

Abstract
The NG2 chondroitin sulfate proteoglycan inhibits axon growth in vitro. Levels of NG2 increase rapidly in the glial scars that form at sites of CNS injury, suggesting that NG2 may inhibit axon regeneration. To determine the functions of NG2, we infused mixtures of neutralizing or non-neutralizing anti-NG2 monoclonal antibodies into the dorsally transected adult rat spinal cord and analyzed the regeneration of ascending mechanosensory axons anatomically. At 1 week after injury, ascending sensory axons in control animals terminated caudal to the lesion within an area containing dense deposits of NG2 immunoreactivity. In animals treated with the neutralizing anti-NG2 antibodies, labeled axons penetrated the caudal border of the lesion and grew into and beyond the lesion center. The low intrinsic growth capacity of adult neurons may also limit the ability of damaged axons to regenerate. To enhance growth, we combined antibody treatment with a peripheral nerve conditioning lesion. After a conditioning lesion and treatment with control, non-neutralizing antibodies, many sensory axons grew into the lesion core. These axons did not grow past the rostral border of the lesion; rather, they grew along the dorsal surface of the spinal cord and within any remaining pieces of the dorsal roots. In contrast, combining a peripheral nerve conditioning lesion with neutralizing anti-NG2 antibodies resulted in sensory axon regeneration past the glial scar and into the white matter rostral to the injury site. The combinatorial approach used here that neutralizes extrinsic inhibition and increases intrinsic growth results in anatomically correct axon regeneration, a prerequisite for functional recovery.
Keywords: NG2, chondroitin sulfate proteoglycans, glial scar, regeneration, sensory axons, spinal cord injury
Introduction
When the brain or spinal cord is injured, damaged axons fail to regrow and establish functional connections with their appropriate target cells. This failure is attributed to a low intrinsic capacity to regenerate, limited amounts of the trophic factors necessary for growth, and molecular features of the environment that suppress regeneration.
The principal environmental feature of the damaged CNS is the glial scar (Reier et al., 1983; Silver and Miller, 2004). This complex tissue acts as a physical and biochemical barrier to axon regeneration. Three different families of macromolecules are implicated in this function. They are the myelin-associated growth inhibitory molecules (Filbin, 2003), the semaphorin family (Pasterkamp et al., 1999, 2001; Moreau-Fauvarque et al., 2003), and several chondroitin sulfate proteoglycans (CSPGs) (Morgenstern et al., 2002).
Members of these molecular families inhibit axon extension in vitro and induce growth cone collapse, but their roles as inhibitors of regeneration in vivo is not fully established. Furthermore, myelin-associated inhibitors may not be available to interact with sprouting axons and inhibit their regeneration as a result of the death of oligodendrocytes after spinal cord injury (SCI) and the rapid clearance of myelin debris by macrophages (Frei et al., 2000; McTigue et al., 2001; Zai and Wrathall, 2005). Despite the increased expression of semaphorins after SCI (Pasterkamp et al., 1999; Frei et al., 2000; Zai and Wrathall, 2005), there is no functional or genetic evidence that these molecules play an inhibitory role.
Growth inhibitory CSPGs are expressed by several different reactive cell types and are associated with myelin (Niederost et al., 1999; Jones et al., 2003b; Tang et al., 2003). CSPG levels increase 24 h after injury and persist for up to 6 months. Treatments that either degrade the glycosaminoglycan (GAG) chains or prevent GAG chain assembly result in the enhanced regeneration of sensory and motor axons (Moon et al., 2001; Bradbury et al., 2002; Grimpe and Silver 2004). Because the removal of GAG chains will alter all species of CSPGs and may disrupt the structure of the extracellular matrix, which species of CSPGs contribute to the inhibitory nature of the glial scar remains an important question.
Here, we used an antibody-mediated neutralization approach to analyze the functions of NG2 after SCI (Schnell and Schwab, 1990). Levels of NG2 increase rapidly after CNS damage, and NG2 inhibits axon growth, repels growing axons, and induces growth cone collapse (Tan et al., 2005). Although these observations suggest that NG2 can contribute to a growth nonpermissive environment, there is scant in vivo functional data. Although an NG2 null mouse does not display enhanced regeneration from descending serotonergic efferents and calcitonin gene-related peptide-positive sensory afferents (de Castro et al., 2005), whether NG2 influences the regeneration of long-distance mechanosensory fibers remains unknown. Using panels of anti-NG2 monoclonal antibodies (mAbs), only some of which reverse the growth inhibitory functions of NG2, we show that neutralizing NG2 promotes the regrowth of mechanosensory afferents into the nonpermissive glial scar core. When combined with a previous peripheral nerve lesion to stimulate the intrinsic growth capacity of the neurons, anti-NG2 treatment results in axons growing past the glial scar and into the white matter rostral to the injury.
Materials and Methods
Animals.
All experiments conform to the university guidelines on the ethical use of animals and were approved by the Institutional Animal Care and Use Committee. Adult female Sprague Dawley rats (∼175–200 g) were anesthetized with a mixture of ketamine and xylazine. After a dorsal laminectomy at thoracic level 8, the dura was resected to expose the spinal cord. Using a microscissor, the spinal cord was transected bilaterally with the lesion extending ventrally to the depth of the central canal (∼1.5 mm). To deliver antibody solutions into the lesion site, a small piece of Gelfoam was soaked in 50 μl of the antibodies and placed over the lesion site (Yick et al., 2000). Animals received either a mixture of control, non-neutralizing anti-NG2 monoclonal antibodies (D31 and 132, 500 μg/ml of each; n = 5) or a mixture of function-neutralizing anti-NG2 monoclonal antibodies (69 and 147, also at 500 μg/ml each; n = 7). The wound was closed with 6-0 sutures, and the animals were kept warm and hydrated until they recovered from the anesthesia. Intensive postoperative care was performed for 72 h, including subcutaneous injections of lactated saline and bladder massage. The animals used in the first set of experiments were allowed to survive for 1 week after the operation.
A second set of animals were treated with chondroitinase ABC (n = 6; Seikagaku, Rockville, MD) or penicillinase control (n = 3; Sigma, St. Louis, MO) enzymes. For both groups, the spinal cord was transected with the methods described above. A pledglet of Gelfoam soaked in 10 μl of 2.5 U/ml of either enzyme solution was placed directly over the lesion site. These animals were allowed to survive for 1 week after the operation. To detect the digestion of the GAG chains of CSPGs in the spinal cord lesion, tissue was stained with a ΔDi-4s antibody (1:2000; Seikagaku), which reacts with the stub of chondroitin 4-sulfate that remains on the core protein.
A third set of animals was subjected to a combined treatment consisting of a peripheral nerve conditioning lesion (Neumann and Woolf, 1999) 1 week before a dorsal spinal cord transection and antibody application. Under anesthesia, the left sciatic nerve was exposed and a ligature was tied distal to the sciatic notch. The nerve was then transected below the ligature. One week later, all rats received a dorsal-over spinal cord lesion as described above and were treated with Gelfoam soaked in control, non-neutralizing anti-NG2 monoclonal antibodies (n = 6) or neutralizing anti-NG2 monoclonal antibodies (n = 7). Postoperative care was performed as above, and the animals were allowed to survive for another 4 weeks.
To demonstrate the presence of exogenous antibody within the lesioned tissue, animals (n = 9) were subjected to the dorsal-over spinal lesions and implanted with Gelfoam pledglets that had been soaked in PBS, normal rabbit IgG, or anti-NG2 monoclonal antibody 132 (1 mg/ml). In some cases, the animals were implanted with Gelfoam pledglets that had been soaked in anti-NG2 monoclonal antibody 132 that had been covalently conjugated with Alexa-Fluor488 (Invitrogen, Carlsbad, CA) according to the instructions of the manufacturer.
Axon tracing.
Four days before the end of each experiment, the animals were reanesthetized, and the left sciatic nerve was exposed. Two microliters of a 1.5% solution of cholera toxin B subunit (CTB) (List Biological Laboratories, Campbell, CA) were injected into the nerve using a hand-held Hamilton syringe equipped with a 32 gauge needle (Bradbury et al., 2002). The solution was injected over 1 min, and the needle was withdrawn over an additional 2 min. The wound was closed by suturing the overlying muscle and skin.
Because incomplete lesions would result in CTB labeling in the dorsal columns at spinal level C2 and in the gracile nucleus, tissue sections from level C2 from each animal were stained with the anti-CTB antibodies. Those animals with evidence of axon sparing (n = 3) were excluded from this study. As positive controls, two animals received an incomplete spinal cord transection lesion at T8 (<1 mm in depth) and two animals received sham surgery before injecting the left sciatic nerve with CTB.
Tissue processing and immunofluorescence staining.
