

Abstract
Executive functions of the brain are believed to require tonic dopamine inputs to the prefrontal cortex (PFC). It is unclear, however, how this background dopamine activity controls synaptic plasticity in the PFC, a possible underlying mechanism of executive functions. Using PFC slices, we show that pairing of dopamine with weak tetanic stimulation, a maneuver that otherwise induces NMDA receptor-independent long-term depression (LTD), induces long-term potentiation (LTP) when “primed” with dopamine. This “priming” occurs through the combined activation of D1 and D2 receptors and requires 12–40 min to develop. Moreover, concurrent synaptic activation of NMDA receptors during priming is necessary for this novel form of LTP. We suggest that a role of background dopamine signals in the PFC is to prevent high-frequency synaptic inputs from abnormally inducing LTD and to secure the induction of LTP.
Keywords: long-term potentiation, long-term depression, dopamine, prefrontal cortex, priming, NMDA receptor
Introduction
Midbrain dopamine neurons exhibit fast phasic and “background” tonic activities (Grace, 1991; Schultz, 2002). The phasic activity is thought to be involved in the encoding of reward-related signals (Reynolds et al., 2001; Schultz, 2002) and promotes long-term potentiation (LTP) in the striatum (Reynolds et al., 2001), hippocampus (Frey et al., 1991), and prefrontal cortex (PFC) (Gurden et al., 1999; Huang et al., 2004). The slow tonic activity supplies ambient concentrations of dopamine in target areas (Floresco et al., 2003), including the PFC (Bassareo and Di Chiara, 1997; Takahata and Moghaddam, 2000). This background presence of dopamine is thought to provide crucial control over higher cognitive functions mediated by the PFC (Akil et al., 1999; Schultz, 1998, 2002).
Lines of animal studies indeed show that manipulations of dopaminergic tone either by direct infusion of dopamine agonists/antagonists in the PFC (Sawaguchi and Goldman-Rakic, 1991; Zahrt et al., 1997; Baldwin et al., 2002; Wang et al., 2004) or by a lesion of ventral tegmental area (Simon et al., 1980) cause significant alterations in executive functions. It is generally thought that the background dopamine level regulates the sensitivity of PFC neurons to event-related inputs (Williams and Goldman-Rakic, 1995). To date, however, the specific cellular mechanisms underlying the effect of tonic background dopamine level on executive functions are still unknown.
An emerging view is that the executive functions depend on lasting neuronal traces formed and stored within the PFC (Dias and Aggleton, 2000; Fuster et al., 2000; Rossi et al., 2001; Simons and Spiers, 2003; Runyan et al., 2004) and that synaptic plasticity in the PFC serves as a mechanistic basis for these neuronal traces (Laroche et al., 2000). Indeed, lasting alterations in discharge rate of PFC neurons were detected during the acquisition of operant spatial discrimination task (Mulder et al., 2003). Furthermore, long-term changes of PFC synaptic transmission were found to accompany a form of behavioral adaptation (Herry and Garcia, 2002).
Here, we hypothesize that tonic background dopamine activity plays a key role in the regulation of PFC synaptic plasticity. Given that dopaminergic projection is removed, the acute PFC slices serve as an excellent model system, which allowed us to mimic background dopaminergic inputs by a “priming” bath-application of dopamine. Previously, we showed that paring tetanic stimulation to bath-application of dopamine (100 μm; 12.5 min) induces long-term depression (LTD) (Otani et al., 1998, 1999). In the present study, to compare with this protocol and to isolate the background stimulation phase, we performed a dopamine double-application protocol. In this protocol, dopamine was applied alone (100 μm; 12.5 min) 40 min before the delivery of the dopamine-tetanic stimulation paring. In the second series of experiments, to supply background dopamine in a more physiologically relevant manner, we continuously applied a lower concentration of dopamine (3 μm) before the pairing. In both cases, surprisingly, the priming of dopamine receptors converted the LTD to LTP.
Materials and Methods
Slice preparation.
All experiments were conducted in accordance with the Policies on the Use of Animals in Neuroscience as approved by the Society for Neuroscience. Male Sprague Dawley rats (23–30 d of age) were decapitated, and their brains were rapidly removed from their skull. Coronal slices containing the prelimbic area of medial frontal cortex (300 μm; 2.2–3.7 mm from the bregma) were sectioned by the use of a Campden vibratome (Campden Instruments, Leics, UK) in chilled (∼0°C) oxygenated (95% O2/5% CO2) artificial CSF (ACSF) composed of the following (in mm): 124 NaCl, 2 KCl, 26 NaHCO3, 1.15 KH2PO4, 1 MgCl2, 2 CaCl2, and 11 d-glucose. The slices were allowed to recover for at least 3 h at room temperature (∼20°C) in continuously oxygenated ACSF. The slices were transferred, one at a time, to a submersion-type recording chamber and perfused with warmed ACSF (28°C) at the rate of 1 ml/min.
By the use of sharp glass micropipettes (GC120F-10; Harvard Apparatus, Holliston, MA) filled with 3 m K-acetate (80–120 MΩ tip resistance), the soma of >170 layer V pyramidal neurons in the prelimbic area was penetrated. Negative currents were initially injected with an Axoclamp 2A amplifier (Molecular Devices, Union City, CA) or a BVC-700A amplifier (Dagan Corporation, Minneapolis, MN). After stabilization of the cells, all or most currents were removed. On average, the cells had a resting membrane potential of −71 ± 0.5 mV, and the potential during the experiments was maintained at −74 ± 0.4 mV. Cells that had a resting membrane potential more negative than −60 mV, a spike height of 60 mV or larger, and an initial input resistance of 30 MΩ or larger were considered healthy. The spike height and the input resistance were re-examined at the end of the experiments. Furthermore, input resistance and bridge balance were often tested during the course of the experiments, with spike height also retested when necessary. Experiments were terminated in cases where cells showed a sign of deterioration (i.e., a constant decline in any of the aforementioned parameters such that >10% deviations from initial values were observed). On average, initial and final spike heights of the accepted cells were 71 ± 0.5 and 70 ± 0.6 mV, respectively. Initial and final input resistances were 54 ± 1.2 and 55 ± 1.3 MΩ, respectively.
Before the experiments, spike discharge mode was routinely examined by applying a depolarizing current step (500 ms) from resting membrane potential. The intensity of the depolarizing step was set at the level where a 30 ms application of the step produced one action potential. Sixty-two percent of the cells were classified as regular spiking, 13% as bursting, and 25% as “adaptation cells” in which initial repetitive spikes ceased abruptly with a strong adaptation. There were no correlations between a discharge mode and the other measurements, including the degree of synaptic plasticity.
Stimulation, recording, and data analysis.
