

Abstract
The transient receptor potential vanilloid 4 (TRPV4) is a primary afferent transducer that plays a crucial role in neuropathic hyperalgesia for osmotic and mechanical stimuli, as well as in inflammatory mediator-induced hyperalgesia for osmotic stimuli. In view of the clinical importance of mechanical hyperalgesia in inflammatory states, the present study investigated the role of TRPV4 in mechanical hyperalgesia induced by inflammatory mediators and the second-messenger pathways involved. Intradermal injection of either the inflammogen carrageenan or a soup of inflammatory mediators enhanced the nocifensive paw-withdrawal reflex elicited by hypotonic or mechanical stimuli in rat. Spinal administration of TRPV4 antisense oligodeoxynucleotide blocked the enhancement without altering baseline nociceptive threshold. Similarly, in TRPV4−/− knock-out mice, inflammatory soup failed to induce any significant mechanical or osmotic hyperalgesia. In vitro investigation showed that inflammatory mediators engage the TRPV4-mediated mechanism of sensitization by direct action on dissociated primary afferent neurons. Additional behavioral observations suggested that multiple mediators are necessary to achieve sufficient activation of the cAMP pathway to engage the TRPV4-dependent mechanism of hyperalgesia. In addition, direct activation of protein kinase A or protein kinase C ϵ, two pathways that mediate inflammation-induced mechanical hyperalgesia, also induced hyperalgesia for both hypotonic and mechanical stimuli that was decreased by TRPV4 antisense and absent in TRPV4−/− mice. We conclude that TRPV4 plays a crucial role in the mechanical hyperalgesia that is generated by the concerted action of inflammatory mediators present in inflamed tissues.
Keywords: pain, mechanotransduction, silent nociceptor, dorsal root ganglion, sensory nerve, inflammatory mediators
Introduction
Transient receptor potential vanilloid 4 (TRPV4), a member of the transient receptor potential family of ligand-gated ion channels (Liedtke et al., 2000; Strotmann et al., 2000), functions as a sensory transducer for osmotic-induced nociception (Alessandri-Haber et al., 2003). In addition, TRPV4 participates in sensation of intense noxious mechanical stimuli in mice (Liedtke and Friedman, 2003; Suzuki et al., 2003a) and in mechanical hyperalgesia in a model of small-fiber painful peripheral neuropathy in rat (Alessandri-Haber et al., 2004). Although TRPV4 plays an important role in the transduction of noxious mechanical stimuli, it apparently does not contribute to detection of threshold-level pain sensation (Liedtke and Friedman, 2003; Suzuki et al., 2003a).
In view of its role in sensation of intense and neuropathic pain, but not in threshold-level pain, we hypothesized that TRPV4-dependent hyperalgesia might be a mechanism specific to pathological pain states, perhaps including inflammatory pain. Inflammatory pain is of particular interest because it is the predominant cause of clinical pain conditions. Confirmation of our hypothesis could have important implications for the treatment of pain because it would identify TRPV4 as a target for the development of a new class of analgesic drugs to treat common pain conditions.
To test our hypothesis, we investigated whether TRPV4 contributes to inflammatory hyperalgesia induced by either the inflammogen carrageenan or by a soup of inflammatory mediators [bradykinin (BK), substance P (SP), prostaglandin E2 (PGE2), serotonin (5-HT), and histamine] (Steen et al., 1996; Kress et al., 1997; Vyklicky et al., 1998; Levine and Reichling, 1999; Linhart et al., 2003). Nociceptive behavioral tests were performed in both rats that received repeated intrathecal injections of antisense oligodeoxynucleotide (ODN) for TRPV4 to transiently downregulate the level of expression of TRPV4 protein and in C57BL/6 mice lacking functional TRPV4 gene. We also performed in vitro experiments to test whether TRPV4 function is altered by direct action of the inflammatory mediators on primary afferent neurons. To elucidate the cellular mechanisms that cause TRPV4-dependent mechanisms to be activated in primary afferents in inflamed tissue, we examined interactions between TRPV4 and two key intracellular second-messenger pathways that mediate inflammatory hyperalgesia, protein kinase A (PKA) and protein kinase C ϵ (PKCϵ) (Levine and Reichling, 1999).
Materials and Methods
Animals.
Experiments were performed on 180–200 g adult male Sprague Dawley rats (Charles River Laboratories, Wilmington, MA) and on C57BL/6 mice lacking functional TRPV4 gene (TRPV4−/− mice) (Liedtke and Friedman, 2003) and TRPV4 wild-type littermates (TRPV4+/+). Experimental protocols were approved by the University of California, San Francisco Committee on Animal Research and conformed to National Institutes of Health guidelines for the use of animals in research.
Drugs.
PGE2, histamine, 5-HT, SP, 8-bromoadenosine 3′, 5′-cAMP (8-Br-cAMP), Rp-adenosine 3′, 5′-cyclic monophosphothioate (Rp-cAMPS), H89 (N-[2-(p-bromocinnamylamino)ethyl]-5-isoquinolinesulfonamide dihydrochloride), and carrageenan were purchased from Sigma (St. Louis, MO). BK was purchased from ICN Biomedicals (Aurora, OH), cAMP-dependent protein kinase A catalytic subunit (PKACS) was purchased from New England Biolabs (Beverly, MA), PKCϵ translocation inhibitor (PKCϵΙ) was purchased from Calbiochem (La Jolla, CA), and HDAPIGYD-protein kinase C activator (pseudoreceptor for activated RACK, ψϵ-RACK) was synthesized by SynPep (Dublin, CA).
For behavioral experiments, stock solutions of SP, BK, histamine, 5-HT, 8-Br-cAMP, PKACS, ψϵ-RACK, and carrageenan were made in 0.9% NaCl. Stock solution of PGE2 was made in 10% ethanol. For all drugs used in behavioral experiments, final experimental concentrations were made in 0.9% NaCl on the day of the experiment (final concentration of ethanol <1%).
For calcium imaging experiments, stock solutions of H89, PKCϵI, BK, and PGE2 were made in 10% dimethylsulfoxide (DMSO), and Rp-cAMPS, histamine, SP, and 5-HT were made in distilled H2O. Final experimental concentrations were made in the isotonic or hypotonic solution (Hypo) on the day of the experiment. Of note, the final concentration of DMSO is 100 μm, and the osmolarity of the solution is not measurably different from the hypotonic solution alone.
Mechanical threshold in rat.
Mechanical nociceptive thresholds were evaluated by the Randall–Sellito paw-withdrawal test with an Ugo Basile algesymeter (Stoelting, Chicago, IL) as described previously (Aley and Levine, 2001; Aley et al., 2001). Baseline mechanical thresholds were recorded as the mean of three measurements (at 10 min intervals) before the injection of hypotonic solution or pharmacological reagents. To induce hyperalgesia, a solution of either carrageenan (1% w/v, 5 μl), inflammatory soup, PGE2 (100 ng/2.5 μl), PGE2 and 5-HT (1 μg each/2.5 μl), 8-Br-cAMP (1 μg/2.5 μl), PKA catalytic subunit (10 U/2.5 μl), or ψϵ-RACK (1 μg/2.5 μl) were injected intradermally into the dorsum of the rat hindpaw, 30 min before behavioral testing. For statistical analysis, each paw was considered to be an independent observation.
For the 8-Br-cAMP dose–response experiment, rats were treated with either TRPV4 antisense or mismatch ODN for 3 d, and, on day 4 after initiation of ODN treatment, mechanical nociceptive thresholds were evaluated before and 15 min after intradermal injection of increasing doses of 8-Br-cAMP (0.1, 0.3, 1, 3, 10, 100, 300, and 1000 ng). The two ODN-treated and control groups were tested in parallel on the same day.
Flinch test in rat.
As described previously (Zheng and Chen, 2001; Zhang et al., 2003; Alessandri-Haber et al., 2004; Houck et al., 2004), rats were acclimated in a transparent observation chamber for 30 min. Rats were then restrained while 10 μl of hypotonic (deionized water, 17 mOsm) or isotonic (0.9% NaCl, 283 mOsm) solution was administered intradermally into the dorsum of the hindpaw via a 30 gauge needle connected to a 100 μl syringe by polyethylene tubing. Immediately after the injection, rats were observed in the chamber for a 5 min period. For experiments involving TRPV4 antisense ODN treatment, the flinch tests were performed on day 4 after initiation of ODN treatment (12 h after the last ODN injection). PGE2 (100 ng/2.5 μl), carrageenan (1%, w/v, 5 μl), the inflammatory soup, 8-Br-cAMP, PKACS, or ψϵ-RACK were injected intradermally 45 min before administration of the hypotonic stimulus, at the same site of injection.
Mechanical nociception in mice.
Mice were acclimated 15–20 min in a transparent box with a metal mesh floor. A calibrated von Frey hair monofilament (Stoelting) was applied through the mesh floor to the plantar skin of the hindpaw. Mechanical threshold was measured as the total number of paw withdrawals in response to a series of five applications (at 3 min intervals) of a 0.17 mN von Frey hair. The response to hyperalgesia-inducing agents was measured 30 min after intraplantar injection of agent.
Dorsal root ganglion cell culture.
L2–L6 dorsal root ganglia (DRGs) were harvested from TRPV4+/+ and TRPV4−/− mice and dissociated and cultured as described previously (Alessandri-Haber et al., 2003). Briefly, dissociated cells were plated on coverslips treated with poly-dl-ornithine (0.1 mg/ml; Sigma) and laminin (5 μg/ml; Invitrogen, Carlsbad, CA), incubated at 37°C in 96.5% air, 3.5% CO2. Neurons were maintained in culture for 3 d in DMEM supplemented with 50–100 ng/ml nerve growth factor, 100 U/ml penicillin/streptomycin, MEM vitamins, and 10% heat-inactivated fetal calf serum (all from Invitrogen).
