

Abstract
A fundamental objective of anesthesia research is to identify the receptors and brain regions that mediate the various behavioral components of the anesthetic state, including amnesia, immobility, and unconsciousness. Using complementary in vivo and in vitro approaches, we found that GABAA receptors that contain the α5 subunit (α5GABAARs) play a critical role in amnesia caused by the prototypic intravenous anesthetic etomidate. Whole-cell recordings from hippocampal pyramidal neurons showed that etomidate markedly increased a tonic inhibitory conductance generated by α5GABAARs, whereas synaptic transmission was only slightly enhanced. Long-term potentiation (LTP) of field EPSPs recorded in CA1 stratum radiatum was reduced by etomidate in wild-type (WT) but not α5 null mutant (α5−/−) mice. In addition, etomidate impaired memory performance of WT but not α5−/− mice for spatial and nonspatial hippocampal-dependent learning tasks. The brain concentration of etomidate associated with memory impairment in vivo was comparable with that which increased the tonic inhibitory conductance and blocked LTP in vitro. The α5−/− mice did not exhibit a generalized resistance to etomidate, in that the sedative-hypnotic effects measured with the rotarod, loss of righting reflex, and spontaneous motor activity were similar in WT and α5−/− mice. Deletion of the α5 subunit of the GABAARs reduced the amnestic but not the sedative-hypnotic properties of etomidate. Thus, the amnestic and sedative-hypnotic properties of etomidate can be dissociated on the basis of GABAAR subtype pharmacology.
Keywords: amnesia, anesthesia, GABAA receptor, tonic inhibition, etomidate, learning and memory
Introduction
General anesthetics are highly lipid-soluble, low-potency compounds that were initially thought to act by nonspecifically perturbing the structure of lipid bilayers. A major advance in the 1980s was the identification of neuronal proteins as anesthetic targets (Franks and Lieb, 1988). Behavioral and neuroimaging studies in humans and animal models have since shown that anesthetics do not nonspecifically depress brain function. Rather, anesthetics cause a compilation of different behavioral endpoints, including amnesia, immobility, and unconsciousness, that are mediated by different brain regions and receptor populations (Campagna et al., 2003). Recently, studies with transgenic and null mutant mice have shown that specific populations of GABAA receptors (GABAARs) contribute to the sedative and immobilizing properties of certain anesthetics (for review, see Rudolph and Antkowiak, 2004). The molecular targets underlying the amnestic properties of anesthetics have been highly elusive but are of great clinical importance and scientific interest. Unintended intraoperative awareness during surgery occurs in one or two cases per 1000 anesthetized patients (Sebel et al., 2004). Because anesthetics are administered to >27 million patients each year, intraoperative awareness has become a major medical concern, as highlighted by a “sentinel alert” released by the Joint Commission on Accreditation of Healthcare Organizations (www.jcaho.org/SentinelEvents/SentinelEventAlert/sea_32.htm). GABAARs that contribute to the amnestic effects of general anesthetics have not been identified previously.
In vitro studies have shown that most intravenous anesthetics, including etomidate, are positive allosteric modulators of GABAAR function (Jurd et al., 2003; Reynolds et al., 2003). In the hippocampus, a brain structure that is involved in learning and memory, GABAARs generate two distinct forms of inhibition (Otis et al., 1991; Bai et al., 2001). Transient IPSCs result from the vesicular release of GABA and the activation of postsynaptic GABAARs, whereas a low-amplitude tonic inhibitory conductance is generated by low concentrations of ambient GABA (Semyanov et al., 2004). In hippocampal pyramidal neurons, GABAARs that generate the tonic and synaptic currents have distinct subunit compositions and pharmacological properties (Yeung et al., 2003; Caraiscos et al., 2004a). We have shown previously that a tonic inhibitory conductance in hippocampal pyramidal neurons is generated primarily by α5 subunit-containing GABAARs (α5GABAARs) (Caraiscos et al., 2004a). The α5 subunit is of particular interest in memory processes, because it is predominantly expressed in the hippocampus (Wainwright et al., 2000). In humans and rodents, only 4% of all GABAARs in the brain but 25% of GABAARs in the hippocampus contain the α5 subunit (Sur et al., 1999). Several lines of evidence suggest that α5GABAARs play a major role in memory processes, because reduced expression of the α5 subunit is associated with better performance of hippocampal-dependent learning tasks (Collinson et al., 2002; Crestani et al., 2002). Also, inverse agonists selective for α5GABAARs have nootropic effects in animal models (Chambers et al., 2003). Here, we test the hypothesis that etomidate, a prototypic intravenous anesthetic, increases the function of α5GABAARs and that this effect contributes to the amnestic but not the sedative-hypnotic properties of this anesthetic.
Materials and Methods
Generation of α5 null mutant (−/−) mice and cell cultures.
The experiments were approved by the Animal Care Committee of the University of Toronto. The generation of the α5−/− mice has been described previously (Collinson et al., 2002). Notably, α5−/− mice exhibit normal life expectancy and normal motor coordination with no overt compensatory changes in other GABAAR subunits. Cultures of hippocampal neurons from α5−/− mice and wild-type (WT) littermates were prepared as previously described (MacDonald et al., 1989) on postnatal day 1. For several experiments, hippocampal cultures were prepared from embryonic Swiss White mice because the number of WT mice available from the heterozygous α5+/− breeding pairs was limited. To increase the amplitude of the tonic current and to facilitate its pharmacological characterization, cultures of dissociated neurons were treated with the GABA-transaminase inhibitor vigabatrin (100 μm), before recording (Wu et al., 2003), although a low-amplitude tonic current can be readily detected in cultured hippocampal neurons without such treatment (Bai et al., 2001).
Electrophysiology.
The concentration-dependent effects of etomidate on tonic and synaptic currents in cultured hippocampal neurons were determined with the whole-cell patch-clamp technique (at 20–23°C). Electrodes were made from borosilicate glass pipettes and fire polished just before use. Currents were recorded with an Axopatch 200A amplifier and headstage (Molecular Devices, Union City, CA) and low-pass filtered at 10 kHz before digitization (Digidata 1200; Molecular Devices). Series resistance and pipette and whole-cell capacitance were cancelled electronically. To monitor series resistance, a hyperpolarizing voltage step of 10 mV was applied. Only cells that demonstrated stable series resistance (<20% change) were used for data analysis. Cells were perfused with a solution containing the following (in mm): 140 NaCl, 2.0 KCl, 1.3 CaCl2, 25 HEPES, and 28 glucose, pH 7.4. Tetrodotoxin (0.3 μm) was added to the extracellular solution to inhibit spontaneous voltage-dependent sodium channel activity. In all experiments, potassium currents were suppressed by dialyzing the cell interior with a CsCl-based internal solution, pH 7.3, that contained the following (in mm): 120 CsCl, 2.0 MgCl2, 1.0 CaCl2, 11 EGTA, 30 HEPES, 2.0 MgATP, and 2.0 tetraethylammonium. The amplitude of the tonic current under control conditions was measured as the difference in the holding current before and during the application of bicuculline methiodide (100 μm). Etomidate (Bedford Laboratories, Bedford, OH) or the vehicle control (35% v/v propylene glycol) at equivalent concentrations was added to the extracellular solution. Solutions were applied to the cell cultures using a multibarrel fast perfusion system.