A bipolar Teflon-coated tungsten stimulating electrode (external diameter, 125 μm; A-M Systems, Carlsborg, WA) was placed on layer I-II of the prelimbic area (immediately interior to the pial surface). The EPSP of ∼10 mV in amplitude was evoked at 0.033 Hz by the application of monophasic constant current square pulses (100 μs duration; A360 stimulus isolator; World Precision Instruments, Sarasota, FL). After acquisition of the baseline responses for a period of at least 20 min, drug application was initiated. All drugs were included in perfusing medium. In a given set of experimental conditions (e.g., those involving the dopamine receptor antagonists), the experiments were always performed in an interleaved manner. To induce long-term plasticity, a train of tetanic stimuli (100 pulses applied at 50 Hz) was delivered four times at 0.1 Hz. All evoked responses were fed to an Axoclamp 2A or BVC-700A amplifier in current-clamp mode and digitized at 10 kHz through a Digidata 1322A interface (Molecular Devices) by the use of Elphy data acquisition program (developed by Dr. G. Sadoc, Institut Alfred Fessard, Centre National de la Recherche Scientifique, Gif-sur-Yvette, France). Individual responses and the changes in the EPSP slope were monitored on-line, whereas the detailed data analysis was performed off-line.
For the analysis, the initial rising slope of the EPSP (1 ms period from its onset, mV/ms), which contains only a monosynaptic component (Hirsch and Crepel, 1990), was calculated for each individual EPSP evoked by the 0.033 Hz test pulses. Changes of the EPSP slope after conditioning/drug application (35–45 min period after conditioning, denoted as “40 min after conditioning”) were expressed as a percentage increase or decrease from the preconditioning baseline level (the 10 min period just before tetanic stimulation or drug application). The percentage values obtained were compared between different experimental groups.
All experiments were performed in the presence of the GABAA antagonist bicuculline methiodide (1 μm). The presence of bicuculline did not disrupt the EPSP for at least 2 h of recording. Percentage changes of the EPSP slope 60 and 120 min after the beginning of bicuculline perfusion were 4.6 ± 7.3 and −2.1 ± 6.9%, respectively (n = 4).
The initial concentration of dopamine was 100 μm in most experiments (with 20 μm ascorbic acid), but this was subject to degradation. We assessed actual concentration of dopamine in our recording chamber at the time of conditioning (i.e., the end of the 12.5 min application), by the use of high-performance liquid chromatography (in collaboration with Dr. J.-P. Tassin, College de France, Paris, France). Our measurement showed that dopamine concentration in the bath was 79 μm. We verified also that the 20 μm level of ascorbic acid does not affect the synaptic responses. Thus, ascorbic acid alone was applied twice for a 12.5 min duration, separated by a 40 min interval, as for our principal dopamine double-application protocol (see Fig. 1d). There were no significant changes in the EPSP slope during and after the ascorbic acid application. Percentage changes of the EPSP slope 40 min after the first and second applications of ascorbic acid were −2.9 ± 4.7% (n = 6) and −1.4 ± 5.2%, respectively (n = 5; recording of one cell was lost just after the second application).
Figure 1.
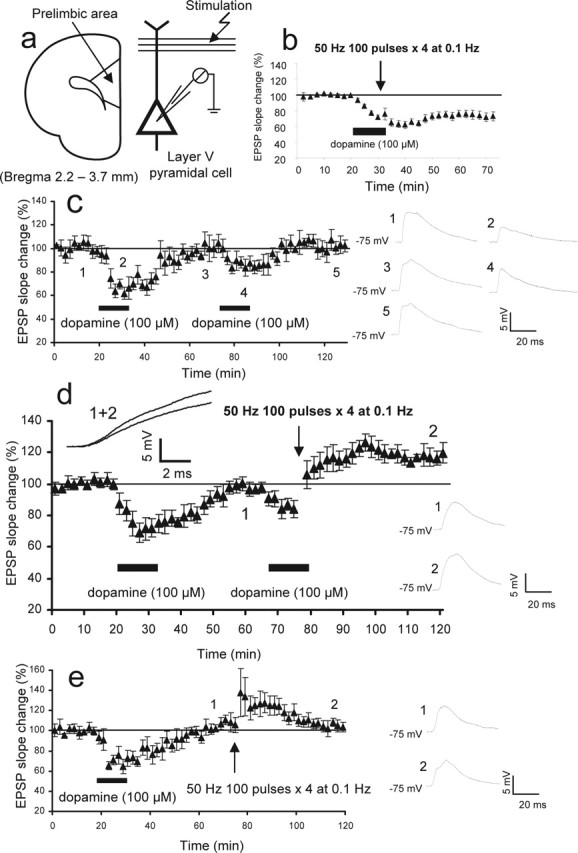
Priming of dopamine receptors facilitates LTP induction in rat PFC neurons. a, The monosynaptic glutamatergic EPSP was evoked by 0.033 Hz test stimuli applied to layer I-II fibers of the PFC slices and recorded at somatic level from layer V pyramidal neurons by the use of sharp micro pipettes. Bicuculline (1 μm) was routinely added in the bathing medium to reduce GABAA inhibition. b, Data taken from our previous experiments (Otani et al., 1999) showing that the delivery of 50 Hz tetanic stimuli (100 pulses, 4 times at 0.1 Hz) in the presence of dopamine (100 μm; 12.5 min) induces LTD of the EPSP (n = 14). This LTD induction is NMDA receptor independent (Otani et al., 1998). Tetanic stimulation alone or dopamine application alone does not induce lasting plasticity (Otani et al., 1998, 1999). c, When dopamine application (100 μm; 12.5 min) is repeated, the transient reduction of the EPSP shows marked habituation (−36 ± 7.6% by the first application vs −16 ± 4.3% by the second application; n = 6; p < 0.05, paired t test). This fact indicates that in slices, hypodopaminergic condition induced homeostatic compensations of the dopamine system. d, Delivery of 50 Hz tetani in association with the second application of dopamine induces LTP (20 ± 3.0% increase 40 min after dopamine/tetani; n = 10; p < 0.00001 compared with the tetani-alone group). Averaged responses taken from the indicated time points (1 and 2) are shown in the insets. The same responses are superimposed with a longer time scale to show the clear difference in the rising slope of the EPSP. e, The second application of dopamine is necessary for LTP. Delivery of 50 Hz stimuli alone, 40 min after the first application of dopamine, induced only decaying potentiation (2.4 ± 4.2% 40 min after tetani; n = 8; p < 0.005). Averaged EPSPs taken from the indicated time points are shown in the insets.
For statistical analysis, a p < 0.05 confidence level was considered significant. Unless otherwise specified, the two-tailed Student’s t test was used. All values are expressed as mean ± SEM. As described previously, morphological identification of pyramidal neurons was routinely performed using biocytin [1.5% loaded in recording electrodes (Otani et al., 1999)]. All identified neurons were classified as layer V pyramidal neurons, based on the shape and location of their cell body and on the long apical dendrite bearing a tuft, which occurs in the superficial layer.
The following drugs were used: bicuculline methiodide (Sigma, St. Quentin Fallavier, France), biocytine (Sigma), dopamine (Sigma), BAPTA (Sigma), R(+)-7-chloro-8-hydroxy-3-methyl-1-phenyl-2,3,4,5-tetrahydro-1H-3-benzazepine hydrochloride (SCH23390) (Tocris Cookson, Bristol, UK), sulpiride (Tocris Cookson), (+)-1-Phenyl-2,3,4,5-tetrahydro-(1H)-3-benzazepine-7,8-diol hydrobromide (SKF38393) (Tocris Cookson), quinpirole (Tocris Cookson), dl-2-amino-5-phosphonopentanoic acid (AP-5) (Tocris Cookson), 6-cyano-7-nitroquinoxaline-2,3-dion (CNQX) (Tocris Cookson), and (RS)-α-methyl-4-carboxyphenylglycine (MCPG) (Tocris Cookson).