Calcium imaging.
Calcium imaging was performed using the fluorescent calcium indicator fura-2 AM between 24 and 72 h after dissociation, as described previously (Alessandri-Haber et al., 2003). Briefly, neurons were loaded with 5 μm fura-2 AM for 20 min in isotonic solution (312 mOsm). Experiments were performed at 20–23°C with the perfusion at a flow rate of 1–2 ml/min. Cells were perfused with isotonic solution for 10 min before the beginning of the recording to allow complete removal of nonhydrolyzed fura ester.
Measurement of the concentration of free calcium ions ([Ca2+]i) was performed by ratiometric imaging with an intensified charge-coupled device (CCD) camera. Fluorescence was excited at 340 and 380 nm, and emitted light was long-pass filtered at 510 nm. The fluorescence ratio, F340/F380, was calculated with Metafluor software (Universal Imaging Corporation, Downington, PA). Calcium calibration was performed with a fura-2 calcium imaging calibration kit (Invitrogen), and apparent free [Ca2+]i was calculated from the equation [Ca2+]i = Kd × ((R − Rmin)/(Rmax − R)) × (F380max/F380min), where Rmin is the ratio at zero free Ca2+, Rmax is the ratio at saturating Ca2+ (e.g., 39 μm), F380max is the fluorescence intensity exciting at 380 nm, for zero Ca2+, and F380min is the fluorescence intensity at saturating free Ca2+.
Given the absence of specific pharmacological blockers of TRPV4, we minimized conductance via other ion channels by using a combination of room temperature HEPES buffer and variation of osmolarity only by modifying d-mannitol concentration. Thus, the standard isotonic solution (312 mOsm) contained the following (in mm): 88 NaCl, 5 KCl, 1 MgCl2, 2.4 CaCl2, 110 d-mannitol, and 10 HEPES, buffered at pH 7.38 with NaOH. The hypotonic solution was adjusted to 212 mOsm (30% hypotonic) by lowering the amount of d-mannitol to 10 mm. Osmolarity and pH were measured before each experiment. The vehicle for the fura-2 AM, DMSO, at its final working dilution did not induce any response in DRG neurons.
To measure the response of DRG neurons to hypotonicity, we averaged the value of the fluorescence ratio when the stimulus-induced increase in [Ca2+]i reached a maximum plateau. This fluorescence ratio was then normalized with respect to the baseline value: fluorescence ratio during hypotonic stimulation divided by resting fluorescence ratio in isotonic solution. The fluorescence ratio was then converted to apparent free [Ca2+]i. At the end of each experiment, a short exposure to solution containing 20 mm KCl was performed to confirm that all cells studied exhibited electrical excitability that is typical of healthy neurons.
For experiments involving sensitization with the inflammatory soup (a combination of 10 μm each BK, SP, histamine, 5-HT, and PGE2), neurons were challenged with a 30% hypotonic solution for 3 min, washed for 6 min with isotonic solution, perfused with an isotonic solution containing the soup for 5 min, and then challenged with 30% hypotonic solution containing the soup for 3 min. Intracellular calcium was calculated from the normalized fluorescence ratio during a 30% hypotonic solution during exposure to the soup divided by the normalized fluorescence ratio in 30% hypotonic solution before exposure to soup.
For experiments testing the effect of inhibitors of PKA or PKCϵ on the hypotonic increase in fluorescence ratio, neurons were first challenged with hypotonic solution for 3 min, rinsed with isotonic solution for 6 min, perfused with isotonic solution containing the inhibitor for 15 min, and challenged with a 30% hypotonic solution containing the inhibitor for 3 min. The effect of the blockers was calculated from the normalized fluorescence ratio during exposure to 30% hypotonic solution in the presence of inhibitor divided by the normalized fluorescence ratio in 30% hypotonic solution before inhibitor.
cAMP ELISA.
Dorsal root ganglia were harvested from rats and dissociated as described above and maintained in culture for 2 d (two ganglia per dish). Cells were washed with culture medium and then incubated for 20 min at 37°C with 100 μm PGE2, 100 μm 5-HT, or PGE2 plus 5-HT (each at 10 μm). The medium was aspirated, and 0.1N HCl was added for 10 min to lyse the cells. Lysates were processed immediately with a cAMP ELISA kit (Assay Designs, Ann Arbor, MI). Four independent experiments were performed.
ODN preparation and administration.
The TRPV4 antisense ODN sequence, 5′-CATCACCAGGATCTGCCATACTG-3′ (Invitrogen), was directed against a unique region of the rat TRPV4 channel (GenBank accession number AF263521). The mismatch ODN sequence was designed by mismatching seven bases (denoted by bold face) of the TRPV4 antisense sequence: 5′-CAACAGGAGGTTCAGGCAAACTG-3′.
ODN was reconstituted in nuclease-free 0.9% NaCl (10 μg/μl) and was administered into the spinal intrathecal space at a dose of 40 μg, once a day for 3 d. As described previously (Alessandri-Haber et al., 2003), for this procedure, rats were anesthetized with 2.5% isoflurane (97.5% O2), a 30 gauge needle was inserted into the subarachnoid space on the midline between L4 and L5 vertebrae, and 20 μl ODN was injected at 1 μl/s by microsyringe.
We have demonstrated previously that TRPV4 is present in sensory nerve fibers, presumably being transported from the cell body toward the peripheral nerve endings, and that our antisense ODN treatment procedure induces a significant and specific decrease in TRPV4 protein expression level in ligated saphenous nerve (Alessandri-Haber et al., 2003, 2004).
Data analysis.
Calcium imaging and behavioral data are presented as mean ± SEM, and comparisons between groups were performed by Student's t test or ANOVA followed by Tukey's multiple comparison post hoc test. Significance was defined as p < 0.05.
Results
TRPV4 contributes to inflammatory hyperalgesia
To investigate whether TRPV4 contributes to inflammatory hyperalgesia, we tested whether the inflammogen carrageenan or a soup of inflammatory mediators (bradykinin, substance P, PGE2, 5-HT, and histamine) would enhance the TRPV4-dependent nociceptive response to an injection of hypotonic solution (10 μl of deionized water) in rat hindpaw. As shown in Figure 1A, carrageenan and inflammatory soup induced, respectively, a 4.6- and a 3.6-fold increase in the number of flinches induced by hypotonicity (3.9 ± 0.3, n = 24 for hypotonicity alone; 18 ± 2, n = 6 after carrageenan; and 14 ± 2, n = 10 after inflammatory soup). To assess the contribution of TRPV4 in this nociceptive test, rats were pretreated with a spinal intrathecal administration of antisense or mismatch ODN for TRPV4, daily for 3 d. Antisense ODN (compared with mismatch) reduced the number of hypotonicity-induced flinches, in the presence of carrageenan or inflammatory soup, by 72 and 43%, respectively (Fig. 1A).
Figure 1.

TRPV4 mediates inflammatory hyperalgesia. A, Inflammatory mediators enhance nocifensive flinching induced by the injection of hypotonic solution. Intradermal injection of a hypotonic solution in rat hindpaw induced nocifensive flinches. Injection of carrageenan (Carr) or inflammatory soup (Soup) before the hypotonic solution induced a 4.6- and a 3.6-fold increase, respectively, in the number of flinches. Treatment with TRPV4 antisense ODN reduced the number of hypotonicity-induced flinches in the presence of carrageenan by 72% (5 ± 1, n = 6 for antisense-treated vs 18 ± 2, n = 6 for mismatch-treated rats; *p < 0.05, unpaired Student's t test) and the ones in the presence of the inflammatory soup by 43% (9 ± 2, n = 6 for antisense-treated vs 16 ± 2, n = 6 for mismatch-treated rats; *p < 0.02, unpaired Student's t test). B, Intradermal injection of carrageenan or inflammatory soup induced a decrease in mechanical nociceptive thresholds in the rat. Spinal intrathecal injection of TRPV4 antisense ODN prevented the mechanical hyperalgesia induced by carrageenan (3.5 ± 3%, n = 6 for antisense-treated vs 26 ± 0.8%, n = 6 for mismatch-treated rats; *p < 0.001, unpaired Student's t test) or by inflammatory soup (−1 ± 4%, n = 6 for antisense-treated vs 30 ± 2%, n = 6 for mismatch-treated rats; *p < 0.0001, unpaired Student's t test). Inset, Treatment with TRPV4 antisense had no effect on mechanical threshold in naive rats (n = 58 for mechanical threshold baseline and n = 20 for each ODN-treated rats; p > 0.05, ANOVA), whereas it prevented the decrease in threshold caused by inflammatory soup or carrageenan.
Intradermal injection of carrageenan or inflammatory soup 20 min before mechanical stimulation of the rat hindpaw also induced hyperalgesia to mechanical stimuli. As shown in Figure 1B, pretreatment with TRPV4 antisense prevented the mechanical hyperalgesia induced by both carrageenan and the inflammatory soup. In contrast, pretreatment with TRPV4 antisense did not affect the baseline nociceptive mechanical threshold in control rats (Fig. 1B, inset).
As an independent test of the role of TRPV4 in mechanical hyperalgesia induced by inflammatory soup, we tested C57BL/6 mice lacking the TRPV4 gene (TRPV4−/−) (Liedtke and Friedman, 2003). As shown in Figure 2A, in the absence of inflammatory soup, the baseline withdrawal response to von Frey hair stimulation was not significantly different between TRPV4−/− mice and wild-type littermates (TRPV4+/+). After intraplantar injection of inflammatory soup, the frequency of paw withdrawal was increased by 2.8-fold in TRPV4+/+ mice (30 ± 5% before and 84 ± 4% after inflammatory soup; n = 10; p < 0.0001, paired Student's t test), whereas it remained unaffected in TRPV4−/− mice (n = 12; p > 0.05, paired Student's t test).