For synaptic currents, peak amplitude, charge transfer [Q, the integrated area under miniature IPSCs (mIPSCs)], and time constant of current decay (τoff) were analyzed. The decay phase was well described by a single exponential equation in the form I(t) = Aoexp (−t/τoff) + C, where I(t) is the current amplitude at any given time t, C is the residual current, and Ao is the current amplitude at time 0. The change in charge transfer (ΔQmIPSC) associated with mIPSC was calculated as we reported previously (Bai et al., 2001) using the following equation: ΔQmIPSC = fdrug × Qdrug − fcon × Qcon, where fdrug and fcon are the frequencies (in Hertz) of mIPSCs, Qdrug and Qcon are the average charge transfer (pC) per mIPSC during drug and control conditions, respectively. Under our experimental conditions, we assumed that the change in charge transfer reflected a proportional change in membrane conductance. The charge transfer per second associated with the tonic current was calculated to be: ΔQTC = ITC × Δt, where ΔQTC is the charge transfer produced by the tonic current, and ITC is the current amplitude at steady state. In additional experiments, the long-term potentiation (LTP) of excitatory potentials was studied using hippocampal slices that were prepared from 6- to 9-month-old male WT and α5−/− mice. After administration of halothane anesthesia, mice were decapitated and their brains were quickly removed and placed in ice-cold oxygenated (95% O2, 5% CO2) artificial CSF (aCSF) (composition in mm: 124 NaCl, 3 KCl, 1.3 MgCl2, 2.6 CaCl2, 1.25 NaH2PO4, 26 NaHCO3, 10 d-glucose) with the osmolarity adjusted to 300–310 mOsm. Slices (350 μm thick) containing transverse sections of the hippocampus were prepared with a vibratome (VT1000E tissue slicer; Leica, Deerfield, IL). After a recovery period of 1 h in the oxygenated aCSF, slices were transferred to a submersion-recording chamber. The CA1 region was isolated from the CA3 region by a surgical cut, and slices were continually perfused with aCSF. Extracellular field EPSPs (fEPSPs) were recorded from the CA1 stratum radiatum neurons using electrodes that contained aCSF. Synaptic responses were evoked by stimulating Schaffer collaterals with 0.8 ms pulses delivered by concentric bipolar stimulating electrodes (Rhodes Medical Instruments, Summerland, CA). Baseline responses were obtained by stimulation at 0.05 Hz using an intensity that yielded a half-maximal field potential slope. LTP was induced by one episode of theta-burst stimulation (TBS) at the same intensity as that used for the baseline response. The TBS protocol consisted of 10 stimulus trains delivered at 5 Hz, each train consisting of four pulses at 100 Hz. TBS was induced after the slice had been perfused with etomidate (1 μm) or the vehicle control for 20 min. A higher concentration of etomidate was used for the hippocampal slices than for the neuronal cultures, because slow diffusion and equilibration means that higher concentrations of lipophilic anesthetics are needed to influence GABAARs in brain slices than in cell culture preparations (Gredell et al., 2004).
Behavioral tests.
All behavioral tests were performed using male age-matched α5−/− and WT mice. The experimenters were blind to mouse genotype and drug treatment.
Contextual fear conditioning.
The conditioning chamber consisted of a Perspex arena with a light mounted in the lid (350 × 200 × 193 mm; Technical and Scientific Equipment, Midland, MI). The floor consisted of stainless steel bars (4 mm diameter, 5 mm apart) that were connected to a computer, which controlled the duration of the test session and the timing, intensity, and duration of the shock. On day 1, single subjects were allowed to explore the chamber for 180 s. They then received three unsignaled foot shocks (duration 2 s, intensity 1 mA) at 60 s intervals. For the drug studies, etomidate (4 mg/kg) or vehicle (propylene glycol in sterile saline at concentration and dose volume equivalent to the etomidate-containing solution) was administered intraperitoneally before the conditioning trial. On day 2, 24 h after the conditioning session, mice were returned to the chamber, and the freezing response was assessed immediately then every 8 s for 8 min. The freezing response was defined as the lack of any movement except that required for breathing. Assessment of the freezing response occurred in the same conditioning chamber in which the mice received the foot shocks.
Morris water maze.
For the Morris water maze probe trial, a circular pool of diameter 1.2 m was filled with tap water (25 ± 2°C) that was made opaque by the addition of a white nontoxic paint. A circular platform with a diameter of 12 cm was placed ∼0.5 cm below the water surface so that it was not visible to the mice. Mice were pretrained for 10 d with four trials each day. In the matching-to-place water maze, the location of the platform was changed daily. Each mouse had 60 s to search for and locate the hidden platform. If the mouse did not locate the platform within 60 s, the experimenter gently guided it to the platform. Each trial ended when the mouse had sat on the platform for 30 s. The intertrial interval was 30 s. The α5−/− mice used in this study have been reported previously to exhibit better performance in the initial acquisition trials of the Morris water maze paradigm (Collinson et al., 2002). We initially tested the effects of etomidate on performance during the acquisition trials in WT and α5−/− mice; however, the results were extremely variable, so this strategy was abandoned (data not shown). Probe trials were subsequently undertaken to test for spatial learning. During the acquisition phase of the probe trials, each mouse was randomly assigned to receive an injection of the vehicle, etomidate (4 mg/kg, i.p.) or ketamine (20 mg/kg, i.p.; Bimeda-MTC Animal Health, Cambridge, Ontario, Canada). Thirty minutes later, the mouse was placed in the water maze. Four acquisition trials were conducted after the injection. The next day, a probe trial was performed to test the ability of the mice to recall the spatial location that previously contained the hidden platform. During the probe trial, the hidden platform was removed from the pool and the mouse was allowed to search for 60 s. The mouse was then promptly rescued, and the trial ended. Mice that learned the correct location of the platform selectively searched in the correct area. The percentage of time spent within the correct area of the pool (four times the diameter of the platform) was used to measure recall. The correct location of the platform represented 16% of the total area of the pool. If no learning occurred, the mouse was expected to spend 16% of the time near the correct location. The vehicle and drugs were both administered twice, each on separate days. The order in which the drug or vehicle was administered was random. The results from the two probe trials were averaged to provide a single value for each mouse, and the effect of etomidate was compared with the effect of the vehicle control. Data records were made with HVS Water 2020 software (VHS Image, Hampton, UK) for off-line analysis. Briefly, a video camera captured the movement of the mouse, and HVS Water 2020 software tracked the mouse in contrast comparison. The time, swim path, and latency of each mouse during a trial were recorded, and the percentage of time spent in the correct region was calculated by the software during analysis.
Rotarod.
Mice were tested on a rotating rod unit (rotarod) to study motor coordination and strength. The mice were trained to walk on a rotarod (Economex; Columbus Instruments, Columbus, OH) revolving at a constant speed of 12 rpm for 120 s consistently. On the test day, one preinjection trial was performed before the animals were treated with vehicle or etomidate. Performance was indicated by the latency to fall from the rotarod at 5, 30, and 60 min after injection. The time the mouse remained on the rotarod was recorded up to a maximum of 120 s.
