

Abstract
An increasing amount of evidence suggests that the family of nuclear factor κB (NF-κB) transcription factors plays an important role in synaptic plasticity and long-term memory formation. The present study investigated the regulation of NF-κB family members p50, p65/RelA, and c-Rel in the hippocampus in response to metabotropic glutamate receptor (mGluR) signaling. Activation of group I metabotropic glutamate receptors (GpI-mGluRs) with the agonist (S)-3,5-dihydroxyphenylglycine (DHPG) resulted in a time-dependent increase in DNA binding activity of p50, p65, and c-Rel in area CA1 of the hippocampus. An antagonist of mGluR5, 2-Methyl-6-(phenylethynyl)pyridine, inhibited the DHPG-induced activation of NF-κB, whereas an antagonist of mGluR1, (S)-(+)-α-amino-4-carboxy-2-methylbenzeneacetic acid, did not. Using a series of inhibitors, we investigated the signaling pathways necessary for DHPG-induced activation of NF-κB and found that they included the phosphatidyl inositol 3-kinase, protein kinase C, mitogen-activated protein kinase kinase, and p38-mitogen-activated protein kinase pathways. To determine the functional significance of mGluR-induced regulation of NF-κB, we measured long-term depression (LTD) of Schaffer-collateral synapses in the hippocampus of c-Rel knock-out mice. Early phase LTD was normal in c-rel−/− mice. However, late-phase LTD (>90 min) was impaired in c-rel−/− mice. The observations of this deficit in hippocampal synaptic plasticity prompted us to further investigate long-term memory formation in c-rel−/− mice. c-rel−/− mice exhibited impaired performance in a long-term passive avoidance task, providing additional evidence for c-Rel in long-term memory formation. These results demonstrate that the NF-κB transcription factor family is regulated by GpI-mGluRs in the hippocampus and that the c-Rel transcription factor is necessary for long-term maintenance of LTD and formation of long-term memory.
Keywords: NF-κB, hippocampus, long-term depression, metabotropic glutamate receptor, c-Rel, memory
Introduction
Persistent forms of synaptic plasticity are thought to contribute to long-term information storage in the nervous system. Transcription is required for consolidation and maintenance of synaptic plasticity in nearly every organism studied, ranging from flies and mollusks to mammals (Milner et al., 1998). However, there is a lack of evidence implicating a role for transcription in one form of synaptic plasticity in the hippocampus, metabotropic glutamate receptor-dependent long-term depression (mGluR-LTD).
mGluR-LTD is induced via activation of group I mGluRs (GpI-mGluRs) and is mechanistically distinct from LTD induced via NMDA receptors (NMDARs) (Bolshakov and Siegelbaum, 1994; Oliet et al., 1997; Kemp and Bashir, 1999; Huber et al., 2000, 2001). mGluR-LTD can be induced through either extended periods of low-frequency stimulation or direct activation of GpI-mGluRs with agonists like (S)-3,5-dihydroxyphenylglycine (DHPG) (Bolshakov and Siegelbaum, 1994; Oliet et al., 1997; Huber et al., 2001). Expression of mGluR-LTD in the adult hippocampus is solely postsynaptic and requires internalization of AMPA receptors (AMPARs) (Snyder et al., 2001; Nosyreva and Huber, 2005). mGluR-LTD is unique in that it requires rapid translation of preexisting dendritic mRNAs for its induction (Huber et al., 2000; Hou and Klann, 2004).
The nuclear factor κB (NF-κB) family of dimeric transcription factors, first discovered in the immune system, appear to play a role in the induction of synaptic plasticity and formation long-term memory (Albensi and Mattson, 2000; Mattson et al., 2000; Kassed et al., 2002; Yeh et al., 2002, 2004; Liou and Hsia, 2003; Meffert et al., 2003; Levenson et al., 2004; Dash et al., 2005). NF-κB subcellular distribution, DNA binding activity, and transcription are regulated by various forms of synaptic activity (Meberg et al., 1996; Suzuki et al., 1997; Albensi and Mattson, 2000; Mattson et al., 2000; Burr and Morris, 2002; Lilienbaum and Israel, 2003; Freudenthal et al., 2004). Moreover, regulation of gene expression by GpI-mGluRs in cortical neurons relies on activation of c-Rel (Pizzi et al., 2005). This suggests that NF-κB could be involved in the induction of a range of transcription-dependent forms of synaptic plasticity in the nervous system, including LTD. In the present study, we provide evidence suggesting that NF-κB is activated by GpI-mGluRs in the hippocampus. Furthermore, we show that loss of the NF-κB family member c-Rel prevents induction of a persistent form of mGluR-LTD.
Materials and Methods
Animals.
Young adult (3–5 weeks of age) mice on a C57BL/6J background strain were used in all experiments. c-Rel mutant animals were generated by targeted replacement of exon 5 (Tumang et al., 1998). The c-Rel colony was maintained by breeding heterozygous (c-rel+/−) animals, yielding littermate c-rel+/+and c-rel−/− experimental subjects. Animals were housed under a 12 h light/dark cycle and allowed access to rodent chow and water ad libitum. Animals were allowed to acclimate to laboratory conditions at least 3 d before use in experiments. All procedures were performed with the approval of the Baylor College of Medicine Institutional Animal Care and Use Committee and according to national guidelines and policies.
Hippocampus slice preparation.
Animals were killed by cervical dislocation. The brain was immersed in ice-cold cutting saline (CS) (in mm: 110 sucrose, 60 NaCl, 3 KCl, 1.25 NaH2PO4, 28 NaHCO3, 0.5 CaCl2, 7 MgCl2, 5 glucose, 0.6 ascorbate) before isolation of the caudal portion containing the hippocampus and entorhinal cortex. Transverse slices (400 μm) were prepared with a Vibratome (Vibratome, St. Louis, MO). During isolation, slices were stored in ice-cold CS. After isolation, cortical tissue was removed, and hippocampal slices were equilibrated in a mixture of 50% CS and 50% artificial CSF (ACSF) (in mm: 125 NaCl, 2.5 KCl, 1.25 NaH2PO4, 25 NaHCO3, 2 CaCl2, 1 MgCl2, 25 glucose) at room temperature (RT). Slices were further equilibrated in 100% ACSF for 45 min at RT, followed by a final incubation in 100% ACSF at 32°C for 1 h. All solutions were saturated with 95%/5% O2/CO2. Slices were treated with the appropriate drugs or vehicle after the last equilibration at 32°C. For experiments investigating the regulation of NF-κB in vitro, slices from six animals were pooled and randomized and divided into two treatment groups: vehicle control and drug treated.
Pharmacologic stimulation of hippocampal slices.
Acute hippocampal slice cultures were maintained in oxygenated (95%/5% O2/CO2) ACSF at 32°C. Activation of GpI-mGluRs was performed by exposing slices to either the GpI-mGluR agonist DHPG (50 μm) or the mGluR5-specific agonist (RS)-2-chloro-5-hydroxyphenylglycine (CHPG; 1 mm) for 10 min. Matched control slices received vehicle (ACSF). To determine which mGluR subtype contributed to observed effects on NF-κB, control experiments assayed the effect of preincubation (5 min) with either the mGluR1 antagonist (S)-(+)-α-amino-4-carboxy-2-methylbenzene-acetic acid (LY 367385; 30 μm) or the mGluR5 antagonist 2-Methyl-6-(phenylethynyl)pyridine (MPEP; 10 μm) on the effect of either DHPG or CHPG. Determination of signaling pathways involved in DHPG-mediated regulation of NF-κB was performed by preincubating slices in one of several drugs that block activation of a specific signaling pathway. The inhibitors used include: (9R,10s,12s)-2,3,9,10,11,12-hexahydro-10-hydroxy-9-methyl-1-oxo-9,12-epoxy-1H-diindolo[1,2,3-fg:3′,2′,1′-kl]pyrrolo[3,4-i][1,6]benzodiazocine-10-carboxylicacid hexyl ester [KT5720; 1 μm, protein kinase A (PKA)], (N-[2-[[[3-(4-chlorophenyl)-2-propenyl]methylamino]methyl]phenyl]-N-(2-hydroxyethyl)-4-methoxybenzenesulfonamide) [KN-93; 10 μm, calcium calmodulin-dependent protein kinase II (CaMKII)], 2-[1-(3-dimethyl-amino-propyl)indol-3-y]-3-(indol-3-yl)maleimide (GF 109,203×) [1 μm; protein kinase C (PKC)], Wortmannin [50 nm; phosphatidyl inositol 3-kinase (PI3-K)], AKT inhibitor IV (3 μm; AKT), 1,4-diamino-2,3-dicyano-1,4-bis[2-amino-phenylthio]butadiene [U0126; 20 μm, mitogen-activated protein kinase kinase (MEK)], and 4-[s-(4-fluorophenyl)-2-[4-(methylsulfonyl)phenyl]-1H-imidazol-4-yl]pyridine [SB 203580; 1 μm, p38 mitogen-activated protein kinase (MAPK)]. KT5720, GF 109,203×, Wortmannin, U0126, and SB 203580 were obtained from Tocris Cookson (Ellisville, MO). All other drugs were obtained from Calbiochem (La Jolla, CA). Regardless of experimental paradigm, at the end of pharmacologic treatment, slices were rinsed six times in normal ACSF, and incubations continued in fresh ACSF.
NF-κB DNA binding activity.
Area CA1 was isolated from hippocampal slices, and nuclear fractions were isolated using commercially available reagents (NE-PER; Pierce, Rockford, IL). DNA binding activity of NF-κB subunits p50, p65, and c-Rel was monitored using a commercially available ELISA-based assay (TransFactor; BD Biosciences, Mountain View, CA) (Pizzi et al., 2005). Briefly, nuclear samples (10 μg) were incubated in an ELISA plate that is coated with oligonucleotides containing an NF-κB consensus regulatory element sequence. The wells were washed and exposed to a primary antibody specific for an individual subunit of NF-κB. Binding of the primary antibody to protein is detected through a chromogenic reaction involving the enzymatic breakdown of 3,3′,5,5′-tetramethylbenzidine via a horseradish peroxidase (HRP)-conjugated secondary antibody. Reactions were quantified spectrophotometrically at a wavelength of 655 nm. For each run, a series of positive and negative controls was performed to ensure specificity of detection of NF-κB DNA binding activity.