Figure 2.

Inflammatory soup does not induce mechanical hyperalgesia in TRPV4−/− mice. A, The baseline withdrawal response frequency to von Frey hair stimulation was not significantly different between TRPV4+/+ and TRPV4−/− mice (n = 10 for TRPV4+/+ and n = 12 for TRPV4−/− mice; *p > 0.05, unpaired Student's t test). However, in the presence of inflammatory soup, the withdrawal response frequency was increased by 2.8-fold in TRPV4+/+ mice (30 ± 5% before and 84 ± 3% after inflammatory soup; n = 10; p < 0.0001, paired Student's t test), whereas the response remained unchanged in TRPV4−/− mice (n = 12; p > 0.05, paired Student's t test). B, Small-diameter DRG neurons from TRPV4+/+ and TRPV4−/− mice were first challenged with a 30% hypotonic solution for 3 min and then challenged with a 30% hypotonic solution containing the inflammatory soup (bradykinin, histamine, serotonin, substance P, and PGE2, 10 μm each). C, In TRPV4+/+ mice, the increase in [Ca2+]i induced by a 30% hypotonic challenge was significantly enhanced in the presence of inflammatory soup (1.7 ± 0.1 μm before and 2.3 ± 0.2 μm after the soup; n = 23; p < 0.05, paired Student's t test). In contrast, in TRPV4−/− mice, the response of DRG neurons to hypotonicity was unaffected (n = 21; p > 0.05, paired Student's t test). Iso, Isotonic solution; caps, capsaicin.
TRPV4 mediates the direct sensitizing action of inflammatory mediators on sensory neurons
To test whether the inflammatory soup-induced enhancement of TRPV4-dependent nociception can be explained by a direct action of the soup on primary afferent nerve endings, we measured calcium ion influx in sensory neurons isolated in vitro. Small-diameter DRG neurons (≤25 μm) were isolated from TRPV4+/+ and TRPV4−/− mice. As shown in Figure 2B, neurons were first challenged with a 30% hypotonic solution (212 mOsm) for 3 min and then challenged with a 30% hypotonic solution containing the inflammatory soup (212 mOsm). At the end of each experiment, neurons were identified as putative nociceptors if application of capsaicin (1 μm, 10 s) induced a significant increase in the concentration of free [Ca2+]i; all neurons included in the analysis were capsaicin responsive. In agreement with our previous results (Alessandri-Haber et al., 2005), hypotonicity induced a significantly smaller increase in [Ca2+]i (∼16%) in neurons from TRPV4−/− mice compared with those from TRPV4+/+ mice (1.46 ± 0.05 μm, n = 21 for TRPV4−/− mice vs 1.7 ± 0.1 μm, n = 23 for TRPV4+/+ mice; p < 0.05, unpaired Student's t test). The difference in the response to hypotonic stimuli between the TRPV4 genotypes is not large but is in agreement with our hypothesis; although TRPV4 participates in osmosensation in DRG neurons, it is not, however, a major osmosensor under physiological conditions. Our current and previous results suggest that TRPV4 plays a central role in the transduction of hypotonic- or hypertonic-induced nociceptive behavior only during inflammatory or neuropathic states (Alessandri-Haber et al., 2003, 2004, 2005).
Indeed, as shown in Figure 2C, inflammatory soup caused a 35% enhancement of the hypotonicity-induced increase in free [Ca2+] in TRPV4+/+ mice (n = 23; p < 0.05, paired Student's t test), whereas the inflammatory soup-induced enhancement was absent in TRPV4−/− mice (n = 21; p > 0.05, paired Student's t test). Moreover, in the presence of inflammatory soup, the [Ca2+]i in neurons from TRPV4+/+ mice was 1.6-fold the [Ca2+]i level in neurons from TRPV4−/− mice (2.3 ± 0.2 μm, n = 21 for TRPV4+/+ mice vs 1.41 ± 0.07, n = 21 for TRPV4−/− mice) (Fig. 2C).
The increase in the [Ca2+]i response to hypotonicity in neurons from TRPV4+/+ mice after a challenge with inflammatory soup was not large, but the response was probably underestimated because of the following: (1) although we record from a subpopulation of DRG neurons (i.e., diameter of ≤25 μm and capsaicin responsive), most of these neurons do not express TRPV4, and (2) in our experimental conditions designed to minimize activation of other ions channels (i.e., HEPES buffer, room temperature, and low sodium concentration), the response of TRPV4 to hypotonicity was probably not maximized. For example, it has been shown that increasing temperature to physiological levels potentiates the response of TRPV4 to hypotonicity or other stimuli (Liedtke et al., 2000; Guler et al., 2002; Gao et al., 2003). Importantly, DRG neurons from TRPV4−/− mice do not show an increase in [Ca2+]i in the presence of inflammatory soup. Therefore, we consider this experiment to be more a qualitative measure (inflammatory soup increases the [Ca2+]i or does not) rather than a quantitative measurement of an increase in [Ca2+]i. These data suggest that the inflammatory mediator-enhanced TRPV4-dependent nociceptive behavior in response to osmotic stimuli is the consequence of the direct action of the mediators on primary afferent neurons.
The combined action of multiple inflammatory mediators is required to engage TRPV4-dependent mechanisms of hyperalgesia
We attempted to identify which mediators are responsible for engaging TRPV4, hypothesizing that some components of the inflammatory soup, when administered alone, would elicit TRPV4-dependent hyperalgesia. As expected, intradermal injection of PGE2 or 5-HT alone induced hyperalgesia to mechanical stimuli (Fig. 3A). However, treatment with TRPV4 antisense had no effect on the PGE2-induced mechanical hyperalgesia (100 ng of PGE2, n = 26 for antisense and mismatch treated) (Fig. 3A). Conceivably, this failure to engage TRPV4-dependent mechanisms of hyperalgesia occurred because the dose of PGE2 was insufficient. Therefore, we investigated the effect of 1 μg of PGE2, a dose that elicits the maximal hyperalgesic effect of this mediator (Khasar et al., 1994). Hyperalgesia induced by 1 μg of PGE2 was somewhat greater compared with that induced by 100 ng of PGE2 (32 ± 0.7%, n = 48 for 100 ng of PGE2 vs 37 ± 1%, n = 12 for 1 μg of PGE2; p < 0.001, unpaired Student's t test). However, treatment with TRPV4 antisense still had no significant effect on the mechanical hyperalgesia induced by the higher dose of PGE2 (Fig. 3A). Similarly, mechanical hyperalgesia induced by 5-HT was also not affected by treatment with TRPV4 antisense (Fig. 3A). We then tested whether the combined administration of PGE2 and 5-HT (each at a submaximal dose of 100 ng) would be sufficient to induce TRPV4-dependent mechanical hyperalgesia, and, as shown on Figure 3A, treatment with TRPV4 antisense prevented the mechanical hyperalgesia (n = 8 for each ODN group; p < 0.05, unpaired Student's t test).
Figure 3.

TRPV4 participation in mechanical hyperalgesia requires a threshold level of cAMP reached only by combined mediators. A, Injection of 1 μg of PGE2 in rat hindpaw induced a slightly greater mechanical hyperalgesia compared with the injection of 100 ng of PGE2 (mean ± SEM; 32 ± 0.7%, n = 48 for 100 ng of PGE2 vs 37 ± 1%, n = 12 for 1 μg of PGE2; p < 0.001, unpaired Student's t test). However, treatment with antisense ODN for TRPV4 did not reduce the100 ng nor the 1 μg PGE2-induced mechanical hyperalgesia (n = 26 for antisense- and mismatch-treated for 100 ng of PGE2 and n = 6 for 1 μg PGE2 antisense- and mismatch-treated rats; p > 0.05, unpaired Student's t test). Injection of 100 ng or 1 μg of 5-HT in rat hindpaw induced a similar level of mechanical hyperalgesia (n = 6; p > 0.05, unpaired Student's t test). Again, treatment with antisense ODN for TRPV4 did not reduce the 5-HT-induced mechanical hyperalgesia (n = 6 for each ODN group; p > 0.05, unpaired Student's t test). In contrast, treatment with antisense ODN for TRPV4 prevented the decrease in paw-withdrawal threshold induced by the coinjection of PGE2 and 5-HT (100 ng each; 0.6 ± 1.5%, n = 8 for antisense-treated and 33 ± 1%, n = 8 for mismatch-treated rats; *p < 0.05, unpaired Student's t test). B, Intraplantar injection of PGE2 or 5-HT (100 ng and 1 μg each) similarly enhanced the withdrawal response frequency to mechanical stimuli in both TRPV4+/+ and TRPV4−/− mice. In TRPV4+/+ mice, the coinjection of PGE2 and 5-HT (100 ng each) induced a 3.9-fold increase in the withdrawal frequency response (23 ± 3%, n = 18 before and 90 ± 4%, n = 6 after coinjection of PGE2 and 5-HT). In contrast, in TRPV4−/− mice, the withdrawal response frequency after the coinjection of PGE2 and 5-HT was not different from baseline (n = 6 after and n = 23 before treatment; *p > 0.05, unpaired Student's t test).
As an independent confirmation, we investigated the withdrawal response frequency of TRPV4+/+ and TRPV4−/− mice to mechanical stimuli after intraplantar injection of PGE2 (100 ng and 1 μg), 5-HT (100 ng and 1 μg), or PGE2 plus 5-HT (100 ng each). Pretreatment with PGE2 or 5-HT (at both concentration) induced a similar 2.8-fold increase in the withdrawal response frequency in both TRPV4+/+ and TRPV4−/− mice (n = 6 for 100 ng and 1 μg of PGE2 and 1 μg of 5-HT; n = 8 for 100 ng of 5-HT; p > 0.05, ANOVA) (Fig. 3B). In contrast, whereas in TRPV4+/+ mice the coinjection of PGE2 and 5-HT at 100 ng each induced a 3.9-fold increase in the withdrawal frequency response, in TRPV4−/−, mice the withdrawal response frequency was not different from baseline. Thus, although PGE2 and 5-HT separately are unable to engage TRPV4, together they can.
