

Abstract
Voltage-gated sodium channel (Nav1) β2 subunits modulate channel gating, assembly, and cell-surface expression in CNS neurons in vitro and in vivo. β2 expression increases in sensory neurons after nerve injury, and development of mechanical allodynia in the spared nerve injury model is attenuated in β2-null mice. Thus, we hypothesized that β2 modulates electrical excitability in dorsal root ganglion (DRG) neurons in vivo. We compared sodium currents (INa) in small DRG neurons from β2+/+ and β2−/− mice to determine the effects of β2 on tetrodotoxin-sensitive (TTX-S) and tetrodotoxin-resistant (TTX-R) Nav1 in vivo. Small-fast DRG neurons acutely isolated from β2−/− mice showed significant decreases in TTX-S INa compared with β2+/+ neurons. This decrease included a 51% reduction in maximal sodium conductance with no detectable changes in the voltage dependence of activation or inactivation. TTX-S, but not TTX-R, INa activation and inactivation kinetics in these cells were slower in β2−/− mice compared with controls. The selective regulation of TTX-S INa was supported by reductions in transcript and protein levels of TTX-S Nav1s, particularly Nav1.7. Low-threshold mechanical sensitivity was preserved in β2−/− mice, but they were more sensitive to noxious thermal stimuli than wild type whereas their response during the late phase of the formalin test was attenuated. Our results suggest that β2 modulates TTX-S Nav1 mRNA and protein expression resulting in increased TTX-S INa and increases the rates of TTX-S Nav1 activation and inactivation in small-fast DRG neurons in vivo. TTX-R INa were not significantly modulated by β2.
Keywords: sodium channel, β subunit, dorsal root ganglion, tetrodotoxin sensitive, nociception, mouse
Introduction
Nav1s are composed of a central, pore-forming α subunit and one or two β subunits that modulate channel expression levels, voltage dependence, and kinetics (Catterall, 2000). β1, β1A, β2, β3, and β4 subunits regulate channel gating, assembly, and cell-surface expression in vitro (Goldin, 1993; Isom et al., 1994; Isom, 2000; Yu et al., 2003; McEwen et al., 2004). β1 and β2 also participate in homophilic (Malhotra et al., 2000) and/or heterophilic (Ratcliffe et al., 2001; Kazarinova-Noyes et al., 2001; McEwen et al., 2004) cell adhesion. β1 and β2 can also interact with extracellular matrix molecules (Srinivasan et al., 1998; Xiao et al., 1999), ankyrin (Srinivasan et al., 1988, 1998; Xiao et al., 1999; Malhotra et al., 2000), and/or receptor tyrosine phosphatase β (Ratcliffe et al., 2000).
The effects of β2 on Nav1 cell-surface expression have been well established in primary CNS neuronal cultures in vitro (Schmidt et al., 1985; Schmidt and Catterall, 1986) and in brains of β2−/− mice in vivo (Chen et al., 2002). In primary CNS neuronal cultures, the expression of β2 results in increased levels of Nav1 at the cell surface. The loss of β2 results in a negative shift in the voltage dependence of Nav1 inactivation as well as significant decreases in INa density in acutely dissociated hippocampal neurons of β2−/− mice (Chen et al., 2002). 3H-saxitoxin binding experiments showed that, although the total cellular level of channels is similar in β2+/+ and β2−/− neurons, there is a 42% reduction in the level of plasma membrane channels in β2−/− neurons, consistent with the observed decreases in INa density. The integral of the optic nerve compound action potential is reduced 30% in β2−/− mice compared with control, and its dependence on stimulus strength is shifted to stronger stimuli, consistent with the reduction in channel cell-surface expression. These data clearly demonstrate that β2 plays a critical role in regulating Nav1 density and functional expression in CNS neurons in vivo.
The purpose of this study was to investigate the role of β2 in sensory dorsal root ganglion (DRG) neurons. β2 expression increases in sensory neurons after nerve injury, and β2−/− mice develop less mechanical allodynia than their wild-type counterparts in the spared nerve injury (SNI) model (Pertin et al., 2005). We hypothesized that β2 may modulate electrical excitability in DRG neurons in vivo. To test this hypothesis, we investigated both tetrodotoxin-sensitive (TTX-S) and tetrodotoxin-resistant (TTX-R) INa in DRG neurons isolated from β2+/+ and β2−/− mice. We report that TTX-S but not TTX-R INas are reduced ∼50% in “small-fast” DRG neurons isolated from β2−/− mice compared with wild-type littermates. In addition, the activation and inactivation kinetics of TTX-S INa are slowed significantly. Behavioral studies show acute thermal hypersensitivity and reduced sensitivity to the inflammatory phase of the formalin test in β2 null mice with no measurable differences in sensitivity to mechanical stimulation. We conclude that β2 plays critical roles in electrical excitability in sensory neurons. Furthermore, β2 may differentially affect subclasses of sensory neurons in the DRG.
Materials and Methods
Preparation of DRG neurons.
The generation of β2−/− (Scn2b null) mice was described previously (Chen et al., 2002). Animals used in the present study were bred from a congenic strain of β2+/− mice that had been backcrossed repeatedly to C57BL/6 for at least 10 generations. All animal experiments were performed in accordance with the guidelines of the University of Michigan Committee on Use and Care of Animals or the Committee on Animal Experimentation of the Canton of Vaud, Lausanne, Switzerland. DRG neurons were acutely dissociated from adult male (8–12 weeks old) β2+/+ or β2−/− littermate mice. Briefly, the mice were killed with 100% CO2 inhalation for 1–2 min in a closed chamber. The vertebral column was then removed and cut longitudinally, and the DRGs on both sides from L4 and L5 spinal cord segments were removed. These ganglia were selected because 98% of sensory fibers in the sciatic nerve have cell bodies in the L4 and L5 DRGs (Swett et al., 1991). The DRGs were placed in minimal essential medium plus glutamine (Invitrogen, Carlsbad, CA) supplemented with 16.5 mm NaHCO3, 28.2 mm glucose, and distilled water or in DMEM/F-12 (Invitrogen) supplemented with 22 mm glucose, to give a final osmolarity of 320 mOsm/l. The medium was filtered through a 0.2 μm filter flask. The DRGs were minced two to three times before being incubated in medium with enzymes in a culture dish at 37°C. The total incubation time was 50 min with enzymes added according to the following protocol: collagenase type II (3 mg/ml; Worthington, Lakewood, NJ) was present for the entire 50 min incubation; DNase type I (0.05 mg/ml; Sigma, St. Louis, MO) and trypsin type I (1 mg/ml; Sigma) were added for the last 20 and 10 min of incubation, respectively. The dish was agitated every 10 min during the incubation. The enzymatic digestion was stopped by the addition of medium with BSA (20 mg/ml) before being replaced with fresh medium (0.5 ml) lacking BSA. Cells remaining in tissue fragments were dispersed into the medium using sterile fire-polished and silicon-coated Pasteur pipettes (6–12 strokes up and down). The cellular suspension was plated on collagen-coated 35 mm culture dishes or glass coverslips and incubated at 37°C in a humidified atmosphere of 95% air plua 5% CO2 for 1 h. Finally, 2 ml of medium supplemented with 10% fetal bovine serum were added. The cells were incubated for 2–10 h before recording, with the first 2 h used to allow cells to settle and adhere to the bottom of the culture dishes or coverslips. The remaining 8 h recording period was sufficiently short enough to minimize changes in electrical properties that may occur in long-term cultures.
Voltage-clamp recording.
Voltage-clamp recordings were performed in the standard whole-cell configuration (Hamill et al., 1981), using an Axopatch 200B voltage-clamp amplifier (Molecular Devices, Union City, CA). The cell capacitance (Cm) was calculated by integrating the area under capacitive transients as described previously (Meza et al., 1994) or read directly from the amplifier. Isolated INa were recorded from single small DRG neurons (12 pF < Cm < 42 pF) at 21°C in the presence of a bath solution that contained the following (in mm): 80 NaCl, 50 choline-Cl, 30 TEA-Cl, 2 CaCl2, 0.2 CdCl2, 10 HEPES, and 5 glucose, pH 7.3 with NaOH. For some cells, the solution contained 40 mm NaCl and 90 mm choline-Cl. Fire-polished patch pipettes were generated from borosilicate glass capillaries (Warner Instruments, Hamden, CT) using a Sutter P-87 puller (Sutter Instruments, Novato, CA) and were filled with an internal solution containing the following (in mm): 70 CsCl, 30 NaCl, 30 TEA-Cl, 10 EGTA, 1 CaCl2, 2 MgCl2, 2 Na2ATP, 0.05 GTP, 10 HEPES, and 5 glucose, pH 7.3 with CsOH. Glass coverslips to which the cells were attached were removed from the incubator and placed into a small-volume recording chamber (∼250 μl). Alternatively, if the cells were plated directly onto a 35 mm culture dish, 1 ml of bath solution was used. All cells were subsequently examined within 10–60 min.
