

Abstract
Synapse formation and maintenance require extensive transsynaptic interactions involving multiple signal transduction pathways. In the cerebellum, Purkinje cells (PCs) receive GABAergic, axo-dendritic synapses from stellate cells and axo-somatic synapses from basket cells, both with GABAA receptors containing the α1 subunit. Here, we investigated the effects of a targeted deletion of the α1 subunit gene on GABAergic synaptogenesis in PCs, using electrophysiology and immunoelectron microscopy. Whole-cell patch-clamp recordings in acute slices revealed that PCs from α10/0 mice lack spontaneous and evoked IPSCs, demonstrating that assembly of functional GABAA receptors requires the α1 subunit. Ultrastructurally, stellate cell synapses on PC dendrites were reduced by 75%, whereas basket cell synapses on the soma were not affected, despite the lack of GABAA-mediated synaptic transmission. Most strikingly, GABAergic terminals were retained in the molecular layer of adult α10/0 mice and formed heterologous synapses with PC spines characterized by a well differentiated asymmetric postsynaptic density. These synapses lacked presynaptic glutamatergic markers and postsynaptic AMPA-type glutamate receptors but contained δ2-glutamate receptors. During postnatal development, initial steps of GABAergic synapse formation were qualitatively normal, and heterologous synapses appeared in parallel with maturation of dendritic spines. These results suggest that synapse formation in the cerebellum is governed by neurotransmitter-independent mechanisms. However, in the absence of GABAA-mediated transmission, GABAergic terminals in the molecular layer apparently become responsive to synaptogenic signals from PC spines and form stable heterologous synapses. In contrast, maintenance of axo-somatic GABAergic synapses does not depend on functional GABAA receptors, suggesting differential regulation in distinct subcellular compartments.
Keywords: synaptogenesis, gene targeting, cerebellum, dendritic spine, parallel fiber, inhibitory transmission
Introduction
The formation of synapses is a complex, multistep process involving reciprocal communication between the presynaptic and postsynaptic elements (Yamaguchi, 2002; Yamagata et al., 2003; Washbourne et al., 2004; Sieburth et al., 2005). Several cell adhesion molecules, such as SynCAM and neurexins, can initiate synapse formation, even in non-neuronal cells (Biederer et al., 2002; Scheiffele, 2003; Yamagata et al., 2003; Chubykin et al., 2005; Sara et al., 2005). Transsynaptic interactions between neurexin and neuroligins contribute to specify the neurotransmitter phenotype of newly formed synapses (Graf et al., 2004; Prange et al., 2004; Washbourne et al., 2004; Chih et al., 2005; Levinson et al., 2005; Nam and Chen, 2005). Finally, members of the L1 family direct the formation of GABAergic synapses on neuronal somata and axon-initial segments (Ango et al., 2004; Saghatelyan et al., 2004) but not on dendrites, suggesting differential mechanisms orchestrating inhibitory synapse formation in distinct subcellular compartments. However, although a match between transmitter and receptors is essential for synaptic function, transmitter synthesis or release is dispensable for synapse formation and aggregation of postsynaptic proteins (Verhage et al., 2000; Varoqueaux et al., 2002; Gally and Bessereau, 2003; Harms and Craig, 2005). Likewise, chronic blockade of neuronal activity or exposure to GABAA receptor antagonists does not affect postsynaptic aggregation of GABAA receptors (Craig et al., 1994; Studler et al., 2002). Transsynaptic communication, nevertheless, is necessary for proper sorting of receptors at postsynaptic sites, as shown by clustering of GABAA receptors opposite glutamatergic terminals in the absence of GABAergic input (Rao et al., 2000; Fritschy and Brünig, 2003; Anderson et al., 2004). Furthermore, several lines of evidence suggest that neuronal activity is required for appropriate maturation of presynaptic terminals (Chattopadhyaya et al., 2004), long-term survival of postsynaptic neurons (Verhage et al., 2000), and maintenance of synaptic contacts (Takeuchi et al., 2005).
In the present study, we investigated the contribution of functional postsynaptic receptors for presynaptic differentiation of GABAergic synapses and correct matching of presynaptic and postsynaptic specializations. To address this issue, we investigated the cerebellum of mutant mice lacking the GABAA receptor α1 subunit gene (Vicini et al., 2001), using electrophysiology and immunoelectron microscopy. Because Purkinje cells (PCs) express almost exclusively α1-GABAA receptors (Laurie et al., 1992a; Persohn et al., 1992), and because this subunit is essential for assembling heteromeric GABAA receptor channels (Sur et al., 2001; Kralic et al., 2002, 2006), we expected that the mutation would cause a loss of GABAA receptors in PCs. We analyzed the function, morphology, and postnatal development of GABAergic synapses in the molecular layer of the cerebellum. The results show that postsynaptic GABAA receptors are essential for the long-term maintenance of GABA synapses formed by stellate cells on PC dendrites, whereas synapses on the soma established by basket cells remain unaffected by the absence of functional postsynaptic receptors.
Materials and Methods
Homozygous GABAA receptor α1 subunit knock-out (α10/0) mice were generated on a mixed C57BL/6J-129Sv/SvJ background at the University of Pittsburgh (Pittsburgh, PA) [see Vicini et al. (2001) for characterization] and bred at the University of Zurich. α10/0 mice and wild-type littermates were obtained by intercrossing heterozygous mutants or homozygotes from first-generation (α1+/+ × α1+/+) and (α10/0 × α10/0) breeding pairs derived from α1+/0 mice. Mutant mice were born at the expected Mendelian ratio, and their phenotype was maintained across generations. All analyses were performed in homozygous mutants and wild-type animals. The experiments had been approved by the local authorities and were performed in accordance with the European Community Council Directive (86/609/EEC) and the institutional guidelines of the Universities of Turin and Zurich.
Electrophysiology.
Mice of both sexes aged 10 d (n = 2) and 18–22 d (n = 12) were deeply anesthetized with enflurane and decapitated. Acute 300-μm-thick parasagittal slices containing the cerebellum were prepared with a vibrating microtome (Microm, Volketswil, Switzerland) and left to recover at least 1 h before measurements (for details, see Marowsky et al., 2004). Whole-cell patch-clamp recordings from visually identified PCs were obtained using borosilicate glass pipettes (Clark, Pangbourne, UK) pulled to 3–4 MΩ resistance. Spontaneous IPSCs and tonic GABAergic currents were recorded in the presence of 4 mm kynurenic acid at a −80 mV holding potential with pipettes filled with a solution containing the following (in mm): 100 CsCl, 2 MgCl2, 0.1 EGTA, 2 MgATP, 0.3 NaGTP, and 40 HEPES (290 mOsm; pH 7.3 adjusted with CsOH). Events were detected off-line (Synaptsoft, Decatur, GA) with a detection threshold at three times the rms noise of an event-free interval. Evoked EPSC/IPSC sequences were recorded with pipettes filled with a solution containing (in mm) 130 K-gluconate, 1 EGTA, 10 HEPES, 5 MgATP, 0.5 NaGTP, and 5 NaCl (290 mOsm; pH, 7.3 adjusted with KOH). Extracellular stimulus electrodes filled with artificial CSF were placed into the granule cell layer in the vicinity of the patched PC, and monopolar stimuli (50 μs, up to 60 μA) were applied. Evoked EPSC/IPSC sequences were measured at holding potentials of −60 and −40 mV. All drugs were purchased from Sigma (Buchs, Switzerland). Statistical significance was assessed using Student's t test.
Immunofluorescence staining.
Juvenile [postnatal day 5 (P5), P7, P10, P15, and P20; n = 4 per genotype and per time point] and adult (>P60; n = 5 per genotype) mice were deeply anesthetized with Nembutal (50 mg/kg, i.p.) and perfused with a fixative containing 4% paraformaldehyde and 0.2% picric acid in 0.15 m phosphate buffer, pH 7.4. The brains were extracted immediately after the perfusion and postfixed in the same solution between 4 h (adults) and 36 h (P5). After cryoprotection in 30% sucrose dissolved in PBS, brains were frozen and cut at 40 μm with a sliding microtome. Free-floating sections were incubated overnight at 4°C in one of the following mixtures of primary antibodies (Table 1) diluted in PBS containing 2% normal goat serum and 0.2% Triton X-100: vesicular inhibitory amino acid transporter (VIAAT)/parvalbumin, vesicular glutamate transporter (VGLUT) 1/calbindin, and VGLUT2/calbindin. Sections were then washed with PBS, incubated for 30 min at room temperature with corresponding secondary antibodies coupled to Alexa 488 (Molecular Probes, Eugene, OR) or Cy3 (Jackson ImmunoResearch, West Grove, PA), washed again, mounted onto gelatizined glass slides, air dried, and coverslipped with fluorescence mounting medium (Dako, Carpinteria, CA).
Table 1.