In addition, intraplantar injection of bradykinin, substance P, or histamine (100 ng each) induced a similar 2.5-fold increase in the withdrawal frequency in TRPV4−/− mice (73 ± 4%, n = 6 for bradykinin; 77 ± 8%, n = 6 for histamine; and 72 ± 5%, n = 8 for substance P). Therefore, we conclude that only the concerted action of more than one inflammatory mediator can engage TRPV4 in mechanical hyperalgesia.
TRPV4 activation appears to require a threshold level of cAMP, attainable only with combined mediators
Mechanical hyperalgesia induced by the soup of inflammatory mediators depends in part on the cAMP/PKA second-messenger pathway (Lotz et al., 1987; Taiwo and Levine, 1988; Taiwo et al., 1989, 1992; Malmberg et al., 1997; Aley and Levine, 1999; Cunha et al., 1999). PGE2 and 5-HT, in particular, also induce hyperalgesia via the cAMP/PKA pathway when administered alone (Taiwo et al., 1989, 1992; Cardenas et al., 1997; Aley and Levine, 1999). Therefore, we hypothesized that the increase in cAMP induced by either PGE2 or 5-HT alone is insufficient to activate TRPV4-dependent mechanisms of hyperalgesia; instead, simultaneous action of multiple inflammatory mediators would be required to achieve sufficient activation of the cAMP pathway to engage TRPV4. Our hypothesis suggests that a threshold level of cAMP must be achieved before TRPV4-dependent hyperalgesia can be produced. Therefore, we tested whether hyperalgesia produced by an exogenous membrane-permeable cAMP analog, 8-Br-cAMP, exhibits such features in its dose–response relationship. As shown in Figure 4A, intradermal injection of 8-Br-cAMP (0.1, 0.3, 1, 3, 10, 100, 300, and 1000 ng) dose dependently induced mechanical hyperalgesia starting at the dose of 3 ng (13 ± 2%; n = 8) and reaching a maximal effect at a dose of 100 ng (34 ± 3%; n = 8). TRPV4 antisense reduced the dose-dependent mechanical hyperalgesia induced by 8-Br-cAMP; the hyperalgesia induced by 100 ng of 8-Br-cAMP was reduced by 38% in antisense-treated compared with mismatch-treated rats (30 ± 3%, n = 8 for mismatch-treated rats; 19 ± 2%, n = 8 for antisense-treated rats; p < 0.05, unpaired Student's t test) (Fig. 4A). Notably, sensitivity to TRPV4 antisense only became evident at the doses of 10–1000 ng. We conclude that a threshold level of cAMP is necessary to engage TRPV4-dependent mechanical hyperalgesia.
Figure 4.
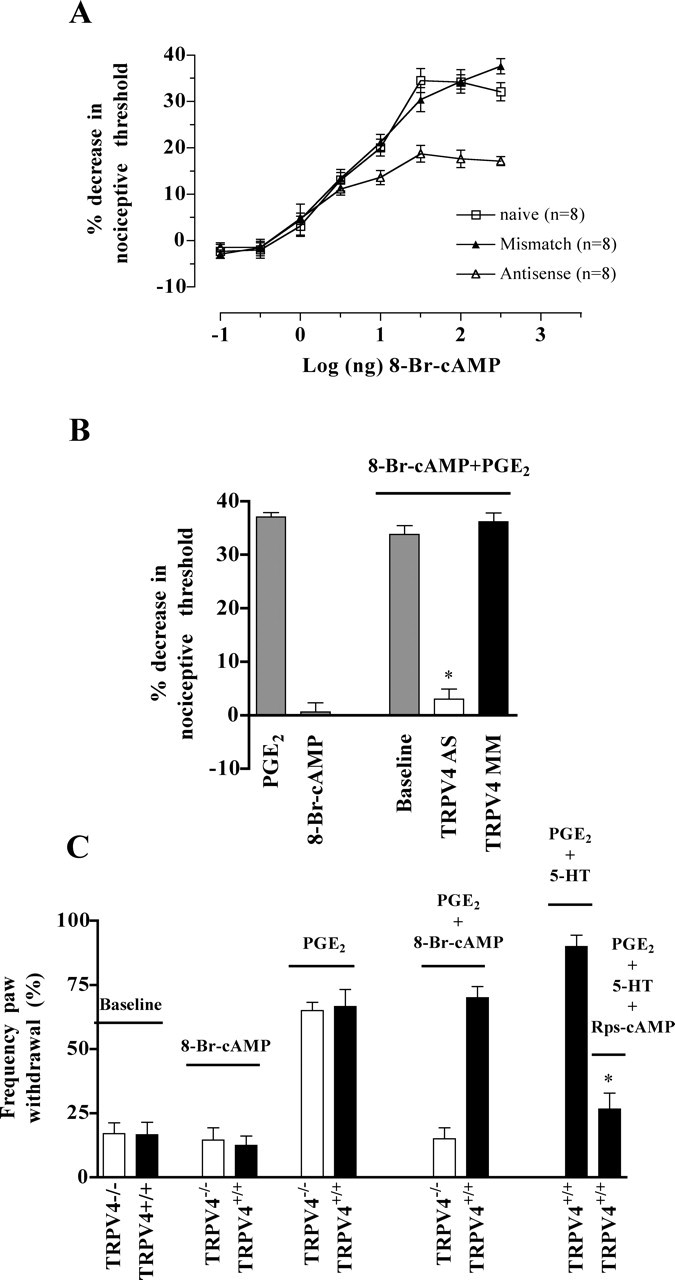
PGE2-induced hyperalgesia switches to a TRPV4-dependent mechanism when a sufficient level of cAMP is reached. A, Rats were treated with either TRPV4 antisense or mismatch ODN for 3 d, and mechanical nociceptive thresholds were evaluated before and 15 min after injection of increasing doses of 8-Br-cAMP (0.1, 0.3, 1, 3, 10, 100, 300, and 1000 ng). The mechanical hyperalgesia induced by 10–1000 ng of 8-Br-cAMP was reduced in TRPV4 antisense-treated compared with mismatch-treated rats. B, Intradermal injection of 1 μg of PGE2 induced a significant decrease in mechanical threshold. The injection of 1 ng of 8-Br-cAMP had no effect on mechanical nociceptive threshold and injection of 1 ng of 8-Br-cAMP 30 min before the injection of 1 μg of PGE2 did not enhance PGE2-induced mechanical hyperalgesia (37 ± 1%, n = 12 for PGE2 alone and 34 ± 2%, n = 12 for 8-Br-cAMP plus PGE2; p > 0.05, unpaired Student's t test). However, in the presence of 8-Br-cAMP, treatment with TRPV4 antisense ODN prevented the PGE2-induced mechanical hyperalgesia (3 ± 2%, n = 12 for antisense-treated vs 36 ± 2%, n = 8 for mismatch-treated rats; *p < 0.0001, unpaired Student's t test). C, Intraplantar injection of 1 ng of 8-Br-cAMP did not affect the withdrawal response frequency in TRPV4+/+ and TRPV4−/− mice and injection of 100 ng of PGE2 induced a threefold increase in the withdrawal response frequency in both TRPV4 genotypes. In contrast, after pretreatment with 1 ng of 8-Br-cAMP, the mechanical hyperalgesia induced by 100 ng of PGE2 was absent in TRPV4−/− mice (n = 12). Furthermore, in the presence of the inhibitor of activation by cAMP of cAMP-dependent protein kinase I and II, Rp-cAMPS, coinjection of PGE2 and 5-HT (100 ng each) failed to increase the withdrawal response frequency in TRPV4+/+ mice (27 ± 6%, n = 12 for PGE2 plus 5-HT plus Rp-cAMPS compared with 70 ± 4%, n = 6 for PGE2 plus 5-HT; *p < 0.05, unpaired Student's t test).
If our hypothesis is correct, then injection of a low dose of 8-Br-cAMP (1 ng) to boost the intracellular level of cAMP activity might enable 1 μg of PGE2 to engage TRPV4-dependent mechanisms of hyperalgesia. As shown in Figure 4, A and B, the 1 ng dose of 8-Br-cAMP alone had no effect on the mechanical nociceptive threshold. In addition, 1 ng of 8-Br-cAMP did not increase the magnitude of PGE2-induced mechanical hyperalgesia (Fig. 4B). However, pretreatment with 1 ng of 8-Br-cAMP caused the hyperalgesia induced by PGE2 to become sensitive to treatment with TRPV4 antisense (3 ± 2%, n = 12 for antisense-treated vs 36 ± 2%, n = 8 for mismatch-treated rats; p < 0.0001, unpaired Student's t test) (Fig. 4B).
Furthermore, 1 ng of 8-Br-cAMP had no effect on the baseline withdrawal response frequency in either TRPV4+/+ or TRPV4−/− mice. In contrast, after pretreatment with 8-Br-cAMP, the PGE2-induced enhancement in the withdrawal response frequency was absent in TRPV4−/− mice (15 ± 4%, n = 12 after pretreatment with 8-Br-cAMP and 65 ± 3%, n = 8 before) (Fig. 4C). In addition, coinjection of the inhibitor of activation by cAMP of cAMP-dependent protein kinase I and II, Rp-cAMPS, with PGE2 and 5-HT (100 ng each) prevented the enhancement in withdrawal response frequency in TRPV4+/+ mice (70 ± 4%, n = 6 for PGE2 plus 5-HT and 27 ± 6%, n = 12 for PGE2 plus 5-HT plus Rp-cAMPS; p < 0.05, unpaired Student's t test) (Fig. 4C).
