

Abstract
Mutations in the genes coding for α-synuclein and parkin cause autosomal-dominant and autosomal-recessive forms of Parkinson's disease (PD), respectively. α-Synuclein is a major component of Lewy bodies, the proteinaceous cytoplasmic inclusions that are the pathological hallmark of idiopathic PD. Lewy bodies appear to be absent in cases of familial PD associated with mutated forms of parkin. Parkin is an ubiquitin E3 ligase, and it may be involved in the processing and/or degradation of α-synuclein, as well as in the formation of Lewy bodies. Here we report the behavioral, biochemical, and histochemical characterization of double-mutant mice overexpressing mutant human A53T α-synuclein on a parkin null background. We find that the absence of parkin does not have an impact on the onset and progression of the lethal phenotype induced by overexpression of human A53T α-synuclein. Furthermore, all major behavioral, biochemical, and morphological characteristics of A53T α-synuclein-overexpressing mice are not altered in parkin null α-synuclein-overexpressing double-mutant mice. Our results demonstrate that mutant α-synuclein induces neurodegeneration independent of parkin-mediated ubiquitin E3 ligase activity in nondopaminergic systems and suggest that PD caused by α-synuclein and parkin mutations may occur via independent mechanisms.
Keywords: Parkinson's disease, parkin, α-synuclein, Lewy bodies, neurodegeneration, animal models
Introduction
Parkinson's disease (PD) is a progressive debilitating neurodegenerative disorder that is characterized by the loss of dopamine (DA) neurons in the substantia nigra, pars compacta (SNpc) (Lang and Lozano, 1998). In addition, there is widespread neurodegeneration throughout the CNS in PD; in fact, loss of DA neurons, which accounts for the major clinical symptoms of PD, is thought to occur only midstage in the disease (Braak et al., 2003). On the cellular level, PD is characterized by the formation of cytoplasmic proteinaceous inclusion bodies called Lewy bodies, and dystrophic neurites containing abnormal protein accumulation, designated as Lewy neurites (Forno, 1996; Lang and Lozano, 1998). These pathological protein aggregates contain a variety of proteins, of which α-synuclein appears to be the major structural component (Spillantini et al., 1997; Goedert, 2001). Accumulation and aggregation of α-synuclein is also observed in other neurodegenerative diseases, including dementia with Lewy bodies, Lewy body variant of Alzheimer's disease, multisystem atrophy, and neurodegeneration with brain iron accumulation type 1. All of these disorders, together with PD, are referred to as α-synucleinopathies (Baba et al., 1998; Irizarry et al., 1998; Neumann et al., 2000; Spillantini and Goedert, 2000; Wirths et al., 2000). α-Synuclein is a natively unfolded cytoplasmic protein of 140 amino acids that appears to play a role in synaptic transmission; however, its physiological function still remains elusive (Goedert, 2001). Point mutations in α-synuclein as well as overexpression of wild-type (WT) α-synuclein through triplication of the chromosomal locus for α-synuclein cause rare forms of autosomal-dominant PD (Polymeropoulos et al., 1997; Kruger et al., 1998; Singleton et al., 2003). Through a variety of studies, including investigations of α-synuclein-overexpressing transgenic mice and invertebrate animal models that develop α-synuclein inclusions and neurodegeneration, it is well established that both WT and mutant α-synuclein have the potential to cause neurodegeneration, depending primarily on the level of expression, among other factors (Feany and Bender, 2000; Masliah et al., 2000; van der Putten et al., 2000; Giasson et al., 2002; Lee et al., 2002; Lakso et al., 2003). In particular, mice overexpressing high levels of mutant human A53T α-synuclein develop an adult-onset neurodegenerative disease with a progressive motor dysfunction, leading to death (Giasson et al., 2002; Lee et al., 2002). However, the mechanism whereby α-synuclein causes neurodegeneration is not known. Neurodegeneration may be initiated by the formation of α-synuclein aggregates composed of protofibrils, oligomers, and/or fibrils (Goedert, 2001). Accompanying the aggregation and deposition of α-synuclein into Lewy bodies/neurites is the marked ubiquitination of these structures, leading to the hypothesis that ubiquitination may play a prominent role in the pathogenesis of α-synucleinopathies (Sharma et al., 2001; Hasegawa et al., 2002; Nonaka et al., 2005). This is further corroborated by the finding that mutations of the ubiquitin E3 ligase, parkin, cause an autosomal-recessive form of PD (Kitada et al., 1998; Zhang et al., 2000). Mutations in the parkin gene lead to the loss of the ubiquitin E3 ligase activity of parkin (Dawson and Dawson, 2003a) and appear to be the most frequent cause of familial PD (Lucking et al., 2000). A number of putative substrates for parkin exist, and the accumulation of one or several of these substrates may account for the neurodegeneration seen in patients with loss-of-function parkin mutations by impairment of ubiquitin-proteasomal degradation (von Coelln et al., 2004a). Notably, α-synuclein may be a parkin substrate. The interaction of parkin with α-synuclein may require posttranslational modification of α-synuclein (Shimura et al., 2001), because native unmodified α-synuclein does not interact with parkin (Chung et al., 2001). Furthermore, overexpression of parkin in rat, Drosophila, and cellular models provides protection against α-synuclein toxicity, suggesting that parkin is involved in the cellular defense mechanism against α-synuclein-induced cell death (Petrucelli et al., 2002; Yang et al., 2003; Yamada et al., 2005). The absence of Lewy bodies in patients with parkin mutations further supports the idea that parkin-mediated ubiquitination may play a role in PD, particularly in the formation of Lewy bodies (Takahashi et al., 1994; Mori et al., 1998). Thus, parkin and α-synuclein may function in a common pathogenic mechanism, leading to the degenerative changes seen in PD. To determine whether parkin is involved in the pathogenesis of α-synucleinopathies in vivo, we generated double-mutant mice overexpressing mutant human A53T α-synuclein on a parkin null background. Here we show that human A53T α-synuclein transgenic mice lacking parkin still develop ubiquitinated α-synuclein inclusions. Importantly, we also show that the absence of parkin does not impact the onset or the progression of neurodegeneration in the human A53T α-synuclein transgenic mice. Together, our results demonstrate that mutant α-synuclein induces neurodegeneration independent of parkin-mediated ubiquitin E3 ligase activity and indicates that mutations in parkin and α-synuclein cause nondopaminergic degeneration through independent pathogenic mechanisms.
Materials and Methods
Generation of double-mutant mice overexpressing human A53T α-synuclein on a parkin null background
The generation of transgenic mice that overexpress human A53T α-synuclein using the mouse prion protein (PrP) promoter, as well as the generation of exon 7-deleted parkin knock-out (KO) mice, has been described previously (Lee et al., 2002; von Coelln et al., 2004b). Parkin null mice were generated by Lexicon Genetics (The Woodlands, TX) as originally described (von Coelln et al., 2004b). Double-mutant mice were generated by crossbreeding these two existing mouse lines (see Fig. 1A). In a first step, parkin null mice were bred to mice heterozygous for the A53T α-synuclein transgene. Parkin heterozygous animals were then bred to double-heterozygous mice generated in the first round of breeding to generate wild-type mice (WT−), parkin null mice (KO−), α-synuclein A53T transgenic mice (WT+), and α-synuclein A53T transgenic mice lacking parkin (KO−), i.e., double-mutant mice. Age-matched littermate mice from the four different strains were used in all experiments. Any potential strain differences were minimized by the examination of littermates from the cross of parkin heterozygous animals with double-mutant heterozygous animals. All procedures involving animals were approved by and conformed to the guidelines of the Institutional Animal Care Committee of Johns Hopkins University.
Figure 1.

Absence of parkin does not change steady-state levels of α-synuclein. A, Breeding strategy to generate parkin null α-synuclein-overexpressing double-mutant mice. F0, F1, and F2 represent the generations of crossbreeding. Percentages represent the expected frequency of each genotype in a sufficiently large number of litters. Het, Heterozygous; TG, transgenic, i.e., A53T α-synuclein-overexpressing mouse. B, Immunoblot analysis of NP 40-soluble lysates from cerebral cortex of 6-month-old mice with or without parkin and with or without overexpressed human A53T α-synuclein was performed using antibodies that recognize both mouse and human α-synuclein (α-syn) and antibodies that recognize only overexpressed human α-synuclein (hu-α-syn). An antibody to actin was used as a loading control, and an antibody to parkin was used to verify the absence of parkin. The expression level of α-synuclein (normalized to actin) was quantified using optical densitometry for overexpressed human A53T α-synuclein (C) and total (mouse and human) α-synuclein (D). Data are expressed as mean ± SEM (n = 3). Statistical analysis was performed by ANOVA, followed by the Student–Newman–Keuls test. n.s., Not statistically significant.
Behavioral analyses
Open-field test was performed in 9-month-old male and female mice from all four genotypes (WT−, KO−, WT+, KO+; n = 6–8 per group) by placing animals into a novel cage. Activity was monitored using the Cage Rack Flex-Field Photobeam Activity System (San Diego Instruments, San Diego, CA), which provides a grid of 4 × 8 infrared beams. The total number of beam breaks over a period of 30 min was recorded and analyzed. For acoustic startle test, 9-month-old male and female mice from all four genotypes (WT−, KO−, WT+, KO+; n = 5–7 per group) were tested for the maximal amplitude of the acoustic startle response in an SR-LAB Startle Reflex System (San Diego Instruments). Mice were placed in the cylindrical enclosure inside the ventilated test box. After 5 min of white background noise, mice were exposed to a series of acoustic stimuli (duration of 40 ms), ranging from 75 to 120 dB, including two 0 dB baseline measurements (absence of stimulus), in a pseudorandom order. Intervals between stimuli were 15 s long. This series of stimuli was repeated six times. The maximal amplitude of each motor response, as detected by a motion sensor, is determined by the software provided by the manufacturer. The average value of the six repeats for each stimulus intensity was calculated and used for statistical analysis of genotype groups.
