

Abstract
Rebound burst activity can be generated in neurons in the subthalamic nucleus (STN) by strong GABAergic inhibitory inputs from the globus pallidus externa (GPe) that is reciprocally connected with the STN. It has been proposed that the rebound burst activity in STN neurons is a key event for generating synchronized rhythmic burst activity in the GPe–STN loop, which may be relevant to the resting tremor in Parkinson’s disease. Here we report that rebound burst firing of STN neurons induces long-lasting bidirectional modifications of GABAergic synaptic transmission in STN neurons themselves. Using the gramicidin perforated-patch clamp technique in the brain slice preparation, we recorded IPSPs from STN neurons during electrical stimulation of the internal capsule. Rebound spikes triggered by hyperpolarizing current pulses were used to induce modification of inhibitory synaptic transmission. We found that long-lasting potentiation of IPSPs could be induced in the neurons exhibiting three or more rebound spikes that had interspike intervals shorter than half of those during base spontaneous activity, whereas long-lasting depression or no change of IPSP amplitude was likely to be observed in neurons that had no rebound burst or two rebound spikes within a burst. The potentiation or depression of IPSPs was associated with a negative or positive shift of reversal potential of IPSPs (EIPSP). The modifications of IPSPs were dependent on activation of postsynaptic voltage-gated calcium channels. This study is the first demonstration that activity-dependent bidirectional modifications of inhibitory synaptic transmission are attributable to bidirectional shifts of EIPSP.
Keywords: inhibitory synapses, plasticity, Cl− reversal potential, Parkinson’s disease, subthalamic nucleus, calcium channels
Introduction
The subthalamic nucleus (STN) is a key structure in the basal ganglia among the subcortical group of structures involved in a variety of behavior, including voluntary movement and movement disorders such as Parkinson’s disease (PD) (Alexander et al., 1990; Wichmann and DeLong, 1996). Unlike the other nuclei in the basal ganglia, which consist of inhibitory GABAergic neurons, the STN is composed of excitatory glutamatergic neurons and thus is considered to be a major driving force of neuronal activity in the basal ganglia circuit (Kitai and Kita, 1987). The STN receives a dense GABAergic innervation from the globus pallidus externa (GPe) (van der Kooy et al., 1981; Smith et al., 1990). The terminals from GPe form symmetrical synaptic contacts predominantly with the proximal dendrites and less frequently with the perikarya and the distal dendrites of the subthalamic cells (Smith et al., 1990).
STN neurons are tonically active and exhibit rhythmic single-spike discharge in vitro (Overton and Greenfield, 1995; Bevan and Wilson, 1999; Beurrier et al., 2000; Xiang et al., 2005). Conversely, they are able to discharge rebound burst firing after the termination of the hyperpolarization of sufficient amplitude (Nakanishi et al., 1987; Bevan et al., 2002). This hyperpolarization could be achieved by strong GABAergic inhibitory inputs from the GPe (Bevan et al., 2002). The GABAergic input-driven rebound burst firing is a critical factor in low-frequency oscillatory activity in the reciprocally connected GPe–STN network, which may be relevant to resting tremor of PD (Plenz and Kitai, 1999). Although it has been well established that the GABAergic input from the GPe plays an important role in regulating STN neuronal activity (Bevan et al., 2002), it is not known how the change in neuronal activity of STN neurons themselves modifies GABAergic synaptic function in the STN. In particular, it is not clear how the rebound burst firing of STN neurons modifies efficacy of inhibitory synapses in STN neurons. The aims of this study were to examine whether the rebound burst activity in STN neurons could induce changes of GABAergic synaptic efficacy to the STN neurons and to determine the mechanism underlying the activity-dependent modification.
Materials and Methods
Slice preparation.
Sagittal slices (300 μm) containing the STN were prepared from Sprague Dawley rats (postnatal day 16–24) of both sexes as described previously (Xiang et al., 2005). Rats were deeply anesthetized (intraperitoneally) with ketamine (100 mg/kg) and xylazine (10 mg/kg) and transcardially perfused with ∼ 20 ml of ice-cold modified artificial CSF (ACSF) oxygenated with 95% O2 and 5% CO2. The modified ACSF was composed of the following (in mm): 230 sucrose, 2.5 KCl, 0.5 CaCl2, 10 MgSO4, 1.25 NaH2PO4, 26 NaHCO3, and 10 d-glucose. The brain was then rapidly removed from the skull, blocked in the sagittal plane, glued to the stage of a vibratome (Vibratome, St. Louis, MO) that was filled with ice-cold modified ACSF, and cut at a thickness of 300 μm. Slices were incubated in ACSF oxygenated continuously with 95% O2 and 5% CO2 at 32°C for 1 h and maintained at room temperature afterward until transferred individually to a recording chamber. The recording chamber was continuously perfused with oxygenated ACSF at 32°C. The ACSF contained the following (in mm): 126 NaCl, 2.5 KCl, 2 CaCl2, 2 MgSO4, 1.25 NaH2PO4, 26 NaHCO3, and 10 d-glucose.
Electrophysiological recordings.