Rats were deeply anesthetized with ketamine and xylazine and transcardially perfused with 250 ml of 0.1 m phosphate buffer (PB) at 37°C, followed by 300 ml of freshly prepared ice-cold paraformaldehyde solution (4% in 0.1 m PB). The spinal cord and hindbrain was removed, postfixed for 2 h at 4°C, and cryoprotected by immersion in 30% sucrose, 0.1 m PB at 4°C. An 8-mm-length block of spinal cord centered on the lesion was frozen in OCT (Triangle Biomedical Sciences, Durham, NC), and 100 serial sections (18 μm thick) in the sagittal plane through the lesion site were cut using a Microm HM505E cryostat and thaw mounted onto gelatinized glass slides. For each animal, a 3-mm-wide block of spinal cord from cervical level 2 was embedded in OCT, and 20-μm-thick sections in the coronal plane were collected. To analyze axon regeneration, 50 alternating sagittal sections from level T8 were immunofluorescently stained with antibodies against CTB to visualize the labeled sensory axons and with antibodies against glial fibrillary acidic protein (GFAP) to visualize the borders of the lesion site. The remaining sections were stained with various combinations of antibodies against NG2, nogoA, or CTB. Immunofluorescence staining was performed as described previously (Levine et al., 1993). The sections were washed in blocking solution [potassium PBS (KPBS), 0.1% Triton X-100, and 4% normal donkey serum) and incubated overnight at 4°C in the primary antibodies diluted in the same blocking solution. After washing in blocking solution, sections were incubated in fluorescent secondary antibodies (Invitrogen) for 90 min at room temperature. Slides were washed in clean KPBS and coverslipped with fluoromount-G (Southern Biotechnology, Birmingham, AL). The following primary antibodies were used: rat anti-MBP (1:100; Serotec, Oxford, UK), mouse anti-S100β (1:1000; Sigma), goat anti-CTB antibody (1:5000; List Biological Laboratories), rabbit anti-GFAP (1:2000; DakoCytomation, Carpinteria, CA), rabbit anti-NG2 (1:1000; Chemicon, Temecula, CA), mouse anti-NG2 (monoclonal antibody 132, 1:2000), mouse anti-ED1 (1:500; Serotec), rabbit anti-nogo (1:500; Alpha Diagnostic, San Antonio, TX), mouse anti-OX42 (1:1000; Serotec), and rabbit anti-fibronectin (1:1000; Sigma). The following secondary antibodies were used: FITC donkey anti-rat (1:500; Jackson ImmunoResearch, West Grove, PA), amino-4-methylcoumarin-3-acetic acid donkey anti-rabbit (1:200; Sigma), Alexa-Fluor594 donkey anti-goat (1:2000), Alexa-Fluor488 donkey anti-mouse (1:2000), and Alexa-Fluor488 donkey anti-rabbit (1:2000). All Alexa-Fluor-conjugated antibodies were obtained from Invitrogen. The sections were examined with a Zeiss (Oberkochen, Germany) Axioplan2 fluorescence microscope, and digital images were captured using a Zeiss Axiocam. Confocal microscopy was performed using a Zeiss LSM510 confocal microscope.
Image analysis.
To analyze and reconstruct the complete appearance of the lesions and the transected/regenerating axons, we used Neurolucida software (MicroBrightField, Colchester, VT). High-resolution (2600 × 2060) digital images of alternating sagittal sections covering a ∼1800 μm width of spinal cord that had been stained with antibodies against CTB and GFAP were aligned and traced. The anti-GFAP staining was used to define the borders of the lesion cavity that was projected onto a tracing of the outline of the spinal cord. All the CTB-labeled axons from each section were traced. The resulting 50 drawings were then aligned and projected to create the reconstructions shown in Figures 4 and 7. Those spinal cord sections that were not traceable because of poor tissue condition, etc. were omitted from each animal. To orient these reconstructions, a traced section taken from the middle of the left dorsal column showing the outline of the lesion cavity was allowed to remain with the composite of all of the CTB-labeled axons. To create the horizontal-plane drawings, these composites were turned on their x-axis, and the caudal/rostral borders in each section was marked and connected. Animated three-dimensional rendering of these drawings are available as supplemental figures (available at www.jneurosci.org as supplemental material).
Figure 4.

Neurolucida projections of the distribution of the CTB-labeled axons at 7 d after treatment with the anti-NG2 antibodies. Digital images of sagittal spinal cord sections were traced using Neurolucida software as described in Material and Methods. Each image is a complete reconstruction showing the distribution of all of the CTB-labeled axons and their orientation relative to the lesion site for each individual animal studied. The shape of the lesion (smooth black line) was traced from a single section taken from the approximate center of the left fasciculus gracilis. A, B, Sagittal orientation. C, D, Horizontal orientation; the jagged lines show the rostral and caudal borders of the lesions, and the straight line denotes the midline of the spinal cord. In all traces, caudal is to the right, and rostral is to the left. A, C, Control, non-neutralizing antibody treatment; B, D, neutralizing antibody treatment. Scale bars, 500 μm.
Figure 7.

Neurolucida projections of the distribution of the CTB-labeled axons after combined peripheral nerve conditioning lesions and antibody treatments. A, C, Treatment with non-neutralizing antibody. B, D, Treatment with neutralizing antibody. A and B are reconstructions in the sagittal plane; C and D are in the horizontal plane as in Figure 3. In A and B, the black line shows the shape of the lesion taken from the approximate center of the left fasciculus gracilis. In C and D, the thin jagged lines denote the rostral and caudal borders of the lesion, and the thick black line denotes the midline of the spinal cord. Each reconstruction shown is from a separate animal. Scale bars, 500 μm.
Quantitative methods.
Lesion size (length in the rostrocaudal axis and depth) was measured from digital images of four alternate sections taken from the approximate center of the left dorsal column that had been immunofluorescently stained as described above. The lesion depth was measured between the dorsal border of the spinal cord tissue and the most ventral point in the lesion cavity. The rostrocaudal axis length was the distance measured between the rostral and caudal GFAP-defined borders surrounding lesion cavity at the median depth (∼0.7 mm). Data from the 7 d and 4 week postinjury survival groups were pooled into treatment and control groups and compared statistically with the Student's t test.
The maximal extent of axon regeneration at 7 d after lesion was determined by measuring the length of the five longest CTB-labeled axons in each animal that received neutralizing anti-NG2 antibodies (total axons measured, n = 35). Axon length was measured from the center of the lesion cavity. The center of the lesion cavity was defined by the approximate midpoint of the rostrocaudal axis in the sagittal reconstruction section taken from the approximate center of the left dorsal column.
The effect of the different antibody treatments on axon growth 7 d after spinal cord transection was assessed by measuring axon density through the lesioned area. The reconstruction drawings described above that show the entirety of the CTB-labeled axons were divided into 25 100-μm-wide regions spanning a distance extending from 1.2 mm rostral to 1.25 mm caudal to the lesion center. The pixel density in each region was measured using MetaMorph software (Universal Imaging Corporation, West Chester, PA), and the values were normalized by setting the region in each animal with the highest pixel density to 100. The data for each individual animal within each group (control, non-neutralizing antibody-treated animals, n = 5; neutralizing antibody-treated animals, n = 7) was expressed as percentage of maximum density, and the means across all of the animals within each group at each point were compared statistically using a one-way repeated-measures ANOVA, followed by the Bonferroni's post hoc analysis. For supplemental Figure 1 (available at www.jneurosci.org as supplemental material), a similar analysis was used to compare the sensory axon growth between neutralizing antibody-treated animals and chondroitinase ABC-treated (n = 6) animals. In addition, similar methods were used for comparing the axon density distribution in animals that received the combination of a peripheral nerve conditioning lesion and non-neutralizing (n = 6) or neutralizing (n = 7) anti-NG2 monoclonal antibodies. In this latter case, however, five 250-μm-wide regions extending rostrally from the lesion center were analyzed using one-way repeated-measures ANOVA, followed by the Bonferroni's post hoc analysis. To measure the number of axons regenerating in the white matter rostral to the lesion site, one-pixel-wide vertical lines were projected onto the reconstructed Neurolucida drawings, and the CTB-labeled axons that intersected these lines were counted and scored for their dorsoventral position. Data was collected at eight 100-μm-spaced intervals between 1000 and 1700 μm. In each animal, these eight measurements were then averaged to represent data at four 200-μm-spaced intervals, which were compared between treatment groups with one-way repeated-measures ANOVA and Bonferroni's post hoc analysis. All statistical tests were performed at the α level of significance of 0.05. Data management and statistical analyses were performed using SigmaStat (version 3.0.1a; Jandel Scientific, Corte Madera, CA) and Microsoft Office Excel 2003 (Microsoft, Seattle, WA) and were graphed as mean ± SEM using SigmaPlot (version 8.02a; Systat Software, Point Richmond, CA).
Figure 1.