Western blot and immunoprecipitation analysis
Expression of α-synuclein (human and mouse), parkin, ubiquitin, and other proteins were determined as described previously (Lee et al., 2002). Briefly, non-ionic detergent-soluble and -insoluble fractions were prepared by homogenization of brain tissue using a Diax 900 homogenizer (Heidolph, Cinnaminson, NJ) in buffer S (10 mm Tris-HCl, pH 7.4, 150 mm NaCl, and 5 mm EDTA) containing protease inhibitors (5 mm PMSF, 10 μg/ml aprotinin, 10 μg/ml leupeptin, and 10 μg/ml pepstatin) and detergent (0.5% Nonidet P-40). The homogenate was centrifuged (5 min at 100,000 × g), and the resulting pellet (P1) and supernatant fraction (soluble fraction) were collected. The P1 was washed once in the same buffer (including rehomogenization and repelleting), and the resulting pellet (P2, non-ionic detergent-insoluble) was solubilized using a Diax 900 homogenizer, followed by sonication and boiling for 5 min, in buffer P containing 1% SDS and 0.5% Na-deoxycholate (i.e., “insoluble” fraction). The protein concentrations of soluble and insoluble fractions were determined using the BCA protein assay (Pierce, Rockford, IL) according to the instructions of the manufacturer. Proteins were separated by SDS-PAGE and electroblotted onto a nitrocellulose membrane (Bio-Rad, Hercules, CA). Immunolabeling was performed using primary antibodies against α-synuclein, as described previously [human specific (Lee et al., 2002); mouse and human, α-synuclein antibody (Syn-1) (BD Biosciences PharMingen, San Jose, CA), parkin (PRK8) (Pawlyk et al., 2003), actin (Sigma, St. Louis, MO), and ubiquitin (DakoCytomation, Carpinteria, CA)], HRP-conjugated secondary antibodies, and ECL solutions (Amersham Biosciences, Piscataway, NJ). Chemiluminescence was visualized using Hyperfilm (Amersham Biosciences). For densitometric analysis, films were scanned and densitometry was performed using the software Fluor Chem version 3.04B (Alpha Innotech, San Leandro, CA). Statistical analysis was performed using the ratios of the densitometric value of each band and its corresponding actin loading control within each genotype group.
For immunoprecipitation of α-synuclein from the insoluble fraction, P2 samples (in buffer P) were diluted 10-fold in buffer S and incubated overnight with 5 μg of anti-α-synuclein antibody Syn-1, before adding 50 μl of Protein G Sepharose 4 FastFlow (1:1 mix in PBS) (Amersham Biosciences) for another 2 h. Sepharose beads were spun down, washed four times in buffer S with increased salt concentration (500 mm NaCl), and incubated in SDS sample buffer at 80°C for 10 min. Immunoblotting was performed as described above, using primary antibodies against ubiquitin (mouse monoclonal) and α-synuclein (rabbit polyclonal) (both from BD Biosciences PharMingen).
Immunohistochemistry and histochemical analysis
Immunohistochemical analysis was performed by perfusing mice intra-aortically with ice-cold PBS, pH 7.4, and 4% paraformaldehyde/PBS, pH 7.4. Brains were removed, postfixed overnight in the same fixative, and paraffin embedded. Sagittal sections of 20 μm thickness were cut, deparaffinized in xylene, and blocked with 10% goat serum/PBS plus 0.2% Triton X-100, before being incubated with primary antibody against α-synuclein (Syn-1), glial fibrillary acidic protein (GFAP) (rabbit polyclonal; DakoCytomation), or ubiquitin (rabbit polyclonal; DakoCytomation), followed by successive incubations with biotin-conjugated anti-rabbit or anti-mouse antibody (goat polyclonal; Jackson ImmunoResearch, West Grove, PA), ABC reagents (Vector Laboratories, Burlingame, CA), and SigmaFast DAB Peroxidase Substrate (Sigma). Some sections were counterstained with cresyl violet, before being dehydrated in graded ethanol, cleared in xylene, and mounted with Permount.
Fluoro Jade B staining was performed according to the protocol of the manufacturer (Histo-Chem, Jefferson, AR). Briefly, brain sections were deparaffinized in xylene and hydrated, followed by 15 min exposure to 0.06% potassium permanganate. Sections were then rinsed in distilled water and exposed for 30 min to a 0.001% Fluoro Jade B solution in 0.1% acetic acid, air dried, and coverslipped using Immumount mounting media (Thermo Shandon, Pittsburgh, PA). For microscopic analysis, an excitation of 488 nm, provided by an argon laser, was used, and the sections were visualized using a Zeiss (Hallbergmoos, Germany) confocal microscope (LSM 510 Meta).
To assess Fluoro Jade B colocalization with α-synuclein and/or GFAP, 30-μm-thick free-floating sagittal brain sections were used from perfusion-fixed frozen brain from all four genotypes. Briefly, brain sections were blocked in 10% donkey serum containing 0.3% Triton X-100 in PBS, pH 7.4, for 30 min at room temperature, followed by incubation with mouse anti-α-synuclein antibody (Syn-1; BD Biosciences PharMingen) at 1:500 or rabbit anti-GFAP antibody (Invitrogen, Carlsbad CA) at 1:500 in 2% donkey serum containing 0.3% Triton X-100 at room temperature for 6 h. Brain slices were washed three times for 10 min in PBS, pH 7.4, at room temperature and with 0.1% donkey serum in PBS for 1 h, followed by incubation with Alexa Fluor-594 donkey anti-rabbit IgG at 1:500 and Alexa Fluor-594 donkey anti-mouse IgG at 1:500 (Invitrogen) overnight at 4°C. Sections were washed in PBS thrice for 5 min each at room temperature. Fluoro Jade B staining was performed on these slices according to the protocol of the manufacturer (Histo-Chem) with slight modification from that we used for paraffin sections. Briefly, brain sections were incubated for 4 min in 0.06% potassium permanganate. Sections were then rinsed in distilled water and exposed for 30 min to a 0.001% Fluoro Jade B solution in 0.1% acetic acid, washed, and incubated with 4′,6-diamidino-2-phenylindole, dilactate (1:1000; Invitrogen) for 10 min and rinsed in distilled water mounted on clean glass slides, air dried, and coverslipped using Immumount mounting media (Thermo Shandon). For microscopic analysis, the sections were visualized using a Zeiss confocal microscope (LSM 510 Meta) equipped with an argon laser.
Quantitation of catecholaminergic and Fluoro Jade B-positive cells
Brain sections of end-stage symptomatic mice (for WT+ and KO+) and age-matched controls for WT− and KO− mice (n = 6–12 for each age group) were processed for tyrosine hydroxylase (TH) and Nissl staining as described previously (von Coelln et al., 2004b). Neurons were counted using the optical fractionator, an unbiased method for cell counting that is not affected by either the volume of reference (SNpc) or the size of the counted elements (neurons) (West, 1993). This method was performed using a computer-assisted image analysis system, consisting of an Axiophot photomicroscope (Zeiss) equipped with a computer-controlled motorized stage, a video camera, and the Stereo Investigator software (MicroBrightField, Williston, VT).
Substantia nigra, pars compacta.
Cell counts of TH-positive neurons of the SNpc were performed as described previously. In brief, immunohistochemistry for TH and Nissl counterstain was performed on every fourth midbrain section throughout the entire extent of SNpc. TH-positive neurons were counted in the left SNpc in these sections. The total number of TH-positive neurons was calculated using the formula described previously for this method (West et al., 1991).
Locus ceruleus.
To count TH-positive locus ceruleus (LC) neurons, TH/Nissl staining was done on every hindbrain section from the rostral part of the pons to the caudal part of the medulla. All LC-containing sections were selected and counted using the optical fractionator, following the same method as described for the SNpc. The LC was identified referring to a commonly used mouse brain atlas (Franklin and Paxinos, 1997).
Fluoro Jade B-positive cells.
Fluoro Jade B-positive cells were counted on sagittal brain sections of brainstem restricted between lateral superior olive rostrally and dorsal reticular medullar nucleus caudally (n = 4 animals per group) using an Axiovision 4.3 cell-counting module (Zeiss).
HPLC electrochemical measurement of catecholamines
To determine the concentration of catecholamines in discrete brain regions by HPLC with electrochemical detection, end-stage symptomatic WT+ and KO+ mice and age-matched WT− and KO− control animals (n = 6–8 per group) were decapitated, brains were quickly removed, and brain regions were dissected. The tissue was weighed and sonicated in 0.2 ml of 0.1 m perchloric acid containing 0.01% EDTA and 25 μg/ml 3,4-dihydroxybenzylamine (Sigma) as an internal standard. After centrifugation (15,000 × g, 10 min, 4°C), 20 μl of the supernatant were injected onto a C-18 reverse-phase Spheri 5, RP-18, 4.6 mm × 25 cm catecholamine column (BASi, West Lafayette, IN). The mobile phase consisted of 0.15 m chloroacetic acid, 0.2 mm EDTA, 0.86 mm sodium octyl sulfate, 4% acetonitrile, and 2.5% tetrahydrofuran, pH 3. The flow rate was kept at 1.5 ml/min. Biogenic amines and their metabolites were detected by an electrochemical detector (Prostar ECD model 370; Varian, Palo Alto, CA), with the working electrode kept at 0.6 V. Data were collected and processed on a Star Chromatography Workstation 5.52 (Varian).
Statistical analysis
Data are expressed as mean ± SEM. Statistical analysis was performed by ANOVA, followed by Student–Newman–Keuls test. Differences were considered significant when p < 0.05.