Open field test.
The sedative properties of etomidate were tested by measuring spontaneous motor activity in an open field. WT and α5−/− were dosed with etomidate (4 or 10 mg/kg) or propylene glycol and then returned to their home cage. Mice were tested for the duration of walking as an index of spontaneous locomotor activity in the open field at 30 and 60 min after injection. Briefly, all mice were tested at the two time points for five consecutive minutes. After placement in the open field, a trained examiner used an event recorder to score the total time spent walking. Odors were controlled between test subjects by wiping the floor and walls of the test chamber with a mild ethanol solution between tests. A heat lamp was placed directly above the open field to accommodate for the hypothermic effects of etomidate.
Loss of righting reflex.
The loss of righting reflex (LORR) was assessed using a classical experimental protocol. The LORR was determined in WT and α5−/− mice for a wide range of etomidate doses (5–20 mg/kg, i.p.). Immediately after injection, the mice were placed in a shoebox cage and observed until they stopped moving. Mice were then placed on their back and scored as anesthetized if they failed to completely right within 30 s. If the mice succeeded in righting themselves three consecutive times, they were scored as awake. All mice were used only once for the LORR assay. Data were analyzed as the percentage of the population that LORR at each dose of etomidate. To determine the dose of etomidate that caused 50% of the maximal response (ED50), the dose–response plot was fit, using nonlinear regression, to the equation: Y = D + [A − D]/[1 + 10((logED50-X) Hill slope)], where D is the minimum response, A is the maximum response, and X is the dose. The time to the LORR was also quantified by measuring the time from the end of the injection to when the mice first demonstrated a LORR.
Brain concentration of etomidate.
HPLC was used to measure the concentration of etomidate in the brains of the mice. The measurements were performed according to a method reported previously (McIntosh and Rajewski, 2001). The brains were collected, weighed, homogenized, and frozen at −80°C. An HPLC unit (Hewlett-Packard 1100 series; Agilent Technologies, Mountain View, CA) was equipped with a variable-wavelength detector. Etomidate analysis was performed using a reverse-phase polaris column (Ansys Technologies, Lake Forest, CA) operating at room temperature. Standard curves were created by adding known amounts of etomidate to blank samples and plotting the peak area against the concentration. The detection wavelength was set at 242 nm. An isocratic mobile phase consisting of 25:25:50 (v/v/v) of acetonitrile, methanol, and 25 mm phosphate buffer, pH 8.1, was used at a flow rate of 1.5 ml/min.
Statistics.
All results are reported as the mean ± SEM. Statistical significance was assessed with the Student's t test or one-way or two-way ANOVA and the Newman–Keul or Bonferroni's post hoc tests, as appropriate.
Results
Etomidate enhanced tonic conductance in pyramidal neurons
The concentration-dependent effects of etomidate on tonic and synaptic currents were first studied using whole-cell currents recorded in pyramidal neurons grown in dissociated cultures. The cell culture preparation permits more effective control of drug concentration and washout of highly lipophilic compounds than is possible with brain slices. The application of a low concentration of etomidate (0.1 μm) to pyramidal neurons from Swiss White mice caused a reversible increase in the tonic conductance (156 ± 24% of control; n = 8; p < 0.05), whereas etomidate (0.1 μm) failed to alter the time course or amplitude of mIPSCs (Fig. 1A, Table 1). Etomidate, applied at a higher concentration (1 μm) further increased the tonic conductance (314 ± 51.4% of control; n = 9; p < 0.05) and prolonged the duration of mIPSCs; however, the inhibitory net charge transfer was 60-fold greater for tonic conductance than for synaptic conductance. This indicated that higher concentrations of etomidate had a markedly greater effect on tonic conductance than on synaptic conductance under the experimental conditions (Fig. 1B).
Figure 1.

Etomidate selectively enhanced the tonic current recorded in cultured hippocampal pyramidal neurons from Swiss White mice. A, Current traces illustrate the effects of etomidate (0.1 and 1 μm) on the tonic current and mIPSCs. B, The increase in net charge transfer by etomidate was greater for the tonic currents than for time-averaged synaptic currents, as summarized in the bar chart. The low concentration of etomidate (0.1 μm) caused no increase in charge transfer associated with the mIPSCs.
Table 1.
Effects of etomidate on spontaneous mIPSCs
| Group | n | Amplitude (pA) | Charge transfer (pA × ms) | Rise (ms) | Decay (ms) | Frequency (Hz) |
|---|---|---|---|---|---|---|
| Swiss | ||||||
| Control | 8 | 32.9 ±3.7 | 641.9 ±90.5 | 1.6 ±0.1 | 21.4 ±3.7 | 3.0 ±0.5 |
| ET 100 nm | 8 | 37.8 ±4.6 | 740.7 ±87.1 | 1.7 ±0.1 | 19.9 ±2.7 | 3.3 ±0.4 |
| ET 1 μm | 8 | 39.7 ±2.7 | 1085.2* ±94.6 | 1.8 ±0.1 | 24.8* ±2.9 | 4.7 ±1.1 |
| WT | ||||||
| Control | 8 | 46.8 ±4.9 | 1219.6 ±185.8 | 2.4 ±0.2 | 26.1 ±2.7 | 0.3 ±0.1 |
| ET 100 nm | 8 | 43.1 ±3.9 | 1339.8 ±188.1 | 2.5 ±0.1 | 32.2 ±3.6 | 0.3 ±0.1 |
| ET 1 μm | 3 | 45.0 ±3.2 | 1813.7 ±153.2 | 2.4 ±0.2 | 52.4* ±8.1 | 0.2 ±0.1 |
| α5−/− | ||||||
| Control | 10 | 44.3 ±5.1 | 1154.6 ±154.6 | 2.5 ±0.2 | 24.8 ±2.4 | 0.2 ±0.1 |
| ET 100 nm | 10 | 43.2 ±3.9 | 1386.8 ±187.6 | 2.3 ±0.1 | 34.8 ±4.2 | 0.1 ±0.0 |
| ET 1 μm | 4 | 55.7 ±3.0 | 2413.4* ±394.1 | 1.9 ±0.1 | 70.6* ±21.8 | 0.2 ±0.1 |
*p < 0.05 compared with control by the one-way ANOVA test. ET, Etomidate.
To determine whether the etomidate-enhanced tonic conductance was generated by α5GABAARs, currents were recorded from neurons obtained from genetically modified mice that lacked the α5 subunit. We anticipated that low concentrations of etomidate would minimally increase the holding current in α5−/− neurons relative to WT neurons, whereas higher concentrations of etomidate would cause similar enhancement of synaptic currents in the two groups. The change in the holding current (rather than the percentage change in the tonic current) was measured, because the tonic current was minimal in α5−/− neurons (Caraiscos et al., 2004a). These studies were also undertaken to determine whether there was a compensatory upregulation of other high-affinity GABAARs, which might be sensitive to low concentrations of etomidate in α5−/− neurons and whether there were differences in the etomidate sensitivity of synaptic currents in WT and α5−/− neurons.