List of antibodies
| Target protein | Species | Dilution | Source |
|---|---|---|---|
| Calbindin | Mouse | 1:5000, IMF | Swant (Bellinzona, Switzerland) |
| GABA (glutaraldehyde conjugate) | Rabbit | 1:1000, EM | Sigma (St. Louis, MO) |
| Glutamate | Rabbit | 1:1000, EM | P. Petrusz (University of North Carolina, Chapel Hill, NC) |
| δ2-Glutamate receptor | Mouse | 1:500, EM | M. Watanabe (Hokkaido University School of Medicine, Sapporo, Japan) |
| AMPA receptor GluR1 | Rabbit | 1:50, EM | Chemicon (Temecula, CA) |
| AMPA receptor GluR2/3 | Rabbit | 1:50, EM | Chemicon |
| Parvalbumin | Mouse | 1:20,000, IMF; 1:5000, EM | Swant |
| VGLUT1 | Rabbit | 1:20,000, IMF; 1:100, EM | Synaptic Systems (Göttingen, Germany) |
| VGLUT2 | Rabbit | 1:5000, IMF; 1:100, EM | Synaptic Systems |
| VIAAT | Rabbit | 1:5000, IMF; 1:1000, EM | Synaptic Systems; Dr. B. Gasnier (Ecole Normale Supérieure, Paris, France) |
IMF, Immunofluorescence; EM, immunoelectron microscopy.
Postembedding immunoelectron microscopy.
Juvenile (P10 and P14; n = 2 per genotype) and adult (n = 5 per genotype) mice were perfused as above with a fixative additionally containing either 2% (fixative 1) or 0.1% (fixative 2) glutaraldehyde, and brains were postfixed overnight. Tissue blocks prepared with fixative 1 were washed in phosphate buffer, postfixed with 1% osmium tetroxide in 0.1 m cacodylate buffer, dehydrated in ethanol, and embedded in Epon 812, as described previously (Giustetto et al., 1998). Ultrathin sections were collected on nickel grids and labeled with antibodies to GABA and glutamate (Table 1) (Phend et al., 1992). In control experiments, specific detection of GABA was ensured by competition of the primary antibodies with 300 μm glutamate and taurine conjugates for 2 h at room temperature (Ottersen et al., 1986). Tissue prepared with fixative 2 (two adult mice per genotype) was cryoprotected (1 and 2 m sucrose in phosphate buffer) and rapidly frozen in liquid propane in a cryofixation unit (KF80; Reichert, Vienna, Austria). Tissue blocks were then transferred to an automatic freeze substitution system (EM-AFS; Leica, Wetzlar, Germany), freeze-substituted with methanol, and embedded in Lowicryl HM20 (Hjelle et al., 1994). Ultrathin sections were collected on adhesive-coated (Electron Microscopy Sciences, Ft. Washington, PA) nickel grids (400 mesh) and processed for the immunogold method (Matsubara et al., 1996; Sassoè-Pognetto and Ottersen, 2000). Sections were labeled either for GABA or VIAAT alone or in combination with antibodies to VGLUT1, VGLUT2, AMPA receptor GluR1 or GluR2/3, or the δ2-glutamate receptor (Table 1). Secondary antibodies (1:20) were goat Fab fragments coupled to 10 or 20 nm colloidal gold particles (British BioCell International, Cardiff, UK).
Pre-embedding immunoelectron microscopy.
This procedure was used for the detection of parvalbumin in tissue from adult mice (fixative 2; n = 2 per gentoype). The cerebellum was dissected and cut into 70 μm parasagittal sections on a vibratome. The sections were cryoprotected in 30% sucrose and repeatedly frozen and thawed to enhance antibody penetration. They were then collected in PBS and processed free-floating as described in detail previously (Giustetto et al., 1998). After incubation at room temperature in primary (72 h) (Table 1) and secondary (goat anti-mouse conjugated to biotin, 1:250; Vector Laboratories, Burlingame, CA) antibodies, the sections were treated with 3,3′-diaminobenzidine, and the reaction product was silver-intensified and gold-toned. Finally, the sections were postfixed with 1% osmium tetroxide, dehydrated in acetone, and flat-embedded in Epon 812 as above.
Data analysis.
Immunofluorescence staining was visualized by confocal microscopy (Zeiss LSM-510 Meta; Jena, Germany) using a 100× objective (numerical aperture, 1.4) and sequential acquisition of separate channels. The pinhole was set to 1.0 Airy unit for each channel, and stacks of 12 consecutive confocal sections (512 × 512) spaced by 0.3–0.5 μm were acquired at a magnification of 60–150 nm/pixel using the full dynamic range of the photomultiplier. For display, images were processed with the image analysis software Imaris (Bitplane, Zurich, Switzerland). Images from both channels were overlaid (maximal intensity projection), and background was subtracted when necessary. For quantification of the density of GABAergic terminals, volumes were reconstructed from 12 confocal images, and individual terminals were identified as isolated objects (pixel size, 100 nm; 1 μm3 minimal apparent volume and minimal intensity >90 on an 8-bit gray scale) and counted automatically (Imaris; Bitplane). Size distribution of VIAAT-positive synaptic profiles was analyzed in single confocal sections (MCID-M5; Imaging Research, St. Catherines, Ontario, Canada). These data were quantified in six to eight sections per animal (n = 3 per genotype; Kolmogorov–Smirnov test).
Ultrathin sections were examined in a JEM-1010 electron microscope (Jeol, Tokyo, Japan) equipped with a side-mounted CCD camera (Mega View III; Soft Imaging System, Münster, Germany). For quantification of immunogold labeling, structures to be analyzed were sampled systematically in several grid squares (1600 μm2) taken from the middle third of the molecular layer from two adult mice per genotype. GABA and glutamate immunogold labeling was assessed in sections embedded in Epon, whereas GluR2/3 labeling was quantified in sections embedded in Lowicryl. The percentage of GABAergic terminals (recognized by the presence of pleomorphic synaptic vesicles and intense immunogold labeling) forming symmetric and asymmetric synaptic junctions in the molecular layer was determined by systematically photographing all GABAergic terminals in four grid squares per animal at a magnification of 60,000× (202 terminals in wild-type and 173 in mutant mice). GABA-positive terminals were readily detected because of the high signal-to-noise ratio of GABA immunogold labeling. The intensity of glutamate immunogold labeling in parallel fiber (PF) terminals, PC spines, and presumptive GABAergic terminals recognized by their morphology (see Results) was assessed by determining the density of gold particles in these structures sampled from 25–30 photographs (magnification, 60,000×) per genotype (n = 2 mutant and 1 wild-type mice). The intensity of GluR2/3 labeling in asymmetric synapses was assessed in α10/0 mice (n = 2) by counting the number of immunogold particles located in the synaptic junction and located within 50 nm of the postsynaptic membrane in a random sample of 278 axo-spinous synapses, 48 asymmetric axo-dendritic synapses, and 66 axo-spinous synapses formed by presumptive GABAergic terminals. Differences in the distribution of immunogold particles in these three populations were analyzed using a nonparametric test (95% confidence intervals; Kaplan-Meier method). Finally, the average number of basket cell terminal profiles making synapses on PC bodies was estimated in single sections processed for GABA immunogold labeling using a sample of 25 cells sectioned through the nucleolus visualized at a magnification of 40,000× in two mice per genotype.
Results
Lack of GABAA receptor-mediated synaptic transmission in PCs of α10/0 mice
To determine the status of inhibitory neurotransmission in PCs of α10/0 mice, spontaneous and evoked synaptic currents were recorded in whole-cell patch-clamp experiments from acute parasagittal slices of the cerebellum (Fig. 1). In PCs from P18–P22 wild-type mice (n = 12 cells), the persistence of spontaneous synaptic currents in the presence of 4 mm kynurenic acid indicated that they were predominantly GABAergic, as reported previously (Konnerth et al., 1990). When a “high-chloride” internal solution was used, spontaneous IPSCs (sIPSCs) could be observed at an average frequency of 20.1 ± 3.4 Hz and a mean amplitude of 168 ± 88.3 pA (n = 12) (Fig. 1B1,B2). The frequency and amplitude of sIPSCs were reduced by >90% (n = 8; p < 0.01) after application of the competitive GABAA receptor antagonist bicuculline (10 μm); additional application of the glycine receptor antagonist strychnine (1 μm) did not affect the small population of sIPSCs that remained (n = 8; p > 0.5). All remaining synaptic activity was blocked by the noncompetitive GABAA receptor antagonist picrotoxin (100 μm; n = 8). Thus, sIPSCs were mediated by GABAA receptors in these cells. No change in the baseline holding current was observed after picrotoxin application, excluding a significant contribution of tonically active GABAA receptors to the recordings.
Figure 1.

