DA on excitability were only blocked by preapplication of ZD7288 (20 μm) or cesium (10 mm) (Figs. 4–6, Table 2) (these chemicals also reduced the DA-induced depolarization of the resting membrane potential in 6 of 11 neurons (ZD7288; baseline, −72.6 ± 1.1 mV; post-DA, −71.7 ± 1.3 mV) and 6 of 9 neurons (Cs+; baseline, −69.9 ± 0.8 mV; post-DA, −69.4 ± 1.0 mV). One shared characteristic of ZD7288 and Cs+ is their ability to block channels responsible for Ih. The results are thus consistent with an effect on excitability through DAergic modulation of Ih. Modulation of Ih is also congruent with the reduction of the apparent efficacy of DA when action potentials are evoked with small-amplitude direct current steps from a depolarized membrane potential (Fig. 2). Furthermore, the DAergic modulation of excitability was not blocked by any other compounds that block K+ channels (Fig. 4, Table 2), Ca2+ channels, or Ca2+-activated K+ channels (Fig. 5, Table 2). To verify that the voltage-dependent reduction of excitability after application of DA was dependent on h-channels, excitability was examined from a range of membrane potentials in the presence of ZD7288. After preapplication of ZD7288, there was no longer a voltage-dependent reduction of excitability (Fig. 6). Additional evidence consistent with a DA-induced enhancement of Ih would include a decrease of input resistance (Fig. 2) and an increase in the hyperpolarization-evoked voltage sag, examined below.
Figure 4.
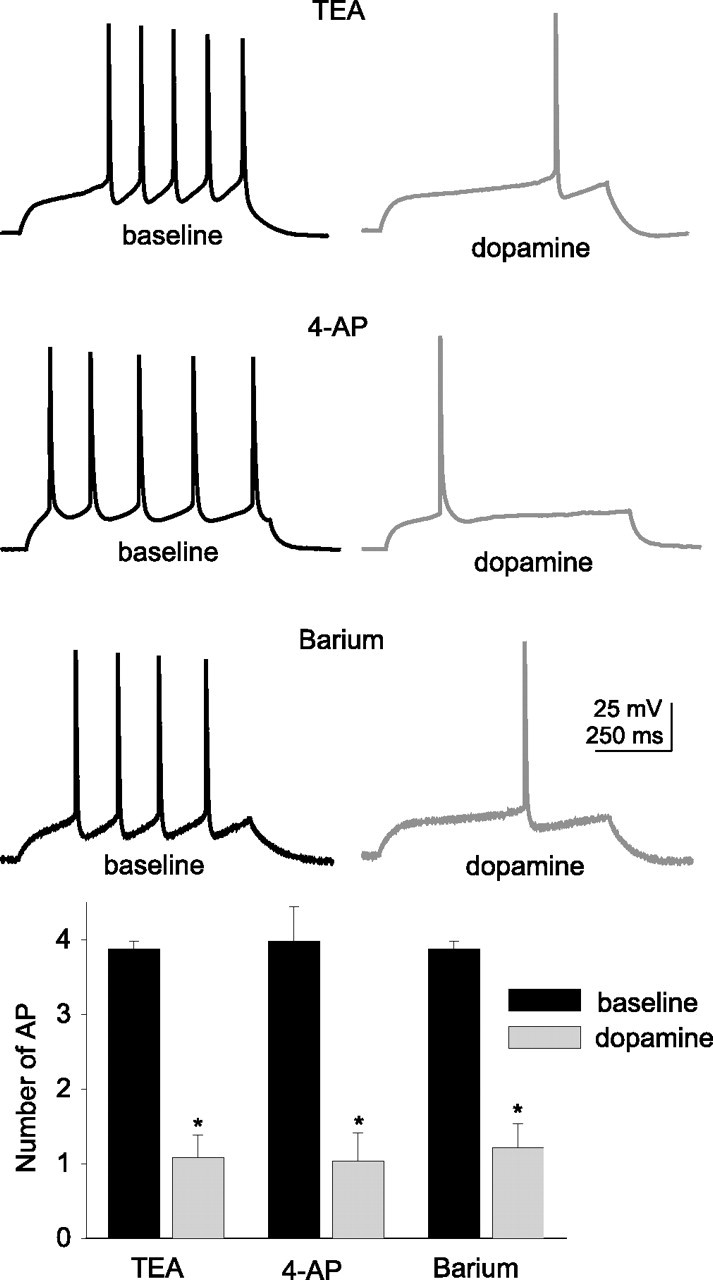
Blockade of select K-channels does not block the actions of dopamine on the excitability of layer V EC pyramidal neurons. Preapplication of TEA (1 mm; n = 6), 4-AP (3 mm; n = 8), or barium (250 μm; n = 7) did not block the DA-induced reduction of excitability of layer V EC pyramidal neurons. These pharmacological agents were applied to the bath at least 15 min before DA was applied. *Significant p value of at least 0.05. Error bars indicate SE.
Figure 5.
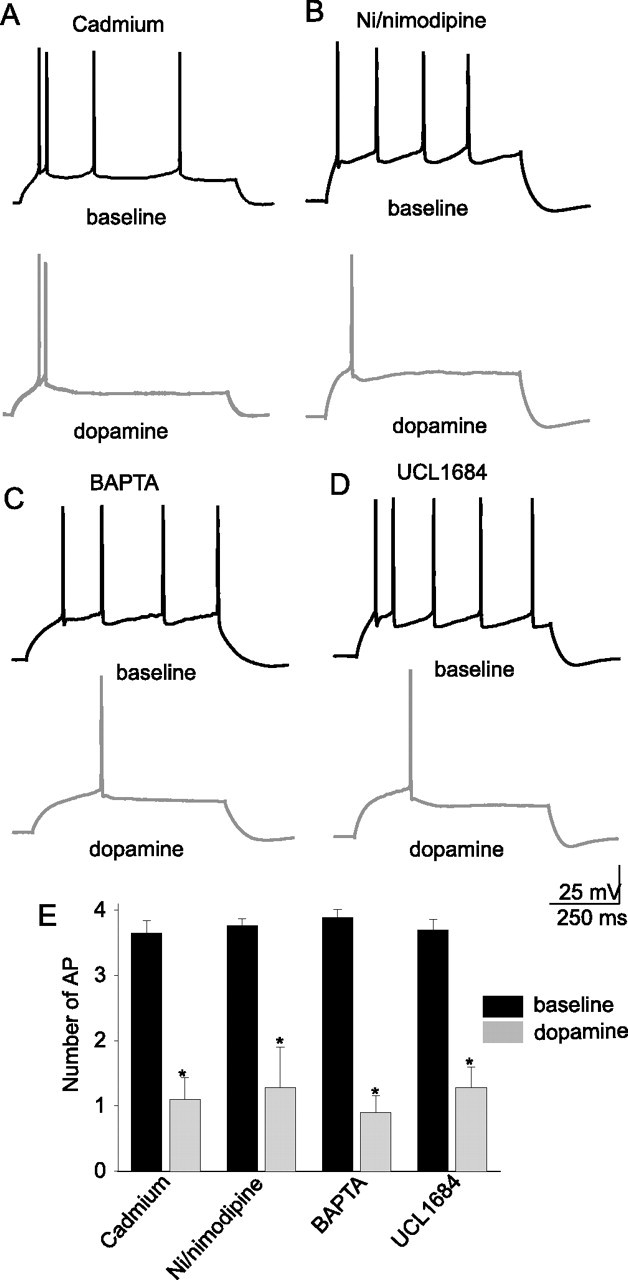
Manipulation of calcium-related events does not block the actions of DA on the excitability of layer V EC pyramidal neurons. Blockade of a subset of voltage-dependent calcium channels by cadmium (200 μm; n = 5) (A) or nickel/nimodipine (50/10 μm) (B) did not attenuate the DA-mediated reduction of neuronal excitability in layer V EC pyramidal neurons. Neither did inclusion of a high concentration of BAPTA (20 μm; n = 5) in the recording pipette (C) or blockade of BK channels by UCL1684 (100 nm; n = 6) (D). *Significant p value of at least 0.05. Error bars indicate SE.
Figure 6.

Blockade of h-channels by nonspecific and specific antagonists reduces the effects of dopamine on excitability of layer V EC pyramidal neurons. Preapplication of cesium (10 mm; n = 9) (A) or ZD7288 (20 μm; n = 11) (B) attenuated the effects of DA (10 μm) on neuronal excitability (C1). This effect was maintained over a range of current injection intensities (C2) and a range of membrane potentials (D), indicating that blockade of Ih abolished the voltage-dependent attenuation of excitability caused by DA. In these experiments, there was even a tendency toward an enhancement of excitability after DA application when h-channels were blocked. Cesium (E) and ZD7288 (F) also blocked the effects of DA on input resistance. G, Group data demonstrate that preapplication of 10 mm cesium (n = 9) or 20 μm ZD7288 (n = 11) attenuated the DA-mediated reduction of input resistance (F). Error bars indicate SE.
Table 2.
Suppression of the DAergic effects on excitability by pharmacological blockade of h-channels
| Drug | Baseline (with drug) | After DA |
|---|---|---|
| K+-channel blockers | ||
| TEA (1 mm; n = 6) | 3.88 ± 0.10 | 1.08 ± 0.30* |
| 4-AP (3 mm; n = 6) | 3.98 ± 0.47 | 1.04 ± 0.37* |
| Ba2+ (250 μm; n = 7) | 3.88 ± 0.10 | 1.21 ± 0.33* |
| Cs+ (10 mm; n = 9) | 3.96 ± 0.10 | 4.38 ± 0.09† |
| Ca2+-channel blockers | ||
| Cd2+ (200 μm; n = 5) | 3.65 ± 0.19 | 1.10 ± 0.33* |
| Nimodipine/Ni2+ (10/50 μm; n = 5) | 3.76 ± 0.11 | 1.28 ± 0.62* |
| Ca2+ chelator | ||
| BAPTA (20 mm; n = 5) | 3.89 ± 0.12 | 0.80 ± 0.26* |
| Ca2+-activated K+- channel blocker | ||
| UCL1684 (100 nm; n= 6) | 3.70 ± 0.16 | 1.28 ± 0.32* |
| h-channel blocker | ||
| ZD7288 (20 μm; n = 12) | 3.76 ± 0.19 | 3.90 ± 0.62† |
* p < 0.05, paired t test.
†Not significant.
The change in input resistance can be a significant contributor to the alterations of excitability. Therefore, we examined which ion channels normally contribute to the apparent input resistance of these neurons close to resting potentials (Table 3). Application of barium (250 μm), TEA (1 mm), Cs+ (10 mm), or ZD7288 (20 μm) all increased the input resistance, measured close to −70 mV. Application of 4-AP (3 mm) produced only a small change of input resistance measured at −70 mV. Application of Ni2+ (50 μm), nimodipine (10 μm), and UCL1684 (100 nm), blockers of Ca2+ currents and Ca2+-activated K+ currents, had no significant effects on the measured input resistance. Thus, of all the compounds that exert a significant effect on excitability, only those that block h-currents, sustained K+ currents, or IRK channels also appeared to alter the input resistance near the resting membrane potential.