These results are consistent with the hypothesis that the switch to a TRPV4-dependent mechanism of hyperalgesia occurs because the increase in cAMP concentration induced by PGE2 plus 5-HT (100 ng each) is higher than that induced by 1 μg of PGE2 or 5-HT alone. Levels of cAMP cannot be measured in primary afferent nerve endings in vivo, but we were able to use immunoassay methods in vitro to measure levels of cAMP accumulation in primary cultures of DRG neurons after exposure to inflammatory mediators.
We tested whether 100 μm PGE2 or 100 μm 5-HT would induce a lesser increase in intracellular cAMP than that induced by PGE2 plus 5-HT at 10 μm each (concentrations chosen to be consistent with our experiments on the effect of inflammatory soup on cultured DRG neurons). Indeed, the increase in the intracellular concentration of cAMP induced by 100 μm PGE2 or 100 μm 5-HT is significantly smaller than that induced by PGE2 plus 5-HT at 10 μm each (16 ± 3%, n = 10 after 100 μm PGE2; 12 ± 6%, n = 5 after 100 μm 5-HT; 25 ± 3%, n = 8 after 10 μm each PGE2 plus 5-HT; p < 0.05). These results suggest that, when a sufficient level of cAMP is reached, PGE2-induced hyperalgesia switches to a TRPV4-dependent mechanism, although the magnitude of hyperalgesia remains unchanged.
PKA appears to mediate cAMP activation of TRPV4-dependent hyperalgesia
An important intracellular target of cAMP is cAMP-dependent PKA, and we hypothesized that TRPV4 is modulated by the cAMP/PKA inflammatory mediator-activated second-messenger pathway. Therefore, we tested whether 8-Br-cAMP (1 μg, 2.5 μl) or PKACS (10 U/2.5 μl) would enhance TRPV4-dependent nociceptive responses in mice.
As shown in Figure 5A, intradermal injection of 8-Br-cAMP and PKACS in the hindpaw induced threefold and fourfold increases, respectively, in the number of flinches induced by hypotonicity in rat. TRPV4 antisense reduced the number of flinches induced by hypotonicity in the presence of 8-Br-cAMP or PKACS, respectively, by 41 and 48% (Fig. 5A).
Figure 5.

TRPV4 participates in cAMP/PKA-mediated hyperalgesia. A, 8-Br-cAMP and PKACS induced, respectively, a threefold and a fourfold increase in the number of flinches induced by hypotonicity in rat (12 ± 1, n = 14 after 8-Br-cAMP, 15 ± 1, n = 10 after PKACS vs 3.9 ± 0.3, n = 24 for hypotonicity alone). Treatment with TRPV4 antisense ODN reduced the number of flinches induced by hypotonicity in the presence of 8-Br-cAMP or PKACS, respectively, by 41% (7.2 ± 0.8, n = 8 for antisense-treated vs 12.2 ± 0.9, n = 8 for mismatch-treated rats; *p < 0.01, unpaired Student's t test) and 48% (8 ± 1, n = 8 for antisense-treated vs 16 ± 2, n = 7 for mismatch-treated rats; *p = 0.0001, unpaired Student's t test). B, Intraplantar injection of PKACS induced a 2.5-fold increase in the withdrawal response frequency in TRPV4+/+ mice (25 ± 6% before and 63 ± 54% in the presence of PKACS; n = 8; *p < 0.001, paired Student's t test). In contrast, PKACS did not affect the withdrawal response frequency in TRPV4−/− mice (n = 18; p > 0.05, paired Student's t test). C, Injection of 8-Br-cAMP or PKACS in rat hindpaw induced a similar degree of mechanical hyperalgesia (35 ± 2%, n = 6 for PKACS vs 35 ± 1%, n = 17 for 8-Br-cAMP; *p > 0.05, unpaired Student's t test). Treatment with TRPV4 antisense ODN attenuated both the 8-Br-cAMP- (4.9 ± 2.6%, n = 8 for antisense-treated vs 35.7 ± 1.3%, n = 8 for mismatch-treated rats; *p < 0.0001, unpaired Student's t test) and the PKACS-induced mechanical hyperalgesia (−3.9 ± 3.8%, n = 8 for antisense-treated vs 32.7 ± 2.5%, n = 8 for mismatch-treated rats; *p < 0.0001, unpaired Student's t test). In contrast, mismatch treatment had no effect on the decrease in mechanical threshold caused by PKACS or 8-Br-cAMP. D, Mean ± SEM of [Ca2+]i for DRG neurons first challenged with a 30% hypotonic solution and then challenged with a 30% hypotonic solution containing either Rp-cAMPS (30 μm) or H89 (10 μm). In TRPV4+/+ mice, the hypotonicity-induced increase in [Ca2+]i was significantly reduced in the presence of Rp-cAMPS or H89 (1.2 ± 0.1 μm, n = 4 for Hypo plus H89, 1.3 ± 0.08 μm, n = 8 for Hypo plus Rp-cAMPS vs 1.7 ± 0.1 μm, n = 12 for Hypo; *p < 0.05, Tukey's multiple comparison test). In contrast, these inhibitors did not significantly affect the response of DRG neurons to hypotonicity in TRPV4−/− mice (p > 0.05, ANOVA and Tukey's multiple comparison test).
We then investigated the withdrawal response frequency of TRPV4+/+ and TRPV4−/− mice to mechanical stimuli before and after injection of PKACS. Pretreatment with PKACS induced a 2.5-fold increase in the withdrawal response frequency in TRPV4+/+ mice (25 ± 6% before and 63 ± 4% in the presence of PKACS; n = 8; p < 0.001, paired Student's t test) (Fig. 5B). In contrast, PKACS had no significant effect on the withdrawal response frequency in TRPV4−/− mice (n = 18 before and after PKACS; p > 0.05, paired Student's t test).
In rat hindpaw, injection of PKACS induced a similar degree of mechanical hyperalgesia as did 8-Br-cAMP (35 ± 2% n = 6 for PKACS vs 35 ± 1%, n = 17 for 8-Br-cAMP; p > 0.05, unpaired Student's t test) (Fig. 5C), and treatment with TRPV4 antisense prevented the mechanical hyperalgesia induced by PKACS (−3.9 ± 3.8%, n = 8 for antisense-treated vs 32.7 ± 2.5%, n = 8 for mismatch-treated rats; p < 0.0001, unpaired Student's t test).
We further investigated whether TRPV4-dependent osmotransduction in dissociated DRG neurons in vitro depended on the cAMP/PKA pathway. DRG neurons (diameter of ≤25 μm) isolated from TRPV4+/+ and TRPV4−/− mice were first challenged with a 30% hypotonic solution and then with a 30% hypotonic solution containing the PKA inhibitors Rp-cAMPS (30 μm) or H89 (10 μm). As shown in Figure 5D, the hypotonic-induced response in TRPV4+/+ neurons was significantly reduced in the presence of Rp-cAMPS or H89 (n = 12 for Hypo; n = 8 Hypo plus Rp-cAMPS; n = 4 for Hypo plus H89; p < 0.05, Tukey's multiple comparison test). In contrast, neither inhibitor had a significant effect on the hypotonic-induced response of TRPV4−/− neurons (n = 16 for Hypo; n = 9 for Hypo plus Rp-cAMPS; n = 7 for Hypo plus H89; p > 0.05, Tukey's multiple comparison test). These findings suggest that TRPV4-mediated mechanisms of hyperalgesia are activated via the cAMP/PKA pathway.
TRPV4 also contributes to PKCϵ-dependent hyperalgesia
The PKCϵ second-messenger pathway is also required for full expression of inflammatory mediator-induced hyperalgesia (Khasar et al., 1994; Cesare et al., 1999). Therefore, we investigated whether PKCϵ-induced enhancement of hypotonic nociception also involves TRPV4-dependent mechanisms. The number of flinches induced by hypotonicity in rat hindpaw was increased by 3.3-fold by pretreatment with the PKCϵ activator ψϵ-RACK (13 ± 2, n = 6 after ψϵ-RACK vs 3.9 ± 0.3, n = 24 before; p < 0.0001, unpaired Student's t test) (Fig. 6A). As shown in Figure 6A, treatment with TRPV4 antisense reduced the number of flinches by 46% compared with mismatch.
Figure 6.

TRPV4 participates in PKCϵ-mediated hyperalgesia. A, The number of flinches induced by hypotonic stimulation was 3.3-fold higher in the presence of the PKCϵ activator ψϵ-RACK (13 ± 2, n = 6 after ψϵ-RACK vs 3.9 ± 0.3, n = 24 before; p < 0.0001, unpaired Student's t test). Treatment with TRPV4 antisense reduced the number of flinches by 46% compared with mismatch-treated rats (5 ± 0.4, n = 6 for antisense-treated vs 11 ± 2, n = 6 for mismatch-treated rats; *p < 0.02, unpaired Student's t test). B, Mean ± SEM of [Ca2+]i. DRG neurons from TRPV4+/+ and TRPV4−/− mice were first challenged with a 30% hypotonic solution and then challenged with a 30% hypotonic solution containing the PKCϵ translocation inhibitor peptide (2 μm). The hypotonicity-induced increase in [Ca2+]i was significantly reduced in the presence of PKCϵ translocation inhibitor peptide in TRPV4+/+ mice (1.3 ± 0.01 μm in the presence of PKCϵ inhibitor vs 1.7 ± 0.1 μm without; n = 13; *p < 0.001, paired Student's t test). In contrast, the inhibitor had no effect on the response of DRG neurons to hypotonicity in TRPV4−/− mice (n = 12; p > 0.05, paired Student's t test). C, Treatment with TRPV4 antisense attenuated the decrease in paw-withdrawal threshold induced by the injection of ψϵ-RACK (1 μg, −0.03 ± 2.10%, n = 6 for antisense-treated vs 28.2 ± 1.7%, n = 6 for mismatch-treated rats; *p < 0.0001, unpaired Student's t test). D, Intraplantar injection of ψϵ-RACK increased the withdrawal response frequency by 2.6-fold in TRPV4+/+ mice (32 ± 5% before and 83 ± 4% in the presence of ψϵ-RACK; n = 12; *p < 0.0001, unpaired Student's t test). In contrast, it did not affect the response in TRPV4−/− mice (n = 14; p > 0.05, unpaired Student's t test).