Etomidate (0.1 μm) increased the holding current in WT neurons (29.7 ± 13.9 pA; n = 7) but not α5−/− neurons (4.2 ± 2.2 pA; n = 7) (p < 0.05) (Fig. 2A,B). No differences were detected in the frequency, amplitude, or time course of mIPSCs between WT and α5−/− neurons in the absence or presence of etomidate (0.1 μm) (Table 1). Thus, low concentrations of etomidate that are reported to be clinically relevant (0.05–0.43 μm) (Rudolph and Antkowiak, 2004) caused a greater enhancement of the tonic current than the mIPSCs. Higher concentrations of etomidate caused a further increase in the holding current and prolonged the mIPSCs; however, this action could be attributed to an indiscriminate effect of etomidate on GABAARs that do not contain the α5 subunit.
Figure 2.

Low concentrations of etomidate increased the holding current in WT but not α5−/− neurons. A, Current traces illustrate the increase in an inward current at different concentrations of etomidate in WT neurons (top trace) and α5−/− neurons (bottom trace). Note the difference in amplitude of the scale bars. B, The bar chart shows that the amplitude of the holding current was greater in WT than in α5−/− neurons. Note that the scale of the y-axis was increased for 1.0 and 10 μm etomidate.
Etomidate reduced LTP in WT but not α5−/− brainslices
LTP is an activity-dependent strengthening of synaptic efficacy at excitatory synapses. LTP is widely regarded as a possible cellular model of learning and memory (Bliss and Collingridge, 1993; Malenka and Bear, 2004). GABAergic inhibition regulates LTP as GABAAR antagonists enhance LTP, whereas positive allosteric modulators reduce LTP and impair memory (Seabrook et al., 1997). Our aim was to determine whether etomidate differentially modulated the LTP of fEPSPs recorded at CA1 pyramidal neurons from WT and α5−/− mice.
As reported previously (Collinson et al., 2002), no differences in LTP were observed between vehicle-treated WT and α5−/− slices immediately after TBS (191 ± 12 vs 208 ± 12%; n = 6 slices per condition; p > 0.05) or 60 min after TBS (170 ± 15 vs 175 ± 11%; n = 6; p > 0.05) (Fig. 3A,B). Etomidate-treated (1 μm) slices from WT mice showed an initial increase in the slope of the fEPSPs (178 ± 16% of control; n = 6 slices) immediately after TBS; however, the increase was not sustained, and the slope of the fEPSPs decreased to 103 ± 13% of control at 60 min (n = 6; p < 0.05). Similarly, the LTP of fEPSPs was initially observed in etomidate-treated (1 μm) slices from α5−/− mice, and the slope of the fEPSPs increased to 192 ± 17% of control (n = 6; p > 0.05) (Fig. 3A,B) immediately after TBS. Unlike the recordings in WT slices, enhancement of the slope of the fEPSPs in α5−/− slices was sustained at 60 min after TBS (168 ± 14% of control; n = 6; p < 0.05) (Fig. 3B) in the presence of etomidate (1 μm). Thus, the attenuation of LTP by etomidate was absent in slices from α5−/− mice.
Figure 3.

Etomidate reduced the LTP of fEPSPs recorded in the CA1 region of hippocampal slices prepared from WT but not α5−/− mice. A, The superimposed traces show averaged fEPSPs recorded from WT mice in the presence of vehicle or etomidate (1 μm) at baseline (1) and 60 min after TBS (2). The time-dependent change in the slope of the fEPSPs recorded in the absence or presence of etomidate (1 μm) is summarized in the graphs. B, Current traces and time-dependent changes in the slope of the fEPSPs for recordings in hippocampal slices from α5−/− mice are shown. Etomidate (1 μm) significantly reduced LTP elicited by 1 s of TBS in pyramidal neurons from WT mice (n = 6) but not α5−/− mice (n = 6) at CA1-Schaffer collateral synapses.
We also measured the amplitude of the tonic current generated by etomidate (1 μm) in CA1 pyramidal neurons in hippocampal slices under voltage-clamp conditions. As observed in recordings from cultured hippocampal neurons, etomidate caused a multiple-fold greater increase in the tonic current in CA1 pyramidal neurons from the WT mice, compared with α5−/− mice (26.4 ± 4.9 pA, n = 10 vs 3.4 ± 1.6 pA, n = 10, respectively; p < 0.05).
Amnestic effects of etomidate are mediated byα5GABAARs
The electrophysiological experiments showed that low concentrations of etomidate enhanced the tonic current and reduced LTP in pyramidal neurons from WT but not α5/− mice. These findings predicted that the amnestic effect of etomidate would be attenuated in α5−/− mice. To study the effects of etomidate on hippocampal-dependent memory, two complementary behavioral assays were performed, a contextual fear conditioning paradigm and the matching-to-place version of the Morris water maze.
Contextual fear conditioning was used to measure the ability of the mice to learn and remember an association between an adverse experience and environmental cues (Fanselow, 1980). WT and α5−/− mice treated with the vehicle exhibited similar freezing behavior (83.1 ± 3.7% of time spent freezing, n = 8 vs 86.9 ± 2.2%, n = 9; p > 0.05) (Fig. 4A). In contrast, etomidate (4 mg/kg, i.p.) reduced the freezing scores in WT but not α5 −/− mice (52.7 ± 5.0%, n = 8 vs 78.8 ± 5.1%, n = 9; p < 0.05) (Fig. 4A). These findings indicate that etomidate impaired the acquisition of contextual fear in WT but not α5−/− mice. To ensure that the reduced etomidate sensitivity exhibited by the α5−/− mice was not a result of a nonspecific insensitivity to general anesthetics, the effect of another anesthetic that does not target GABAARs was tested. The dissociative anesthetic ketamine is thought to cause amnesia by inhibiting the NMDA subtype of glutamate receptors. As shown previously, the vehicle had no effect in WT and α5−/− mice (76.9 ± 5.5% of time spent in freezing behavior, n = 7 vs 82.2 ± 4.6%, n = 7; p > 0.05), whereas ketamine (20 mg/kg, i.p.) caused impairment in both WT and α5−/− mice, as shown by a similar reduction in freezing scores (WT mice, 34.8 ± 8.7% of the time, n = 8, p < 0.05; α5−/− mice, 35.8 ± 5.7% of the time, n = 8, p < 0.05) (Fig. 4A,B).
Figure 4.