Table 3.
Blockade of DAergic effects on input resistance by blockers of h-channels
| Drug | Baseline (with drug) | After DA |
|---|---|---|
| K+-channel blockers | ||
| TEA (1 mm; n = 6) | 157.1 ± 6.2 | 141.9 ± 5.8* |
| 4-AP (3 mm; n = 8) | 121.5 ± 5.3 | 109.7 ± 5.7* |
| Ba2+ (250 μm; n = 7) | 144.1 ± 4.9 | 131.4 ± 4.2* |
| Cs+ (10 mm; n = 9) | 176.8 ± 4.4 | 178.1 ± 5.1† |
| Ca2+-channel blockers | ||
| Cd2+ (200 μm; n = 5) | 110.6 ± 6.1 | 92.7 ± 5.4* |
| Nimodipine/Ni2+ (10/50 μm; n = 5) | 108.0 ± 5.0 | 95.0 ± 4.8* |
| Ca2+ chelator | ||
| BAPTA (20 μm; n = 5) | 117.2 ± 6.3 | 107.9 ± 6.1* |
| Ca2+-activated K+-channel blocker | ||
| UCL1684 (100 nm; n = 6) | 103.3 ± 4.8 | 92.5 ± 4.9* |
| h-channel blocker | ||
| ZD7288 (20 μm; n = 11) | 144.2 ± 4.2 | 149.7 ± 4.2† |
* p < 0.05, paired t test.
†Not significant.
The effects of DA on input resistance measured at −70 mV were blocked by ZD7288 (20 μm) or by cesium (10 mm), but not by any of the other compounds listed above (Fig. 3, Table 3), indicating that the effects of DA on the apparent input resistance were likely attributable to modulation of h-channels. Furthermore, these results provide additional evidence for an association between the effects of DA on RN and on excitability. In addition, the effects of DA on the membrane time constant were blocked by ZD7288 (20 μm; baseline with ZD7288, 70.8 ± 11.4 ms; post-DA with ZD7288, 73.9 ± 13.9 ms; n = 11; t = 0.907; p = 0.399; paired t test).
To further investigate the target of DAergic modulation observed in this study, other electrophysiological parameters were compared before and after DA. The transient Na+ current (INa) was monitored by measuring the peak amplitude and rise time of the action potential peak before and after DA. There was no significant difference in the action potential amplitude, although many neurons displayed a tendency for an increase (baseline, 104.8 ± 1.1 mV; post-DA, 105.7 ± 1.0 mV; n = 27; t = 0.27; p = 0.788; df = 26; paired t test). The action potential rise time did not significantly change (baseline, 96.8 ± 5.53; post-DA, 99.8 ± 5.77; t = −0.75; df = 17; p = 0.46). There was no significant change in the action potential threshold after application of DA (baseline, −38.3 ± 0.8 mV; post-DA, −39.0 ± 0.8 mV; n = 18; t = 1.89; p = 0.075; df = 17; paired t test).
To examine currents that underlie the afterhyperpolarization potential (AHP), both the fAHP evoked by a single action potential and the sAHP evoked by a train of action potentials were measured before and after DA. The amplitude of the fAHP decreased after application of DA (Fig. 7A) (baseline, 10.5 ± 0.6 mV; post-DA, 9.6 ± 0.5 mV; n = 27; t = 2.76; p = 0.011; df = 26; paired t test). A reduction of fAHP would not be expected to cause the decrease of excitability observed after application of DA. In conjunction with the pharmacological evidence that demonstrates that the effects of DA were not reduced when BK channels were blocked, this suggests that BK channels are unlikely to be the target of DA in its actions that reduce excitability. The amplitude of the sAHP, measured from a membrane potential of near −55 mV, was also decreased after application of DA (Fig. 7B) (baseline, 8.4 ± 0.9 mV; post-DA, 5.9 ± 0.8 mV; n = 10; t = −2.99; p = 0.015; df = 9; paired t test), as seen in other studies (Malenka and Nicoll, 1986; Pedarzani and Storm, 1995a). This sAHP was reduced in the presence of UCL1684 (100 nm; baseline, 8.1 ± 0.9 mV; post-UCL, 1684 2.1 ± 0.6 mV; n = 4) or Ni2+ (50 μm; baseline, 8.7 ± 1.1 mV; post-Ni2+, 2.6 ± 1.2 mV; n = 4). To ensure that Ih was not contaminating our measure of the effects of DA on the sAHP, the sAHP was also examined in the presence of ZD7288 (20 μm). DA still reduced the amplitude of the sAHP when Ih was blocked by ZD7288 (baseline, 7.7 ± 0.8 mV; post-DA, 5.1 ± 1.1 mV; n = 5; p = 0.022; df = 4; paired t test).
Figure 7.
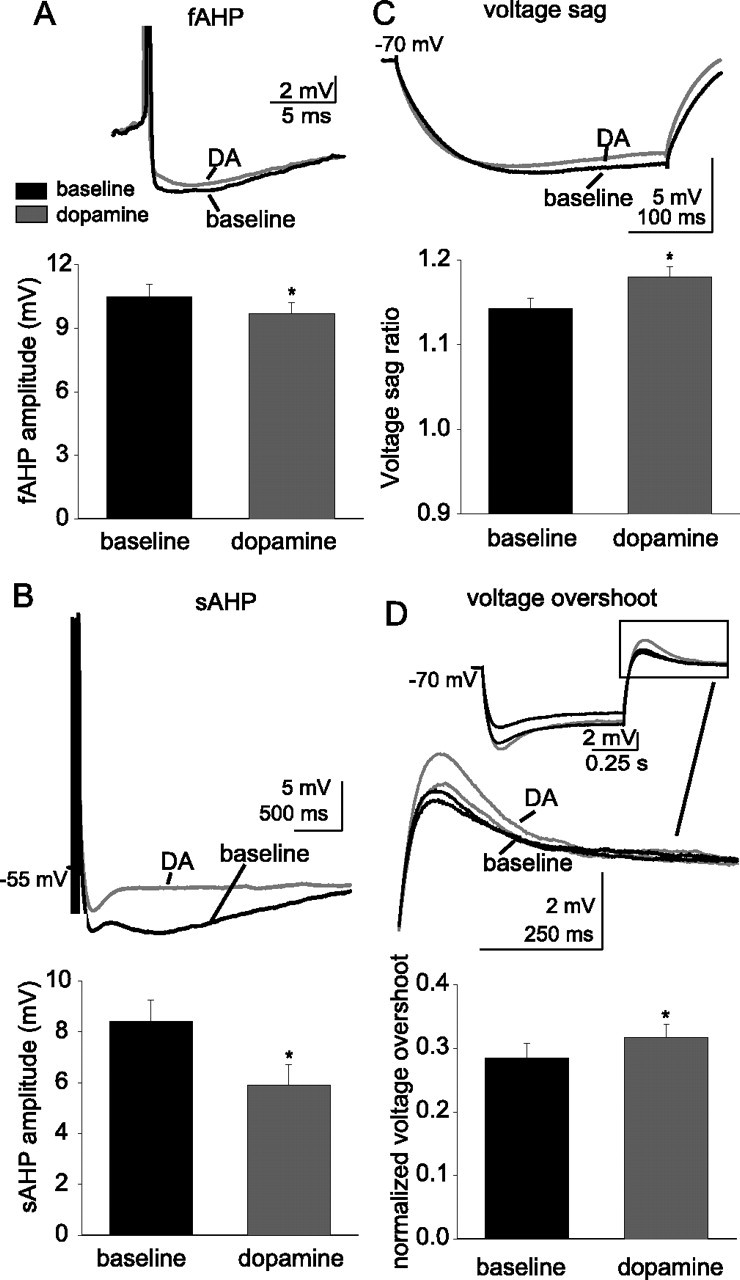
Application of dopamine enhances voltage sag in response to a hyperpolarizing current step. Application of 10 μm DA reduced the amplitude of the fAHP evoked by a single action potential (A) (n = 27) and reduced the amplitude of the sAHP evoked by a train of action potentials (B) (n = 10). Therefore, it is unlikely that changes in fAHP or sAHP could account for the DA-induced reduction of excitability. The voltage sag that can be observed after hyperpolarization of the membrane with current steps was enhanced after DA application (C) (n = 32), measured as the ratio between the voltage deflection at steady-state and peak voltage changes. Additionally, the voltage overshoot observed after cessation of the current step was also enhanced after application of DA (D) (n = 23). The black traces are baseline conditions, and the red traces are after DA application. *Significant p value of at least 0.05. Error bars indicate SE.
The voltage sag during prolonged hyperpolarizing direct current steps and the rebound overshoot after cessation of the current step are hallmarks of the presence of Ih. To obtain independent, nonpharmacological evidence for a role of h-channels, the voltage sag ratio in response to a prolonged (700 ms) hyperpolarizing direct current step and the normalized voltage rebound were examined. The voltage sag persisted in the presence of barium (250 μm), so it is unlikely to be attributable to M-currents or KIRs in these neurons (Armstrong et al., 1982; Sakmann and Trube, 1984; Gibor et al., 2004; Prole and Marrion, 2004). The voltage sag ratio was increased after application of DA (Fig. 7C) (baseline, 1.14 ± 0.01; post-DA, 1.19 ± 0.01; n = 32; t = −2.37; p = 0.02; df = 31; paired t test). The peak amplitude of the normalized rebound voltage deflection was also enhanced after DA application (Fig. 7D) (baseline, 0.29 ± 0.02; post-DA, 0.32 ± 0.02; n = 23; t = −2.65; df = 22; p = 0.015; paired t test).
A subset of key experiments was replicated at more physiological temperatures (∼34°C). At this temperature, similar effects of DA were still observed. Thus, DA diminished the number of action potentials evoked from equivalent amplitudes of current injection (baseline number of action potentials, 4.0 ± 0.2; post-DA, 1.53 ± 0.7; n = 5; t = 3.52; df = 4; p = 0.024; paired t test), and increased the voltage sag (baseline voltage sag ratio, 1.16 ± 0.01; post-DA, 1.21 ± 0.01; n = 5; t = 2.37; df = 4; p < 0.01; paired t test). Furthermore, at this temperature, the effects of DA on excitability were blocked by ZD7288 (20 μm; baseline number of action potentials, 3.6 ± 0.3; post-DA, 3.9 ± 0.41; n = 6; t = 2.80; p = 0.038; df = 5; paired t test). Overall, these data are consistent with the conclusion that DA receptors decrease membrane excitability in these neurons predominantly via an increase in h-currents.