Western blotting.
Loading buffer was added (final concentration, 6.25 mm Tris, pH 6.8, 2% SDS, 10% glycerol, 1.25% 2-mercaptoethanol, 0.1% bromophenol blue), and samples were incubated at RT for 20 min before SDS-PAGE. Samples were run on a discontinuous polyacrylamide gel consisting of either an 8, 12, or 15% acrylamide resolving gel, depending on the molecular weight of the protein of interest, and a 4% acrylamide stacking gel. After one-dimensional PAGE, proteins were transferred to polyvinylidene difluoride membranes for immunoblotting. Membranes were blocked in 3% BSA in TTBS (in mm: 150 NaCl, 20 Tris, pH 7.5, 0.05% Tween 20) for 45 min at RT. Membranes were incubated in primary antibodies overnight at 4°C and in HRP-conjugated anti-rabbit secondary antibodies (Jackson ImmunoResearch, West Grove, PA) for 2.5 h at RT. Immunolabeling of membranes was detected via chemiluminescence (Amersham Biosciences, Piscataway, NJ; SuperSignal, Pierce). Luminescence was recorded with Blue Light film (ISCBioExpress, Kaysville, UT), digitized (Epson Perfection 1240U; Epson, Long Beach, CA), and integrated densities of each band quantified with NIH ImageJ. Several exposures were captured for each immunoblot to ensure that all densitometry was performed on images taken in the linear exposure range. Primary antibodies used were as follows: Phospho-PDK1S241 (1:1000), Phospho-RafS259 (1:1000), Phospho-AKTS473 (1:1000), Phospho-AKTT308 (1:1000), total AKT (1:1000), Phospho-ERK1/2T202/Y204 (1:2000) (Cell Signaling Technology, Beverly, MA); total extracellular signal-regulated kinase 1/2 (ERK1/2; 1:2000), p50/p105 (1:2000), mGluR1 (1:2000), mGluR5 (1:2000) (Upstate Biotechnology, Lake Placid, NY); Phospho-p38T180/Y182 (1:1000), total p38 (1:1000) (Calbiochem, San Diego, CA); p65/RelA (1:2000; Chemicon, Temecula, CA); and c-Rel (1:50; Santa Cruz Biotechnology, Santa Cruz, CA).
Real-time, reverse transcription-quantitative PCR.
Area CA1 and dentate gyrus were isolated from the hippocampus and immediately frozen on dry ice. RNA was isolated from tissue using Trizol (Invitrogen, Carlsbad, CA). Real-time, reverse transcription-quantitative PCR (QPCR) was performed using the Chromo4 real-time PCR system (Bio-Rad, Waltham, MA), iScript One-Step reagents (Bio-Rad), and Taqman-based real-time probes. Real-time probes for p50 and p65 were purchased directly from Applied Biosystems (assay ID- p50/p105: Mm00476361_m1, p65/RelA: Mm00501346_m1; Applied Biosystems, Foster City, CA). Custom probes were designed to measure c-Rel (forward primer, ttcaatgtgggtgaacagca; reverse primer, tatttggggcacggttatca; Taqman probe, tcacaactgctctgcctcccattgt) and β-tubulin 4 (forward primer, tggacgagatggagttcacc; reverse primer, gctttccctaacctgcttgg; Taqman probe, gaggaggaagagggcgaggatgagg). Equal amounts of RNA were loaded in each reaction, and all samples were assayed in triplicate. C(t) values were set using c-rel+/+ samples. Samples were reanalyzed if C(t) values of replicates varied more than ±5%. Equal loading was monitored by assaying levels of β-tubulin 4.
Slice electrophysiology.
Electrophysiology was performed in an interface chamber (Fine Science Tools, Foster City, CA). Oxygenated ACSF (95%/5% O2/CO2) was warmed (30°C; TR-100 temperature controller; Fine Science Tools) and perfused into the recording chamber at a rate of 1 ml/min. Electrophysiological traces were amplified (model 1800 amplifier; A-M Systems, Sequim, WA), digitized, and stored (Digidata models 1200 and 1320A with Clampex software; Molecular Devices, Sunnyvale, CA). Extracellular stimuli were administered (model 2200 stimulus isolator; A-M Systems) on the border of area CA3 and CA1 along the Schaffer-collaterals using enameled, bipolar platinum-tungsten (92:8%) electrodes. Field EPSPs (fEPSPs) were recorded in stratum radiatum with an ACSF-filled glass recording electrode (1–3 MΩ). The relationship between fiber volley and fEPSP slopes over various stimulus intensities (0.5–15 V, 25 nA to 1.5 μA) was used to assess baseline synaptic transmission. All subsequent experimental stimuli were set to an intensity that evoked an fEPSP that had a slope of 50% of the maximum fEPSP slope. Paired-pulse facilitation was measured at various interstimulus intervals (20, 50, 100, 200, 300 ms). LTD was induced by exposing slices to DHPG (50 μm) for 10 min. Synaptic efficacy was monitored 20 min before and 2 h after induction of LTD by recording fEPSPs every 20 s (traces were averaged for every 2 min interval). Simultaneous recordings were made from c-rel+/+ and c-rel−/− littermate slices in the same chamber, minimizing variability attributable to development or slice microenvironment. The experimenter performing the electrophysiology was blind to genotype or treatment.
Minianalysis.
Mice were anesthetized using isoflurane and killed by decapitation. Brains were removed and placed in ice-cold dissecting solution (modified ACSF; in mm: 220 sucrose, 2.5 KCl, 1.25 NaH2PO4, 25 NaHCO3, 0.5 CaCl2, 7.0 MgCl2, 7.0 glucose). Horizontal brain slices containing the hippocampus (350 μm) were obtained using a Leica (Nussloch, Germany) VT1000S vibratome and placed in holding chambers containing room temperature oxygenated ACSF. For whole-cell patch-clamp recordings of miniature EPSCs, slices were placed into a submerged recording chamber and perfused with heated (32°C) and oxygenated ACSF containing 1 μm tetrodotoxin (Tocris Cookson) and 10 μm picrotoxin (Sigma) at a rate of 2 ml/min. Hippocampal pyramidal neurons were visualized using an Olympus BX51W1 inverted microscope equipped with infrared-differential interference contrast optics. Electrodes were pulled using TW150–4 glass (World Precision Instruments, Sarasota, FL) on a Sutter Instruments (Novato, CA) P-97 microelectrode puller and were filled with the following (in mm): 135 K+-gluconate, 5 NaCl, 2 MgCl2, 10 HEPES, 0.6 EGTA, 4 Na2ATP, 0.4 NaGTP, pH 7.4. Electrodes had resistances of 3–4 MΩ, and series resistance values ranged from 6–29 MΩ. Resting membrane potentials were corrected for junction potential and ranged from −67.9 to −85.0 mV (c-rel+/+, n = 9, −78.5 ± 1.4 mV; c-rel−/−, n = 9, −75.4 ± 1.6 mV; p < 0.2; unpaired t test). Input resistance values ranged from 56.9 to 230.2 MΩ (c-rel+/+, n = 9, 109.3 ± 20.5 MΩ; c-rel−/−, n = 9, 93.3 ± 13.4 MΩ; p < 0.6; unpaired t test). All cells were held at −70 mV, and miniature EPSCs were recorded for 5 min from each cell using Clampex (Molecular Devices). Cumulative amplitudes and interevent intervals were analyzed using the Mini Analysis Program (version 6.0.3; Synaptosoft, Decatur, GA). Statistical analysis was performed using the Kolmogorov–Smirnov goodness of fit test. The experimenter performing minianalysis was blind to genotype.
Passive avoidance.
Animals were placed into the “light” side of a two-chamber shuttle box (Med Associates, St. Albans, VT). Latency to step through a doorway into a “dark” side was recorded. During the training day, once animals moved into the dark side, the door was closed and animals received a mild electric shock (2 s; 0.5 mA) 5 s after entry. Animals were removed from the apparatus 12–15 s after receiving the shock. Testing sessions were capped at 300 s; no animal required >35 s to enter the dark side of the chamber during the training session. The experimenter performing passive avoidance experiments was blind to genotype.
Results
Activation of NF-κB by mGluR5
We determined whether nuclear DNA binding activity of various NF-κB subunits (p50, p65, c-Rel) was regulated in response to activation of GpI-mGluRs. Hippocampal slices were exposed to the GpI-mGluR agonist DHPG (50 μm, 10 min). Nuclear DNA binding activity of p50, p65, and c-Rel was maximally increased 50 min after DHPG treatment in area CA1 (Fig. 1A) (p < 0.01 for p50, p65, and c-Rel). DNA binding activity returned to basal levels 110 min after treatment (Fig. 1A). These results suggest that activation of GpI-mGluRs induced a time-dependent modulation of NF-κB DNA binding activity.
Figure 1.
