

Abstract
The neuropeptide corticotropin-releasing factor (CRF) is believed to play a role in a number of psychiatric conditions, including anxiety disorders and depression. In the present study, male Sprague Dawley rats were used to examine the behavioral effects of altering dopamine transmission on CRF-enhanced startle, a behavioral assay believed to reflect stress- or anxiety-like states. Systemic administration of the selective dopamine D1 receptor antagonist SCH 23390 [R(+)-7-chloro-8-hydroxy-3-methyl-1-phenyl-2,3,4,5-tetrahydro-1H-3-benzazepine hydrochloride] (0, 0.01, 0.05, 0.1, 0.5 mg/kg) dose dependently blocked the effect of CRF (1 μg, i.c.v.) on startle at doses that had no effect on baseline startle response. Immunohistochemical studies showed that most CRF-containing cells in the dorsolateral division of the bed nucleus of the stria terminalis (BSTld), part of the critical brain area mediating CRF-enhanced startle, are surrounded by a dense plexus of tyrosine hydroxylase (TH)-positive fibers. Intra-BSTld injections of the retrograde tracer Fluorogold (FG) into the TH field identified neurons in the major dopaminergic areas (A8-A10), but not the major noradrenergic areas [A5, A6 (locus ceruleus), A7], as a significant source of TH-positive innervation. The majority of FG-filled cells double-labeled for TH were found in the dorsocaudal A10 cell group (A10dc) located in the periaqueductal gray area. Together, these data suggest that neuronal regulation of the BSTld by specific dopaminergic pathways and receptors may be an important mechanism for controlling CRF-dependent moods and affective states. These data also suggest that compounds with D1 receptor antagonist properties might have anxiolytic-like effects that could be useful for treating conditions associated with hyperactive CRF systems.
Keywords: anxiety, CRH, BNST, SCH 23390, startle, D1
Introduction
Stress has been implicated in both the development and manifestation of such psychiatric illnesses as anxiety and depression (Heim and Nemeroff, 1999), highly comorbid disorders (Kessler et al., 2005) that may share common elements of an underlying pathophysiology (Boyer, 2000). As such, the neural substrates activated by stress have become the focus of numerous clinical and preclinical studies (Vermetten and Bremner, 2002a,b). Central to many of these studies is the role of the neuropeptide corticotropin-releasing factor (CRF) in mediating both the neuroendocrine and CNS responses to stress. In animals, brain CRF levels are increased under stress (Chappell et al., 1986), and it is well established that intracerebroventricular infusion of CRF elicits many of the same endocrine, physiological, and behavioral changes evoked by a variety of stressors (Dunn and Berridge, 1990).
As with CRF, catecholamine [i.e., norepinephrine (NE) and dopamine (DA)] levels are increased in the brain after stress (Finlay et al., 1995). Although many reports have suggested that NE-CRF interactions might play a role in the adaptive and maladaptive responses to stress (Gold and Chrousos, 2002), less is known about the interaction between DA and CRF in mediating stress effects. Potential sites of convergence between these systems may exist within the central nucleus of the amygdala (CeA) and the bed nucleus of the stria terminalis (BST), interconnected structures of the extended amygdala that share similar cytoarchitecture and afferent/efferent connections (Alheid et al., 1995). Both areas have a high density of CRF-containing neurons (Cassell et al., 1986; Moga et al., 1989) and receive dopaminergic inputs that make direct synaptic contact with CRF neurons (Phelix et al., 1992; Eliava et al., 2003).
In the present study, we used the enhancement of the acoustic startle reflex by CRF to study DA-CRF interactions in a behavioral assay believed to reflect stress- or anxiety-like states (Davis et al., 1997). CRF-enhanced startle is blocked by CRF antagonists (Schulz et al., 1996) and by drugs that modulate GABAergic systems such as the anxiolytic drug chlordiazepoxide (Swerdlow et al., 1986) or the neurosteroid tetrahydroprogesterone (Toufexis et al., 2004). To date, the involvement of other neurotransmitter systems in CRF-mediated behaviors has not been fully explored. Hence, one goal of the present study was to examine the behavioral effects of treatment with the selective dopamine D1 receptor antagonist SCH 23390 on CRF-enhanced startle. Although studies have shown that both the CeA and BST may be part of the neural circuitry underlying CRF-enhanced startle, only direct infusions of CRF into the BST (Lee and Davis, 1997), and not the CeA (Liang et al., 1992a), have been shown to mimic the effects of intracerebroventricular CRF. Intra-BST infusion of a CRF antagonist has also been shown to reduce CRF-enhanced startle (Lee and Davis, 1997), providing additional evidence that the BST is a critical brain area involved in the stress-like effects of CRF on this behavior. Based on these data, a second goal of the present study was to determine the source of dopaminergic innervation to the BST using retrograde-tracing and immunohistochemical techniques.
Materials and Methods
Animals
The animals were male Sprague Dawley rats (Charles River, Raleigh, NC) weighing ∼400 g and housed in group cages of four rats each until surgery. Animals were maintained on a 12 h light/dark cycle (lights on at 7:00 A.M.) with food and water continuously available. All animal procedures were approved by the Institutional Animal Care and Use Committee of McLean Hospital in strict accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Behavioral studies
Apparatus.
Matching and testing of animals was conducted in four identical startle cages consisting of 19 × 9 × 14 cm Plexiglas and wire-mesh cages attached to a load-cell platform. Both the startle cages and platform were contained within a 69 × 36 × 42 cm ventilated sound-attenuating isolation box with the inside temperature monitored and maintained at ∼20°C (Med Associates, Georgia, VT). Cage movement resulted in displacement of a transducer in the platform where the resultant voltage was amplified and digitized on a scale of 0 to ±2000 U by an analog-to-digital converter card interfaced to a personal computer. Startle amplitude was proportional to the amount of cage movement and defined as the maximum peak-to-peak voltage that occurred during the first 200 ms after onset of the startle stimulus. Constant wide-band background noise (60 dB; 10–20 kHz) and 50 ms startle stimuli (1–32 kHz white noise; 5 ms rise/decay) were generated by an audio stimulator (Med Associates) and delivered through speakers located 7 cm behind the startle cage. The calibration, presentation, and sequencing of all stimuli were under the control of the personal computer using specially designed software (Med Associates).
Intracerebroventricular cannulation.
Rats were anesthetized with Nembutal (50 mg/kg, i.p.) and placed in a Kopf stereotaxic instrument (model 900; Kopf Instruments, Tujunga, CA) with blunt ear bars. The skin was retracted, and a hole was drilled in the skull above the lateral ventricle. Stainless-steel guide cannulas (23 gauge; Plastics One, Roanoke, VA) with an internal dummy stylet extending 1.5 mm beyond the guide cannula tip were lowered into the brain using the following coordinates: −0.8 mm caudal to bregma, +1.3 mm lateral to the midline, −3.5 mm ventral to dura. Three stainless-steel screws (size 0–80; Small Parts, Miami Lakes, FL) were also placed in the skull to anchor the guide cannula, and Loctite adhesive (Loctite, Newington, CT) and dental acrylic (Stoelting, Wood Dale, IL) were used to cement the cannula in place. Animals were placed under a heating lamp, and after recovery, the rats were singly housed in plastic cages (45 × 24 × 20 cm) with wood-shaving bedding.
Procedure
All behavioral tests were conducted between 10:00 A.M. and 4:00 P.M. with counterbalanced treatment groups to control for variations in diurnal rhythms that might affect startle (Frankland and Ralph, 1995).
Matching.