Currents were low-pass filtered at 5 kHz with a four-pole Bessel filter and digitally sampled at 20 or 40 kHz. Capacitive transients were canceled with the amplifier circuitry, and linear leakage currents were digitally subtracted on-line with P/4 routines (Armstrong and Bezanilla, 1977). The use of the transient cancellation feature on the amplifier provided estimates for Cm and series resistance. The Cm estimated in this way was similar to that estimated by the integrating method (see above). Patch electrodes had resistances of 0.8–2.5 MΩ, and the series resistance was typically in the range 1–5 MΩ. When appropriate, this was reduced by 40–60% using the compensation circuit of the amplifier. The holding potential was always −80 mV. Recordings were performed using pClamp 8 and 9 software (Molecular Devices).
To analyze the voltage dependence of channel activation, the sodium conductance (GNa) was calculated. Peak current data for each cell were divided by the respective driving force (Vm − Vrev), plotted against Vm, and fit to a Boltzmann equation of the following form:
where Gmax is the maximum GNa, V1/2 is the voltage at which 50% of the Nav1 are activated, and k is the slope of the curve. Steady-state inactivation was measured by applying a double-pulse protocol, consisting of a 500 ms prepulse ranging from −120 to 20 mV (in 5 and 10 mV increments), followed by a test pulse to 0 mV. Each data set (a plot of peak INa during the 0 mV test pulse vs prepulse voltage) was fit with the summation of two Boltzmann equations of the following form:
where F1 and F2 are the fractions of the first and second components of inactivation, respectively. The most negative component (component 1) results from the TTX-S INa whereas the other results from TTX-R INa. V1/2 is the potential at which half of the INa was inactivated, and k is the slope factor for each component. The sum of both fractions is the calculated maximum INa (F1 + F2 = Imax). Data points were then normalized with respect to Imax to obtain the inactivation curve. An alternate calculation method, yielding similar results, is included in the on-line supplemental material (available at www.jneurosci.org).
To examine the rate of channel recovery from inactivation, a protocol was designed comprising a 500 ms prepulse to −120 mV, followed by a test pulse to 0 mV, and then returning to −120 mV for a variable time period (0.25, 0.5, 1, 2, 4, 6, 8, 10, 20, 30, 40, 50, 75, 100, 200, 300, 400, 500, and 750 ms) before application of a second test pulse to 0 mV. The INa amplitude from the second 0 mV pulse was divided by the amplitude of the corresponding first test pulse to obtain the fraction of INa recovered after the recovery time. The data were fit with a double-exponential equation of the following form:
where INa p2/INa p1 is the fraction of current recovered; f1 and f2 are the fractions of the fast and slow recovery components, respectively; t is recovery time; and τ1 and τ2 are the time constants for each recovery component.
Analysis of electrophysiological data.
Data were analyzed using pClamp 8 and 9 (Molecular Devices) and SigmaPlot 7 (SPSS, Chicago, IL). The statistical significance of differences between mean values for β2+/+ and β2−/− neurons was evaluated by Student’s unpaired t test, with p < 0.050 considered significant. Results are presented as means ± SEM.
Real-time reverse transcription-PCR.
Unilateral L3–L5 DRGs from β2+/+ and β2−/− mice were rapidly dissected and collected in RNA-later solution (Qiagen AG, Basel, Switzerland). Each individual sample consisted of a pool of six DRGs dissected from two animals. Total RNA was isolated from each individual sample using the RNeasy Mini kit (Qiagen AG) with a DNase step (RNase free DNase set; Qiagen AG) on the column. After RNA quality and quantity were assessed by electrophoresis and spectrophotometry, 1.5 μg of RNA for each sample was reverse transcribed using Omniscript reverse transcriptase following the manufacturer’s instructions (Qiagen AG). Beacon Designer 3.0 software (Primer Biosoft International, Palo Alto, CA) was used to design primer and probe sequences (Table 1) according to SYBR green specifications (Vandesompele et al., 2002). Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was chosen as an endogenous control to normalize expression levels of the different Nav1 α and β subunits. Real-time PCRs were performed in a 20 μl total volume containing 50 ng of cDNA, 300 nm of each primer, 10 μl of 2× iQ SYBR green mix containing nucleotides, iTaq DNA polymerase, SYBR green, and fluorescein (Bio-Rad, Reinach, Switzerland) using the MyiQ Single Color Real-Time PCR Detection System (Bio-Rad). The amplification protocol was as follows: 3 min at 95°C, 45 cycles of 10 s at 95°C for denaturation, and 45 s at 60°C for annealing and extension; specificity was assessed using a DNA melting curve by measuring fluorescence during gradual temperature increments (0.5°C) from 55 to 95°C.
Table 1.
Primer and probe sequences used for real-time RT-PCR
| Gene | Primer pairs 5′–3′ |
|---|---|
| Nav1.1 | Fw AACAAGCTTCATTCACATACAATAAG |
| Rev AGGAGGGCGGACAAGCTG | |
| Nav1.2 | Fw GGGAACGCCCATCAAAGAAG |
| Rev ACGCTATCGTAGGAAGGTGG | |
| Nav1.3 | Fw GGGTGTTGGGTGAGAGTGGAG |
| Rev AATGTAGTAGTGATGGGCTGATAAGAG | |
| Nav1.6 | Fw AGCAAAGACAAACTGGACGATACC |
| Rev CACTTGAACCTCTGGACACAACC | |
| Nav1.7 | Fw TCCTTTATTCATAATCCCAGCCTCAC |
| Rev GATCGGTTCCGTCTCTCTTTGC | |
| Nav1.8 | Fw ACCGACAATCAGAGCGAGGAG |
| Rev ACAGACTAGAAATGGACAGAATCACC | |
| Nav1.9 | Fw TGAGGCAACACTACTTCACCAATG |
| Rev AGCCAGAAACCAAGGTACTAATGATG | |
| β1 | Fw GTGTATCTCCTGTAAGCGTCGTAG |
| Rev ATTCTCATAGCGTAGGATCTTGACAA | |
| β2 | Fw GGCCACGGCAAGATTTACCT |
| Rev CACCAAGATGACCACAGCCA | |
| β3 | Fw ACTGAAGAGGCGGGAGAAGAC |
| Rev GGTGAGGAAGACCAGGAGGATG | |
| β4 | Fw CCCTTGGTGTAGAAACTAAGCAGAG |
| Rev CAGAAGCGAGTCAGTCAGATACG | |
| GAPDH | Fw TCCATGACAACTTTGGCATTG |
| Rev CAGTCTTCTGGGTGGCAGTGA |
Fw, Forward; Rev, reverse.
To determine the profile of expression of different Nav1 subunits (Nav1.1, Nav1.2, Nav1.3, Nav1.6, Nav1.7, Nav1.8, and Nav1.9 and β1, β2, β3, and β4) in β2−/− and β2+/+ mice, a pool of three samples of each cDNA was used. PCR was performed in triplicate.
The level of expression of Nav1 α and β subunits detected in β2−/− and β2+/+ mice was determined using four individual samples of total RNA from each genotype; each amplification was performed in triplicate for each target mRNA. The efficiency of amplification was determined by serial dilution of starting DNA, and standard curves were constructed from the respective mean critical threshold (CT) value for GAPDH and Nav1 transcripts. The relative expression of each target gene was calculated based on real-time PCR efficiencies and the threshold value of the unknown sample versus the standard sample (Pfaffl, 2001).
Western blot analysis of DRG membrane preparations.
DRGs were removed and stored in ice-cold dissection medium. After a brief centrifugation at 4°C, the supernatant was discarded and the DRGs were rinsed two times in Tris-EGTA buffer (50 mm Tris, pH 8.0 with NaOH, and 10 mm EGTA) containing Complete protease inhibitors (Roche, Indianapolis, IN) at twice the recommended concentration. The DRGs were homogenized and centrifuged at 3000 × g for 5 min at 4°C to remove nuclei. The supernatant was ultra-centrifuged at 4°C for 10 min at 195,000 × g, and the pellets resuspended in 60 μl of Tris-EGTA buffer containing Complete protease inhibitors. One microliter of the suspension was used for protein assay. Equal amounts of protein (100 μg) were loaded on 4–15% polyacrylamide gradient gels and separated by SDS-PAGE. Proteins were then transferred to nitrocellulose as described previously (McEwen et al., 2004). Blots were probed with specific Nav1 α subunit antibodies. Anti-Nav1.1 (Chemicon, Temecula, CA) was used at a 1:500 dilution, anti-Nav1.7 (Chemicon) was used at a 1:500 dilution, and anti-Nav1.6 (Neuromab) was used at a 1:100 dilution. All blots were subsequently probed with anti-α-tubulin diluted 1:5000 (Cedarlane Laboratories, Hornby, Ontario, Canada). The immune signal from this housekeeping protein was used to control for loading differences. Densitometric measurement of the immunoreactive Nav1 α bands was performed using Scion (Frederick, MD) Image. Each Nav1 band was first normalized to the corresponding α-tubulin band for each lane on the gel. Nav1 expression levels in the β2−/− lanes were then calculated as a percentage of the corresponding wild-type levels, using the α-tubulin normalized β2+/+ Nav1 levels as 100%.