Gramicidin-based perforated-patch recordings were made from visually identified STN neuron soma. The pores formed by gramicidin are selectively permeable to monovalent cations and small uncharged molecules such as water but not permeable to Cl− and intracellular macromolecules (Myers and Haydon, 1972; Kyrozis and Reichling, 1995). Thus, physiological intracellular concentration of Cl− remains undisturbed, and measurement of reversal potential of GABAA receptor-mediated responses is of little bias (Ulrich and Huguenard, 1997; Bevan et al., 2000). An Olympus Optical (Lake Success, NY) BX50WI upright microscope equipped with a low-power objective (4×) was used to identify the STN, and a 40× water immersion objective coupled with Hoffman optics and infrared video was used to visualize individual STN neurons. An Axopatch 1D amplifier (Molecular Devices, Palo Alto, CA) was used for perforated-patch recordings. Patch pipettes were prepared from borosilicate glass (Sutter Instruments, Novato, CA) using a Flaming-Brown micropipette puller (model P-97; Sutter Instruments). The pipette solution contained the following (in mm): 106 K-MeSO4, 25 KCl, 1 MgCl2, 0.025 CaCl2, 10 HEPES, and 0.1 EGTA. The pH of the pipette solution was adjusted to 7.3 with 1 m KOH, and osmolarity was adjusted to ∼295 mOsm. Gramicidin was added to the pipette solution to a final concentration of 20–30 μg/ml immediately before use. Patch pipettes had resistances of 2–4 MΩ when filled with the above solution. Stable series resistances (50–70 MΩ) were usually obtained 30–50 min after forming a gigaohm seal and compensated electronically on-line. IPSPs were recorded from STN neurons at a subthreshold hyperpolarized membrane potential (−65 to −70 mV) in the presence of ionotropic glutamate receptor antagonists 6,7-dinitroquinoxaline-2,3-dione (DNQX) (20 μm) and CGP 37849 [dl-(E)-2-amino-4-methyl-5-phosphono-3-pentenoic acid] (5 μm) and GABAB receptor antagonist CGP 55845 [(2S)-3-[[(15)-1-(3,4-dichlorophenyl)ethyl]amino-2-hydroxypropyl)(phenylmethyl)phosphinic acid] (5 μm). Electrical stimulation was applied to the internal capsule every 10 s through a concentric bipolar stimulating electrode (Frederick Haer Company, Bowdoinham, ME), and stimulation intensity ranged from 10 to 50 μA with 100 μs in duration. Rebound spikes were triggered by hyperpolarizing current pulses (−200 to −250 pA, 600 ms in duration, repeated 10 times at 0.1 Hz) with 0 holding current. ClampFit (Molecular Devices), Origin (Microcal, Northampton, MA), and MiniAnalysis (Synaptosoft, Decatur, GA) were used for data analysis. Spikes after the termination of the hyperpolarized current pulse and having interspike intervals shorter than those during base spontaneous activity were termed as rebound activity, rebound firing, or rebound spikes in this study. We defined a rebound burst as the one that had two or more rebound spikes that had interspike intervals shorter than half of those during base spontaneous activity.
Drugs.
NMDA and AMPA/kainite receptor antagonists CGP 37849 and DNQX and GABAB receptor antagonist CGP 55845 and SR 95531 (gabazine) [6-imino-3-(4-methoxyphenyl)-1(6H)-pyridazinebutanoic acid hydrobromide] were obtained from Tocris Cookson (Ellisville, MO). They were made as concentrated stock solutions in water or DMSO and stored at −20°C. Stocks were thawed and diluted to final concentrations in ACSF and applied through a multibarreled microperfusion pipette (inner diameter, ∼200 μm) placed within 0.5 mm from the recorded neuron. Gramicidin was obtained from Sigma (St. Louis, MO) and dissolved in DMSO (20–30 mg/ml) and diluted in the pipette solution at the final concentration immediately before use.
Statistics.
Group data were presented as mean ± SEM. The means of paired data were compared using the nonparametric Wilcoxon’s matched pairs test. p < 0.05 was considered to be statistically significant.
Results
Modification of GABAergic synaptic transmission by rebound burst activity in STN neurons
First, we examined whether rebound burst activity could modify GABAergic synaptic transmission in STN neurons. Gramicidin-based perforated-patch current-clamp recordings were made from STN neurons. IPSPs were evoked by stimulation of the internal capsule at a subthreshold hyperpolarized membrane potential (−65 to −70 mV) in the presence of ionotropic glutamate receptor antagonists DNQX (20 μm) and CGP 37849 (5 μm) and GABAB receptor antagonist CGP 55845 (5 μm). After baseline IPSPs were recorded at the subthreshold membrane potential for 6–10 min, rebound spikes were generated by hyperpolarizing current pulses (−200 to −250 pA, 600 ms in duration, repeated 10 times at 0.1 Hz) with 0 holding current. Recording of IPSPs was resumed at the same subthreshold membrane potential as before the rebound activity. We found that, 15–20 min after triggering rebound spikes, the amplitude of IPSPs increased by >20% of the control value in 9 of 20 cells (Fig. 1A,B1), decreased by at least 20% of the control value in 6 of 20 cells (Fig. 2A,B1), and had <20% change in 5 cells. The potentiation or depression of IPSPs could last as long as the recording permitted (up to 65 min after rebound activity). To confirm that the evoked IPSPs were mediated by GABAA receptors, in some cases, we applied the GABAA antagonist gabazine to the recording chamber at the conclusion of the experiments. As illustrated in Figure 1, A and B, the response was abolished after gabazine application. The input resistance (RN) was monitored during the experiment by applying a hyperpolarizing current pulse (−10 pA) before the synaptic stimulation (Figs. 1A, 2A). Only recordings without significant change of RN were included in the final analysis (Figs. 1A,B3, 2A,B3). These data indicated that changes in IPSP amplitude after rebound spikes were not attributable to the alteration of RN.