Reactivity of the anti-NG2 monoclonal antibodies. A, The indicated antibodies were used in either immunoblotting or immunoprecipitation experiments as described in Materials and Methods. NG2, The extracellular domain of the NG2 core protein produced as a myc/his fusion protein (200–300 ng/lane); M, CNS myelin (25 μg protein/lane); SC, spinal cord extracts. For immunoblotting, 60–70 μg of protein was electrophoresed per lane, and, for immunoprecipitation, antibody was incubated with 150 μg of protein extract. The horizontal arrows indicate the electrophoretic mobility of the NG2 core protein. B–F, To ensure that the monoclonal antibodies are delivered into the injured spinal cord, adult rat spinal cords were dorsally transected and treated with Gelfoam soaked in 50 μl of anti-NG2 mAb 132 (1 μg/μl). The animals were killed after 3 d and processed for immunofluorescence. B, mAb 132 diffuses through the lesion site and binds to blood vessels and small process-bearing cells. Staining with donkey anti-mouse Alexa-Fluor594. C, Gelfoam infusion of PBS vehicle reveals no specific staining with anti-mouse Alexa-Fluor594. D–F, High-power view of the region inside the dotted box in C. D, Gelfoam-infused mAb 132 is visualized using donkey anti-mouse Alexa-Fluor594. E, Staining of the same tissue section with rabbit anti-NG2 and donkey anti-rabbit Alexa-Fluor488. F, Merged image of D and E. An OPC-like cell is in the center of the image, and a blood vessel in the top right corner is noted by a white arrow. Scale bars: B, C, 200 μm; D–F, 100 μm.
Antibodies and biochemical methods.
The derivation and specificity of the four monoclonal antibodies used here have been described previously (Ughrin et al., 2003).
Both immunoblotting and immunoprecipitation were used to assess the reactivity of these antibodies against proteins found in rat spinal cord. For the immunoprecipitations, normal rat spinal cord was homogenized in 0.15 m NaCl, 10 mm Tris, pH 8.0, 1% Nonidet P-40 (NP-40), 2 mm phenylmethylsulfonyl fluoride, and 1 μg/ml leupeptin, and the homogenate was centrifuged at 100,000 × g for 30 min at 4°C. mAb D31 or 147 was added to the supernatants, which were incubated at 4°C for 2 h before collection of the immune complexes on protein G-Sepharose (Sigma). After washing, the immunoprecipitated proteins were separated on 6% polyacrylamide-SDS gels and electrophoretically transferred to nitrocellulose, and any NG2-like proteins were identified by blotting with rabbit anti-NG2 antibody (1 μg/ml) and ECL detection (Amersham Biosciences, Arlington Heights, IL). For immunoblotting, either the total NP-40 extract of spinal cord or a PBS-soluble extract (also a 100,000 × g supernatant) was used. All primary antibodies were used at 1 μg/ml. CNS myelin was prepared as described previously (Norton and Poduslo, 1973). As a positive control in these experiments, we used the extracellular domain of NG2 with a myc/6xhis tail (Ughrin et al., 2003).
Results
The NG2 core protein consists of a large extracellular domain, a single membrane-spanning domain, and a short cytoplasmic tail (Nishiyama et al., 1991). There is one chondroitin sulfate side chain, most likely attached to serine 999 (Stallcup and Dahlin-Huppe, 2001). The extracellular domain can be divided into three subdomains based on the predicted secondary structure. We previously generated a series of domain-specific monoclonal antibodies and characterized the ability of these antibodies to neutralize the inhibitory functions of NG2 using in vitro axon growth and growth cone collapse assays (Ughrin et al., 2003). mAb 69 is directed against domain 1 and neutralizes domain1-Fc fusion proteins. This antibody had no neutralizing effect on growth inhibition caused by MAG. mAb 147 is directed against domain 3 and has neutralizing activity, whereas mAb 132, also domain 3 specific, has no neutralizing activity. A fourth mAb, D31.20, is directed against domain 2 and has no neutralizing activity. These antibodies all react with recombinant NG2 and with the NG2 core protein present in extracts of normal adult rat spinal cord (Fig. 1A). mAb 132 recognizes NG2 on Western blots and does not react with any proteins present in CNS myelin. This antibody also precipitates NG2 from spinal cord extracts (data not shown). mAb 147 recognizes the NG2 core protein and several lower-molecular-weight species on Western blots. These are likely to be NG2-degradation products because they are sometimes observed on Western blots with the other anti-NG2 mAbs. Trace amounts of these putative breakdown products are present in myelin. This antibody also immunoprecipitates a single anti-NG2 reactive polypeptide with an electrophoretic mobility identical to that of recombinant NG2. mAb 69 also binds to NG2 on Western blots but does not precipitate NG2 efficiently. mAb D31.10 effectively precipitates NG2 from spinal cord extracts but fails to recognize NG2 on Western blots. The ability of all four mAbs to bind to NG2 within extracts of spinal cord demonstrates that they are effective tools for perturbing NG2 function in vivo.
After injury, NG2 is present on cell surfaces and as a shed extracellular protein (Levine, 1994; Asher et al., 2005). In previous studies, we showed that, whereas mAb 69 (anti-D1) can neutralize the inhibitory effects of cell surface NG2, a combination of mAb 69 and mAb 147 (anti-D3) was required to neutralize effectively the inhibitory effects of a soluble protein comprising the entire extracellular domain of NG2 (Ughrin et al., 2003). Therefore, to test the functions of NG2, we infused combinations of either the two neutralizing (69 and 147) or the two non-neutralizing antibodies (132 and D31.10) into the lesioned spinal cord using diffusion from small Gelfoam pledglets as described in Materials and Methods. To confirm that antibody was present in the damaged tissue, we treated lesioned animals with mAb 132 and then stained the sections with an Alexa-Fluor-conjugated anti-mouse antibody. The antibody was present within the lesion core and bound to small process-bearing cells within the lesion penumbra (Fig. 1B). Antibody was also detected on blood vessels. Other than autofluorescence emanating from macrophages within the lesion core, there was no staining of cellular elements when the animals were treated with Gelfoam soaked in PBS (Fig. 1C). The antibody-positive, small process-bearing cells had the multipolar appearance characteristic of NG2-expressing cells in the adult rat spinal cord (Dawson et al., 2003). We confirmed that the infused antibody bound to cells expressing NG2 by double staining the tissue with an Alexa-Fluor-conjugated anti-mouse antibody and with rabbit anti-NG2 and an appropriately labeled secondary antibody. As shown in Figure 1D–F, the process-bearing cells and the blood vessels that bound the infused antibody were also stained with the rabbit anti-NG2 antibody. Antibody was present at the lesion site and not in tissue sections of spinal cord taken 2 cm from the damaged area (data not shown). Some animals were also treated with anti-NG2 antibodies that had been covalently labeled with Alexa-Fluor488. Antibody was detected at 4, 5, and 7 d after lesion but not at 12 and 14 d after injury (data not shown). Thus, diffusion out of Gelfoam pledglets provides a means of administering reagents to the injured spinal cord throughout a 1 week postinjury time period. Moreover, the antibodies recognized and bound to the NG2 proteoglycan expressed in both normal and injured spinal cord.
After a dorsal-over hemisection of spinal cord at level T8, a complex V-shaped glial scar formed. The lesion core was filled with NG2-positive cells (Figs. 2, 3). This NG2-positive cell population includes oligodendrocyte progenitor cells (OPCs), meningeal cells, endothelial cells, and macrophages (Jones et al., 2002; Tang et al., 2003). It is possible that some of the NG2-positive cells are non-myelinating Schwann cells, although the expression of NG2 by Schwann cells remains controversial (Martin et al., 2001; Schneider et al., 2001; Morgenstern et al., 2003).
Figure 2.
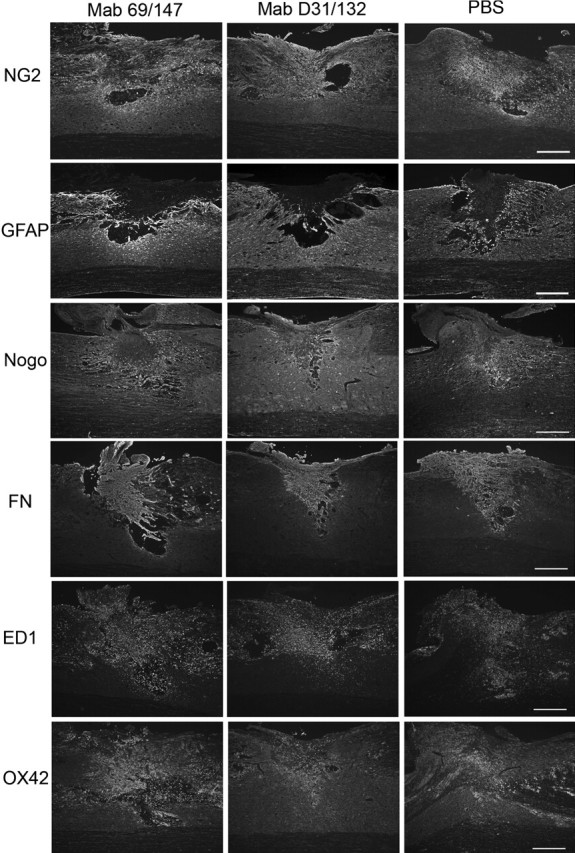
Appearance of the glial scar at 7 d after spinal cord transection and treatment with PBS, control, non-neutralizing anti-NG2 antibodies (D31/132), or neutralizing antibodies (69/147). Sagittal sections were stained with antibodies against NG2, GFAP, nogo, fibronectin (FN), ED1, and OX42. Scale bars, 500 μm.
Figure 3.