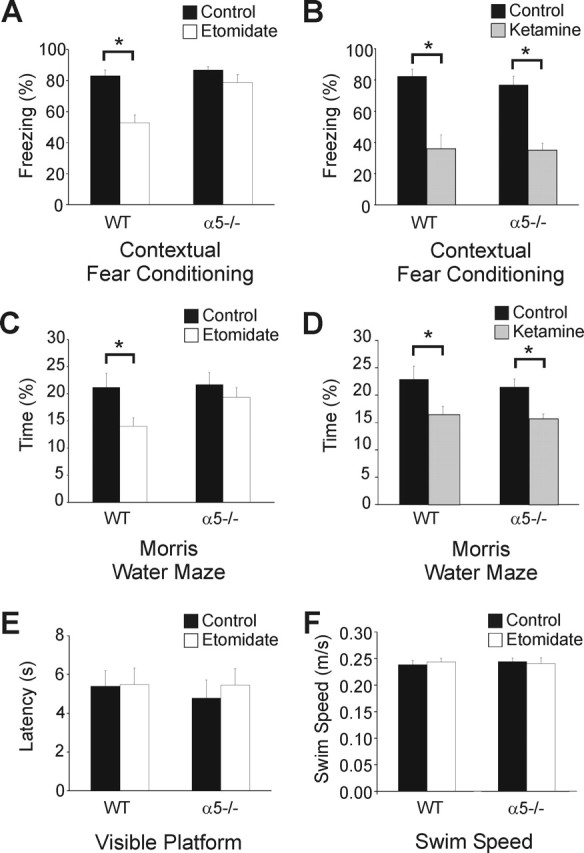
Etomidate impaired contextual fear conditioning in WT but not α5−/− mice. A, The bar graphs show the effects of vehicle (black) and etomidate (white; 4 mg/kg, i.p.) on the freezing scores (mean ± SEM). The scores were reduced in the WT but not the α5−/− mice, which indicates impairment of memory acquisition after the administration of etomidate. B, Compared with vehicle control (black), ketamine (white; 20 mg/kg, i.p., gray) caused a similar reduction in freezing scores, which indicates a similar impairment in fear conditioning for the same context. C, Spatial learning was impaired by etomidate (4 mg/kg, i.p.) in WT but not α5−/− mice. The Morris water maze probe trial showed that etomidate (black) reduced the amount of time the mice spent in the area where the platform had been located the previous day. Impaired memory retrieval was shown by WT but not α5−/− mice. D, In contrast, impairment by ketamine did not depend on the mouse genotype. E, No differences were detected in the visible platform trial (E) or swim speed (F) between the two genotypes (WT control, 0.24 ± 0.01 m/s, n = 16 vs WT etomidate, 0.24 ± 0.01 m/s, n = 15, p > 0.05; α5−/− control, 0.24 ± 0.02 m/s, n = 15 vs α5−/− etomidate, 0.24 ± 0.2 m/s, n = 15, p > 0.05).
The Morris water maze was next used as an independent test of spatial learning. Etomidate (4 mg/kg, i.p.) impaired memory performance in WT mice, as shown by the reduced percentage of time that mice spent swimming in the area that had previously contained the hidden platform (control, 21.2 ± 2.6% of time spent in the correct location vs etomidate, 14.0 ± 1.6%, n = 16; p < 0.05) (Fig. 4C). The reduction in performance was not exhibited by the etomidate-treated α5−/− mice (control, 21.7 ± 2.3% vs etomidate, 19.3 ± 1.8%, n = 16; p > 0.05) (Fig. 4C). We also tested the effects of ketamine on the ability of the mice to locate the platform during a probe trial. Ketamine (20 mg/kg, i.p.) caused a similar impairment of spatial learning in WT mice (control, 22.1 ± 2.7% vs ketamine, 15.4 ± 2.2%, n = 17; p < 0.05) and α5−/− mice (control, 21.5 ± 2.1 vs 15.0 ± 1.8%; n = 15; p < 0.05) (Fig. 4D).
Visible platform trials were performed to test for possible genotype differences in motivational factors, perceptual and motor abilities, and nonspecific effects of etomidate. The platform that was usually submerged was raised to 0.5 cm above the waterline and marked with a brightly colored flag. The mice were injected with vehicle or etomidate (4 mg/kg, i.p.) 30 min before the visible platform trial. There were no differences in the latency to locate the platform between the two genotypes, in the absence or presence of etomidate (WT control, 5.4 ± 0.8 s, n = 8 vs WT etomidate, 5.5 ± 0.9 s, n = 8, p > 0.05; α5−/− control, 4.8 ± 0.9 s, n = 8 vs α5−/− etomidate, 5.5 ± 0.8 s, n = 8, p > 0.05) (Fig. 4E). Also, no difference in mean swimming speed was detected between the genotypes during pretraining, the acquisition trial (WT control, 0.23 ± 0.01 m/s, n = 16 vs WT etomidate, 0.22 ± 0.01 m/s, n = 15, p > 0.05; α5−/− control, 0.20 ± 0.02 m/s, n = 15 vs α5−/− etomidate, 0.20 ± 0.2 m/s, n = 15, p > 0.05) or the probe trials (Fig. 4F). The lack of difference in swim speed (in the absence or presence of etomidate) for both groups confirmed the procedural ability of the mice to perform the task.
Sedative-hypnotic effects of etomidate are not mediated byα5GABAARs
Performance on the rotarod measures sensorimotor control and coordination and is thought to depend primarily on neuronal circuits in the cerebellum and spinal cord. To ensure there were no differences in sensorimotor function in WT and α5−/− mice at the dose of etomidate and time points that were selected for the behavioral experiments, we examined impairment of walking on the rotarod. Five minutes after the injection of etomidate, both groups showed a similar reduction in the latency to fall off the rotarod (Fig. 5A). No impairment of performance was detected 30 min after etomidate (4 mg/kg, i.p.), which was the dose selected for the fear conditioning and Morris water maze experiments.
Figure 5.

Etomidate impairment of motor coordination, spontaneous motor activity, and LORR was not increased in α5−/− mice. A, Etomidate (4 mg/kg) caused impairment of motor performance in the 12 rpm rotarod test. One preinjection trial was performed on the test day before the mice were treated with etomidate (WT, n = 16; α5−/−, n = 15). The results are expressed as the latency to fall off the rotarod (mean ± SEM). Both groups had impaired responses 5 min after injection (*p > 0.05, one-way ANOVA) but not at 30 or 60 min. B, Spontaneous locomotor activity (walking) was reduced by etomidate as shown for the open field test. No difference in baseline activity was observed between WT and α5−/− mice after injection of the vehicle control. Etomidate (4 mg/kg and 10 mg/kg, i.p.) caused a concentration-dependent reduction in locomotion that was similar in WT and α5−/− mice. C, Dose–response analysis for etomidate causing the LORR is shown. The data points represent the percentage of the WT (filled square) or α5−/− (open square) mouse populations that exhibited LORR at the dose indicated on the abscissa. Overlapping data points for the two genotypes are represented by a third symbol (filled circle). Mice were tested at doses of 5, 7.5, 10, 12.5, 15, and 20 mg/kg, and a total of 69 mice were studied. The fitted curves were generated using a sigmoidal equation that provided the following values: WT ED50 = 9.6 mg/kg ± 1.1, Hill slope parameter h = 5.8 ± 1.8; α5−/− ED50 = 9.2 ± 1.1, h = 5.7 ± 1.5, p < 0.05. D, The latency to LORR recorded from the time of etomidate injection. Error bars show the mean ± SEM. Each data point represents six to eight mice, and no differences were detected between WT and α5−/− mice. The time to LORR was significantly different at all doses of etomidate (ANOVA; p < 0.05) with the exception of 10 versus 12.5 mg/kg.