We then investigated whether PKCϵ contributes to TRPV4-mediated osmotransduction in DRG neurons. Neurons isolated from TRPV4+/+ and TRPV4−/− mice were first challenged with a 30% hypotonic solution and then with a 30% hypotonic solution containing the PKCϵ translocation inhibitor peptide PKCϵΙ (2 μm). As shown in Figure 6B, the [Ca2+]i was significantly reduced in the presence of PKCϵΙ in TRPV4+/+ mice (1.3 ± 0.1 μm in the presence of PKCϵΙ vs 1.7 ± 0.1 μm without; n = 13; p < 0.001, paired Student's t test). In contrast, PKCϵΙ had no significant effect on the response of DRG neurons to hypotonicity in TRPV4−/− mice (n = 12; p > 0.05; paired Student's t test).
We next investigated whether TRPV4 participates in PKCϵ-induced mechanical hyperalgesia (Fig. 6C). Treatment with antisense ODN for TRPV4 attenuated the mechanical hyperalgesia induced by the injection of ψϵ-RACK in the rat hindpaw (−0.03 ± 2.10%, n = 6 for antisense-treated vs 28.2 ± 1.7%, n = 6 for mismatch-treated rats; p < 0.0001, unpaired Student's t test).
To confirm that PKCϵ-induced hyperalgesia involves a TRPV4 mechanism, we tested whether ψϵ-RACK-induced hyperalgesia is diminished in TRPV4 knock-out mice. As shown in Figure 6D, ψϵ-RACK increased the withdrawal response frequency by 2.6-fold in TRPV4+/+ mice (32 ± 5% without and 83 ± 4% in the presence of ψϵ-RACK; n = 12; p < 0.0001, paired Student's t test). In contrast, ψϵ-RACK had no significant effect on the withdrawal response frequency in TRPV4−/− mice (n = 14; p > 0.05, paired Student's t test). These findings suggest that PKCϵ-mediated component of hyperalgesia also involves a TRPV4 mechanism.
Discussion
After tissue injury, inflammatory mediators such as 5-HT, histamine, PGE2, and bradykinin directly sensitize primary afferent neurons, resulting in hyperalgesia. Nociceptors then display a lowered threshold of activation, an increased spontaneous activity, and an increased response to a suprathreshold stimulus (Levine and Reichling, 1999; Millan, 1999). It has been demonstrated that inflammatory mediators increase nociceptor excitability by modulating ion channels, including transducers (for review, see Millan, 1999).
We demonstrate here for the first time, using two different models (i.e., transient downregulation of the level of expression of TRPV4 protein in the rat and TRPV4 knock-out mice) that, after exposure to inflammatory mediators, TRPV4 plays a crucial role in mechanical hyperalgesia. Because of the difference in size and behavior between rat and mice, different behavioral tests were performed to measure the mechanical hyperalgesia induced by inflammatory mediators in the hindpaws of the two species. In both species, we demonstrate that the cAMP/PKA and the PKCϵ second-messenger pathways can engage TRPV4-dependent mechanical hyperalgesia. Our results suggest that the concerted action of inflammatory mediators is necessary to engage TRPV4 in mechanical hyperalgesia (i.e., each of the five inflammatory mediators tested alone does not engage TRPV4). Moreover, we show that at least two inflammatory mediators (i.e., PGE2 and 5-HT) can act synergistically through cAMP to engage TRPV4-dependent mechanical hyperalgesia. However, the PKCϵ pathway also participates in inflammatory mediator-induced hyperalgesia (Khasar et al., 1994; Cesare et al., 1999; Hucho et al., 2005), and the existence of crosstalk or interaction between the cAMP/PKA and PKC pathways has been demonstrated in sensory neurons (Sugita et al., 1997; Gu and Huang, 1998). In addition, the activation of PKC has been shown to potentiate the effect of PKA on sodium ion channels (Cantrell et al., 2002) and on the cystic fibrosis transmembrane conductance regulator channel (Chen et al., 2004). Therefore, we do not exclude a synergistic action of the inflammatory mediators through a cAMP- and PKCϵ-dependent interaction, and additional experiments will be conducted to investigate this hypothesis.
Using DRG neurons isolated in vitro, we provide evidence that the inflammatory mediator-induced mechanism of TRPV4-dependent hyper-responsiveness may reside entirely within the sensory neuron. However, we cannot rule out that TRPV4 located in non-neuronal cells in the skin (i.e., keratinocytes) might make some additional contribution to the hyperalgesia.
Of note, it was demonstrated recently that thermal hyperalgesia induced by carrageenan was significantly reduced in TRPV4−/− mice (Todaka et al., 2004). In addition, we demonstrated recently that TRPV4 also plays a major role in chemotherapy-induced mechanical hyperalgesia (Alessandri-Haber et al., 2004). Together with the present results, these observations suggest that TRPV4 has a central role in pathological pain induced by a variety of stimulus modalities rather than in normal discriminatory pain sensation.
Our data suggest that TRPV4 contributes specifically to pathological pain conditions because its activation requires relatively high levels of intracellular cAMP. It appears that such levels are not easily achieved by the action of a single inflammatory mediator, but that concerted action of multiple mediators (as would occur in inflamed tissues) is required. Consistent with this suggestion, it was shown that, although administration of cAMP analogs can lower mechanical thresholds in single cutaneous afferent of rat skin in vitro (Kress et al., 1996), PGE2 alone cannot (Lang et al., 1990).
TRPV4 is thought to be a transducer of noxious hypo-osmotic (Alessandri-Haber et al., 2003) and mechanical (Liedtke and Friedman, 2003; Suzuki et al., 2003a) stimuli. TRPV4 is expressed in small-diameter DRG neurons and in free nerve endings in the skin (Suzuki et al., 2003b). TRPV4 is a nonselective cation channel with an outwardly rectifying current–voltage relationship (Liedtke et al., 2000; Strotmann et al., 2000; Nilius et al., 2001; Voets et al., 2002) that lacks the hallmarks of a voltage-gated channel (Papazian et al., 1991; Mannikko et al., 2002; Lecar et al., 2003). Thus, the role of TRPV4 is most likely to contribute to hyperalgesia by enhancing transduction rather than by contributing directly to voltage-gated action potential currents.
TRPV4 is engaged by two key intracellular second-messenger pathways of inflammatory hyperalgesia
PKC has been shown to be involved in TRPV4 function in vitro (Gao et al., 2003; Xu et al., 2003), but this is the first report of a functional interaction between TRPV4 and a specific PKC isozyme. Interestingly, mice lacking either the PKCϵ gene (Khasar et al., 1999) or the TRPV4 gene (Suzuki et al., 2003a) exhibited diminished nociception in the acetic acid writhing test. Importantly, stimulation of the cAMP pathway can lead to PKCϵ translocation (Graness et al., 1997; Hucho et al., 2005). We have shown previously that, in sensitized nociceptors, the response of TRPV4 to mechanical and osmolar stimuli depend partly on Src tyrosine kinase phosphorylation (Alessandri-Haber et al., 2004, 2005). Of note, cAMP/PKA and PKCϵ phosphorylation pathways can both directly interact with the Src tyrosine kinase family (Ping et al., 1999, 2002; Purcell and Carew, 2001; Stork and Schmitt, 2002).
Although a putative PKA phosphorylation site in the TRPV4 N-terminal region has been described previously (Nilius et al., 2004), this is the first study reporting a functional interaction between TRPV4 and the PKA pathway. We demonstrate that PGE2-induced hyperalgesia to hypotonic stimuli is inhibited by 50% after treatment with antisense ODN for TRPV4 (Alessandri-Haber et al., 2003). Interestingly, we demonstrate that, to engage TRPV4 in inflammatory-induced mechanical hyperalgesia, a threshold level of cAMP is necessary. However, when this threshold is reached, mechanical hyperalgesia induced by a inflammatory mediator, such as PGE2, clearly switches from a TRPV4-independent mechanism to a completely TRPV4-dependent mechanism. This suggests that, although high levels of cAMP engage TRPV4 in mechanical hyperalgesia, inactivation of TRPV4-independent mechanisms must also occur. At this time, it is difficult to formulate a detailed hypothesis for the underlying cellular mechanism. Many G-protein-coupled receptors for hormones can signal through different G-proteins. For example, although most of the functions of the β2-adrenergic receptor are mediated through Gs-protein and the cAMP/PKA pathway, this receptor can also couple to Gi-protein, which is cAMP-independent and initiates other signaling events (Xiao et al., 1995; Daaka et al., 1997). Similarly, in the heart, lower concentrations (10 nm) of epinephrine can activate Gs (10 nm epinephrine), whereas higher concentrations (1–100 μm) activate Gi pathways (Heubach et al., 2004). In fact, we have demonstrated that the activation of a Gi/Ras/extracellular signal-regulated kinase signaling pathway produces inflammatory pain that is independent of PKA or PKCϵ pathways (Aley et al., 2001). Future experiments will further address this question.
Is TRPV4 involved in the function of “silent” nociceptors?