A–C, Lack of spontaneous (A, B) or evoked (C) IPSCs in PCs from α10/0 mice (A1, A2, C1) compared with wild-type mice (B1, B2, C2) in whole-cell patch-clamp recordings performed in acute slices in the presence of 4 mm kynurenic acid. A, Representative traces from low (A1) and high-temporal (A2) resolution illustrating the absence of detectable currents in mutant mice; neither bicuculline, strychnine, nor picrotoxin caused a shift in the holding current. B, Example of traces from wild-type mice depicting the high frequency of spontaneous currents and their suppression by bicuculline and picrotoxin. Note that coapplication of strychnine in the presence of bicuculline produced no further effect on spontaneous IPSCs. C, The absence of evoked IPSCs in mutants is demonstrated in a PC held at −60 mV (gray) and at −40 mV (black). In wild type (C2), a typical sequence of EPSC/IPSC is evident; in α10/0 (C1), only the EPSC is observed. D, Control recording of a cortical pyramidal cell held at −60 mV (gray) and at −40 mV (black) in a slice from a mutant mouse, demonstrating the presence of a typical sequence of EPSC/IPSC. wt, Wild type; cereb., cerebellum.
No sIPSCs were detected in recordings from PCs in P18–P22 α10/0 mice (n = 18 cells) (Fig. 1A1,A2). The cells did not react to application of bicuculline (10 μm), strychnine (1 μm), or picrotoxin (100 μm) (n = 10). Synaptic transmission in PCs was analyzed further in EPSC/IPSC sequences evoked with extracellular stimulus electrodes. With a low-chloride-containing internal solution, EPSCs were detected as inward currents at holding potentials of −60 mV, whereas IPSCs were measured as outward currents at −40 mV. In slices from wild-type mice, EPSCs (peak amplitude, 223 ± 58 pA; n = 4) were immediately followed by IPSCs (peak amplitude, −218 ± 29 pA; n = 4) (Fig. 1C2). In slices from α10/0 mice, the average EPSC amplitude was 216 ± 115 pA (n = 6). However, no evoked outward current was ever observed at a holding potential of −40 mV; instead, an inward current of 160 ± 82 pA (n = 6) was measured (Fig. 1C1). Under these conditions, spontaneous EPSCs were not different between genotypes, whereas no spontaneous IPSCs were detected in slices from α10/0 mice (data not shown). Likewise, no GABAA receptor-mediated responses could be detected in PCs recorded from two young mutant mice (P10; n = 5), whereas spontaneous and evoked kynurenic acid-sensitive EPSCs could readily be observed (data not shown).
As a control, recordings from cortical pyramidal cells in α10/0 mice revealed both evoked and spontaneous IPSCs (mean EPSC amplitude, 191 ± 32 pA; mean IPSC amplitude, −204 ± 18 pA; n = 3), showing that the inhibitory deficit is not ubiquitous (Fig. 1D). Again, similar results were obtained at P10. In summary, neither tonic inhibition nor phasic IPSCs were apparent in PCs of α10/0 mice.
Abnormal morphology of GABAergic terminals in the molecular layer
As reported previously (Kralic et al., 2005, 2006), the cerebellum of α10/0 mice had a normal cytoarchitecture, and the morphology of PC bodies, dendrites, and spines, as visualized by parvalbumin or calbindin immunofluorescence, was indistinguishable from wild type (Fig. 2). The complete absence of postsynaptic GABAergic inhibition in PCs afforded a unique opportunity to test whether GABAergic terminals and synapses are affected in α10/0 mice. Confocal microscopy imaging of VIAAT immunofluorescence revealed the presence of GABAergic terminals in both genotypes, which appeared larger in α10/0 mice, notably in the inner molecular layer, just above the PC layer (Fig. 2A,B). Using double staining with parvalbumin to label PC dendrites, VIAAT-positive terminals frequently were apposed to PC proximal dendrites and surrounded PC somata in both genotypes (Fig. 2D,E). They appeared more heterogeneous in size in mutant mice; a significant difference in the mean cross-section area was evident by cumulative probability distribution analysis (Fig. 2C) (p < 0.005; Kolmogorov–Smirnov test). However, the numerical density of terminals, as determined quantitatively in the middle of the molecular layer, was unchanged between genotypes (494 ± 24 terminals/10,000 μm3 in wild-type mice versus 510 ± 44 in mutant mice; n = 3 per genotype; NS). As a control, the distribution and staining pattern of glutamatergic terminals (Hioki et al., 2003) was apparently not affected, as verified by visual assessment for PFs with VGLUT1− (data not shown) and climbing fibers (CFs) with VGLUT2 immunoreactivity (Fig. 2F,G).
Figure 2.
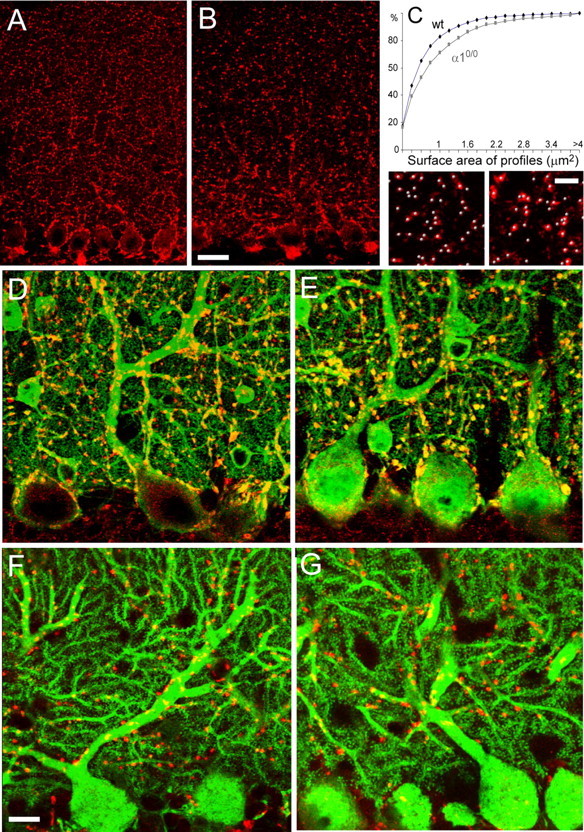
Abnormal morphology of GABAergic but not glutamatergic terminals in the molecular layer of α10/0 mice. A, B, Confocal microscopy images of VIAAT immunofluorescence in wild-type (A) and α10/0 (B) adult mice. Each panel shows a single confocal layer. In α10/0 mice, VIAAT-positive terminals appear larger in the deeper molecular layer. C, Cumulative size distribution analysis of VIAAT-positive terminals revealing that mutants have a significantly greater proportion of large terminals (Kolmogorov–Smirnov test, p < 0.005; n = 3 per genotype). The bottom panels depict the automatic identification of VIAAT-positive terminals (gray beads) in three-dimensional reconstruction images used for quantification of terminal density. No significant difference was found in mutant compared with wild-type (wt) mice (see text). D, E, Double immunofluorescence staining for parvalbumin (green) and VIAAT (red) in the cerebellum of adult wild-type (D) and α10/0 (E) mice (stack of 12 confocal sections spaced by 0.3 μm) depicting the presence of numerous terminals on the soma and proximal dendrites of PCs. The terminals appear yellow because they are colabeled for parvalbumin and VIAAT. Note that the VIAAT-positive terminals appear larger in mutant mice. F, G, Double immunofluorescence staining for calbindin (green) and VGLUT2 (red) in the cerebellum of adult wild-type (F) and α10/0 (G) mice (stack of 5 confocal sections spaced by 0.4 μm); the morphology of PC dendrites and spines is similar in both genotypes. CF terminals labeled for VGLUT2 retain their characteristic distribution along dendrites. Scale bars, 20 μm.
Presence of heterologous synapses in the molecular layer of α10/0 mice
Ultrastructurally, GABAergic presynaptic profiles were identified by immunogold labeling for GABA (Fig. 3A–C). In wild-type mice, these terminals formed symmetric synapses apposed to a dendritic shaft or a cell body (data not shown). Such synapses were also seen in mutant mice (Fig. 3A,B). The GABA labeling intensity, judged by the density of gold particles, was similar in both genotypes. A quantitative analysis in sections from two mice per genotype showed, unexpectedly, that 74% of GABAergic terminals forming a synapse in the molecular layer of mutant mice were associated with PC spines containing a characteristic asymmetric postsynaptic specialization (Fig. 3C; see Fig. 3G for quantification). The GABA-positive terminals establishing asymmetric junctions were unusually large and contacted several spines (up to nine in a single ultrathin section). They were distributed throughout the molecular layer, in good correlation with the immunofluorescence data (Fig. 2B). The GABAergic phenotype of these terminals forming heterologous synapses was confirmed by immunogold labeling for VIAAT, which was readily observed in large profiles forming asymmetric synapses with multiple PC spines (Fig. 3D). As expected, VIAAT immunogold labeling also was prominent in terminals forming symmetric synapses, such as basket cell terminals surrounding the cell body of PC (data not shown), and it was absent from glutamatergic structures.