One week after surgery, rats were given a test session to familiarize them to handling, the apparatus, and the startle stimuli. Animals with excessively high or low startle responses (<50, >350 startle units) were not used, because this typically reflects abnormal sensorimotor responding or untoward effects of the surgery. In addition, to examine any effect of R(+)-7-chloro-8-hydroxy-3-methyl-1-phenyl-2,3,4,5-tetrahydro-1H-3-benzazepine hydrochloride (SCH 23390) on the startle reflex itself, it was important to establish experimental groups with equivalent levels of baseline startle before testing. To do this, rats were placed in the cages and given a 5 min acclimation period followed by the presentation of two habituating startle stimuli [100 dB, 30 s interstimulus interval (ISI)]. Rats were then presented with 100 startle stimuli at each of three different intensities (95, 100, and 105 dB) in a semirandom order with a 30 s ISI. Rats were then matched into the different treatment groups having equivalent levels of startle averaged across the three startle intensities (Table 1).
Table 1.
Mean startle amplitude (cage displacement units ± SE) and group size for animals matched into the different treatment conditions
| Intracerebroventricular infusions | Systemic injectionsDose of SCH 23390 (mg/kg, s.c.) |
||||
|---|---|---|---|---|---|
| Vehicle (saline) | 0.01 | 0.05 | 0.1 | 0.5 | |
| Vehicle (aCSF) | 211 ± 27 | 210 ± 21 | 206 ± 32 | 217 ± 24 | 217 ± 24 |
| (n = 7) | (n = 8) | (n = 8) | (n = 7) | (n = 7) | |
| CRF (1 μg) | 214 ± 19 | 213 ± 34 | 212 ± 31 | 212 ± 23 | 210 ± 25 |
| (n = 14) | (n = 8) | (n = 8) | (n = 11) | (n = 7) | |
Testing.
Two days later, rats were returned to the startle testing room and received subcutaneous injections of either vehicle (VEH) (0.9% saline) or SCH 23390 (0.01, 0.05, 0.1, or 0.5 mg/kg, s.c.; Sigma, St Louis, MO). Rats were then placed in individual plastic cages, and their dummy stylettes were removed and replaced with infusion cannulas (30 gauge, 1.5 mm projection from the tip of the guide cannula; Plastics One) attached to Hamilton microsyringes (10 μl) by polyethylene tubing. A Harvard Apparatus (model 22; Harvard Apparatus, Holliston, MA) infusion pump was used to deliver 2 μl of either vehicle [artificial CSF (aCSF); Harvard Bioscience, Holliston, MA] or CRF (0.5 μg/μl; American Peptide Company, Sunnyvale, CA) directly into the lateral ventricle at a rate of 1 μl/min for 2 min. The infusion cannulas were left in place for 1 min after the infusion and then removed and the dummy stylettes replaced. Rats were immediately placed in the startle cages and given a 5 min acclimation period followed by presentation of two habituating startle stimuli (100 dB, 30 s ISI). Rats were then presented with 400 startle stimuli at each of three different intensities (95, 100, and 105 dB) in a semirandom order with a 30 s ISI for a total test session of 200 min.
Histology.
At the end of the experiment, the animals were overdosed with pentobarbitol (130 mg/kg, i.p.) and perfused intracardially with 0.9% saline (200 ml) followed by 4% paraformaldehyde (500 ml). The brains were removed and stored for 3–4 d in a 30% sucrose/0.1 m PBS, pH 7.4, solution and subsequently cut in 40 μm coronal sections. Sections were mounted on microscope slides, stained with cresyl violet, and coverslipped with Permount (Fisher Scientific, Pittsburgh, PA). Verification of accurate cannula placement in the lateral ventricle was assessed under a light microscope by an observer blind to the treatment conditions. As a result, three animals with misplaced cannulas (cannula tips had entered the lateral septum) were excluded from the study. These omissions are accounted for in the matching data, and group size is presented in Table 1.
Statistical analysis.
Startle amplitude data were expressed as the mean averaged across the three startle-eliciting intensities across time. The effect of SCH 23390 on both CRF-enhanced startle (six treatment groups including a VEH-VEH and VEH-CRF group) and baseline startle (five treatment groups including a VEH-VEH group) was evaluated using two separate two-way ANOVA with treatment group as a between-subjects factor and blocks of time (four 50 min blocks) as a within-subjects factor. Subsequent multiple comparisons for significant differences between the VEH-VEH and VEH-CRF groups and every other treatment group at each block of time were made using Dunn's test (Bonferroni-adjusted multiple t test).
Anatomical studies
We used the nomenclature, delineation of structures, and stereotaxic reference system of Paxinos and Watson (1997). Although we used tyrosine hydroxylase (TH)-immunohistochemistry as a general marker for catecholaminergic neurons, the location of both dopaminergic and noradrenergic neurons has been mapped thoroughly (Armstrong et al., 1982; Hokfelt et al., 1984). Hence, major dopaminergic [A8-A10, including the A10 dorsorostral (A10dr) and A10 dorsocaudal (A10dc) group] and noradrenergic areas (A5-A7) were also referred to by their grouping after Hokfelt et al. (1984).
Fluorescent immunohistochemistry.
Double-labeling of CRF-containing cells and TH-positive fibers was performed on 20 μm sections through the BST. All incubations were done on a rocker at room temperature. The sections were preincubated in antibody medium (2% normal donkey serum, 1% bovine serum albumin, 0.3% Triton X-100 in 0.1 m PBS) for 2 h followed by incubation for 24 h with primary antibodies; goat polyclonal antibody against CRF (1:1000; Santa Cruz Biotechnology, Santa Cruz, CA) and rabbit polyclonal antibody against TH (1:4000; Chemicon, Temecula, CA) were diluted in antibody medium. The sections were washed with 0.1 m PBS and incubated for 2 h with two different secondary antibodies (minimum species cross-reactivity; Jackson ImmunoResearch, West Grove, PA): fluorescein isothiocyanate (FITC)-conjugated donkey anti-goat and tetramethyl rhodamine isothiocyanate (TRITC)-conjugated donkey anti-rabbit (1:400 both antibodies in antibody medium). Sections were mounted on microscope slides and coverslipped with DPX Mountant [distyrene, a plasticizer (tricresyl phosphate), and xylene (Fluka, Milwaukee, WI)] for fluorescence microscopy and observed with a Zeiss Axioscope 2 (Zeiss, Oberkochen, Germany) under appropriate filters for FITC (excitation/emission peaks, 492/520) and TRITC fluorescence (excitation/emission peaks, 550/570). Still-frame images were captured with a digital camera (AxioCam; Zeiss) interfaced with a Macintosh G4 computer and analyzed for double labeling of CRF-positive cells and TH-positive fibers in the BST using Adobe Photoshop software (Adobe Systems, San Jose, CA).
Fluorogold injections.
Rats were anesthetized with Nembutal (50 mg/kg, i.p.) and placed in a Kopf stereotaxic instrument with blunt ear bars. The skin was retracted, and a hole was drilled in the skull for injections of the retrograde tracer Fluorogold (FG) (0.4% in 0.1 m cacodylic acid; Fluorochrome, Englewood, CO) aimed at the dorsolateral division of the bed nucleus of the stria terminalis (BSTld) (unilaterally) using a 20° mediolateral angled approach (to avoid the lateral ventricle) and the following coordinates: −0.2 mm posterior to bregma, +3.5 mm lateral to the midline, −6.0 mm ventral to dura. Iontophoretic injections were made by passing an alternating current (4 μA; 7 s on, 7 s off for 2 min) through the solution contained in a glass micropipette (15 μm tip diameter). After the injection, the pipette was left in place for 10 min to avoid leakage after removal of the pipette. The skull hole was filled with sterile Gelfoam (Pharmacia and Upjohn, Kalamazoo, MI) and the incision was closed with wound clips (Stoelting). One week later, the rats were overdosed with pentobarbitol (130 mg/kg, i.p.) and perfused intracardially with 0.9% saline (200 ml) followed by 2% paraformaldehyde, 0.05% gluteraldehyde, and 0.2% picric acid in 0.1 m PBS (500 ml). After the perfusion, the brains were removed and stored for 3–4 d in a 30% sucrose/0.1 m PBS solution. Subsequently, the brains were cut serially in 20 μm coronal sections and collected in 0.1 m PBS. Alternate sections through the BST were mounted on microscope slides, coverslipped with DPX Mountant for fluorescence microscopy, and observed with a Zeiss Axioscope 2 under appropriate filters for FG fluorescence (excitation/emission peaks, 323/408 nm).