Behavioral tests.
Eight-week-old male β2−/− or β2+/+ mice were habituated to the environment, the tester, and the apparatus for at least 2 weeks before testing. All behavioral testing was performed by an observer blinded to the mouse genotype.
The hot-plate assay was conducted by placing the animals (n = 9 in each group) on the hot-plate surface set at varying temperatures (49, 52, and 55°C) (Cao et al., 1998). The latency of response was determined by a clear hindpaw lick. The cutoff was adjusted for each temperature to avoid tissue damage: 60 s for 49°C, 30 s for 52°C, and 20 s for 55°C.
The tail-flick assay was conducted using a tail-flick analgesia meter (Columbus Instrument, Columbus, OH), and the mice were gently restrained in a conic plastic cloth. The latency of response was recorded manually at two different light-beam intensities (4 and 7; n = 4 in each group) with a cutoff at 20 s (Wilson and Mogil, 2001).
Mechanical sensitivity assessment was performed by applying an ascending series of non-noxious Von Frey monofilaments (Stoelting, Wood Dale, IL) to the plantar surface of each hindpaw (n = 9 in each group). For this purpose, mice were placed on an elevated platform with a delicate wire netting floor. The withdrawal threshold (in grams) was defined as the lowest force that evoked a brisk withdrawal response to at least 2 of 10 stimuli (Suter et al., 2003).
For the formalin test, 10 μl of 5% formalin (formaldehyde; Sigma, St. Louis, MO) was injected subcutaneously in the left hindpaw (n = 6 in each group). The time the animal spent shaking/flinching and licking the injected paw was recorded for each 5 min interval, from the injection time up to 80 min (Wei et al., 2001).
Analysis of behavioral data.
Data are represented as mean ± SEM. Differences between groups were compared using one- or two-way ANOVA for unpaired variables, followed by post hoc Bonferroni’s correction when appropriate. Von Frey series present logarithmic differences between hairs, and logarithmic-transformed values were used for the analysis, enabling ANOVA tests (Suter et al., 2003). Statistical analyses were performed using JMP statistical software (version 5.01; SAS Institute, Cary, NC). A p value ≤0.05 was considered statistically significant.
Results
Identification and definition of neuronal size
Using criteria described in previous studies (Abdulla and Smith, 2001, 2002), DRG neurons were first assigned to “small,” “medium,” or “large” groups on the basis of their Cm: <42, 42–72, and >72 pF, respectively (β2+/+ cells, n = 89; β2−/− cells, n = 85). There were no differences in the proportions of small, medium, and large cells between the two strains (data not shown). We focused this study on small neurons, with a mean Cm of 24.9 ± 1.7 pF (n = 35) for β2+/+ neurons and 23.9 ± 1.4 pF (n = 32) for β2−/− neurons. Assuming a specific Cm of 1 μF/cm2 (Hille, 2001), and that the cells have a spherical shape without invaginations, cell-surface area and diameter can be estimated. Thus, the mean Cm corresponds to cell diameters of 28.1 and 27.6 μm, respectively, for β2+/+ and β2−/− neurons. Neurons with diameters <30 μm were considered nociceptive neurons, or type C cells (Study and Kral, 1996; Flake et al., 2004).
Total INa, the sum of TTX-S and TTX-R INa (Roy and Narahashi, 1992; Rush et al., 1998; Abdulla and Smith, 2002; Dib-Hajj et al., 2002), was recorded using a series of depolarizing voltage commands from a holding potential of −80 mV. To explore the voltage dependence of INa in DRG neurons of β2+/+ and β2−/− mice, we took advantage of the previously described activation and inactivation properties of peripheral nerve Nav1 (Cummins and Waxman, 1997; Akopian et al., 1996, 1999). Thus, a current–voltage (I–V) protocol with a 500 ms prepulse to −120 or −50 mV, followed by a test pulse from −100 to +40 mV was applied, with steps of 5 and 10 mV, waiting 10 s between each step (Fig. 1A, inset). When the I–V protocol with a prepulse to −120 mV was applied, the total INa was obtained (Fig. 1A, top traces). A second I–V protocol was subsequently applied to the same cell, but with a prepulse to −50 mV, to inactivate TTX-S INa and thus record only the TTX-R component (Fig. 1A, middle traces). Finally, the TTX-S component was obtained by digitally subtracting the data obtained with the second protocol from the first (Fig. 1A, bottom traces). Similar results were obtained using 300 nm TTX on some cells (data not shown). This protocol has the advantage of obtaining separate I–V relationships for both TTX-S and TTX-R INa in the absence of TTX, thus saving time and reducing cell deterioration. The two I–V curves allow us to classify the small neurons into two subgroups: small-fast and “small-slow” DRG neurons. When the maximum amplitude of TTX-S INa was >70% of the total INa, the cells were placed in the small-fast subgroup (Abdulla and Smith, 2002). These cells made up 49% (17 of 35) of the β2+/+ small cell population and 53% (17 of 32) of the β2−/− small neurons. Cells placed in the second subgroup had TTX-R INa >70% of the total INa. These cells made up 46% (16 of 35) of the β2+/+ and 41% (13 of 32) of the β2−/− small cells. INa in the 6% of cells remaining from the total cell population, two cells of each genotype, did not clearly fall into either category. Thus, these cells were not included in our analysis.
Figure 1.

Current–voltage relationships. A, Protocol for separation of TTX-R and TTX-S INa. A 500 ms prepulse to −120 or −50 mV was applied before a 50 ms test pulse from −100 to 40 mV with steps of 5 or 10 mV (inset). Currents evoked from one β2−/− small-fast DRG neuron by test pulses from −50 to 0 mV are shown. Both TTX-S and TTX-R INa were apparent after the −120 mV prepulse (top traces); only TTX-R INa were obtained after the −50 mV prepulse (middle traces), and the TTX-S component was obtained (bottom traces) by digitally subtracting the TTX-R INa from the total INa B, Average peak INa density–voltage relationships for TTX-R INa (circles) and TTX-S INa (squares) of small-fast DRG neurons (means ± SEM), β2+/+ (closed symbols; n = 15), or β2−/− (open symbols; n = 14). Smooth lines are I–V curves generated using the Boltzmann fit parameters of the respective activation curves. Inset, I–V curves of total INa from the same cells as in A, β2+/+ (closed symbols) and β2−/− (open symbols). C, Similar to B, but for small-slow neurons, β2+/+ (closed symbols; n = 14), or β2−/− (open symbols; n = 11).
The absence of β2 results in reduced TTX-S INa
Plots of peak INa density versus command voltage for small-fast and small-slow DRG neurons are shown in Figure 1, B and C, respectively. For both β2+/+ (n = 15) and β2−/− (n = 14) small-fast DRG neurons, the main INa was TTX-S, as expected by definition. The activation threshold for this INa was between −55 and −50 mV, and the maximum inward INa fell between −30 and −20 mV. For both β2+/+ (n = 14) and β2−/− (n = 11) small-slow neurons, the main INa was TTX resistant, detectable between −40 and −30 mV with maximal inward INa at approximately −10 mV. All currents measured displayed a reversal potential (Vrev) of ∼25 mV, corresponding to the calculated equilibrium potential for sodium ions under these recording conditions (ENa = 25 mV). The I–V curves for small-fast neurons from β2−/− mice show a significant reduction in TTX-S INa density compared with those neurons isolated from β2+/+ mice (Fig. 1B). This reduction can also be clearly observed directly in the total INa (Fig. 1B, inset).
To better compare the voltage dependence of channel activation, the sodium conductance (GNa) was calculated as described in Materials and Methods. For β2+/+ and β2−/− small-slow DRGs neurons (Fig. 2C,D), V1/2 and k were similar for both TTX-R and TTX-S INa; only Gmax for TTX-S INa showed a reduction of ∼29% in β2−/− neurons compared with β2+/+ neurons, however this reduction was not significant (p = 0.665). For small-fast cells, the mean value of V1/2 and k were also similar for TTX-R and TTX-S INa between groups (Fig. 2B). The Gmax for TTX-R INa was ∼30% smaller in β2−/− cells compared with β2+/+ (Fig. 2A, dashed lines), but, again, this difference was not significant (p = 0.083). In contrast, the Gmax for TTX-S INa measured in β2−/− neurons was ∼51% smaller than that observed in β2+/+ cells (Fig. 2A, solid lines), and this reduction was statistically significant (p = 0.001). These results suggest that β2 subunits regulate cell-surface levels of TTX-S but not TTX-R Nav1 in DRG neurons. Alternatively, it is possible that β2 alters TTX-S Nav1 single-channel conductance; however, based on our previous results (Isom et al., 1995b; Chen et al., 2002), we propose that the former is the most likely mechanism of β2 action.
Figure 2.