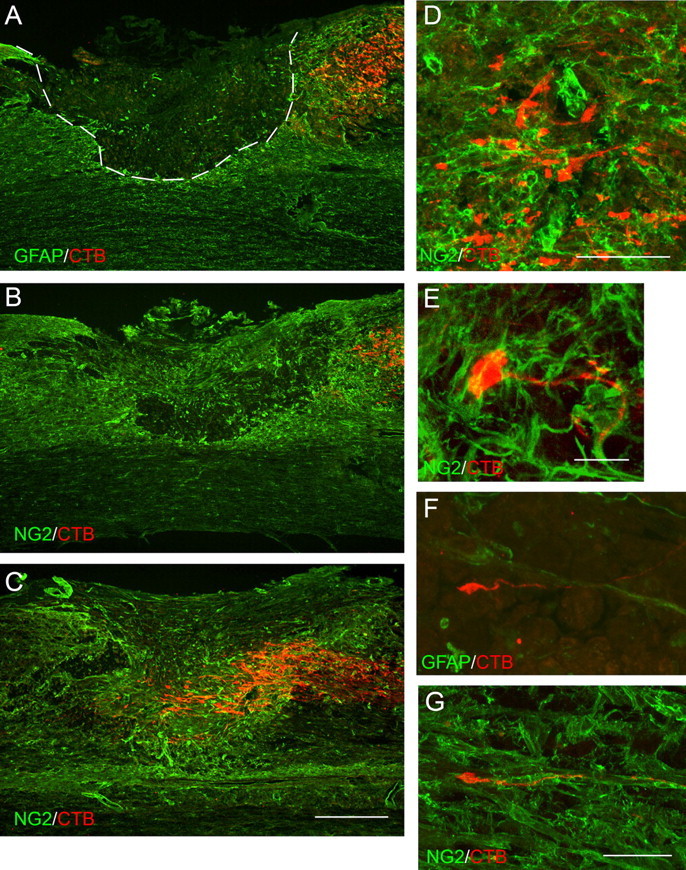
Neutralizing anti-NG2 monoclonal antibodies promote the regeneration of ascending mechanosensory axons. Adult rats were subjected to dorsal-over transection lesions at spinal level T8 and treated with either control, non-neutralizing anti-NG2 antibodies or neutralizing antibodies. The images shown are sagittal sections oriented so that caudal is to the right and rostral is to the left. A–C are taken from similar mediolateral positions in the spinal cord, slightly to the left of the central canal. A, Control, non-neutralizing antibody treatment. CTB-labeled axons (red) terminate caudal to the edge of the lesion, which is defined by anti-GFAP staining (green). The dashed line separates the central core from the lesion penumbra. B, Control, non-neutralizing antibody treatment. Note that anti-NG2 immunoreactivity is present within and around lesion cavity. Axons end in the NG2-rich penumbra. C, Axon distribution in animals treated with neutralizing anti-NG2 antibodies (red, CTB; green, NG2). Axons grow into and past the center of the lesion core, within regions containing high levels of anti-NG2 immunoreactivity. D, Nonregenerating axons (red) end in the NG2-enriched region (green) with irregular profiles and enlarged, bulb-like endings. E, A dystrophic end bulb (red) from a non-neutralizing antibody-treated animal is enmeshed in NG2-positive cellular processes (green). F, An axon with a growth cone-like ending growing in the lesion cavity; neutralizing antibody treatment. G, A regenerating axon growing along an NG2-positive small-diameter blood vessel; neutralizing antibody treatment. Scale bars: A–C, 500 μm; D, F, G, 100 μm; E, 20 μm. E–G are confocal micrographs.
To determine whether the infused antibodies had effects on scar formation, wound healing, and the cellular invasions that can occur after SCI, we compared the distribution of several cellular antigens in injured spinal cords that had been treated with PBS, neutralizing monoclonal anti-NG2 antibodies, or non-neutralizing antibodies for 7 d (Fig. 2). In all three treatment groups, increased NG2 immunoreactivity was present in the lesion core and the surrounding area. This surround contained reactive astrocytes and nogo-positive cellular elements. Large numbers of fibronectin-positive meningeal cells filled the lesion cavities. ED1-positive macrophages and OX42-positive microglia were present throughout the lesion site, in both the central core and the penumbra. No differences were observed in the distribution of these cellular antigens across the three treatment groups. The general size and shape of the lesions did not vary with the different antibody treatments (Table 1). These data suggest that the antibodies had no differential effects on the behavior of glial cells, meningeal cells, and macrophages and that the process of wound healing did not vary across the different treatments.
Table 1.
Lesion dimensions
| Treatment | Lesion length (μm) | Lesion depth (μm) |
|---|---|---|
| 7 d survival, non-neutralizing antibodies | 1252 ± 623 | 1590 ± 442 |
| 7 d survival, neutralizing antibodies | 1469 ± 556 | 1539 ± 637 |
| 28 d survival, non-neutralizing antibodies | 1792 ± 491 | 1459 ± 415 |
| 28 d survival, neutralizing antibodies | 1815 ± 520 | 1407 ± 340 |
Short-term treatment with neutralizing antibodies promotes regrowth
To observe axon distribution at the antibody-treated lesion sites, we injected CTB into the left sciatic nerve at 3 d after lesion and, 4 d later, killed the animals and prepared the tissue for immunofluorescence staining. Cholera toxin is taken up predominantly by medium- and large-diameter axons and transported transganglionically into the dorsal columns and brainstem (Lamotte et al., 1991). In control animals treated with non-neutralizing anti-NG2 antibodies, labeled mechanosensory axons ended within the NG2-rich penumbra region ∼400–600 μm caudal to the lesion center (Fig. 3A,B). These axons had irregular trajectories and dystrophic end bulbs that appear embedded in a meshwork of NG2-positive processes (Fig. 3D,E). When the lesioned animals were treated for 7 d with a mixture of neutralizing anti-NG2 antibodies, the labeled axons grew past the caudal penumbra and into the lesion center (Fig. 3C). Single axons often extended along GFAP-positive fibers and ended in hook-shaped growth cone-like endings (Fig. 3F). Some axons grew in association with blood vessels (Fig. 3G).
The data presented above demonstrate that treatment of acute spinal cord injuries with antibodies that neutralize NG2 function can promote the regeneration of ascending mechanosensory axons. It is possible, however, that some of the labeled axons we observed were surgically spared from the lesion, pushed aside during the lesioning, or grew around rather than through the usually nonpermissive glial scar (Steward et al., 2003). Potential surgical sparing was assessed by cutting and staining coronal sections through the cervical spinal cord with the anti-cholera toxin antibodies. No labeled axonal profiles were observed (data not shown). To determine whether the axons were growing around or through the damaged area, we reconstructed the trajectories of the CTB-labeled axons within a 2.4-mm-long span of the spinal cord centered on the lesion site using Neurolucida software as described in Materials and Methods. These reconstructions reveal the complete trajectories of the labeled axons into and through the lesioned area, the borders of which can be visualized by anti-GFAP immunostaining. Figure 4 shows such reconstructions for each individual case treated with either neutralizing or non-neutralizing antibodies (for more complete three-dimensional rendering of this data, see supplemental movies 1–4, available at www.jneurosci.org as supplemental material). In the control cases treated with non-neutralizing antibodies, most of the axons stopped within the penumbra and failed to enter the lesion core. In contrast, after treatment with neutralizing antibodies, axons grew through the central core region of the scar. When it was possible to resolve single axons, they had relatively straight trajectories with few branches. Regenerating axons were present throughout the dorsoventral extent of the scar and were not preferentially distributed under the lesion core. Figure 4, C and D, shows horizontal projections of these reconstructions for a typical control and experimental case. In the control antibody case shown, the axons approached but did not enter the central core. After treatment with the neutralizing antibodies, the axons grew through the lesion site. Some of the axons appeared to be growing within the contralateral dorsal funiculus, but there is not extensive growth around the injury site. These data show that the glial scar functions as a barrier to axon regeneration and that the treatment of lesioned animals with antibodies that neutralize NG2-induced growth inhibition can promote axon growth into that barrier.
To quantitatively compare the different treatments, we measured axon density within a 2.4-mm-long strip of spinal cord aligned around the lesion center. As shown in Figure 5, in animals treated with control, non-neutralizing antibodies, only a small number of axons grew to within 300 μm caudal to the lesion center. After treatment with neutralizing antibodies, labeled axons grew past the caudal penumbra and entered the lesion core. Some grew past the center of the core. The extent of regeneration promoted by the neutralizing anti-NG2 antibodies is comparable with the regeneration observed after treating the injured spinal cords with chondroitinase ABC (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). We also measured the five longest regenerating axons in each of the neutralizing antibody-treated animals (seven animals for a total of 35 axons); mean length past the lesion center was 478.5 ± 116.2 μm. This distance is appropriate for axon growth during a 1 week survival period (Steward et al., 2003).
Figure 5.

Axon density caudal and rostral to the lesion center from spinal cord-injured animals that were treated with either control, non-neutralizing (n = 5) or neutralizing anti-NG2 monoclonal antibodies (n = 7). Data shown is the mean ± SEM (∗p < 0.05, ∗∗p < 0.001, one-way repeated-measures ANOVA with Bonferroni's post hoc tests).