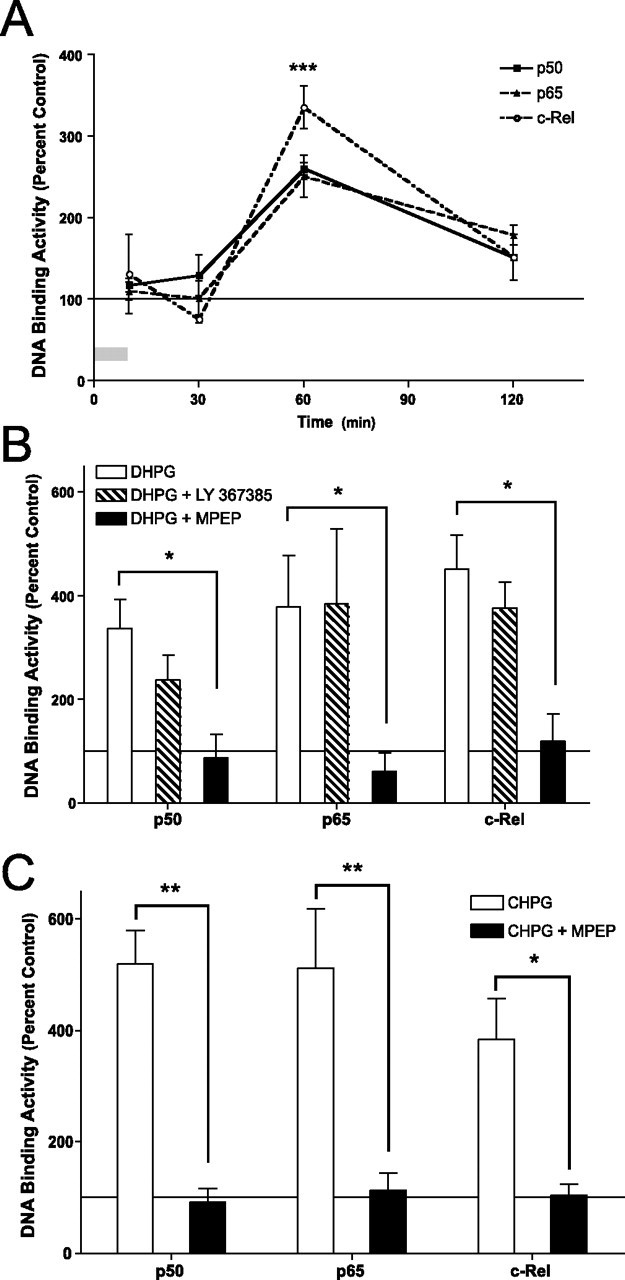
mGluR5 can activate NF-κB in area CA1 of the hippocampus. Nuclear DNA binding activity of NF-κB subunits p50, p65, and c-Rel was measured in area CA1 of transverse hippocampal slices (400 μm) in response to activation of GpI-mGluRs. A, Hippocampal slices were treated with DHPG (50 μm; 10 min), and DNA binding activity was measured in area CA1 of the hippocampus at various times after treatment. DHPG induced significant, time-dependent increases in nuclear DNA binding activity for p50 (F(3,13) = 34; p < 0.0001), p65 (F(3,13) = 11; p < 0.002), and c-Rel (F(3,13) = 16; p < 0.0005). The asterisk indicates significant difference (*p < 0.01) relative to the first time point as determined by Dunnett’s post hoc test. B, Slices were pretreated with either an mGluR1 antagonist (LY 367385, 30 μm) or an mGluR5 antagonist (MPEP, 10 μm) before treatment with DHPG (50 μm; 10 min). Nuclear DNA binding activity was measured 50 min after the end of DHPG treatment. LY 367385 had no effect on DHPG-induced increases in NF-κB DNA binding activity. MPEP significantly decreased DHPG-induced activation of NF-κB (F(4,53) = 11; p < 0.0001). The asterisk indicates significant difference (*p < 0.05) relative to DHPG-treated slices as determined by post hoc Bonferroni comparisons. C, To further confirm a role for mGluR5 in activation of NF-κB, slices were treated with the mGluR5 agonist CHPG (1 mm; 10 min). Significant activation of p50, p65, and c-Rel were observed 50 min after the end of treatment (F(2,23) = 38; p < 0.001). Pretreatment with MPEP (10 μm) blocked CHPG-induced increases in p50 (p < 0.01), p65 (p < 0.01), and c-Rel (p < 0.05). Asterisks indicate significant differences as determined by post hoc Bonferroni comparisons. For all panels, error bars indicate SEM. Percentage change was calculated based on DNA binding activity measured from matched control slices treated with vehicle.
The family of mGluRs is comprised of three major groups (Conn and Pin, 1997). GpI-mGluRs comprise mGluR1 and mGluR5 (Nakanishi, 1992). We determined the specific receptor subtype that was responsible for mediating the GpI-mGluR-induced activation of the NF-κB transcription factor family using a pharmacologic approach. Treatment of slices with DHPG (50 μm; 10 min) led to a robust activation of the NF-κB transcription factor family 50 min after treatment (Fig. 1B) (p < 0.0001). The mGluR1-specific antagonist LY 367385 (30 μm) had no effect on activation of the NF-κB transcription factor family (Fig. 1B). In contrast, the mGluR5-specific antagonist MPEP (10 μm) blocked the DHPG-induced activation of NF-κB (Fig. 1B) (p < 0.05 for p50, p65, and c-Rel). These results suggest that GpI-mGluR-induced activation of NF-κB occurs through mGluR5 but not mGluR1.
To further test the involvement of mGluR5 in activation of NF-κB, slices were treated with the mGluR5-specific agonist CHPG (1 mm; 10 min). CHPG led to a robust activation of the NF-kB transcription factor family 50 min after treatment (Fig. 1C) (p < 0.0001). MPEP (10 μm) completely blocked CHPG-induced activation of NF-κB (Fig. 1C) (p < 0.01 for p50 and p65; p < 0.05 for c-Rel). Considered together with the results above and previous studies (Pizzi et al., 2005), these results indicate that GpI-mGluR-induced activation of NF-κB is mediated primarily through mGluR5.
Signaling pathways coupling GpI-mGluRs with NF-κB
Several different signaling pathways could couple GpI-mGluRs with activation of the NF-κB transcription factor family. We first determined which signaling molecules were activated in area CA1 of the hippocampus in response to activation of GpI-mGluRs. Previous studies indicated that activation of GpI-mGluRs engages the PI3-K- phosphoinositide-dependent kinase-1 (PDK1)-AKT signaling cascade (Hou and Klann, 2004). In concert with these results, treatment of hippocampal slices with DHPG resulted in significant increases in levels of phospho-PDK1 (Fig. 2A) (t = 2.2; df = 12; p < 0.05) and phospho-AKT (Fig. 2A) (t = 2.3; df = 11; p < 0.05). Once activated, both PDK1 and AKT can engage various components of the ras signaling pathway. We observed significant increases in phospho-Raf1 (p < 0.05) and phospho-ERK2 (p < 0.05), indicative of engagement of ras-mediated signaling (Fig. 2A). Several studies suggest that the p38-MAPK signaling cascade is engaged by GpI-mGluR activation, and that p38-MAPK may play a role in mGluR-dependent synaptic plasticity (Bolshakov et al., 2000; Rush et al., 2002; Gallagher et al., 2004; Huang et al., 2004). We observed a significant increase in levels of phospho-p38 MAPK in area CA1 after stimulation with DHPG (Fig. 2A) (p < 0.05). Together, these results suggest that the PI3-K-PDK1-AKT, Ras-Raf-MEK-ERK, and p38-MAPK signaling cascades are all activated in response to GpI-mGluR stimulation.
Figure 2.

GpI-mGluR-mediated signaling activates PDK1, AKT, Raf-1, ERK2, and p38-MAPK. The signaling pathways that were activated by DHPG in area CA1 of the hippocampus were determined by Western blotting using phospho-specific antibodies. Equal loading of protein was confirmed by blotting for total protein and actin (data not shown). A, Levels of phospho-PDK1, AKT, Raf-1, ERK2, and p38-MAPK were significantly increased (p < 0.05) immediately after treatment with DHPG (50 μm; 10 min). Representative Western blots are shown above summary densitometry. C indicates samples from matched control slices receiving vehicle; E indicates slices receiving DHPG. Arrows indicate a band specific for a particular signaling molecule. Each bar represents the average of 11 replicates. B, No significant differences in levels of phospho-PDK1, AKT, Raf-1, ERK2, or p38-MAPK were observed 50 min after DHPG treatment. Each bar represents the average of nine replicates. C, DHPG-induced activation of ERK2, but not p38 MAPK, was inhibited by pretreatment with the PKC inhibitor GF 109,203× (1 μm; F(1,8) = 11; p < 0.05). Each bar represents the average of four to six replicates. D, DHPG-induced activation of ERK2, but not p38 MAPK, was inhibited by pretreatment with the PI3-K inhibitor Wortmannin (50 nm; F(1,8) = 7.8; p < 0.05). Each bar represents the average of four to six replicates. In A and B, asterisks indicate significant difference from vehicle controls as determined by one-sample t test. In C and D, asterisks indicate significant difference between groups as determined by two-way ANOVA with repeated measures. Error bars indicate SEM.
Activation of GpI-mGluRs led to engagement of both the ERK and p38 MAPK pathways. Some studies have suggested that these two pathways are engaged through distinct signaling mechanisms (Bolshakov et al., 2000; Rush et al., 2002; Huang et al., 2004). We determined whether GpI-mGluRs were coupled to ERK and p38 through either PI3-K- or PKC-dependent signaling pathways. Pretreatment of hippocampal slices with the PKC inhibitor GF 109,203× (1 μm; 10 min) before DHPG exposure (50 μm; 10 min) blocked activation of ERK2 but had no effect on activation of p38 MAPK in area CA1 and the dentate gyrus of the hippocampus (Fig. 2C) (p < 0.05). Similarly, pretreatment of slices with the PI3-K inhibitor Wortmannin (50 nm; 10 min) inhibited DHPG-induced activation of ERK2 but not p38 MAPK (Fig. 2D) (p < 0.05). These results indicate that DHPG-induced activation of ERK2 requires PI3-K and PKC, whereas DHPG-induced activation of p38 MAPK is independent of PI3-K and PKC, consistent with previous observations (Hou and Klann, 2004; Huang et al., 2004).
Our results and previous studies indicate that several signaling pathways are activated in response to GpI-mGluR signaling, including the PI3-K, Ras, and p38-MAPK pathways (Fig. 2A) (Gallagher et al., 2004; Hou and Klann, 2004). Through a series of pharmacologic experiments, we determined which signaling pathways play a role in coupling regulation of NF-κB with GpI-mGluR activation. Inhibiting PI3-K with the specific inhibitor Wortmannin (50 nm) completely blocked DHPG-induced regulation of NF-κB (Fig. 3A–C) (p50, p < 0.05; p65, c-Rel, p < 0.01). Interestingly, pharmacologic inhibition of AKT activation (AKT Inhibitor IV; 3 μm) had no significant effect on DHPG-induced activation of NF-κB (Fig. 3A–C). Thus, PI3-K, but not AKT, is necessary for GpI-mGluR-mediated activation of NF-κB in area CA1 of the hippocampus.