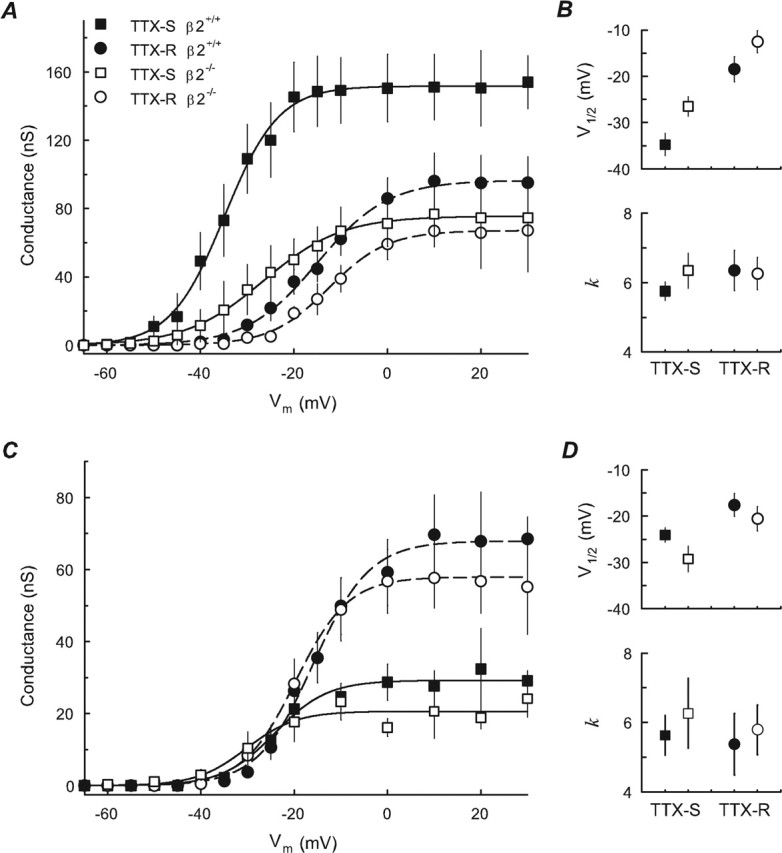
Voltage dependence of activation. A, Activation curve of peak sodium conductance: TTX-R (circles) and TTX-S (squares) obtained from the same small-fast cells as in Figure 1B, β2+/+ (closed symbols), and β2−/− (open symbols). Smooth lines are fits to a Boltzmann function for TTX-R (dashed lines) and TTX-S (solid lines) currents, respectively. B, Midpoint potential (V1/2) and slope factor (k) of fitted activation curves of small-fast cells. C, D, Same small-slow cells as in Figure 1C with symbols as in A and B. Error bars indicate SEM.
β2 does not affect the voltage dependence of INa inactivation
Steady-state inactivation was measured as described in Materials and Methods. An example of the INa obtained from a typical small-fast β2+/+ neuron in response to a test pulse to 0 mV is shown in the inset to Figure 3A. In this example, as in practically all small-fast DRG neurons tested, the fast INa (TTX-S) is inactivated at more negative voltages than the slow INa (TTX-R). The mean of the individual curves are shown in Figure 3A for small-fast neurons and in Figure 3C for small-slow neurons. The corresponding V1/2 and k are compared in Figure 3, B and D. The voltage dependence of TTX-S and TTX-R INa inactivation in small β2−/− neurons was nearly identical to that of TTX-S and TTX-R INa of small β2+/+ neurons (Fig. 3), therefore the β2 subunit does not regulate the INa voltage dependence in small DRG neurons.
Figure 3.
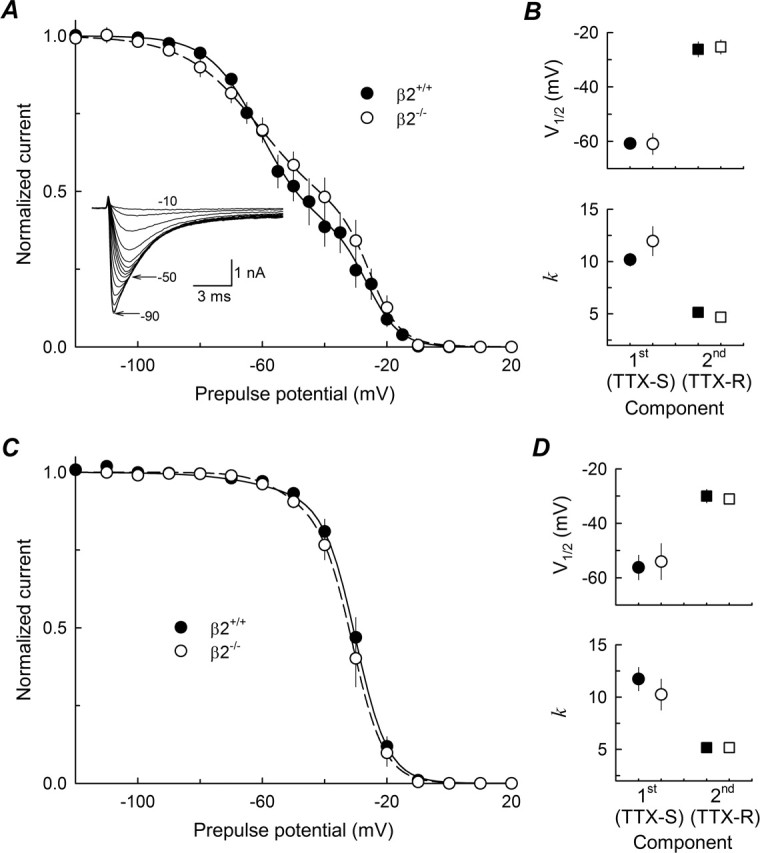
Voltage dependence of inactivation. A, Peak INa at 0 mV, normalized to its maximal value (inset), as a function of voltage during a 500 ms prepulse. INa were measured from β2+/+ (n = 13; closed circles) and β2−/− (n = 10; open circles) small-fast DRG neurons. Each data set was fit with a double Boltzmann function (lines). The inset is an example of INa at 0 mV, from one small-fast β2+/+ cell, after 500 ms prepulses from −90 to −10 mV. B, Parameters of fitted inactivation curves shown in A; circles represent the first component (TTX-S), and squares represent the second component (TTX-R). C, D, Inactivation curves and parameters, respectively, for small-slow β2+/+ (n = 12) and β2−/− (n = 8) neurons; symbols are as in A and B. Error bars indicate SEM.
According to our classification of cells into small-fast and small-slow based on the proportion of TTX-S INa to that of TTX-R INa, the average values of F1 and F2 (the proportion of TTX-S and TTX-R INa) are close to 0.7 and 0.3, respectively, for both β2+/+ and β2−/− small-fast cells. For small-slow cells, F1 and F2 are ∼0.1 and 0.9, respectively. Thus, the main INa for small-fast cells is TTX-S, whereas for small-slow cells it is TTX-R.
Effects of β2 on INa kinetics
The rate of channel recovery from inactivation was measured as described in Materials and Methods. A typical set of INa traces obtained using this protocol is shown in the inset of Figure 4. In agreement with previous reports, the recovery from inactivation curve shows two components: the TTX-R INa shows fast recovery, and the TTX-S INa shows slow recovery (Cummins and Waxman, 1997; Rush et al., 1998). Time constants for β2+/+ were τ1 = 1.3 ± 0.2 ms and τ2 = 95.3 ± 17.8 ms (n = 6), and time constants for β2−/− were τ1 = 1.5 ± 0.5 ms and τ2 = 83.3 ± 29.4 ms (n = 6); the fraction of TTX-S INa was 0.88 ± 0.03 and 0.80 ± 0.12 for β2+/+ and β2−/−, respectively. There were no significant differences between groups.
Figure 4.

Recovery from inactivation. The average time course of recovery from inactivation for total INa from small-fast neurons β2+/+ (closed symbols; n = 6) and β2−/− (open symbols; n = 5) is shown. The data were fit with a double exponential (lines) with the following results: for β2+/+, τ1 = 1.3 ± 0.2 ms and τ2 = 95.3 ± 17.8 ms; for β2−/−, τ1 = 1.5 ± 0.5 ms and τ2 = 83.3 ± 29.4 ms. Inset, A representative record from one β2+/+ cell shows INa obtained from recovery intervals of 0.25–30 ms to −120 mV. Error bars indicate SEM.
Superimposition of INa obtained from small-fast β2+/+ and β2−/− neurons suggested different rates of channel activation and inactivation (Fig. 5A). To evaluate the activation kinetics, two points on each time course were measured: the time to achieve 50% of the maximum current after onset of the test pulse to 0 mV (T1/2 peak), and the time to achieve the peak INa with the same test pulse (Tpeak). Comparing both groups of small-fast neurons, these times were significantly different (Fig. 5B) with T1/2peak and Tpeak for β2−/− (n = 12), 60 and 69% longer than β2+/+ (n = 16) neurons, respectively. To evaluate the inactivation kinetics for the same INa, the decaying phase of the INa was fit using a double-exponential function, obtaining two inactivation time constants (τfast and τslow). Only τfast was affected by the loss of β2 expression (Fig. 5C): τfast = 1.25 ± 0.17 ms for β2+/+ neurons compared with 2.13 ± 0.23 ms for β2−/− neurons, an increase of ∼70%. In contrast, τslow was the same for both groups (Fig. 5C). We performed similar analyses for small-slow β2+/+ (n = 11) and β2−/− (n = 9) neurons. Neither activation nor inactivation kinetics were statistically different (data not shown).