The sedative and hypnotic properties of etomidate were studied as a positive control by measuring spontaneous motor activity and the LORR, respectively. These behavioral endpoints were unlikely to be mediated by hippocampal neurons or α5GABAARs (Nelson et al., 2002). The sedative properties of etomidate were studied with an open field test, which examines spontaneous locomotor activity after injection of etomidate or vehicle control. Locomotion was similar in WT and α5−/− mice 30 min after the vehicle (Fig. 5B). Etomidate (4 and 10 mg/kg) caused a similar reduction in spontaneous movement at 30 min in WT and α5−/− mice as shown in Figure 5B.
It is widely accepted that a surrogate experimental measure for anesthetic-induced unconsciousness is the LORR (Rudolph and Antkowiak, 2004). The ability of a wide range of etomidate doses to cause the LORR was determined for WT and α5−/− mice. A dose–response plot for LORR showed that the dose of etomidate that caused half the maximal response was similar in WT and α5−/− mice (9.6 and 9.2 mg/kg, n = 35 and 34, respectively; p < 0.05). The time to LORR was also measured after injection of the various doses of etomidate; no differences were detected between the WT and α5−/− mice. Together, the above results show that the α5−/− mice do not exhibit a resistance to etomidate for the behavioral endpoints used to measure sedation and loss of consciousness.
The brain concentration of etomidate was similar in WT and α5−/−mice
To determine whether differences in the absorption or metabolism contributed to the reduced sensitivity of α5−/− mice to the amnestic effect of etomidate, the brain concentrations of etomidate were measured using HPLC. Etomidate (4 mg/kg, i.p.) was administered and the mice were killed at the same time point (30 min) as for the fear conditioning and Morris water maze experiments. No differences were detected in the brain concentration of etomidate in WT and α5−/− neurons (0.74 ± 0.21 μm, n = 4 vs 0.79 ± 0.33 μm, n = 4; p > 0.05). No etomidate was detected in the brains of vehicle-treated WT (n = 4) or α5−/− mice (n = 4). Thus, differences in the behavioral performance of WT and α5−/− mice could not be attributed to a difference in the brain concentration of etomidate. Moreover, the concentration of etomidate that caused amnesia in mice in vivo was similar to the etomidate concentration that preferentially enhanced the tonic conductance in vitro and reduced LTP (Fig. 1B,C).
Discussion
The absence of the α5 subunit resulted in decreased sensitivity to etomidate for impairment of learning and memory performance. The reduced sensitivity of α5−/− mice to etomidate could not be attributed to differences in drug pharmacokinetics, because brain concentrations of etomidate were similar in WT and α5−/− mice. The resistance of the α5−/− mice was specific for memory performance, because deletion of the α5 subunit did not influence the effect of etomidate on the LORR or spontaneous motor activity. Also, sensorimotor function, coordination, motor learning, perception, and motivation were similar in WT and α5−/− mice, in the absence and presence of etomidate, as evidenced by the rotarod and open field tests. These results provide evidence for a selective population of GABAARs that contributes to the amnestic but not sedative-hypnotic effects of a general anesthetic. The findings support the hypothesis that etomidate acts on different GABAAR subtypes, located in different neuronal circuits, to generate the multiple components of the anesthetic state (Eger et al., 1997).
Electrophysiological studies showed that a low concentration of etomidate primarily enhanced a tonic conductance in pyramidal neurons, whereas higher etomidate concentrations further increased the tonic conductance and prolonged the duration of mIPSCs in WT and α5−/− neurons. The free aqueous concentration of etomidate associated with amnesia in the mammalian brain has been estimated at ∼0.43 μm (Rudolph and Antkowiak, 2004). This value is based on the concentration of etomidate that produces immobility in half of subjects (1.5 μm) and studies that suggest amnesia occurs at ∼20–40% of the immobilizing concentration of anesthetics (Dwyer et al., 1992). The brain concentration of etomidate in the WT and α5−/− mice, measured at the dose and time point selected for the Morris water maze acquisition trial and fear conditioning, was 0.77 μm. A similar concentration of etomidate predominantly enhanced the tonic inhibitory conductance in vitro.
The above results are consistent with our previous report that showed that low concentrations of the inhaled anesthetic isoflurane selectively enhanced a tonic but not synaptic conductance in cultured hippocampal neurons (Caraiscos et al., 2004b). Isoflurane was used in the previous study, because the concentration of an inhaled anesthetic in the CNS, which causes specific behavioral effects, can be relatively accurately determined (Hemmings et al., 2005). Inhaled anesthetics readily diffuse from the alveoli into the capillary blood and equilibrate such that, at steady state, the concentration in the end-expired gas would be equivalent to the concentration in the CNS. Thus, the concentration of the inhaled anesthetic in the brain can be accurately estimated by measuring the concentration in the exhaled gas. Results from a previous in vitro study (Caraiscos et al., 2004b) stimulated the hypothesis that potentiation of α5GABAAR contributes to the amnestic properties of anesthetics. In the present study, we used both electrophysiological and behavioral assays and selected an injectable anesthetic rather than isoflurane because of the obvious difficulties in administering an inhaled anesthetic during behavioral testing. This is particularly true for the Morris water maze, a paradigm that is widely used for investigating the pharmacologic aspects of memory and learning. The results show that activation of α5GABAA receptors contributes to the amnestic effects of etomidate. Moreover, the amnestic and sedative-hypnotic effects of an anesthetic can be dissociated on the basis of the pharmacologic properties of the GABAA receptor subunits.
To further examine the cellular mechanisms underlying memory impairment by etomidate, EPSPs were recorded in CA1 stratum radiatum. In the absence of etomidate, pyramidal neurons in hippocampal slices showed no differences in the induction or maintenance of LTP in WT relative to α5−/− mice as reported previously (Collinson et al., 2002). The induction of LTP by high-frequency stimuli and maximal fEPSP strength were similar in WT and α5−/− mice (Collinson et al., 2002). The authors also reported no differences in the ability of low-frequency stimuli to activate fEPSPs in CA1 or the dentate gyrus; however, the ability of paired-pulse stimuli to facilitate the amplitude of synaptic potentials was significantly increased in α5−/− mice. Increased paired-pulse facilitation was specific to the CA1 region and was not observed in the dentate gyrus, where α5GABAARs are not highly expressed. Our results showed that etomidate generated a large tonic conductance and inhibited LTP in WT but not α5−/− slices, an observation that supports the hypothesis that LTP is necessary for learning and memory. The molecular mechanisms underlying this altered excitability by α5GABAARs and modulation of LTP by other classes of GABAergic compounds, including benzodiazepines, will be the subjects of future studies.
Genetic, pharmacological, and electrophysiological studies using mouse models have previously implicated α5GABAARs in learning and memory processes. A knock-in strain of transgenic mice that expressed an α5 subunit point mutation (H105R) showed an unexpected reduction in α5GABAAR expression in pyramidal neurons and improved performance for hippocampal-dependent learning tasks (Crestani et al., 2002). Trace fear conditioning but not delayed conditioning or contextual conditioning was facilitated in the α5(H105R) mice. The α5−/− null mutant mice used in this study have been reported previously to exhibit improved performance in the initial acquisition trials of the Morris water maze (Collinson et al., 2002). We initially tested the effects of etomidate on performance during the acquisition trials in WT and α5−/− mice. The results were highly variable (data not shown). The probe trial, which is often considered the true criterion for the acquisition of the Morris water maze task, showed a consistent difference between genotypes only in the presence of etomidate. The normal performance of the mice in the visible platform task and their impaired performance in the hidden platform task are highly consistent with a deficit in learning and memory caused by etomidate in the WT but not α5−/− mice. To complement the Morris water maze studies, we also examined fear conditioning. We and others report no difference in the baseline performance for contextual fear conditioning (Collinson et al., 2002). However, a reduction in contextual fear was evident after administration of etomidate in WT mice but not α5−/− mice. Contextual fear was reduced similarly in both groups by ketamine, which indicates that the resistance of α5−/− mice to etomidate cannot be attributed to a general resistance to neurodepressive drugs.