Some sections were further processed to determine the location of the FG deposit in relation to the CRF-containing cells in the BSTld. Sections were incubated in 0.6% hydrogen peroxide in 0.1 m PBS for 30 min to eliminate endogenous peroxidases, followed by washes in 0.1 m PBS. To reduce nonspecific binding, the sections were preincubated in antibody medium for 2 h followed by incubation for 24 h with a goat polyclonal antibody against CRF (1:1000; Santa Cruz Biotechnology) diluted in antibody medium. The sections were washed with PBS and incubated for 1 h with a donkey anti-goat biotinylated secondary antibody (1:400; minimum species cross-reactivity; Jackson ImmunoResearch). The sections were washed with PBS and incubated for 30 min with the avidin-biotin complex (Vector Laboratories, Burlingame, CA). The sections were then incubated for 5 min in 3,3′-diaminobenzidine/H2O2 (Sigma Fast; Sigma) as a chromagen for visualization of CRF-containing cells in the BSTld. Sections were mounted on microscope slides and coverslipped with Permount.
Sections determined to have discrete FG deposits restricted to the BSTld were further processed for the detection of retrogradely labeled FG-filled cells and TH-positive cells in the major dopaminergic and noradrenergic areas. TH fluorescent immunohistochemistry was performed as described above. Still-frame images of the A8 [retrorubral field (RRF)], A9 [substantia nigra pars compacta (SNc)], A10 [ventral tegmental area (VTA)], and dorsal A10 [located in the periaqueductal gray (PAG)] dopamine cell-containing areas and the A5, A6 [locus ceruleus (LC)], and A7 noradrenergic areas were captured and analyzed for quantification of double-labeled FG and TH-positive cells using Adobe Photoshop software.
Results
Behavioral studies
Effect of SCH 23390 on CRF-enhanced startle
Figure 1A illustrates the effect of different doses of SCH 23390 on the enhancement of startle by CRF. A two-way ANOVA with treatment as a between-subjects factor and blocks of time (1–4) as a within-subjects factor revealed a significant main effect of treatment (F(5,49) = 4.3; p < 0.005), block (F(3,49) = 18.1; p < 0.0001), and a significant treatment by block interaction (F(15,147) = 4.6; p < 0.0001). Multiple comparisons showed no significant differences between groups during the first 50 min block of time, although the startle-enhancing effect of CRF does have its initial onset toward the latter half of this block of time (∼35–40 min) (Fig. 1C). Additional comparisons showed that startle was significantly elevated in the VEH-CRF group compared with the VEH-VEH group across the next three blocks of time (p ≤ 0.05), having its biggest effect during the first half of block 3 (between 100 and 125 min) (Fig. 1C). Startle was also significantly elevated by CRF in animals treated with either the 0.01 or 0.5 mg/kg dose of SCH 23390 during the second and third blocks of time, indicating a lack of effect of the lowest and highest doses of the D1 antagonist on CRF-enhanced startle during this peak-effect time. In contrast, animals treated with the 0.1 mg/kg dose of SCH 23390 showed a dramatic reduction in CRF-enhanced startle; startle levels were not significantly different from the VEH-VEH group but were significantly different from the VEH-CRF group across the second and third blocks of time and attenuated during the fourth block of time. Animals treated with the 0.05 mg/kg dose of SCH 23390 showed a similar statistically significant reduction in CRF-enhanced startle over the third block of time, but CRF-enhanced startle was only attenuated by this dose of SCH 23390 at the second and fourth blocks of time.
Figure 1.

Behavioral studies. A, Effect of pretreatment (subcutaneous) with the dopamine D1 receptor antagonist SCH 23390 (SCH; doses in mg/kg) on the enhancement of startle by CRF (1 μg, i.c.v.). There was a significant dose-dependent reduction in CRF-enhanced startle by SCH 23390 (0.05 and 0.1 mg/kg) during peak CRF effects on startle; data are collapsed across blocks of 50 min for statistical comparisons. B, Effect of SCH 23390 on baseline startle responding. Baseline startle was not significantly different from vehicle controls (VEH-VEH group) at any dose of SCH 23390 (SCH) except for the high dose (0.5 mg/kg) during the first 50 min block of time. C, D, Full time course data are expressed as the percentage difference from the VEH-VEH group for the effect of SCH 23390 (SCH) on CRF-enhanced startle (C) and baseline startle (D). *p < 0.05, **p < 0.005, ***p < 0.0005 compared with VEH-VEH group; †p < 0.05, ††p < 0.005 compared with VEH-CRF group.
Effect of SCH 23390 on startle alone
Figure 1B illustrates the effect of different doses of SCH 23390 on startle in rats given intracerebroventricular infusions of vehicle (aCSF) instead of CRF. A two-way ANOVA with treatment as a between-subjects factor and blocks of time as a within-subjects factor revealed only a significant main effect of block (F(3,32) = 3.79; p < 0.05). Multiple comparisons after significant main effect revealed that baseline startle was significantly reduced by the 0.5 mg/kg dose of SCH 23390 during the first block of time. The suppression of startle by this dose of SCH 23390 wears off over the course of the test session (Fig. 1D) and has no significant effect on startle across the next three blocks of time. The important result to note here is that both the 0.05 and 0.1 mg/kg doses of SCH 23390, doses that clearly reduce the CRF effect on startle, have no effect on startle by themselves.
Anatomical studies
CRF-TH immunohistochemistry in the BST
Brain sections through the BST were double immunolabeled with antibodies against human/rat CRF plus the 20 kDa rat TH enzyme and visualized with antigen-specific fluorescent probes. CRF neurons (Fig. 2A) were found exclusively in the dorsolateral division of the BST [also referred to as the oval nucleus by some investigators (Ju and Han, 1989)] in the noncolchicine-treated rats used in this study. Moderate staining of CRFergic fibers and puncta below the anterior commissure (AC) in the ventral BST was also seen, but CRF neurons were not observed in this subdivision of the BST. In the same sections, TH-positive fibers constituted a dense plexus of innervation in the BSTld clearly outlining neurons in this area (Fig. 2B). These TH fibers are most likely dopaminergic given that previous studies have shown a lack of labeling for dopamine β-hydroxylase (DBH; a marker of noradrenergic neurons) in the BSTld (Hornby and Piekut, 1989; Freedman and Cassell, 1994; Phelix et al., 1994). As shown in the merged image of Figure 2C, most of the cells embedded in the TH fiber field were CRF-containing neurons, thus demonstrating a close relationship between these two neurotransmitter/neuropeptide systems in the BSTld.
Figure 2.
