

Abstract
RNA interference (RNAi), which allows selective gene silencing, has been proposed for functional genomic analysis and for the treatment of human disease. However, induction of RNAi in mammalian cells by expression of double-stranded RNA can activate innate antiviral response pathways that perturb off-target gene expression. The activation and functional consequences of these effects in neurons are unknown. We find that expression of subsets of short hairpin RNAs (shRNAs) in rat hippocampal pyramidal neurons can have off-target effects that reduce the complexity of dendritic arbors and trigger the loss of dendritic spines. Morphological changes are accompanied by electrophysiological perturbations in passive membrane properties and a decrease in the number and strength of excitatory and inhibitory synapses. These perturbations depend on the shRNA sequence and are independent of the identity of the targeted protein. Our results indicate that off-target effects of RNAi severely perturb neuronal structure and function and may lead to the functional withdrawal of affected cells from the brain circuitry.
Keywords: RNA interference, short hairpin RNA, interferon response, synaptic transmission, dendritic spine, off-target effects
Introduction
RNA interference (RNAi) is an evolutionarily conserved process of sequence-specific post-transcriptional gene silencing that is triggered by double-stranded RNA (dsRNA) (Fire et al., 1998; Hannon, 2002). In plants, Caenorhabditis elegans, and Drosophila, RNAi is specifically and efficiently induced by expression of long dsRNA with sequence homology to the targeted mRNA. In mammalian cells, similar strategies trigger the interferon response pathway, which is thought to be part of the cellular viral defense mechanism (Samuel, 2001). To avoid these “off-target” effects, short interfering RNAs (∼20 bp), or short palindromic dsRNAs that form hairpins (shRNAs), are used to silence specific mRNAs in mammalian cells (Elbashir et al., 2001). However, even these short forms of dsRNA can activate an innate cellular immunity response (Bridge et al., 2003; Sledz et al., 2003; Sledz and Williams, 2004) and trigger reactive effects, including alterations in gene transcription and protein translation (Persengiev et al., 2004).
Specific knockdown of synaptic proteins in mammalian neurons poses additional challenges. Because many synaptic proteins have long half-lives and are present in large intracellular reserve pools, their effective knockdown requires maintained and robust RNAi induction (Sui et al., 2002). Furthermore, endogenous microRNAs regulate neurite outgrowth (Vo et al., 2005) and dendritic spine morphology (Schratt et al., 2006). Thus, activation of RNAi could compete for endogenous cellular machinery and indirectly alter synapse structure and function.
Here we examine the off-target effects of shRNA expression in hippocampal pyramidal neurons in rat organotypic slice cultures. We find that expression of shRNA against luciferase, whose coding region is not found in the rat genome, triggers dramatic loss of dendritic spines and simplification of dendritic arbors. The effects are rapid, require signaling through dsRNA-dependent protein kinase (PKR), and result in activation of interferon target genes. In addition, the morphological changes are accompanied by a loss of excitatory and inhibitory synapses. Induction of RNAi against endogenous proteins can lead to similar effects in a manner that depends on the shRNA sequence and not the targeted transcript. Our results demonstrate the complexity of interpreting synaptic changes after induction of RNAi. They highlight the need to use a combination of control experiments that should include rescue of RNAi-mediated effects by reintroducing the targeted protein (Chih et al., 2005) and, when possible, a combination of RNAi knockdown and other approaches (Hering and Sheng, 2003; Dunah et al., 2005).
Materials and Methods
All experimental protocols were conducted according to National Institutes of Health and Harvard Medical School guidelines.
Vectors.
Expression plasmids for shRNA were generated from a dual-promoter CMV-EGFP/U6-shRNA vector (Tavazoie et al., 2005). The following sequences were used to make shRNAs vectors (vector name, target, sequence, reference): shLUCI, firefly Photinus pyralis luciferase, CGTACGAATACTTCGATTgagctcAATCGAAGTATTCCGCGTACGcttttt (Elbashir et al., 2001; Persengiev et al., 2004); shMORA, MORF4L1, GTAAAGATTCCTGAAGAGCgagctcGCTCTTCAGGAATCTTTAC-ctttttg and shMORB, MORF4L1, GATGGTGGCAGTACCAGTGgagctcCACTGGTACTGCCACCATCctttttg (Bridge et al., 2003); shEGFP, EGFP, GGCGATGCCACCTACGGCAAGctcgagCTTGCCGTAGG-TGGCATCGCCctttttg (Sui et al., 2002). The luciferase reporter vector consisted of pGL3 with μMx2 promoter (Asano et al., 2003). All enzymes were purchased from New England Biolabs (Beverly, MA).