Modulation of dendritic excitability
Several studies from other brain regions indicate that most of pyramidal neuronal h-channels are preferentially located in the apical dendrites, HCN channels are found on the apical dendrites of pyramidal neurons (Lorincz et al., 2002; Notomi and Shigemoto, 2004), and h-currents are greater in dendritic recordings, compared with somatic recordings (Magee, 1998; Berger et al., 2001; Shah et al., 2004). To examine whether DA modulates dendritic h-channels, direct voltage recordings were obtained from the apical dendrites of layer V EC neurons, at a distance of up to 225 μm from the soma. The amplitude of the voltage sag in response to hyperpolarizing step current injection was more prominent in dendritic recordings (Fig. 8A) (normalized voltage sag at the soma, 1.14 ± 0.01; n = 32; in the dendrites, 1.20 ± 0.02; n = 25; t = −2.7; p = 0.007; df = 55; t test), with an apparent progression with distance. Application of DA (10 μm) enhanced the amplitude of the voltage sag (Fig. 8B) (baseline normalized voltage sag, 1.19 ± 0.03; post-DA, 1.26 ± 0.06; t = −2.35; p = 0.04; df = 9; paired t test; the effect size was greater in the dendrites than in the soma, at 0.07 vs 0.05, or an effect size ∼30% greater), and decreased the apparent input resistance (Fig. 8B). In conjunction with this increased voltage sag was a decrease in the dendritic excitability, as measured by the number of action potentials evoked by a fixed amplitude of current injection (Fig. 8C) (baseline, 3.96 ± 0.31 action potentials; post-DA, 0.42 ± 0.15 action potentials; t = 13.7; p < 0.0001; df = 11; paired t test). The amplitude of the effect of DA was greater in the dendrites compared with the soma (3.54 vs 2.43 action potentials). Furthermore, although there was no significant difference in the baseline number of action potentials (dendrites, 3.96 ± 0.31 action potentials; soma, 3.86 ± 0.23 action potentials; df = 37; t = 0.770; p = 0.446; t test), after application of DA there was a significantly lower number of action potentials in the dendrites compared with the soma (dendrites, 0.42 ± 0.15 action potentials; soma, 1.22 ± 0.21 action potentials; df = 37; t = −2.41; p = 0.021). Thus, the increased amplitude of the voltage sag in the dendrites, in conjunction with the greater effect of DA on excitability in the dendrites, may indicate that there is a preferential distribution of h-channels in the apical dendrites of these neurons.
Figure 8.

Dopamine modulates dendritic excitability and voltage sag. A, The amplitude of the voltage sag in response to a prolonged hyperpolarizing step current, a reflection of h-channel activation, increases at more distal apical dendritic recording sites. This plot depicts somatic recordings (at 0 μm; mean ± 2 SDs) and the sag ratio recorded from the apical dendrite at variable distances from the soma. B, Application of DA (10 μm) increases the amplitude of the voltage sag (n = 10; 150–225 μm from the soma). The amplitude of this effect is greater in the dendritic recordings than in the somatic recordings. C, In parallel with the enhanced voltage sag, the excitability of the dendrites is also reduced after application of DA (n = 12; 150–225 μm from the soma). The black traces are baseline conditions, and the red traces are after DA application. *Significant p value of at least 0.05. Error bars indicate SE.
Excitability: DA receptor subtype and second messenger system
To determine which DA receptors are involved in the reduction of excitability and input resistance, two approaches were used: specific agonists for DA D1 or D2 receptors were used, and specific antagonists for DA D1 or D2 receptors were applied at least 10 min before application of DA. Application of the DA D1 agonist SKF81297 (10 μm) mimicked the effects of DA, decreasing the excitability of layer V EC pyramidal neurons (Fig. 9A) (baseline, 3.27 ± 0.48 action potentials; post-SKF81297, 1.47 ± 0.39 action potentials; n = 6; t = 4.58; p = 0.0059; df = 5; paired t test). In addition, preapplication of the DA D1 antagonist SCH23390 (5 μm) prevented the DA-induced decrease in excitability usually observed with application of 10 μm DA (Fig. 9B) (baseline SCH23390, 3.70 ± 0.37 action potentials; post-DA, 3.93 ± 0.43; n = 5; t = −2.18; p = 0.117; df = 4; paired t test). The effects of DA were not mimicked by the DA D2 agonist quinpirole (10 μm), which tended to increase the excitability of layer V EC pyramidal neurons (Fig. 9C) (baseline, 3.51 ± 0.18 action potentials; post-quinpirole, 4.14 ± 0.18 action potentials; n = 6; t = −2.22; p = 0.077; df = 5; paired t test). Preapplication of the DA D2 antagonist sulpiride (5–10 μm) did not significantly reduce the effects of DA (10 μm) on excitability (Fig. 9D) (baseline sulpiride, 3.76 ± 0.31 action potentials; post-DA, 3.00 ± 0.51; n = 5; t = 2.26; p = 0.87; df = 4; paired t test). This is consistent with a DA D1 receptor-mediated effect of DA in reducing layer V EC neuronal excitability.
Figure 9.
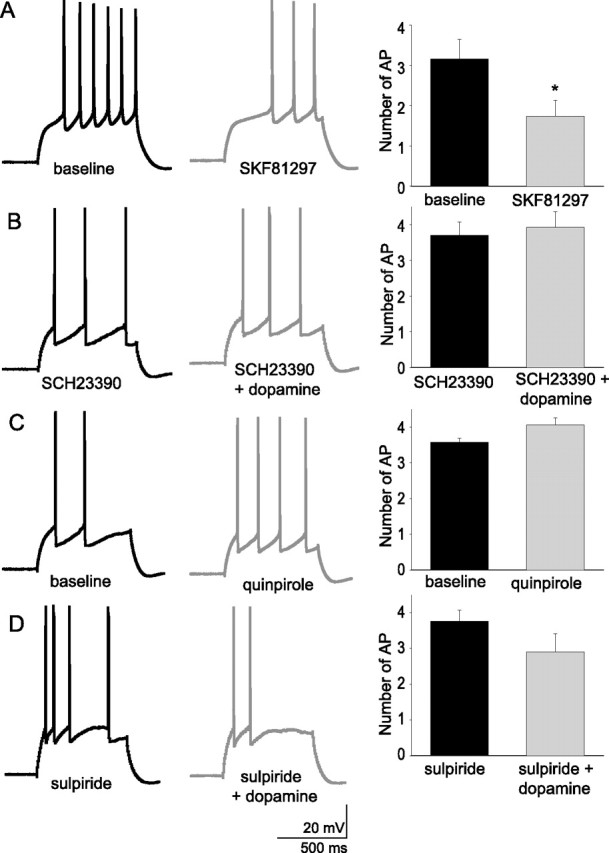
The actions of dopamine on the excitability of layer V EC pyramidal neurons is likely mediated by activation of D1 receptors. Application of the DA D1 agonist SKF81297 (10 μm; n = 6) mimicked the effects of dopamine on excitability (A), and the effects of DA were attenuated by preapplication of the DA antagonist SCH23390 (5 μm; n = 5) (B). The effects of DA were not mimicked by the DA D2 agonist quinpirole (10 μm; n = 6) (C), and the effects of DA were not blocked by preapplication of the DA D2 antagonist sulpiride (5–10 μm; n = 5) (D). *Significant p value of at least 0.05. Error bars indicate SE.
Traditionally, DA receptors are thought to affect cellular events by modulation of cAMP levels via G-protein-mediated alteration of adenylyl cyclase activity. To determine whether the effects of DA are mediated via the cAMP pathway, activators and inhibitors of the cAMP pathway were used. The effects of DA (10 μm) were greatly attenuated when the blocker of adenylyl cyclase, SQ22536, (1 mm) was included in the recording pipette, suggesting that the effects of DA were likely mediated by DA-induced activation of adenylyl cyclase (Fig. 10) (baseline, 3.65 ± 0.20 action potentials; post-DA, 3.19 ± 0.30 action potentials; n = 6; t = 1.48; p = 0.20; df = 5; paired t test). It is known that cAMP can exert actions directly on ion channels or can activate protein kinase A, which phosphorylates many ion channels (Gershon et al., 1992; Braha et al., 1993; Pedarzani and Storm, 1993; Drain et al., 1994; Cantrell et al., 1997; Smith and Goldin, 1997). To determine whether cAMP was responsible for, or mimicked the effects of DA, forskolin (50 μm) or 8-Br-cAMP (100–250 μm) were bath-applied. Neither forskolin nor 8-Br-cAMP mimicked the effects of DA (Fig. 10) (baseline, 3.4 ± 0.3 action potentials; post-forskolin, 5.6 ± 0.5 action potentials; n = 10; t = −4.58; p < 0.001; df = 9; paired t test; baseline, 3.78 ± 0.10 action potentials; post-8-Br-cAMP, 3.96 ± 0.27 action potentials; n = 5; t = −0.78; p = 0.48; df = 4; paired t test). However, when KT5720 (1.2 μm; a PKA inhibitor) was included in the recording pipette, 8-Br-cAMP (100–250 μm) now mimicked the action of DA (Fig. 10B) (baseline, 3.80 ± 0.21 action potentials; post-8-Br-cAMP, 1.63 ± 0.47 action potentials; n = 6; t = 4.12; p = 0.0092; df = 5; paired t test), and the excitatory effect of forskolin was greatly attenuated (Fig. 10B) (baseline, 3.9 ± 0.3 action potentials; post-forskolin, 3.3 ± 0.3 action potentials; n = 6; t = 1.44; p = 0.22; df = 5; paired t test; forskolin without KT5720, 5.6 ± 0.5; n = 10; t = 3.53; df = 15; p = 0.003; Student's t test). This indicates that cAMP may exert PKA-dependent and PKA-independent actions on excitability, effects that enhance and reduce excitability, respectively. Furthermore, inclusion of KT5720 (1.2 μm) in the recording pipette did not block the effects of DA (10 μm) on excitability (Fig. 10B) (baseline, 3.86 ± 0.18 action potentials; post-DA, 1.36 ± 0.45 action potentials; n = 7; t = 6.36; p = 0.0007; df = 6; paired t test), indicating that PKA is not necessary for the suppressive actions of DA on excitability. This is consistent with the hypothesis that DA alters excitability of layer V EC neurons by activation of adenylyl cyclase, and subsequent direct, or at least non-PKA-dependent actions, of cAMP on h-channels.
Figure 10.