Results
LTP induction is facilitated by dopamine priming
The glutamatergic monosynaptic EPSP was evoked by layer I-II afferent stimulation and recorded from the soma of layer V pyramidal neurons in PFC slices prepared from juvenile rats (23–30 d of age) (Fig. 1a). Perfusion of the PFC slices with a relatively high concentration of dopamine (100 μm) for an extended period of time (12.5 min) resulted in a transient decrease of synaptic efficacy that lasted no >30 min (Fig. 1c) (first dopamine application, n = 6) (Otani et al., 1998, 1999). The dopamine application alone did not induce lasting synaptic changes unlike in the hippocampus (Huang and Kandel, 1995; Sajikumar and Frey, 2004). When dopamine was applied for the second time, however, the depression of the EPSP was significantly smaller (Fig. 1c) (−36 ± 7.6% change by the first application vs −16 ± 4.3% by the second application; p < 0.05, paired t test). Although the molecular mechanism underlying this dopamine-induced acute reduction of the EPSP is unknown, the clear habituation indicates that the low dopamine condition in slices had induced a homeostatic compensation in the dopamine system (Grace, 1991). Indeed, in PFC slices, a significant amount of endogenous release of dopamine may not be present unless residual axons receive high-frequency input (Young and Yang, 2005). It is then thought that the second application of dopamine better models physiological dopamine effects than the first application.
As shown in Figure 1b, when the first application of dopamine was paired with a weak tetanic stimulation (50 Hz; 100 pulses; four times at 0.1 Hz), an NMDA receptor-independent LTD was induced (−25 ± 6.0% at 40 min; n = 14) (Otani et al., 1998, 1999). This weak tetanus itself induced no lasting plasticity (0.1 ± 1.2% at 40 min; n = 12; data not shown) (Otani et al., 1998, 1999). In the next series of experiments, we applied dopamine first without concurrent application of the 50 Hz stimuli (Fig. 1d). When the dopamine-induced acute depression had fully reversed (40 min later), dopamine was reapplied in an identical manner. This second application of dopamine was paired with the same weak tetanic stimulation described above. Remarkably, this procedure induced LTP (Fig. 1d). The EPSP slope was 20 ± 3.0% above baseline 40 min after dopamine and tetani (n = 10; p < 0.00001 compared with the tetanus-alone control group, which showed 0.1 ± 1.2% change at 40 min; two-tailed t test). The second application of dopamine was necessary for the induction of LTP. When omitted, the 50 Hz tetani induced only decaying potentiation (Fig. 1e) (2.4 ± 4.2% at 40 min; n = 8; p < 0.005; compared with the group depicted in Fig. 1d).
Two sets of control experiments were performed. First, to exclude the possibility that prolonged dialysis of the postsynaptic cells facilitated the induction of LTP, we performed the dialysis of postsynaptic neurons for an extended period of time (>1 h recording) before the application of dopamine (Fig. 2a; see legend for more details of the method). Under this condition, the 50 Hz tetani still induced a small but significant LTD (−8.2 ± 4.9%; n = 6; p < 0.0002 compared with the group depicted in Fig. 1d, and p < 0.05 compared with the tetanus-alone control group). Second, one may argue that a smaller synaptic depression (habituation) induced by the second application of dopamine may lead to the induction of LTP. To exclude this possibility, we applied 10–30 μm (rather than 100 μm) of dopamine, again after a prolonged postsynaptic dialysis (Fig. 2b). These concentrations of dopamine induced acute depression (−16 ± 5.2%) similar in degree to that induced by the second application of 100 μm dopamine (−16 ± 4.3%). However, delivery of 50 Hz tetani in the presence of the 10–30 μm dopamine induced LTD, but not LTP (−15 ± 6.8%; n = 5; p < 0.0005 compared with the group depicted in Fig. 1d, and p < 0.01 compared with the tetanus-alone control group). From these experiments, we conclude that for the induction of LTP, both preapplication of dopamine and the pairing of a second application of dopamine with weak tetanic stimulation are necessary. We will refer to the prestimulation of dopamine receptors as priming and the pairing of dopamine application with the 50 Hz tetani as “induction.”
Figure 2.
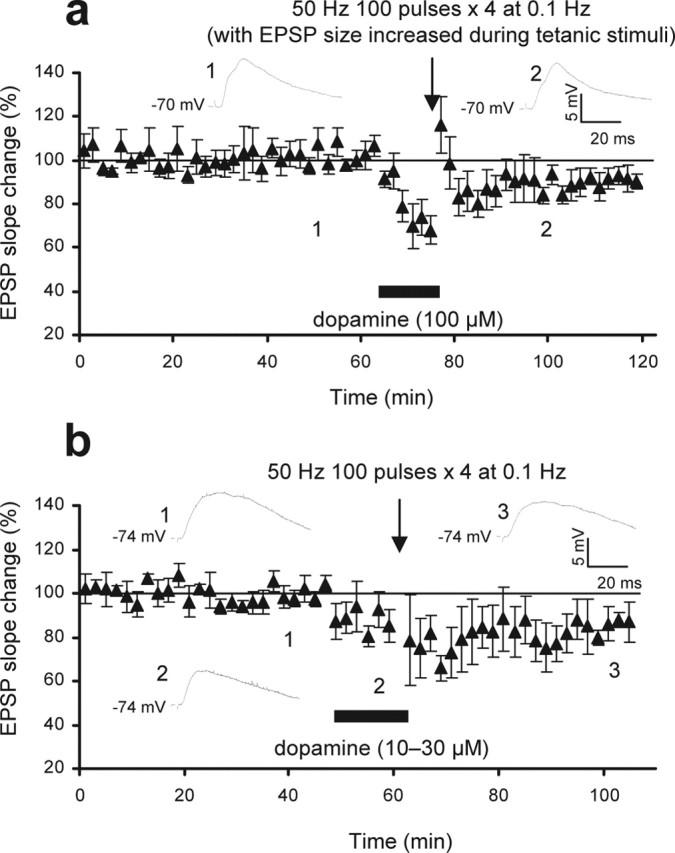
Experiments to establish the dopamine-facilitated LTP. a, In this set of experiments (n = 6), the dialysis of postsynaptic cells during the long preconditioning response recording (>1 h) (Fig. 1d) was controlled. In addition, we equally controlled the smaller acute synaptic depression (habituation) seen with the second application of dopamine (Fig. 1, compare b and d). Thus, after long-term recording of baseline responses at 0.033 Hz, dopamine (100 μm; 12.5 min) was identically applied. Just before the delivery of tetani, the EPSP slope was increased to baseline level by increasing the stimulus intensity. Under this condition, 50 Hz stimuli did not induce LTP (−8.2 ± 4.9%; p < 0.0002). Averaged EPSPs taken from indicated time points are shown in the insets. b, In this set of experiments, we controlled the smaller acute synaptic depression (habituation) seen with the second dopamine (Fig. 1d) by the use of lower concentrations of dopamine (10–30 μm; 12.5 min; n = 5). Therefore, again after a long baseline recording (to control postsynaptic dialysis), the application of these concentrations of dopamine induced a reduction in the EPSP slope (−16 ± 5.2%) similar to that induced by the second application of 100 μm dopamine (−16 ± 5.7%) (Fig. 1d). Delivery of 50 Hz stimuli in the presence of 10–30 μm dopamine, however, induced LTD (−15 ± 6.8%; n = 5; p < 0.0001). Averaged EPSPs taken from indicated time points are shown in the insets.