Figure 1.

Long rebound bursts induce long-lasting potentiation of IPSPs in STN neurons. A, Representative traces of IPSPs during control (a), 45 min after rebound bursts (b), and after application of GABAA receptor antagonist gabazine (1 mm, 10 μl dropped into the recording chamber) (c). Each trace is an average of 10–15 responses. The downward deflection before IPSPs was a membrane potential response during hyperpolarization current injection for monitoring change of input resistance. B, Time courses of IPSP amplitude (B1), membrane potential (B2), and normalized input resistance (B3) during control, after triggering 10 rebound bursts, and applying gabazine for the cell in A. Inset, A representative rebound burst triggered by hyperpolarizing current pulse (−200 pA and 600 ms in duration).
Figure 2.
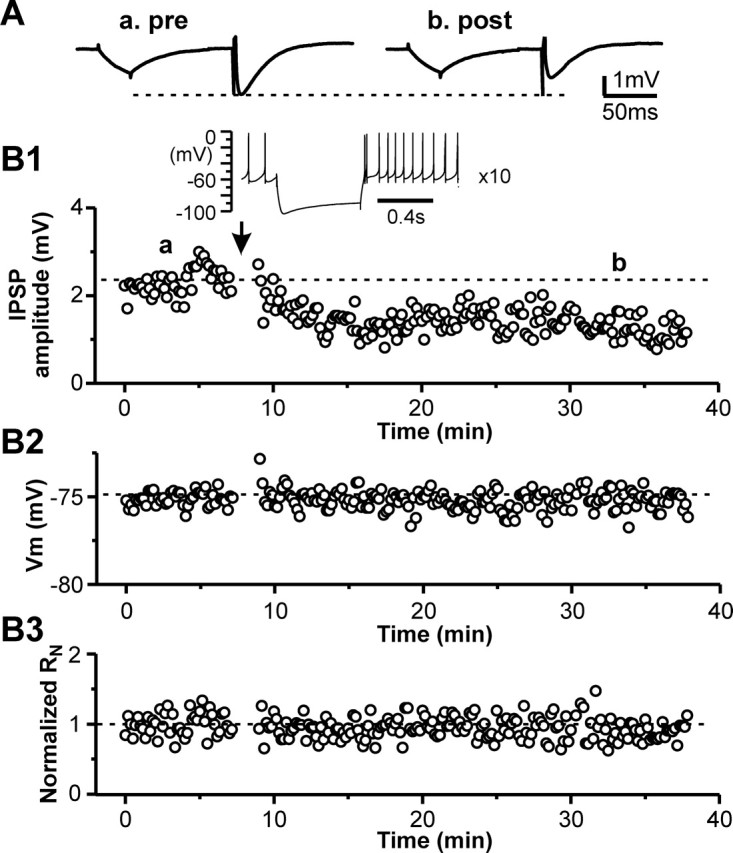
Short rebound bursts induce long-lasting depression of IPSPs in STN neurons. A, Representative traces of IPSPs during control (a) and 20 min after rebound activity (b). Each trace is an average of 10–15 responses. B, Time courses of IPSP amplitude (B1), membrane potential (B2), and normalized input resistance (B3) during control and after triggering rebound activity 10 times for the cell in A. Inset, A representative rebound activity triggered by hyperpolarizing current pulse (−200 pA and 600 ms in duration), showing two spikes within a rebound burst.
To analyze the relationship between the IPSP modification and the rebound activity, we defined a rebound burst as one that contained one or more interspike intervals that were shorter than half of those during base spontaneous activity and plotted the change of IPSP amplitude versus the number of spikes within a burst. As illustrated in Figure 3A, a potentiation was more likely to be induced in the neurons that exhibited three or more spikes within a rebound burst, whereas depression or no change of IPSP amplitude was more likely to be observed in the neurons that had no rebound burst or two spikes within a burst. There was a significant correlation between the percentage change in synaptic efficacy and the number of rebound spikes (r = 0.529; n = 20; p < 0.05, Pearson’s test). The relationship between the amount of change in synaptic efficacy and the rebound activity was readily appreciated when the data were divided into two groups based on the number of spikes within a rebound burst, one including neurons that exhibited three or more spikes within a rebound burst and the other including those showing no rebound bursts or two spikes within a burst. When plotted as cumulative probability histograms, the two data groups showed remarkable difference (Fig. 3B). Kolmogorov–Smirnov test revealed that the difference was statistically significant (p < 0.005). For cells with three or more spikes within a rebound burst, the IPSP amplitude was significantly increased after the rebound burst (140.3 ± 10.8% of the control value; n = 10; p < 0.01) with no significant changes in RN (106.3 ± 8.2% of control value; n = 10; p > 0.2;). In contrast, for cells showing no rebound burst or two spikes within a burst, the IPSP amplitude was significantly decreased after the rebound spikes (81.7 ± 17.9% of the control value; n = 10; p < 0.03) with no significant changes in RN (98.3.0 ± 11.6% of control value; n = 10; p > 0.5).
Figure 3.