Results
Steady-state levels of α-synuclein are independent of the expression of parkin
To generate mice that overexpress human A53T α-synuclein on a parkin null background, we pursued a crossbreeding strategy of two existing genetically modified mouse lines. Mice in which both alleles of the parkin gene have been inactivated by targeted disruption of exon 7 (parkin KO mice) (von Coelln et al., 2004b) were bred to transgenic mice overexpressing human A53T α-synuclein driven by the mouse PrP promoter (Lee et al., 2002). In two successive breeding steps, cohorts of littermates with or without endogenous parkin expression and with or without transgenic A53T α-synuclein expression were generated in ratios consistent with Mendelian principles (Fig. 1A). To minimize potential strain effects, parkin heterozygous animals were bred to double-heterozygous mice generated in the first round of breeding to generate WT−, KO−, WT+, and KO−, i.e., double-mutant mice. Age-matched littermate mice from the four different strains were used in all experiments. Genotypes were identified by PCR analysis of tail DNA as described previously (Lee et al., 2002; von Coelln et al., 2004b).
The amount of mutant human A53T α-synuclein present in the brains of transgenic animals correlates with disease onset and survival (Lee et al., 2002), and, moreover, the E3 ligase activity of parkin has been implicated in the degradation of α-synuclein (Shimura et al., 2001). Accordingly, we first determined the amount of endogenous and overexpressed α-synuclein in mice lacking parkin by immunoblot analysis and subsequent densitometry (Fig. 1). We find that lack of parkin does not affect the levels of endogenous mouse α-synuclein (in nontransgenic mice) (Fig. 1B,D), total (endogenous plus transgenic) α-synuclein (Fig. 1B,D), or overexpressed human A53T α-synuclein (Fig. 1B,C) compared with parkin WT mice. Thus, the steady-state levels of both endogenous α-synuclein and mutant human A53T α-synuclein are independent of the expression of parkin.
Behavioral deficits in human A53T α-synuclein transgenic mice are not altered by the absence of parkin
Human A53T α-synuclein transgenic mice have early behavioral abnormalities that are characterized by hyperactivity in open-field testing, possibly attributable to derangements in dopaminergic neurotransmission (Lee et al., 2002). To determine whether the absence of parkin influences these early behavioral abnormalities, we conducted open-field testing in a cohort of 9-month-old mice of each relevant genotype (Fig. 2A). Human A53T α-synuclein transgenic mice show significantly increased activity regardless of the presence or absence of parkin (Fig. 2A).
Figure 2.
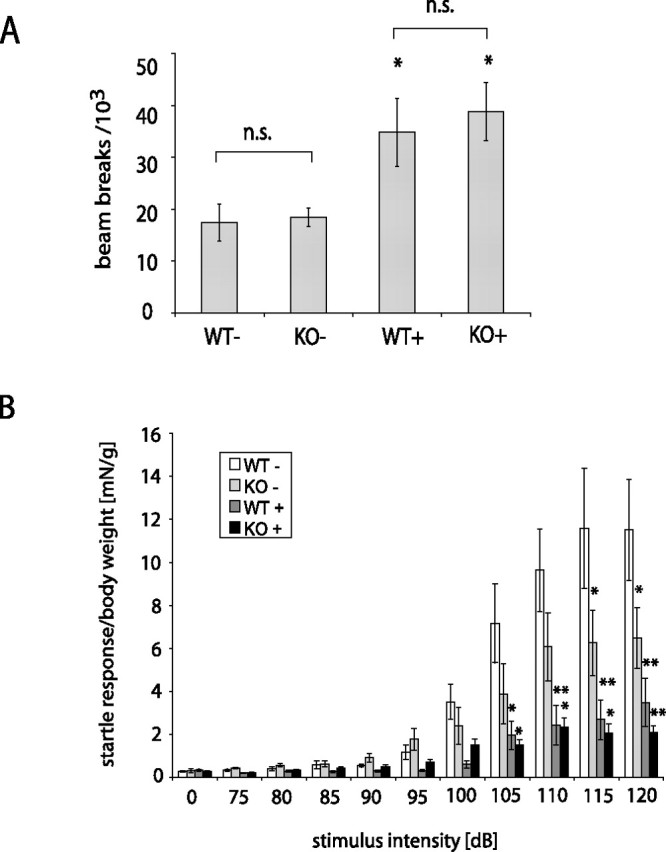
Behavioral deficits in mice overexpressing human A53T α-synuclein are not altered by the absence of parkin. A, The Open-field test (n = 6–8) shows hyperactivity of transgenic mice in the presence and absence of parkin. B, Acoustic startle analysis (n = 5–7) reveals reduced startle in parkin null mice and a reduction of the startle response in A53T α-synuclein-overexpressing mice, as well as in double-mutant mice. Data are expressed as mean ± SEM. Statistical analysis was performed by ANOVA, followed by the Student–Newman–Keuls test (*p < 0.05; **p < 0.01). n.s., Not statistically significant.
Parkin KO mice show a marked deficit in acoustic startle (von Coelln et al., 2004b) (Fig. 2B). Therefore, we determined whether the transgenic expression of human A53T α-synuclein alters the startle response in parkin null mice. As shown previously, parkin KO mice have reduced startle response compared with WT littermate controls (Fig. 2B). Surprisingly, overexpression of human A53T α-synuclein leads to a marked reduction of the acoustic startle response (Fig. 2B). The absence of parkin in human A53T α-synuclein transgenic mice has no significant impact on the acoustic startle response (Fig. 2B). Together, these results indicate that the behavioral abnormalities observed in both A53T α-synuclein transgenic mice and parkin KO mice occur independently of each other.
Age at onset and progression of the lethal phenotype induced by overexpression of human A53T α-synuclein are independent of parkin
Mice overexpressing human A53T α-synuclein develop an adult-onset phenotype with rapidly progressive motor dysfunction leading to death (Lee et al., 2002). To determine the influence of parkin on this α-synuclein-induced disease, we examined the length of survival and disease progression of transgenic mice overexpressing human A53T α-synuclein on both a parkin wild-type and parkin null background (Fig. 3). Human A53T α-synuclein transgenic mice lacking parkin are indistinguishable from transgenic mice on parkin WT background. Specifically, both mice show impaired extension of hindlimbs when suspended by the tail, altered gait, coarse coat attributable to improper grooming, bradykinesia, impaired reach, poor righting reflexes, and dystonic posturing near the end stage of disease (data not shown). The mean survival time of human A53T α-synuclein transgenic mice (12.4 months) is similar to that of transgenic mice lacking parkin (12.6 months) (Fig. 3). Furthermore, progression of disease (measured as number of days between first observable signs of disease to terminal loss of postural reflexes) is not significantly different in human A53T α-synuclein transgenic mice compared with parkin KO transgenic mice (data not shown). Therefore, parkin has no impact on the onset and progression of the lethal phenotype induced by overexpression of human A53T α-synuclein in mice.
Figure 3.

Progression of the lethal phenotype induced by overexpression of human A53T α-synuclein is independent of parkin. Survival curves of transgenic mice on a parkin WT and KO background, respectively, were generated by following cohorts of both genotypes (WT+, n = 22; KO+, n = 38) until mice had to be killed because of terminal disease.
Absence of parkin does not alter the accumulation of α-synuclein and the formation of ubiquitin-positive inclusions in human A53T α-synuclein mutant mice
Despite the lack of behavioral and neurological differences between parkin WT and KO mice overexpressing human A53T α-synuclein, parkin may modulate cellular neuropathology associated with the expression of mutant human α-synuclein. In human A53T α-synuclein transgenic mice, neurological abnormalities are accompanied by abnormal neuronal accumulation of α-synuclein and ubiquitin (Giasson et al., 2002; Lee et al., 2002). To determine whether these cellular lesions are affected by a lack of parkin expression, immunohistochemical analysis was performed on brain sections from animals of each genotype in the late stage of disease (or healthy age-matched nontransgenic animals). Labeling with human-specific anti-α-synuclein antibodies reveals neuronal accumulation of human α-synuclein in a number of brain regions, including the brainstem, cerebellum, and spinal cord, regardless of the presence or absence of parkin (Fig. 4A–F).
Figure 4.

Abnormal accumulation of human α-synuclein and ubiquitin in transgenic mice overexpressing human A53T α-synuclein is not affected by a lack of parkin expression. Paraffin-embedded brain sections from parkin WT/A53T α-synuclein transgenic mice (A, C, E, G, L, N) or parkin null A53T α-synuclein transgenic mice (B, D, F, H, I–K, M, O) were immunolabeled for human α-synuclein (A–F) or ubiquitin (G–O). Shown are representative images from pons (A, B, I, J), deep cerebellar nuclei (C, D, K), spinal cord (E, F, N, O) cortex (G, H), and red nucleus (L, M). Abnormal accumulation of human α-synuclein or ubiquitin in cell bodies (arrows) and processes (arrowheads) associated with the expression of mutant human A53T α-synuclein were not significantly affected by the lack of parkin expression. Scale bars: A–H, J, K, O, 50 μm; I, L–N, 100 μm.
Previous characterization of the A53T α-synuclein transgenic mice indicated that the neuronal accumulation of ubiquitin was a prominent indicator of cellular pathology (Lee et al., 2002, 2004). In brain sections from both parkin WT and KO α-synuclein transgenic mice, immunohistochemical analysis shows prominent accumulation of ubiquitin in neuronal cell bodies and neurites in all affected regions (Fig. 4G–O), including brainstem (Fig. 4I,J), deep cerebellar nuclei (Fig. 4K), midbrain (Fig. 4L,M), spinal cord (Fig. 4N,O), and selected hypothalamic regions. In addition, ubiquitin accumulation is seen occasionally in cortical neurons (Fig. 4G,H). Age-matched nontransgenic animals with and without parkin did not show accumulation of human α-synuclein and ubiquitin (data not shown).