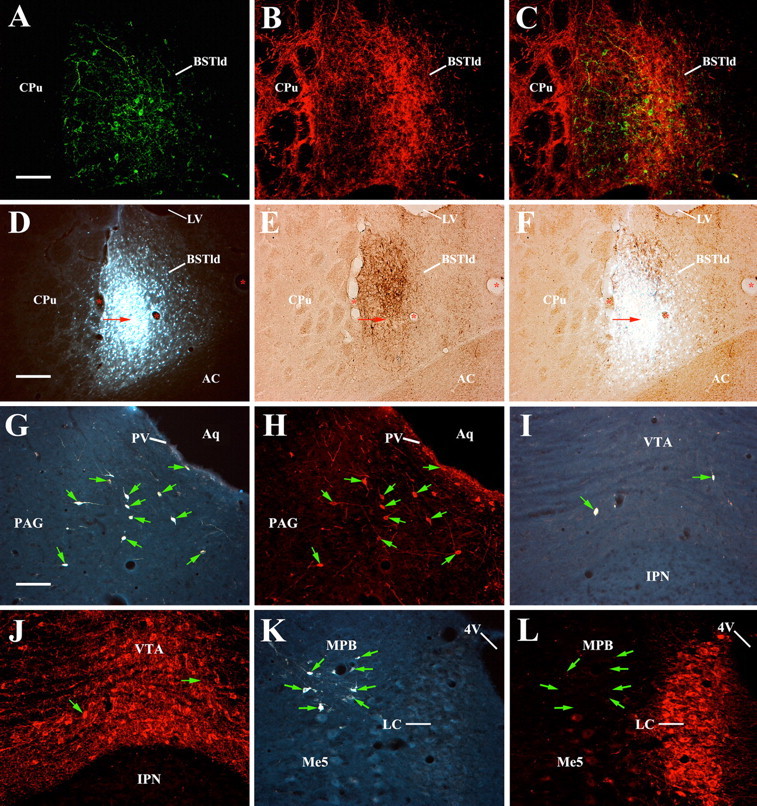
Anatomical studies. A, Coronal section (200×) of FITC-labeled CRF neurons in the BSTld. B, Same section showing TRITC-labeled TH-positive fibers in the BSTld. C, Merged image of A and B shows strong colocalization of CRF neurons with TH-positive fibers in the BSTld. To determine the source of the TH-positive innervation to the BSTld, discrete deposits of the retrograde tracer FG were made. D, Coronal section (100×) of an FG deposit in the BSTld; the deposit center is marked with an arrow. E, Light-microscope image of the adjacent brain section to that shown in D; the section was processed to determine the location of the FG deposit in relation to CRF neurons (brown-labeled cells) in the BSTld. F, Merged image of D and E shows the FG deposit to be restricted to the CRF-dense part of the BSTld with little spread into the adjacent caudate–putamen (CPu). The lateral ventricle (LV), AC, and prominent blood vessels (marked with asterisks) were used to align the two images for merging. G, Coronal section (200×) of retrogradely labeled FG-filled cells in the A10dc PAG area. H, Same section showing that most FG-filled cells in the PAG were also TH positive (indicated by arrows in G and H). I, Fewer FG-filled cells were seen in sections through the VTA, of which most were also TH positive (J). K, Coronal section (200×) showing retrogradely labeled cells at the level of the LC. FG-filled cells were not found in the LC but were seen in the adjacent medial parabrachial nucleus (MPB). L, Same section from K showing the location of the FG-filled cells (marked by arrows) in relation to TH-positive neurons (known to be noradrenergic) in the LC. Aq, Aqueduct of Sylvius; IPN, interpeduncular nucleus; 4V, fourth ventricle; Me5, mesencephalic 5 nucleus. Scale bars: A–C, G–L, 50 μm; D–F, 100 μm.
FG deposits in the BSTld
Two rats had discrete FG deposits restricted to the BSTld; a representative image from one of these cases is shown in Figure 2D. The centers of the deposits were in the BSTld at or just rostral to the crossing of the AC (Fig. 3). In both cases, the spread of the FG deposits was approximately equivalent in size (∼200 μm3) and was confined to the BSTld with complete sparing of the lateral caudate–putamen, the medial subnucleus of the BST, or the AC itself. To confirm that the FG deposit was located in and around CRF neurons of the BSTld and would presumably be taken up by afferents that contact CRF neurons, adjacent brain sections were processed for CRF immunoreactivity. As shown in Figure 2E, the small core of necrosis indicates that the center of the FG iontophoretic deposit was indeed located within the CRF neuron-dense part of the BSTld, the area of heaviest TH-positive innervation (Fig. 2C). Although the deposit appears to be located ventral to the CRF neuronal field, this may be an artifact caused by the deposit itself. This notion is supported by comparing the extent of CRF labeling from the contralateral side where no FG deposit was made. Here, CRF labeling extends more ventrally, almost to the AC (data not shown), suggesting that there is reduced CRF expression on the deposit side caused by necrosis associated with the FG deposit itself (Dado et al., 1990). Although iontophoretic delivery of FG through a glass micropipette with a small tip diameter helps to reduce gross lesion/mechanical disruption effects, it has been reported that the principle mechanism of FG take-up is through damaged fibers (Schmued and Fallon, 1986), although diffusion of FG into nerve terminals or undamaged axons of passage may also be possible (Dado et al., 1990; Wessendorf, 1991). Accordingly, a small lesion such as that indicated in Figure 2E may actually facilitate retrograde labeling of BSTld afferents. The caveat to such a process, however, is that fibers of passage through the BSTld may also take-up FG leading to false-positive results. As such, anterograde tracing studies from brain nuclei identified as BSTld afferent sources (see below) would be warranted to complement these FG retrograde tracing studies.
Figure 3.
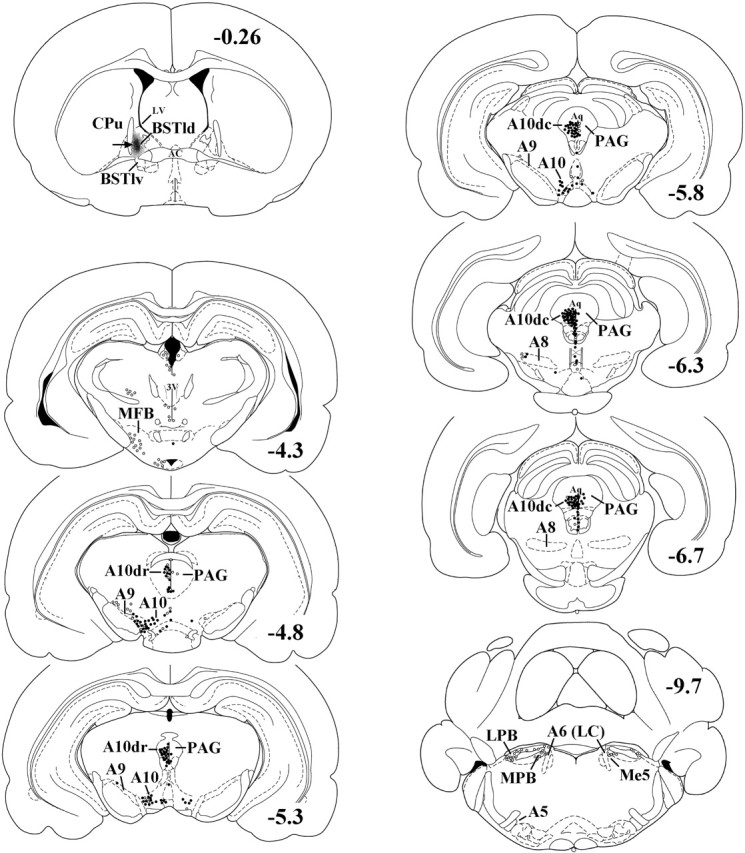
Representative illustration of the location of an FG deposit (black area marked by an arrow) in the BSTld. FG-filled cells (open circles) and FG-filled cells double-labeled for TH (filled circles) were found throughout the major dopaminergic areas (A8-A10) but not the major noradrenergic areas (A5-A7). Neurons in the A10dc area were the greatest source of TH-positive innervation to the BSTld. A significant number of FG-filled and double-labeled neurons was also seen in the PV (Fig. 2G,H) surrounding the aqueduct of Sylvius (Aq). For clarity of the illustration, mapping of the PV efferents to the BSTld has been omitted. Coronal sections were taken from the atlas of Paxinos and Watson (1997) and are in millimeters caudal to bregma. BSTlv, Ventrolateral bed nucleus of the stria terminalis; CPu, caudate–putamen; MFB, medial forebrain bundle; LPB, lateral parabrachial nucleus; MPB, medial parabrachial nucleus; Me5, mesencephalic 5 nucleus; 3V, third ventricle; LV, lateral ventricle.