Figure 5.

Activation and inactivation kinetics of INa at 0 mV. A, Normalized INa evoked by a test pulse to 0 mV from a holding potential of −80 mV. INa are shown from a typical small-fast β2+/+ cell and a typical small-fast β2−/− cell. B, Time to achieve 50 and 100% activation. The data were obtained from small-fast β2+/+ (n = 16) and small-fast β2−/− (n = 18) neurons. C, Time constants of INa inactivation for the same cells as in B. The two time constants of inactivation (τfast and τslow) were obtained by fitting the decay phase of the INa with a double-exponential function. ∗p < 0.05, Significantly different from β2+/+. Error bars indicate SEM.
Nav1 mRNA levels are regulated by β2
To begin to investigate the molecular basis for the observed reduction in TTX-S INa in β2−/− neurons, we measured Nav1 α and β subunit mRNA levels in DRGs from both wild-type and null mice. The profile of expression and relative abundance of Nav1 subunit transcripts (Nav1.1, Nav1.2, Nav1.3, Nav1.6, Nav1.7, Nav1.8, and Nav1.9 and β1, β2, β3, and β4) (Table 1) were evaluated independently in β2+/+ and β2−/− mice by real-time reverse transcription (RT)-PCR. Nav1 subunit mRNA levels were normalized to the highest Nav1 subunit transcript expressed in β2+/+ DRGs: Nav1.7 for α subunits and β1 for β subunits (Fig. 6). In both genotypes, the order of the Nav1 subunit transcript expression levels was the same, with the exception of β2 in the null mice: Nav1.7 > Nav1.8 > Nav1.9 > Nav1.6 > Nav1.1 > Nav1.3 > Nav1.2 and β1 > β4 > β3 > β2. In β2+/+ DRGs, the Nav1.7 transcript was the most abundant. It was twice as abundant as Nav1.6, ∼7 times more abundant than Nav1.3 or Nav1.1, and ∼25 times more abundant than Nav1.2, all TTX-S channels. In addition, Nav1.7 was more abundant than transcripts for the TTX-R channels Nav1.8 and Nav1.9 (Fig. 6A). For mRNA of β subunits, β1 was 6–10 times more abundant than the other β subunits (Fig. 6B). Interestingly, β2 appeared to be the lowest expressed β subunit mRNA; however, as shown above, it plays a major functional role in DRG neurons.
Figure 6.

Expression levels of Nav1 mRNAs in DRGs. A, Transcript levels of Nav1 α subunits expressed in β2+/+ (filled bars) and β2−/− (open bars) DRGs; all levels are normalized to Nav1.7 levels measured in β2+/+ DRGs. B, Normalized transcript levels of β subunits to β1 expressed in β2+/+ DRGs. Data are mean ± SEM from a real-time reverse transcription-PCR experiment performed in triplicate using a mix of three samples.
In β2−/− DRGs, the profile of α and β subunit expression did not change, with the exception of β2. However, the abundance of some subunit transcripts did change, notably Nav1.7 (p = 0.043) (Fig. 6). Experiments were then performed to determine the ratios of specific subunit mRNA expression levels in null versus wild-type neurons. β2+/+ and β2−/− DRGs were assessed by real-time RT-PCR and normalized to GAPDH. The ratios of Nav1 α and β subunit transcript levels in β2−/−/β2+/+ DRGs are shown in Figure 7. The expression levels of three TTX-S Nav1 transcripts, Nav1.3, Nav1.6 and Nav1.7, were significantly reduced (p < 0.05) 20–25% by the β2 null mutation (Fig. 7A); similar reductions were found for β3 and β4 mRNAs (Fig. 7B), whereas no changes were observed for the other subunits.
Figure 7.

Effect of the β2 null mutation on Nav1 mRNA levels. A, Ratio of β2−/−/β2+/+ transcript levels for α subunit mRNAs. B, Ratio of β2−/−/β2+/+ transcript levels for β subunit mRNAs. Black bars, mRNA level does not change; gray bars, mRNA level is reduced. ∗p < 0.05.
TTX-S Nav1 protein is regulated by β2
Western blot analyses of membrane preparations of DRG neurons for the TTX-S α subunits Nav1.7, Nav1.6, and Nav1.1, all normalized to α-tubulin as a housekeeping protein loading control, were performed to determine whether the observed changes in mRNA expression were reflected at the level of protein. Changes in mRNA levels are not always reflected by altered protein expression, with Nav-β2 subunits serving as an example of this phenomenon (Malhotra et al., 2001; Pertin et al., 2005). We observed that Nav1.1 and Nav1.7 protein levels were reduced in β2−/− DRG neurons compared with β2+/+, whereas the levels of Nav1.6 did not appear to change (Fig. 8A). Similar results to those shown in the figure were obtained from three or four independent experiments. Immunoreactive bands were normalized to α-tubulin by densitometry, and changes in β2−/− levels were expressed as a percentage of wild-type levels for each Nav1 (Fig. 8B). These calculations showed that Nav1.1 (p = 0.082; n = 3) and Nav1.7 (p < 0.001; n = 3) were reduced in the null DRGs compared with wild type. The values for Nav1.6 were not significantly different (p = 0.23; n = 4). We propose that the observed reduction in mRNA and protein levels of Nav1.7 (and possibly the reduction in Nav1.1 protein expression) may underlie the reduction in TTX-S INa measured in β2−/− neurons.
Figure 8.

Reduction in TTX-S Nav1 protein levels in β2 null neurons. A, Equal aliquots of DRG protein homogenates were separated by SDS-PAGE, transferred to nitrocellulose, and probed with specific Nav1 α subunit antibodies as indicated. All blots were subsequently probed with anti-α-tubulin to control for sample loading. The α-tubulin blot corresponding to the Nav1.7 blot is shown as an example. B, Immunoreactive bands were quantified using densitometry. Each band density was first normalized to its corresponding α-tubulin signal, and β2−/− levels for each Nav1 were expressed as a percentage of β2+/+ levels. For Nav1.1 and Nav1.7: #p < 0.1; ∗p < 0.05; n = 3. There was no significant change for Nav1.6 (n = 4). Error bars represent SEM.
β2−/− mice show increased thermal but not mechanical sensitivity
Because TTX-S Nav1s are involved in nociceptive transmission, we hypothesized that β2−/− mice would exhibit altered responses to noxious stimuli. We showed previously that β2 expression is upregulated in sensory neurons in neuropathic pain and that development of mechanical allodynia in the SNI model is attenuated in β2−/− mice compared with wild type (Pertin et al., 2005). To determine whether β2 also plays a role in acute pain pathways, we compared the responses of the β2−/− mice to acute thermal and mechanical stimuli with those of their wild-type littermates. In the hot-plate test at 49°C (Fig. 9A), β2+/+ mice exhibited a latency of response of 34.9 ± 3.0 s. In contrast, β2−/− mice displayed a significantly shorter response latency of 27.4 ± 1.5 s (p < 0.05; n = 9 in each group). At higher temperatures, no difference was observed between groups (p ≥ 0.05). To confirm the hot-plate test results and to determine whether thermal hypersensitivity in β2−/− mice was induced by spinal processes, we evaluated the simple tail-flick reflex response to a radiant heat beam focused on the tail. At a low-intensity setting (4) of the tail-flick analgesia meter (Fig. 9B), the latency was significantly shorter in β2−/− mice (5.5 ± 1.1 s) compared with β2+/+ mice (9.1 ± 1.2 s) (p < 0.01; n = 4 in each group). At a higher intensity (7), a difference between the two groups was not discernible (p ≥ 0.05) (Fig. 9B). To investigate whether compensatory upregulation of heat-sensing genes occurred in the β2−/− mice and thus could be responsible for the observed thermal hypersensitivity, we measured the levels of TRPV1 and TRPV2 in DRGs isolated from β2+/+ and β2−/− mice. No differences were detected between the two genotypes (TRPV1: ratio β2−/−/β2+/+ was 0.95, p = 0.57; TRPV2: ratio β2−/−/β2+/+ was 0.99, p = 0.95).
Figure 9.

Noxious heat sensitivity. A, In the hot-plate test at 49°C, the latency response was decreased in β2−/− mice compared with β2+/+ animals (n = 9 in each group; ∗ p < 0.05). At higher temperatures (52 and 55°C), no statistically significant difference was observable. B, Tail-flick test. Values represent the latency response from the heat source. The latency decreased significantly in β2−/− mice compared with β2+/+ (n = 4 in each group; ∗p < 0.01). No differences were observed at higher intensities. Error bars indicate SEM.
Mechanical withdrawal threshold responses to a series of calibrated monofilaments applied to both paws in both groups were also recorded. Deletion of β2 did not modify the animals’ response, and we did not observe any significant differences between groups (p ≥ 0.05; n = 9 in each group) (Fig. 10). For β2+/+ mice, the values were 0.246 ± 0.05 g (left hindpaw) and 0.249 ± 0.07 g (right hindpaw) compared with 0.235 ± 0.05 g (left hindpaw) and 0.283 ± 0.08 g (right hindpaw) for β2−/− mice.