Relationship between the change in IPSP amplitude and the rebound spike activity. A1, Plot of the change in IPSP amplitude and the number of rebound spikes that had interspike intervals shorter than half of those during the base spontaneous activity. Data were fit by a second-degree polynomial function (curve). A2, Part of the plot in the dashed box in A1 was expanded, demonstrating a nearly linear relationship between the change in IPSP amplitude and the number of spikes within a rebound burst. B, Cumulative probability histograms for cells with three or more spikes within a rebound burst (filled symbols) and those with two or no rebound burst spikes (open symbols). p < 0.005, Kolmogorov–Smirnov test.
Changes in reversal potential of GABAA receptor-mediated IPSPs as a mechanism for modification of inhibitory synaptic transmission
To determine whether changes in the reversal potential of IPSP (EIPSP) are responsible for modifying GABAergic synaptic transmission, we recorded IPSPs in STN neurons at various membrane potentials (Vm) before and 20–40 min after rebound activity (Fig. 4A1,B1) and plotted the IPSP amplitude against the membrane potential (Fig. 4A2,B2). A linear regression line was used to fit the linear portion of data close to the EIPSP, and the intercept of the regression line with the abscissa was taken as EIPSP (Fig. 4A2,B2). Negative shifts of EIPSP were found in the neurons exhibiting the potentiation of IPSPs (−79.9 ± 1.2 mV in control and −83.1 ± 1.1 mV after potentiation; n = 9; p < 0.002) (Fig. 4A2,C1), whereas positive shifts of EIPSP were associated with the neurons showing depression (−80.6 ± 1.5 mV in control and −78.4 ± 1.8 mV after depression; n = 6; p < 0.04) (Fig. 4B2,C1). To ensure that EIPSP measurement with gramicidin-loaded pipette was stable over time, we measured EIPSP twice at an interval of 20–30 min without eliciting rebound activity in five cells. The result indicated no significant difference between the two measurements (79.0 ± 1.9 vs 78.7 ± 1.9 mV; n = 5; p > 0.4)
Figure 4.
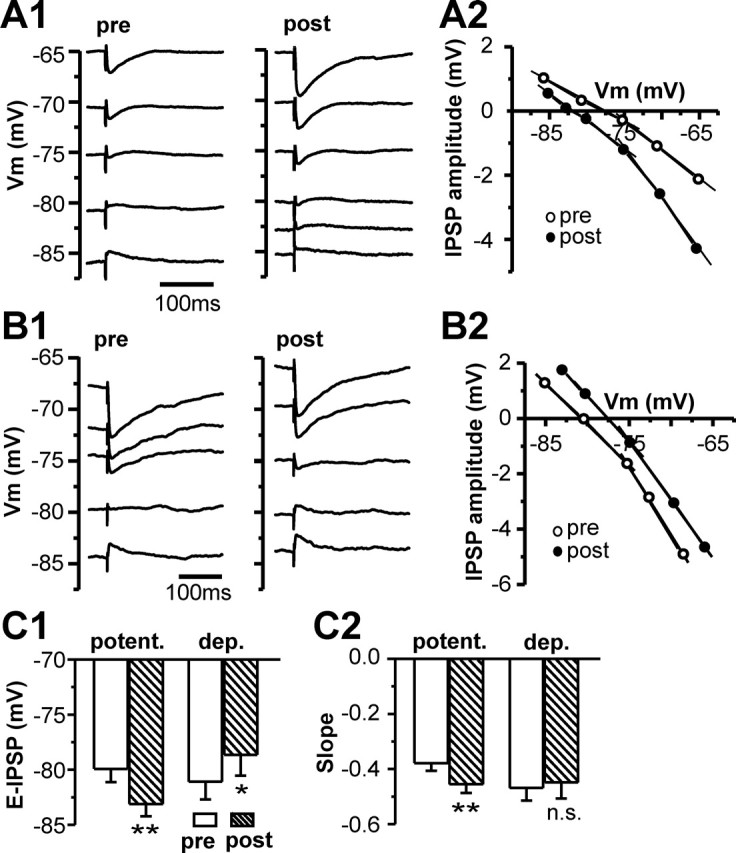
Bidirectional shifts of the EIPSP are associated with long-lasting modification of inhibitory synaptic transmission in STN neurons. A, B, Samples of average IPSPs recorded from two STN neurons showing long-lasting potentiation (A) or depression (B) at various membrane potentials before (pre) and 25 min after (post) rebound bursts (A1, B1) and plots of the IPSP amplitude–Vm relationship (A2, B2). C, Summary of EIPSP (C1) and the slope of the IPSP–Vm relationship at the membrane potential from −65 and −75 mV (C2) before (pre) and after (post) rebound spikes for nine cells that displayed long-lasting potentiation (potent.) and six cells that exhibited long-lasting depression (dep.). *p < 0.03, **p < 0.02. n.s. indicates p > 0.1.
In addition to the shifts of EIPSP associated with IPSP modification, at the range of −65 to −75 mV, the slope of the IPSP–Vm relationship became steeper after IPSP potentiation (−0.39 ± 0.06 in control and −0.45 ± 0.06 after potentiation; n = 9; p < 0.01) (Fig. 4C2), but the slope did not significantly change for the cells that showed depression (−0.38 ± 0.04 in control and −0.36 ± 0.05 after depression; n = 6; p > 0.1) (Fig. 4C2). The increased slope of the IPSP–Vm relationship suggested that an increase in synaptic conductance might also contribute to the increase of synaptic efficacy.