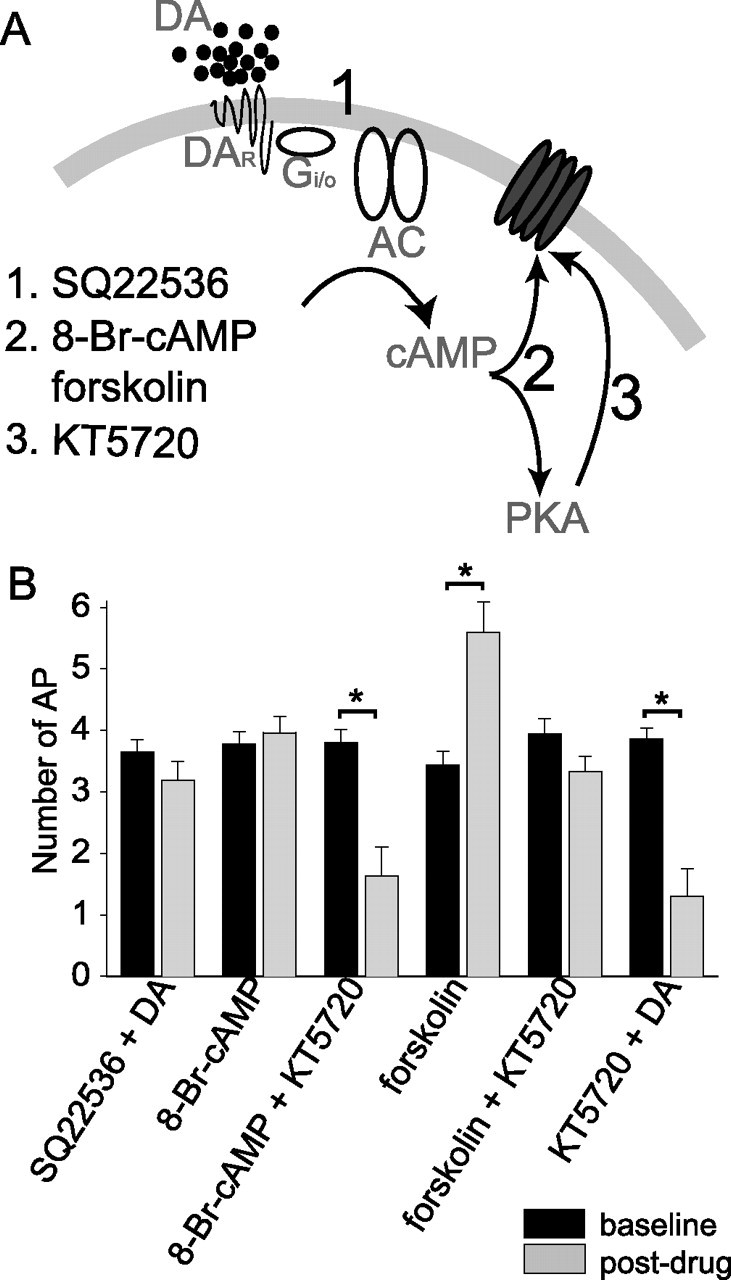
The actions of dopamine on the excitability of layer V EC pyramidal neurons appear to be mediated by a cAMP-dependent, but PKA-independent pathway. A, To examine the second messenger system that may be involved, 1 mm SQ22536 was included in the pipette to block the activation of adenylyl cyclase by G-proteins, 100–250 μm 8-Br-cAMP or 50 μm forskolin was used to mimic DA-mediated enhancement of adenylyl cyclase, or 1.2 μm KT5720 was included in the pipette to block activation of PKA by cAMP. B, The effects of DA on excitability were blocked when SQ22536 was included in the pipette (n = 6), indicating that a G-protein-mediated cascade is activated by DA. Neither 8-Br-cAMP (n = 5) nor forskolin (n = 10) alone mimic the effects of DA, yet they did mimic the effects of DA when the PKA blocker KT5720 was included in the pipette (8-Br-cAMP, n = 6; forskolin, n = 6). This may indicate that there are pathways in these neurons by which cAMP can act independently of PKA, and furthermore, this pathway exerts a similar action as DA. Finally, the effects of DA on excitability were not occluded by inclusion of KT5720 in the pipette (n = 7), indicating, along with the previous results, that the actions of DA on excitability are likely mediated by direct cAMP-mediated modulation of h-channels, or other protein. *Significant p value of at least 0.05. Error bars indicate SE.
Modulation of synaptic summation
To investigate the functional effects of DA-mediated modulation of h-channels on EPSP integration, we stimulated trains of EPSPs (3–9 mV EPSPs; 10 stimuli at 50 ms intervals) before and after applying DA. In these experiments, the concentration of CNQX was decreased to 0.1–1 μm (a minimal amount of CNQX was necessary to avoid epileptiform activity evoked by synaptic stimulation when GABAA receptors were blocked; blockade of GABAA receptors may artificially prolong the duration of the EPSP, and therefore the contribution of h-channels to synaptic integration). EPSPs measured at −70 mV displayed only a small degree of temporal summation (Fig. 11A). Application of 10 μm DA further reduced EPSP summation (Fig. 10A) (baseline, 1.4 ± 0.2; post-DA, 1.2 ± 0.3; n = 6; t = 2.49; p = 0.05; df = 5; paired t test) without a significant effect on EPSP amplitude or paired pulse facilitation (baseline amplitude, 5.6 ± 1.5 mV; post-DA, 4.9 ± 1.0 mV; n = 6; t = 0.99; p = 0.37; df = 5; paired t test; baseline paired-pulse facilitation, 1.56 ± 0.12; post-DA, 1.52 ± 0.11; n = 6; t = 0.90; p = 0.41; df = 5; paired t test).
Figure 11.
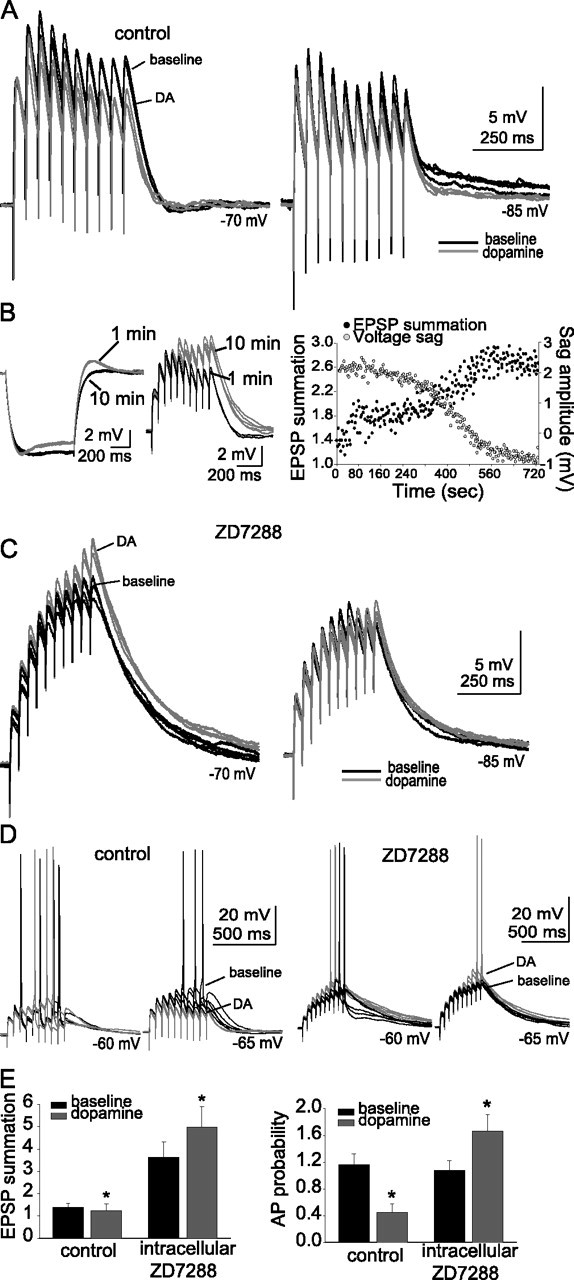
Dopaminergic modulation of EPSP summation and APs evoked by EPSPs depends on h-channels. A, EPSPs displayed a small degree of temporal summation at rest and at hyperpolarized potentials (black traces). Application of DA caused an additional reduction in summation of EPSPs (red traces; n = 6). B, Inclusion of 20 μm ZD7288 in the recording pipette caused a gradual decrease in the voltage sag measured during a small hyperpolarizing current step and a co-occurring enhancement of EPSP summation. This was quantified as ratio of the amplitude of the last EPSP to the first EPSP, as exemplified in the traces and plot of data from this neuron. C, When 20 μm ZD7288 was included in the recording pipette, summation was greatly enhanced compared with control neurons (n = 7; black traces). Furthermore, application of DA resulted in an increase of EPSP summation (red traces; n = 7). D, When the membrane potential is depolarized with constant DC, EPSPs can summate enough to evoke action potentials. In control conditions, application of DA resulted in a reduction in the probability of an action potential evoked by a train of EPSPs (n = 5; left traces). When ZD7288 was included in the recording pipette, application of DA resulted in an increase of the probability of action potentials evoked by EPSPs (right traces; n = 7) The group data are quantified in E (probability measured from EPSPs evoked at −65 mV in this plot). The black traces are baseline conditions, and the red traces are after DA application. *Significant p value of at least 0.05. Error bars indicate SE.
To examine the role of h-channels in the effects of DA on EPSP summation and EPSP–action potential coupling, 20 μm ZD7288 was included in the recording pipette. Inclusion of ZD7288 in the pipette resulted in a gradual decrease in the measured voltage sag accompanied by an increase in the summation of EPSPs after initiation of whole-cell recordings (Fig. 11B). Control neurons displayed a stable sag ratio over a similar time course (1.13 ± 0.03 at ∼1 min; 1.12 ± 0.03 after ∼10 min; p > 0.05; n = 7) and EPSP summation (1.3 ± 0.5 at ∼1 min; 1.4 ± 0.4 after ∼10 min; p > 0.05; n = 7). In contrast to control conditions, application of DA (10 μm) in the presence of intracellular ZD7288 resulted in an increase in the summation of EPSPs (Fig. 11C) (baseline EPSP summation, 3.6 ± 0.7; post-DA, 4.5 ± 0.9; n = 7; t = −2.74; p = 0.03; df = 6; paired t test). As above, there was no significant effect of DA on EPSP amplitude or paired-pulse facilitation when ZD7288 was included in the recording pipette (baseline EPSP amplitude, 4.5 ± 0.9 mV; post-DA, 4.4 ± 0.6 mV; n = 7; t = 0.59; p = 0.59; df = 6; paired t test; baseline paired-pulse facilitation, 1.8 ± 0.3; post-DA, 1.7 ± 0.2; n = 7; t = 0.16; p = 0.88; df = 6; paired t test).
In addition, when summation of a train of EPSPs was great enough to result in action potential firing, DA reduced the probability of action potential firing (Fig. 11D) (baseline action potential probability, 1.16 ± 0.13 AP/train; post-DA, 0.45 ± 0.16 AP/train; n = 5; t = 10.96; p < 0.001; df = 4; paired t test; EPSPs evoked from −65 mV were used for this comparison). In contrast, when ZD7288 was included in the recording pipette, the probability of action potential firing evoked by EPSP trains was increased (Fig. 11D) (baseline action potential probability, 1.1 ± 0.1 AP/train; post-DA, 1.7 ± 0.3 AP/train; n = 5; t = −3.76; p = 0.02; df = 4; paired t test). Thus, it appears that DA can reduce the summation of a train of large EPSPs, and reduce EPSP–action potential coupling, in a manner that depends on h-channels.