Figure 10.

Basal mechanical sensitivity. Withdrawal mechanical thresholds were similar in β2+/+ and β2−/− animals (n = 9 in each group; p > 0.05). Error bars indicate SEM.
β2−/− mice show reduced response to inflammatory pain
We next performed an extended formalin test to determine the role of β2 in this model of acute and inflammatory pain (Tjolsen et al., 1992; Wei et al., 2001). During the initial phase of acute pain, the responses of β2−/− and β2+/+ mice were similar (Fig. 11). After the initial phase, an early second phase from 10 to 55 min and a later second phase from 55 to 80 min have been described previously (Wei et al., 2001). These late phases are considered to be models of inflammatory pain (Tjolsen et al., 1992). During the late phase, the behavioral response of β2−/− mice was significantly attenuated when compared with β2+/+ mice (Fig. 11).
Figure 11.
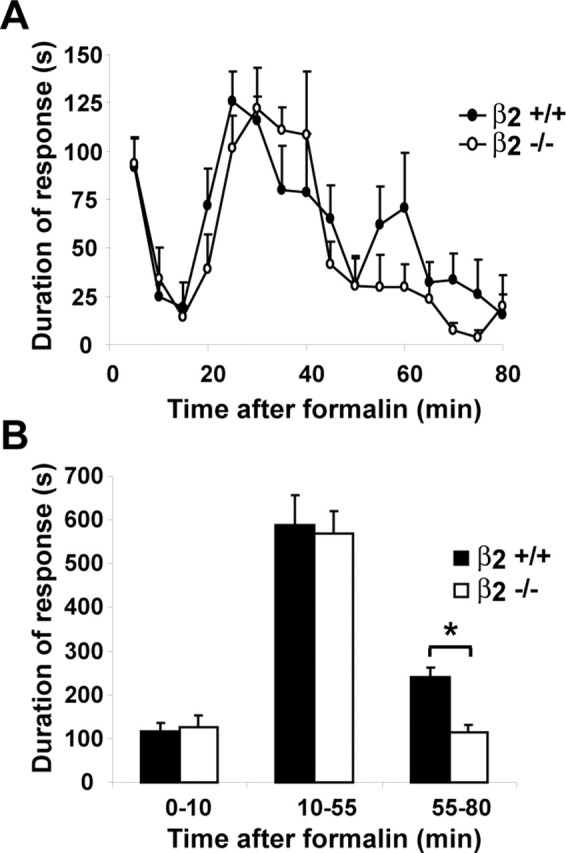
Formalin test. A, Time course of the formalin response after intraplantar injection of 10 μl of 5% formalin. B, Cumulative formalin response to the initial phase (0–10 min), early second phase (10–55 min), and late second phase (55–80 min). β2+/+, filled bars or symbols; β2−/−, open bars or symbols. n = 6 per group; ∗p < 0.05. Error bars indicate SEM.
Discussion
Nav1s in sensory neurons control membrane excitability and contribute to the transmission of nociceptive information to the spinal cord. Both TTX-S and TTX-R Nav1s are expressed in DRG neurons (Baker and Wood, 2001), as are β1, β1A, β2, β3, and β4 (Black et al., 1996; Kazen-Gillespie et al., 2000; Yu et al., 2003). The α subunit cDNAs express functional Nav1 in heterologous expression systems. However, for TTX-S α subunits, the currents characteristic of these channels expressed in isolation are quite different from native currents. Coexpression of the β subunits with these channels results in shifts in the voltage dependence of activation and inactivation, changes in channel modal gating behavior resulting in increases in the rates of inactivation and recovery from inactivation (Isom et al., 1994), and increases in channel expression at the plasma membrane as assessed by 3H-saxitoxin binding (Isom et al., 1995a; Kazarinova-Noyes et al., 2001; McEwen et al., 2004). The β subunit-mediated effects on TTX-R Nav1 expressed in heterologous systems are not well understood (Sangameswaran et al., 1996; Malhotra et al., 2001; Vijayaragavan et al., 2001, 2004), and the functional effects of Nav β subunits on α are dependent on the recipient cell type in vitro (Chen et al., 2002; Meadows and Isom, 2005). Thus, the use of in vivo models is critical to the understanding of their physiological roles. The present studies using β2 null mice make important and novel contributions to elucidating the function of β2 in electrical signal transduction in sensory neurons and are the first report of the differential effects of β2 on TTX-S versus TTX-R channels.
In the present study, we compared INa in DRG neurons isolated from β2+/+ and β2−/− mice to determine the effects of β2 on TTX-S and TTX-R Nav1s in vivo. Small-fast DRG neurons acutely isolated from β2−/− mice showed significant decreases in TTX-S but not TTX-R INa compared with DRG neurons isolated from wild-type littermates. This decrease was not a result of changes in the voltage dependence of activation or inactivation of TTX-S Nav1s. TTX-S, but not TTX-R, INa activation and inactivation kinetics in small-fast DRG neurons were significantly slower in β2−/− mice compared with β2+/+. Our results predict that β2 expression results in increased levels of TTX-S Nav1 mRNA and protein expression, particularly Nav1.7, increased levels of TTX-S Nav1 cell-surface expression, and increased rates of TTX-S INa activation and inactivation in small-fast DRG neurons in vivo. TTX-R INa in small-slow and small-fast DRG neurons are insensitive to modulation by β2.
Interestingly, the β2 null mutation affects TTX-S INa in small-slow versus small-fast DRG neurons differently. TTX-S INa are dramatically reduced in small-fast neurons but are essentially unaffected in small-slow neurons. A possible explanation for this observation is that small-slow and small-fast DRG neurons may express vastly different levels of β2 protein. Alternatively, perhaps these two neuronal populations express different profiles of TTX-S Nav1s that are differentially regulated by β2 in vivo. Previously, we reported that β2 protein expression is low in normal DRG neurons, although immunocytochemical staining could be detected in small, medium, and large cells (Pertin et al., 2005). Unfortunately, this low level of β2 staining precluded our ability to perform a more complete analysis of differential expression in small neurons in the present study. Because β2 mRNA levels are poor predictors of β2 protein levels (Malhotra et al., 2001; Pertin et al., 2005) and because evidence suggests that posttranscriptional regulation of β2 may be critical to β2 protein expression in the DRG (Pertin et al., 2005), single-cell RT-PCR experiments would not be helpful in understanding the relative levels of β2 peptides in small-slow versus small-fast cells. Until a more sensitive antibody can be developed, this issue will be difficult to address. Nevertheless, it is intriguing to consider that, as reported recently by the Waxman group (Rush et al., 2006), ion channel mutations can have opposing physiological effects in various neuronal cell types as a result of cell background differences, including different repertoires of ion channels within the two type of neurons.
Nav1s play a major role in pain through determination of membrane excitability in central and peripheral neurons. The threshold for repetitive firing of sensory neurons, and thus ectopic activity, is primarily determined by the density of Nav1s in the plasma membrane. If excitability is suppressed (e.g., by a reduction in cell-surface Nav1s below a certain threshold level), the neuron can no longer respond to depolarizing stimuli by generating pacemaker activity (Devor, 2006). TTX-S Nav1s play roles in inflammatory, visceral, and neuropathic pain. This has been directly demonstrated by administration of low doses of TTX locally to DRGs (Lyu et al., 2000) or systemically (Marcil et al., 2006) in rodents. According to these studies, nanomolar doses of TTX decrease pain behavior in the late inflammatory (but not early acute) phase of the formalin test, in the writhing test of visceral pain, in mechanical allodynia in the Chung model of neuropathic pain (Kim and Chung, 1992), and in mechanical allodynia and thermal hyperalgesia in the Seltzer model of neuropathic pain (Seltzer et al., 1990). TTX administered at these low doses had no detectable effects on action potential conduction despite exerting significant behavioral effects. Thus, we predicted that β2−/− mice, which have an ∼50% reduction in expression of TTX-S INa in small-fast DRG neurons, would have reduced levels of firing in response to depolarizing stimuli and thus reduced inflammatory and neuropathic pain responses with little effect on action potential propagation, similar to the effects of local anesthetic, membrane-stabilizing drugs (Devor, 2006). Indeed, the allodynic response to the SNI model of neuropathic pain was reduced in β2−/− mice, although basal mechanical sensitivity was not altered before nerve injury (Pertin et al., 2005). The behavior of β2−/− mice in the early acute phase of the formalin test was not different from wild-type animals; however, the response of these mice to the late, inflammatory phase was attenuated, consistent with the TTX experiments and similar to results in Nav1.7 null mice (Nassar et al., 2004). Action potential conduction velocity remains intact in β2−/− mice (Chen et al., 2002), consistent with the reported effects of nanomolar TTX. The response to low-threshold mechanical stimulation with von Frey hairs was not affected by the β2 null mutation. These results are consistent with the observation that TTX does not block mechanically activated currents in sensory neurons (Drew et al., 2004) and that Nav1.7 null mice show no differences in their response to von Frey hairs (Nassar et al., 2004).