Retrograde labeling of TH-positive afferents to the BSTld
FG deposits restricted to the BSTld retrogradely labeled cells in the major dopaminergic, but not noradrenergic, areas under investigation. The majority of FG-filled cells were found within the PAG area, where dopamine neurons of the A10dr and A10dc group are located (Hokfelt et al., 1984). A high number of large, multipolar neurons with thick apical dendrites and extensive processes were found to be double labeled for FG and TH (Fig. 2G,H). In addition, a high number of small, round/oval-shaped neurons located on or near the ventral surface of the aqueduct of Sylvius (as described by Flores et al., 2004) were also double labeled for FG and TH. Given their different morphology and relative confinement to the periventricular (PV) surface of the aqueduct, we did not include them in A10dr/A10dc cell counts (Table 2) but counted them as a separate group. A moderate number of double-labeled cells was found in the A10/VTA, and only a few double-labeled cells were found in the A9/SNc. Very few FG-filled or double-labeled cells were found in the A8/RRF, and almost no FG-filled cells were found in the A7, A6/LC, or A5 noradrenergic areas. The distribution of FG-filled cells and those that were double labeled for TH, from a representative animal, is presented in Figure 3. Cell counts in each of these areas (average of two animals) are presented in Table 2 and show that neurons in the A10dc/PAG are the greatest source of TH-positive innervation to the BSTld. Because A10dc neurons do not contain DBH (Flores et al., 2004), it is presumed that these TH-positive neurons are dopaminergic.
Table 2.
Quantification of Fluorogold-filled cells (FG+) and those double labeled for TH (TH+ and FG+) in catecholaminergic cell groups after FG deposits in the BSTld
| Brain area | # sections examined | Atlas sections (mm caudal to bregma) | # cells FG+ | # cells TH+ & FG+ | %TH |
|---|---|---|---|---|---|
| Dopaminergic | |||||
| MFB | 5 | −4.3 to −4.5 | 11 | 0 | 0% |
| PV | 47 | −4.8 to −6.72 | 118 | 70 | 59% |
| A10dr | 20 | −4.8 to −5.6 | 42 | 33 | 79% |
| A10dc | 23 | −5.8 to −6.72 | 143 | 140 | 98% |
| A10 (VTA) | 36 | −4.8 to −6.3 | 74 | 60 | 81% |
| A9 (SNc) | 37 | −4.8 to −6.3 | 13 | 12 | 92% |
| A8 (RRF) | 13 | −6.3 to −7.04 | 4 | 2 | 50% |
| Noradrenergic | |||||
| A7 | 2 | −8.72 to −8.8 | 0 | 0 | 0% |
| A6 (LC) | 10 | −9.16 to −10.3 | 2 | 0 | 0% |
| A5 | 10 | −9.16 to −10.3 | 0 | 0 | 0% |
MFB, Medial forebrain bundle.
Other afferent connections to the BSTld
For the purposes of this study, we were interested in identifying the sources of TH-positive innervation of the BSTld and confined our search to major dopaminergic and noradrenergic brain areas. We did observe, however, several other areas that had heavy retrograde labeling, including the paraventricular thalamic nucleus, the central nucleus of the amygdala, the posterior basolateral and basomedial nuclei of the amygdala, the amygdalopiriform transition area, the ventral and lateral hypothalamic areas, and the parabrachial nucleus. The identification of FG-filled cells in these areas is consistent with other reports describing afferent input to the BSTld (Weller and Smith, 1982).
Discussion
In rats, an increase in startle amplitude by intracerebroventricular CRF has been demonstrated reliably in many laboratories (Weber and Richardson, 2001; Conti et al., 2002; Servatius et al., 2005), is predominantly mediated by CRF1 receptors (Jones et al., 1998), and has been shown to be mostly independent of hypothalamic–pituitary–adrenal axis activity (Lee et al., 1994) and autonomic (sympathetic) nervous system activation (Liang et al., 1992b). Here, we report for the first time that dopaminergic neurotransmission plays an important role in CRF-enhanced startle. Animals treated with the selective dopamine D1 receptor antagonist SCH 23390 showed a significant dose-dependent reduction of the startle-enhancing effects of CRF. Importantly, the doses that reduced CRF-enhanced startle (0.05 and 0.1 mg/kg) did not affect baseline startle response. Thus, the effect of SCH 23390 on CRF-enhanced startle appears to reflect a true interaction between dopamine and CRF systems rather than a potential artifact of the effects of SCH 23390 on sensorimotor processes per se (e.g., catalepsy, auditory impairments). It is also clear that SCH 23390 significantly reduced, but did not fully block, CRF-enhanced startle, suggesting a D1-independent component to CRF-enhanced startle. Other neurotransmitter systems activated by intracerebroventricular CRF, but not affected by D1 receptor-mediated neurotransmission, might intersect (directly or indirectly) with the startle reflex arc to facilitate the behavior. In addition, intracerebroventricular CRF could have direct, D1-independent effects on the brainstem circuitry mediating startle. This seems likely based on a previous study showing a startle-enhancing effect of direct infusions of CRF into the nucleus reticularis pontis caudalis (PnC) (Birnbaum and Davis, 1998), an obligatory nucleus mediating the startle reflex (Lee et al., 1996). A hypothetical framework for understanding D1-dependent and -independent effects on CRF-enhanced startle is illustrated in Figure 4 and described in detail below.
Figure 4.

A model of DA-CRF interactions underlying CRF effects on startle. Based on the results of the present study, together with other anatomical, electrophysiological, and behavioral studies (see Discussion for details), we proposed a BST-PAG feedforward circuit (adapted from Koob, 1999) that could mediate CRF effects on startle and account for the blockade of CRF-enhanced startle by SCH 23390. Activation of this circuit may be initiated by intracerebroventricular (icv) CRF (iC) effects on CRF neurons in the BSTld or A10dc DA neurons in the PAG. D1 receptor-dependent, CRF-mediated effects could exit this feedforward loop and be transmitted to the primary acoustic startle pathway in the brainstem via a direct projection from the lateral BST to the PnC. Accordingly, blockade of D1 receptors in the BST by SCH 23390 may sufficiently interrupt this feedforward activation leading to a reduction in CRF-enhanced startle. As reflected in our behavioral data, the small D1-independent effect of intracerebroventricular CRF on startle may be mediated by a direct effect of CRF on neurons in the PnC. CRN, Cochlear root neurons.
It is presently unclear to us why the SCH 23390 blockade is absent at the highest dose (0.5 mg/kg). Although SCH 23390 is a potent and selective blocker of dopamine D1 receptors (Iorio et al., 1983), at higher doses (>1.5 mg/kg) it has been shown to interact with serotonin 5-HT2 receptors in some brain areas (Bischoff et al., 1986). It is possible that the 0.5 mg/kg dose of SCH 23390 used in the present study had nonspecific effects at other receptors or other DA receptor subtypes that might account for the inverted-U profile seen in the present study. It is interesting to note that this putative nonspecific effect of SCH 23390 (0.5 mg/kg) appears to wear off at ∼150 min, thereby unmasking a reduction of CRF-enhanced startle that lasts for the remainder of the test session.
The results from our anatomical studies extend the findings from a recent study examining the origin of dopaminergic innervation to nuclei of the extended amygdala in female Wistar albino rats (Hasue and Shammah-Lagnado, 2002). Both the present study and the study by Hasue and Shammah-Lagnado (2002) show a heavy projection from neurons in the A10dc/PAG area to the BSTld. However, Hasue and Shammah-Lagnado (2002) reported that only 43% of A10dc/PAG neurons were double labeled for FG and TH, whereas we found that 98% of A10dc/PAG neurons were double labeled. Although animal gender and strain may account for the differences, inconsistencies between studies may have to do with the size of the FG deposit in the BSTld. Because we had a much more discrete FG deposit in the BSTld (∼200 μm spread in our study versus ∼600 μm spread in theirs), primarily restricted to the part of the BSTld innervated by TH-positive fibers, our overall number of retrogradely labeled cells was much lower. The larger FG deposit in the study by Hasue and Shammah-Lagnado (2002) may have been taken up by BST afferents outside the TH-positive innervation field, thus leading to a larger number of retrogradely labeled neurons and an overall lower proportion of neurons double labeled for FG and TH, as reported for the A10dc.