Involvement of dopamine D1 and D2 receptors in LTP
With a novel LTP protocol established, we then determined the subtype of dopamine receptors involved in this LTP. All pharmacological experiments hereafter were performed in interleaved manner. First, the D1 antagonist SCH23390 (1 μm; n = 6) (Fig. 3a) or the D2 antagonist sulpiride (10 μm; n = 6) (Fig. 3b) was bath-applied during the priming phase. Neither antagonist changed baseline EPSP nor the degree of acute depression induced by dopamine (−32 ± 9.7% with SCH23390; −27 ± 11% with sulpiride; p > 0.3 compared with the acute depression induced by the first dopamine in Fig. 1d). Forty minutes after drug application, dopamine was applied for the second time and paired to 50 Hz tetani. In both antagonist groups, LTP was blocked (0.5 ± 5.8%, p < 0.01 and −8.3 ± 5.2%, p < 0.0002, respectively, compared with Fig. 1d) (see Fig. 3e for summary graph), suggesting that both D1 and D2 receptors must be prestimulated (primed) to facilitate a later induction of LTP. Second, SCH23390 (1 μm; n = 7) (Fig. 3c) or sulpiride (10 μm; n = 7) (Fig. 3d) was applied during the induction phase. In both groups, LTP was blocked (−8.6 ± 4.8%, p < 0.0001 and −13 ± 7.1%, p < 0.002, compared with Fig. 1d) (see Fig. 3f for summary graph). Thus, induction also requires the concurrent activation of D1 and D2 receptors.
Figure 3.
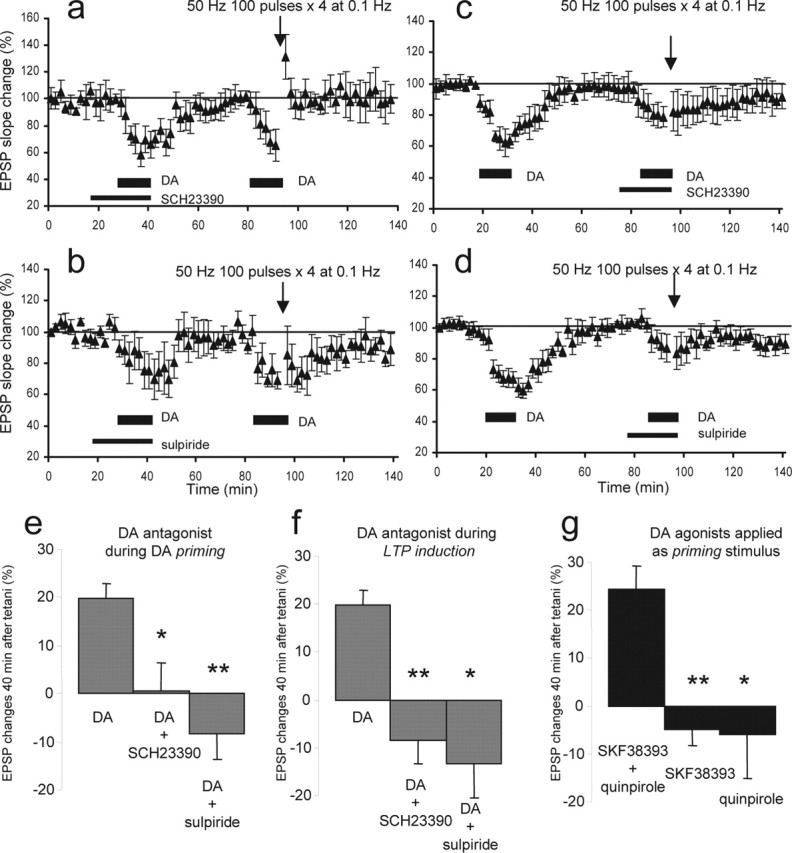
Concurrent activation of dopamine D1 and D2 receptors is necessary for both priming and induction of the LTP. a, The presence of D1 receptor antagonist SCH23390 (1 μm) from 10 min before the first application of dopamine (100 μm; 12.5 min) until the end of the application blocked LTP by the second dopamine–50 Hz tetani paring (0.5 ± 5.8% change 40 min after the second dopamine/tetani; n = 6; p < 0.01). b, The presence of D2 antagonist sulpiride (10 μm) during the first application of dopamine also blocked the LTP by the second dopamine–tetani pairing (−8.3 ± 5.2%; n = 6; p < 0.0002). c, The presence of SCH23390 (1 μm) from 10 min before the second application of dopamine until the end of the application and tetani blocked the induction of LTP (−8.6 ± 4.8%; n = 7; p < 0.0001). d, The presence of sulpiride (10 μm) during the second application of dopamine and tetani also blocked the LTP (−13 ± 7.1%; n = 7; p < 0.002). e, Summary graph showing the EPSP slope changes 40 min after the second dopamine (DA)–tetani paring in three different priming conditions. Left (DA), The condition in which dopamine alone was applied as priming stimulus and 20% LTP was induced (n = 10) (see Fig. 1d). Middle (DA+SCH23390), The group depicted in Figure 3a (n = 6) in which D1 receptors were blocked by SCH23390 during the first application of dopamine and LTP was absent (p < 0.01). Right (DA+sulpiride), The group depicted in b (n = 6) in which D2 receptors were blocked by sulpiride during the first application of dopamine and LTP was absent (p < 0.0002). f, Summary graph showing the EPSP slope changes 40 min after the second dopamine–tetani paring in three different induction conditions. Left (DA), The condition in which dopamine alone was applied as priming stimulus and 20% LTP was induced (n = 10) (Fig. 1d). Middle (DA+SCH23390), The group depicted in c (n = 7) in which SCH23390 (1 μm) was present during the second application of dopamine and tetani. LTP was absent. Right (DA+sulpiride), The group depicted in d (n = 7) in which sulpiride (10 μm) was present during the second application of dopamine and tetani. LTP was also absent. g, Summary graph showing the EPSP slope changes 40 min after the second dopamine–tetani paring in three different agonist conditions. Left (SKF38393+quinpirole), D1 agonist SKF38393 (25 μm) and D2 agonist quinpirole (25 μm) were applied together for 12.5 min as priming stimulus (n = 6). Forty minutes after the agonist application, 50 Hz tetani were delivered in the presence of dopamine (100 μm). There was LTP (24 ± 5.0% 40 min after the dopamine–tetani paring). Middle (SKF38393), D1 agonist SKF38393 (25–50 μm) was applied for 12.5 min as priming stimulus (n = 8). Forty minutes after the SKF38393 application, 50 Hz tetani were delivered in the presence of dopamine (100 μm). There was no LTP (−4.8 ± 3.4% at 40 min; n = 8; p < 0.001 compared with the SKF+quinpirole group). Right (quinpirole), D2 agonist quinpirole (25 μm) was applied as priming stimulus (n = 7), and 40 min after quinpirole, 50 Hz tetani were delivered in the presence of dopamine (100 μm). There was no LTP (−5.9 ± 9.3% at 40 min; n = 7; p < 0.02). Error bars represent SEM.