Involvement of voltage-gated calcium channels in modification of inhibitory synaptic transmission
Because voltage-gated calcium channels are likely to be activated during rebound activity, we examined the effect of the high voltage-gated L-type calcium channel blocker nimodipine and the low voltage-gated T-type calcium channel blocker mibefradil on the rebound activity-induced modification of GABAergic synaptic transmission. Nimodipine (20 μm) had no effect on baseline IPSP amplitude (102.5 ± 2.4% of control value; n = 10; p > 0.1). In the presence of nimodipine, regardless of the number of spikes within a rebound burst, only depression or no change was observed after the rebound activity. A representative experiment was shown in Figure 5, A and B, in which long-lasting depression was induced in an STN cell that exhibited four spikes within a rebound burst. The relationship between the change in IPSP amplitude and the number of spikes within a rebound burst was plotted in Figure 5C. There was no significant correlation between the percentage change in synaptic efficacy and the number of spikes within a burst (r = −0.262; n = 12; p > 0.4, Pearson’s test), which was in high contrast to the result obtained in the absence of the calcium channel blocker (ref. Fig. 3A). When the data were divided into two groups on the basis of the spike number within a rebound burst and plotted as cumulative probability histograms, there was no significant difference between the two datasets (p > 0.5, Kolmogorov–Smirnov test) (Fig. 5D). The depression induced in the presence of nimodipine was also associated with a shift of EIPSP toward more positive levels (−79.2 ± 1.0 mV before and −74.1 ± 1.6 mV after rebound spikes; n = 9; p < 0.02). The result suggested that Ca2+ influx through L-type Ca2+ channels was pivotal for the rebound activity-induced potentiation.
Figure 5.
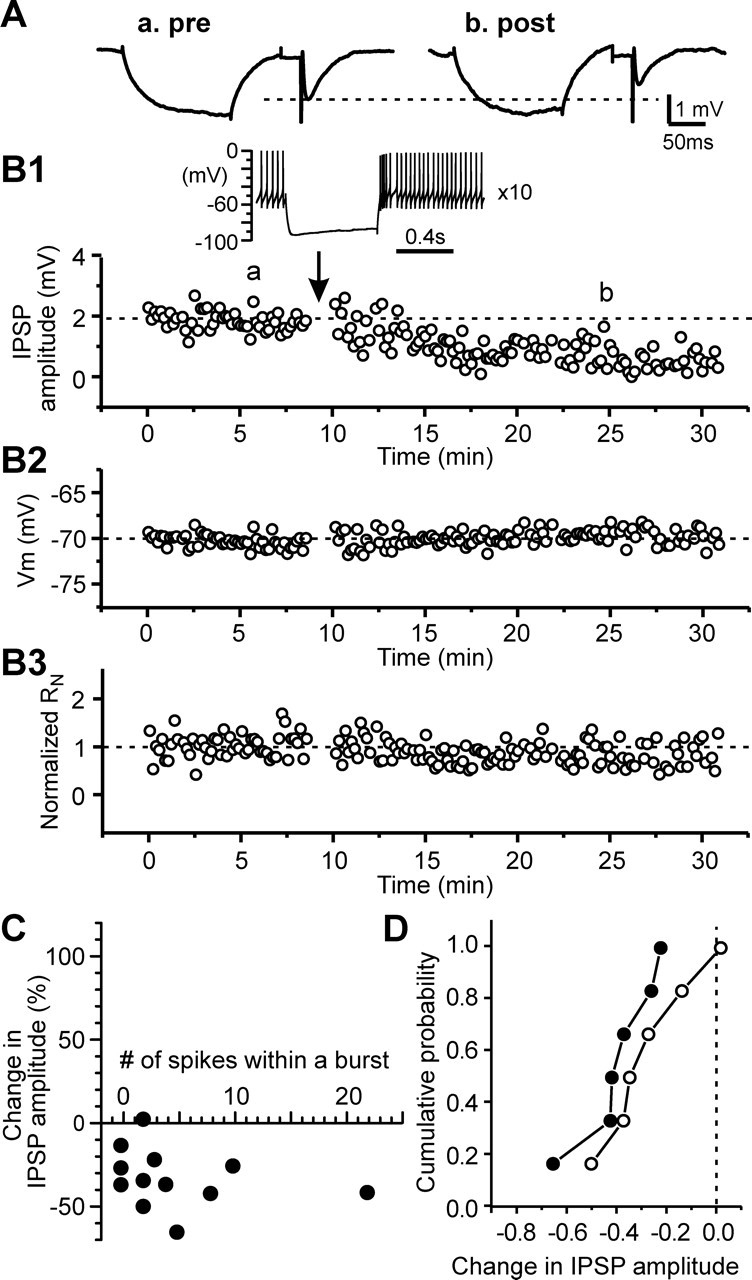
Rebound bursts induce long-lasting depression of IPSPs in the presence of L-type calcium channel blocker. A, Average traces of IPSPs during control (a) and after rebound bursts (b) taken from an STN cell in the presence of 20 μm nimodipine. B, Time courses of IPSP amplitude (B1), membrane potential (B2), and normalized input resistance (B3) during control and after triggering rebound burst 10 times for the cell in A. Inset, A rebound burst triggered by hyperpolarizing current pulse (−250 pA and 600 ms in duration), showing four spikes within a rebound burst. C, Plot of the change in IPSP amplitude and the number of rebound spikes that had interspike intervals shorter than half of those during the base spontaneous activity. D, Cumulative probability histograms for cells with three or more rebound spikes (filled symbols) and those with two or no rebound burst spikes (open symbols). p > 0.5, Kolmogorov–Smirnov test.