Despite discrepancies between the present study and that of Hasue and Shammah-Lagnado (2002), both studies are important in that they identify a novel A10dc-BST projection that may be involved in the neurobiology of stress. Along these lines, several reports have shown that the BST and, more generally, the extended amygdala, is involved in a number of stress- and anxiety-like behaviors (Walker et al., 2003), including those directly mediated by CRF (Erb and Stewart, 1999; Jasnow et al., 2004) and DA (Lamont and Kokkinidis, 1998; de la Mora et al., 2005). Together, these data suggest that DA-CRF interactions in the extended amygdala could be an important mechanism in the control of mood states. If true, an imbalance or breakdown in the allostatic state between these two systems within the extended amygdala [putatively linked to chronic stress exposure (Schulkin et al., 1994; McEwen, 2000)] could play a role in mood and/or anxiety disorders or represent a potential target for treatment of these psychiatric illnesses (Spedding et al., 2003).
A major aim of the research in our laboratory has been to identify the neural circuits and mechanisms underlying CRF-mediated stress- and anxiety-like behaviors. The BST is rich in CRF-containing neurons (Ju et al., 1989) and CRF1 receptors (Wynn et al., 1984), and chemical lesions of the BST (comprising both the dorsal and ventral BST) completely block CRF-enhanced startle (Lee and Davis, 1997). Although intra-BST CRF only partially mimics the intracerebroventricular CRF effects on startle, and intra-BST infusion of a CRF antagonist does not completely block CRF-enhanced startle, the results from Lee and Davis (1997) strongly suggest that the BST is a critical site mediating CRF-enhanced startle.
Putative activation of CRF neurons in the BSTld by exogenous CRF would increase CRF release in BSTld efferent targets such as the PAG (Gray and Magnuson, 1992). The PAG has low to moderate levels of CRF1 receptors (DeSouza et al., 1985; Smagin et al., 2001) but high expression levels of CRF1 and CRF2 mRNA, particularly in the area of the dorsal raphe (Chalmers et al., 1995; Van Pett et al., 2000), where many A10dc neurons are found (Herbert, 1992), including those that project to the BSTld (present study). CRF release in the PAG, or exogenous CRF itself, could drive A10dc neurons, consistent with the observation that CRF has an excitatory effect on PAG neurons (Bowers et al., 2003). This would facilitate DA release from A10dc neurons via ascending projections to the BSTld to sustain or enhance the activity of CRF neurons through D1-mediated DA neurotransmission. The observation that the BST has moderate to high levels of dopamine D1 receptors (Savasta et al., 1986; Scibilia et al., 1992) and receives heavy dopaminergic inputs (present study) that make direct synaptic contact with CRF neurons (Phelix et al., 1992) supports the idea that DA plays a major role in controlling the activity of CRF neurons in the BSTld.
We hypothesize that the reduction of CRF-enhanced startle by SCH 23390 seen in the present study may result from a blockade of D1 receptors in the BSTld. An obvious strategy to complement the present studies would be to examine whether intra-BST microinjections of SCH 23390 are sufficient to block CRF-enhanced startle. Although such studies are planned for the future, they are complicated by the fact that the specific subnucleus of the BST that must be targeted (BSTld) is located directly below the lateral ventricles. Alternative methods (such as an angled approach for the infusion cannulas) must be developed to ensure that microinjections into the BSTld can affect a significant proportion of this subnucleus without diffusing into the adjacent ventricles. An additional complication of such studies is that the intracerebroventricular cannula must now be implanted intracisternally (because it would occupy the same space on the skull as the intra-BST cannulas), such as that done in the study by Lee and Davis (1997). Although these future studies are technically challenging, they are an important step to determine the brain substrates involved in these systemic drug effects.
According to our model, D1 receptor-dependent, CRF-mediated effects could exit this feedforward circuit and be transmitted to the primary acoustic startle pathway in the brainstem via a direct projection from the lateral BST to the PnC (Rosen et al., 1991). CRF-immunoreactive fibers (Merchenthaler, 1984; Sakanaka et al., 1987) and CRF1 receptors (moderate levels) (Smagin et al., 2001) are both found in the PnC, and the observation that infusions of CRF into the PnC can itself enhance startle (Birnbaum and Davis, 1998) suggests that this area is sensitive to the effects of CRF. An excitatory drive from the BST to the PnC alone is likely not sufficient to account for the entire CRF-enhanced startle effect, however, because intra-PnC CRF produces a much smaller enhancement of startle (∼50% increase from baseline) than that seen with intracerebroventricular CRF (∼200% increase from baseline). Along these lines, it is possible that the small SCH 23390-insenstive component of CRF-enhanced startle seen in the present study (∼50% increase from controls) may reflect excitatory effects of exogenous CRF directly on PnC neurons (and would thus be independent of D1-mediated effects occurring within our proposed BST-PAG circuit).
The results of the present report add to a substantial literature studying the link between stress, CRF, and psychiatric disorders. In general, these studies have shown that increases in CRF-neurotransmission have anxiety- and depressive-like effects, whereas decreases in CRF-neurotransmission have anxiolytic- and antidepressive-like effects (Steckler and Holsboer, 1999). Together, these preclinical findings complement reports from clinical studies where an association between hyperactive CRF systems and some types of anxiety (e.g., posttraumatic stress disorder) and depression (e.g., major depression) have been found (Mitchell, 1998; Arborelius et al., 1999). Although we are unaware of any studies that have used SCH 23390 in clinical trials, the results of our study suggest that compounds with D1 receptor antagonist properties might have anxiolytic-like effects that could be useful in treating conditions associated with hyperactive CRF systems.
Footnotes
This work was supported by The Stanley Medical Research Institute (B.M.C.) and National Institute of Mental Health Grant MH63266 (W.A.C.).
References
- Alheid G, de Olmos JS, Beltramino CA (1995). Amygdala and extended amygdala. In: The rat nervous system (Paxinos G, ed.) pp. 443–494. New York: Academic.