Based on the above experiments, one may argue that LTP was blocked by the SCH23390 or sulpiride applied during the priming phase because of insufficient washout of these antagonists at the time of induction. To eliminate this possibility, we used dopamine agonists. If both D1 and D2 receptors are necessary for priming, a combined application of D1 and D2 agonists as priming stimulus should facilitate LTP, whereas a lone application of either agonist should fail to facilitate LTP. This LTP after the combined application could not be explained by the presence of residual agonists at the time of induction, because the additional receptor stimulation by residual agonists in the presence of 100 μm dopamine is unlikely to convert LTD to LTP. Thus, as the priming stimulus, D1 agonist SKF38393 (25–50 μm; n = 8), D2 agonist quinpirole (25 μm; n = 7), or SKF38393 plus quinpirole (25 and 25 μm; n = 6) were bath-applied for 12.5 min. Forty minutes after this application, a dopamine application was paired with 50 Hz stimuli. LTP could only be induced after the combined SKF38393 plus quinpirole application (Fig. 3g) (24 ± 5.0% at 40 min; p < 0.001 compared with tetanus-alone control group). We conclude that priming involves the activation of both D1 and D2 receptors.
Involvement of NMDA receptors in priming and induction
Blockade of NMDA receptors within the PFC retards operant learning (Baldwin et al., 2002). Moreover, abuse with NMDA receptor antagonist phencyclidine disrupts PFC-dependent cognitive function (Egerton et al., 2005). Assuming that NMDA receptor-dependent synaptic plasticity is important for PFC-dependent cognitive processes, we tested the role of the NMDA receptors in specific phases (i.e., priming or induction) of the PFC LTP. First, we applied the NMDA receptor antagonist dl-AP-5 (100 μm) during priming (Fig. 4a) (n = 7). AP-5 caused a slight reduction in baseline EPSP, but this effect was transient, as verified in separate neurons (−1.0 ± 6.4%, 40 min after washout; n = 7). The presence of AP-5 during priming nullified the priming effect. Therefore, pairing of the second dopamine perfusion with 50 Hz tetani resulted in LTD instead of LTP (−18 ± 6.3%; p < 0.0001 compared with the group depicted in Fig. 1d). Next, we applied AP-5 during the induction phase (Fig. 4c,d) (n = 9). In this case, interestingly, five cells showed clear LTP (31 ± 4.1%) (Fig. 4c), and the other four showed degrees of LTD (−31 ± 17%) (Fig. 4d). There was no difference in morphological characteristics and membrane properties between these two groups of neurons. Importantly, this AP-5 effect could generate a net “no-LTP” result in field recording. This may have occurred in two previous studies in which AP-5 blocked induction of PFC LTP (Gurden et al., 1999; Huang et al., 2004). Together, these data suggest that NMDA receptor-mediated synaptic transmission is necessary for priming and contributes to induction in a subpopulation of PFC neurons.
Figure 4.

The formation of priming is NMDA receptor dependent, but induction of LTP can be NMDA receptor independent. a, The presence of dl-AP-5 (100 μm) from 10 min before the first application of dopamine until the end of application completely blocked the induction of LTP by the second dopamine–tetani paring. Rather, LTD was induced by the paring (−18 ± 6.3%, 40 min after the second dopamine/tetani; n = 7; p < 0.0001). b, Left, Example traces recorded from a representative cell showing that dopamine (100 μm; 12.5 min) transiently reduced isolated NMDA receptor-mediated EPSP but did not potentiate it. The NMDA receptor-mediated EPSP (evoked to 0.033 Hz test pulses) was isolated by the addition of CNQX (5 μm) and bicuculline (1 μm) in the bath and by slightly increasing the stimulus intensity. Right, Summary graph (n = 6) showing that dopamine application acutely reduces the NMDA receptor-mediated EPSP (−48 ± 7.9% reduction) and that the reduction returns to baseline level in 40 min (−2.1 ± 6.5%). c, In five of nine cells tested, LTP was clearly induced by the second dopamine–tetani paring in the presence of dl-AP-5 (31 ± 4.1%, 40 min after the second dopamine/tetani; n = 5). d, In the remaining four cells, however, the second dopamine–tetani paring in the presence of dl-AP-5 resulted in LTD (−31 ± 17%; n = 4). DA, Dopamine. Error bars represent SEM.
Many forms of synaptic plasticity in various brain regions depend on metabotropic glutamate receptors (mGluRs). We therefore tested whether the present form of LTP also requires the activation of mGluRs. When the common mGluR antagonist (RS)-MCPG (300 μm) was present during the induction phase, LTP was blocked (−26 ± 7.7, p < 0.0001 compared with the group depicted in Fig. 1d; n = 6) (Fig. 5). Thus, the LTP induction is mGluR dependent. The presence of MCPG during the priming phase also blocked LTP (2.3 ± 7.0%; n = 7; p < 0.05 compared with Fig. 1d; data not shown). However, the mGluR involvement in priming remains inconclusive at this stage, because it is possible that MCPG is not fully washed out by the time of induction. When 50 Hz stimulation was paired with dopamine perfusion 40 min after sole application of MCPG (300 μm; 22.5 min), the LTD induction that normally occurs after pairing (Otani et al., 1998) was found to be blocked (3.3 ± 11%; n = 5; data not shown). This suggests that there may be residual MCPG at the time of induction, which may block induction of plasticity (Otani et al., 1999).
Figure 5.

Induction of LTP requires the activation of mGluRs. The presence of MCPG (300 μm) from 10 min before the second application of dopamine until the end of the application and tetani blocked the induction of LTP and rather resulted in LTD (−27 ± 7.7%, 40 min after second dopamine and tetani; p < 0.0001; n = 6). The presence of MCPG during priming also blocked LTP (2.3 ± 7.0%; n = 7; p < 0.05; data not shown). However, the involvement of mGluRs during priming cannot be firmly concluded, because MCPG applied during priming may not be completely washed out by the time of induction (see Results, Involvement of NMDA receptors in priming and induction).
Postsynaptic mechanisms of the dopamine-facilitated LTP
A number of experiments were performed to address the cellular mechanisms underlying the dopamine-primed, NMDA receptor-dependent LTP. First, to determine the induction locus, hyperpolarizing currents (to −110 mV) were applied through recording electrodes during the application of 50 Hz tetani to counteract tetanus-induced postsynaptic depolarization (n = 6) (Fig. 6a). Under this condition, the LTP was blocked (−5.5 ± 7.1%; p < 0.002 compared with the group depicted in Fig. 1d). Second, the Ca2+ chelator BAPTA (50–100 mm in recording electrodes) was injected into postsynaptic sites before the experiments (n = 6) (Fig. 6b). The application of BAPTA also blocked LTP (−4.0 ± 8.5%; p < 0.01 compared with Fig. 1d). Together, these results suggest that the induction of this novel form of LTP requires both postsynaptic depolarization during tetanus and postsynaptic increases in Ca2+ level. Next, we tested whether dopamine primes the PFC LTP by potentiating the NMDA receptor-mediated EPSP (Zheng et al., 1999; Seamans et al., 2001). To test this, NMDA receptor-mediated EPSP was pharmacologically isolated (Fig. 4b, left traces), and dopamine was identically applied (100 μm; 12.5 min). The dopamine caused a rapid and transient reduction, but not potentiation, of the NMDA receptor-mediated EPSP (n = 6) (Fig. 4b). We conclude that the priming effect of dopamine is not caused by a potentiation of NMDA receptors.