In addition to the more detailed regional morphological analysis, the overall distribution and abundance of ubiquitin pathology throughout the CNS was examined in two mice from each relevant genotype and schematically summarized (Fig. 5). Ubiquitin-positive inclusion are absent in brains from nontransgenic animals. Notably, there are no obvious differences in the distribution and regional density of ubiquitin-positive inclusions in human A53T α-synuclein transgenic mice with or without parkin (Fig. 5). Together, these results suggest that the pathological accumulation of α-synuclein, as well as the formation of ubiquitinated inclusions induced by overexpression of human A53T α-synuclein in this mouse model of synucleinopathy, both occur independently of the ubiquitin E3 ligase activity of parkin.
Figure 5.

Anatomical distribution of pathological protein inclusions in α-synuclein transgenic mice is unaltered by the absence of parkin. Sagittal brain sections and transversal spinal cord sections of mice killed at the terminal stage of α-synuclein-induced disease (or age-matched for WT− and KO−) were analyzed regarding the distribution of ubiquitin-immunoreactive protein inclusions. Each black dot represents one immunoreactive neuronal cell on a typical brain section of an affected animal. 1, Olfactory bulb; 2, cerebral cortex; 3, corpus callosum; 4, striatum; 5, anterior commissure; 6, nucleus accumbens; 7, third ventricle; 8, hippocampus; 9, thalamus; 10, hypothalamus; 11, superior colliculus; 12, inferior colliculus; 13, mesencephalon; 14, substantia nigra, pars compacta; 15, pons; 16, facial nerve; 17, medulla oblongata; 18, deep cerebellar nuclei; 19, cerebellar cortex; 20, olfactory tubercle; 21, globus pallidus; 22, amygdala; 23, substantia nigra, pars reticulata; 24, ventral horn; 25, central canal; 26, dorsal horn.
Neurodegeneration in human A53T α-synuclein mutant mice is unaltered by the absence of parkin
The extent of neurodegeneration was examined in the affected brain regions of human A53T α-synuclein mutant mice with or without parkin at a terminal stage of α-synuclein-induced disease. Astrocytic activation, characterized by upregulated expression of GFAP, serves as an indirect indicator of overall neuronal abnormality in A53T α-synuclein transgenic mice (Lee et al., 2002). Therefore, the distribution of GFAP was analyzed by immunohistochemistry (Fig. 6). In the late stage of disease, human A53T α-synuclein transgenic mice with or without parkin show severe astroglial reaction in a number of brain regions, including midbrain (Fig. 6C,D), brainstem (Fig. 6G,H), cerebellum, and spinal cord (Fig. 6K,L), whereas GFAP-immunoreactive cells are sparse in the same regions in nontransgenic mice (Fig. 6A,B,E,F,I,J). In contrast, telencephalic and diencephalic regions in α-synuclein transgenic mice are relatively free of abnormal GFAP immunoreactivity (data not shown). Overall, the distribution of astroglial reaction is not affected by the absence of parkin, and it is comparable with that reported previously for human A53T α-synuclein transgenic mice (Lee et al., 2002).
Figure 6.
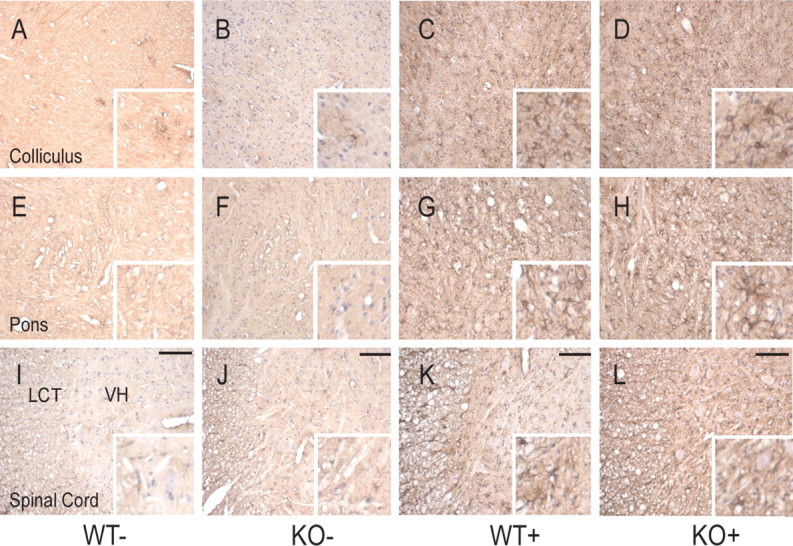
The pathology-associated astrocytic response in A53T α-synuclein transgenic mice is not affected by the lack of parkin expression in parkin KO/α-synuclein transgenic double-mutant mice. Paraffin-embedded brain sections were immunostained for GFAP. Representative sections from colliculus (A–D), pons (E, F), and cervical spinal cord (I–L) are shown in overview and detail (inset). Scale bars, 50 μm. A normal pattern of astrocytic staining (with sparse GFAP-positive cells) is observed in nontransgenic mice with or without parkin (A, B, E, F, I, J). In clinically affected A53T α-synuclein transgenic mice (C, G, K), GFAP staining reveals a significant astrocytic response. The pattern of GFAP staining is not obviously affected by the lack of parkin expression (D, H, L). Note that, in spinal cord, activation of astrocytes is seen in both the lateral column tracts (LCT) and in ventral horn (VH).
As a direct marker of neurodegeneration, we stained brain sections with the fluorescent marker Fluoro Jade B, which has been shown to specifically stain cells that undergoing degeneration (Schmued and Hopkins, 2000). Human A53T α-synuclein transgenic mice show a large number of Fluoro Jade B-positive cells throughout their brainstem, regardless of the absence or presence of parkin, whereas nontransgenic animals do not show any Fluoro Jade B-positive staining (Fig. 7A). To assess whether Fluoro Jade B colocalizes with neurons having α-synuclein inclusions and/or reactive glia characterized by upregulated expression of GFAP in A53T α-synuclein transgenic mice with or without parkin, immunohistochemical analysis was performed. Fluoro Jade B colocalizes or is in close apposition with GFAP-positive reactive glia in the pathologically affected brainstem region of A53T α-synuclein transgenic mice (supplemental Fig. 1A, available at www.jneurosci.org as supplemental material). Fluoro Jade B also colocalizes partially with cells harboring α-synuclein inclusions and also other cell types that do not stain positive for α-synuclein (supplemental Fig. 1B, available at www.jneurosci.org as supplemental material), suggesting that Fluoro Jade B stains both neurons positive for α-synuclein inclusions and reactive glia. Colocalization of Fluoro Jade B with GFAP and/or α-synuclein in human A53T α-synuclein transgenic mice was unaffected regardless of the presence or absence of parkin. Quantification of Fluoro Jade B-positive cells shows equal numbers of degenerating cells in the brainstem of A53T α-synuclein mutant mice with and without parkin (Fig. 7B). In summary, neurodegeneration induced by overexpression of mutant human A53T α-synuclein does not differ in parkin null mice compared with parkin WT mice.
Figure 7.
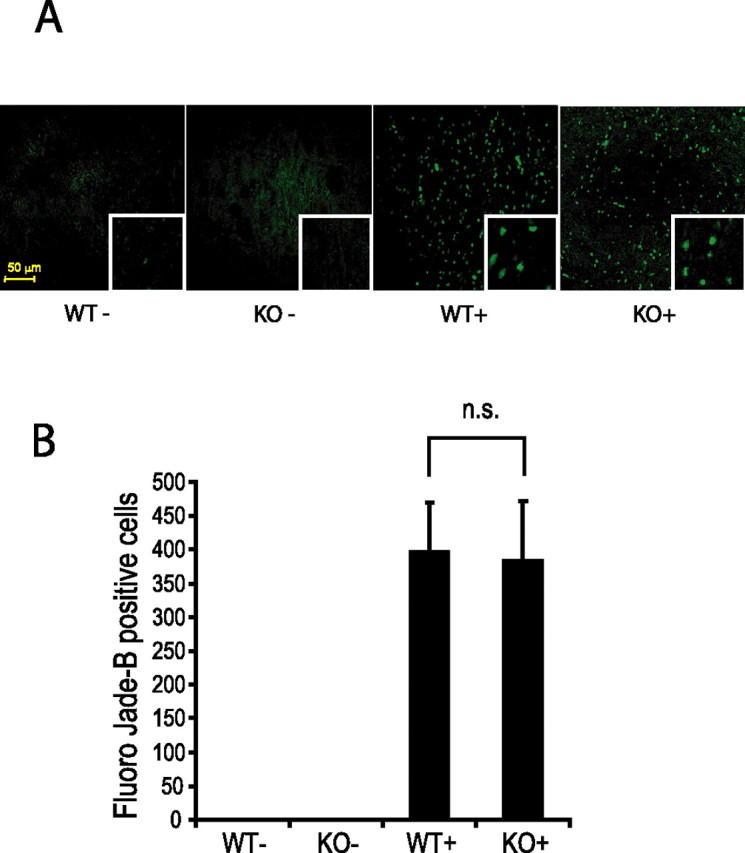
Neurodegeneration in A53T α-synuclein transgenic mice as visualized by Fluoro Jade B staining is unaltered by the absence of parkin. Sagittal brain sections at the level of brainstem of mice killed at the terminal stage of α-synuclein-induced disease (or age-matched for WT− and KO−) were analyzed regarding the quantity and distribution of Fluoro Jade B-positive cells. A, Fluoro Jade B-positive cells are abundant in α-synuclein transgenic mice regardless of the presence of parkin. B, Quantification of Fluoro Jade B-positive cells. Data are expressed as mean ± SEM (n = 4). Statistical analysis was performed by ANOVA, followed by the Student–Newman–Keuls test. Scale bar, 50 μm. n.s., Not statistically significant.