To exclude a potential presynaptic effect of DA, as well as to examine modulation of inputs to the dendrites, α-currents were injected directly to the apical dendrite (150–225 μm from the soma) of layer V EC neurons in an attempt to mimic synaptic inputs. These current injections were applied in a manner similar to synaptic stimulation above (10 α-current injections at a 50 ms interstimulus interval), in the presence of synaptic blockers (see Materials and Methods). The amplitude of the baseline current injection was regulated to evoke a single αPSP with an amplitude near 5 mV. Trains of current injection evoked a train of αPSPs that displayed a small degree of summation (Fig. 12A). Application of DA greatly reduced the amplitude of summation (Fig. 12A) (baseline ratio of 10th PSP over first PSP, 1.32 ± 0.07; post-DA, 1.18 ± 0.05; t = 4.1; p = 0.007; df = 6; paired t test), with minimal effects on the amplitude of a single αPSP (baseline, 6.0 ± 0.6 mV; post-DA, 5.8 ± 0.6 mV). This was accompanied by a decrease in the probability of action potentials evoked by summation of αPSPs (Fig. 12B) (baseline probability, 1.24 ± 0.13 action potentials per αPSP train; post-DA, 0.16 ± 0.05 action potentials; t = 9.41; p < 0.0001; df = 11; paired t test). Thus, the effects of DA on summation of synaptic inputs can be readily explained by postsynaptic modulation that corresponds to effects of DA on apical dendrites of layer V EC pyramidal neurons.
Figure 12.

Dopamine modulates αPSP summation and APs in the apical dendrite. A, Injection of α-shaped currents into the dendrites of layer V EC neurons (150–225 μm from the soma) results in αPSPs that summate similarly to EPSPs (black traces). Application of DA (10 μm; n = 7) reduces the amount of summation (red traces; quantified as the ratio of the 10th PSP to the 1st PSP). B, When the amplitude of α-current injection is great enough or the membrane is depolarized a small amount, αPSPs summate enough to evoke action potentials (black traces). Application of DA (10 μm; n = 12) reduces the probability of an action potential evoked by summation of αPSPs. The black traces are baseline conditions, and the red traces are after DA application. *Significant p value of at least 0.05. Error bars indicate SE.
Discussion
The DA system is known to have a potent impact on functions that depend on the EC, such as recall and consolidation of memory (Ardenghi et al., 1997; Izquierdo et al., 1998; Barros et al., 2001; Liu et al., 2004). Our data demonstrate that DA may influence these EC-dependent behaviors, in part, by direct modulation of EC layer V pyramidal neurons. One means by which DA modulates these neurons is via modulation of h-channels.
Application of DA caused a decrease of the excitability of layer V EC pyramidal neurons. This action of DA displayed a unique voltage dependence indicative of the participation of voltage-gated ion channels. Our data indicate that several voltage-dependent ion channels can regulate the excitability of layer V EC pyramidal neurons, including TEA- and 4-AP-sensitive K+ channels, and h-channels. However, our data are consistent with DAergic modulation of h-channels, and not these other channels, in the regulation of excitability. After application of DA, several indicators of h-channel activity were enhanced, including voltage sag and rebound voltage overshoot. The accompanying decrease of RN and faster membrane time constant indicate enhancement of a conductance that is active near Vrest. Furthermore, the suppressive effects of DA were reduced or reversed at depolarized potentials, and were pharmacologically blocked by ZD7288 and cesium. Thus, although h-currents were not directly measured, and voltage sag is only an indirect measure of h-channel activity, it is likely that h-channels underlie the effects of DA on excitability. Other ion channels are unlikely to account for the observed effects, because none of the K+ or Ca2+ channel blockers used attenuated the actions of DA.
Although our evidence indicates that modulation of h-channels may be the main process by which DA alters excitability in these neurons, it is unlikely to be the only channel modulated by DA. Specifically, when h-channels were inactivated by depolarization or pharmacologically blocked, an increase, not a decrease, of excitability was observed after application of DA. Potential explanations include an increase of INa or the observed decreases in the fast and slow AHPs. Indeed, other studies demonstrate a modulation of Na+ channel activity by DA (Surmeier et al., 1992; Cepeda et al., 1995; Schiffmann et al., 1995; Zhang et al., 1998; Cantrell et al., 1999a,b; Aizman et al., 2000; Hayashida and Ishida, 2004; Hu et al. 2005). However, previous studies of pyramidal neurons in the prefrontal cortex and hippocampus demonstrate a D1-mediated decrease of peak Na+ currents (Cantrell et al., 1999a,b; Maurice et al., 2001). In this study, there was little evidence for a similar modulation of layer V EC pyramidal neurons. The trends toward increased action potential amplitude, faster rise time, and more hyperpolarized threshold potential suggest an increase of INa. Although it is possible that D1 DAergic downregulation of Na channels contributes to the reduction of excitability observed here, it appears that, if anything, an enhancement of Na+ channel activity, potentially through D2 receptors (Surmeier et al., 1992), may also occur in these neurons.
Previous studies have examined the actions of DA on EC neurons (Pralong et al., 1993; Behr et al., 2000), but focused on the effects of DA on synaptic inputs, and did not examine excitability, basic membrane properties, or ionic mechanisms of modulation in a detailed manner. However, other studies indicate that DA can modulate h-channels in some neurons (Jiang et al., 1993; Harris-Warrick et al., 1995; Vargas and Lucero, 1999; Storm et al., 2000; Wu and Hablitz, 2005). In EC layer V pyramidal neurons, our data suggest that DAergic modulation of h-channels is PKA independent and may involve direct cAMP-mediated modulation of h channels, as has been observed in other studies (DiFrancesco and Mangoni, 1994; Pedarzani and Storm, 1995b; Luthi and McCormick, 1999; Storm et al., 2000; Chen et al., 2001; Wainger et al., 2001; Accili et al., 2002) [this direct modulation of h-channels by cAMP, along with the relatively slow kinetics of the voltage sag, may indicate that the h-channels are composed of HCN2 subunits (Kaupp and Seifert, 2001; Wainger et al., 2001; Wang et al., 2001), or HCN1 and HCN2 subunits (Chen et al., 2001), both of which are expressed at high levels in neocortex (Monteggia et al., 2000; Notomi and Shigemoto, 2004)]. It is initially surprising that neither 8-Br-cAMP nor forskolin exerts the same effect on excitability as DA, because the effects of DA were cAMP dependent. However, the broad actions of global elevations of cAMP on kinases and ion channels compared with the potentially localized non-PKA-dependent actions of DA may explain this difference.
Although modulation of membrane excitability is a key parameter for understanding the effects of DA on EC pyramidal neuronal function, it is usually synaptic inputs that will ultimately drive pyramidal neuronal firing. Previous studies have examined the modulation of EPSPs to layer V EC neurons (Pralong and Jones, 1993; Behr et al., 2000) and have indicated that DA may modulate single EPSPs. We examined the effects of h-channels on temporal integration using intracellular perfusion of ZD7288 into the recorded neuron. Intracellular perfusion of ZD7288 did not change the paired-pulse ratio, nor was there a difference between the paired-pulse ratio of perfused and control neurons, consistent with a predominantly postsynaptic effect. During the course of the intracellular perfusion, summation of EPSPs was enhanced in close temporal association with a decrease of voltage sag. Although DA reduced the summation of EPSPs in control neurons, DA enhanced summation and EPSP-induced action potential firing in ZD7288-perfused neurons. This indicates that h-channels can potently modulate synaptic integration in these neurons, similar to other pyramidal cells (Magee, 1998, 1999; Nolan et al., 2004; Shah et al., 2004; Fan et al., 2005). Furthermore, DAergic suppression of EPSP integration depends, at least in part, on h-channel activation. This modulation of EPSP integration would strongly influence EC function. It is unclear how DA enhanced EPSP summation when h-channels are blocked, but potential means include enhancement of Na+ channel activity, as previously predicted (Rosenkranz and Johnston, 2003), and indicated by the tendency for increased indices of Na+ channel activity seen above, or a reduction in the sAHP, which can modulate synaptic integration (Lancaster et al., 2001; Faber et al., 2005).
Most h-channels are thought to be located on the dendrites (Magee, 1998; Berger et al., 2001; Lorincz et al., 2002; Notomi and Shigemoto, 2004; Shah et al., 2004). Thus, h-channels may be in an optimal location to modulate synaptic integration in layer V EC pyramidal neurons. The subcellular localization of DA receptors in the EC is not yet clearly characterized. However, it has been observed that most DAergic synapses appear to contact dendritic shafts or spines (Erickson et al., 2000). Others have observed a laminar distribution of D1 receptors in the EC consistent with modulation of dendritic inputs (Richfield et al., 1989; Goldsmith and Joyce, 1994). Thus, D1 receptors are in a key position to modulate synaptic inputs through effects on h-channels. Most of the recordings in this study were performed at the soma, which may underestimate the dendritic modulation by DA because of the potential electrotonic remoteness of dendritic events. However, the recordings obtained from dendritic locations confirmed that similar modulation occurs in the dendrites, and may in fact be more potent. This relatively preferential modulation of dendritic excitability compared with somatic excitability may result in a temporary disconnect between dendritic synaptic input and somatic action potential firing when the neuron is near resting membrane potentials. However, this disconnect could be reversed when the neuron is depolarized by large or coordinated synaptic inputs or depolarized states.
The voltage-dependent manner of DAergic modulation is expected to filter the effects of synaptic inputs that arrive when the neuron is at resting membrane potentials. However, when in a depolarized state, DA may enhance neuronal responses to inputs. Thus, near resting membrane potentials, h-channels are active, and modulation of these channels by DA will reduce excitability. When the membrane potential is depolarized, the contribution of h-channels will be minimized, and the contribution of other channels will become more apparent, such as DAergic reduction of channels that underlie the sAHP, causing an enhancement of excitability. Layer V pyramidal neurons of the EC display transitions between a hyperpolarized and depolarized state in vivo (J. A. Rosenkranz and A. A. Grace, unpublished data). The effects of DA on excitability and synaptic integration would thus differ between the two states, based in part on modulation of h-channels. A role for h-channels is also indicated in theta rhythmicity (Dickson et al., 2000a; Hu et al., 2002; Kitayama et al., 2002; Kocsis and Li, 2004). Theta rhythmicity, which may be generated in the EC (Alonso and Garcia-Austt, 1987; Schmitz et al., 1998; Kocsis et al., 1999), is hypothesized to influence memory formation and recall (Mizumori et al., 1990; O'Keefe, 1993; Buzsaki, 1996; Dickson et al., 2000b; Judge and Hasselmo, 2004). It is possible that modulation of theta rhythmicity by DAergic actions on h-channels may underlie some of the effects of DA on memory processes. Patients with schizophrenia, in addition to displaying EC anatomical disruptions (Falkai et al., 1988; Arnold et al., 1991; Akil and Lewis, 1997; Krimer et al., 1997; Joyal et al., 2002; Prasad et al., 2004), often display enhancements of theta rhythmicity (Ford et al., 1986; Merrin et al., 1989; Miyauchi et al., 1990; Czobor and Volavka, 1992; Omori et al., 1995; Fehr et al., 2001) as well as abnormalities of the DAergic system in the EC (Goldsmith et al., 1997; Akil et al., 2000). Abnormal modulation of EC physiology by DA may thus underlie some of the symptoms of schizophrenia.