Figure 3.
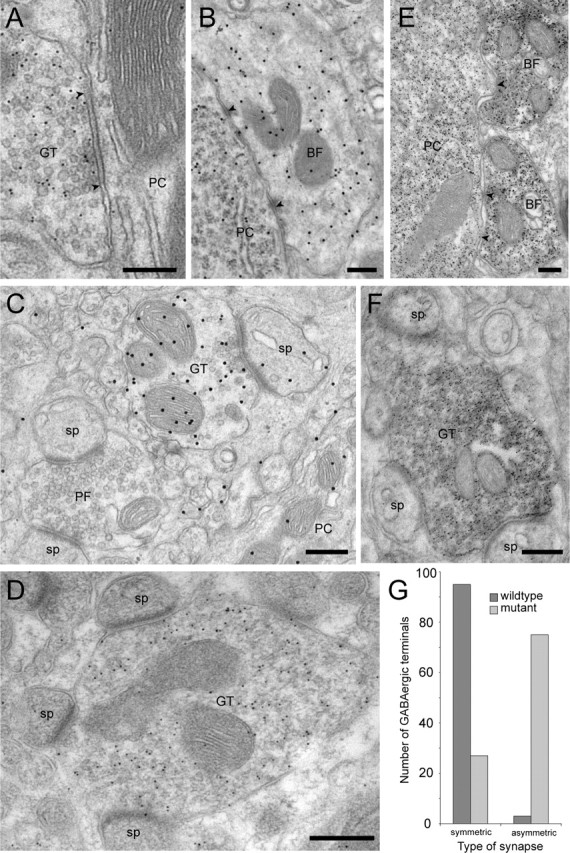
Ultrastructural characterization of GABAergic terminals in the molecular layer of α10/0 mice. A, A typical GABA-positive terminal (10 nm gold particles) makes a symmetric synapse (arrowheads) on a PC dendrite. B, Normal morphology of a basket cell terminal labeled for GABA (20 nm gold particles) contacting a PC soma; arrowheads mark two small symmetric synapses. C, A GABA-positive terminal (labeled with 20 nm gold particles) makes a heterologous, asymmetric synapse with a PC spine. In the vicinity, an unlabeled PF terminal makes asymmetric synapses with two spines. A portion of a PC dendrite is visible in the bottom right corner. D, VIAAT-positive terminal (10 nm gold particles) making heterologous, asymmetric synapses with four PC spines in the plane of the section; note the large size of such aberrant terminals. E, Normal morphology and parvalbumin labeling of two basket cell terminals making symmetric synapses (arrowheads) on a PC body, which is also parvalbumin positive. F, Pre-embedding immunoperoxidase staining for parvalbumin in a terminal making heterologous asymmetric synapses with three PC spines. G, Quantification of the distribution of GABA-positive profiles (postembedding immunogold labeling with 10 nm gold particles) forming symmetric or asymmetric synapses in ultrathin sections of wild-type and mutant mice (n = 2 animals per genotype). Note that GABA-positive profiles forming asymmetric synapses represent the large majority of synaptic profiles in mutant mice. Scale bars, 300 nm. GT, GABAergic terminal; BF, basket cell terminal; sp, spine.
The formation of heterologous synapses by GABAergic terminals was selective for the molecular layer, because symmetric synapses on PC somata (Fig. 3B) were not affected by the mutation. The average number of basket cell terminals surrounding PC bodies ranged between 17.5 and 19 in wild-type mice and between 19 and 20.5 in α10/0 mice (n = 25 cells sampled in two animals per genotype). Similarly, asymmetric synapses established by PFs and CFs retained their normal morphology, confirming that no major reorganization of excitatory inputs to PCs occurs in α10/0 mice. These results suggest that, in the absence of functional synaptic GABAA receptors in PCs, GABAergic terminals are impaired selectively in the molecular layer and form heterologous synapses with PC spines.
Stellate cells provide most of the inhibitory input to PC dendrites. To determine whether “aberrant” terminals originate from these cells, immunostaining for parvalbumin, which is strongly expressed in stellate cells, was performed. Numerous terminals forming heterologous synapses in the molecular layer were labeled for parvalbumin (Fig. 3F), although unlabeled terminals were also detected, possibly because of the limited penetration of antibodies. The normal morphology and distribution of basket cell terminals in mutant mice, which also express parvalbumin, was confirmed by visual assessment in this material (Fig. 3E).
Heterologous synapses are not glutamatergic
Immunogold labeling for glutamate (Fig. 4A,B) and its vesicular transporters, VGLUT1 and VGLUT 2 (Fig. 4C-D), was then used to determine whether profiles forming heterologous synapses express a dual GABAergic/glutamatergic phenotype. To distinguish the neurotransmitter pool from the metabolic pool of glutamate, the density of immunogold particles was compared quantitatively in a population of glutamatergic presynaptic profiles (PF terminals), in PC spines, and in profiles forming heterologous synapses identified by the presence of at least two synaptic contacts with spines (Fig. 4A). Although the latter sample could include some CF or PF terminals, the results show that the average immunogold labeling in these aberrant profiles was similar to that of PC spines (Fig. 4B) and less than half that of PF terminals, suggesting that aberrant terminals do not use glutamate for neurotransmission. Furthermore, the intensity of glutamate labeling in PF terminals was comparable in mutant and wild type (Fig. 4F), indicating that no major compensation takes place at this level. These findings were confirmed by the observation that VGLUT2 immunogold labeling was present exclusively in CF terminals (Fig. 4C) and VGLUT1 labeling was present exclusively in PF terminals (Fig. 4D,D′). Double labeling with VIAAT directly demonstrated the mutual exclusion of vesicular glutamate and GABA transporters within single terminals in both genotypes (Fig. 4C,D).
Figure 4.
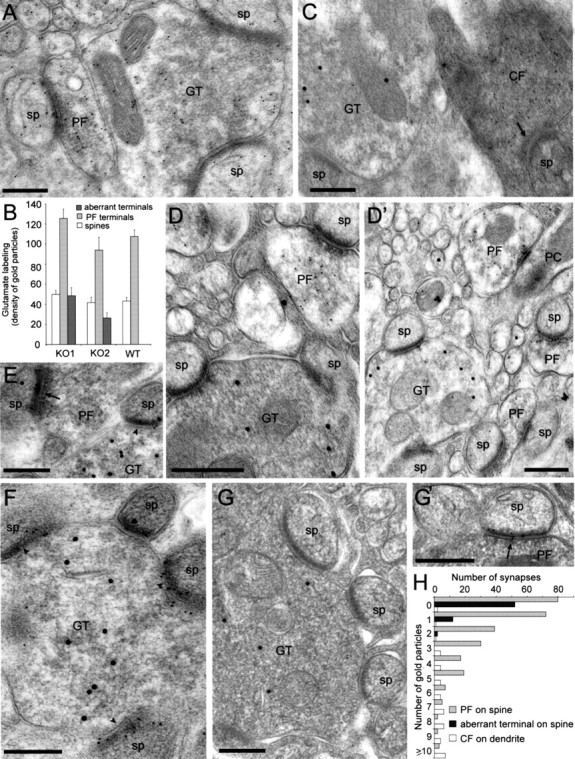
Absence of presynaptic glutamatergic markers in large terminals forming heterologous synapses with spines carrying δ2-type, but not AMPA-type, glutamate receptors in α10/0 mice. A, Differential labeling intensity for glutamate (10 nm gold particles) in GABAergic and glutamatergic profiles, as illustrated for weakly labeled large terminals contacting two spines and adjacent PF terminals, which contain a higher density of gold particles. Note that PC spines contain a similar low labeling as the aberrant terminal. B, Quantification of the density of glutamate immunogold labeling in PF terminals compared with aberrant axon terminals and PC spines in two mutants and one wild-type (WT) mouse. In both mutants, the density of gold particles in aberrant terminals is comparable to that of PC spines and ∼60% lower than in PF terminals sampled on the same grids. Note that in PFs and PC spines, the labeling density is similar in the three animals sampled. Error bars indicate SE. C, Segregated labeling of VIAAT (20 nm gold particles) in a terminal making a heterologous asymmetric synapse on a spine and VGLUT2 (10 nm gold particles) in a CF terminal recognized by its electron-dense ultrastructure, also making an asymmetric synapse with a spine (arrow). D, D′, Segregated labeling of VIAAT (20 nm gold particles) in large terminals making heterologous asymmetric synapses with spines and VGLUT1 (10 nm gold particles) in neighboring PF terminals. E, Labeling of δ2-glutamate receptors (10 nm gold particles) in spines that establish synapses with either a PF terminal (arrowhead) or a GABA-positive terminal (20 nm gold particles; arrow). F, Identification of δ2-glutamate receptors (10 nm gold particles) in the postsynaptic density of spines contacted by an aberrant terminal labeled for VIAAT (20 nm gold particles). G, G′, Lack of GluR2/3 subunit labeling in asymmetric synapses formed by an aberrant terminal labeled for VIAAT (20 nm gold particles), in contrast to those formed by PF terminals (arrowhead in G′). H, Differential distribution of immunogold particles for the GluR2/3 subunit in asymmetric synapses formed by PFs on spines, CFs on dendrites, and aberrant terminals on spines; data are pooled from two α10/0 mice. Scale bars, 300 nm. GT, Terminal; sp, spine.