Overexpression of mutant human A53T α-synuclein and absence of parkin do not show synergistic effects on catecholaminergic brainstem neurons
PD is characterized by the relatively selective loss of DA neurons in the SNpc, and the loss of norepinephrine (NE) neurons in the LC. The NE neurotransmitter system originating from the LC is impaired in parkin null mice (von Coelln et al., 2004b). Accordingly, we conducted unbiased stereological cell counts of TH-positive neurons in the SNpc and LC neurons in brains of transgenic mice (with and without parkin) in the late stage of disease and in the same regions of healthy age-matched nontransgenic animals (with and without parkin). As expected, parkin null mice show significantly reduced numbers of TH-positive LC neurons. However, expression of A53T mutant human α-synuclein did not lead to additional reduction in the number of LC neurons in parkin null mice. There was no loss of LC neurons in A53T α-synuclein transgenic mice with wild-type parkin. Furthermore, no significant differences in the number of TH-positive neurons in the SNpc are observed among the various genotypes (Table 1). Moreover, HPLC electrochemical analysis of striatal DA and its metabolites did not show any significant differences among the various genotypes. Analysis of NE levels in the spinal cord, a target region of noradrenergic LC neurons, shows a trend toward a decrement of NE in parkin null mice, but there are no significant differences in NE levels among the various genotypes (Table 2).
Table 1.
Stereologic analysis of SNpc and LC neurons
| Genotype | SNpc |
LC |
||
|---|---|---|---|---|
| Cell count | % | Cell count | % | |
| WT− | 7361 ± 263 | 100.0 | 1310 ± 71 | 100.0 |
| KO− | 7328 ± 209 | 99.6 | 954 ± 53* | 73.7* |
| WT+ | 7357 ± 237 | 100.0 | 1141 ± 94 | 87.0 |
| KO+ | 7335 ± 236 | 99.7 | 1016 ± 79* | 77.5* |
Stereological analysis of the number of TH-positive neurons in the SNpc and LC in parkin WT and null mice with and without A53T α-synuclein overexpression. Data are expressed as mean ± SEM (n = 4–5 for SNpc and n = 8–12 for LC) and as ratio of the respective mean to WT− mean in percentage. Statistical analysis was performed by ANOVA, followed by the Student–Newman–Keuls test;*p < 0.05.
Table 2.
HPLC analysis of catecholamines
| Genotype | Striatum |
Spinal cord |
||||||
|---|---|---|---|---|---|---|---|---|
| DA |
DOPAC |
HVA |
NE |
|||||
| ng/mg | % | ng/mg | % | ng/mg | % | ng/mg | % | |
| WT− | 18.3 ± 0.5 | 100.0 | 1.68 ± 0.12 | 100.0 | 1.52 ± 0.10 | 100.0 | 0.54 ± 0.03 | 100.0 |
| KO− | 18.1 ± 0.5 | 98.9 | 1.77 ± 0.09 | 105.2 | 1.74 ± 0.12 | 114.9 | 0.45 ± 0.02 | 84.3 |
| WT+ | 19.7 ± 0.5 | 107.9 | 1.63 ± 0.10 | 96.9 | 1.77 ± 0.16 | 116.8 | 0.52 ± 0.04 | 96.2 |
| KO+ | 18.7 ± 0.5 | 102.4 | 1.54 ± 0.17 | 91.9 | 1.40 ± 0.11 | 92.5 | 0.52 ± 0.02 | 96.2 |
HPLC analysis of catecholamines in distinct brain regions of parkin WT and null mice with and without A53T α-synuclein overexpression. Data are expressed as mean ± SEM (n = 6–8) and as ratio of the respective mean to WT− mean in percentage. Statistical analysis was performed by ANOVA, revealing no significant differences among group means. DOPAC, 3,4-Dihydroxy-phenylacetic acid; HVA, homovanillic acid.
Accumulation of insoluble α-synuclein species and ubiquitination of α-synuclein are independent of the E3 ligase activity of parkin
The formation of pathological neuronal accumulation of α-synuclein in transgenic mice is associated with the accumulation of detergent-insoluble A53T α-synuclein species in biochemical fractionation assays (Giasson et al., 2002; Lee et al., 2002). In addition, A53T α-synuclein might be ubiquitinated in pathological inclusions in human A53T α-synuclein transgenic mice (Sampathu et al., 2003). To determine whether the abnormal accumulation and ubiquitination of insoluble A53T α-synuclein is altered because of the absence of parkin, the pathologically affected brainstem of parkin WT and KO transgenic mice in the late stage of A53T α-synuclein-induced disease was fractionated into non-ionic detergent-soluble and -insoluble fractions (Fig. 8). Immunoblot analysis of these fractions indicates that there is no alteration in the amount of α-synuclein in either the soluble or the insoluble fraction of pathologically affected tissue from α-synuclein transgenic mice with and without parkin (Fig. 8A, top, arrowhead). Moreover, the insoluble high-molecular-weight species of α-synuclein (Fig. 8A, top, arrows) as well as the proteolytically processed fragments of α-synuclein (Fig. 8A, top, double-lined arrow) from parkin KO A53T α-synuclein transgenic mice form a pattern indistinguishable from that observed in parkin WT A53T α-synuclein transgenic mice (Li et al., 2005).
Figure 8.
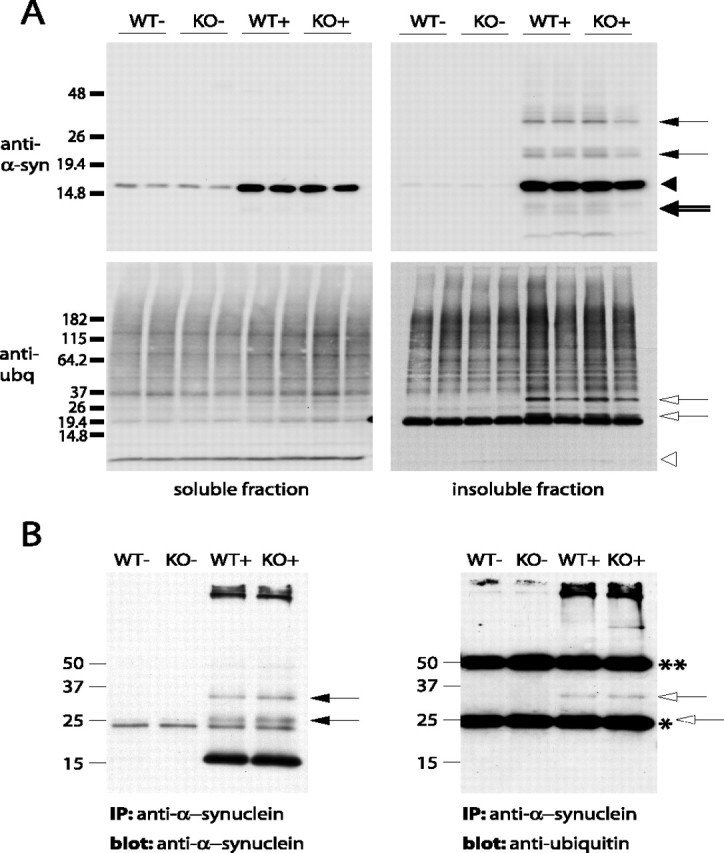
Accumulation of insoluble α-synuclein, proteolytic processing, and ubiquitination of α-synuclein are parkin independent. Brainstem lysates of terminally ill (or age-matched control) mice were separated into non-ionic detergent-soluble and -insoluble fractions. Immunoblotting was performed using antibodies against α-synuclein and ubiquitin of both soluble and insoluble fraction (A) and of immunoprecipitated (IP) α-synuclein (B). Filled arrows, Mono- and di-ubiquitinated α-synuclein species. Filled arrowhead, α-Synuclein monomer, unmodified. Double-lined arrow, Proteolytically processed α-synuclein fragments. Open arrows, Bands corresponding to the mono- and di-ubiquitinated α-synuclein species. Open arrowhead, Ubiquitin monomer. Dotted open arrow, Mono-ubiquitinated α-synuclein species covered by the IgG light chain after immunoprecipitation. *IgG light chain; **IgG heavy chain. Molecular weight markers are indicated.
We also monitored total ubiquitination in both the soluble and insoluble fraction (Fig. 8A, bottom). Although there is an overall increase in ubiquitinated proteins in the insoluble fraction of mice overexpressing human A53T α-synuclein compared with nontransgenic control samples, we observed no significant difference in the overall level of ubiquitination in α-synuclein transgenic mice in the presence or absence of parkin (Fig. 8A, bottom). Two specific protein bands are especially prominent, with molecular weights matching the predicted molecular weight of mono- and di-ubiquitinated α-synuclein (Fig. 8A, bottom, open arrows) and corresponding to the molecular weight of the two most abundant higher-molecular-weight species of α-synuclein (Fig. 8A, top, arrows). It has been speculated previously that these bands correspond to ubiquitinated forms of α-synuclein (Sampathu et al., 2003). To determine whether ubiquitination of α-synuclein is altered in the absence of parkin, we immunoprecipitated α-synuclein from the insoluble fraction of brainstem of end-stage diseased human A53T α-synuclein transgenic mice with and without parkin and performed immunoblots for α-synuclein and ubiquitin (Fig. 8B). In addition to the α-synuclein monomer and a nonspecific 20 kDa band (present in all lanes), the α-synuclein blot shows the same two prominent higher-molecular-weight bands for α-synuclein transgenic animals as described above (Fig. 8B, left, arrows). In the ubiquitin blot, a band corresponding to the upper one of these two bands is, presumably, the di-ubiquitinated form of α-synuclein (Fig. 8B, right, open arrow). The lower band, however, is masked by the IgG light chain (Fig. 8B, right, dotted arrow). Importantly, there is no discernible difference between the samples of human A53T α-synuclein transgenic mice with or without parkin. Together, these results show that, although α-synuclein is ubiquitinated in pathological detergent-insoluble inclusions in mice overexpressing human A53T α-synuclein, parkin has no effect on the solubility, proteolytic processing, or ubiquitination of α-synuclein. In conclusion, these results establish that parkin-mediated ubiquitin E3 ligase activity does not participate in the pathogenesis of neurodegeneration induced by mutant human A53T α-synuclein. Moreover, these results suggest that other ubiquitin E3 ligases play more important roles in the ubiquitination of α-synuclein.