Combining anti-NG2 treatment with a peripheral nerve conditioning lesion
The recovery of neuronal function after SCI may require long-distance axon regeneration. The data presented above show that short-term treatment with anti-NG2 antibodies can promote axon growth into the nonpermissive scar region and that, after a 7 d survival period, the axons had grown several hundred micrometers. Continued growth of axons rostral to the lesion site is likely to be limited by the low intrinsic capacity of DRG neurons to support long-distance regeneration after SCI, as well as the presence of growth-inhibitory molecules. One established method for increasing the intrinsic growth state of DRG neurons is the peripheral nerve conditioning lesion paradigm described by Richardson and Issa (1984). A lesion of the sciatic nerve 2 or 7 d before a dorsal column lesion results in axon sprouting and growth at the lesion site (Neumann and Woolf, 1999; Neumann et al., 2002). This growth, however, is often ectopic, with the axons extending along the dorsal surface of the cord, within any remaining pieces of dorsal root, and within the gray matter ventral and lateral to the dorsal columns. We wondered therefore whether combining treatments that increase the intrinsic growth state of the neurons with the neutralizing antibodies might allow axons to grow long distances within the dorsal columns rostral to the lesion site. Figures 6 and 7 show the results of experiments in which rats were subjected to a complete transection of the left sciatic nerve 1 week before lesioning the dorsal spinal cord and implanting antibody-containing Gelfoam pledglets. When treated with control, non-neutralizing antibodies, lesion-primed mechanosensory axons grew past the caudal penumbra and entered the central core region of the glial scar (Fig. 6A). Once in the core region, they turned dorsally and laterally, often growing over the dorsal surface of the cord and into any remaining pieces of dorsal root (Fig. 6B). Although these axons extended for distances as long as 4 mm within the dorsal roots and the dorsal root entry zone, they failed to reenter the CNS. Lesion-primed axons growing in the CNS did not extend within the white matter rostral to the injury site. These observations are consistent with previous studies (Neumann and Woolf, 1999; Neumann et al., 2002; Steinmetz et al., 2005) and emphasize that the non-neutralizing antibodies have no direct effects on axon behavior.
Figure 6.
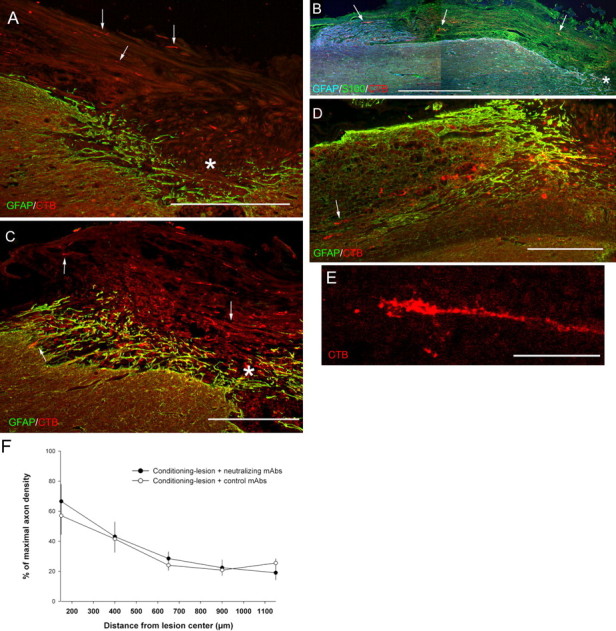
Combining neutralizing anti-NG2 antibody treatment with a peripheral nerve conditioning lesion promotes sensory axon growth into the white matter rostral to the lesion site. Section orientation and staining is the same as in Figure 2. All images are from animals that survived 28 d after spinal cord lesion. A, After a peripheral nerve conditioning lesion and treatment with non-neutralizing antibodies, CTB-labeled axons enter the lesion core (asterisk) and grow along the dorsal surface of the spinal cord, often within remaining pieces of dorsal roots (arrows). B, Control antibody-treated sensory axons (arrows, red) grow into dorsal roots that contain S100β-positive Schwann cells (green) but appear to stop as they enter the dorsal root entry zone, a region containing GFAP immunoreactivity (blue). The asterisk marks the lesion cavity. C, After a peripheral nerve conditioning lesion and treatment with neutralizing anti-NG2 antibodies, numerous axons grow into the lesion site (arrows and asterisk as in A). D, Neutralizing antibody treatment; lesion-primed axons have regenerated past the rostral edge of the lesion border and into the degenerating white matter rostral to the lesion. The arrow points to a growth cone within the white matter. E, An axon from an animal treated with the neutralizing antibodies that has grown within the white matter ∼4 mm rostral to the lesion center with an elongated growth cone-like ending. F, Measurements of axon density rostral to the lesion center in the conditioning-lesioned and antibody-treated animals. There are no significant differences in axon density rostral to the lesion site across antibody treatments (p ≥ 0.08–1.0, one-way ANOVA and Bonferroni's post hoc test). Data shown are the mean ± SEM. Scale bars: A–D, 500 μm; E, 50 μm. E is a confocal micrograph.
When neutralizing anti-NG2 antibodies were applied to the injured spinal cords of lesion-primed animals, the pattern of axon growth was different (Figs. 6, 7). As in control cases, the labeled mechanosensory axons entered the core region of the glial scar (Fig. 6C). In addition to turning dorsally and laterally, axons grew through the core region and entered the white matter rostral to the lesion (Fig. 6C,D). The regenerating axons had relatively straight-appearing axon shafts and growth cone-like endings (Fig. 6E). In some cases, individual axons could be followed for as long as 6.8 mm past the center of the lesion site. There were no significant differences in axon density rostral to the lesion site between treatment groups (Fig. 6F), but there were differences in the distribution of the regenerating axons (see below). As in the 7 d-treated cases described above, we stained coronal sections from spinal level C2 for cholera toxin B subunit immunoreactivity to rule out potential axon sparing. No labeled axons were found in the cases included for analysis here.
The patterns of axon growth after lesion priming and antibody treatment can best be seen in the reconstructions shown in Figure 7. Reconstruction in the sagittal plane (Fig. 7A) shows the preference of lesion-primed and control antibody-treated axons to grow dorsally. The horizontal plane (Fig. 7C) reveals a Y-shaped pattern as axons enter the scar region but then avoid and grow around the central core. After treatment with the neutralizing antibodies, although many axons still grow preferentially on the dorsal surface of the cord, a significant fraction of the axons enter and extend within the white matter (Fig. 7B,D). We measured the fraction of the labeled axons within the white matter rostral to the lesion site (Fig. 8A). After treatment with neutralizing antibodies, more than one-third of the labeled axons grow within the white matter rostral to the lesion site. CTB-labeled axons growing in the degenerating white matter rostral to the lesion site are shown in Figure 8B. Thus, once axons that have been stimulated to grow by a peripheral nerve conditioning lesion pass through the glial scar barrier, they are capable of long-distance growth within their normal pathways.
Figure 8.
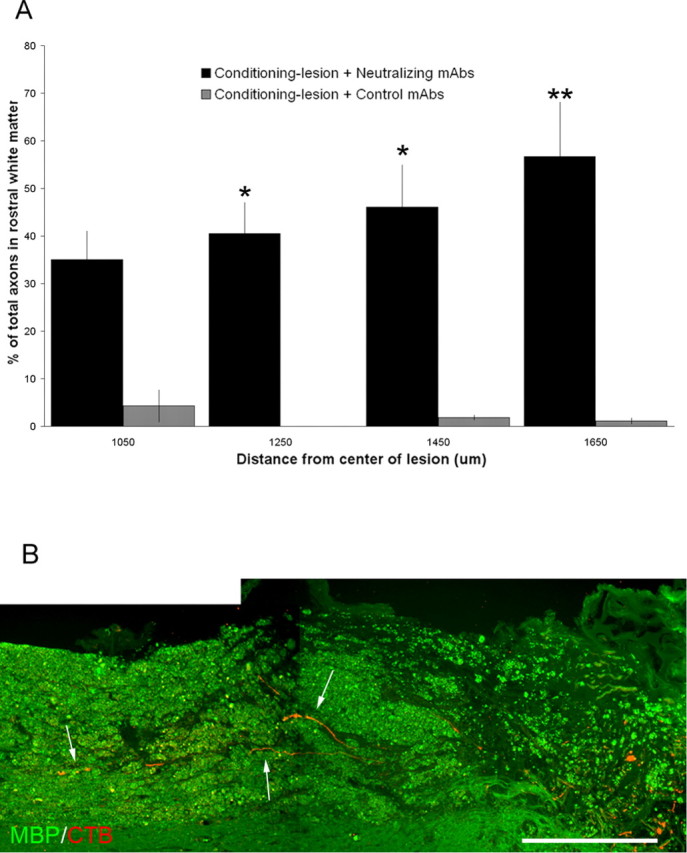
Quantitation of the effects of combined peripheral nerve conditioning lesions and antibody treatments. A, Axon growth in the white matter rostral to the lesion. Axon number was counted and the axons were scored for being either in the white matter or along the dorsal surface of the cord. Data shown are the mean ± SEM (∗p < 0.05, ∗∗p < 0.001, one-way repeated-measures ANOVA with Bonferroni's post hoc tests). B, The arrows point to regenerating mechanosensory axons (red) growing in the degenerating white matter rostral to the lesion site (green, anti-myelin basic protein stain). Scale bar, 500 μm.