- Arborelius L, Owens MJ, Plotsky PM, Nemeroff CB (1999). The role of corticotropin-releasing factor in depression and anxiety disorders. J Endocrinol 160:1–12. [DOI] [PubMed] [Google Scholar]
- Armstrong DM, Ross CA, Pickel VM, Joh TH, Reis DJ (1982). Distribution of dopamine-, noradrenaline-, and adrenaline-containing cell bodies in the rat medulla oblongata: demonstrated by the immunocytochemical localization of catecholamine biosynthetic enzymes. J Comp Neurol 212:173–187. [DOI] [PubMed] [Google Scholar]
- Birnbaum SG, Davis M (1998). Modulation of the acoustic startle reflex by infusion of corticotropin-releasing hormone into the nucleus reticularis pontis caudalis. Brain Res 782:318–323. [DOI] [PubMed] [Google Scholar]
- Bischoff S, Heinrich M, Sonntag JM, Krauss J (1986). The D-1 dopamine receptor antagonist SCH 23390 also interacts potently with brain serotonin (5-HT2) receptors. Eur J Pharmacol 129:367–370. [DOI] [PubMed] [Google Scholar]
- Bowers LK, Swisher CB, Behbehani MM (2003). Membrane and synaptic effects of corticotropin-releasing factor on periaqueductal gray neurons of the rat. Brain Res 981:52–57. [DOI] [PubMed] [Google Scholar]
- Boyer P (2000). Do anxiety and depression have a common pathophysiological mechanism? Acta Psychiatr Scand Suppl 102:24–29. [PubMed] [Google Scholar]
- Cassell MD, Gray TS, Kiss JZ (1986). Neuronal architecture in the rat central nucleus of the amygdala: a cytological, hodological and immunocytochemical study. J Comp Neurol 246:478–499. [DOI] [PubMed] [Google Scholar]
- Chalmers DT, Lovenberg TW, De Souza EB (1995). Localization of novel corticotropin-releasing factor receptor (CRF2) mRNA expression to specific subcortical nuclei in rat brain: comparison with CRF1 receptor mRNA expression. J Neurosci 15:6340–6350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chappell PB, Smith MA, Kilts CD, Bissette G, Ritchie J, Anderson C, Nemeroff CB (1986). Alterations in corticotropin-releasing factor-like immunoreactivity in discrete rat brain regions after acute and chronic stress. J Neurosci 6:2908–2914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conti LH, Murry JD, Ruiz MA, Printz MP (2002). Effects of corticotropin-releasing factor on prepulse inhibition of the acoustic startle response in two rat strains. Psychopharmacology (Berl) 161:296–303. [DOI] [PubMed] [Google Scholar]
- Dado RJ, Burstein R, Cliffer KD, Giesler GJ Jr (1990). Evidence that Fluoro-Gold can be transported avidly through fibers of passage. Brain Res 533:329–333. [DOI] [PubMed] [Google Scholar]
- Davis M, Walker DL, Lee Y (1997). Amygdala and bed nucleus of the stria terminalis: differential roles in fear and anxiety measured with the acoustic startle reflex. Philos Trans R Soc Lond B Biol Sci 352:1675–1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Mora MP, Cárdenas-Cachón L, Vázquez-Garcia M, Crespo-Ramirez M, Jacobsen K, Höistad M, Agnati L, Fuxe K (2005). Anxiolytic effects of intra-amygdaloid injection of the D1 antagonist SCH23390 in the rat. Neurosci Lett 377:101–105. [DOI] [PubMed] [Google Scholar]
- DeSouza EB, Insel TR, Perrin MH, Rivier J, Vale WW, Kuhar MJ (1985). Corticotropin-releasing factor receptors are widely distributed within the rat central nervous system: an autoradiographic study. J Neurosci 5:3189–3203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn AJ, Berridge CW (1990). Physiological and behavioral responses to corticotropin-releasing factor administration: Is CRF a mediator of anxiety or stress responses. Brain Res Brain Res Rev 15:71–100. [DOI] [PubMed] [Google Scholar]
- Eliava M, Yilmazer-Hanke D, Asan E (2003). Interrelations between monoaminergic afferents and corticotropin-releasing factor-immunoreactive neurons in the rat central amygdaloid nucleus: ultrastructural evidence for dopaminergic control of amygdaloid stress systems. Histochem Cell Biol 120:183–197. [DOI] [PubMed] [Google Scholar]
- Erb S, Stewart J (1999). A role for the bed nucleus of the stria terminalis, but not the amygdala, in the effects of corticotropin-releasing factor on stress-induced reinstatement of cocaine seeking. J Neurosci 19:1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finlay JM, Zigmond MJ, Abercrombie ED (1995). Increased dopamine and norepinephrine release in medial prefrontal cortex induced by acute and chronic stress: effects of diazepam. Neuroscience 64:619–628. [DOI] [PubMed] [Google Scholar]
- Flores JA, Banoua FE, Galan-Rodriguez B, Fernandez-Espejo E (2004). Opiate anti-nociception is attenuated following lesions of large dopamine neurons of the periaqueductal grey: critical role for D1 (not D2) dopamine receptors. Pain 110:205–214. [DOI] [PubMed] [Google Scholar]
- Frankland PW, Ralph MR (1995). Circadian modulation in the rat acoustic startle circuit. Behav Neurosci 109:43–48. [DOI] [PubMed] [Google Scholar]
- Freedman LJ, Cassell MD (1994). Distribution of dopaminergic fibers in the central division of the extended amygdala of the rat. Brain Res 633:243–252. [DOI] [PubMed] [Google Scholar]
- Gold PW, Chrousos GP (2002). Organization of the stress system and its dysregulation in melancholic and atypical depression: high vs low CRH/NE states. Mol Psychiatry 7:254–275. [DOI] [PubMed] [Google Scholar]
- Gray TS, Magnuson DJ (1992). Peptide immunoreactive neurons in the amygdala and the bed nucleus of the stria terminalis project to the midbrain central gray in the rat. Peptides 13:451–460. [DOI] [PubMed] [Google Scholar]
- Hasue RH, Shammah-Lagnado SJ (2002). Origin of the dopaminergic innervation of the central extended amygdala and accumbens shell: a combined retrograde tracing and immunohistochemical study in the rat. J Comp Neurol 454:15–33. [DOI] [PubMed] [Google Scholar]
- Heim C, Nemeroff CB (1999). The impact of early adverse experiences on brain systems involved in the pathophysiology of anxiety and affective disorders. Biol Psychiatry 46:1509–1522. [DOI] [PubMed] [Google Scholar]
- Herbert H (1992). Evidence for projections from medullary nuclei onto serotonergic and dopaminergic neurons in the midbrain dorsal raphe nucleus of the rat. Cell Tissue Res 270:149–156. [DOI] [PubMed] [Google Scholar]
- Hokfelt T, Martensson R, Bjorklund A, Kleinau S, Goldstein M (1984). Distribution maps of tyrosine-hydroxylase-immunoreactive neurons in the rat brain. In: Handbook of chemical neuroanatomy. Classical transmitters in the CNS, part I (Bjorklund A, Hokfelt T, eds) pp. 277–379. Amsterdam: Elsevier Science.
- Hornby PJ, Piekut DT (1989). Opiocortin and catecholamine input to CRF-immunoreactive neurons in rat forebrain. Peptides 10:1139–1146. [DOI] [PubMed] [Google Scholar]
- Iorio LC, Barnett A, Leitz FM, Houser VP, Korduba CA (1983). SCH 23390, a potential benzazepine anti-psychotic with unique interactions on dopaminergic systems. J Pharmacol Exp Ther 226:462–468. [PubMed] [Google Scholar]
- Jasnow AM, Davis M, Huhman KL (2004). Involvement of central amygdalar and bed nucleus of the stria terminalis corticotropin-releasing factor in behavioral response to social defeat. Behav Neurosci 118:1052–1061. [DOI] [PubMed] [Google Scholar]
- Jones DNC, Kortekaas R, Slade PD, Middlemiss DN, Hagan JJ (1998). The behavioral effects of corticotropin-releasing factor-related peptides in rats. Psychopharmacology (Berl) 138:124–132. [DOI] [PubMed] [Google Scholar]
- Ju G, Han Z (1989). Coexistence of corticotropin releasing factor and neurotensin within oval nucleus in the bed nuclei of the stria terminalis. Neurosci Lett 99:246–250. [DOI] [PubMed] [Google Scholar]
- Ju G, Swanson LW, Simerly RB (1989). Studies on the cellular architecture of the bed nuclei of the stria terminalis in the rat: II. Chemoarchitecture. J Comp Neurol 280:603–621. [DOI] [PubMed] [Google Scholar]
- Kessler RC, Chiu WT, Demler O, Walters EE (2005). Prevalence, severity, and comorbidity of 12-month DSM-IV disorders in the National Comorbidity Survey Replication. Arch Gen Psychiatry 62:617–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koob GF (1999). Corticotropin-releasing factor, norepinephrine, and stress. Biol Psychiatry 46:1167–1180. [DOI] [PubMed] [Google Scholar]
- Lamont EW, Kokkinidis L (1998). Infusion of the dopamine D1 receptor antagonist SCH 23390 into the amygdala blocks fear expression in a potentiated startle paradigm. Brain Res 795:128–136. [DOI] [PubMed] [Google Scholar]
- Lee Y, Davis M (1997). Role of the hippocampus, bed nucleus of the stria terminalis and amygdala in the excitatory effect of corticotropin releasing hormone (CRH) on the acoustic startle reflex. J Neurosci 17:6434–6446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y, Schulkin J, Davis M (1994). Effect of corticosterone on the enhancement of the acoustic startle reflex by corticotropin releasing factor (CRF). Brain Res 666:93–98. [DOI] [PubMed] [Google Scholar]
- Lee Y, Lopez DE, Meloni EG, Davis M (1996). A primary acoustic startle circuit: obligatory role of cochlear root neurons and the nucleus reticularis pontis caudalis. J Neurosci 16:3775–3789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang KC, Melia KR, Campeau S, Falls WA, Miserendino MJD, Davis M (1992a). Lesions of the central nucleus of the amygdala, but not of the paraventricular nucleus of the hypothalamus, block the excitatory effects of corticotropin releasing factor on the acoustic startle reflex. J Neurosci 12:2313–2320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang KC, Melia KR, Miserendino MJD, Falls WA, Campeau S, Davis M (1992b). Corticotropin-releasing factor: long-lasting facilitation of the acoustic startle reflex. J Neurosci 12:2303–2312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McEwen BS (2000). Allostasis and allostatic load: implications for neuropsychopharmacology. Neuropsychopharmacology 22:108–124. [DOI] [PubMed] [Google Scholar]
- Merchenthaler I (1984). Corticotropin releasing factor (CRF)-like immunoreactivity in the rat central nervous system. Extrahypothalamic distribution. Peptides 5:53–69. [DOI] [PubMed] [Google Scholar]
- Mitchell AJ (1998). The role of corticotropin releasing factor in depressive illness: a critical review. Neurosci Biobehav Rev 22:635–651. [DOI] [PubMed] [Google Scholar]
- Moga MM, Saper CB, Gray TS (1989). Bed nucleus of the stria terminalis: cytoarchitecture, immunohistochemistry, and projection to the parabrachial nucleus in the rat. J Comp Neurol 283:315–332. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C (1997). In: The rat brain in stereotaxic coordinates Ed 3 New York: Academic.