Figure 3.
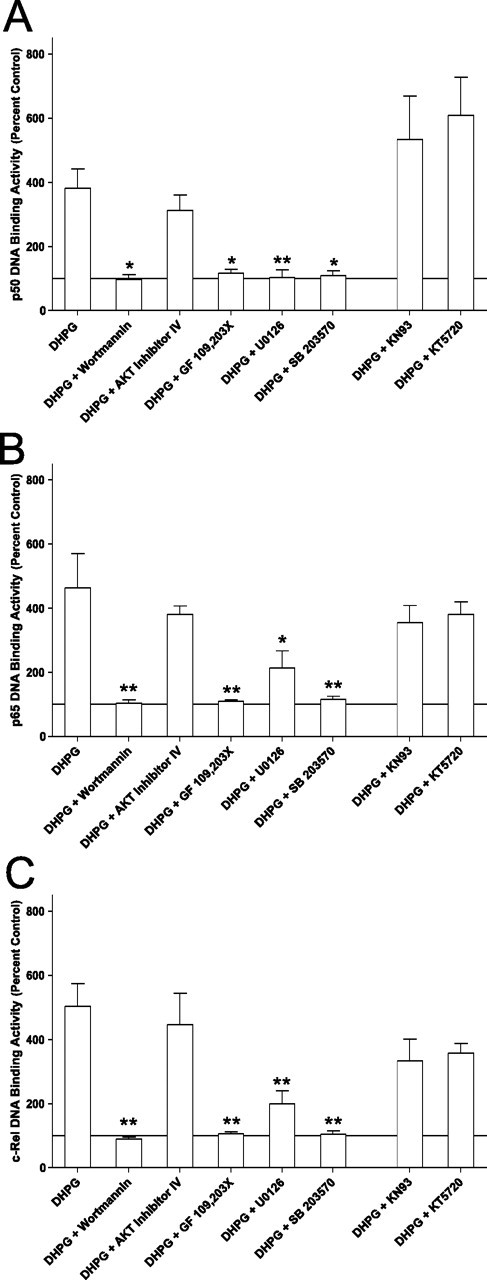
PI3-K, PKC, MEK, and p38-MAPK are necessary for DHPG-induced activation of NF-κB. To determine which signaling pathways were necessary for GpI-mGluR-dependent activation of NF-κB, slices were pretreated with various inhibitors of signaling pathways before treatment with DHPG. The panels represent summary data for p50 (F(7,43) = 9; p < 0.0001) (A), p65 (F(7,43) = 6; p < 0.0001) (B), or c-Rel (F(7,43) = 7; p < 0.0001) (C). KN-93 (CaMKII; 10 μm), KT5720 (PKA, 1 μm), and AKT inhibitor IV (AKT, 3 μm) had no significant effect on DHPG-induced activation of NF-κB. Wortmannin (PI3-K, 50 nm), GF 109,203× (PKC, 1 μm), U0126 (MEK, 20 μm), and SB 203580 (p38-MAPK, 1 μm) significantly inhibited DHPG-induced activation of NF-κB. In each panel, bars represent the mean of four to eight replicates. Error bars are SEM. Asterisks indicate significant difference from DHPG alone (*p < 0.05; **p < 0.01) as determined by Dunnett’s multiple comparison test.
The Ras signaling pathway can be activated by a number of upstream molecules, including PKC. Several studies have implicated the atypical PKCζ in activation of NF-κB in the nervous system (Macdonald et al., 1999; Sanz et al., 1999, 2000; Esteve et al., 2002). Complete inhibition of the DHPG-induced activation of NF-κB was observed in the presence of the PKC inhibitor GF 109,203× (Fig. 3A–C) (1 μm; p50, p < 0.05; p65, c-Rel, p < 0.01). One downstream target of the Ras signaling pathway is MEK, the kinase responsible for activation of ERK. Using the MEK inhibitor U0126 (20 μm), DHPG-induced activation of NF-κB was completely inhibited (Fig. 3A–C) (p50, c-Rel, p < 0.01; p65, p < 0.05). These results suggest that the PKC-Ras-MEK-ERK signaling pathway is necessary for coupling GpI-mGluR signaling to activation of NF-κB in the nervous system.
The final signaling pathway that was activated by DHPG in our biochemical experiments was p38-MAPK. A previous study implicated p38-MAPK, activated by the Rap-1 small GTPase coupled to the βγ subunit of the GpI-mGluR, in induction of mGluR-LTD in the hippocampus (Huang et al., 2004). Inhibition of p38-MAPK with SB 203580 (1 μm) resulted in complete blockade of the DHPG-induced activation of NF-κB in area CA1 (Fig. 3A–C) (p50, p < 0.05; p65, c-Rel, p < 0.01). This suggests that p38-MAPK is also involved in coupling GpI-mGluR signaling with activation of NF-κB in the hippocampus.
In addition to the three signaling pathways we found to be activated by mGluR-mediated signaling, we investigated the role of two additional pathways that have been implicated in activation of NF-κB. Previous studies have shown that Ca2+ and CaMKII can activate NF-κB in the nervous system (Cruise et al., 2000; Lilienbaum and Israel, 2003; Meffert et al., 2003). Blocking CaMKII activity with the inhibitor KN-93 (10 μm) had no significant effect on GpI-mGluR-mediated activation of p50, p65, or c-Rel (Fig. 3A–C). Likewise, although some evidence exists that suggests the cAMP-dependent protein kinase (PKA) may participate in activation of NF-κB (Baeuerle and Henkel, 1994; Min et al., 2004), the PKA inhibitor KT5720 (1 μm) had no effect on DHPG-induced activation of p50, p65, or c-Rel in area CA1 of the hippocampus (Fig. 3A–C). These results suggest that CaMKII and PKA do not play a significant role in coupling GpI-mGluR signaling to activation of NF-κB in the hippocampus.
A role for c-Rel in GpI-mGluR long-term depression
Several studies have shown that activation of GpI-mGluRs has significant effects on the synaptic state of hippocampal Schaffer-collateral synapses. For example, activation of GpI-mGluRs can modulate induction of synaptic plasticity (van Dam et al., 2004). Additionally, direct stimulation of GpI-mGluRs with DHPG or low-frequency synaptic activity in the presence of an NMDAR antagonist can induce a robust, long-lasting, and protein synthesis-dependent form of LTD (Huber et al., 2000; Hou and Klann, 2004). Our results indicate that stimulation of GpI-mGluRs leads to activation of NF-κB, suggesting that NF-κB transcription factors may play a role in the induction of mGluR-LTD.
We used a knock-out mouse model lacking the c-Rel transcription factor (c-Rel−/−) (Tumang et al., 1998) to test the hypothesis that an NF-κB transcription factor may be important for induction of mGluR-dependent synaptic plasticity in area CA1 of the hippocampus.c-relb/b mice had no change in expression of either p50 or p65 mRNA or protein in area CA1 (Fig. 4A,C) or the dentate gyrus (Fig. 4B,D) of the hippocampus, suggesting that loss of c-Rel was not compensated for by upregulation of other classical NF-aB subunits. Expression of mGluR1 and mGluR5 in area CA1 (Fig. 5A) (p < 0.8) and the dentate gyrus (Fig. 5B) (p < 0.5) was unaffected by loss of c-Rel, indicating that expression of GpI-mGluRs was normal in c-rel−/− mice. Moreover, DHPG-induced activation of both ERK2 and p38 MAPK in area CA1 of the hippocampus was unaffected by loss of c-Rel (Fig. 5C) (p < 1.0). Considered together, these results indicate that loss of c-Rel has no significant effect on expression of the p50 or p65 NF-κB subunits, expression of GpI-mGluRs, or levels of GpI-mGluR-mediated activation of ERK2 or p38 MAPK, suggesting that loss of c-Rel has no effect on the capacity for GpI-mGluR-mediated signaling in the hippocampus.
Figure 4.

c-rel−/− mice have normal levels of p50 and p65 mRNA and protein. Area CA1 and the dentate gyrus of c-rel+/+, c-rel+/−, and c-rel−/− animals was isolated from each hemisphere and processed for RNA or protein extraction. A, No significant differences in expression of p50 or p65 mRNA were observed using real-time reverse transcription-QPCR in area CA1 (p50 and p65 c-rel+/+ vs c-rel−/−; p > 0.05 as determined by Bonferroni posttests). Expression of c-Rel mRNA in c-rel+/− animals was ∼50% of c-rel+/+ animals and was significantly decreased in c-rel−/− animals (F(2,14) = 4; p < 0.05). B, No significant differences in expression of p50 or p65 mRNA were observed using real-time reverse transcription-QPCR in the dentate gyrus (p50 and p65 c-rel+/+ vs c-rel−/−, p > 0.05 as determined by Bonferroni posttests). Expression of c-Rel mRNA in c-rel+/− animals was ∼50% of c-rel+/+ animals and was significantly decreased in c-rel−/− animals (F(2,14) = 8; p < 0.001). C, Analysis of tissue derived from the same animals used in A revealed a significant difference in expression of c-Rel between c-rel+/+ and c-rel−/− animals (F(2,64) = 5; p < 0.05). No significant differences were observed in expression of p50 or p65 protein (p > 0.05; post hoc Bonferroni analysis). D, Analysis of tissue derived from the same animals used in B revealed a significant difference in expression of c-Rel between c-rel+/+ and c-rel−/− animals (F(2,58) = 4; p < 0.05). No significant differences were observed in expression of p50 or p65 protein (p > 0.05; post hoc Bonferroni analysis). In C and D, representative Western blots are shown above summary densitometry. Arrows next to blots indicate immunoreactive band specific for p50, p65, or c-Rel, and c-Rel genotype is indicated by either +/+ (wild type), +/− (heterozygous), or −/− (null). In all panels, error bars indicate SEM. The asterisk indicates significant difference (*p < 0.05) from c-rel+/+ as determined by post hoc Bonferroni comparisons.