Footnotes
This work was supported by National Institutes of Health Grants MH48432 (D.J.) and MH67460 (J.A.R.). We thank Dr. Randy Chitwood for extremely useful advice and discussion, and Eric Lee for expert histological processing of tissue.
References
- Accili EA, Proenza C, Baruscotti M, DiFrancesco D (2002). From funny current to HCN channels: 20 years of excitation. News Physiol Sci 17:32–37. [DOI] [PubMed] [Google Scholar]
- Alonso A, Garcia-Austt E (1987). Neuronal sources of theta rhythm in the entorhinal cortex of the rat. II. Phase relations between unit discharges and theta field potentials. Exp Brain Res 67:502–509. [DOI] [PubMed] [Google Scholar]
- Agrawal N, Hamam BN, Magistretti J, Alonso A, Ragsdale DS (2001). Persistent sodium channel activity mediates subthreshold membrane potential oscillations and low-threshold spikes in rat entorhinal cortex layer V neurons. Neuroscience 102:53–64. [DOI] [PubMed] [Google Scholar]
- Agrawal N, Alonso A, Ragsdale DS (2003). Increased persistent sodium currents in rat entorhinal cortex layer V neurons in a post-status epilepticus model of temporal lobe epilepsy. Epilepsia 44:1601–1604. [DOI] [PubMed] [Google Scholar]
- Aizman O, Brismar H, Uhlen P, Zettergren E, Levey AI, Forssberg H, Greengard P, Aperia A (2000). Anatomical and physiological evidence for D1 and D2 dopamine receptor colocalization in neostriatal neurons. Nat Neurosci 3:226–230. [DOI] [PubMed] [Google Scholar]
- Akil M, Lewis DA (1997). Cytoarchitecture of the entorhinal cortex in schizophrenia. Am J Psychiatry 154:1010–1012. [DOI] [PubMed] [Google Scholar]
- Akil M, Edgar CL, Pierri JN, Casali S, Lewis DA (2000). Decreased density of tyrosine hydroxylase-immunoreactive axons in the entorhinal cortex of schizophrenic subjects. Biol Psychiatry 47:361–370. [DOI] [PubMed] [Google Scholar]
- Ardenghi P, Barros D, Izquierdo LA, Bevilaqua L, Schroder N, Quevedo J, Rodrigues C, Madruga M, Medina JH, Izquierdo I (1997). Late and prolonged post-training memory modulation in entorhinal and parietal cortex by drugs acting on the cAMP/protein kinase A signalling pathway. Behav Pharmacol 8:745–751. [DOI] [PubMed] [Google Scholar]
- Armstrong CM, Swenson RP Jr, Taylor SR (1982). Block of squid axon K channels by internally and externally applied barium ions. J Gen Physiol 80:663–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold SE, Hyman BT, Van Hoesen GW, Damasio AR (1991). Some cytoarchitectural abnormalities of the entorhinal cortex in schizophrenia. Arch Gen Psychiatry 48:625–632. [DOI] [PubMed] [Google Scholar]
- Barros DM, Mello e Souza T, De David T, Choi H, Aguzzoli A, Madche C, Ardenghi P, Medina JH, Izquierdo I (2001). Simultaneous modulation of retrieval by dopaminergic D1, beta-noradrenergic, serotonergic-1A and cholinergic muscarinic receptors in cortical structures of the rat. Behav Brain Res 124:1–7. [DOI] [PubMed] [Google Scholar]
- Behr J, Gloveli T, Schmitz D, Heinemann U (2000). Dopamine depresses excitatory synaptic transmission onto rat subicular neurons via presynaptic D1-like dopamine receptors. J Neurophysiol 84:112–119. [DOI] [PubMed] [Google Scholar]
- Berger T, Larkum ME, Luscher HR (2001). High I(h) channel density in the distal apical dendrite of layer V pyramidal cells increases bidirectional attenuation of EPSPs. J Neurophysiol 85:855–868. [DOI] [PubMed] [Google Scholar]
- Braha O, Edmonds B, Sacktor T, Kandel ER, Klein M (1993). The contributions of protein kinase A and protein kinase C to the actions of 5-HT on the L-type Ca2+ current of the sensory neurons in Aplysia. J Neurosci 13:1839–1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buzsaki G (1996). The hippocampo-neocortical dialogue. Cereb Cortex 6:81–92. [DOI] [PubMed] [Google Scholar]
- Cantrell AR, Smith RD, Goldin AL, Scheuer T, Catterall WA (1997). Dopaminergic modulation of sodium current in hippocampal neurons via cAMP-dependent phosphorylation of specific sites in the sodium channel alpha subunit. J Neurosci 17:7330–7338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantrell AR, Scheuer T, Catterall WA (1999a). Voltage-dependent neuromodulation of Na+ channels by D1-like dopamine receptors in rat hippocampal neurons. J Neurosci 19:5301–5310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantrell AR, Tibbs VC, Westenbroek RE, Scheuer T, Catterall WA (1999b). Dopaminergic modulation of voltage-gated Na+ current in rat hippocampal neurons requires anchoring of cAMP-dependent protein kinase. J Neurosci 19:1-6RC21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cepeda C, Chandler SH, Shumate LW, Levine MS (1995). Persistent Na+ conductance in medium-sized neostriatal neurons: characterization using infrared videomicroscopy and whole cell patch-clamp recordings. J Neurophysiol 74:1343–1348. [DOI] [PubMed] [Google Scholar]
- Chen S, Wang J, Siegelbaum SA (2001). Properties of hyperpolarization-activated pacemaker current defined by coassembly of HCN1 and HCN2 subunits and basal modulation by cyclic nucleotide. J Gen Physiol 117:491–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper DC, Moore SJ, Staff NP, Spruston N (2003). Psychostimulant-induced plasticity of intrinsic neuronal excitability in ventral subiculum. J Neurosci 23:9937–9946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czobor P, Volavka J (1992). Level of haloperidol in plasma is related to electroencephalographic findings in patients who improve. Psychiatry Res 42:129–144. [DOI] [PubMed] [Google Scholar]
- Descarries L, Lemay B, Doucet G, Berger B (1987). Regional and laminar density of the dopamine innervation in adult rat cerebral cortex. Neuroscience 21:807–824. [DOI] [PubMed] [Google Scholar]
- Dickson CT, Magistretti J, Shalinsky MH, Fransen E, Hasselmo ME, Alonso A (2000a). Properties and role of I(h) in the pacing of subthreshold oscillations in entorhinal cortex layer II neurons. J Neurophysiol 83:2562–2579. [DOI] [PubMed] [Google Scholar]
- Dickson CT, Magistretti J, Shalinsky M, Hamam B, Alonso A (2000b). Oscillatory activity in entorhinal neurons and circuits. Mechanisms and function. Ann NY Acad Sci 911:127–150. [DOI] [PubMed] [Google Scholar]
- DiFrancesco D, Mangoni M (1994). Modulation of single hyperpolarization-activated channels (i(f)) by cAMP in the rabbit sino-atrial node. J Physiol (Lond) 474:473–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diop L, Gottberg E, Briere R, Grondin L, Reader TA (1988). Distribution of dopamine D1 receptors in rat cortical areas, neostriatum, olfactory bulb and hippocampus in relation to endogenous dopamine contents. Synapse 2:395–405. [DOI] [PubMed] [Google Scholar]
- Dolcos F, LaBar KS, Cabeza R (2005). Remembering one year later: role of the amygdala and the medial temporal lobe memory system in retrieving emotional memories. Proc Natl Acad Sci USA 102:2626–2631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong Y, White FJ (2003). Dopamine D1-class receptors selectively modulate a slowly inactivating potassium current in rat medial prefrontal cortex pyramidal neurons. J Neurosci 23:2686–2695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong Y, Cooper D, Nasif F, Hu XT, White FJ (2004). Dopamine modulates inwardly rectifying potassium currents in medial prefrontal cortex pyramidal neurons. J Neurosci 24:3077–3085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drain P, Dubin AE, Aldrich RW (1994). Regulation of Shaker K+ channel inactivation gating by the cAMP-dependent protein kinase. Neuron 12:1097–1109. [DOI] [PubMed] [Google Scholar]
- Eder C, Heinemann U (1996). Potassium currents in acutely isolated neurons from superficial and deep layers of the juvenile rat entorhinal cortex. Pflügers Arch 432:637–643. [DOI] [PubMed] [Google Scholar]
- Eder C, Klee R, Heinemann U (1996). Modulation of A-currents by [K+]o in acutely isolated pyramidal neurones of juvenile rat entorhinal cortex and hippocampus. NeuroReport 7:1565–1568. [DOI] [PubMed] [Google Scholar]
- Egorov AV, Heinemann U, Muller W (2002a). Differential excitability and voltage-dependent Ca2+ signalling in two types of medial entorhinal cortex layer V neurons. Eur J Neurosci 16:1305–1312. [DOI] [PubMed] [Google Scholar]
- Egorov AV, Hamam BN, Fransen E, Hasselmo ME, Alonso AA (2002b). Graded persistent activity in entorhinal cortex neurons. Nature 420:173–178. [DOI] [PubMed] [Google Scholar]
- Erickson SL, Sesack SR, Lewis DA (2000). Dopamine innervation of monkey entorhinal cortex: postsynaptic targets of tyrosine hydroxylase-immunoreactive terminals. Synapse 36:47–56. [DOI] [PubMed] [Google Scholar]
- Faber ES, Delaney AJ, Sah P (2005). SK channels regulate excitatory synaptic transmission and plasticity in the lateral amygdala. Nat Neurosci 8:635–641. [DOI] [PubMed] [Google Scholar]
- Falkai P, Bogerts B, Rozumek M (1988). Limbic pathology in schizophrenia: the entorhinal region—a morphometric study. Biol Psychiatry 24:515–521. [DOI] [PubMed] [Google Scholar]
- Fallon JH, Koziell DA, Moore RY (1978). Catecholamine innervation of the basal forebrain. II. Amygdala, suprarhinal cortex and entorhinal cortex. J Comp Neurol 180:509–532. [DOI] [PubMed] [Google Scholar]
- Fan Y, Fricker D, Brager DH, Chen X, Lu HC, Chitwood RA, Johnston D (2005). Activity-dependent decrease of excitability in rat hippocampal neurons through increases in I(h). Nat Neurosci 8:1542–1551. [DOI] [PubMed] [Google Scholar]
- Fehr T, Kissler J, Moratti S, Wienbruch C, Rockstroh B, Elbert T (2001). Source distribution of neuromagnetic slow waves and MEG-delta activity in schizophrenic patients. Biol Psychiatry 50:108–116. [DOI] [PubMed] [Google Scholar]