The results reported here have implications for human memory processes. In humans and rodents, α5GABAARs exhibit similar structural, kinetic, and pharmacological properties and similar patterns of expression (Faure-Halley et al., 1993; Barria et al., 1997; Sur et al., 1999; Wainwright et al., 2000). Compounds that selectively reduce α5GABAAR function in animal models are currently being investigated in humans as potentially nootropic compounds (Chambers et al., 2003). Our findings suggest that drugs that selectively increase the function of α5GABAARs may also have a therapeutic role. There is a need for drugs that cause amnesia without sedation or unconsciousness. Such compounds may be of value for patients whose condition is too unstable to allow adequate doses of currently available anesthetics or to prevent the formation of unpleasant memories. Furthermore, our results raise the intriguing possibility of a genetic basis for some cases of intraoperative awareness in humans. Unpleasant recall of intraoperative events can occur despite a depth of anesthesia that is associated with lack of movement (Sandin et al., 2000). Polymorphisms occurring for the human α5 subunits are associated with a reduction in receptor function (Papadimitriou et al., 2001a,b). Moreover, the α5 subunit is downregulated under certain pathological conditions, including epilepsy (Houser and Esclapez, 2003). A reduction in number or function of α5GABAARs may predispose patients to intraoperative awareness. Alternatively, it is possible that anesthetic effects on α5GABAARs contribute to the postoperative cognitive and memory dysfunction that occurs in ∼25% of elderly patients (Moller et al., 1998). Animal studies have shown that subtle long-term deficits in memory performance persisted for weeks after general anesthesia (Culley et al., 2003, 2004). Drugs that selectively reduce α5GABAAR function may have benefits in the memory disorders that occur after general anesthesia.
In conclusion, mice with a null mutation of the α5 subunit of the GABAAR exhibited reduced sensitivity to the amnestic but not the sedative-hypnotic effects of etomidate. Amnesia was likely caused by a direct interaction between etomidate and α5GABAARs rather than an indirect effect of α5 subunit deletion, because memory impairment by ketamine was similar in α5−/− and WT mice. The results contribute growing evidence that α5GABAARs play a central role in learning and memory processes. Additionally, increased tonic inhibition generated by α5GABAAR may contribute to amnesia caused by GABAergic drugs.
Footnotes
*V.Y.C. and L.J.M. contributed equally to this work.
This work was supported by a Canada Research Chair, Canadian Institutes of Health Research grant, and Premier's Research Excellence Award (B.A.O.). K.A.W. and N.C. are employed by Merck Sharpe and Dohme Limited. B.A.O. is an advisor to Merck Sharpe and Dohme Limited. We sincerely thank Drs. James Sonner and Michael Laskin (Department of Anesthesia, University of California, San Francisco, CA) for performing the HPLC measurements of etomidate concentrations.
References
- Bai D, Zhu G, Pennefather P, Jackson MF, MacDonald JF, Orser BA (2001). Distinct functional and pharmacological properties of tonic and quantal inhibitory postsynaptic currents mediated by γ-aminobutyric acidA receptors in hippocampal neurons. Mol Pharmacol 59:814–824. [DOI] [PubMed] [Google Scholar]
- Barria A, Derkach V, Soderling T (1997). Identification of the Ca2+/calmodulin-dependentprotein kinase II regulatory phosphorylation site in the α-amino-3-hydroxyl-5-methyl-4-isoxazole-propionate-type glutamate receptor. J Biol Chem 272:32727–32730. [DOI] [PubMed] [Google Scholar]
- Bliss TV, Collingridge GL (1993). A synaptic model of memory: long-term potentiation in the hippocampus. Nature 361:31–39. [DOI] [PubMed] [Google Scholar]
- Campagna JA, Miller KW, Forman SA (2003). Mechanisms of actions of inhaled anesthetics. N Engl J Med 348:2110–2124. [DOI] [PubMed] [Google Scholar]
- Caraiscos VB, Elliott EM, You-Ten KE, Cheng VY, Belelli D, Newell JG, Jackson MF, Lambert JJ, Rosahl TW, Wafford KA, MacDonald JF, Orser BA (2004a). Tonic inhibition in mouse hippocampal CA1 pyramidal neurons is mediated by α5 subunit-containing γ-aminobutyric acid type A receptors. Proc Natl Acad Sci USA 101:3662–3667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caraiscos VB, Newell JG, You-Ten KE, Elliott EM, Rosahl TW, Wafford KA, MacDonald JF, Orser BA (2004b). Selective enhancement of tonic GABAergic inhibition in murine hippocampal neurons by low concentrations of the volatile anesthetic isoflurane. J Neurosci 24:8454–8458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers MS, Atack JR, Broughton HB, Collinson N, Cook S, Dawson GR, Hobbs SC, Marshall G, Maubach KA, Pillai GV, Reeve AJ, MacLeod AM (2003). Identification of a novel, selective GABAA α5 receptor inverse agonist which enhances cognition. J Med Chem 46:2227–2240. [DOI] [PubMed] [Google Scholar]
- Collinson N, Kuenzi FM, Jarolimek W, Maubach KA, Cothliff R, Sur C, Smith A, Otu FM, Howell O, Atack JR, McKernan RM, Seabrook GR, Dawson GR, Whiting PJ, Rosahl TW (2002). Enhanced learning and memory and altered GABAergic synaptic transmission in mice lacking the α5 subunit of the GABAA receptor. J Neurosci 22:5572–5580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crestani F, Keist R, Fritschy JM, Benke D, Vogt K, Prut L, Bluthmann H, Mohler H, Rudolph U (2002). Trace fear conditioning involves hippocampal α5 GABAA receptors. Proc Natl Acad Sci USA 99:8980–8985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Culley DJ, Baxter M, Yukhananov R, Crosby G (2003). The memory effects of general anesthesia persist for weeks in young and aged rats. Anesth Analg 96:1004–1009. [DOI] [PubMed] [Google Scholar]
- Culley DJ, Baxter MG, Yukhananov R, Crosby G (2004). Long-term impairment of acquisition of a spatial memory task following isoflurane-nitrous oxide anesthesia in rats. Anesthesiology 100:309–314. [DOI] [PubMed] [Google Scholar]