Discussion
Whether parkin-mediated ubiquitination of α-synuclein underlies the pathogenesis of synucleinopathies is still a subject of debate. There are several fundamental observations that, together, appear to suggest a key role for parkin in the formation of α-synuclein-containing inclusions, including Lewy bodies. First, Lewy bodies contain α-synuclein and ubiquitin as major components. Second, parkin-associated PD is characterized by the absence of Lewy bodies. Third, most disease-associated parkin mutations are loss-of-function mutations. It has therefore been hypothesized that parkin ubiquitinates one or several proteins to target them for degradation, which then, in idiopathic PD, accumulate and form inclusion bodies attributable to dysfunction of the proteasome (Dawson and Dawson, 2003b). In particular, the finding that parkin may interact with and ubiquitinate an O-glycosylated form of α-synuclein supports a model of pathogenesis in which α-synuclein and parkin both contribute to the same pathogenic pathway of dysfunctional protein degradation (Shimura et al., 2001). In this scenario, parkin is responsible for targeting the potentially toxic α-synuclein to the proteasome for degradation or, alternatively, under certain conditions for inclusion body formation. In the context of this model, the fact that parkin-associated parkinsonism is characterized by an early age of onset supports the notion that parkin-mediated inclusion body formation represents a protective strategy used by neurons that is not available in the absence of this protein (Lotharius and Brundin, 2002). A similar hypothesis has been put forward for the pathogenesis of polyglutamine diseases, such as spinocerebellar ataxia-1, in which deletion of the E3 ligase E6-AP reduces the frequency of ubiquitinated nuclear inclusions, whereas accelerating pathology induced by expression of a mutant form of ataxin-1 (Cummings et al., 1999). Based on this observation, sequestration into inclusion bodies of proteins that are resistant to degradation is viewed as part of the cellular defense mechanism rather than a detrimental event in the pathogenesis (Ciechanover and Brundin, 2003). According to the model outlined above, parkin-associated PD and idiopathic PD would essentially share the same pathogenic mechanism, with the lack of parkin resulting in the loss of a potential protective mechanism, leading to early-onset disease. Such a protective effect of parkin is also suggested by a number of recent studies, in which we and others have shown that overexpression of parkin ameliorates α-synuclein-induced toxicity in models ranging from cell culture to Drosophila and rat (Petrucelli et al., 2002; Yang et al., 2003; Chung et al., 2004; Yamada et al., 2005). The protective effect of parkin in these models may be related to its role as a “multipurpose neuroprotective agent” (Feany and Pallanck, 2003) and be mediated through complex mechanisms that are yet to be fully understood. Such mechanisms might possibly involve the interaction of parkin with synphilin-1 and the proteasome rather than a role as E3 ubiquitin ligase for α-synuclein, because parkin fails to influence α-synuclein levels in these models.
A unifying concept like this, in which parkin acts as detoxifying agent for α-synuclein, predicts that the inactivation of the parkin gene in mice overexpressing mutant human A53T α-synuclein will result in an earlier onset of disease as well as the absence (or a significantly reduced number) of α-synuclein- and ubiquitin-positive inclusions. However, with the data presented here, we provide evidence that the absence of parkin does not have an impact on either of these two crucial parameters in a mouse model of α-synuclein-induced disease. Immunohistochemical analysis of affected brain regions of α-synuclein-overexpressing mice lacking parkin shows formation of proteinaceous inclusions containing α-synuclein and ubiquitin that are indistinguishable in terms of morphology, distribution, and abundance from those inclusions found in brains of α-synuclein transgenic mice on a parkin WT background. Furthermore, markers for neurodegeneration such as increased GFAP immunoreactivity and Fluoro Jade B staining show no difference between α-synuclein transgenic mice with or without parkin expression. Consistently, the survival curves of both groups of mice do not differ significantly, nor is there a difference regarding the speed of disease progression or the clinical phenotype. Thus, an unknown gain of function is probably the pathogenic event in synucleinopathies as opposed to aggregation and ubiquitination of α-synuclein.
Our study also provides considerable insight pertaining to the questions of ubiquitination and degradation of α-synuclein. There have been controversial results on how α-synuclein is degraded in the cell as part of the physiological turnover of proteins, a question that might also prove critical to understanding why α-synuclein is sequestered into inclusion bodies rather than being degraded in neurons of patients suffering from idiopathic PD. Evidence for proteasomal degradation, possibly independent of ubiquitination, as well as for autophagic degradation by lysosomes has been reported previously (Bennett et al., 1999; Tofaris et al., 2001; Cuervo et al., 2004). Recently, it was suggested that the cochaperone CHIP (C terminus of Hsp70-interacting protein) might act as a switch between both degradation pathways (Shin et al., 2005). The observation that parkin may polyubiquitinate an O-glycosylated form of α-synuclein supports the concept of proteasomal degradation of α-synuclein, with parkin as the critical E3 ligase (Shimura et al., 2001). In this study, we show in an in vivo model of synucleinopathy that the steady-state levels of α-synuclein (endogenous mouse and overexpressed human protein) are independent of the presence or absence of parkin, strongly arguing against a critical role for parkin in α-synuclein catabolism under physiological as well as pathological conditions. As to the question whether ubiquitination is crucial to inclusion body formation, a significant percentage of α-synuclein-containing inclusion bodies in human disease are ubiquitin negative, raising doubts about the necessity of ubiquitination for protein sequestration into Lewy body-like structures and fueling speculations that proteins other than α-synuclein might actually be ubiquitinated in Lewy bodies (Spillantini et al., 1998; Gomez-Tortosa et al., 2000). There has been significant (albeit indirect) evidence that mono- and di-ubiquitinated species of α-synuclein are present in the insoluble fraction of brain extracts from patients with Lewy body dementia (Hasegawa et al., 2002; Sampathu et al., 2003). In this study, we provide direct evidence that α-synuclein itself is ubiquitinated in a mouse model of synucleinopathy. Immunoprecipitated α-synuclein is immunopositive for ubiquitin, with a molecular weight corresponding to a di-ubiquitinated form of α-synuclein. Furthermore, the immunoprecipitation results reveal high-molecular-weight species that stain positive for α-synuclein as well as ubiquitin, which may represent polyubiquitinated α-synuclein species. Most importantly, the presence of the mono- and di-ubiquitinated forms of α-synuclein, as well as the polyubiquitinated species, is unaltered by the presence or absence of parkin, clearly arguing against parkin as the E3 ligase that ubiquitinates α-synuclein.
Previously, we reported loss of LC neurons and reduced levels of NE in parkin-deficient mice, as well as a marked reduction of the acoustic startle response (von Coelln et al., 2004b). In the present study, we show that parkin null mice and parkin null α-synuclein-overexpressing double-mutant mice both show a significant loss of LC neurons. However, there is no significant difference between these two genotypes regarding the number of LC neurons, neither is there between α-synuclein transgenic mice and WT controls. HPLC analysis also fails to show an additional deficit of the noradrenergic system resulting from overexpression of mutant α-synuclein. The previously reported impairment of the acoustic startle response in parkin KO mice was confirmed. Surprisingly, α-synuclein-overexpressing mice (with or without parkin) show an even greater reduction of acoustic startle. Because these mice do not show loss of LC neurons, there has to be a different explanation than a deficit of the central noradrenergic system, which we had suggested previously in parkin-deficient mice (von Coelln et al., 2004b). There are a number of potential mechanisms that could account for the reduced acoustic startle response (Hoffman and Ison, 1980). It is conceivable that the neurodegeneration that occurs in the α-synuclein-transgenic mice, particularly in the brainstem and spinal cord, causes this deficit. In addition, we did not exclude the possibility that α-synuclein-overexpressing mice have hearing deficits, which would account for the impaired acoustic startle response. Significant hearing deficits have been excluded in parkin null mice, as reported previously (von Coelln et al., 2004b). Importantly, our behavioral data does not show any interference of the effects of parkin deficiency on α-synuclein overexpression. Therefore, the finding that α-synuclein transgenic mice have impaired acoustic startle does not influence our overall conclusion of this study. The in-depth analysis of this phenomenon requires future study that may help to understand which neuronal systems are mainly affected in this mouse model of synucleinopathy. Together, our results from parkin KO α-synuclein-overexpressing double-mutant mice compared with parkin KO mice do not show any additional deficit of the noradrenergic system resulting from overexpression of mutant α-synuclein. We therefore provide evidence that, at least in the animal model used here, there does not seem to be an interaction between parkin and α-synuclein relevant to parkin-related pathogenesis in the central noradrenergic system, which is affected in human PD. Also vice versa, there is no detectable influence of parkin deficiency on the phenotype induced by overexpression of human A53T α-synuclein.
There are, however, certain limitations to the animal models used for this study. Notably, neither parkin null mice nor human A53T α-synuclein transgenic mice develop substantial deficits of substantia nigra DA neurons, a feature that is characteristic of both synuclein- and parkin-associated parkinsonian syndromes in humans. Furthermore, there is extensive spinal cord pathology in α-synuclein-overexpressing mice, which might be an essential factor determining lethality, whereas the spinal cord is not affected in human PD. It is therefore conceivable that substantia nigra pathology would develop at a later stage of disease in our double-mutant mice, if only they survived long enough for such a manifestation to occur. Given the severity and rapid progression of the α-synuclein-induced phenotype in the transgenic mice used in our experiments, this remains a possibility that will prove very difficult to test. Along these lines, we cannot exclude the possibility that there may be nigral pathways for cell survival that are parkin sensitive, whereas certain spinal neuron survival pathways may be parkin insensitive, in which case the latter neuronal population might still determine the course of disease in α-synuclein-overexpressing mice, regardless of the absence or presence of parkin. However, we believe that it is quite remarkable that the absence of parkin in the α-synuclein transgenic mice does not lead to the loss of dopaminergic neurons. Indeed, our original hypothesis was that the absence of parkin would lead to a dopaminergic phenotype in the α-synuclein transgenic mice. Moreover, the absence of parkin fails to alter the neuropathologic abnormalities in the hindbrain/brainstem of the α-synuclein transgenic mice, areas that are affected early in human PD (Braak et al., 2003). Thus, the fact that lack of parkin does not result in an alteration of the α-synuclein-induced phenotype in both affected and nonaffected brain areas argues in favor of our conclusion that parkin does not play a crucial role in α-synuclein metabolism.