- Feldon J, Weiner I (1991). The latent inhibition model of schizophrenic attention disorder. Haloperidol and sulpiride enhance rats' ability to ignore irrelevant stimuli. Biol Psychiatry 29:635–646. [DOI] [PubMed] [Google Scholar]
- Ford MR, Goethe JW, Dekker DK (1986). EEG coherence and power in the discrimination of psychiatric disorders and medication effects. Biol Psychiatry 21:1175–1188. [DOI] [PubMed] [Google Scholar]
- Gershon E, Weigl L, Lotan I, Schreibmayer W, Dascal N (1992). Protein kinase A reduces voltage-dependent Na+ current in Xenopus oocytes. J Neurosci 12:103743–3752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibor G, Yakubovich D, Peretz A, Attali B (2004). External barium affects the gating of KCNQ1 potassium channels and produces a pore block via two discrete sites. J Gen Physiol 124:83–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldsmith SK, Joyce JN (1994). Dopamine D2 receptor expression in hippocampus and parahippocampal cortex of rat, cat, and human in relation to tyrosine hydroxylase immunoreactive fibers. Hippocampus 4:354–373. [DOI] [PubMed] [Google Scholar]
- Goldsmith SK, Shapiro RM, Joyce JN (1997). Disrupted pattern of D2 dopamine receptors in the temporal lobe in schizophrenia. A postmortem study. Arch Gen Psychiatry 54:649–658. [DOI] [PubMed] [Google Scholar]
- Gorelova N, Seamans JK, Yang CR (2002). Mechanisms of dopamine activation of fast-spiking interneurons that exert inhibition in rat prefrontal cortex. J Neurophysiol 88:3150–3166. [DOI] [PubMed] [Google Scholar]
- Gulledge AT, Jaffe DB (2001). Multiple effects of dopamine on layer V pyramidal cell excitability in rat prefrontal cortex. J Neurophysiol 86:586–595. [DOI] [PubMed] [Google Scholar]
- Haist F, Bowden Gore J, Mao H (2001). Consolidation of human memory over decades revealed by functional magnetic resonance imaging. Nat Neurosci 4:1139–1145. [DOI] [PubMed] [Google Scholar]
- Hamam BN, Amaral DG, Alonso AA (2002). Morphological and electrophysiological characteristics of layer V neurons of the rat lateral entorhinal cortex. J Comp Neurol 451:45–61. [DOI] [PubMed] [Google Scholar]
- Harris-Warrick RM, Coniglio LM, Levini RM, Gueron S, Guckenheimer J (1995). Dopamine modulation of two subthreshold currents produces phase shifts in activity of an identified motoneuron. J Neurophysiol 74:1404–1420. [DOI] [PubMed] [Google Scholar]
- Hayashida Y, Ishida AT (2004). Dopamine receptor activation can reduce voltage-gated Na+ current by modulating both entry into and recovery from inactivation. J Neurophysiol 92:3134–3141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henze DA, Gonzalez-Burgos GR, Urban NN, Lewis DA, Barrionuevo G (2000). Dopamine increases excitability of pyramidal neurons in primate prefrontal cortex. J Neurophysiol 84:2799–2809. [DOI] [PubMed] [Google Scholar]
- Hermann A, Gorman AL (1981). Effects of 4-aminopyridine on potassium currents in a molluscan neuron. J Gen Physiol 78:63–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman DA, Magee JC, Colbert CM, Johnston D (1997). K+ channel regulation of signal propagation in dendrites of hippocampal pyramidal neurons. Nature 387:869–875. [DOI] [PubMed] [Google Scholar]
- Hu H, Vervaeke K, Storm JF (2002). Two forms of electrical resonance at theta frequencies, generated by M-current, h-current and persistent Na+ current in rat hippocampal pyramidal cells. J Physiol (Lond) 545:783–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu XT, Dong Y, Zhang XF, White FJ (2005). Dopamine D2 receptor-activated Ca2+ signaling modulates voltage-sensitive sodium currents in rat nucleus accumbens neurons. J Neurophysiol 93:1406–1417. [DOI] [PubMed] [Google Scholar]
- Hyman BT, Van Hoesen GW, Damasio AR, Barnes CL (1984). Alzheimer's disease: cell-specific pathology isolates the hippocampal formation. Science 225:1168–1170. [DOI] [PubMed] [Google Scholar]
- Izquierdo I, Medina JH, Izquierdo LA, Barros DM, de Souza MM, Mello e Souza T (1998). Short- and long-term memory are differentially regulated by monoaminergic systems in the rat brain. Neurobiol Learn Mem 69:219–224. [DOI] [PubMed] [Google Scholar]
- Jiang ZG, Pessia M, North RA (1993). Dopamine and baclofen inhibit the hyperpolarization-activated cation current in rat ventral tegmental neurones. J Physiol (Lond) 462:753–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joyal CC, Laakso MP, Tiihonen J, Syvalahti E, Vilkman H, Laakso A, Alakare B, Rakkolainen V, Salokangas RK, Hietala J (2002). A volumetric MRI study of the entorhinal cortex in first episode neuroleptic-naive schizophrenia. Biol Psychiatry 51:1005–1007. [DOI] [PubMed] [Google Scholar]
- Judge SJ, Hasselmo ME (2004). Theta rhythmic stimulation of stratum lacunosum-moleculare in rat hippocampus contributes to associative LTP at a phase offset in stratum radiatum. J Neurophysiol 92:1615–1624. [DOI] [PubMed] [Google Scholar]
- Kapur N, Brooks DJ (1999). Temporally-specific retrograde amnesia in two cases of discrete bilateral hippocampal pathology. Hippocampus 9:247–254. [DOI] [PubMed] [Google Scholar]
- Kaupp UB, Seifert R (2001). Molecular diversity of pacemaker ion channels. Annu Rev Physiol 63:235–257. [DOI] [PubMed] [Google Scholar]
- Kitayama M, Taguchi T, Miyata H, Matsuda Y, Yamauchi T, Kogure S (2002). The extracellular current blocking effect of cesium chloride on the theta wave in the rabbit hippocampal CA1 region. Neurosci Lett 334:45–48. [DOI] [PubMed] [Google Scholar]
- Kocsis B, Li S (2004). In vivo contribution of h-channels in the septal pacemaker to theta rhythm generation. Eur J Neurosci 20:2149–2158. [DOI] [PubMed] [Google Scholar]
- Kocsis B, Bragin A, Buzsaki G (1999). Interdependence of multiple theta generators in the hippocampus: a partial coherence analysis. J Neurosci 19:6200–6212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotzbauer PT, Trojanowsk JQ, Lee VM (2001). Lewy body pathology in Alzheimer's disease. J Mol Neurosci 17:225–232. [DOI] [PubMed] [Google Scholar]
- Krimer LS, Herman MM, Saunders RC, Boyd JC, Hyde TM, Carter JM, Kleinman JE, Weinberger DR (1997). A qualitative and quantitative analysis of the entorhinal cortex in schizophrenia. Cereb Cortex 7:732–739. [DOI] [PubMed] [Google Scholar]
- Kroner S, Rosenkranz JA, Grace AA, Barrionuevo G (2005). Dopamine modulates excitability of basolateral amygdala neurons in vitro. J Neurophysiol 93:1598–1610. [DOI] [PubMed] [Google Scholar]
- Lancaster B, Hu H, Ramakers GM, Storm JF (2001). Interaction between synaptic excitation and slow afterhyperpolarization current in rat hippocampal pyramidal cells. J Physiol (Lond) 536:809–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavenex P, Amaral DG (2000). Hippocampal-neocortical interaction: a hierarchy of associativity. Hippocampus 10:420–430. [DOI] [PubMed] [Google Scholar]
- Lavin A, Grace AA (2001). Stimulation of D1-type dopamine receptors enhances excitability in prefrontal cortical pyramidal neurons in a state-dependent manner. Neuroscience 104:335–346. [DOI] [PubMed] [Google Scholar]
- Liu Z, Richmond BJ, Murray EA, Saunders RC, Steenrod S, Stubblefield BK, Montague DM, Ginns EI (2004). DNA targeting of rhinal cortex D2 receptor protein reversibly blocks learning of cues that predict reward. Proc Natl Acad Sci USA 101:12336–12341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorincz A, Notomi T, Tamas G, Shigemoto R, Nusser Z (2002). Polarized and compartment-dependent distribution of HCN1 in pyramidal cell dendrites. Nat Neurosci 5:1185–1193. [DOI] [PubMed] [Google Scholar]
- Luthi A, McCormick DA (1999). Modulation of a pacemaker current through Ca2+-induced stimulation of cAMP production. Nat Neurosci 2:634–641. [DOI] [PubMed] [Google Scholar]
- Magee JC (1998). Dendritic hyperpolarization-activated currents modify the integrative properties of hippocampal CA1 pyramidal neurons. J Neurosci 18:7613–7624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magee JC (1999). Dendritic Ih normalizes temporal summation in hippocampal CA1 neurons. Nat Neurosci 2:508–514. [DOI] [PubMed] [Google Scholar]
- Malenka RC, Nicoll RA (1986). Dopamine decreases the calcium-activated afterhyperpolarization in hippocampal CA1 pyramidal cells. Brain Res 379:210–215. [DOI] [PubMed] [Google Scholar]
- Maurice N, Tkatch T, Meisler M, Sprunger LK, Surmeier DJ (2001). D1/D5 dopamine receptor activation differentially modulates rapidly inactivating and persistent sodium currents in prefrontal cortex pyramidal neurons. J Neurosci 21:2268–2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer ML, Westbrook GL (1983). A voltage-clamp analysis of inward (anomalous) rectification in rat sensory ganglion neurons. J Physiol (Lond) 375:327–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormick DA, Huguenard JR (1992). A model of the electrophysiological properties of thalamocortical relay neurons. J Neurophysiol 68:1384–1400. [DOI] [PubMed] [Google Scholar]
- Merrin EL, Floyd TC, Fein G (1989). EEG coherence in unmedicated schizophrenic patients. Biol Psychiatry 25:60–66. [DOI] [PubMed] [Google Scholar]
- Miyauchi T, Tanaka K, Hagimoto H, Miura T, Kishimoto H, Matsushita M (1990). Computerized EEG in schizophrenic patients. Biol Psychiatry 28:488–494. [DOI] [PubMed] [Google Scholar]
- Mizumori SJ, Barnes CA, McNaughton BL (1990). Behavioral correlates of theta-on and theta-off cells recorded from hippocampal formation of mature young and aged rats. Exp Brain Res 80:365–373. [DOI] [PubMed] [Google Scholar]