Discussion
The repair of the damaged spinal cord may require the long-distance regrowth of damaged axons and the reestablishment of appropriate synapses. After treatment with antibodies that bind to and neutralize specifically the NG2 proteoglycan, medium- and large-diameter mechanosensory axons grow into the growth nonpermissive environment of the glial scar and, when combined with a previous peripheral nerve conditioning lesion, grow past the scar and into the degenerating white matter rostral to the lesion site. These findings show that the accumulations of NG2 at injury sites are a significant factor in the failure of axon regeneration and emphasize the utility of combinatorial treatments that target both the environment through which axons must regrow and the intrinsic growth state of the injured neuron.
Because CSPGs are increased after injury and can inhibit or repel growing neurons in vitro, much attention has been focused on their functions (Snow et al., 1990; McKeon et al., 1991; Moon et al., 2001; Tang et al., 2003). After SCI, levels of NG2 increase rapidly. This increase has been estimated to be twofold (Tang et al., 2003) but may be considerably higher (Camand et al., 2004). Levels of neurocan and versican, two other growth-inhibitory CSPGs, also increase after SCI, but the time course of expression and their spatial distribution differs from that of NG2. Neurocan is initially decreased in the region bordering the lesion and then slowly increases, reaching maximum levels in the lesion penumbra at ∼2 weeks after injury (Jones et al., 2003a). Versican V2 accumulates slowly in the regions surrounding the lesion, not in the central core (Asher et al., 2002; Jones et al., 2003a). Myelin antigens are usually removed from lesion sites by the actions of macrophages (Stichel et al., 1995), and nogoA protein and mRNA are reduced or absent from the lesion core (Wang et al., 2002; Hunt et al., 2003). Immediately after transection, sensory axons in the dorsal columns retract for ∼300 μm and, over the next 24 h, sprout and attempt to regenerate (Kerschensteiner et al., 2005). Thus, when damaged sensory axons attempt to regrow, they do so in areas containing increased levels of NG2 but reduced levels of other inhibitory molecules.
To test the functions of the elevated levels of NG2, we infused antibodies using small Gelfoam pledglets. This method is an effective short-term means of administering agents to the injured spinal cord (Yick et al., 2000). After a 7 d treatment, regenerating axons traverse the accumulations of NG2 within the caudal borders of the lesion, enter the core of the scar, and grow past the lesion center. The failure to detect CTB-labeled axons in the cervical spinal cord rules out axon sparing, and the three-dimensional reconstructions eliminate the possibilities that axons were pushed aside during the lesioning process or grow around and under the damaged area as often happens with other treatments (Bradbury et al., 1999). The effectiveness of the antibodies suggests that NG2 plays a major inhibitory role during the early stages of recovery from SCI.
We used a previous peripheral nerve conditioning lesion to stimulate the intrinsic growth state of centrally lesioned axons (Richardson and Issa, 1984; Neumann and Woolf, 1999). After control treatments, lesion-primed axons grow ectopically, frequently into any remaining pieces of dorsal roots. Growth into the white matter rostral to the lesion site is rare. Thus, the increased growth state promoted by a peripheral nerve conditioning lesion is insufficient to overcome the inhibitory and repulsive properties of the glial scar. This is consistent with the observation that lesion-primed sensory axons fail to grow onto spots of aggrecan in vitro and into semaphorin-containing areas of the glial scar (Pasterkamp et al., 2001; Steinmetz et al., 2005). After treatment with the neutralizing anti-NG2 antibodies, however, at least 40% of the lesion-primed axons that have entered the scar core continue to grow past the rostral border of the lesion and extend within the white matter. Because cholera toxin labels predominantly the large- and medium-diameter sensory axons that grow in the fasciculus gracilis, the regenerating axons are regrowing in their normal pathway. Such anatomically correct axon regeneration may be prerequisite for functional recovery.
The conclusion that NG2 is a significant factor in growth prevention by the glial scar is at odds with recent observations that axons can grow onto transplanted NG2-positive fibroblasts (Jones et al., 2003b). Growth inhibition by CSPGs is highly context dependent, and the manner in which CSPGs are presented to neurons, the type of neuron tested, and the presence, absence, and concentrations of different extracellular matrix and cell adhesion molecules can all affect neuronal behavior (Dou and Levine 1995; Snow et al., 1996; Garwood et al., 1999). For example, regenerating axons grow along NG2-positive blood vessels but stop when they contact other types of NG2-expressing cells (Rezajooi et al., 2004). It will be important in future studies to understand how other molecules, particularly the semaphorins, may modulate the effects of NG2 on injured neurons (Shearer et al., 2003; Kantor et al., 2004).
A recent study found no evidence of axon regeneration after SCI in an NG2 null mouse (de Castro et al., 2005). Because that study looked only at serotonergic and CGRP-containing axons, not the long-distance projecting mechanosensory afferents examined here, a direct comparison of the results is not possible. de Castro et al. (2005) found that, at 8 weeks after injury in both wild-type and NG2−/− mice, serotonergic fibers penetrated the scar for only 50 μm. When sagittal sections through the lesion site were examined, approximately twice as many sections in the wild-type mice contained serotonergic fibers compared with the NG2 null mice. Without complete reconstructions of the axon trajectories, such as was done here, it is difficult to know whether the increased incidence of fibers in the wild-type mice is attributable to increased sprouting or because the axons are growing around the repulsive NG2-positive lesion core.
The lack of concordance between the antibody treatments here and genetic models for studying NG2 function is not unusual. Genetic deletion of MAG does not promote regeneration, although MAG is a potent inhibitor of axon growth in vitro (Bartsch et al. 1995). Infusion of the anti-nogo antibody IN-1 promotes the regeneration of lesioned corticospinal tract axons in both rodents and primates (Schnell and Schwab, 1990; Fouad et al., 2004). However, in three different mice strains null for nogo, there is no sprouting and regeneration of corticospinal tract axons, modest sprouting, or sprouting and regeneration in young mice only (Kim et al., 2003; Simonen et al., 2003; Zheng et al., 2003). MAG, nogo, and oligodendrocyte myelin glycoprotein signal through a receptor complex consisting of the nogo receptor, p75, and LINGO (Bandtlow and Dechant, 2004). Genetic deletion of the nogo receptor does not foster regeneration, but the transgenic expression of a soluble receptor antagonist does (Li et al., 2005; Zheng et al., 2005). Why genetic targeting of known growth-inhibitory molecules often fails but pharmacological targeting succeeds is unknown. This disparity may reflect inherent limitations of genetic approaches when studying complex phenomena such as spinal injury and its repair.
The positive effects of the neutralizing anti-NG2 antibodies raise the question of how the antibodies work. We considered several possibilities. First, the antibodies could directly promote axon growth from adult DRG neurons. When applied to isolated adult DRG neurons, however, the antibodies had no effect on basal growth over a 48 h period (A. T. Rorai and J. M. Levine, unpublished observations). Second, NG2 could be present in a complex with other growth-inhibitory molecules. Binding of the antibody to NG2 would then block multiple proteins. Although we could not detect the growth-inhibitory molecules neurocan, phosphacan, and semaphorin5A in anti-NG2 immunoprecipitates of spinal cord lysates (J. M. Levine, unpublished observations), we cannot rule out associations of NG2 with other modifiers of axon growth, especially extracellular matrix-associated molecules (Burg et al., 1996). The neutralizing and non-neutralizing antibodies used here bind to both the membrane-associated and secreted forms of NG2 (Ughrin. et al., 2003), and the data in Figure 1 show that the infused antibodies bind to NG2-expressing cells. The simplest explanation then is that the antibodies bind to NG2 on both the cell surface and in the extracellular matrix and prevent an interaction of critical regions of the NG2 core protein with the sprouting growth cones of damaged neurons. Although we cannot exclude that the antibody directly effects NG2-expressing cells within the lesions, we observed no differences in scar size and appearance across the different treatments. Given the barrier functions of NG2, it remains possible that antibody treatment leads to the increased invasion of the damaged tissue by macrophages and other cell types. Whether an increased inflammatory response at the site of axon transection would be beneficial for regenerating axons is uncertain (Lacroix et al., 2002; Cafferty et al., 2004; Steinmetz et al., 2005).
Glial scar-associated axon growth-inhibitory molecules are only one aspect of the problem of regeneration and repair after SCI. In addition to blocking or neutralizing these molecules, repair may require providing trophic support, preventing secondary damage, and encouraging the remyelination of spared or regenerating nerve fibers. Recently, combinatorial approaches that address multiple aspects of SCI have been shown to have beneficial effects in several different animal models (Lu et al., 2003; Nikulina et al., 2004; Pearse et al., 2004; Fouad et al., 2005; Steinmetz et al., 2005). Targeting the accumulations of NG2 within the glial scar may be a useful addition to these combinatorial strategies.