Another important point is the possibility of compensatory effects through upregulation of other E3 ligases or alternative pathways of protein degradation or disposal in parkin KO mice. However, we recently reported accumulation of the parkin substrate p38/JTV-1 not only in brains of patients suffering from autosomal-recessive juvenile parkinsonism caused by parkin mutations but also in brainstem tissue of parkin-deficient mice (Ko et al., 2005). In this analysis, all other parkin substrates described to date did not show a significant increase in protein levels compared with WT control tissue. Although we cannot conclusively exclude compensatory mechanisms (e.g., upregulation of other E3 ligases), the apparent lack of such a compensatory mechanism for p38/JTV-1 suggests that the other parkin substrates, including α-synuclein, may either not be authentic parkin substrates or parkin may serve an alternative function, such as lysine 63 (K63)-linked ubiquitination (Lim et al., 2005), which serves nondegradative roles.
In conclusion, the lack of any observable synergistic effect or interference of parkin deficiency on the one hand and overexpression of human A53T α-synuclein on the other hand suggests that the pathogenesis of parkin-associated neurodegeneration and α-synuclein-induced neurodegeneration (e.g., in idiopathic PD) particularly in nondopaminergic systems may be fundamentally different. According to this model, the absence of Lewy bodies in parkin-associated parkinsonism does not reflect the inability of ailing neurons to protect themselves by sequestering toxic proteins into inclusion bodies but may simply reflect a mechanism of disease independent of α-synuclein and possibly even independent of protein accumulation altogether. Recent evidence from human subjects with parkin deficiency supports the concept that parkin plays no role in α-synuclein metabolism (Miller et al., 2005). Our data presented here suggest that parkin- and synuclein-associated forms of parkinsonism particularly in nondopaminergic systems might in fact constitute two entirely different disease entities, with many shared clinical features but essentially independent pathogenesis.
Footnotes
*R.v.C. and B.T. contributed equally to this work.
R. von Coelln's present address: Department of Neurodegenerative Disease, Hertie Institute for Clinical Brain Research, University of Tuebingen, 72076 Tuebingen, Germany.
K. L. Lim's present address: Neurodegeneration Research Laboratory, National Neuroscience Institute of Singapore, 11, Jalan Tan Tock Seng, Singapore 308433.
R. Saffary's present address: Department of Biology, University of Maryland School of Medicine, Baltimore, MD 21201.
This work was supported by National Institutes of Health–National Institute of Neurological Disorders and Stroke Grants NS 38377, NS 48206, and NS 38065, the Lee Martin Trust, and the Sylvia Nachlas Trust. T.M.D. is the Leonard and Madlyn Abramson Professor in Neurodegenerative Diseases.
References
- Baba M, Nakajo S, Tu PH, Tomita T, Nakaya K, Lee VM, Trojanowski JQ, Iwatsubo T (1998). Aggregation of alpha-synuclein in Lewy bodies of sporadic Parkinson's disease and dementia with Lewy bodies. Am J Pathol 152:879–884. [PMC free article] [PubMed] [Google Scholar]
- Bennett MC, Bishop JF, Leng Y, Chock PB, Chase TN, Mouradian MM (1999). Degradation of alpha-synuclein by proteasome. J Biol Chem 274:33855–33858. [DOI] [PubMed] [Google Scholar]
- Braak H, Del Tredici K, Rub U, de Vos RA, Jansen Steur EN, Braak E (2003). Staging of brain pathology related to sporadic Parkinson's disease. Neurobiol Aging 24:197–211. [DOI] [PubMed] [Google Scholar]
- Chung KK, Zhang Y, Lim KL, Tanaka Y, Huang H, Gao J, Ross CA, Dawson VL, Dawson TM (2001). Parkin ubiquitinates the alpha-synuclein-interacting protein, synphilin-1: implications for Lewy-body formation in Parkinson disease. Nat Med 7:1144–1150. [DOI] [PubMed] [Google Scholar]
- Chung KK, Thomas B, Li X, Pletnikova O, Troncoso JC, Marsh L, Dawson VL, Dawson TM (2004). S-nitrosylation of parkin regulates ubiquitination and compromises parkin's protective function. Science 304:1328–1331. [DOI] [PubMed] [Google Scholar]
- Ciechanover A, Brundin P (2003). The ubiquitin proteasome system in neurodegenerative diseases: sometimes the chicken, sometimes the egg. Neuron 40:427–446. [DOI] [PubMed] [Google Scholar]
- Cuervo AM, Stefanis L, Fredenburg R, Lansbury PT, Sulzer D (2004). Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science 305:1292–1295. [DOI] [PubMed] [Google Scholar]
- Cummings CJ, Reinstein E, Sun Y, Antalffy B, Jiang Y, Ciechanover A, Orr HT, Beaudet AL, Zoghbi HY (1999). Mutation of the E6-AP ubiquitin ligase reduces nuclear inclusion frequency while accelerating polyglutamine-induced pathology in SCA1 mice. Neuron 24:879–892. [DOI] [PubMed] [Google Scholar]
- Dawson TM, Dawson VL (2003a). Rare genetic mutations shed light on the pathogenesis of Parkinson disease. J Clin Invest 111:145–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson TM, Dawson VL (2003b). Molecular pathways of neurodegeneration in Parkinson's disease. Science 302:819–822. [DOI] [PubMed] [Google Scholar]
- Feany MB, Bender WW (2000). A Drosophila model of Parkinson's disease. Nature 404:394–398. [DOI] [PubMed] [Google Scholar]
- Feany MB, Pallanck LJ (2003). Parkin: a multipurpose neuroprotective agent? Neuron 38:13–16. [DOI] [PubMed] [Google Scholar]
- Forno LS (1996). Neuropathology of Parkinson's disease. J Neuropathol Exp Neurol 55:259–272. [DOI] [PubMed] [Google Scholar]
- Franklin KBJ, Paxinos G (1997). In: The mouse brain in stereotaxic coordinates San Diego: Academic.
- Giasson BI, Duda JE, Quinn SM, Zhang B, Trojanowski JQ, Lee VM (2002). Neuronal alpha-synucleinopathy with severe movement disorder in mice expressing A53T human alpha-synuclein. Neuron 34:521–533. [DOI] [PubMed] [Google Scholar]
- Goedert M (2001). Alpha-synuclein and neurodegenerative diseases. Nat Rev Neurosci 2:492–501. [DOI] [PubMed] [Google Scholar]
- Gomez-Tortosa E, Newell K, Irizarry MC, Sanders JL, Hyman BT (2000). Alpha-synuclein immunoreactivity in dementia with Lewy bodies: morphological staging and comparison with ubiquitin immunostaining. Acta Neuropathol (Berl) 99:352–357. [DOI] [PubMed] [Google Scholar]
- Hasegawa M, Fujiwara H, Nonaka T, Wakabayashi K, Takahashi H, Lee VM, Trojanowski JQ, Mann D, Iwatsubo T (2002). Phosphorylated alpha-synuclein is ubiquitinated in alpha-synucleinopathy lesions. J Biol Chem 277:49071–49076. [DOI] [PubMed] [Google Scholar]
- Hoffman HS, Ison JR (1980). Reflex modification in the domain of startle. I. Some empirical findings and their implications for how the nervous system processes sensory input. Psychol Rev 87:175–189. [PubMed] [Google Scholar]
- Irizarry MC, Growdon W, Gomez-Isla T, Newell K, George JM, Clayton DF, Hyman BT (1998). Nigral and cortical Lewy bodies and dystrophic nigral neurites in Parkinson's disease and cortical Lewy body disease contain alpha-synuclein immunoreactivity. J Neuropathol Exp Neurol 57:334–337. [DOI] [PubMed] [Google Scholar]
- Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, Yokochi M, Mizuno Y, Shimizu N (1998). Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 392:605–608. [DOI] [PubMed] [Google Scholar]
- Ko HS, von Coelln R, Sriram SR, Kim SW, Chung KK, Pletnikova O, Troncoso J, Johnson B, Saffary R, Goh EL, Song H, Park BJ, Kim MJ, Kim S, Dawson VL, Dawson TM (2005). Accumulation of the authentic parkin substrate aminoacyl-tRNA synthetase cofactor, p38/JTV-1, leads to catecholaminergic cell death. J Neurosci 25:7968–7978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruger R, Kuhn W, Muller T, Woitalla D, Graeber M, Kosel S, Przuntek H, Epplen JT, Schols L, Riess O (1998). Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson's disease. Nat Genet 18:106–108. [DOI] [PubMed] [Google Scholar]
- Lakso M, Vartiainen S, Moilanen AM, Sirvio J, Thomas JH, Nass R, Blakely RD, Wong G (2003). Dopaminergic neuronal loss and motor deficits in Caenorhabditis elegans overexpressing human alpha-synuclein. J Neurochem 86:165–172. [DOI] [PubMed] [Google Scholar]
- Lang AE, Lozano AM (1998). Parkinson's disease. First of two parts. N Engl J Med 339:1044–1053. [DOI] [PubMed] [Google Scholar]