Hippocampal slice cultures.
Slice cultures were prepared as described previously (Stoppini et al., 1991) from postnatal day 7 Sprague Dawley pups (Charles River Laboratories, Wilmington, MA) (Tavazoie et al., 2005). Biolistic gene transfer (Lo et al., 1994) was used for transfection at 3 d in vitro (DIV). Pyramidal neurons expressing enhanced green fluorescent protein (EGFP) were identified by their characteristic morphology and were imaged 4, 7, or 14 d after transfection. The amount of EGFP-expressing vector (either control or shRNA vector) was kept constant (60 μg of DNA/12.5 mg of gold of 1.6 μm diameter) to ensure similar fluorescence intensity.
Electrophysiology.
Hippocampal slice cultures were superfused at room temperature with artificial CSF (ACSF) containing the following (in mm): 127 NaCl, 25 NaHCO3, 1.25 Na2HPO4, 2.5 KCl, 2 CaCl2, 1 MgCl2, 25 glucose, saturated with 95% O2/5% CO2. Whole-cell voltage-clamp recordings were performed from transfected pyramidal neurons (green fluorescence and visible gold particle in the soma). Pipettes (3–5 MΩ) contained two different internal solutions that yielded similar mean EPSC frequencies and amplitudes. Solution A contained the following (in mm): 100 CsCl, 35 CsF, 10 HEPES, 10 EGTA, pH 7.3 (290 mOsm). Solution B contained the following (in mm): 120 cesium methane-sulfonate, 10 HEPES, 10 EGTA, 4 MgCl2, 0.4 NaGTP, 4 MgATP, 10 phosphocreatine, pH 7.3 (290 mOsm). Alexa594 (20 μm; Invitrogen, Eugene, OR) was added to the internal solution to fill the cell and confirm its identity. Miniature EPSCs (mEPSCs) were recorded for 250–500 s using an Axopatch 200B amplifier (Molecular Devices, Union City, CA), filtered at 2 kHz, and digitized at 10 kHz. Series resistance (8–19 MΩ), input resistance, and membrane capacitance were calculated from a −5 mV step. Recording was discontinued if series resistance increased >20 MΩ. mEPSC frequency and amplitude were calculated in Igor Pro (WaveMetrics, Lake Oswego, OR) by identifying events based on derivative and amplitude threshold. All chemicals were obtained from Sigma (St. Louis, MO) or Tocris (Ellisville, MO).
Imaging and morphological analyses.
Image collection and analysis were performed as described previously (Tavazoie et al., 2005) using a custom two-photon laser-scanning microscope and software. Slices were superfused with ACSF, and transfected pyramidal neurons were identified based on green fluorescence and characteristic morphology. For spine analyses, image stacks consisting of 10–32 sections (512 × 512 pixels, 64 × 64 μm) spaced 1 μm apart were collected from the apical dendritic tree. Three to five stacks were taken from each neuron at this magnification plus one image stack at a lower magnification (field, 280 μm) for the analyses of branching and dendritic length. Morphological analysis was performed using custom software written in MatLab 6.5 (MathWorks, Natick, MA).
Luciferase assay.
Dissociated hippocampal neurons were prepared from embryonic day 19 Sprague Dawley rats as described by Banker and Goslin (1998) with some modifications: hippocampi were digested in ∼7.5 mg of papain (Worthington Biochemical, Lakewood, NJ) with traces of l-cysteine in 5 ml of dissociation media (in mm: 10 kyneurinic acid, 100 MgCl2, 100 HEPES in HBSS), and cells were plated directly onto previously coated (poly-d-Lysine, 1 mg/ml), tissue culture-treated, plastic 24-well plates at 50,000–60,000 cells/well in plating media. The plating medium contained MEM (catalog #11095-080), 1 mm pyruvic acid (Sigma), 33.2 mm glucose (Sigma), and 10% horse serum. The medium was replaced with growth medium (2% B27 supplement, 2 mm l-glutamine, 50 U/ml penicillin, 50 μg/ml streptomycin in Neurobasal media) 4 h later, and one-third of its volume was replaced twice per week. After 5–6 DIV, cells were cotransfected (Lipofectamine-2000) with three vectors: shRNA or control EGFP, reporter vector (mx2::firefly luciferase), and normalizing vector (CMV::Renilla luciferase) at 0.45, 1.35, and 0.15 μg per well, respectively. After 24 h, cells were lysated and luciferase activity was measured using the Dual-Luciferase Reporter system (Promega, Madison, WI). Each condition was run in triplicate, and experiments were repeated three to four times. All materials for tissue culture were purchased from Invitrogen (Carlsbad, CA) unless noted.