Next, we investigated whether PC spines postsynaptic to GABA-positive terminals express glutamate receptors. Double-immunogold labeling revealed that the δ2-glutamate receptor, which is selectively located at PF synapses on PC spines (Takayama et al., 1995), was present in the vast majority of spines apposed to both PFs and aberrant terminals labeled for GABA (Fig. 4E) or VIAAT (Fig. 4F), as determined by visual inspection in sections from two mice per genotype. This result suggests that the expression and postsynaptic targeting of δ2-glutamate receptors is not dependent on the presynaptic transmitter phenotype. In contrast, labeling for the AMPA-type glutamate receptor GluR2/3 subunit revealed that asymmetric synapses formed by VIAAT-positive, aberrant terminals were devoid of GluR2/3 labeling (Fig. 4G), whereas the majority of those formed by PF terminals were labeled (Fig. 4G′). A quantitative analysis was therefore performed in single-labeled sections from two α10/0 mice (Fig. 4H). A significant difference was found in the distribution of GluR2/3 immunogold labeling in heterologous synapses (81% of which were not labeled), and only 2% contained a maximum of two immunogold particles and asymmetric synapses formed by PFs either on spines (62% contained between 1 and 11 gold particles) or on dendritic shafts (96% contain between 1 and 13 gold particles) (Fig. 4H). A weak labeling for GluR1 was detected in both genotypes, located occasionally in asymmetric synapses formed by PFs.
GABAergic synapses are correctly assembled at initial stages of synaptogenesis
The results so far reveal in α10/0 mice a differential alteration of GABAergic terminals formed by stellate and basket cells onto PC dendrites and cell bodies, respectively. To determine whether the absence of GABAA receptor-mediated neurotransmission affects maturation of GABAergic terminals and formation of symmetric synapses in the molecular layer or alters synapse maintenance in the adult cerebellum, we assessed by visual inspection the development of GABAergic synapses during ontogeny. The results are based on visual observation of sections from four animals per age and per genotype. Littermates derived from heterozygous breedings were used to minimize variability. VIAAT-positive terminals were seen first at P5 in the nascent molecular layer (data not shown), and their density increased rapidly thereafter following the maturation of PC dendrites. At P7, PCs became strongly immunoreactive for parvalbumin, as seen in all animals, but only few dendritic spines were evident. GABAergic terminals typically were apposed to PC dendrites and somata, as seen by double immunofluorescence with parvalbumin in wild-type (Fig. 5A,A′) and in α10/0 (Fig. 5B,B′) mice. Clear evidence for axo-dendritic appositions could readily be obtained by three-dimensional reconstruction of high-magnification images (Fig. 5A′,B′). At P21, GABAergic innervation of PC somata and proximal dendrites was extensive in both genotypes (Fig. 5C,D). The distribution of VIAAT-positive terminals in the molecular layer was similar in both genotypes, although they appeared slightly larger in mutants, notably in the vicinity of PC somata. Altogether, these qualitative observations suggest that the development of GABAergic innervation of PC somata and proximal dendrites is not impaired in mutant mice.
Figure 5.
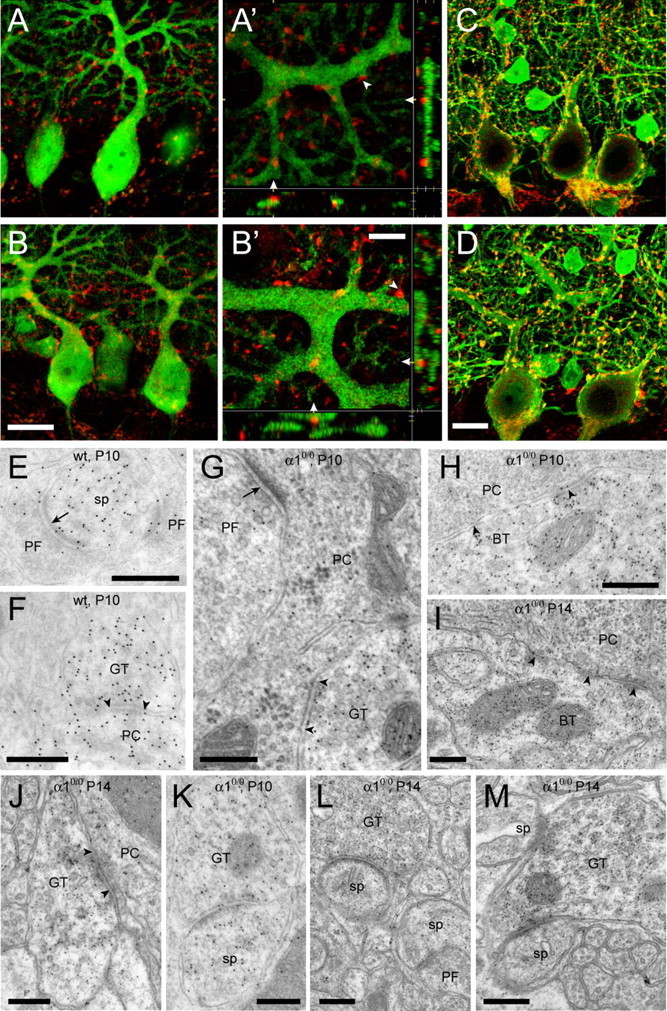
Normal formation of GABAergic synapses and delayed appearance of heterologous synapses in the developing cerebellum of α10/0 mice. A, B, Double immunofluorescence staining for parvalbumin (green) and VIAAT (red) in the cerebellum of P7 wild-type (A, A′) and mutant (B, B′) mice. At low magnification, VIAAT terminals are evident in the deep molecular layer and appear closely apposed to PC dendrites in both genotypes (A, B). At high magnification, three-dimensional reconstructions of confocal image stacks confirm the direct apposition of VIAAT-positive terminals onto PC dendrites (arrowheads; A′, wild type; B′, α10/0 mouse; P7). These panels depict x–y, y–z, and x–z projections of up to 12 confocal layers spaced by 0.3 μm; the side panels show the corresponding cross section at the level indicated by the white triangles. C, D, At P21, the GABAergic innervation of PCs appears similar to that seen in adult mice (see Fig. 2), with VIAAT-positive terminals being larger in mutant (D) than in wild-type (C) mice. E–M, Postembedding immunoelectron microscopy for GABA in P10 wild-type (E, F), P10 α10/0 (G, H, K), and P14 α10/0 (I, J, L, M) mice. Note that PC spines are strongly labeled for GABA at P10 (E, F) and much less so at P14 (L, M). Asymmetric synapses formed by GABA-negative, presumptive PFs onto spines and dendrites are evident in both genotypes (E, G; arrows); in addition, axo-dendritic symmetric synapses formed by GABA-positive terminals are present at both P10 and P14 (F, G, J; arrowheads). Likewise, basket cell terminals (BT) forming symmetric synapses are readily recognized in mutant mice (H, I). K, A heterologous, asymmetric synapse formed by a GABA-positive terminal with a spine, also labeled for GABA, in a section from a P10 α10/0 mouse. L, M, These structures became more evident at P14, with the presence of GABAergic terminals forming asymmetric synapses either with a single spine (L) or with multiple spines (M). Note that synaptic contacts between PF terminals and spines were present in the same sections (L). Scale bars: (in B, D) A–D, 20 μm; E–M, 300 nm. wt, Wild type; sp, spine; GT, GABA-positive terminal.
To determine whether GABAergic terminals form symmetric synapses onto developing PC dendrites and somata, postembedding immunogold labeling for GABA was performed in P10 and P14 mice (n = 2 per age and per genotype) (Fig. 5E–M). At P10, a prominent staining for GABA was observed in PC dendrites and spines, whereas PF terminals were immunonegative, as expected (Fig. 5E,G). Basket cell terminals around PC somata were readily recognized in both mice of each genotype, but the synaptic contacts were small and less discernable than in older animals (Fig. 5H). Axo-dendritic, symmetric synapses formed by terminals labeled for GABA were rare but could be observed in both mice of each genotype (Fig. 5F,G). In α10/0 mice, only a single heterologous synaptic contact between a GABAergic terminal and a spine, both strongly labeled for GABA (Fig. 5K), could be detected in sections from two P10 animals (in a sample of >200 GABA-labeled terminals examined in sections from two animals). At P14, mature axo-somatic (Fig. 5I) and axo-dendritic (Fig. 5J) symmetric synapses were evident in both α10/0 mutant and wild-type mice. Spine density was much increased in the molecular layer, and, in mutant mice, GABA-positive terminals forming asymmetric synapses with spines were readily observed (Fig. 5L,M) besides unlabeled PF terminals making similar synapses (Fig. 5L), as expected. These results suggest that the initial steps of GABAergic synapse formation are unperturbed in α10/0 mice, although heterologous synapses appear in parallel with spine differentiation.