An unexpected finding in our study was a normal thermal sensitivity of β2−/− mice at high temperatures (52–55°C), whereas at a lower temperature (49°C), β2−/− mice displayed relative heat hypersensitivity in the hot-plate test. Similarly, in the tail-flick test, β2−/− mice were more responsive to low-intensity stimulation. RT-PCR experiments showed that this response was not attributable to compensatory upregulation of TRPV1 or TRPV2 in the null DRGs, although we cannot rule out other genes involved in thermal sensation. Heat sensing involves thermal C- and A-fiber nociceptors, with a distribution of the high-threshold (52°C) TRPV2 sensor in larger DRG neurons (Greffrath et al., 2003; Patapoutian et al., 2003; Shimosato et al., 2005). It is thus possible that in this very subset of DRG neurons, the expression of Nav1s is different and the deletion of β2 may alter proprieties and produce a hyperexcitable phenotype for lower-threshold nociceptors (Rush et al., 2006). On the other hand, the hyperexcitable phenotype may be related to central alteration of pain signaling. Experiments in cats have demonstrated that the dominant effect of noxious heat on dorsal horn interneurons is inhibitory (Schomburg et al., 2000). For instance, one may suggest that spinal interneurons in the null animals present reduced cell-surface TTX-S Nav1 expression and thus reduced excitability. However, because nociception ultimately involves peripheral and central neurons in ascending and descending pathways, the ultimate answer to this question must wait for the future development of tissue-specific β2 null mice that will allow the study of β2 subunits in individual neuronal populations.
We propose that the major functional role of Nav β2 subunits in vivo is to regulate TTX-S Nav1 α subunit insertion into the plasma membrane in both CNS and PNS neurons, thus critically regulating neuronal excitability. β2−/− mice, under homeostatic conditions, are able to maintain normal patterns of neuronal firing. However, in models of inflammatory or neuropathic pain, in which induction of peripheral firing and central sensitization are required, our results suggest the density of plasma membrane Nav1 is insufficient and peripheral bursts can neither be evoked nor maintained.
Footnotes
*I.D. and L.L.I. contributed equally to this work.
This work was supported by National Multiple Sclerosis Society Research Grant RG-2882 (L.L.I.), Michigan Life Sciences Corridor Grant 001654 (L.L.I.), Gastrointestinal Peptide Research Center of the University of Michigan Grant 2P30DK34933 (L.F.L.-S.), National Institutes of Health Grant R01DK056997 (J.W.), Swiss National Science Foundation Grant 31000-112577 (I.D.), and the Pierre Mercier Science Foundation (I.D.). We thank Heather O’Malley, Audrey Speelman, Anne-Frederique Bourquin, and Temugin Berta for expert technical assistance and Dr. Kathryn E. Rogers for helpful scientific discussions.
References
- Abdulla FA, Smith PA (2001). Axotomy- and autotomy-induced changes in Ca2+ and K+ channel currents of rat dorsal root ganglion neurons. J Neurophysiol 85:644–658. [DOI] [PubMed] [Google Scholar]
- Abdulla FA, Smith PA (2002). Changes in Na(+) channel currents of rat dorsal root ganglion neurons following axotomy and axotomy-induced autotomy. J Neurophysiol 88:2518–2529. [DOI] [PubMed] [Google Scholar]
- Akopian AN, Sivilotti L, Wood JN (1996). A tetrodotoxin-resistant voltage-gated sodium channel expressed by sensory neurons. Nature 379:257–262. [DOI] [PubMed] [Google Scholar]
- Akopian AN, Souslova V, England S, Okuse K, Ogata N, Ure J, Smith A, Kerr BJ, McMahon SB, Boyce S, Hill R, Stanfa LC, Dickenson AH, Wood JN (1999). The tetrodotoxin-resistant sodium channel SNS has a specialized function in pain pathways. Nat Neurosci 2:541–548. [DOI] [PubMed] [Google Scholar]
- Armstrong C, Bezanilla F (1977). Inactivation of the sodium channel. II. Gating current experiments. J Gen Physiol 70:567–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker MD, Wood JN (2001). Involvement of Na+ channels in pain pathways. Trends Pharmacol Sci 22:27–31. [DOI] [PubMed] [Google Scholar]
- Black JA, Dib-Hajj S, McNabola K, Jeste S, Rizzo MA, Kocsis JD, Waxman SG (1996). Spinal sensory neurons express multiple sodium channel alpha-subunit mRNAs. Brain Res Mol Brain Res 43:117–131. [DOI] [PubMed] [Google Scholar]
- Cao YQ, Mantyh PW, Carlson EJ, Gillespie AM, Epstein CJ, Basbaum AI (1998). Primary afferent tachykinins are required to experience moderate to intense pain. Nature 392:390–394. [DOI] [PubMed] [Google Scholar]
- Catterall WA (2000). From ionic currents to molecular mechanisms: the structure and function of voltage-gated sodium channels. Neuron 26:13–25. [DOI] [PubMed] [Google Scholar]
- Chen C, Bharucha B, Chen Y, Westenbroek RE, Brown A, Malhotra JD, Jones D, Avery C, Gillespie PJ III, Kazen-Gillespie KA, Saunders TL, Macdonald RL, Ransom B, Scheuer T, Catterall WA, Isom LL (2002). Reduced sodium channel density, altered voltage dependence of inactivation, and increased susceptibility to seizures in mice lacking sodium channel β2 subunits. Proc Natl Acad Sci USA 99:17072–17077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummins TR, Waxman SG (1997). Downregulation of tetrodotoxin-resistant sodium currents and upregulation of a rapidly repriming tetrodotoxin-sensitive sodium current in small spinal sensory neurons after nerve injury. J Neurosci 17:3503–3514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devor M (2006). Sodium channels and mechanisms of neuropathic pain. J Pain 7:S3–S12. [DOI] [PubMed] [Google Scholar]
- Dib-Hajj S, Black JA, Cummins TR, Waxman SG (2002). NaN/Nav1.9: a sodium channel with unique properties. Trends Neurosci 25:253–259. [DOI] [PubMed] [Google Scholar]
- Drew LJ, Rohrer DK, Price MP, Blaver KE, Cockayne DA, Cesare P, Wood JN (2004). Acid-sensing ion channels ASIC2 and ASIC3 do not contribute to mechanically activated currents in mammalian sensory neurones. J Physiol (Lond) 556:691–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flake NM, Lancaster E, Weinreich D, Gold MS (2004). Absence of an association between axotomy-induced changes in sodium currents and excitability in DRG neurons from the adult rat. Pain 109:471–480. [DOI] [PubMed] [Google Scholar]
- Goldin AL (1993). Accessory subunits and sodium channel inactivation. Curr Opin Neurobiol 3:272–277. [DOI] [PubMed] [Google Scholar]
- Greffrath W, Binzen U, Schwarz ST, Saaler-Reinhardt S, Treede RD (2003). Co-expression of heat sensitive vanilloid receptor subtypes in rat dorsal root ganglion neurons. NeuroReport 14:2251–2255. [DOI] [PubMed] [Google Scholar]
- Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ (1981). Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflügers Arch 391:85–100. [DOI] [PubMed] [Google Scholar]
- Hille B (2001). In: Ion channels of excitable membranes Ed 3 Sunderland, MA: Sinauer Associates.