Mechanical hyperalgesia depends in part on recruitment of previously “silent” nociceptors (Handwerker et al., 1991; Kress et al., 1992; Schmelz et al., 1994; Schmidt et al., 1995; Millan, 1999). Thus, in the setting of inflammation or peripheral neuropathy, mechano-insensitive (silent) nociceptors become responsive (Martin et al., 1987; Habler et al., 1988; Schaible and Schmidt, 1988a; McMahon and Koltzenburg, 1990; Schmidt et al., 1995, 2000). Interestingly, hyperalgesia to tonic mechanical stimulation is closely correlated with the discharge pattern of mechano-insensitive C-fibers (Adriaensen et al., 1984; Schmidt et al., 1995, 2000; Andrew and Greenspan, 1999). Additionally, an active contribution of silent C-mechano-insensitive fibers in chronic pain was demonstrated recently (Orstavik et al., 2003).
The cellular mechanisms responsible for the evanescent nature of silent nociceptors are unknown. Our observations suggest that, under normal conditions, TRPV4 may be inactive as a mechanical transducer but that it may be activated by the combined action of multiple inflammatory mediators or in neuropathy (Alessandri-Haber et al., 2004). Conceivably, primary afferent nerve fibers that express TRPV4 as their only mechanical transducer would exhibit properties of silent nociceptors, becoming mechanically responsive only in pathological conditions. Indeed, in parallel to our observations, inflammation induced by carrageenan or by inflammatory soup activates silent nociceptors (Schaible and Schmidt, 1985, 1988a; Xu et al., 2000), but PGE2 alone is insufficient (Mizumura et al., 1987; Schaible and Schmidt, 1988b; Lang et al., 1990; Grubb et al., 1991; Handwerker et al., 1991; Kress et al., 1992).
Conclusion
This work confirms the hypothesis that TRPV4 plays a crucial role in the development of hyperalgesia in inflamed tissue. That a relatively high level of cAMP is required to activate TRPV4-dependent mechanisms of hyperalgesia may explain why this transducer makes little contribution to baseline nociception in normal tissue. Its predominant contribution to pathological pain raises the possibility that TRPV4 could present a promising target for the development of a novel class of analgesics that would selectively affect pathological pain.
Footnotes
This work was supported by National Institutes of Health Grant NS053880. We thank Drs. Wolfgang Liedtke and Jeffrey Friedman for kindly providing the TRPV4 knock-out mice.
References
- Adriaensen H, Gybels J, Handwerker HO, Van Hees J (1984). Nociceptor discharges and sensations due to prolonged noxious mechanical stimulation—a paradox. Hum Neurobiol 3:53–58. [PubMed] [Google Scholar]
- Alessandri-Haber N, Yeh JJ, Boyd AE, Parada CA, Chen X, Reichling DB, Levine JD (2003). Hypotonicity induces TRPV4-mediated nociception in rat. Neuron 39:497–511. [DOI] [PubMed] [Google Scholar]
- Alessandri-Haber N, Dina OA, Yeh JJ, Parada CA, Reichling DB, Levine JD (2004). Transient receptor potential vanilloid 4 is essential in chemotherapy-induced neuropathic pain in the rat. J Neurosci 24:4444–4452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alessandri-Haber N, Joseph E, Dina OA, Liedtke W, Levine JD (2005). TRPV4 mediates pain-related behavior induced by mild hypertonic stimuli in the presence of inflammatory mediator. Pain 118:70–79. [DOI] [PubMed] [Google Scholar]
- Aley KO, Levine JD (1999). Role of protein kinase A in the maintenance of inflammatory pain. J Neurosci 19:2181–2186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aley KO, Levine JD (2001). Rapid onset pain induced by intravenous streptozotocin in the rat. J Pain 2:146–150. [DOI] [PubMed] [Google Scholar]
- Aley KO, Martin A, McMahon T, Mok J, Levine JD, Messing RO (2001). Nociceptor sensitization by extracellular signal-regulated kinases. J Neurosci 21:6933–6939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrew D, Greenspan JD (1999). Peripheral coding of tonic mechanical cutaneous pain: comparison of nociceptor activity in rat and human psychophysics. J Neurophysiol 82:2641–2648. [DOI] [PubMed] [Google Scholar]
- Cantrell AR, Tibbs VC, Yu FH, Murphy BJ, Sharp EM, Qu Y, Catterall WA, Scheuer T (2002). Molecular mechanism of convergent regulation of brain Na+ channels by protein kinase C and protein kinase A anchored to AKAP-15. Mol Cell Neurosci 21:63–80. [DOI] [PubMed] [Google Scholar]
- Cardenas CG, Del Mar LP, Cooper BY, Scroggs RS (1997). 5-HT4 receptors couple positively to tetrodotoxin-insensitive sodium channels in a subpopulation of capsaicin-sensitive rat sensory neurons. J Neurosci 17:7181–7189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cesare P, Dekker LV, Sardini A, Parker PJ, McNaughton PA (1999). Specific involvement of PKC-epsilon in sensitization of the neuronal response to painful heat. Neuron 23:617–624. [DOI] [PubMed] [Google Scholar]
- Chen Y, Button B, Altenberg GA, Reuss L (2004). Potentiation of effect of PKA stimulation of Xenopus CFTR by activation of PKC: role of NBD2. Am J Physiol Cell Physiol 287:C1436–C1444. [DOI] [PubMed] [Google Scholar]
- Cunha FQ, Teixeira MM, Ferreira SH (1999). Pharmacological modulation of secondary mediator systems—cyclic AMP and cyclic GMP—on inflammatory hyperalgesia. Br J Pharmacol 127:671–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daaka Y, Luttrell LM, Lefkowitz RJ (1997). Switching of the coupling of the β2-adrenergic receptor to different G proteins by protein kinase A. Nature 390:88–91. [DOI] [PubMed] [Google Scholar]
- Gao X, Wu L, O'Neil RG (2003). Temperature-modulated diversity of TRPV4 channel gating: activation by physical stresses and phorbol ester derivatives through protein kinase C-dependent and -independent pathways. J Biol Chem 278:27129–27137. [DOI] [PubMed] [Google Scholar]
- Graness A, Adomeit A, Ludwig B, Muller WD, Kaufmann R, Liebmann C (1997). Novel bradykinin signalling events in PC-12 cells: stimulation of the cAMP pathway leads to cAMP-mediated translocation of protein kinase Cϵ. Biochem J 327:147–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grubb BD, Birrell GJ, McQueen DS, Iggo A (1991). The role of PGE2 in the sensitization of mechanoreceptors in normal and inflamed ankle joints of the rat. Exp Brain Res 84:383–392. [DOI] [PubMed] [Google Scholar]
- Gu Y, Huang LY (1998). Cross-modulation of glycine-activated Cl− channels by protein kinase C and cAMP-dependent protein kinase in the rat. J Physiol (Lond) 506:331–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guler AD, Lee H, Iida T, Shimizu I, Tominaga M, Caterina M (2002). Heat-evoked activation of the ion channel, TRPV4. J Neurosci 22:6408–6414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habler HJ, Janig W, Koltzenburg M (1988). A novel type of unmyelinated chemosensitive nociceptor in the acutely inflamed urinary bladder. Agents Actions 25:219–221. [DOI] [PubMed] [Google Scholar]
- Handwerker HO, Kilo S, Reeh PW (1991). Unresponsive afferent nerve fibres in the sural nerve of the rat. J Physiol (Lond) 435:229–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heubach JF, Ravens U, Kaumann AJ (2004). Epinephrine activates both Gs and Gi pathways, but norepinephrine activates only the Gs pathway through human β2-adrenoceptors overexpressed in mouse heart. Mol Pharmacol 65:1313–1322. [DOI] [PubMed] [Google Scholar]
- Houck CS, Khodorova A, Reale AM, Strichartz GR, Davar G (2004). Sensory fibers resistant to the actions of tetrodotoxin mediate nocifensive responses to local administration of endothelin-1 in rats. Pain 110:719–726. [DOI] [PubMed] [Google Scholar]
- Hucho TB, Dina OA, Levine JD (2005). Epac mediates a cAMP-to-PKC signaling in inflammatory pain: an isolectin B4+ neuron-specific mechanism. J Neurosci 25:6119–6126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khasar SG, Ho T, Green PG, Levine JD (1994). Comparison of prostaglandin E1- and prostaglandin E2-induced hyperalgesia in the rat. Neuroscience 62:345–350. [DOI] [PubMed] [Google Scholar]
- Khasar SG, Lin YH, Martin A, Dadgar J, McMahon T, Wang D, Hundle B, Aley KO, Isenberg W, McCarter G, Green PG, Hodge CW, Levine JD, Messing RO (1999). A novel nociceptor signaling pathway revealed in protein kinase Cϵ mutant mice. Neuron 24:253–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kress M, Koltzenburg M, Reeh PW, Handwerker HO (1992). Responsiveness and functional attributes of electrically localized terminals of cutaneous C-fibers in vivo and in vitro. J Neurophysiol 68:581–595. [DOI] [PubMed] [Google Scholar]
- Kress M, Rodl J, Reeh PW (1996). Stable analogues of cyclic AMP but not cyclic GMP sensitize unmyelinated primary afferents in rat skin to heat stimulation but not to inflammatory mediators, in vitro. Neuroscience 74:609–617. [DOI] [PubMed] [Google Scholar]
- Kress M, Reeh PW, Vyklicky L (1997). An interaction of inflammatory mediators and protons in small diameter dorsal root ganglion neurons of the rat. Neurosci Lett 224:37–40. [DOI] [PubMed] [Google Scholar]
- Lang E, Novak A, Reeh PW, Handwerker HO (1990). Chemosensitivity of fine afferents from rat skin in vitro. J Neurophysiol 63:887–901. [DOI] [PubMed] [Google Scholar]
- Lecar H, Larsson HP, Grabe M (2003). Electrostatic model of S4 motion in voltage-gated ion channels. Biophys J 85:2854–2864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine JD, Reichling D (1999). In: Peripheral mechanisms of inflammatory pain (Wall PD, Melzack R, Bonica JJ, eds) New York: Churchill Livingston.