Because rebound burst firing in STN neurons is generated in part by activation of low-threshold T-type Ca2+ channels, Ca2+ influx through T-type channels might contribute to the induction of IPSP depression when L-type Ca2+ channels were blocked. To test this possibility, we examined the effect of a combination of nimodipine and a T-type Ca2+ channel blocker mibefradil on the induction of IPSP modification. In the presence of nimodipine (20 μm) and mibefradil (10 μm), no STN neurons displayed more than two rebound spikes that had interspike intervals shorter than half of those during spontaneous firing, and no modification of IPSP amplitude was observed after the rebound activity (102.8 ± 3.2% of the control; n = 6; p > 0.5) (Fig. 6), and neither did EIPSP change after the rebound activity (−76.5 ± 0.6 vs −76.1 ± 0.6 mV; n = 6; p > 0.05). To assess the effect of blocking T-type current alone on IPSP modification, we performed a similar experiment in the presence of mibefradil alone. As with the application of a combination of mibefradil and nimodipine, no STN neurons exhibited more than two rebound spikes that had interspike intervals shorter that half of those during spontaneous activity (n = 3). After the rebound activity, two of the three STN cells showed depression of IPSP amplitude by >20% (Fig. 7), and one cell did not show modification. The depression induced in the presence of mibefradil was also accompanied by a positive shift of EIPSP (−79.5 to −76.1 mV for one and −75.2 to −72.0 mV for the other STN cell). Together, these data indicated that Ca2+ influx through voltage-gated calcium channels during rebound activity played critical roles in modifications of inhibitory synaptic transmission in STN neurons.
Figure 6.

Application of a combination of L-type and T-type calcium blockers prevents the induction of rebound activity-induced long-lasting modification of IPSPs. A, Average traces of IPSPs during control (a) and after rebound activity (b) taken from an STN cell in the presence of 20 μm nimodipine and 10 μm mibefradil. B, Time courses of IPSP amplitude (B1), membrane potential (B2), and normalized input resistance (B3) during control and after triggering rebound activity 10 times for the cell in A. Inset, A rebound activity triggered by hyperpolarizing current pulse (−200 pA and 600 ms in duration), showing that no rebound spikes had interspike intervals shorter than half of those during base spontaneous activity.
Figure 7.

Long-lasting depression induced in the presence of T-type calcium channel blocker. A, Average traces of IPSPs during control (a) and after rebound activity (b) taken from an STN cell in the presence of 10 μm mibefradil. B, Time courses of IPSP amplitude (B1), membrane potential (B2), and normalized input resistance (B3) during control and after triggering rebound activity 10 times for the cell in A. Inset, A rebound activity triggered by hyperpolarizing current pulse (−200 pA and 600 ms in duration), showing that no rebound spikes had interspike intervals shorter than half of those during base spontaneous activity.
Discussion
The present study demonstrated that synaptic responses to inhibitory inputs in STN neurons were dynamically modified by rebound burst activity. The direction of modification depended on the number of spikes within a rebound burst. Long-lasting increase in inhibitory synaptic efficacy tended to be induced in the neurons exhibiting three or more rebound spikes that had interspike intervals shorter than half of those during base spontaneous activity, whereas long-lasting decrease or no change in IPSP amplitude was likely to be observed in the neurons that had no rebound burst or two rebound spikes within a burst. The modification of inhibitory synaptic efficacy was mainly attributable to the shift of the reversal potential of IPSPs. The potentiation and depression of IPSPs were associated with a negative and positive shift of EIPSP, respectively. This is the first demonstration that the activity-dependent bidirectional modification of the inhibitory synaptic efficacy is attributable to the shifts of EIPSP. This bidirectional regulation of GABAergic signaling is likely to play a significant role in information processing in the basal ganglia.
In principle, changes in the efficacy of inhibitory synaptic transmission could be accounted for by the alteration in amount of neurotransmitter released from presynaptic terminals and alterations in responsiveness of postsynaptic receptors or in the reversal potential of GABAA receptor-mediated responses. Long-lasting potentiation and depression of GABAergic synaptic transmission have been reported in several other brain regions. In the dorsomedial and rostral nuclei of the solitary tract and neonatal hippocampus, long-lasting plasticity of inhibitory synaptic transmission was attributed to presynaptic mechanism (Glaum and Brooks, 1996; Caillard et al., 1999; Grabauskas and Bradley, 1999). In contrast, changes in postsynaptic GABAA receptor function were indicated in the plasticity of inhibitory synapses in neocortical pyramidal cells, cerebellar Purkinje cells, and the neurons in the deep cerebellar nuclei (Morishita and Sastry, 1996; Aizenman et al., 1998; Ouardouz and Sastry, 2000; Holmgren and Zilberter, 2001). In addition, a positive shift of the reversal potential of GABAA receptor-mediated responses has been reported recently to be associated with inhibitory synaptic modification induced by brain-derived neurotrophic factor, coincident presynaptic and postsynaptic activity, or prolonged postsynaptic spiking in hippocampal cultures and acute slices (Wardle and Poo, 2003; Woodin et al., 2003; Fiumelli et al., 2005).