Figure 5.
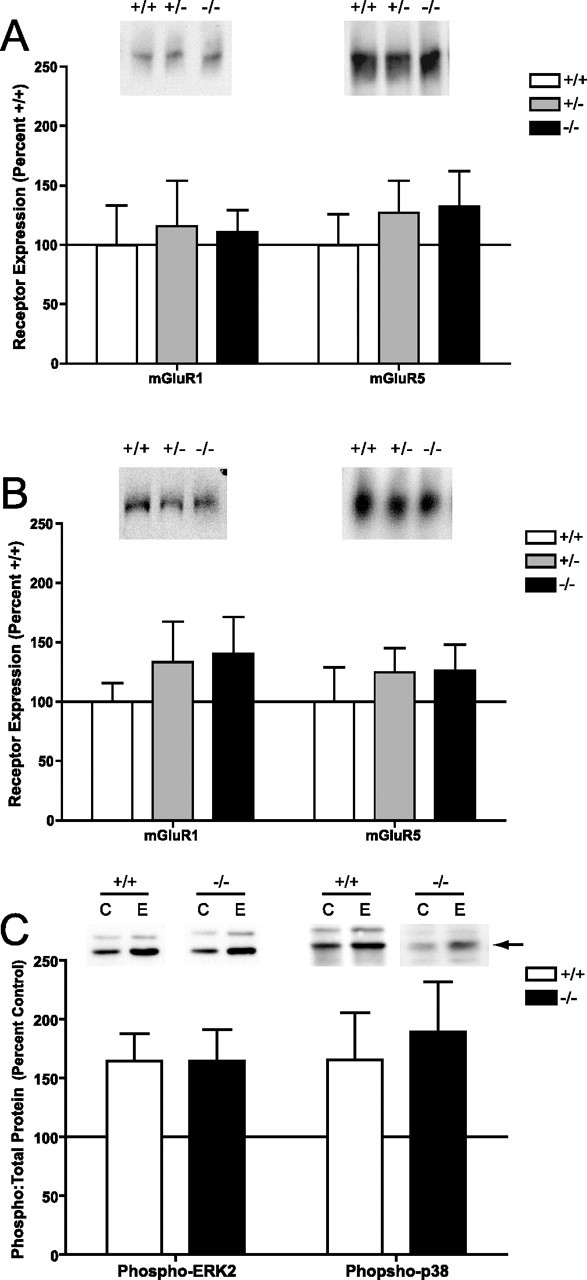
Expression of GpI-mGluRs and induction of MAPK is normal in c-rel−/− animals. Levels of GpI-mGluRs and activation of MAPK was measured in the hippocampus from c-rel−/− animals. A, No significant differences in expression of mGluR1 or mGluR5 were observed in area CA1 (F(2,40) = 0.3; p < 0.8). B, No significant differences in expression of mGluR1 or mGluR5 were observed in the dentate gyrus (F(2,38) = 0.8; p < 0.5). C, No significant differences in DHPG-induced activation of ERK2 or p38 MAPK were observed in area CA1 (F(1,16) = 0.003; p < 1.0). In all panels, representative Western blots are shown above summary densitometry. Genotype and experimental treatment (C, control; E, DHPG) are indicated above blots. Error bars indicate SEM.
To assess basal synaptic function in the hippocampus, two measures of Schaffer-collateral synaptic transmission were made in groups of littermate c-rel+/+ and c-rel−/− mice. Input/output curves for c-rel−/− mice were significantly smaller than those from c-rel+/+ mice, suggesting a reduction in basal synaptic transmission in c-rel−/− mice (Fig. 6A) (p < 0.0001). Paired-pulse facilitation in c-rel+/+ and c-rel−/− mice was comparable, suggesting the reduction in synaptic transmission was most likely not a result of a reduction in neurotransmitter release (Fig. 6B) (p < 0.7). To further investigate the decrease in synaptic transmission, analysis of miniature, spontaneous EPSCs was performed using whole-cell patch clamp of CA1 pyramidal neurons. No significant differences were observed in spontaneous event amplitude (Fig. 6C) (p < 1.0) or interevent interval (Fig. 6D) (p = 0.05), suggesting that postsynaptic and presynaptic function, respectively, in c-rel−/− animals is normal. In addition, no significant differences were observed in resting membrane potential (Fig. 6E) (p < 0.2) or input resistance (Fig. 6F) (p < 0.6), indicating that loss of c-Rel has no effect on the basal biophysical properties of CA1 pyramidal neurons. Collectively, these results suggest that the modest reduction in hippocampal synaptic transmission observed in c-rel−/− mice is attributable to a decrease in the number, but not efficacy, of synaptic inputs into area CA1.
Figure 6.
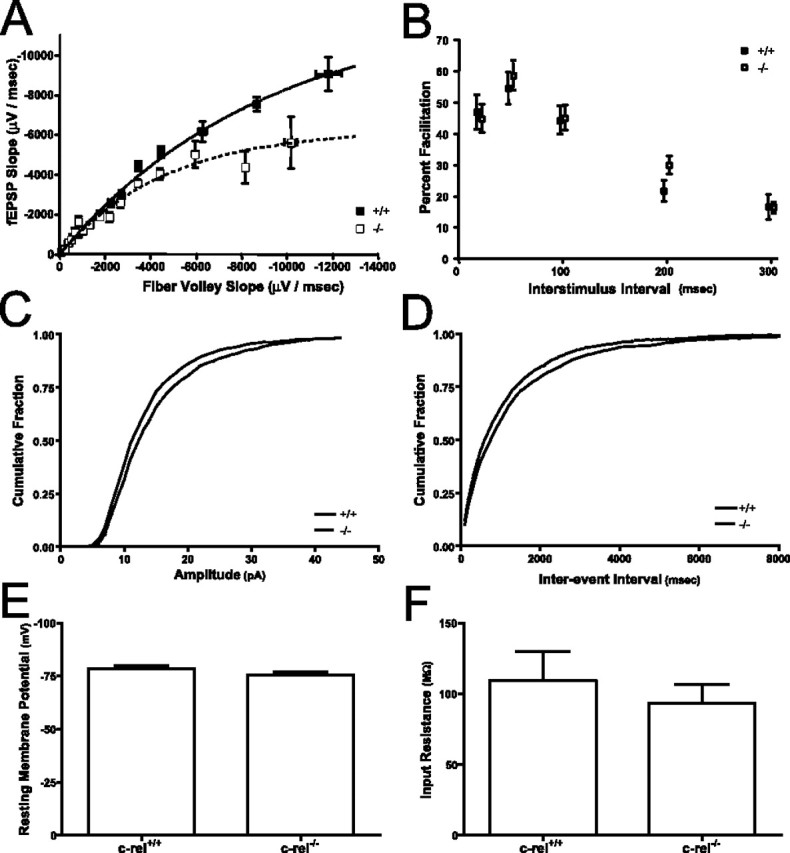
Loss of c-Rel on hippocampal synaptic transmission. Synaptic transmission was assessed at Schaffer-collateral synapses of c-rel+/+ and c-rel−/− littermate animals. A, Synaptic transmission as determined by the ratio of fEPSP slope (postsynaptic depolarization) versus fiber volley slope (presynaptic depolarization). Animals lacking c-Rel had significantly reduced synaptic transmission relative to wild-type littermates (F(2,28) = 34; p < 0.0001). B, No significant differences were seen in paired-pulse facilitation measured at several interstimulus intervals (20, 50, 100, 200, 300 ms) between c-rel+/+ and c-rel−/− littermates (F(1,62) = 0.3; p < 0.7). C, Minianalysis of Schaffer-collateral synapses revealed no significant difference in spontaneous event amplitude (D = 0.02; n = 560; p < 1.0). D, Minianalysis of Schaffer-collateral synapses revealed no significant difference in spontaneous interevent interval (D = 0.05; n = 719; p < 0.1). E, No significant differences were observed in resting membrane potential of pyramidal neurons in area CA1 of the hippocampus (t = 1.5; df = 16; p < 0.2). F, No significant differences were observed in input resistance of pyramidal neurons in area CA1 of the hippocampus (t = 0.7; df = 16; p < 0.6). In all panels, error bars indicate SEM.
To further assess synaptic function in c-rel−/− mice, we measured GpI-mGluR-dependent LTD in area CA1 of the hippocampus. Application of DHPG (50 μm; 10 min) to the hippocampus led to a rapid and robust decrease in synaptic efficacy in both c-rel+/+ and c-rel−/− mice (Fig. 7A), indicating that the ability to detect and acutely respond to GpI-mGluR stimulation was normal in c-rel−/− mice, providing a physiologic confirmation of our biochemical studies of GpI-mGluR-induced activation of ERK2 and p38 MAPK (Fig. 5C). Levels of synaptic depression remained qualitatively normal in c-rel−/− mice for ∼70 min after induction, at which point synaptic efficacy gradually increased to predepression levels (Fig. 7A). These data indicate a derangement in induction of persistent mGluR-LTD in c-rel−/− mice and suggest c-Rel as a transcription factor that is important for either induction and/or maintenance of a late phase of mGluR-dependent LTD in the hippocampus.
Figure 7.