Discussion
The present results reveal major alterations of GABAergic synapses in PCs in the absence of detectable GABAA receptor function. Although initial steps of synapse formation on PC proximal dendrites and cell bodies appear unaffected in α10/0 mice, as assessed by visual inspection, the majority of GABAergic terminals in the molecular layer of adult mice form heterologous, asymmetric synapses with spines expressing δ2-type, but lacking AMPA-type, glutamate receptors. As a consequence, the number of GABAergic synapses on PC dendrites is strongly reduced in adult animals, whereas perisomatic synapses are retained. Therefore, α1-GABAA receptors in PCs are required to prevent formation of heterologous synapses and for long-term maintenance of axo-dendritic, but not axo-somatic, synapses. Furthermore, differentiation of the postsynaptic density in spines and expression of δ2-glutamate receptors are independent of the presynaptic partner, whereas AMPA receptors require glutamatergic transmission for synaptic targeting. Thus, the differentiation and long-term maintenance of presynaptic and postsynaptic elements in the cerebellum is primarily cell autonomous.
In agreement with previous studies of acutely dissociated PCs (Sur et al., 2001; Kralic et al., 2005), α1 subunit gene deletion results in complete loss of spontaneous and evoked IPSCs in PCs recorded from juvenile mice. No such alteration was observed in the cerebral cortex, as expected (Bosman et al., 2005). Furthermore, no picrotoxin-sensitive tonic currents were detected to compensate for the absence of synaptic GABAA receptors in PCs. GABAB receptors are abundant in PC spines (Fritschy et al., 1999; Kulik et al., 2002), where they modulate type 1 metabotropic glutamate receptors independently of GABA (Hirono et al., 2001; Tabata et al., 2004). Although activation of GABAB receptors could have been conceivable at heterologous synapses, our patch-clamp recordings provided no evidence for GABAB-mediated events in PCs from mutant mice (data not shown). Therefore, GABAB receptors do not seem to compensate for the loss of GABAA receptors.
PCs start expressing α1-GABAA receptors during late fetal stages (Laurie et al., 1992b; Fritschy et al., 1994), before the development of basket and stellate cell synapses (McLaughlin et al., 1975; Ango et al., 2004; Takayama and Inoue, 2005), and are not known to express other α subunit variants during ontogeny (Laurie et al., 1992b). These findings are confirmed here by the absence of detectable spontaneous and evoked IPSCs in PCs from P10 α10/0 mice. The initial formation of morphologically normal GABAergic synapses in juvenile mutant mice indicates that the absence of GABAA receptor-mediated signaling does not impair synaptogenesis. Furthermore, the normal distribution of glutamatergic terminals positive for VGLUT1 and VGLUT2 showed that the formation of glutamatergic synapses is also not affected in the absence of GABAergic transmission. In particular, the activity-dependent elimination of supernumerary CFs (Rabacchi et al., 1992; Hashimoto et al., 2001) likely takes place during development of α10/0 mice, suggesting that global network properties are primarily normal in the maturing molecular layer.
Ultrastructurally, the presence of heterologous synapses between presynaptic GABAergic terminals and postsynaptic spines carrying a well differentiated postsynaptic density is a striking morphological feature in the molecular layer of mutant mice, which was seen in all animals examined. Prominent labeling for GABA and VIAAT and the absence of presynaptic markers of glutamatergic transmission demonstrates the GABAergic nature of these terminals. Differentiation of the postsynaptic density in spines contacted by GABAergic terminals therefore occurs independently of the nature of the presynaptic partner. However, AMPA and δ2-glutamate receptors exhibit a strikingly different dependence on glutamatergic transmission for synaptic targeting and/or maintenance. The near complete absence of GluR2/3 subunit labeling selectively in heterologous synapses indicates that AMPA receptors are regulated by presynaptic signals. A global downregulation is excluded by the labeling seen opposite PF terminals. A switch between GluR2, the main AMPA receptor subunit expressed by adult PCs (Lambolez et al., 1992; Martin et al., 1993), and GluR1 is also unlikely, in view of the low labeling for the latter subunit in both genotypes. Our results therefore exclude a major change in AMPA receptor expression to compensate for the lack of GABAergic function in PCs and underscore the importance of glutamatergic transmission for synaptic targeting of AMPA receptors. The function of δ2-glutamate receptors remains elusive. During development, their presence in PC spines is regulated by PF terminals (Takayama et al., 1997). In turn, these receptors are required for synapse stabilization and long-term preservation of PF terminals (Takeuchi et al., 2005). Our results indicate that δ2-glutamate receptors are stabilized also at heterologous synapses formed by GABAergic terminals, strengthening the concept that several aspects of synapse formation and maintenance are transmitter independent.
Heterologous synapses have been reported previously in the cerebellar cortex of reeler mice, which lack granule cells and, consequently, PF terminals (Wilson et al., 1981), and in nodding mice, characterized by a delayed maturation of PC spines (Sotelo, 1990). In α10/0 mice, the appearance of heterologous synapses paralleled the formation of dendritic spines, suggesting that these structures provide a synaptogenic signal for GABAergic terminals that is sufficient for synapse formation in the absence of GABAergic transmission. Thus, there might be a competition between PC dendrites and spines for synapse formation with stellate cell axons. GABAA-mediated synaptic transmission is crucial for maintaining correctly matched symmetric synapses and avoiding the formation of heterologous synapses. The fact that GABAA receptors are dispensable for initial synapse formation finds a clear parallel at the neuromuscular junction, where the postsynaptic apparatus can initially develop in myotubes devoid of acetylcholine receptors, although the receptors are needed for the full differentiation of the synaptic junction (Marangi et al., 2001).
The formation of heterologous synapses on dendritic spines affects symmetric GABAergic synapses on PC dendritic shafts but not somata. This finding reveals a principal difference in the maintenance of the two types of synapses. However, a marker distinguishing between stellate and basket cell terminals would be required to know which cell type makes the axo-dendritic synapses found in adult mice. Basket cell terminals preferentially express the hyperpolarization-activated cyclic nucleotide-gated cation channel subunit HCN1 (Lujan et al., 2005). HCN1 staining of the basket cell pinceau targeting the axon-initial segment was unaffected in α10/0 mice, but it was not detectable in individual terminals on proximal dendrites (our unpublished observations), precluding any conclusion about their distribution in mutant mice. However, because the remaining symmetric synapses on PC dendrites were distributed across the entire thickness of the molecular layer, it seems rather unlikely that they represent ectopic basket cell synapses. Therefore, the localization, rather than the origin, of axo-dendritic synapses determines their limited stability in α10/0 mice
Several mechanisms can explain the differential requirement of GABAA receptors for long-term stabilization of axo-dendritic and axo-somatic synapses. It is possible that terminals in the molecular layer are subject to more intense competition with dendritic spines than basket cell terminals targeted toward PC somata. In addition, recent evidence demonstrated the role of ankyrin G for proper targeting and differentiation of basket cell synapses on the soma and axon-initial segment of PCs (Ango et al., 2004). Transsynaptic interactions involving adhesion molecules might be sufficient for long-term maintenance of axo-somatic synapses. Another possible mechanism involves differential signaling by the neurexin–neuroligin complex. As mentioned in the Introduction, these proteins have been implicated in synapse formation and transmitter specification. In particular, neuroligin 2 is selectively clustered in GABAergic synapses in the adult brain (Varoqueaux et al., 2004) and contributes to their formation during development by binding to presynaptic β-neurexin (Graf et al., 2004; Chih et al., 2005). In PCs, neuroligin 2 is present in somatic and dendritic GABAergic synapses (Varoqueaux et al., 2004), suggesting at first that it does not account for the differential maintenance of basket and stellate cell synapses in α10/0 mice. However, recent studies revealed that isoforms of neuroligins interacting with both α- and β-neurexins play distinct roles in synapse formation and maintenance (Boucard et al., 2005). To resolve these issues, it will be important to analyze the subcellular distribution of neuroligin 2 in PCs in relation to synaptogenesis in α10/0 mice.
Phenotypically, α10/0 mice represent a novel genetic model of essential tremor, which responds well to drugs used in patients affected by this disorder (Kralic et al., 2005). Although the cause of the tremor in mutant mice is not established, the deficit in GABAergic transmission affecting PCs might play a key role. Indeed, in contrast to α10/0 mice, destruction of Golgi cells, leading to hyperactivity of granule cells, causes severe ataxia and impairment of motor coordination (Watanabe et al., 1998), whereas increased CF activity potentiates GABAergic inhibition and induces impairment in motor learning and motor coordination (Ohtsuki et al., 2004). The present study suggests that essential tremor is caused by a subtle deficit affecting cerebellar output, which might potentially be corrected therapeutically by increasing GABAergic inhibition onto PCs.