- Isom LL (2000). Pathobiology of visceral pain: molecular mechanisms and therapeutic implications, I. Cellular and molecular biology of sodium channel β-subunits: therapeutic implications for pain? Am J Physiol Gastrointest Liver Physiol 278:G349–G353. [DOI] [PubMed] [Google Scholar]
- Isom LL, De Jongh KS, Catterall WA (1994). Auxiliary subunits of voltage-gated ion channels. Neuron 12:1183–1194. [DOI] [PubMed] [Google Scholar]
- Isom LL, Scheuer T, Brownstein AB, Ragsdale DS, Murphy BJ, Catterall WA (1995a). Functional co-expression of the β1 and type IIA α subunits of sodium channels in a mammalian cell line. J Biol Chem 270:3306–3312. [DOI] [PubMed] [Google Scholar]
- Isom LL, Ragsdale DS, De Jongh KS, Westenbroek RE, Reber BF, Scheuer T, Catterall WA (1995b). Structure and function of the β2 subunit of brain sodium channels, a transmembrane glycoprotein with a CAM motif. Cell 83:433–442. [DOI] [PubMed] [Google Scholar]
- Kazarinova-Noyes K, Malhotra JD, McEwen DP, Mattei LN, Berglund EO, Ranscht B, Levinson SR, Schachner M, Shrager P, Isom LL, Xiao Z-C (2001). Contactin associates with Na+ channels and increases their functional expression. J Neurosci 21:7517–7525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazen-Gillespie KA, Ragsdale DS, D’Andrea MR, Mattei LN, Rogers KE, Isom LL (2000). Cloning, localization, and functional expression of sodium channel β1A subunits. J Biol Chem 275:1079–1088. [DOI] [PubMed] [Google Scholar]
- Kim SH, Chung JM (1992). An experimental model for peripheral neuropathy produced by segmental spinal nerve ligation in the rat. Pain 50:355–363. [DOI] [PubMed] [Google Scholar]
- Lyu YS, Park SK, Chung K, Chung JM (2000). Low dose of tetrodotoxin reduces neuropathic pain behaviors in an animal model. Brain Res 871:98–103. [DOI] [PubMed] [Google Scholar]
- Malhotra JD, Kazen-Gillespie K, Hortsch M, Isom LL (2000). Sodium channel β subunits mediate homophilic cell adhesion and recruit ankyrin to points of cell-cell contact. J Biol Chem 275:11383–11388. [DOI] [PubMed] [Google Scholar]
- Malhotra JD, Chen C, Rivolta I, Abriel H, Malhotra R, Mattei LN, Brosius FC, Kass RS, Isom LL (2001). Characterization of sodium channel α and β subunits in rat and mouse cardiac myocytes. Circulation 103:1303–1310. [DOI] [PubMed] [Google Scholar]
- Marcil J, Walczak JS, Guindon J, Ngoc AH, Lu S, Beaulieu P (2006). Antinociceptive effects of tetrodotoxin (TTX) in rodents. Br J Anaesth 96:761–768. [DOI] [PubMed] [Google Scholar]
- McEwen DP, Meadows LS, Chen C, Thyagarajan V, Isom LL (2004). Sodium channel β1 subunit-mediated modulation of Nav1.2 currents and cell surface density is dependent on interactions with contactin and ankyrin. J Biol Chem 279:16044–16049. [DOI] [PubMed] [Google Scholar]
- Meadows LS, Isom LL (2005). Sodium channels as macromolecular complexes: implications for inherited arrhythmia syndromes. Cardiovasc Res 67:448–458. [DOI] [PubMed] [Google Scholar]
- Meza U, Avila G, Felix R, Gomora JC, Cota G (1994). Long-term regulation of calcium channels in clonal pituitary cells by epidermal growth factor, insulin, and glucocorticoids. J Gen Physiol 104:1019–1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nassar MA, Stirling LC, Forlani G, Baker MD, Matthews EA, Dickenson AH, Wood JN (2004). Nociceptor-specific gene deletion reveals a major role for Nav1.7 (PN1) in acute and inflammatory pain. Proc Natl Acad Sci USA 101:12706–12711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patapoutian A, Peier AM, Story GM, Viswanath V (2003). ThermoTRP channels and beyond: mechanisms of temperature sensation. Nat Rev Neurosci 4:529–539. [DOI] [PubMed] [Google Scholar]
- Pertin M, Ji RR, Berta T, Powell AJ, Karchewski L, Tate SN, Isom LL, Woolf CJ, Gilliard N, Spahn DR, Decosterd I (2005a). Upregulation of the voltage-gated sodium channel β2 subunit in neuropathic pain models: characterization of expression in injured and non-injured primary sensory neurons. J Neurosci 25:10970–10980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfaffl MW (2001). A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res 29:e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratcliffe CF, Qu Y, McCormick KA, Tibbs VC, Dixon JE, Scheuer T, Catterall WA (2000). A sodium channel signaling complex: modulation by associated receptor protein tyrosine phosphatase β. Nat Neurosci 3:437–444. [DOI] [PubMed] [Google Scholar]
- Ratcliffe CF, Westenbrook RE, Curtis R, Catterall WA (2001). Sodium channel β1 and β3 subunits associate with neurofascin through their extracellular immunoglobulin-like domain. J Cell Biol 154:427–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy ML, Narahashi T (1992). Differential properties of tetrodotoxin-sensitive and tetrodotoxin-resistant sodium channels in rat dorsal root ganglion neurons. J Neurosci 12:2104–2111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rush AM, Brau ME, Elliott AA, Elliott JR (1998). Electrophysiological properties of sodium current subtypes in small cells from adult rat dorsal root ganglia. J Physiol (Lond) 511:771–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rush AM, Dib-Hajj SD, Liu S, Cummins TR, Black JA, Waxman SG (2006). A single sodium channel mutation produces hyper- or hypoexcitability in different types of neurons. Proc Natl Acad Sci USA 103:8245–8250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sangameswaran L, Delgado SG, Fish LM, Koch BD, Jakeman LB, Stewart GR, Sze P, Hunter JC, Eglen RM, Herman RC (1996). Structure and function of a novel voltage-gated, tetrodotoxin-resistant sodium channel specific to sensory neurons. J Biol Chem 271:5953–5956. [DOI] [PubMed] [Google Scholar]
- Schmidt J, Rossie S, Catterall WA (1985). A large intracellular pool of inactive Na+ channel alpha subunits in developing rat brain. Proc Natl Acad Sci USA 82:4847–4851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt JW, Catterall WA (1986). Biosynthesis and processing of the alpha subunit of the voltage-sensitive sodium channel in rat brain neurons. Cell 46:437–445. [DOI] [PubMed] [Google Scholar]
- Schomburg ED, Jankowska E, Wiklund Fernstrom K (2000). Nociceptive input to spinal interneurones in reflex pathways from group II muscle afferents in cats. Neurosci Res 38:447–450. [DOI] [PubMed] [Google Scholar]
- Seltzer Z, Dubner R, Shir Y (1990). A novel behavioral model of neuropathic pain disorders produced in rats by partial sciatic nerve injury. Pain 43:205–218. [DOI] [PubMed] [Google Scholar]
- Shimosato G, Amaya F, Ueda M, Tanaka Y, Decosterd I, Tanaka M (2005). Peripheral inflammation induces up-regulation of TRPV2 expression in rat DRG. Pain 119:225–232. [DOI] [PubMed] [Google Scholar]
- Srinivasan J, Schachner M, Catterall WA (1998). Interaction of voltage-gated sodium channels with the extracellular matrix molecules tenascin-C and tenascin-R. Proc Natl Acad Sci USA 95:15753–15757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasan Y, Elmer L, Davis J, Bennett V, Angelides K (1988). Ankyrin and spectrin associate with voltage-dependent sodium channels in brain. Nature 333:177–180. [DOI] [PubMed] [Google Scholar]
- Study RE, Kral MG (1996). Spontaneous action potential activity in isolated dorsal root ganglion neurons from rats with a painful neuropathy. Pain 65:235–242. [DOI] [PubMed] [Google Scholar]
- Suter MR, Papaloizos M, Berde CB, Woolf CJ, Gilliard N, Spahn DR, Decosterd I (2003). Development of neuropathic pain in the rat spared nerve injury model is not prevented by a peripheral nerve block. Anesthesiology 99:1402–1408. [DOI] [PubMed] [Google Scholar]
- Swett JE, Torigoe Y, Elie VR, Bourassa CM, Miller PG (1991). Sensory neurons of the rat sciatic nerve. Exp Neurol 114:82–103. [DOI] [PubMed] [Google Scholar]
- Tjolsen A, Berge OG, Hunskaar S, Rosland JH, Hole K (1992). The formalin test: an evaluation of the method. Pain 51:5–17. [DOI] [PubMed] [Google Scholar]
- Vandesompele J, De Paepe A, Speleman F (2002). Elimination of primer-dimer artifacts and genomic coamplification using a two-step SYBR green I real-time RT-PCR. Anal Biochem 303:95–98. [DOI] [PubMed] [Google Scholar]
- Vijayaragavan K, O’Leary ME, Chahine M (2001). Gating properties of Nav1.7 and Nav1.8 peripheral nerve sodium channels. J Neurosci 21:7909–7918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vijayaragavan K, Powell AJ, Kinghorn IJ, Chahine M (2004). Role of auxiliary beta1-, beta2-, and beta3-subunits and their interaction with Na(v)1.8 voltage-gated sodium channel. Biochem Biophys Res Commun 319:531–540. [DOI] [PubMed] [Google Scholar]
- Wei F, Wang GD, Kerchner GA, Kim SJ, Xu HM, Chen ZF, Zhuo M (2001). Genetic enhancement of inflammatory pain by forebrain NR2B overexpression. Nat Neurosci 4:164–169. [DOI] [PubMed] [Google Scholar]
- Wilson SG, Mogil JS (2001). Measuring pain in the (knockout) mouse: big challenges in a small mammal. Behav Brain Res 125:65–73. [DOI] [PubMed] [Google Scholar]
- Xiao Z-C, Ragsdale DS, Malhotra JD, Mattei LN, Braun PE, Schachner M, Isom LL (1999). Tenascin-R is a functional modulator of sodium channel β subunits. J Biol Chem 274:26511–26517. [DOI] [PubMed] [Google Scholar]
- Yu FH, Westenbroek RE, Silos-Santiago I, McCormick KA, Lawson D, Ge P, Ferriera H, Lilly J, DiStefano PS, Catterall WA, Scheuer T, Curtis R (2003). Sodium channel β4, a new disulfide-linked auxiliary subunit with similarity to β2. J Neurosci 23:7577–7585. [DOI] [PMC free article] [PubMed] [Google Scholar]