Figure 6.
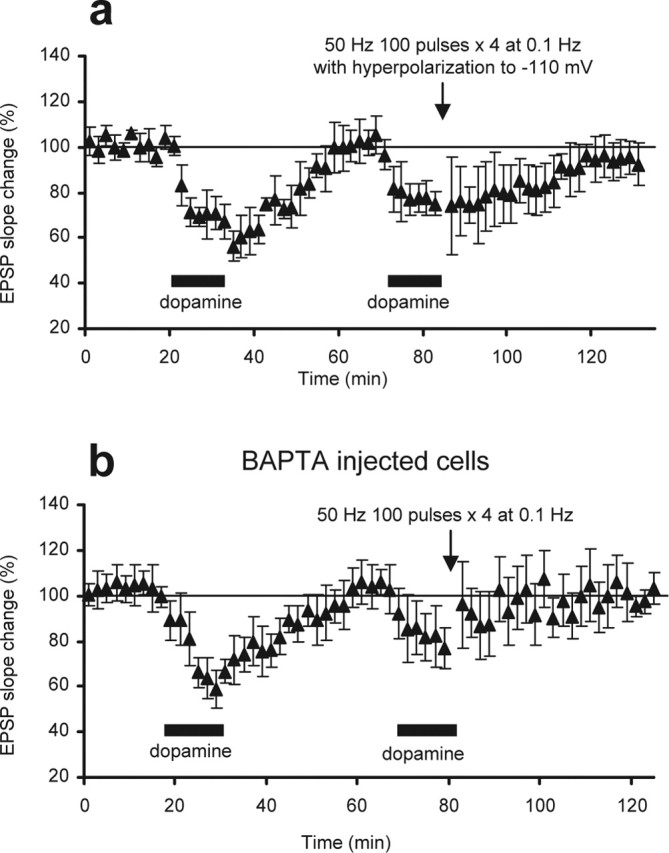
Postsynaptic locus of dopamine-facilitated LTP. a, LTP induction requires postsynaptic depolarization. During the delivery of 50 Hz tetani in the presence of the second dopamine, postsynaptic membrane was hyperpolarized to −110 mV to counteract tetanus-induced depolarization. Under this condition, LTP induction was blocked (−5.5 ± 7.1%, 40 min after the second dopamine–tetani paring; n = 6; p < 0.002). b, LTP induction requires postsynaptic increases of [Ca2+]. The Ca2+ chelator BAPTA (50–100 mm included in recording pipettes) was injected into postsynaptic sites before the experiments. Diffusion of BAPTA, which was verified by a block of Ca2+-dependent spike-train adaptation, did not change baseline EPSP. Under this condition, dopamine-facilitated LTP was blocked (−4.0 ± 8.5% at 40 min; n = 6; p < 0.01).
Time-dependent facilitation of LTP by continuous application of low concentration of dopamine
Thus far, we used the spaced (40 min interval) double-application of a relatively high concentration of dopamine to provide background dopamine signal and induce LTP. Using brain microdialysis, a method that detects only dopamine increases resulting from tonic release (Floresco et al., 2003), Bassareo and Di Chiara (1997) have indeed shown large and repeated augmentations of dopamine concentration in rat PFC during goal-directed behavior (∼100% increases with 120 min interval). Furthermore, the double-application method was ideal to isolate dopamine signal specifically involved in LTD-LTP conversion (priming). However, an alternative and perhaps more physiologically relevant way of providing background dopamine signal is through a continuous perfusion of a low concentration of dopamine. Therefore, in the final set of experiments, we continuously applied dopamine at 3 μm in the bath. This concentration was chosen because this was approximately the lowest concentration of exogenously applied dopamine to induce visible changes in the slope of the evoked EPSP (i.e., a small decrease or increase; the average change was 5.2 ± 5.0% at 12.5 min after the beginning of perfusion, n = 8, and −3.7 ± 8.6% at the end of the experiments, n = 7) (Fig. 7a, open circles). Interestingly, the continuous perfusion of 3 μm dopamine before 50 Hz tetani (for 40 min) was sufficient to promote LTP (32 ± 14%, 40 min after tetani; n = 7; p < 0.025 compared with the above no-tetanus group) (Fig. 7a, black triangles). In contrast, a 12.5 min perfusion before the tetani was insufficient for LTP (2.1 ± 7.4%; n = 10; p > 0.4 compared with the above no-tetanus group, and p < 0.05 compared with the 40 min perfusion group) (Fig. 7a, gray triangles). These results suggest that the priming effect requires 12.5–40 min to develop. Our preliminary experiments (in preparation) show that this LTP also requires the activation of both D1 and D2 receptors and NMDA receptors. Interestingly (Fig. 7b), when 50 Hz stimulation was paired with 100 μm dopamine superimposed on the background concentration of 3 μm dopamine, the tetanus equally induced LTP but not LTD (24 ± 14%; n = 7; p < 0.005 compared with the LTD group depicted in Fig. 1b). Thus, LTD (Fig. 1b) was converted to LTP (Fig. 7b) also by the continuous application of a low concentration of dopamine.
Figure 7.
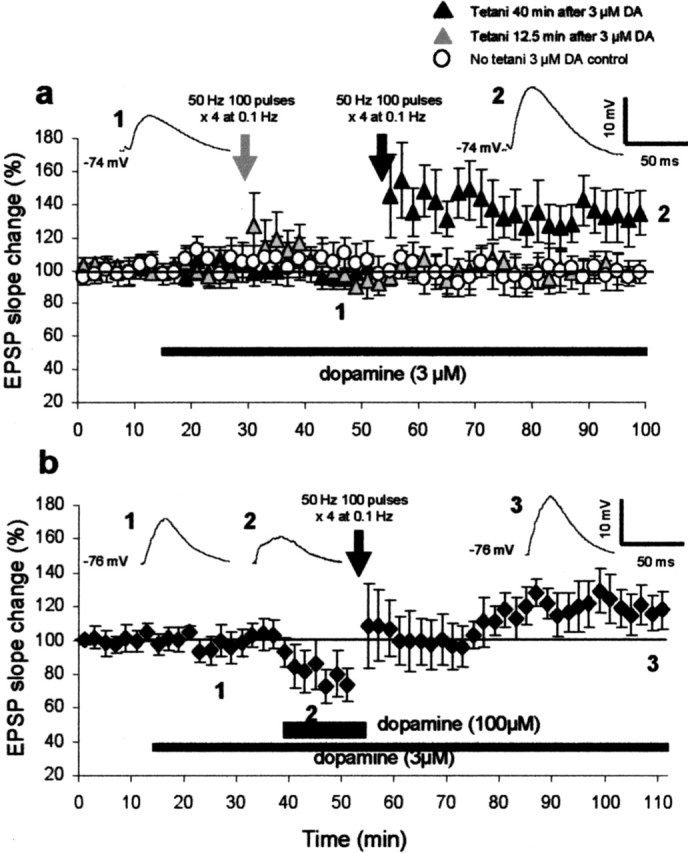
Continuous presence of low concentration of dopamine (3 μm) for 40 min before tetani facilitates the induction of LTP and prevents the induction of LTD. a, In the first group (gray triangles), 3 μm dopamine was perfused in the bath for 12.5 min before the delivery of 50 Hz tetani. No LTP was induced (2.1 ± 7.4%, 40 min after tetani; n = 10). In the second group (black triangles), 3 μm dopamine was perfused for 40 min before the tetani delivery. In this case, the 50 Hz tetani induced LTP (32 ± 14%; p < 0.05; n = 7). Averaged EPSPs taken from the indicated time points are shown in the insets for this LTP group. In the third group (open circles), 3 μm dopamine was perfused without delivery of tetani for the entire duration of the experiments (n = 8). b, The 3 μm dopamine was identically applied as above, and 12.5 min before the delivery of 50 Hz tetani, dopamine concentration was increased to 100 μm until the end of the tetani. Under this condition, the tetani–100 μm dopamine paring induced LTP (24 ± 14%, 40 min after tetani; n = 7; p < 0.005) but not LTD. Averaged EPSPs taken from the indicated time points are shown in the insets.