Footnotes
This work was supported by grants from the National Institutes of Health, the Christopher Reeve Paralysis Foundation, and the New York State Spinal Cord Injury Research Board.
References
- Asher RA, Morgenstern DA, Shearer MC, Adcock KH, Pesheva P, Fawcett JW (2002). Versican is upregulated in CNS injury and is a product of oligodendrocyte lineage cells. J Neurosci 23:2225–2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asher RA, Morgenstern DA, Properzi F, Nishiyama A, Levine JM, Fawcett JW (2005). Two separate metalloproteinase activities are responsible for the shedding and processing of the NG2 proteoglycan in vitro. Mol Cell Neurosci 29:82–96. [DOI] [PubMed] [Google Scholar]
- Bandtlow C, Dechant G (2004). From cell death to neuronal regeneration, effects of the p75 neurotrophin receptor depend on interactions with partner subunits. Sci STKE 2004:pe24. [DOI] [PubMed] [Google Scholar]
- Bartsch U, Bandtlow CE, Schnell L, Bartsch S, Spillmann AA, Rubin BP, Hillenbrand R, Montag D, Schwab ME, Schachner M (1995). Lack of evidence that myelin-associated glycoprotein is a major inhibitor of axonal regeneration in the CNS. Neuron 15:1375–1381. [DOI] [PubMed] [Google Scholar]
- Bradbury EJ, Khemani S, Von R, King Priestley JV, McMahon SB (1999). NT-3 promotes growth of lesioned adult rat sensory axons ascending in the dorsal columns of the spinal cord. Eur J Neurosci 11:3873–3883. [DOI] [PubMed] [Google Scholar]
- Bradbury EJ, Moon LD, Popat RJ, King VR, Bennett GS, Patel PN, Fawcett JW, McMahon SB (2002). Chondroitinase ABC promotes functional recovery after spinal cord injury. Nature 416:636–640. [DOI] [PubMed] [Google Scholar]
- Burg MA, Tillet E, Timpl R, Stallcup WB (1996). Binding of the NG2 proteoglycan to type VI collagen and other extracellular matrix molecules. J Biol Chem 271:26110–26116. [DOI] [PubMed] [Google Scholar]
- Cafferty WB, Gardiner NJ, Das P, Qiu J, McMahon SB, Thompson SW (2004). Conditioning injury-induced spinal axon regeneration fails in interleukin-6 knock-out mice. J Neurosci 24:4432–4443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camand E, Morel MP, Faissner A, Sotelo C, Dusart I (2004). Long-term changes in the molecular composition of the glial scar and progressive increase of serotoninergic fibre sprouting after hemisection of the mouse spinal cord. Eur J Neurosci 20:1161–1176. [DOI] [PubMed] [Google Scholar]
- Dawson MR, Polito A, Levine JM, Reynolds R (2003). NG2-expressing glial progenitor cells: an abundant and widespread population of cycling cells in the adult rat CNS. Mol Cell Neurosci 24:476–488. [DOI] [PubMed] [Google Scholar]
- de Castro R Jr, Tajrishi R, Claros J, Stallcup WB (2005). Differential responses of spinal axons to transection: influence of the NG2 proteoglycan. Exp Neurol 192:299–309. [DOI] [PubMed] [Google Scholar]
- Dou CL, Levine JM (1995). Differential effects of glycosaminoglycans on neurite growth on laminin and L1 substrates. J Neurosci 15:8053–8066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filbin MT (2003). Myelin-associated inhibitors of axonal regeneration in the adult mammalian CNS. Nat Rev Neurosci 4:703–713. [DOI] [PubMed] [Google Scholar]
- Fouad K, Klusman I, Schwab ME (2004). Regenerating corticospinal fibers in the marmoset (Callitrix jacchus) after spinal cord lesion and treatment with the anti-Nogo-A antibody IN-1. Eur J Neurosci 20:2479–2482. [DOI] [PubMed] [Google Scholar]
- Fouad K, Schnell L, Bunge MB, Schwab ME, Liebscher T, Pearse DD (2005). Combining Schwann cell bridges and olfactory-ensheathing glia grafts with chondroitinase promotes locomotor recovery after complete transection of the spinal cord. J Neurosci 25:1169–1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frei E, Klusman I, Schnell L, Schwab ME (2000). Reactions of oligodendrocytes to spinal cord injury: cell survival and myelin repair. Exp Neurol 163:373–380. [DOI] [PubMed] [Google Scholar]
- Garwood J, Schnadelbach O, Clement A, Schutte K, Bach A, Faissner A (1999). DSD-1-proteoglycan is the mouse homolog of phosphacan and displays opposing effects on neurite outgrowth dependent on neuronal lineage. J Neurosci 19:3888–3899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimpe B, Silver J (2004). A novel DNA enzyme reduces glycosaminoglycan chains in the glial scar and allows microtransplanted dorsal root ganglia axons to regenerate beyond lesions in the spinal cord. J Neurosci 24:1393–1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt D, Coffin RS, Prinjha RK, Campbell G, Anderson PN (2003). Nogo-A expression in the intact and injured nervous system. Mol Cell Neurosci 24:1083–1102. [DOI] [PubMed] [Google Scholar]
- Jones LL, Yamaguchi Y, Stallcup WB, Tuszynski MH (2002). NG2 is a major chondroitin sulfate proteoglycan produced after spinal cord injury and is expressed by macrophages and oligodendrocyte progenitors. J Neurosci 22:2792–2803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones LL, Margolis RU, Tuszynski MH ((2003a)). The chondroitin sulfate proteoglycans neurocan, brevican, phosphacan, and versican are differentially regulated following spinal cord injury. Exp Neurol 182:399–411. [DOI] [PubMed] [Google Scholar]
- Jones LL, Sajed D, Tuszynski MH (2003b). Axonal regeneration through regions of chondroitin sulfate proteoglycan deposition after spinal cord injury: a balance of permissiveness and inhibition. J Neurosci 23:9276–9288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kantor DB, Chivatakarn O, Peer KL, Oster SF, Inatani M, Hansen MJ, Flanagan JG, Yamaguchi Y, Stretavan DW, Giger RJ, Kolodkin AL (2004). Semaphorin 5A is a bifunctional axon guidance cue regulated by heparan and chondroitin sulfate proteoglycans. Neuron 44:961–976. [DOI] [PubMed] [Google Scholar]
- Kerschensteiner M, Schwab ME, Lichtman JW, Misgeld T (2005). In vivo imaging of axonal degeneration and regeneration in the injured spinal cord. Nat Med 11:572–577. [DOI] [PubMed] [Google Scholar]
- Kim JE, Li S, GrandPre T, Qiu D, Strittmatter SM (2003). Axon regeneration in young adult mice lacking Nogo-A/B. Neuron 38:187–199. [DOI] [PubMed] [Google Scholar]
- Lacroix S, Chang L, Rose-John S, Tuszynski MH (2002). Delivery of hyper-interkeukin-6 to the injured spinal cord increases neutrophil and macrophage infiltration and inhibits axon growth. J Comp Neurol 454:213–238. [DOI] [PubMed] [Google Scholar]
- Lamotte CC, Kapadia SE, Shapiro CM (1991). Central projections of the sciatic, saphenous, median, and ulnar nerves of the rat demonstrated by transganglionic transport of choleragenoid-HRP (B-HRP) and wheat germ agglutinin-HRP (WGA-HRP). J Comp Neurol 311:546–562. [DOI] [PubMed] [Google Scholar]
- Levine JM (1994). Increased expression of the NG2 chondroitin-sulfate proteoglycan after brain injury. J Neurosci 14:4716–4730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine JM, Stincone F, Lee YS (1993). Development and differentiation of glial precursor cells in the rat cerebellum. Glia 7:307–321. [DOI] [PubMed] [Google Scholar]
- Li S, Kim JE, Budel S, Hampton TG, Strittmatter SM (2005). Transgenic inhibition of Nogo-66 receptor function allows axonal sprouting and improved locomotion after spinal injury. Mol Cell Neurosci 29:26–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu P, Jones LL, Snyder EY, Tuszynski MH (2003). Neural stem cells constitutively secrete neurotrophic factors and promote extensive host axonal growth after spinal cord injury. Exp Neurol 181:115–129. [DOI] [PubMed] [Google Scholar]
- Martin S, Levine AK, Chen ZJ, Uhgrin Y, Levine JM (2001). Deposition of the NG2 proteoglycan at nodes of Ranvier in the peripheral nervous system. J Neurosci 21:8119–8128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKeon RJ, Schreiber RC, Rudge JS, Silver J (1991). Reduction of neurite outgrowth in a model of glial scarring following CNS injury is correlated with expression of inhibitory molecules on reactive astrocytes. J Neurosci 11:3398–3411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McTigue DM, Wei P, Stokes BT (2001). Proliferation of NG2-positive cells and altered oligodendrocyte numbers in the contused rat spinal cord. J Neurosci 21:3392–3400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon LDF, Asher RA, Rhodes KE, Fawcett JW (2001). Regeneration of CNS axons back to their original target following treatment of adult rat brain with chondroitinase ABC. Nat Neurosci 4:465–466. [DOI] [PubMed] [Google Scholar]