Footnotes
This work was supported by grants from the Swiss National Science Foundation (31-63901.00 to J.-M.F. and 631-066012 to K.E.V.) and the Italian MIUR (FIRB RBNE019J7C-005 to M.S.-P.). We thank G. Homanics for providing α10/0 mice and P. Petrusz, B. Gasnier, O. P. Ottersen, and M. Watanabe for generous gifts of antibodies.
References
- Anderson TR, Shah PA, Benson DL (2004). Maturation of glutamatergic and GABAergic synapse composition in hippocampal neurons. Neuropharmacology 47:694–705. [DOI] [PubMed] [Google Scholar]
- Ango F, Di Cristo G, Higashiyama H, Bennett V, Wu P, Huang ZJ (2004). Ankyrin-based subcellular gradient of neurofascin, an immunoglobulin family protein, directs GABAergic innervation at Purkinje axon initial segment. Cell 119:257–272. [DOI] [PubMed] [Google Scholar]
- Biederer T, Sara Y, Mozhayeva M, Atasoy D, Liu X, Kavalali ET, Sudhof TC (2002). SynCAM, a synaptic adhesion molecule that drives synapse assembly. Science 297:1525–1531. [DOI] [PubMed] [Google Scholar]
- Bosman LWJ, Heinen K, Spijker S, Brussaard AB (2005). Mice lacking the major adult GABAA receptor subtype have normal number of synapses, but retain juvenile IPSC kinetics until adulthood. J Neurophysiol 94:338–346. [DOI] [PubMed] [Google Scholar]
- Boucard AA, Chubykin AA, Comoletti D, Taylor P, Sudhof TC (2005). A splice code for trans-synaptic cell adhesion mediated by binding of neuroligin 1 to alpha- and beta-neurexins. Neuron 48:229–236. [DOI] [PubMed] [Google Scholar]
- Chattopadhyaya B, Di Cristo G, Higashiyama H, Knott GW, Kuhlman SJ, Welker E, Huang ZJ (2004). Experience and activity-dependent maturation of perisomatic GABAergic innervation in primary visual cortex during a postnatal critical period. J Neurosci 24:9598–9611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chih B, Engelman H, Scheiffele P (2005). Control of excitatory and inhibitory synapse formation by neuroligins. Science 307:1324–1328. [DOI] [PubMed] [Google Scholar]
- Chubykin AA, Liu X, Comoletti D, Tsigelny I, Taylor P, Sudhof TC (2005). Dissection of synapse induction by neuroligins: effect of a neuroligin mutation associated with autism. J Biol Chem 280:22365–22374. [DOI] [PubMed] [Google Scholar]
- Craig AM, Blackstone CD, Huganir RL, Banker G (1994). Selective clustering of glutamate and γ-aminobutyric acid receptors opposite terminals releasing the corresponding neurotransmitters. Proc Natl Acad Sci USA 91:12373–12377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fritschy JM, Brünig I (2003). Formation and plasticity of GABAergic synapses: physiological mechanisms and pathophysiological implications. Pharmacol Ther 98:299–323. [DOI] [PubMed] [Google Scholar]
- Fritschy JM, Paysan J, Enna A, Mohler H (1994). Switch in the expression of rat GABAA-receptor subtypes during postnatal development: an immunohistochemical study. J Neurosci 14:5302–5324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fritschy JM, Meskenaite V, Weinmann O, Honer M, Benke D, Mohler H (1999). GABAB-receptor splice variants GB1a and GB1b in rat brain: developmental regulation, cellular distribution, and extrasynaptic localization. Eur J Neurosci 11:761–768. [DOI] [PubMed] [Google Scholar]
- Gally C, Bessereau JL (2003). GABA is dispensable for the formation of junctional GABA receptor clusters in Caenorhabditis elegans. J Neurosci 23:2591–2599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giustetto M, Kirsch J, Fritschy JM, Cantino D, Sassoè-Pognetto M (1998). Localisation of the clustering protein gephyrin at GABAergic synapses in the main olfactory bulb of the rat. J Comp Neurol 395:231–244. [DOI] [PubMed] [Google Scholar]
- Graf ER, Zhang X, Jin SX, Linhoff MW, Craig AM (2004). Neurexins induce differentiation of GABA and glutamate postsynaptic specializations via neuroligins. Cell 119:1013–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harms KJ, Craig AM (2005). Synapse composition and organization following chronic activity blockade in cultured hippocampal neurons. J Comp Neurol 490:72–84. [DOI] [PubMed] [Google Scholar]
- Hashimoto K, Ichikawa R, Takechi H, Inoue Y, Aiba A, Sakimura K, Mishina M, Hashikawa T, Konnerth A, Watanabe M, Kano M (2001). Roles of glutamate receptor δ2 subunit (GluRδ2) and metabotropic glutamate receptor subtype 1 (mGluR1) in climbing fiber synapse elimination during postnatal cerebellar development. J Neurosci 21:9701–9712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hioki H, Fujiyama F, Taki K, Tomioka R, Furuta T, Tamamaki N, Kaneko T (2003). Differential distribution of vesicular glutamate transporters in the rat cerebellar cortex. Neuroscience 117:1–6. [DOI] [PubMed] [Google Scholar]
- Hirono M, Yoshioka T, Konishi S (2001). GABAB receptor activation enhances mGluR-mediated responses at cerebellar excitatory synapses. Nat Neurosci 4:1207–1216. [DOI] [PubMed] [Google Scholar]
- Hjelle OP, Chaudhry FA, Ottersen OP (1994). Antisera to glutathione: characterization and immunocytochemical application to the rat cerebellum. Eur J Neurosci 6:793–804. [DOI] [PubMed] [Google Scholar]
- Konnerth A, Llano I, Armstrong CM (1990). Synaptic currents in cerebellar Purkinje cells. Proc Natl Acad Sci USA 87:2662–2665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kralic JE, O'Buckley TK, Khisti RT, Hodge CJ, Homanics GE, Morrow AL (2002). GABAA receptor α1 subunit deletion alters receptor subtype assembly, pharmacological and behavioral responses to benzodiazepines and zolpidem. Neuropharmacology 43:685–694. [DOI] [PubMed] [Google Scholar]
- Kralic JE, Criswell HE, Ostermann JL, O'Buckley TK, Wilkie ME, Matthews DA, Hamre K, Breese GR, Homanics GE, Morrow AL (2005). Genetic essential tremor in γ-aminobutyric acid A receptor α1 subunit knockout mice. J Clin Invest 115:774–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kralic JE, Sidler C, Parpan F, Homanics G, Morrow AL, Fritschy JM (2006). Compensatory alteration of inhibitory synaptic circuits in thalamus and cerebellum of GABAA receptor α1 subunit knockout mice. J Comp Neurol 495:408–421. [DOI] [PubMed] [Google Scholar]
- Kulik A, Nakadate K, Nyiri G, Notomi T, Malitschek B, Bettler B, Shigemoto R (2002). Distinct localization of GABAB receptors relative to synaptic sites in the rat cerebellum and ventrobasal thalamus. Eur J Neurosci 15:291–307. [DOI] [PubMed] [Google Scholar]
- Lambolez B, Audinat E, Bochet P, Crepel F, Rossier J (1992). AMPA receptor subunits expressed by single Purkinje cells. Neuron 9:247–258. [DOI] [PubMed] [Google Scholar]
- Laurie DJ, Seeburg PH, Wisden W (1992a). The distribution of 13 GABAA receptor subunit mRNAs in the rat brain. II. Olfactory bulb and cerebellum. J Neurosci 12:1063–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurie DJ, Wisden W, Seeburg PH (1992b). The distribution of thirteen GABAA receptor subunit mRNAs in the rat brain. III. Embryonic and postnatal development. J Neurosci 12:4151–4172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levinson JN, Chery N, Huang K, Wong TP, Gerrow K, Kang R, Prange O, Wang YT, El-Husseini A (2005). Neuroligins mediate excitatory and inhibitory synapse formation: involvement of PSD-95 and neurexin-1β in neuroligin induced synaptic specificity. J Biol Chem 280:17312–17319. [DOI] [PubMed] [Google Scholar]
- Lujan R, Albasanz JL, Shigemoto R, Juiz JM (2005). Preferential localization of the hyperpolarization-activated cyclic nucleotide-gated cation channel subunit HCN1 in basket cell terminals of the rat cerebellum. Eur J Neurosci 21:2073–2082. [DOI] [PubMed] [Google Scholar]
- Marangi PA, Forsayeth JR, Mittaud P, Erb-Vogtli S, Blake DJ, Moransard M, Sander A, Fuhrer C (2001). Acetylcholine receptors are required for agrin-induced clustering of postsynaptic proteins. EMBO J 20:7060–7073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marowsky A, Fritschy JM, Vogt KE (2004). Functional mapping of GABAA receptor subtypes in the amygdala. Eur J Neurosci 20:1280–1289. [DOI] [PubMed] [Google Scholar]