The present study shows that a negative or positive shift of the reversal potential of IPSPs is associated with the long-lasting inhibitory synaptic potentiation or depression, respectively, and that Ca2+ influx through voltage-gated calcium channels plays an important role in bridging the rebound activity and the EIPSP shift. It has been reported that induction of long-lasting plasticity of inhibitory synaptic transmission requires an increase in the postsynaptic intracellular calcium concentration ([Ca2+]i) (Kano et al., 1992; Komatsu, 1996; McLean et al., 1996; Morishita and Sastry, 1996; Aizenman et al., 1998; Caillard et al., 1999; Ouardouz and Sastry, 2000; Holmgren and Zilberter, 2001; Woodin et al., 2003; Fiumelli et al., 2005). In some cases, the plasticity could be initiated solely by the postsynaptic activity and subsequent Ca2+ influx (Kano et al., 1992; Komatsu, 1996; Morishita and Sastry, 1996; Aizenman et al., 1998; Caillard et al., 1999; Ouardouz and Sastry, 2000; Fiumelli et al., 2005). In other cases, both the activation of inhibitory synapses and the postsynaptic depolarization were required to achieve the long-lasting plasticity (Holmgren and Zilberter, 2001;Woodin et al., 2003). In the deep cerebellar nuclei, rebound burst spikes could induce bidirectional plasticity of inhibitory synaptic transmission (Aizenman et al., 1998). This plasticity depends on an increase in postsynaptic [Ca2+]i, but the underlying mechanism is not clear. Conversely, in rat hippocampal cultures and acute slices, Ca2+-dependent long-lasting inhibitory synaptic depression was associated with a positive shift of Cl− reversal potential and attributable to downregulation of the function of the K-Cl cotransporter KCC2 (Woodin et al., 2003; Fiumelli et al., 2005). In these studies, blockade of L-type Ca2+ channels prevented the activity-dependent positive shift of Cl− reversal potential (Woodin et al., 2003; Fiumelli et al., 2005). In contrast, our data indicated that blockade of L-type channel alone only prevented the induction of IPSP potentiation associated with the negative shift of EIPSP, but did not block the depression associated with the positive shift of EIPSP. Based on our results that only long-last depression is induced by rebound activity in the presence of the L-type or T-type channel blocker and that no modification is induced in the presence of a combination of L-type and T-type channel blockers, we argue that, in STN neurons, a large increase in Ca2+ influx associated with more spikes within a rebound burst leads to long-lasting potentiation of IPSPs attributable to a negative shift of EIPSP, whereas a small rise of Ca2+ influx associated with rebound spikes devoid of bursts or fewer spikes within a burst leads to the long-lasting depression attributable to a positive shift of EIPSP. It still remains to be determined what intracellular signaling cascades link the Ca2+ signal and the bidirectional shift of EIPSP and how changes in K-Cl cotransporter activity are involved in the bidirectional modification in STN neurons.
In addition to the negative shift of EIPSP associated with the potentiation, a significant increase in the slope of the IPSP–Vm relationship was also noted after the potentiation. This suggested that an increase in synaptic conductance might also contribute to enhanced synaptic efficacy. Increased synaptic conductance can be attributable to an increase in neurotransmitter release from presynaptic terminals or an increase in postsynaptic GABAA receptor functions. This question remains to be further investigated.
The reciprocal connection between GABAergic neurons in the GPe and glutamatergic neurons in the STN has been proposed as neural substrate for the rhythmic burst activity relevant to PD (Plenz and Kitai, 1999). Under the parkinsonian condition, the synchronized burst activity of GPe neurons could provide strong inhibitory inputs to STN neurons and may drive STN neurons to fire rebound bursts. The sustained rebound bursts could cause potentiation of inhibitory responses in a subset of STN neurons, which, in turn, could lead to more excitation in GPe neurons. Thus, the activity-dependent modification of inhibitory responses in STN neurons might be involved in pathophysiology of PD.
Footnotes
This work was supported by an institutional fund of the Center for Neurobiology of Brain Diseases. We thank Dr. D. L. Armbruster for editing the previous version of this manuscript.