- Moreau-Fauvarque C, Kumanogoh A, Camand E, Jaillard C, Barbin G, Boquet I, Love C, Jones EY, Kikutani H, Lubetzki C, Dusart I, Chedotal A (2003). The transmembrane semaphorin Sema4D/CD100, an inhibitor of axonal growth, is expressed on oligodendrocytes and upregulated after CNS lesion. J Neurosci 23:9229–9239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgenstern DA, Asher RA, Fawcett JW (2002). Chondroitin sulphate proteoglycans in the CNS injury response. Prog Brain Res 137:313–332. [DOI] [PubMed] [Google Scholar]
- Morgenstern DA, Asher RA, Naidu M, Carlstedt T, Levine JM, Fawcett JW (2003). Expression and glycanation of the NG2 proteoglycan in developing, adult, and damaged peripheral nerve. Mol Cell Neurosci 24:787–802. [DOI] [PubMed] [Google Scholar]
- Neumann S, Woolf CJ (1999). Regeneration of dorsal column fibers into and beyond the lesion site following adult spinal cord injury. Neuron 23:83–91. [DOI] [PubMed] [Google Scholar]
- Neumann S, Bradke F, Tessier-Lavigne M, Basbaum AI (2002). Regeneration of sensory axons within the injured spinal cord induced by intraganglionic cAMP elevation. Neuron 34:885–893. [DOI] [PubMed] [Google Scholar]
- Niederost BP, Zimmermann DR, Schwab ME, Bandtlow CE (1999). Bovine CNS myelin contains neurite growth-inhibitory activity associated with chondroitin sulfate proteoglycans. J Neurosci 19:8979–8989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikulina E, Tidwell JL, Dai HN, Bregman BS, Filbin MT (2004). The phosphodiesterase inhibitor rolipram delivered after a spinal cord lesion promotes axonal regeneration and functional recovery. Proc Natl Acad Sci USA 101:8786–8790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishiyama A, Dahlin KJ, Prince JT, Johnstone SR, Stallcup WB (1991). The primary sequence of NG2, a novel membrane spanning proteoglycan. J Cell Biol 114:359–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norton WT, Poduslo SE (1973). Myelination in rat brain: method of myelin isolation. J Neurochem 21:749–757. [DOI] [PubMed] [Google Scholar]
- Pasterkamp RJ, Giger RJ, Ruitenberg MJ, Holtmaat AJ, De Wit J, De Winter F, Verhaagen J (1999). Expression of the gene encoding the chemorepellent semaphorin III is induced in the fibroblast component of neural scar tissue formed following injuries of adult but not neonatal CNS. Mol Cell Neurosci 13:143–166. [DOI] [PubMed] [Google Scholar]
- Pasterkamp RJ, Anderson PN, Verhaagen J (2001). Peripheral nerve injury fails to induce growth of lesioned ascending dorsal column axons into spinal cord scar tissue expressing the axon repellent Semaphorin3A. Eur J Neurosci 13:457–471. [DOI] [PubMed] [Google Scholar]
- Pearse DD, Pereira FC, Marcillo AE, Bates ML, Berrocal YA, Filbin MT, Bunge MB (2004). cAMP and Schwann cells promote axonal growth and functional recovery after spinal cord injury. Nat Med 10:610–616. [DOI] [PubMed] [Google Scholar]
- Reier PJ, Stensaas LJ, Guth L (1983). The astrocytic scar as an impediment to regeneration in the central nervous system. In: Spinal cord reconstruction (Kao CC, Bunge RP, Reier PJ, eds) pp. 163–195. New York: Raven.
- Rezajooi K, Pavlides M, Winterbottom J, Stallcup WB, Hamlyn PJ, Lieberman AR, Anderson PN (2004). NG2 proteoglycan expression in the peripheral nervous system: upregulation following injury and comparison with CNS lesions. Mol Cell Neurosci 25:572–584. [DOI] [PubMed] [Google Scholar]
- Richardson PM, Issa VM (1984). Peripheral injury enhances central regeneration of primary sensory neurones. Nature 309:791–793. [DOI] [PubMed] [Google Scholar]
- Schneider S, Bosse F, D'Urso D, Muller H, Sereda MW, Nave K, Niehaus A, Kempf T, Schnolzer M, Trotter J (2001). The AN2 prptein is a novel marker for the Schwann cell lineage expressed by immature and unmyelinating Schwann cells. J Neurosci 21:920–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnell L, Schwab ME (1990). Axonal regeneration in the rat spinal cord produced by an antibody against myelin-associated neurite growth inhibitors. Nature 343:269–272. [DOI] [PubMed] [Google Scholar]
- Shearer MC, Niclou SP, Brown D, Asher RA, Holtmaat AJ, Levine JM, Verhaagen J, Fawcett JW (2003). The astrocyte/meningeal cell interface is a barrier to neurite outgrowth which can be overcome by manipulation of inhibitory molecules or axonal signalling pathways. Mol Cell Neurosci 24:913–925. [DOI] [PubMed] [Google Scholar]
- Silver J, Miller JH (2004). Regeneration beyond the glial scar. Nat Rev Neurosci 5:146–156. [DOI] [PubMed] [Google Scholar]
- Simonen M, Pedersen V, Weinmann O, Schnell L, Buss A, Ledermann B, Christ F, Sansig G, van der Putten H, Schwab ME (2003). Systemic deletion of the myelin-associated outgrowth inhibitor Nogo-A improves regenerative and plastic responses after spinal cord injury. Neuron 38:201–211. [DOI] [PubMed] [Google Scholar]
- Snow DM, Lemmon V, Carrino DA, Calpan AI, Silver J (1990). Sulfated proteoglycans in astroglial barriers inhibit neurite outgrowth in vitro. Exp Neurol 109:111–130. [DOI] [PubMed] [Google Scholar]
- Snow DM, Brown EM, Letourneau PC (1996). Growth cone behavior in the presence of soluble chondroitin sulfate proteoglycan (CSPG), compared to behavior on CSPG bound to laminin or fibronectin. Int J Dev Neurosci 14:331–349. [DOI] [PubMed] [Google Scholar]
- Stallcup WB, Dahlin-Huppe K (2001). Chondroitin sulfate and cytoplasmic domain- dependent membrane targeting of the NG2 proteoglycan promote retraction fiber formation and cell polarization. J Cell Sci 114:2315–2325. [DOI] [PubMed] [Google Scholar]
- Steinmetz MO, Horn KP, Tom VJ, Miller JH, Busch SA, Nair D, Silver DJ, Silver J (2005). Chronic enhancement of the intrinsic growth capacity of sensory neurons combined with the degradation of inhibitory proteoglycans allows functional regeneration of sensory axons through the dorsal root entry zone in the mammalian spinal cord. J Neurosci 25:8066–8076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steward O, Zheng B, Tessier-Lavigne M (2003). False resurrections: distinguishing regenerated from spared axons in the injured central nervous system. J Comp Neurol 459:1–8. [DOI] [PubMed] [Google Scholar]
- Stichel CC, Wunderlich G, Schwab ME, Muller HW (1995). Clearance of myelin constituents and axonal sprouting in the transected postcommissural fornix of the adult rat. Eur J Neurosci 7:401–411. [DOI] [PubMed] [Google Scholar]
- Tan AM, Zhang W, Levine JM (2005). NG2: A component of the glial scar that inhibits axon growth. J Anat 207:717–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang X, Davies JE, Davies SJ (2003). Changes in distribution, cell associations, and protein expression levels of NG2, neurocan, phosphacan, brevican, versican V2, and tenascin-C during acute to chronic maturation of spinal cord scar tissue. J Neurosci Res 71:427–444. [DOI] [PubMed] [Google Scholar]
- Ughrin YM, Chen ZJ, Levine JM (2003). Multiple regions of the NG2 proteoglycan inhibit neurite growth and induce growth cone collapse. J Neurosci 23:175–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Chun SJ, Treloar H, Vartanian T, Greer CA, Strittmatter SM (2002). Localization of Nogo-A and Nogo-66 receptor proteins at sites of axon-myelin and synaptic contact. J Neurosci 22:5505–5515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yick LW, Wu W, So KF, Yip HK, Shum DK (2000). Chondroitinase ABC promotes axonal regeneration of Clarke's neurons after spinal cord injury. NeuroReport 11:1063–1067. [DOI] [PubMed] [Google Scholar]
- Zai LJ, Wrathall JR (2005). Cell proliferation and replacement following contusive spinal cord injury. Glia 50:247–257. [DOI] [PubMed] [Google Scholar]
- Zheng B, Ho C, Li S, Keirstead H, Steward O, Tessier-Lavigne M (2003). Lack of enhanced spinal regeneration in Nogo-deficient mice. Neuron 38:213–224. [DOI] [PubMed] [Google Scholar]
- Zheng B, Atwal J, Ho C, Case L, He XL, Garcia KC, Steward O, Tessier-Lavigne M (2005). Genetic deletion of the Nogo receptor does not reduce neurite inhibition in vitro or promote corticospinal tract regeneration in vivo. Proc Natl Acad Sci USA 102:1205–1210. [DOI] [PMC free article] [PubMed] [Google Scholar]