- Martin LJ, Blackstone CD, Levey AI, Huganir RL, Price DL (1993). AMPA glutamate receptor subunits are differentially distributed in rat brain. Neuroscience 53:327–358. [DOI] [PubMed] [Google Scholar]
- Matsubara A, Laake JH, Davanger S, Usami S, Ottersen OP (1996). Organization of AMPA receptor subunits at a glutamate synapse: a quantitative immunogold analysis of hair cell synapses in the rat organ of Corti. J Neurosci 16:4457–4467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin BJ, Wood JG, Saito K, Roberts E, Wu JY (1975). The fine structural localization of glutamate decarboxylase in developing axonal processes and presynaptic terminals of rodent cerebellum. Brain Res 85:355–371. [DOI] [PubMed] [Google Scholar]
- Nam CI, Chen L (2005). Postsynaptic assembly induced by neurexin-neuroligin interaction and neurotransmitter. Proc Natl Acad Sci USA 102:6137–6142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohtsuki G, Kawaguchi SY, Mishina M, Hirano T (2004). Enhanced inhibitory synaptic transmission in the cerebellar molecular layer of the GluRδ2 knock-out mouse. J Neurosci 24:10900–10907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ottersen OP, Storm-Mathisen J, Madsen S, Skumlien S J. S (1986). Evaluation of the immunocytochemical method for amino acids. Med Biol 64:147–158. [PubMed] [Google Scholar]
- Persohn E, Malherbe P, Richards JG (1992). Comparative molecular neuroanatomy of cloned GABAA receptor subunits in the rat CNS. J Comp Neurol 326:193–216. [DOI] [PubMed] [Google Scholar]
- Phend KD, Weinberg RJ, Rustioni A (1992). Techniques to optimize post-embedding single and double staining for amino acid neurotransmitters. J Histochem Cytochem 40:1011–1020. [DOI] [PubMed] [Google Scholar]
- Prange O, Wong TP, Gerrow K, Wang YT, El-Husseini AE (2004). A balance between excitatory and inhibitory synapses is controlled by PSD-95 and neuroligin. Proc Natl Acad Sci USA 101:13915–13920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabacchi S, Bailly Y, Delhaye-Bouchaud N, Mariani J (1992). Involvement of the N-methyl d-aspartate (NMDA) receptor in synapse elimination during cerebellar development. Science 256:1823–1825. [DOI] [PubMed] [Google Scholar]
- Rao A, Cha EM, Craig AM (2000). Mismatched appositions of presynaptic and postsynaptic components in isolated hippocampal neurons. J Neurosci 20:8344–8353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saghatelyan AK, Nikonenko AG, Sun M, Rolf B, Putthoff P, Kutsche M, Bartsch U, Dityatev A, Schachner M (2004). Reduced GABAergic transmission and number of hippocampal perisomatic inhibitory synapses in juvenile mice deficient in the neural cell adhesion molecule L1. Mol Cell Neurosci 26:191–203. [DOI] [PubMed] [Google Scholar]
- Sara Y, Biederer T, Atasoy D, Chubykin A, Mozhayeva MG, Sudhof T, Kavalali ET (2005). Selective capability of SynCAM and neuroligin for functional synapse assembly. J Neurosci 25:260–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sassoè-Pognetto M, Ottersen OP (2000). Organization of ionotropic glutamate receptors at dendrodendritic synapses in the rat olfactory bulb. J Neurosci 20:2192–2201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheiffele P (2003). Cell-cell signaling during synapse formation in the CNS. Annu Rev Neurosci 26:485–508. [DOI] [PubMed] [Google Scholar]
- Sieburth D, Ch'ng Q, Dybbs M, Tavazoie M, Kennedy S, Wang D, Dupuy D, Rual JF, Hill DE, Vidal M, Ruvkun G, Kaplan JM (2005). Systematic analysis of genes required for synapse structure and function. Nature 436:510–517. [DOI] [PubMed] [Google Scholar]
- Sotelo C (1990). Cerebellar synaptogenesis: what we can learn from mutant mice. J Exp Biol 153:225–249. [DOI] [PubMed] [Google Scholar]
- Studler B, Fritschy JM, Brünig I (2002). GABAergic and glutamatergic terminals differentially influence the organization of GABAergic synapses in rat cerebellar granule cells in vitro. Neuroscience 114:123–133. [DOI] [PubMed] [Google Scholar]
- Sur C, Wafford KA, Reynolds DS, Hadingham KL, Bromidge F, Macaulay A, Collinson N, O'Meara G, Howell O, Newman R, Myers J, Atack JR, Dawson GR, McKernan RM, Whiting PJ, Rosahl TW (2001). Loss of the major GABAA receptor subtype in the brain is not lethal in mice. J Neurosci 21:3409–3418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabata T, Araishi K, Hashimoto K, Hashimotodani Y, van der Putten H, Bettler B, Kano M (2004). Ca2+ activity at GABAB receptors constitutively promotes metabotropic glutamate signaling in the absence of GABA. Proc Natl Acad Sci USA 101:16952–16957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takayama C, Inoue Y (2005). Developmental expression of GABA transporter-1 and -3 during formation of the GABAergic synapses in the mouse cerebellar cortex. Dev Brain Res 158:41–49. [DOI] [PubMed] [Google Scholar]
- Takayama C, Nakagawa S, Watanabe M, Mishina M, Inoue Y (1995). Light- and electron-microscopic localization of the glutamate receptor channel δ2 subunit in the mouse Purkinje cell. Neurosci Lett 188:89–92. [DOI] [PubMed] [Google Scholar]
- Takayama C, Nakagawa S, Watanabe M, Kurihara H, Mishina M, Inoue Y (1997). Altered intracellular localization of the glutamate receptor channel δ2 subunit in weaver and reeler Purkinje cells. Brain Res 745:231–242. [DOI] [PubMed] [Google Scholar]
- Takeuchi T, Miyazaki T, Watanabe M, Mori H, Sakimura K, Mishina M (2005). Control of synaptic connection by glutamate receptor δ2 in the adult cerebellum. J Neurosci 23:2146–2156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varoqueaux F, Sigler A, Rhee JS, Brose N, Enk C, Reim K, Rosenmund C (2002). Total arrest of spontaneous and evoked synaptic transmission but normal synaptogenesis in the absence of Munc13-mediated vesicle priming. Proc Natl Acad Sci USA 99:9037–9042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varoqueaux F, Jamain S, Brose N (2004). Neuroligin 2 is exclusively localized to inhibitory synapses. Eur J Cell Biol 83:449–456. [DOI] [PubMed] [Google Scholar]
- Verhage M, Maia AS, Plomp JJ, Brussaard AB, Heeroma JH, Vermeer H, Toonen RF, Hammer RE, van den Berg TK, Missler M, Geuze HJ, Sudhof TC (2000). Synaptic assembly of the brain in the absence of neurotransmitter secretion. Science 287:864–869. [DOI] [PubMed] [Google Scholar]
- Vicini S, Ferguson C, Prybylowski K, Kralic J, Morrow AL, Homanics GE (2001). GABAA receptor α1 subunit deletion prevents developmental changes of inhibitory synaptic currents in cerebellar neurons. J Neurosci 21:3009–3016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Washbourne P, Dityatev A, Scheiffele P, Biederer T, Weiner JA, Christopherson KS, El-Husseini A (2004). Cell adhesion molecules in synapse formation. J Neurosci 24:9244–9249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe D, Inokawa H, Hashimoto K, Suzuki N, Kano M, Shigemoto R, Hirano T, Toyama K, Kaneko S, Yokoi M, Moriyoshi K, Suzuki M, Kobayashi K, Nagatsu T, Kreitman RJ, Pastan I, Nakanishi S (1998). Ablation of cerebellar Golgi cells disrupts synaptic integration involving GABA inhibition and NMDA receptor activation in motor coordination. Cell 95:17–27. [DOI] [PubMed] [Google Scholar]
- Wilson L, Sotelo C, Caviness VS (1981). Heterologous synapses upon Purkinje cells in the cerebellum of the reeler mutant mouse: an experimental light and electron microscopic study. Brain Res 213:63–82. [DOI] [PubMed] [Google Scholar]
- Yamagata M, Sanes JR, Weiner JA (2003). Synaptic adhesion molecules. Curr Opin Cell Biol 15:621–632. [DOI] [PubMed] [Google Scholar]
- Yamaguchi Y (2002). Glycobiology of the synapse: the role of glycans in the formation, maturation, and modulation of synapses. Biochim Biophys Acta 1573:369–376. [DOI] [PubMed] [Google Scholar]