References
- Aizenman CD, Manis PB, Linden DJ (1998). Polarity of long-term synaptic gain change is related to postsynaptic spike firing at a cerebellar inhibitory synapse. Neuron 21:827–835. [DOI] [PubMed] [Google Scholar]
- Alexander GE, Crutcher MD, DeLong MR (1990). Basal ganglia-thalamocortical circuits: parallel substrates for motor, oculomotor, “prefrontal” and “limbic” functions. Prog Brain Res 85:119–146. [PubMed] [Google Scholar]
- Beurrier C, Bioulac B, Hammond C (2000). Slowly inactivating sodium current (INaP) underlies single-spike activity in rat subthalamic neurons. J Neurophysiol 83:1951–1957. [DOI] [PubMed] [Google Scholar]
- Bevan MD, Wilson CJ (1999). Mechanisms underlying spontaneous oscillation and rhythmic firing in rat subthalamic neurons. J Neurosci 19:7617–7628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bevan MD, Wilson CJ, Bolam JP, Magill PJ (2000). Equilibrium potential of GABAA current and implications for rebound burst firing in rat subthalamic neurons in vitro. J Neurophysiol 83:3169–3172. [DOI] [PubMed] [Google Scholar]
- Bevan MD, Magill PJ, Hallworth NE, Bolam JP, Wilson CJ (2002). Regulation of the timing and pattern of action potential generation in rat subthalamic neurons in vitro by GABA-A IPSPs. J Neurophysiol 87:1348–1362. [DOI] [PubMed] [Google Scholar]
- Caillard O, Ben Ari Y, Gaiarsa J-L (1999). Long-term potentiation of GABAergic synaptic transmission in neonatal rat hippocampus. J Physiol (Lond) 518:109–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiumelli H, Cancedda L, Poo MM (2005). Modulation of GABAergic transmission by activity via postsynaptic Ca2+-dependent regulation of KCC2 function. Neuron 48:773–786. [DOI] [PubMed] [Google Scholar]
- Glaum SR, Brooks PA (1996). Tetanus-induced sustained potentiation of monosynaptic inhibitory transmission in the rat medulla: evidence for a presynaptic locus. J Neurophysiol 76:30–38. [DOI] [PubMed] [Google Scholar]
- Grabauskas G, Bradley RM (1999). Potentiation of GABAergic synaptic transmission in the rostral nucleus of the solitary tract. Neuroscience 94:1173–1182. [DOI] [PubMed] [Google Scholar]
- Holmgren CD, Zilberter Y (2001). Coincident spiking activity induces long-term changes in inhibition of neocortical pyramidal cells. J Neurosci 21:8270–8277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kano M, Rexhausen U, Dreessen J, Konnerth A (1992). Synaptic excitation produces a long-lasting rebound potentiation of inhibitory synaptic signals in cerebellar Purkinje cells. Nature 356:601–604. [DOI] [PubMed] [Google Scholar]
- Kitai ST, Kita H (1987). Anatomy and physiology of the subthalamic nucleus: a driving force of the basal ganglia. In: The basal ganglia. II. Structure and function: current concepts (Carpenter MB, Jayaraman A, eds) pp. 357–373. New York: Plenum.
- Komatsu Y (1996). GABAB receptors, monoamine receptors, and postsynaptic inositol trisphosphate-induced Ca2+ release are involved in the induction of long-term potentiation at visual cortical inhibitory synapses. J Neurosci 16:6342–6352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyrozis A, Reichling DB (1995). Perforated-patch recording with gramicidin avoids artifactual changes in intracellular chloride concentration. J Neurosci Methods 57:27–35. [DOI] [PubMed] [Google Scholar]
- McLean HA, Caillard O, Ben Ari Y, Gaiarsa JL (1996). Bidirectional plasticity expressed by GABAergic synapses in the neonatal rat hippocampus. J Physiol (Lond) 496:471–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morishita W, Sastry BR (1996). Postsynaptic mechanisms underlying long-term depression of GABAergic transmission in neurons of the deep cerebellar nuclei. J Neurophysiol 76:59–68. [DOI] [PubMed] [Google Scholar]
- Myers VB, Haydon DA (1972). Ion transfer across lipid membranes in the presence of gramicidin A. II. The ion selectivity. Biochim Biophys Acta 274:313–322. [DOI] [PubMed] [Google Scholar]
- Nakanishi H, Kita H, Kitai ST (1987). Electrical membrane properties of rat subthalamic neurons in an in vitro slice preparation. Brain Res 437:35–44. [DOI] [PubMed] [Google Scholar]
- Ouardouz M, Sastry BR (2000). Mechanisms underlying LTP of inhibitory synaptic transmission in the deep cerebellar nuclei. J Neurophysiol 84:1414–1421. [DOI] [PubMed] [Google Scholar]
- Overton PG, Greenfield SA (1995). Determinants of neuronal firing pattern in the guinea-pig subthalamic nucleus: an in vivo and in vitro comparison. J Neural Transm Park Dis Dement Sect 10:41–54. [DOI] [PubMed] [Google Scholar]
- Plenz D, Kitai ST (1999). A basal ganglia pacemaker formed by the subthalamic nucleus and external globus pallidus. Nature 400:677–682. [DOI] [PubMed] [Google Scholar]
- Smith Y, Bolam JP, Von Krosigk M (1990). Topographical and synaptic organization of the GABA-containing pallidosubthalamic projection in the rat. Eur J Neurosci 2:500–511. [DOI] [PubMed] [Google Scholar]
- Ulrich D, Huguenard JR (1997). GABAA-receptor-mediated rebound burst firing and burst shunting in thalamus. J Neurophysiol 78:1748–1751. [DOI] [PubMed] [Google Scholar]
- van der Kooy D, Hattori T, Shannak K, Hornykiewicz O (1981). The pallido-subthalamic projection in rat: anatomical and biochemical studies. Brain Res 204:253–268. [DOI] [PubMed] [Google Scholar]
- Wardle RA, Poo MM (2003). Brain-derived neurotrophic factor modulation of GABAergic synapses by postsynaptic regulation of chloride transport. J Neurosci 23:8722–8732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wichmann T, DeLong MR (1996). Functional and pathophysiological models of the basal ganglia. Curr Opin Neurobiol 6:751–758. [DOI] [PubMed] [Google Scholar]
- Woodin MA, Ganguly K, Poo MM (2003). Coincident pre- and postsynaptic activity modifies GABAergic synapses by postsynaptic changes in Cl− transporter activity. Neuron 39:807–820. [DOI] [PubMed] [Google Scholar]
- Xiang Z, Wang L, Kitai ST (2005). Modulation of spontaneous firing in rat subthalamic neurons by 5-HT receptor subtypes. J Neurophysiol 93:1145–1157. [DOI] [PubMed] [Google Scholar]