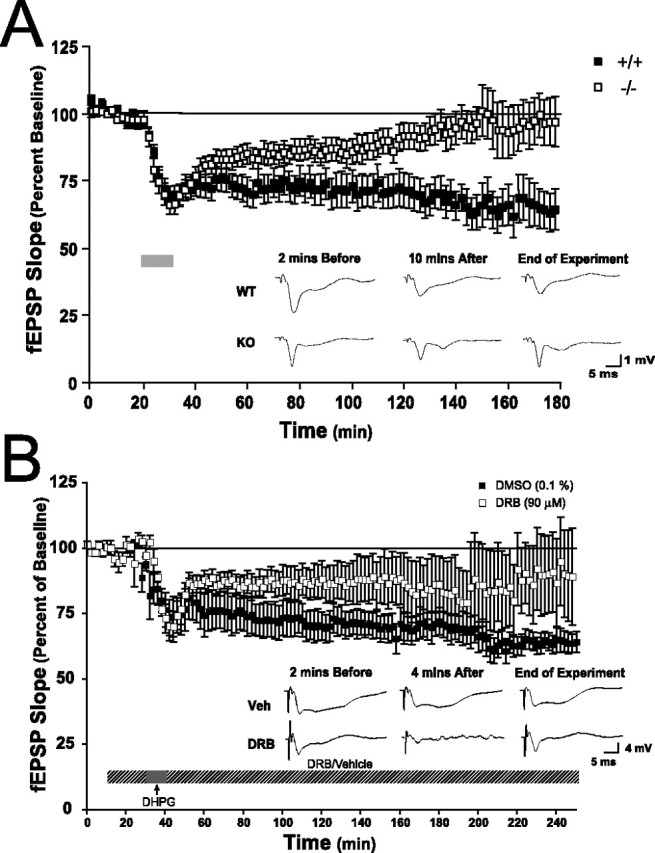
Impaired hippocampal long-term depression in c-rel−/− animals. mGluR-LTD was induced at Schaffer-collateral synapses by treatment with DHPG (50 μm, 10 min; gray bar). A, Acute synaptic depression was identical between slices from c-rel+/+ and c-rel−/− littermates. LTD was similar between c-rel+/+ and c-rel−/− animals for ∼90 min, when synaptic depression in slices from c-rel−/− animals decayed to basal levels of synaptic efficacy (F(1,89) = 340; p < 0.0001). Representative traces 2 min before, 10 min after, and 150 min after DHPG treatment are shown below summary electrophysiology. Calibration: 1 mV, 5 ms. B, Pretreatment of slices with the transcription inhibitor DRB (90 μm; 20 min) attenuated induction of mGluR-LTD (F(1,19) = 214; p < 0.001). In all panels, error bars indicate SEM.
Previous studies have demonstrated that transcription-dependent forms of LTD can be induced at Schaffer-collateral synapses in the hippocampus (Kauderer and Kandel, 2000). Impaired induction of mGluR-LTD in the hippocampus of c-rel−/− mice (Fig. 7A) coupled with normal expression of GpI-mGluRs and normal MAPK signaling (Fig. 5) suggested the existence of a transcription-dependent late phase of mGluR-LTD. To investigate whether mGluR-LTD in the hippocampus was sensitive to inhibitors of transcription, mGluR-LTD was induced in hippocampal slices that were pretreated with the transcription inhibitor 5,6-dichloro-1-β-d-ribofuranosylbenzimidazole (DRB; 90 μm). Early induction of LTD was normal in slices exposed to either the vehicle (0.1% DMSO) or DRB, suggesting that transcription is not required for induction and early expression of mGluR-LTD in the hippocampus (Fig. 7B). However, LTD was impaired and decayed to basal levels 3.5 h after induction in slices treated with DRB (Fig. 7B) (p < 0.001). These results suggest that long-lasting forms of mGluR-LTD are sensitive to the transcription inhibitor DRB.
Impaired long-term memory formation in c-rel−/− animals
Synaptic plasticity is the leading candidate cellular mechanism for storage of long-term memory in the nervous system. Thus, it is possible that c-rel−/− mice may have impaired capacity for long-term memory formation. We determined performance of c-rel+/+ and c-rel−/− animals in a passive avoidance task. In this amygdala- and hippocampus-dependent task, animals learn to avoid entry into a dark environment (Isaacson and Wickelgren, 1962; Ambrogi Lorenzini et al., 1997; Irvine et al., 2005). Performance of both c-rel+/+ and c-rel−/− littermates was identical during the training day, indicating that c-rel−/− animals have an equivalent motivational desire to move from a well-lit environment to a dark environment (Fig. 8). However, when latency to enter the dark half of the shuttle box was assessed 24 h after training, c-rel−/− animals exhibited a significant decrease relative to their c-rel+/+ littermates (Fig. 8) (p < 0.05), indicating an impaired capacity for long-term memory formation. Previous studies of memory formation in c-rel−/− animals indicated that formation of amygdala-dependent cued fear conditioning is normal (Levenson et al., 2004). Therefore, we conclude that the deficit in formation of long-term passive avoidance memory observed in c-rel−/− animals is a result of a deficit in hippocampus-dependent long-term memory formation.
Figure 8.

Impaired performance in a passive avoidance task in c-rel−/− animals. Passive avoidance was assessed in a two-chamber shuttle box. During the training day, c-rel+/+ and c-rel−/− animals performed equally, moving into the dark side of the chamber within 15 s of placement into the shuttle box. When tested 24 h after training, c-rel−/− animals exhibited a significantly lower latency to enter the dark side compared with c-rel+/+ littermates (U = 159; p < 0.05; n+/+ = 15; n−/− = 36), indicating reduced capacity for formation of long-term passive avoidance memory.
Discussion
The present study provides evidence that signaling through GpI-mGluRs can regulate the NF-κB family of transcription factors in the hippocampus. In addition, our study provides the first evidence that c-Rel, an NF-κB transcription factor, is required for persistence of mGluR-LTD in the hippocampus. Together, these results are the first to link GpI-mGluR-induced long-term depression in the hippocampus to the function of a transcription factor.
Several studies suggest that long-term changes in neural function induced through GpI-mGluR signaling require transcription. Signaling through GpI-mGluRs induces and/or modulates persistent forms of transcription-dependent synaptic plasticity and long-term memory (Riedel et al., 1994; Balschun et al., 1999; Maciejak et al., 2003; Manahan-Vaughan and Braunewell, 2005; Naie and Manahan-Vaughan, 2005). Moreover, activation of GpI-mGluRs and/or induction of LTD in the hippocampus regulate the phosphorylation of two transcription factors, cAMP response element-binding protein and Elk-1 (Choe and Wang, 2002; Mao and Wang, 2002; Thiels et al., 2002; Boulware et al., 2005). Our findings that signaling through GpI-mGluRs activates several members of the NF-κB transcription factor family and that maintenance of mGluR-LTD requires c-Rel extends previous observations implicating GpI-mGluRs in transcription-dependent synaptic plasticity and long-term memory.
Previous studies have not implicated transcription in the induction, expression, or maintenance of hippocampal mGluR-LTD (Huber et al., 2000). This is in contrast to NMDA-R-dependent LTD at hippocampal Schaffer-collateral synapses, LTD of cerebellar Purkinje neurons, and to numerous studies documenting a requirement for transcription in long-term facilitation of the Aplysia sensorimotor synapse and long-term potentiation (LTP) of the hippocampal Schaffer-collateral synapse (Montarolo et al., 1986; Nguyen et al., 1994; Linden, 1996; Kauderer and Kandel, 2000; Karachot et al., 2001). One possible explanation for an apparent lack of effect of transcription inhibitors on mGluR-LTD could be that the initial studies of mGluR-LTD monitored synaptic efficacy for only 90 min after induction of LTD. In studies of LTP, no effect of transcription inhibitors was observed until 2 h after induction (Nguyen et al., 1994). In our experiments, significant increases in NF-κB DNA binding activity did not occur until 50 min after the end of DHPG treatment (Fig. 1A), and significant decay of LTD in c-rel−/− mice did not begin until ∼90 min after induction (Fig. 7A). Maximal differences in LTD were observed 2 h after induction (Fig. 7A). Thus, early experiments on the role of transcription in hippocampal mGluR-LTD did not rule out a role for transcription in a late phase of LTD with a similar temporal profile as the transcription-dependent late-phase of LTP. Using the transcription inhibitor DRB, we observed a diminution in mGluR-LTD in the hippocampus 2–3 h after its induction (Fig. 7B).
It should be noted that the effect of DRB (Fig. 7B) does not phenocopy the effect that loss of c-Rel has on mGluR-LTD (Fig. 7A). The discrepancy between loss of c-Rel and treatment with DRB could be attributable to an incomplete cessation of transcription by DRB because of either dose or partial diffusion into the tissue slice or an effect of c-Rel on nontranscription-dependent processes necessary for induction or expression of mGluR-LTD. One possibility is that global knock-out of c-Rel during all phases of development could lead to neurodevelopmental defects that prevent the induction or expression of mGluR-LTD. There are no reports of gross neurodevelopmental defects in c-Rel knock-out mice (Tumang et al., 1998; Levenson et al., 2004). NF-κB/Rel-mediated transcription is not detectable in the nervous system until shortly after embryonic day 12 in the rhombencephalon (Schmidt-Ullrich et al., 1996). NF-κB/Rel-mediated transcription is not detectable until after birth in the cortex [postnatal day 6 (P6)], cerebellum (P8), and hippocampus (P15) (Schmidt-Ullrich et al., 1996). All of these results suggest that NF-κB/Rel-mediated transcription is not necessary for neural development.
Another possible effect that loss of c-Rel could have is disruption of the expression of proteins involved in the induction or expression of mGluR-LTD. For example, expression of glutamate receptors or signaling molecules could be altered in c-rel−/− mice. Expression of mGluR1 and mGluR5 are normal in the hippocampus of c-rel−/− mice (Fig. 5A,B). Activation of both ERK2 and p38 MAPK by DHPG is normal in c-rel−/− mice (Fig. 5C). Moreover, the acute physiologic response to DHPG and subsequent short-term (<60 min) synaptic depression is essentially unchanged in c-rel−/− animals (Fig. 7A). If the signaling pathways responsible for induction of mGluR-LTD were deranged, then one would expect a derangement in MAPK signaling and an immediate loss of mGluR-LTD (Huber et al., 2001; Gallagher et al., 2004; Hou and Klann, 2004). The temporal manifestation of the deficit in mGluR-LTD in c-rel−/− animals suggests that processes responsible for long-term maintenance and/or expression of LTD were specifically affected and not induction (Nguyen et al., 1994).