- Phelix CF, Liposits Z, Paull WK (1992). Monoamine innervation of bed nucleus of stria terminalis: an electron microscopic investigation. Brain Res Bull 28:949–965. [DOI] [PubMed] [Google Scholar]
- Phelix CF, Liposits Z, Paull WK (1994). Catecholamine-CRF synaptic interaction in a septal bed nucleus: afferents of neurons in the bed nucleus of the stria terminalis. Brain Res Bull 33:109–119. [DOI] [PubMed] [Google Scholar]
- Rosen JB, Hitchcock JM, Sananes CB, Miserendino MJD, Davis M (1991). A direct projection from the central nucleus of the amygdala to the acoustic startle pathway: anterograde and retrograde tracing studies. Behav Neurosci 105:817–825. [DOI] [PubMed] [Google Scholar]
- Sakanaka M, Shibasaki T, Lederis K (1987). Corticotropin-releasing factor-like immunoreactivity in the rat brain as revealed by a modified cobalt-glucose oxidase-diaminobenzidine method. J Comp Neurol 260:256–298. [DOI] [PubMed] [Google Scholar]
- Savasta M, Dubois A, Scatton B (1986). Autoradiographic localization of D1 dopamine receptors in the rat brain with [3H]SCH 23390. Brain Res 375:291–301. [DOI] [PubMed] [Google Scholar]
- Schmued LC, Fallon JH (1986). Fluoro-Gold: a new fluorescent retrograde axonal tracer with numerous unique properties. Brain Res 377:147–154. [DOI] [PubMed] [Google Scholar]
- Schulkin J, McEwen BS, Gold PW (1994). Allostasis, amygdala, and anticipitory angst. Neurosci Biobehav Rev 18:385–396. [DOI] [PubMed] [Google Scholar]
- Schulz DW, Mansbach RS, Sprouse J, Braselton JP, Collins J, Corman M, Dunaiskis A, Faraci S, Schmidt AW, Seeger T, Seymour P, Tingley FD, Winston EN, Chen YL, Heym J (1996). CP-154,526: a potent and selective nonpeptide antagonist of corticotropin releasing factor receptors. Proc Natl Acad Sci USA 93:10477–10482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scibilia RJ, Lachowicz JE, Kilts CD (1992). Topographic nonoverlapping distribution of D1 and D2 dopamine receptors in the amygdaloid nuclear complex of the rat brain. Synapse 11:146–154. [DOI] [PubMed] [Google Scholar]
- Servatius RJ, Beck KD, Moldow RL, Salameh G, Tumminello TP, Short KR (2005). A stress-induced anxious state in male rats: corticotropin-releasing hormone induces persistent changes in associative learning and startle reactivity. Biol Psychiatry 57:865–872. [DOI] [PubMed] [Google Scholar]
- Smagin GN, Heinrichs SC, Dunn AJ (2001). The role of CRH in behavioral responses to stress. Peptides 22:713–724. [DOI] [PubMed] [Google Scholar]
- Spedding M, Neau I, Harsing L (2003). Brain plasticity and pathology in psychiatric disease: sites of action for potential therapy. Curr Opin Pharmacol 3:1–8. [DOI] [PubMed] [Google Scholar]
- Steckler T, Holsboer F (1999). Corticotropin-releasing hormone receptor subtypes and emotion. Biol Psychiatry 46:1480–1508. [DOI] [PubMed] [Google Scholar]
- Swerdlow NR, Geyer MA, Vale WW, Koob GF (1986). Corticotropin-releasing factor potentiates acoustic startle in rats: blockade by chlordiazepoxide. Psychopharmacology (Berl) 88:147–152. [DOI] [PubMed] [Google Scholar]
- Toufexis DJ, Davis C, Hammond A, Davis M (2004). Progesterone attenuates corticotropin-releasing factor-enhanced but not fear-potentiated startle via the activity of its neuroactive metabolite, allopregnanolone. J Neurosci 24:10280–10287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Pett K, Viau V, Bittencourt JC, Chan RKW, Li H-Y, Arias C, Prins GS, Perrin MH, Vale W, Sawchenko PE (2000). Distribution of mRNAs encoding CRF receptors in brain and pituitary of rat and mouse. J Comp Neurol 428:191–212. [DOI] [PubMed] [Google Scholar]
- Vermetten E, Bremner JD (2002a). Circuits and systems in stress. II. Applications to neurobiology and treatment in posttraumatic stress disorder. Depress Anxiety 16:14–38. [DOI] [PubMed] [Google Scholar]
- Vermetten E, Bremner JD (2002b). Circuits and systems in stress. I. Preclinical studies. Depress Anxiety 15:126–147. [DOI] [PubMed] [Google Scholar]
- Walker DL, Toufexis DJ, Davis M (2003). Role of the bed nucleus of the stria terminalis versus the amygdala in fear, stress, and anxiety. Eur J Pharmacol 463:199–216. [DOI] [PubMed] [Google Scholar]
- Weber M, Richardson R (2001). Centrally administered corticotropin-releasing hormone and peripheral injections of strychnine hydrochloride potentiate the acoustic startle response in preweanling rats. Behav Neurosci 115:1273–1282. [DOI] [PubMed] [Google Scholar]
- Weller KL, Smith DA (1982). Afferent connections to the bed nucleus of the stria terminalis. Brain Res 232:255–270. [DOI] [PubMed] [Google Scholar]
- Wessendorf M (1991). Fluoro-Gold: composition, and mechanism of uptake. Brain Res 553:135–148. [DOI] [PubMed] [Google Scholar]
- Wynn PC, Hauger RL, Holmes MC, Millan MA, Catt KJ, Aguilera G (1984). Brain and pituitary receptors for corticotropin releasing factor: localization and differential regulation after adrenalectomy. Peptides 5:1077–1084. [DOI] [PubMed] [Google Scholar]