Statistics.
Data are expressed as mean ± SEM. Statistical differences were determined by the Kolmogorov–Smirnov test when analyzing cumulative distributions and by an unpaired t test or ANOVA when comparing mean values.
Results
We examined the off-target effects of RNAi activation in postmitotic mammalian neurons by expressing an shRNA with sequence homology to nucleotides 153–173 of the coding region of P. pyralis luciferase (shLUCI), a region with no homology in the mammalian genome. shRNA with this sequence efficiently silences exogenous luciferase in mammalian cells (Elbashir et al., 2001) and activates an interferon-like response in HeLa S3 cells (Persengiev et al., 2004). shLUCI expression was driven by the U6 RNA polymerase III promoter in a dual promoter vector that also contains cytomegalovirus (CMV)-driven EGFP.
Retraction of dendrites and spines by shRNA targeting luciferase
Pyramidal neurons in organotypic hippocampal brain slice cultures transfected with control EGFP or shLUCI were identified by their characteristic morphology (Fig. 1). Dendrites of control neurons transfected with EGFP were highly branched (37 ± 3.2 dendritic segments; n = 12), whereas those transfected with shLUCI displayed reduced dendritic complexity (28 ± 2.6 segments; n = 11; p < 0.05) (Fig. 1A,C). No difference was observed in the mean length of the dendritic segments (control, 45 ± 1.4 μm; shLUCI, 42 ± 1.6 μm; n = 444–307 segments). shLUCI-expressing neurons also had reduced dendritic spine density at 4 d post-transfection (DPT) compared with controls (control: 0.69 ± 0.04 μm−1, n = 14; shLUCI: 0.37 ± 0.03 μm−1, n = 16; 47 ± 4% reduction; p < 0.01) (Fig. 1D). Spine loss progressed with continued shLUCI expression, reaching 54 ± 6 and 88 ± 5% reductions in spine density at 7 DPT (n = 13) and 14 DPT (n = 7), respectively (Fig. 1F). During this period, spine density in control neurons increased, indicating that shLUCI expression triggered the retraction of existing spines and suppressed the formation of new spines. Spine loss was accompanied by slight shortening of dendritic spines from 0.91 ± 0.01 μm in controls to 0.82 ± 0.01 μm in neurons expressing shLUCI at 4 DPT (n = 2107 and 1582; p < 0.05) (Fig. 1E). The shortening of dendritic spines was observed at every time point analyzed, indicating a persistent change in spine morphology (Fig. 1G). Reducing the concentration of shLUCI by ∼60% also resulted in loss of dendritic spines (0.28 ± 0.04 μm−1 at 7 DPT; n = 9), indicating that even the minimal amount of vector required for efficient transfection triggers off-target effects.
Figure 1.