There is an increasing body of evidence that the NF-κB transcription factor family plays an important role in long-term information storage in the brain. Several studies have suggested that NF-κB plays a role in synaptic plasticity. NF-κB transcription factors are activated by a wide dynamic range of synaptic activity in the brain (Meberg et al., 1996; Meffert et al., 2003). Pre-exposure to NF-κB decoy oligonucleotides blocks induction of LTP in the hippocampus and amygdala (Albensi and Mattson, 2000; Yeh et al., 2002). Additionally, a series of experiments investigating the cytokine tumor necrosis factor (TNF), a powerful activator of the NF-κB system in nearly every tissue examined, have shown that exposure to exogenous TNFα inhibits induction of LTP and that genetic knock-out of TNF receptors inhibits induction of LTD (Tancredi et al., 1992; Cunningham et al., 1996; Albensi and Mattson, 2000). Our results extend these findings by demonstrating a specific member of the NF-κB transcription factor family, c-Rel, plays a direct role in the induction of mGluR-LTD in the hippocampus.
Synaptic plasticity is one of the leading candidate cellular mechanisms for storage of long-term memory in the nervous system. Several studies have implicated the NF-κB system in the formation of long-term memory. Infusion of NF-κB decoy oligonucleotides into the amygdala blocks formation of fear-potentiated startle (Yeh et al., 2002, 2004), and infusion into the hippocampus blocks formation of spatial memory (Dash et al., 2005). Simultaneous knock-out of the NF-κB family member p65/RelA and TNF-R1 blocks formation of hippocampus-dependent spatial memory but not striatal-dependent working memory (Meffert et al., 2003). In the current and previous studies, we provide several lines of evidence that knock-out of c-Rel inhibits formation of hippocampus-dependent long-term memory but does not affect amygdala-dependent long-term memory formation (Fig. 8) (Levenson et al., 2004). All of these results indicate that NF-κB plays a role in long-term memory formation and that two members of the NF-κB family, p65/RelA and c-Rel, play important roles in formation of hippocampus-dependent long-term memory.
The function of NF-κB makes it a unique transcription factor. NF-κB was the first transcription factor to be localized to synaptic regions in the CNS (Korner et al., 1989). Several subsequent studies have confirmed that latent NF-κB dimers are present in synaptic regions, and that once activated, NF-κB translocates from the synapse to the nucleus (Kaltschmidt et al., 1993, 1995; Meberg et al., 1996; Cruise et al., 2000; Wellmann et al., 2001; Meffert et al., 2003; Scholzke et al., 2003). This suggests that the NF-κB transcription factor family may act as a synapse-to-nucleus messenger in the nervous system. What is the specific role of NF-κB in the context of synaptic plasticity? Previous reports suggest that NF-κB-mediated transcription in general is required for induction of LTP (Albensi and Mattson, 2000; Yeh et al., 2002), and we present data suggesting that c-Rel specifically is necessary for maintenance of mGluR-LTD (Fig. 7A). We propose the hypothesis that the NF-κB system is involved in the induction and/or maintenance of persistent, transcription-dependent forms of synaptic plasticity in general in the nervous system. Thus, in their role as a plasticity-inducing transcription factor potentially controlling bidirectional changes in synaptic strength, the NF-κB family could play a unique role in facilitating the storage of several different kinds of memory in the nervous system.
Footnotes
K. J. O’Riordan’s present address: Department of Pharmacology, University of Wisconsin School of Medicine and Public Health, Madison, WI 53706.
L. Malone’s present address: Center for Learning and Memory, University of Texas–Austin, Austin, TX 78735.
J. D. Sweatt’s present address: Department of Neurobiology, University of Alabama–Birmingham, Birmingham, AL 35294.
This work was supported by National Institute of Mental Health Grant MH57014 (J.D.S.). We thank E. Klann and F. Lubin for helpful scientific discussions.
References
- Albensi BC, Mattson MP (2000). Evidence for the involvement of TNF and NF-kappaB in hippocampal synaptic plasticity. Synapse 35:151–159. [DOI] [PubMed] [Google Scholar]
- Ambrogi Lorenzini CG, Baldi E, Bucherelli C, Sacchetti B, Tassoni G (1997). Analysis of mnemonic processing by means of totally reversible neural inactivations. Brain Res Brain Res Protoc 1:391–398. [DOI] [PubMed] [Google Scholar]
- Baeuerle PA, Henkel T (1994). Function and activation of NF-kappa B in the immune system. Annu Rev Immunol 12:141–179. [DOI] [PubMed] [Google Scholar]
- Balschun D, Manahan-Vaughan D, Wagner T, Behnisch T, Reymann KG, Wetzel W (1999). A specific role for group I mGluRs in hippocampal LTP and hippocampus-dependent spatial learning. Learn Mem 6:138–152. [PMC free article] [PubMed] [Google Scholar]
- Bolshakov VY, Siegelbaum SA (1994). Postsynaptic induction and presynaptic expression of hippocampal long-term depression. Science 264:1148–1152. [DOI] [PubMed] [Google Scholar]
- Bolshakov VY, Carboni L, Cobb MH, Siegelbaum SA, Belardetti F (2000). Dual MAP kinase pathways mediate opposing forms of long-term plasticity at CA3-CA1 synapses. Nat Neurosci 3:1107–1112. [DOI] [PubMed] [Google Scholar]
- Boulware MI, Weick JP, Becklund BR, Kuo SP, Groth RD, Mermelstein PG (2005). Estradiol activates group I and II metabotropic glutamate receptor signaling, leading to opposing influences on cAMP response element-binding protein. J Neurosci 25:5066–5078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burr PB, Morris BJ (2002). Involvement of NMDA receptors and a p21Ras-like guanosine triphosphatase in the constitutive activation of nuclear factor-kappa-B in cortical neurons. Exp Brain Res 147:273–279. [DOI] [PubMed] [Google Scholar]
- Choe ES, Wang JQ (2002). Regulation of transcription factor phosphorylation by metabotropic glutamate receptor-associated signaling pathways in rat striatal neurons. Neuroscience 114:557–565. [DOI] [PubMed] [Google Scholar]
- Conn PJ, Pin JP (1997). Pharmacology and functions of metabotropic glutamate receptors. Annu Rev Pharmacol Toxicol 37:205–237. [DOI] [PubMed] [Google Scholar]
- Cruise L, Ho LK, Veitch K, Fuller G, Morris BJ (2000). Kainate receptors activate NF-kappaB via MAP kinase in striatal neurones. NeuroReport 11:395–398. [DOI] [PubMed] [Google Scholar]
- Cunningham AJ, Murray CA, O’Neill LA, Lynch MA, O’Connor JJ (1996). Interleukin-1 beta (IL-1 beta) and tumour necrosis factor (TNF) inhibit long-term potentiation in the rat dentate gyrus in vitro. Neurosci Lett 203:17–20. [DOI] [PubMed] [Google Scholar]
- Dash PK, Orsi SA, Moore AN (2005). Sequestration of serum response factor in the hippocampus impairs long-term spatial memory. J Neurochem 93:269–278. [DOI] [PubMed] [Google Scholar]
- Esteve PO, Chicoine E, Robledo O, Aoudjit F, Descoteaux A, Potworowski EF, St-Pierre Y (2002). Protein kinase C-zeta regulates transcription of the matrix metalloproteinase-9 gene induced by IL-1 and TNF-alpha in glioma cells via NF-kappa B. J Biol Chem 277:35150–35155. [DOI] [PubMed] [Google Scholar]
- Freudenthal R, Romano A, Routtenberg A (2004). Transcription factor NF-kappaB activation after in vivo perforant path LTP in mouse hippocampus. Hippocampus 14:677–683. [DOI] [PubMed] [Google Scholar]
- Gallagher SM, Daly CA, Bear MF, Huber KM (2004). Extracellular signal-regulated protein kinase activation is required for metabotropic glutamate receptor-dependent long-term depression in hippocampal area CA1. J Neurosci 24:4859–4864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou L, Klann E (2004). Activation of the phosphoinositide 3-kinase-Akt-mammalian target of rapamycin signaling pathway is required for metabotropic glutamate receptor-dependent long-term depression. J Neurosci 24:6352–6361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang CC, You JL, Wu MY, Hsu KS (2004). Rap1-induced p38 mitogen-activated protein kinase activation facilitates AMPA receptor trafficking via the GDI.Rab5 complex. Potential role in (S)-3,5-dihydroxyphenylglycene-induced long term depression. J Biol Chem 279:12286–12292. [DOI] [PubMed] [Google Scholar]
- Huber KM, Kayser MS, Bear MF (2000). Role for rapid dendritic protein synthesis in hippocampal mGluR-dependent long-term depression. Science 288:1254–1257. [DOI] [PubMed] [Google Scholar]
- Huber KM, Roder JC, Bear MF (2001). Chemical induction of mGluR5- and protein synthesis-dependent long-term depression in hippocampal area CA1. J Neurophysiol 86:321–325. [DOI] [PubMed] [Google Scholar]
- Irvine EE, Vernon J, Giese KP (2005). AlphaCaMKII autophosphorylation contributes to rapid learning but is not necessary for memory. Nat Neurosci 8:411–412. [DOI] [PubMed] [Google Scholar]
- Isaacson RL, Wickelgren WO (1962). Hippocampal ablation and passive avoidance. Science 138:1104–1106. [DOI] [PubMed] [Google Scholar]
- Kaltschmidt C, Kaltschmidt B, Baeuerle PA (1993). Brain synapses contain inducible forms of the transcription factor NF-kappa B. Mech Dev 43:135–147. [DOI] [PubMed] [Google Scholar]