- Liedtke W, Friedman JM (2003). Abnormal osmotic regulation in trpv4−/− mice. Proc Natl Acad Sci USA 100:13698–13703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liedtke W, Choe Y, Marti-Renom MA, Bell AM, Denis CS, Sali A, Hudspeth AJ, Friedman JM, Heller S (2000). Vanilloid receptor-related osmotically activated channel (VR-OAC), a candidate vertebrate osmoreceptor. Cell 103:525–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linhart O, Obreja O, Kress M (2003). The inflammatory mediators serotonin, prostaglandin E2 and bradykinin evoke calcium influx in rat sensory neurons. Neuroscience 118:69–74. [DOI] [PubMed] [Google Scholar]
- Lotz M, Carson DA, Vaughan JH (1987). Substance P activation of rheumatoid synoviocytes: neural pathway in pathogenesis of arthritis. Science 235:893–895. [DOI] [PubMed] [Google Scholar]
- Malmberg AB, Brandon EP, Idzerda RL, Liu H, McKnight GS, Basbaum AI (1997). Diminished inflammation and nociceptive pain with preservation of neuropathic pain in mice with a targeted mutation of the type I regulatory subunit of cAMP-dependent protein kinase. J Neurosci 17:7462–7470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mannikko R, Elinder F, Larsson HP (2002). Voltage-sensing mechanism is conserved among ion channels gated by opposite voltages. Nature 419:837–841. [DOI] [PubMed] [Google Scholar]
- Martin HA, Basbaum AI, Kwiat GC, Goetzl EJ, Levine JD (1987). Leukotriene and prostaglandin sensitization of cutaneous high-threshold C- and A-δ mechanonociceptors in the hairy skin of rat hindlimbs. Neuroscience 22:651–659. [DOI] [PubMed] [Google Scholar]
- McMahon SB, Koltzenburg M (1990). Novel classes of nociceptors: beyond Sherrington. Trends Neurosci 13:199–201. [DOI] [PubMed] [Google Scholar]
- Millan MJ (1999). The induction of pain: an integrative review. Prog Neurobiol 57:1–164. [DOI] [PubMed] [Google Scholar]
- Mizumura K, Sato J, Kumazawa T (1987). Effects of prostaglandins and other putative chemical intermediaries on the activity of canine testicular polymodal receptors studied in vitro. Pflügers Arch 408:565–572. [DOI] [PubMed] [Google Scholar]
- Nilius B, Prenen J, Wissenbach U, Bodding M, Droogmans G (2001). Differential activation of the volume-sensitive cation channel TRP12 (OTRPC4) and volume-regulated anion currents in HEK-293 cells. Pflügers Arch 443:227–233. [DOI] [PubMed] [Google Scholar]
- Nilius B, Vriens J, Prenen J, Droogmans G, Voets T (2004). TRPV4 calcium entry channel: a paradigm for gating diversity. Am J Physiol Cell Physiol 286:C195–C205. [DOI] [PubMed] [Google Scholar]
- Orstavik K, Weidner C, Schmidt R, Schmelz M, Hilliges M, Jorum E, Handwerker H, Torebjork E (2003). Pathological C-fibres in patients with a chronic painful condition. Brain 126:567–578. [DOI] [PubMed] [Google Scholar]
- Papazian DM, Timpe LC, Jan YN, Jan LY (1991). Alteration of voltage-dependence of Shaker potassium channel by mutations in the S4 sequence. Nature 349:305–310. [DOI] [PubMed] [Google Scholar]
- Ping P, Zhang J, Zheng YT, Li RC, Dawn B, Tang XL, Takano H, Balafanova Z, Bolli R (1999). Demonstration of selective protein kinase C-dependent activation of Src and Lck tyrosine kinases during ischemic preconditioning in conscious rabbits. Circ Res 85:542–550. [DOI] [PubMed] [Google Scholar]
- Ping P, Song C, Zhang J, Guo Y, Cao X, Li RC, Wu W, Vondriska TM, Pass JM, Tang XL, Pierce WM, Bolli R (2002). Formation of protein kinase Cϵ-Lck signaling modules confers cardioprotection. J Clin Invest 109:499–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purcell AL, Carew TJ (2001). Modulation of excitability in Aplysia tail sensory neurons by tyrosine kinases. J Neurophysiol 85:2398–2411. [DOI] [PubMed] [Google Scholar]
- Schaible HG, Schmidt RF (1985). Effects of an experimental arthritis on the sensory properties of fine articular afferent units. J Neurophysiol 54:1109–1122. [DOI] [PubMed] [Google Scholar]
- Schaible HG, Schmidt RF (1988a). Time course of mechanosensitivity changes in articular afferents during a developing experimental arthritis. J Neurophysiol 60:2180–2195. [DOI] [PubMed] [Google Scholar]
- Schaible HG, Schmidt RF (1988b). Excitation and sensitization of fine articular afferents from cat's knee joint by prostaglandin E2. J Physiol (Lond) 403:91–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmelz M, Schmidt R, Ringkamp M, Handwerker HO, Torebjork HE (1994). Sensitization of insensitive branches of C nociceptors in human skin. J Physiol 480:389–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt R, Schmelz M, Forster C, Ringkamp M, Torebjork E, Handwerker H (1995). Novel classes of responsive and unresponsive C nociceptors in human skin. J Neurosci 15:333–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt R, Schmelz M, Torebjork HE, Handwerker HO (2000). Mechano-insensitive nociceptors encode pain evoked by tonic pressure to human skin. Neuroscience 98:793–800. [DOI] [PubMed] [Google Scholar]
- Steen KH, Steen AE, Kreysel HW, Reeh PW (1996). Inflammatory mediators potentiate pain induced by experimental tissue acidosis. Pain 66:163–170. [DOI] [PubMed] [Google Scholar]
- Stork PJ, Schmitt JM (2002). Crosstalk between cAMP and MAP kinase signaling in the regulation of cell proliferation. Trends Cell Biol 12:258–266. [DOI] [PubMed] [Google Scholar]
- Strotmann R, Harteneck C, Nunnenmacher K, Schultz G, Plant TD (2000). OTRPC4, a nonselective cation channel that confers sensitivity to extracellular osmolarity. Nat Cell Biol 2:695–702. [DOI] [PubMed] [Google Scholar]
- Sugita S, Baxter DA, Byrne JH (1997). Modulation of a cAMP/protein kinase A cascade by protein kinase C in sensory neurons of Aplysia. J Neurosci 17:7237–7244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki M, Mizuno A, Kodaira K, Imai M (2003a). Impaired pressure sensation in mice lacking TRPV4. J Biol Chem 278:22664–22668. [DOI] [PubMed] [Google Scholar]
- Suzuki M, Watanabe Y, Oyama Y, Mizuno A, Kusano E, Hirao A, Ookawara S (2003b). Localization of mechanosensitive channel TRPV4 in mouse skin. Neurosci Lett 353:189–192. [DOI] [PubMed] [Google Scholar]
- Taiwo YO, Levine JD (1988). Characterization of the arachidonic acid metabolites mediating bradykinin and noradrenaline hyperalgesia. Brain Res 458:402–406. [DOI] [PubMed] [Google Scholar]
- Taiwo YO, Bjerknes LK, Goetzl EJ, Levine JD (1989). Mediation of primary afferent peripheral hyperalgesia by the cAMP second messenger system. Neuroscience 32:577–580. [DOI] [PubMed] [Google Scholar]
- Taiwo YO, Heller PH, Levine JD (1992). Mediation of serotonin hyperalgesia by the cAMP second messenger system. Neuroscience 48:479–483. [DOI] [PubMed] [Google Scholar]
- Todaka H, Taniguchi J, Satoh JI, Mizuno A, Suzuki M (2004). Warm temperature-sensitive transient receptor potential vanilloid 4 (TRPV4) plays an essential role in thermal hyperalgesia. J Biol Chem 279:35133–35138. [DOI] [PubMed] [Google Scholar]
- Voets T, Prenen J, Vriens J, Watanabe H, Janssens A, Wissenbach U, Bodding M, Droogmans G, Nilius B (2002). Molecular determinants of permeation through the cation channel TRPV4. J Biol Chem 277:33704–33710. [DOI] [PubMed] [Google Scholar]
- Vyklicky L, Knotkova-Urbancova H, Vitaskova Z, Vlachova V, Kress M, Reeh PW (1998). Inflammatory mediators at acidic pH activate capsaicin receptors in cultured sensory neurons from newborn rats. J Neurophysiol 79:670–676. [DOI] [PubMed] [Google Scholar]
- Xiao RP, Ji X, Lakatta EG (1995). Functional coupling of the β2-adrenoceptor to a pertussis toxin-sensitive G protein in cardiac myocytes. Mol Pharmacol 47:322–329. [PubMed] [Google Scholar]
- Xu F, Satoh E, Iijima T (2003). Protein kinase C-mediated Ca2+ entry in HEK 293 cells transiently expressing human TRPV4. Br J Pharmacol 140:413–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu GY, Huang LY, Zhao ZQ (2000). Activation of silent mechanoreceptive cat C and Aδ sensory neurons and their substance P expression following peripheral inflammation. J Physiol (Lond) 528:339–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang XY, Bai ZT, Chai ZF, Zhang JW, Liu Y, Ji YH (2003). Suppressive effects of BmK IT2 on nociceptive behavior and c-Fos expression in spinal cord induced by formalin. J Neurosci Res 74:167–173. [DOI] [PubMed] [Google Scholar]
- Zheng JH, Chen J (2001). Differential roles of spinal neurokinin 1/2 receptors in development of persistent spontaneous nociception and hyperalgesia induced by subcutaneous bee venom injection in the conscious rat. Neuropeptides 35:32–44. [DOI] [PubMed] [Google Scholar]