- Monteggia LM, Eisch AJ, Tang MD, Kaczmarek LK, Nestler EJ (2000). Cloning and localization of the hyperpolarization-activated cyclic nucleotide-gated channel family in rat brain. Brain Res Mol Brain Res 81:129–139. [DOI] [PubMed] [Google Scholar]
- Murray EA, Baxter MG, Gaffan D (1998). Monkeys with rhinal cortex damage or neurotoxic hippocampal lesions are impaired on spatial scene learning and object reversals. Behav Neurosci 112:1291–1303. [DOI] [PubMed] [Google Scholar]
- Neher E (1992). Correction for liquid junction potentials in patch clamp experiments. Methods Enzymol 207:123–131. [DOI] [PubMed] [Google Scholar]
- Nicoll RA (1988). The coupling of neurotransmitter receptors to ion channels in the brain. Science 241:545–551. [DOI] [PubMed] [Google Scholar]
- Nolan MF, Malleret G, Dudman JT, Buhl DL, Santoro B, Gibbs E, Vronskaya S, Buzsaki G, Siegelbaum SA, Kandel ER, Morozov A (2004). A behavioral role for dendritic integration: HCN1 channels constrain spatial memory and plasticity at inputs to distal dendrites of CA1 pyramidal neurons. Cell 119:719–732. [DOI] [PubMed] [Google Scholar]
- Notomi T, Shigemoto R (2004). Immunohistochemical localization of Ih channel subunits, HCN1–4, in the rat brain. J Comp Neurol 471:241–276. [DOI] [PubMed] [Google Scholar]
- O'Keefe J (1993). Hippocampus, theta, and spatial memory. Curr Opin Neurobiol 3:917–924. [DOI] [PubMed] [Google Scholar]
- Omori M, Koshino Y, Murata T, Murata I, Nishio M, Sakamoto K, Horie T, Isaki K (1995). Quantitative EEG in never-treated schizophrenic patients. Biol Psychiatry 38:305–309. [DOI] [PubMed] [Google Scholar]
- Pape HC, McCormick DA (1989). Noradrenaline and serotonin selectively modulate thalamic burst firing by enhancing a hyperpolarization-activated current. Nature 340:715–718. [DOI] [PubMed] [Google Scholar]
- Pedarzani P, Storm JF (1993). PKA mediates the effects of monoamine transmitters on the K+ current underlying the slow spike frequency adaptation in hippocampal neurons. Neuron 11:1023–1035. [DOI] [PubMed] [Google Scholar]
- Pedarzani P, Storm JF (1995a). Dopamine modulates the slow Ca2+-activated K+ current IAHP via cyclic AMP-dependent protein kinase in hippocampal neurons. J Neurophysiol 74:2749–2753. [DOI] [PubMed] [Google Scholar]
- Pedarzani P, Storm JF (1995b). Protein kinase A-independent modulation of ion channels in the brain by cyclic AMP. Proc Natl Acad Sci USA 92:11716–11720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pralong E, Jones RS (1993). Interactions of dopamine with glutamate- and GABA-mediated synaptic transmission in the rat entorhinal cortex in vitro. Eur J Neurosci 5:760–767. [DOI] [PubMed] [Google Scholar]
- Prasad KM, Patel AR, Muddasani S, Sweeney J, Keshavan MS (2004). The entorhinal cortex in first-episode psychotic disorders: a structural magnetic resonance imaging study. Am J Psychiatry 161:1612–1619. [DOI] [PubMed] [Google Scholar]
- Prole DL, Marrion NV (2004). Ionic permeation and conduction properties of neuronal KCNQ2/KCNQ3 potassium channels. Biophys J 86:1454–1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richfield EK, Young AB, Penney JB (1989). Comparative distributions of dopamine D-1 and D-2 receptors in the cerebral cortex of rats, cats, and monkeys. J Comp Neurol 286:409–426. [DOI] [PubMed] [Google Scholar]
- Richter H, Heinemann U, Eder C (2000). Hyperpolarization-activated cation currents in stellate and pyramidal neurons of rat entorhinal cortex. Neurosci Lett 281:33–36. [DOI] [PubMed] [Google Scholar]
- Rosenkranz JA, Johnston D (2003). Dopamine receptor activation modulates synaptic integration in layer V neurons of the entorhinal cortex. Soc Neurosci Abstr 29:573.5. [Google Scholar]
- Ruob C, Elsner J, Weiner I, Feldon J (1997). Amphetamine-induced disruption and haloperidol-induced potentiation of latent inhibition depend on the nature of the stimulus. Behav Brain Res 88:35–41. [DOI] [PubMed] [Google Scholar]
- Sakmann B, Trube G (1984). Voltage-dependent inactivation of inward-rectifying single-channel currents in the guinea-pig heart cell membrane. J Physiol (Lond) 347:659–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiffmann SN, Lledo PM, Vincent JD (1995). Dopamine D1 receptor modulates the voltage-gated sodium current in rat striatal neurones through a protein kinase A. J Physiol (Lond) 483:95–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz D, Gloveli T, Behr J, Dugladze T, Heinemann U (1998). Subthreshold membrane potential oscillations in neurons of deep layers of the entorhinal cortex. Neuroscience 85:999–1004. [DOI] [PubMed] [Google Scholar]
- Segal M, Barker JL (1984). Rat hippocampal neurons in culture: potassium conductances. J Neurophysiol 51:1409–1433. [DOI] [PubMed] [Google Scholar]
- Shah MM, Anderson AE, Leung V, Lin X, Johnston D (2004). Seizure-induced plasticity of h channels in entorhinal cortical layer III pyramidal neurons. Neuron 44:495–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith RD, Goldin AL (1997). Phosphorylation at a single site in the rat brain sodium channel is necessary and sufficient for current reduction by protein kinase A. J Neurosci 17:6086–6093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spain WJ, Schwindt PC, Crill WE (1987). Anomalous rectification in neurons from cat sensorimotor cortex. J Neurophysiol 57:1555–1576. [DOI] [PubMed] [Google Scholar]
- Squire LR, Stark CE, Clark RE (2004). The medial temporal lobe. Annu Rev Neurosci 27:279–306. [DOI] [PubMed] [Google Scholar]
- Steffenach HA, Witter M, Moser MB, Moser EI (2005). Spatial memory in the rat requires the dorsolateral band of the entorhinal cortex. Neuron 45:301–313. [DOI] [PubMed] [Google Scholar]
- Storm JF, Pedarzani P, Haug TM, Winther T (2000). Modulation of K+ channels in hippocampal neurons: transmitters acting via cyclic AMP enhance the excitability of hippocampal neurons through kinase-dependent and -independent modulation of AHP- and h-channels. In: Slow synaptic responses and modulation (Kuba K, Higashida H, Brown DA, Yoshioka T, eds) pp. 78–92. Tokyo: Springer.
- Surmeier DJ, Eberwine J, Wilson CJ, Cao Y, Stefani A, Kitai ST (1992). Dopamine receptor subtypes colocalize in rat striatonigral neurons. Proc Natl Acad Sci USA 89:10178–10182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swerdlow NR, Stephany N, Wasserman LC, Talledo J, Sharp R, Auerbach PP (2003). Dopamine agonists disrupt visual latent inhibition in normal males using a within-subject paradigm. Psychopharmacology (Berl) 169:314–320. [DOI] [PubMed] [Google Scholar]
- Sybirska E, Davachi L, Goldman-Rakic PS (2000). Prominence of direct entorhinal-CA1 pathway activation in sensorimotor and cognitive tasks revealed by 2-DG functional mapping in nonhuman primate. J Neurosci 20:5827–5834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarazi FI, Tomasini EC, Baldessarini RJ (1999). Postnatal development of dopamine D1-like receptors in rat cortical and striatolimbic brain regions: an autoradiographic study. Dev Neurosci 21:43–49. [DOI] [PubMed] [Google Scholar]
- Thompson S (1982). Aminopyridine block of transient potassium current. J Gen Physiol 80:1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng KY, O'Donnell P (2004). Dopamine-glutamate interactions controlling prefrontal cortical pyramidal cell excitability involve multiple signaling mechanisms. J Neurosci 24:5131–5139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vargas G, Lucero MT (1999). Dopamine modulates inwardly rectifying hyperpolarization-activated current (Ih) in cultured rat olfactory receptor neurons. J Neurophysiol 81:149–158. [DOI] [PubMed] [Google Scholar]
- Wainger BJ, DeGennaro M, Santoro B, Siegelbaum SA, Tibbs GR (2001). Molecular mechanism of cAMP modulation of HCN pacemaker channels. Nature 411:805–810. [DOI] [PubMed] [Google Scholar]
- Wang J, Chen S, Siegelbaum SA (2001). Regulation of hyperpolarization-activated HCN channel gating and cAMP modulation due to interactions of COOH terminus and core transmembrane regions. J Gen Physiol 118:237–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiner DM, Levey AI, Sunahara RK, Niznik HB, O'Dowd BF, Seeman P, Brann MR (1991). D1 and D2 dopamine receptor mRNA in rat brain. Proc Natl Acad Sci USA 88:1859–1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West AR, Grace AA (2002). Opposite influences of endogenous dopamine D1 and D2 receptor activation on activity states and electrophysiological properties of striatal neurons: studies combining in vivo intracellular recordings and reverse microdialysis. J Neurosci 22:294–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Hablitz JJ (2005). Cooperative activation of D1 and D2 dopamine receptors enhances a hyperpolarization-activated inward current in layer I interneurons. J Neurosci 25:6322–6328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoyama C, Okamura H, Nakajima T, Taguchi J, Ibata Y (1994). Autoradiographic distribution of [3H]YM-09151–2, a high-affinity and selective antagonist ligand for the dopamine D2 receptor group, in the rat brain and spinal cord. J Comp Neurol 344:121–136. [DOI] [PubMed] [Google Scholar]
- Zagotta WN, Brainard MS, Aldrich RW (1988). Single-channel analysis of four distinct classes of potassium channels in Drosophila muscle. J Neurosci 8:4765–4779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X-F, Hu X-T, White FJ (1998). Whole-cell plasticity in cocaine withdrawal: reduced sodium currents in nucleus accumbens neurons. J Neurosci 18:488–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou FM, Hablitz JJ (1999). Dopamine modulation of membrane and synaptic properties of interneurons in rat cerebral cortex. J Neurophysiol 81:967–976. [DOI] [PubMed] [Google Scholar]