- Kaltschmidt C, Kaltschmidt B, Baeuerle PA (1995). Stimulation of ionotropic glutamate receptors activates transcription factor NF-kappa B in primary neurons. Proc Natl Acad Sci USA 92:9618–9622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karachot L, Shirai Y, Vigot R, Yamamori T, Ito M (2001). Induction of long-term depression in cerebellar Purkinje cells requires a rapidly turned over protein. J Neurophysiol 86:280–289. [DOI] [PubMed] [Google Scholar]
- Kassed CA, Willing AE, Garbuzova-Davis S, Sanberg PR, Pennypacker KR (2002). Lack of NF-kappaB p50 exacerbates degeneration of hippocampal neurons after chemical exposure and impairs learning. Exp Neurol 176:277–288. [DOI] [PubMed] [Google Scholar]
- Kauderer BS, Kandel ER (2000). Capture of a protein synthesis-dependent component of long-term depression. Proc Natl Acad Sci USA 97:13342–13347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemp N, Bashir ZI (1999). Induction of LTD in the adult hippocampus by the synaptic activation of AMPA/kainate and metabotropic glutamate receptors. Neuropharmacology 38:495–504. [DOI] [PubMed] [Google Scholar]
- Korner M, Rattner A, Mauxion F, Sen R, Citri Y (1989). A brain-specific transcription activator. Neuron 3:563–572. [DOI] [PubMed] [Google Scholar]
- Levenson JM, Choi S, Lee SY, Cao YA, Ahn HJ, Worley KC, Pizzi M, Liou HC, Sweatt JD (2004). A bioinformatics analysis of memory consolidation reveals involvement of the transcription factor c-Rel. J Neurosci 24:3933–3943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lilienbaum A, Israel A (2003). From calcium to NF-kappa B signaling pathways in neurons. Mol Cell Biol 23:2680–2698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linden DJ (1996). A protein synthesis-dependent late phase of cerebellar long-term depression. Neuron 17:483–490. [DOI] [PubMed] [Google Scholar]
- Liou HC, Hsia CY (2003). Distinctions between c-Rel and other NF-kappaB proteins in immunity and disease. Bioessays 25:767–780. [DOI] [PubMed] [Google Scholar]
- Macdonald NJ, Perez-Polo JR, Bennett AD, Taglialatela G (1999). NGF-resistant PC12 cell death induced by arachidonic acid is accompanied by a decrease of active PKCζ and nuclear factor κB. J Neurosci Res 57:219–226. [DOI] [PubMed] [Google Scholar]
- Maciejak P, Taracha E, Lehner M, Szyndler J, Bidzinski A, Skorzewska A, Wislowska A, Zienowicz M, Plaznik A (2003). Hippocampal mGluR1 and consolidation of contextual fear conditioning. Brain Res Bull 62:39–45. [DOI] [PubMed] [Google Scholar]
- Manahan-Vaughan D, Braunewell KH (2005). The metabotropic glutamate receptor, mGluR5, is a key determinant of good and bad spatial learning performance and hippocampal synaptic plasticity. Cereb Cortex 15:1703–1713. [DOI] [PubMed] [Google Scholar]
- Mao L, Wang JQ (2002). Interactions between ionotropic and metabotropic glutamate receptors regulate cAMP response element-binding protein phosphorylation in cultured striatal neurons. Neuroscience 115:395–402. [DOI] [PubMed] [Google Scholar]
- Mattson MP, Culmsee C, Yu Z, Camandola S (2000). Roles of nuclear factor kappaB in neuronal survival and plasticity. J Neurochem 74:443–456. [DOI] [PubMed] [Google Scholar]
- Meberg PJ, Kinney WR, Valcourt EG, Routtenberg A (1996). Gene expression of the transcription factor NF-kappa B in hippocampus: regulation by synaptic activity. Brain Res Mol Brain Res 38:179–190. [DOI] [PubMed] [Google Scholar]
- Meffert MK, Chang JM, Wiltgen BJ, Fanselow MS, Baltimore D (2003). NF-kappa B functions in synaptic signaling and behavior. Nat Neurosci 6:1072–1078. [DOI] [PubMed] [Google Scholar]
- Milner B, Squire LR, Kandel ER (1998). Cognitive neuroscience and the study of memory. Neuron 20:445–468. [DOI] [PubMed] [Google Scholar]
- Min KJ, Yang MS, Jou I, Joe EH (2004). Protein kinase A mediates microglial activation induced by plasminogen and gangliosides. Exp Mol Med 36:461–467. [DOI] [PubMed] [Google Scholar]
- Montarolo PG, Goelet P, Castellucci VF, Morgan J, Kandel ER, Schacher S (1986). A critical period for macromolecular synthesis in long-term heterosynaptic facilitation in Aplysia. Science 234:1249–1254. [DOI] [PubMed] [Google Scholar]
- Naie K, Manahan-Vaughan D (2005). Pharmacological antagonism of metabotropic glutamate receptor 1 regulates long-term potentiation and spatial reference memory in the dentate gyrus of freely moving rats via N-methyl-d-aspartate and metabotropic glutamate receptor-dependent mechanisms. Eur J Neurosci 21:411–421. [DOI] [PubMed] [Google Scholar]
- Nakanishi S (1992). Molecular diversity of glutamate receptors and implications for brain function. Science 258:597–603. [DOI] [PubMed] [Google Scholar]
- Nguyen PV, Abel T, Kandel ER (1994). Requirement of a critical period of transcription for induction of a late phase of LTP. Science 265:1104–1107. [DOI] [PubMed] [Google Scholar]
- Nosyreva ED, Huber KM (2005). Developmental switch in synaptic mechanisms of hippocampal metabotropic glutamate receptor-dependent long-term depression. J Neurosci 25:2992–3001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliet SH, Malenka RC, Nicoll RA (1997). Two distinct forms of long-term depression coexist in CA1 hippocampal pyramidal cells. Neuron 18:969–982. [DOI] [PubMed] [Google Scholar]
- Pizzi M, Sarnico I, Boroni F, Benarese M, Steimberg N, Mazzoleni G, Dietz GP, Bahr M, Liou HC, Spano PF (2005). NF-kappaB factor c-Rel mediates neuroprotection elicited by mGlu5 receptor agonists against amyloid beta-peptide toxicity. Cell Death Differ 12:761–772. [DOI] [PubMed] [Google Scholar]
- Riedel G, Wetzel W, Reymann KG (1994). (R,S)-alpha-methyl-4-carboxyphenylglycine (MCPG) blocks spatial learning in rats and long-term potentiation in the dentate gyrus in vivo. Neurosci Lett 167:141–144. [DOI] [PubMed] [Google Scholar]
- Rush AM, Wu J, Rowan MJ, Anwyl R (2002). Group I metabotropic glutamate receptor (mGluR)-dependent long-term depression mediated via p38 mitogen-activated protein kinase is inhibited by previous high-frequency stimulation and activation of mGluRs and protein kinase C in the rat dentate gyrus in vitro. J Neurosci 22:6121–6128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanz L, Sanchez P, Lallena MJ, Diaz-Meco MT, Moscat J (1999). The interaction of p62 with RIP links the atypical PKCs to NF-kappaB activation. EMBO J 18:3044–3053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanz L, Diaz-Meco MT, Nakano H, Moscat J (2000). The atypical PKC-interacting protein p62 channels NF-kappaB activation by the IL-1-TRAF6 pathway. EMBO J 19:1576–1586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt-Ullrich R, Memet S, Lilienbaum A, Feuillard J, Raphael M, Israel A (1996). NF-kappaB activity in transgenic mice: developmental regulation and tissue specificity. Development 122:2117–2128. [DOI] [PubMed] [Google Scholar]
- Scholzke MN, Potrovita I, Subramaniam S, Prinz S, Schwaninger M (2003). Glutamate activates NF-kappaB through calpain in neurons. Eur J Neurosci 18:3305–3310. [DOI] [PubMed] [Google Scholar]
- Snyder EM, Philpot BD, Huber KM, Dong X, Fallon JR, Bear MF (2001). Internalization of ionotropic glutamate receptors in response to mGluR activation. Nat Neurosci 4:1079–1085. [DOI] [PubMed] [Google Scholar]
- Suzuki T, Mitake S, Okumura-Noji K, Yang JP, Fujii T, Okamoto T (1997). Presence of NF-kappaB-like and IkappaB-like immunoreactivities in postsynaptic densities. NeuroReport 8:2931–2935. [DOI] [PubMed] [Google Scholar]
- Tancredi V, D’Arcangelo G, Grassi F, Tarroni P, Palmieri G, Santoni A, Eusebi F (1992). Tumor necrosis factor alters synaptic transmission in rat hippocampal slices. Neurosci Lett 146:176–178. [DOI] [PubMed] [Google Scholar]
- Thiels E, Kanterewicz BI, Norman ED, Trzaskos JM, Klann E (2002). Long-term depression in the adult hippocampus in vivo involves activation of extracellular signal-regulated kinase and phosphorylation of Elk-1. J Neurosci 22:2054–2062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tumang JR, Owyang A, Andjelic S, Jin Z, Hardy RR, Liou ML, Liou HC (1998). c-Rel is essential for B lymphocyte survival and cell cycle progression. Eur J Immunol 28:4299–4312. [DOI] [PubMed] [Google Scholar]
- van Dam EJ, Kamal A, Artola A, de Graan PN, Gispen WH, Ramakers GM (2004). Group I metabotropic glutamate receptors regulate the frequency-response function of hippocampal CA1 synapses for the induction of LTP and LTD. Eur J Neurosci 19:112–118. [DOI] [PubMed] [Google Scholar]
- Wellmann H, Kaltschmidt B, Kaltschmidt C (2001). Retrograde transport of transcription factor NF-kappa B in living neurons. J Biol Chem 276:11821–11829. [DOI] [PubMed] [Google Scholar]
- Yeh SH, Lin CH, Lee CF, Gean PW (2002). A requirement of nuclear factor-kappaB activation in fear-potentiated startle. J Biol Chem 277:46720–46729. [DOI] [PubMed] [Google Scholar]
- Yeh SH, Lin CH, Gean PW (2004). Acetylation of nuclear factor-kappaB in rat amygdala improves long-term but not short-term retention of fear memory. Mol Pharmacol 65:1286–1292. [DOI] [PubMed] [Google Scholar]