- Lee HJ, Khoshaghideh F, Patel S, Lee SJ (2004). Clearance of α-synuclein oligomeric intermediates via the lysosomal degradation pathway. J Neurosci 24:1888–1896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MK, Stirling W, Xu Y, Xu X, Qui D, Mandir AS, Dawson TM, Copeland NG, Jenkins NA, Price DL (2002). Human α-synuclein-harboring familial Parkinson's disease-linked Ala-53 → Thr mutation causes neurodegenerative disease with α-synuclein aggregation in transgenic mice. Proc Natl Acad Sci USA 99:8968–8973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, West N, Colla E, Pletnikova O, Troncoso JC, Marsh L, Dawson TM, Jakala P, Hartmann T, Price DL, Lee MK (2005). Aggregation promoting C-terminal truncation of α-synuclein is a normal cellular process and is enhanced by the familial Parkinson's disease-linked mutations. Proc Natl Acad Sci USA 102:2162–2167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim KL, Chew KC, Tan JM, Wang C, Chung KK, Zhang Y, Tanaka Y, Smith W, Engelender S, Ross CA, Dawson VL, Dawson TM (2005). Parkin mediates nonclassical, proteasomal-independent ubiquitination of synphilin-1: implications for Lewy body formation. J Neurosci 25:2002–2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lotharius J, Brundin P (2002). Pathogenesis of Parkinson's disease: dopamine, vesicles and alpha-synuclein. Nat Rev Neurosci 3:932–942. [DOI] [PubMed] [Google Scholar]
- Lucking CB, Durr A, Bonifati V, Vaughan J, De Michele G, Gasser T, Harhangi BS, Meco G, Denefle P, Wood NW, Agid Y, Brice A (2000). Association between early-onset Parkinson's disease and mutations in the parkin gene. French Parkinson's Disease Genetics Study Group. N Engl J Med 342:1560–1567. [DOI] [PubMed] [Google Scholar]
- Masliah E, Rockenstein E, Veinbergs I, Mallory M, Hashimoto M, Takeda A, Sagara Y, Sisk A, Mucke L (2000). Dopaminergic loss and inclusion body formation in alpha-synuclein mice: implications for neurodegenerative disorders. Science 287:1265–1269. [DOI] [PubMed] [Google Scholar]
- Miller DW, Crawley A, Gwinn-Hardy K, Lopez G, Nussbaum R, Cookson MR, Singleton AB, Hardy J, Dogu O (2005). Unaltered alpha-synuclein blood levels in juvenile Parkinsonism with a parkin exon 4 deletion. Neurosci Lett 374:189–191. [DOI] [PubMed] [Google Scholar]
- Mori H, Kondo T, Yokochi M, Matsumine H, Nakagawa-Hattori Y, Miyake T, Suda K, Mizuno Y (1998). Pathologic and biochemical studies of juvenile parkinsonism linked to chromosome 6q. Neurology 51:890–892. [DOI] [PubMed] [Google Scholar]
- Neumann M, Adler S, Schluter O, Kremmer E, Benecke R, Kretzschmar HA (2000). Alpha-synuclein accumulation in a case of neurodegeneration with brain iron accumulation type 1 (NBIA-1, formerly Hallervorden-Spatz syndrome) with widespread cortical and brainstem-type Lewy bodies. Acta Neuropathol (Berl) 100:568–574. [DOI] [PubMed] [Google Scholar]
- Nonaka T, Iwatsubo T, Hasegawa M (2005). Ubiquitination of alpha-synuclein. Biochemistry 44:361–368. [DOI] [PubMed] [Google Scholar]
- Pawlyk AC, Giasson BI, Sampathu DM, Perez FA, Lim KL, Dawson VL, Dawson TM, Palmiter RD, Trojanowski JQ, Lee VM (2003). Novel monoclonal antibodies demonstrate biochemical variation of brain parkin with age. J Biol Chem 278:48120–48128. [DOI] [PubMed] [Google Scholar]
- Petrucelli L, O'Farrell C, Lockhart PJ, Baptista M, Kehoe K, Vink L, Choi P, Wolozin B, Farrer M, Hardy J, Cookson MR (2002). Parkin protects against the toxicity associated with mutant alpha-synuclein: proteasome dysfunction selectively affects catecholaminergic neurons. Neuron 36:1007–1019. [DOI] [PubMed] [Google Scholar]
- Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, Stenroos ES, Chandrasekharappa S, Athanassiadou A, Papapetropoulos T, Johnson WG, Lazzarini AM, Duvoisin RC, Di Iorio G, Golbe LI, Nussbaum RL (1997). Mutation in the alpha-synuclein gene identified in families with Parkinson's disease. Science 276:2045–2047. [DOI] [PubMed] [Google Scholar]
- Sampathu DM, Giasson BI, Pawlyk AC, Trojanowski JQ, Lee VM (2003). Ubiquitination of alpha-synuclein is not required for formation of pathological inclusions in alpha-synucleinopathies. Am J Pathol 163:91–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmued LC, Hopkins KJ (2000). Fluoro-Jade B: a high affinity fluorescent marker for the localization of neuronal degeneration. Brain Res 874:123–130. [DOI] [PubMed] [Google Scholar]
- Sharma N, McLean PJ, Kawamata H, Irizarry MC, Hyman BT (2001). Alpha-synuclein has an altered conformation and shows a tight intermolecular interaction with ubiquitin in Lewy bodies. Acta Neuropathol (Berl) 102:329–334. [DOI] [PubMed] [Google Scholar]
- Shimura H, Schlossmacher MG, Hattori N, Frosch MP, Trockenbacher A, Schneider R, Mizuno Y, Kosik KS, Selkoe DJ (2001). Ubiquitination of a new form of alpha-synuclein by parkin from human brain: implications for Parkinson's disease. Science 293:263–269. [DOI] [PubMed] [Google Scholar]
- Shin Y, Klucken J, Patterson C, Hyman BT, McLean PJ (2005). The co-chaperone carboxyl terminus of Hsp70-interacting protein (CHIP) mediates alpha-synuclein degradation decisions between proteasomal and lysosomal pathways. J Biol Chem 280:23727–23734. [DOI] [PubMed] [Google Scholar]
- Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, Hulihan M, Peuralinna T, Dutra A, Nussbaum R, Lincoln S, Crawley A, Hanson M, Maraganore D, Adler C, Cookson MR, Muenter M, Baptista M, Miller D, Blancato J, et al (2003). alpha-Synuclein locus triplication causes Parkinson's disease. Science 302:841. [DOI] [PubMed] [Google Scholar]
- Spillantini MG, Goedert M (2000). The alpha-synucleinopathies: Parkinson's disease, dementia with Lewy bodies, and multiple system atrophy. Ann NY Acad Sci 920:16–27. [DOI] [PubMed] [Google Scholar]
- Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M (1997). Alpha-synuclein in Lewy bodies. Nature 388:839–840. [DOI] [PubMed] [Google Scholar]
- Spillantini MG, Crowther RA, Jakes R, Hasegawa M, Goedert M (1998). α-Synuclein in filamentous inclusions of Lewy bodies from Parkinson's disease and dementia with lewy bodies. Proc Natl Acad Sci USA 95:6469–6473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi H, Ohama E, Suzuki S, Horikawa Y, Ishikawa A, Morita T, Tsuji S, Ikuta F (1994). Familial juvenile parkinsonism: clinical and pathologic study in a family. Neurology 44:437–441. [DOI] [PubMed] [Google Scholar]
- Tofaris GK, Layfield R, Spillantini MG (2001). alpha-synuclein metabolism and aggregation is linked to ubiquitin-independent degradation by the proteasome. FEBS Lett 509:22–26. [DOI] [PubMed] [Google Scholar]
- van der Putten H, Wiederhold KH, Probst A, Barbieri S, Mistl C, Danner S, Kauffmann S, Hofele K, Spooren WP, Ruegg MA, Lin S, Caroni P, Sommer B, Tolnay M, Bilbe G (2000). Neuropathology in mice expressing human α-synuclein. J Neurosci 20:6021–6029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Coelln R, Dawson VL, Dawson TM (2004a). Parkin-associated Parkinson's disease. Cell Tissue Res 318:175–184. [DOI] [PubMed] [Google Scholar]
- von Coelln R, Thomas B, Savitt JM, Lim KL, Sasaki M, Hess EJ, Dawson VL, Dawson TM (2004b). Loss of locus coeruleus neurons and reduced startle in parkin null mice. Proc Natl Acad Sci USA 101:10744–10749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West MJ (1993). New stereological methods for counting neurons. Neurobiol Aging 14:275–285. [DOI] [PubMed] [Google Scholar]
- West MJ, Slomianka L, Gundersen HJ (1991). Unbiased stereological estimation of the total number of neurons in the subdivisions of the rat hippocampus using the optical fractionator. Anat Rec 231:482–497. [DOI] [PubMed] [Google Scholar]
- Wirths O, Weickert S, Majtenyi K, Havas L, Kahle PJ, Okochi M, Haass C, Multhaup G, Beyreuther K, Bayer TA (2000). Lewy body variant of Alzheimer's disease: alpha-synuclein in dystrophic neurites of A beta plaques. NeuroReport 11:3737–3741. [DOI] [PubMed] [Google Scholar]
- Yamada M, Mizuno Y, Mochizuki H (2005). Parkin gene therapy for alpha-synucleinopathy: a rat model of Parkinson's disease. Hum Gene Ther 16:262–270. [DOI] [PubMed] [Google Scholar]
- Yang Y, Nishimura I, Imai Y, Takahashi R, Lu B (2003). Parkin suppresses dopaminergic neuron-selective neurotoxicity induced by Pael-R in Drosophila. Neuron 37:911–924. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Gao J, Chung KK, Huang H, Dawson VL, Dawson TM (2000). Parkin functions as an E2-dependent ubiquitin-protein ligase and promotes the degradation of the synaptic vesicle-associated protein, CDCrel-1. Proc Natl Acad Sci USA 97:13354–13359. [DOI] [PMC free article] [PubMed] [Google Scholar]