Discussion
Using PFC slices prepared from juvenile rats, we discovered that prestimulation of D1 and D2 dopamine receptors (priming) converts NMDA receptor-independent LTD (Otani et al., 1998) to LTP. This conversion depends on the activation of NMDA receptors during the priming process. NMDA receptor involvement in the induction phase of this LTP was not as clear; however, it was clearly shown that induction requires mGluR activation. The dopamine priming was achieved by two different protocols: (1) phased exposure (12.5 min) to a relatively high concentration of dopamine (100 μm); and (2) the continuous application of a low concentration of dopamine (3 μm). With the latter method, we showed that priming requires 12.5–40 min to develop.
It is widely believed that levels of tonic background dopamine concentration regulate cognitive functions (Grace, 1991; Schultz, 1998, 2002; Akil et al., 1999; Goto and Grace, 2005). A recent theoretical study suggested that novelty-related increases in tonic dopamine inputs in the hippocampus facilitate LTP and memory encoding (Lisman and Grace, 2005). However, previous experimental studies in the PFC (Gurden et al., 1999; Huang et al., 2004), striatum (Reynolds et al., 2001), and hippocampus (Frey et al., 1991) limited their focus to possible effects of fast phasic release of dopamine on LTP. Although rapid release of dopamine during brief high-frequency input does facilitate synaptic plasticity in the PFC (Young and Yang, 2005; our observation), no studies have as yet explored the role of the background presence of dopamine in synaptic plasticity. Using PFC slices, which are mostly deprived of tonic dopamine inputs, we thus revealed that the background dopamine signal permits the induction of LTP in the PFC. This may explain why LTP can be easily induced by tetanus alone in the intact PFC (Gurden et al., 1999), where basal levels of dopamine are tonically maintained (Takahata and Moghaddam, 2000). We discovered furthermore that the priming by dopamine requires concurrent low-frequency stimulation of NMDA receptors (Fig. 4a). Previously, NMDA receptors in the PFC were implicated in the generation of delay period activity (Brunel and Wang, 2001) as well as induction of LTP (Gurden et al., 1999; Huang et al., 2004). Our result thus adds an important and novel role for the NMDA receptors in PFC plasticity; that is, a preparatory regulation for dopamine-dependent induction of LTP.
In our in vitro procedure, at least 4 h elapsed between slice preparation and the first bath-application of dopamine. Because dopamine receptors are tonically stimulated in the intact brain, this time course suggests that the priming triggered in vivo fades during the 4 h of the in vitro incubation (Fig. 1, compare b and d). Such an extremely low dopamine condition may not occur in the intact PFC, but reduced levels of tonic dopamine inputs may indeed be present in the PFC of schizophrenics (Akil et al., 1999). We showed equally that the lack of baseline activation of NMDA receptors converts LTP to LTD (Fig. 4a). Again, in schizophrenic PFC, NMDA receptor dysfunction may exist (Sokolov, 1998). In this regard, we add that chronic administration of NMDA receptor antagonists disrupts PFC-dependent cognitive function (Egerton et al., 2005).
Together, we suggest that a pathological conversion of LTP to LTD may occur in the PFC when the level of basal stimulation of dopamine and NMDA receptors is low. Such an abnormal conversion would disrupt long-term neuronal representations within the PFC and impair the context-dependent selection of action repertories, a cognitive component necessary for the realization of goal-directed behavior (Fuster et al., 2000; Miller and Cohen, 2001; Otani, 2002). The slow and durable increases of extracellular dopamine concentration (Bassareo and Di Chiara, 1997; Mingote et al., 2004), together with the baseline NMDA receptor activation, may prevent abnormal LTD induction and secure event-related high-frequency inputs to induce LTP.
Footnotes
This work was supported by the French Minister of Research, the Centre National de la Recherche Scientifique, and the University of Paris VI. We are grateful to Drs. Bai Lu, Cliff Abraham, Yukiori Goto, Bogdan Kolomiets, Therese Cronin, Tim Bliss, John Lisman, David Lewis, Julie Frey, and Matthijs Feenstra for their useful comments.