- Dwyer R, Bennett HL, Eger EI, Heilbron D (1992). Effects of isoflurane and nitrous oxide in subanesthetic concentrations on memory and responsiveness in volunteers. Anesthesiology 77:888–898. [DOI] [PubMed] [Google Scholar]
- Eger EI, Koblin DD, Harris RA, Kendig JJ, Pohorille A, Halsey MJ, Trudell JR (1997). Hypothesis: inhaled anesthetics produce immobility and amnesia by different mechanisms at different sites. Anesth Analg 84:915–918. [DOI] [PubMed] [Google Scholar]
- Fanselow MS (1980). Conditioned and unconditional components of post-shock freezing. Pavlov J Biol Sci 15:177–182. [DOI] [PubMed] [Google Scholar]
- Faure-Halley C, Graham D, Arbilla S, Langer SZ (1993). Expression and properties of recombinant α1β2γ2 and α5β2γ2 forms of the rat GABAA receptor. Eur J Pharmacol 246:283–287. [DOI] [PubMed] [Google Scholar]
- Franks NP, Lieb WR (1988). Volatile general anaesthetics activate a novel neuronal K+ current. Nature 333:662–664. [DOI] [PubMed] [Google Scholar]
- Gredell JA, Turnquist PA, MacIver MB, Pearce RA (2004). Determination of diffusion and partition coefficients of propofol in rat brain tissue: implications for studies of drug action in vitro. Br J Anaesth 93:810–817. [DOI] [PubMed] [Google Scholar]
- Hemmings HC Jr, Akabas MH, Goldstein PA, Trudell JR, Orser BA, Harrison NL (2005). Emerging molecular mechanisms of general anesthetic action. Trends Pharmacol Sci 26:503–510. [DOI] [PubMed] [Google Scholar]
- Houser CR, Esclapez M (2003). Downregulation of the α5 subunit of the GABAA receptor in the pilocarpine model of temporal lobe epilepsy. Hippocampus 13:633–645. [DOI] [PubMed] [Google Scholar]
- Jurd R, Arras M, Lambert S, Drexler B, Siegwart R, Crestani F, Zaugg M, Vogt KE, Ledermann B, Antkowiak B, Rudolph U (2003). General anesthetic actions in vivo strongly attenuated by a point mutation in the GABAA receptor β3 subunit. FASEB J 17:250–252. [DOI] [PubMed] [Google Scholar]
- MacDonald JF, Mody I, Salter MW (1989). Regulation of N-methyl-d-aspartate receptors revealed by intracellular dialysis of murine neurones in culture. J Physiol (Lond) 414:17–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIntosh MP, Rajewski RA (2001). A simple and efficient high performance liquid chromatographic assay for etomidate in plasma. J Pharm Biomed Anal 24:689–694. [DOI] [PubMed] [Google Scholar]
- Malenka RC, Bear MF (2004). LTP and LTD: an embarrassment of riches. Neuron 44:5–21. [DOI] [PubMed] [Google Scholar]
- Moller JT, Cluitmans P, Rasmussen LS, Houx P, Rasmussen H, Canet J, Rabbitt P, Jolles J, Larsen K, Hanning CD, Langeron O, Johnson T, Lauven PM, Kristensen PA, Biedler A, van Beem H, Fraidakis O, Silverstein JH, Beneken JE, Gravenstein JS (1998). Long-term postoperative cognitive dysfunction in the elderly ISPOCD1 study. ISPOCD investigators. International Study of Post-Operative Cognitive Dysfunction. Lancet 351:857–861. [DOI] [PubMed] [Google Scholar]
- Nelson LE, Guo TZ, Lu J, Saper CB, Franks NP, Maze M (2002). The sedative component of anesthesia is mediated by GABAA receptors in an endogenous sleep pathway. Nat Neurosci 5:979–984. [DOI] [PubMed] [Google Scholar]
- Otis TS, Staley KJ, Mody I (1991). Perpetual inhibitory activity in mammalian brain slices generated by spontaneous GABA release. Brain Res 545:142–150. [DOI] [PubMed] [Google Scholar]
- Papadimitriou G, Dikeos D, Daskalopoulou E, Karadima G, Avramopoulos D, Contis C, Stefanis C (2001a). Association between GABAA receptor α5 subunit gene locus and schizophrenia of a later age of onset. Neuropsychobiology 43:141–144. [DOI] [PubMed] [Google Scholar]
- Papadimitriou GN, Dikeos DG, Karadima G, Avramopoulos D, Daskalopoulou EG, Stefanis CN (2001b). GABAA receptor β3 and α5 subunit gene cluster on chromosome 15q11–q13 and bipolar disorder: a genetic association study. Am J Med Genet 105:317–320. [DOI] [PubMed] [Google Scholar]
- Reynolds DS, Rosahl TW, Cirone J, O'Meara GF, Haythornthwaite A, Newman RJ, Myers J, Sur C, Howell O, Rutter AR, Atack J, Macaulay AJ, Hadingham KL, Hutson PH, Belelli D, Lambert JJ, Dawson GR, McKernan R, Whiting PJ, Wafford KA (2003). Sedation and anesthesia mediated by distinct GABAA receptor isoforms. J Neurosci 23:8608–8617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudolph U, Antkowiak B (2004). Molecular and neuronal substrates for general anaesthetics. Nat Rev Neurosci 5:709–720. [DOI] [PubMed] [Google Scholar]
- Sandin RH, Enlund G, Samuelsson P, Lennmarken C (2000). Awareness during anaesthesia: a prospective case study. Lancet 355:707–711. [DOI] [PubMed] [Google Scholar]
- Seabrook GR, Easter A, Dawson GR, Bowery BJ (1997). Modulation of long-term potentiation in CA1 region of mouse hippocampal brain slices by GABAA receptor benzodiazepine site ligands. Neuropharmacology 36:823–830. [DOI] [PubMed] [Google Scholar]
- Sebel PS, Bowdle TA, Ghoneim MM, Rampil IJ, Padilla RE, Gan TJ, Domino KB (2004). The incidence of awareness during anesthesia: a multicenter United States study. Anesth Analg 99:833–839. [DOI] [PubMed] [Google Scholar]
- Semyanov A, Walker MC, Kullmann DM, Silver RA (2004). Tonically active GABAA receptors: modulating gain and maintaining the tone. Trends Neurosci 27:262–269. [DOI] [PubMed] [Google Scholar]
- Sur C, Fresu L, Howell O, McKernan RM, Atack JR (1999). Autoradiographic localization of α5 subunit-containing GABAA receptors in rat brain. Brain Res 822:265–270. [DOI] [PubMed] [Google Scholar]
- Wainwright A, Sirinathsinghji DJ, Oliver KR (2000). Expression of GABAA receptor α5 subunit-like immunoreactivity in human hippocampus. Brain Res Mol Brain Res 80:228–232. [DOI] [PubMed] [Google Scholar]
- Wu Y, Wang W, Richerson GB (2003). Vigabatrin induces tonic inhibition via GABA transporter reversal without increasing vesicular GABA release. J Neurophysiol 89:2021–2034. [DOI] [PubMed] [Google Scholar]
- Yeung JY, Canning KJ, Zhu G, Pennefather P, MacDonald JF, Orser BA (2003). Tonically activated GABAA receptors in hippocampal neurons are high-affinity, low-conductance sensors for extracellular GABA. Mol Pharmacol 63:2–8. [DOI] [PubMed] [Google Scholar]