References
- Akil M, Pierri JN, Whitehead RE, Edgar CL, Mohila C, Sampson AR, Lewis DA (1999). Lamina-specific alterations in the dopamine innervation of the prefrontal cortex in schizophrenic subjects. Am J Psychiat 156:1580–1589. [DOI] [PubMed] [Google Scholar]
- Baldwin AE, Sadeghian K, Kelley AE (2002). Appetitive instrumental learning requires coincident activation of NMDA and dopamine D1 receptors within the medial prefrontal cortex. J Neurosci 22:1063–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassareo V, Di Chiara G (1997). Differential influence of associative and nonassociative learning mechanisms on the responsiveness of prefrontal and accumbal dopamine transmission to food stimuli in rats fed ad libitum. J Neurosci 17:851–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunel N, Wang X-J (2001). Effects of neuromodulation in a cortical network model of object working memory dominated by recurrent inhibition. J Comput Neurosci 11:63–85. [DOI] [PubMed] [Google Scholar]
- Dias R, Aggleton JP (2000). Effects of selective excitotoxic prefrontal lesions on acquisition of nonmatching- and matching-to-place in the T-maze in the rat: differential involvement of the prelimbic and anterior cingulate cortices in providing behavioural flexibility. Eur J Neurosci 12:4457–4466. [DOI] [PubMed] [Google Scholar]
- Egerton A, Reid L, McKerchar CE, Morris BJ, Pratt JA (2005). Impairment in perceptual attentional set-shifting following PCP administration: a rodent model of set-shifting deficits in schizophrenia. Psychopharmacol 179:77–84. [DOI] [PubMed] [Google Scholar]
- Floresco SB, West AR, Ash B, Moore H, Grace AA (2003). Afferent modulation of dopamine neuron firing differentially regulates tonic and phasic dopamine transmission. Nat Neurosci 6:968–973. [DOI] [PubMed] [Google Scholar]
- Frey U, Matthies H, Reymann KG (1991). The effect of dopaminergic D1 receptor blockade during tetanization on the expression of long-term potentiation in the rat CA1 region in vitro. Neurosci Lett 129:111–114. [DOI] [PubMed] [Google Scholar]
- Fuster JM, Bodner M, Kroger JK (2000). Cross-modal and cross-temporal association in neurons of frontal cortex. Nature 405:347–351. [DOI] [PubMed] [Google Scholar]
- Goto Y, Grace AA (2005). Dopaminergic modulation of limbic and cortical drive of nucleus accumbens in goal-directed behaviour. Nat Neurosci 8:805–812. [DOI] [PubMed] [Google Scholar]
- Grace A (1991). Phasic versus tonic dopamine release and the modulation of dopamine system responsivity: a hypothesis for the eiology of schizophrenia. Neuroscience 41:1–24. [DOI] [PubMed] [Google Scholar]
- Gurden H, Tassin JP, Jay TM (1999). Integrity of the mesocortical dopaminergic system is necessary for complete expression of in vivo hippocampal–prefrontal cortex long-term potentiation. Neuroscience 94:1019–1027. [DOI] [PubMed] [Google Scholar]
- Herry C, Garcia R (2002). Prefrontal cortex long-term potentiation, but not long-term depression, is associated with the maintenance of extinction of learned fear in mice. J Neurosci 22:577–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirsch JC, Crepel F (1990). Use-dependent changes in synaptic efficacy in rat prefrontal neurons in vitro. J Physiol (Lond) 427:31–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang YY, Kandel ER (1995). D1/D5 receptor agonists induce a protein synthesis-dependent late potentiation in the CA1 region of the hippocampus. Proc Natl Acad Sci USA 92:2446–2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang YY, Simpson E, Kellendonk C, Kandel ER (2004). Genetic evidence for the bidirectional modulation of synaptic plasticity in the prefrontal cortex by D1 receptors. Proc Natl Acad Sci USA 101:3236–3241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laroche S, Davis S, Jay TM (2000). Plasticity at hippocampal to prefrontal cortex synapses: dual roles in working memory and consolidation. Hippocampus 10:438–446. [DOI] [PubMed] [Google Scholar]
- Lisman JE, Grace AA (2005). The hippocampal-VTA loop: controlling the entry of information into long-term memory. Neuron 46:1–11. [DOI] [PubMed] [Google Scholar]
- Miller EK, Cohen JD (2001). An integrative theory of prefrontal cortex function. Annu Rev Neurosci 24:167–202. [DOI] [PubMed] [Google Scholar]
- Mingote S, de Bruin LPC, Feenstra MGP (2004). Noradrenaline and dopamine efflux in the prefrontal cortex in relation to appetitive classical conditioning. J Neurosci 24:2475–2480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulder AB, Nordquist RE, Ôrgüt O, Pennartz CMA (2003). Learning-related changes in response patterns of prefrontal neurons during instrumental conditioning. Behav Brain Res 146:77–88. [DOI] [PubMed] [Google Scholar]
- Otani S (2002). Memory trace in prefrontal cortex: theory for the cognitive switch. Biol Rev Cam Phil Soc 77:563–577. [DOI] [PubMed] [Google Scholar]
- Otani S, Blond O, Desce J-M, Crepel F (1998). Dopamine facilitates long-term depression of glutamatergic transmission in rat prefrontal cortex. Neuroscience 85:669–676. [DOI] [PubMed] [Google Scholar]
- Otani S, Auclair N, Desce JM, Roisin M-P, Crépel F (1999). Dopamine receptors and groups I and II mGluRs cooperate for long-term depression induction in rat prefrontal cortex through converging postsynaptic activation of MAP kinases. J Neurosci 19:9788–9802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds JN, Hyland BI, Wickens JR (2001). A cellular mechanism of reward related learning. Nature 413:67–70. [DOI] [PubMed] [Google Scholar]
- Rossi S, Cappa SF, Babiloni C, Pasqualetti P, Miniussi C, Carducci F, Babiloni F, Rossini PM (2001). Prefrontal cortex in long-term memory: an “interference” approach using magnetic stimulation. Nat Neurosci 4:948–952. [DOI] [PubMed] [Google Scholar]
- Runyan JD, Moore AN, Dash PK (2004). A role for prefrontal cortex in memory storage for trace fear conditioning. J Neurosci 24:1288–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sajikumar S, Frey JU (2004). Late-associativity, synaptic tagging, and the role of dopamine during LTP and LTD. Neurobiol Learn Mem 82:12–25. [DOI] [PubMed] [Google Scholar]
- Sawaguchi T, Goldman-Rakic PS (1991). D1 dopamine receptors in prefrontal cortex: involvement in working memory. Science 251:947–950. [DOI] [PubMed] [Google Scholar]
- Schultz W (1998). Predictive reward signal of dopamine neurons. J Neurophysiol 80:1–27. [DOI] [PubMed] [Google Scholar]
- Schultz W (2002). Getting formal with dopamine and reward. Neuron 36:241–263. [DOI] [PubMed] [Google Scholar]
- Seamans JK, Durstewitz D, Christie BR, Stevens CF, Sejnowski TJ (2001). Dopamine D1/D2 receptor modulation of excitatory synaptic inputs to layer V prefrontal cortex neurons. Proc Natl Acad Sci USA 98:301–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon H, Scatton B, Le Moal M (1980). Dopamine A10 neurones are involved in cognitive function. Nature 286:150–151. [DOI] [PubMed] [Google Scholar]
- Simons JS, Spiers HJ (2003). Prefrontal and medial temporal lobe interactions in long-term memory. Nat Rev Neurosci 4:637–648. [DOI] [PubMed] [Google Scholar]
- Sokolov BP (1998). Expression of NMDAR1, GluR1, GluR7, and KA1 glutamate receptor mRNAs is decreased in frontal cortex of “neuroleptic-free” schizophrenics: evidence on reversible up-regulation by typical neuroleptics. J Neurochem 71:2454–2464. [DOI] [PubMed] [Google Scholar]
- Takahata R, Moghaddam B (2000). Target-specific glutamate regulation of dopamine neurons in the ventral tegmental area. J Neurochem 75:1775–1778. [DOI] [PubMed] [Google Scholar]
- Wang M, Vijayraghavan S, Goldman-Rakic PS (2004). Selective D2 receptor actions on the functional circuitry of working memory. Science 303:853–856. [DOI] [PubMed] [Google Scholar]
- Williams GV, Goldman-Rakic PS (1995). Modulation of memory fields by dopamine D1 receptors in prefrontal cortex. Nature 376:572–575. [DOI] [PubMed] [Google Scholar]
- Young CE, Yang CR (2005). Dopamine D1-like receptor modulates layer- and frequency-specific short-term synaptic plasticity in rat prefrontal cortical neurons. Eur J Neurosci 21:3310–3320. [DOI] [PubMed] [Google Scholar]
- Zahrt J, Taylor JR, Mathew RG, Arnsten AF (1997). Supranormal stimulation of D1 dopamine receptors in the rodent prefrontal cortex impairs spatial working memory performance. J Neurosci 17:8528–8535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng P, Zhang XX, Bunney BS, Shi WX (1999). Opposite modulation of cortical N-methyl-d-aspartate receptor-mediated responses by low and high concentrations of dopamine. Neuroscience 91:527–535. [DOI] [PubMed] [Google Scholar]