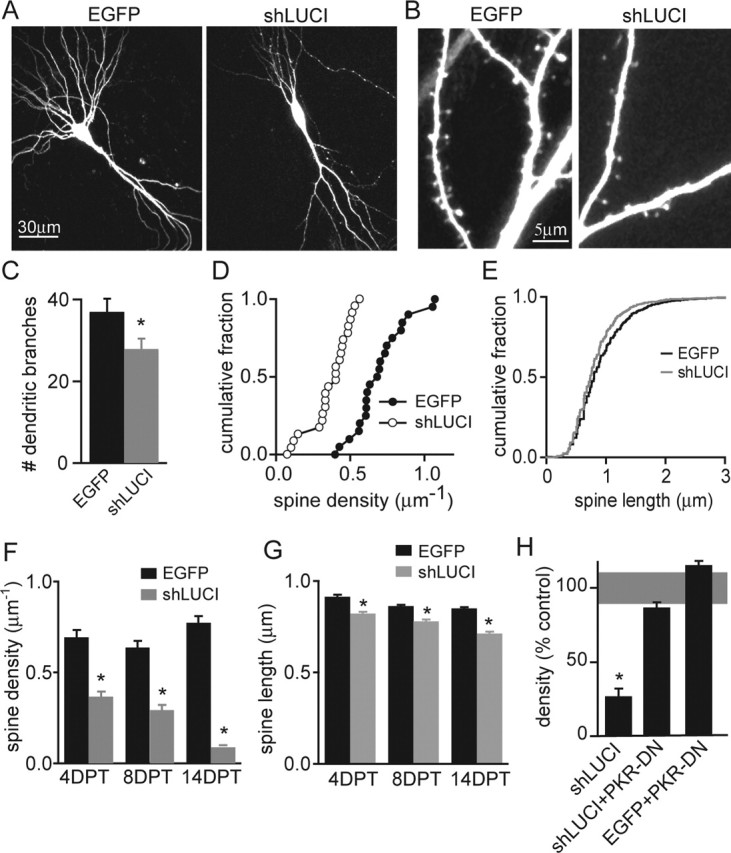
shRNA expression induces retraction of dendrites and spines. A, Hippocampal pyramidal neurons in organotypic slice cultures transfected with control EGFP vector (left) or shLUCI (right) imaged at 7 DPT. B, Representative images of apical dendrites from neurons expressing EGFP (left) or shLUCI (right) vectors. C, Average number of dendritic branches for EGFP (black) and shLUCI (gray) neurons at 7 DPT. D, Cumulative distribution of spine density at 4 DPT for neurons transfected with EGFP (filled circles) and shLUCI (open circles) (n = 14–16). E, Cumulative distribution of lengths of apical dendritic spines at 4 DPT for neurons transfected with EGFP (black) and shLUCI (gray) (n = 2107 and 1582 spines, respectively). F, Summary data of average spine densities at 4, 7, and 14 DPT for EGFP (black bars) and shLUCI (gray bars) neurons (n = 7 and 16). G, Summary data of average spine length at 4, 7, and 14 DPT for EGFP (black bars) and shLUCI (gray bars) neurons (n = 7 and 16). H, Spine density relative to EGFP controls for neurons expressing shLUCI or coexpressing a dominant-negative mutant of PKR (PKR-DN) with shLUCI or EGFP at 7 DPT (n = 8–15). The shaded gray region shows 2 × SEM for control spine density. ∗p < 0.05 compared with control.
Small interfering RNAs (siRNAs) can activate PKR in other cell types (Bridge et al., 2003; Sledz et al., 2003). We tested whether the morphological perturbations induced by shLUCI required signaling through PKR by coexpression of shLUCI and a dominant-negative mutant of PKR (PKR-DN) (Takizawa et al., 2002). Expression of PKR-DN alone had little effect on spine density (115 ± 3%; n = 4) but prevented the spine loss when cotransfected with shLUCI (87 ± 3%; n = 5) (Fig. 1H).
Off-target actions on synaptic transmission
In hippocampal pyramidal neurons, each dendritic spine typically houses the postsynaptic components of a single glutamatergic synapse. Thus, decreased spine density is likely to reflect a loss of synapses. To assay for electrophysiological changes, whole-cell voltage-clamp recordings were made from shLUCI-transfected neurons and their untransfected neighbors in organotypic slices at 7 DPT. Cell capacitance of shLUCI-transfected neurons was reduced by ∼50% compared with controls (control: 186 ± 12 pF, n = 22; shLUCI: 95 ± 9 pF n = 27; p < 0.01), and the input resistance was increased by ∼30% (control, 201 ± 24 MΩ; shLUCI, 294 ± 29 MΩ; p < 0.05). These changes are consistent with a loss of membrane area. To monitor the number and strength of synapses, recordings were made of spontaneous mEPSCs in the presence of 1 μm TTX and 20 μm bicuculline to block voltage-gated sodium channels and inhibitory GABAA receptors, respectively. The frequency of mEPSCs, which reflects the number of excitatory synapses made onto the neuron, was 0.38 ± 0.09 Hz in control neurons and 0.14 ± 0.04 Hz in shLUCI neurons (n = 12–15; p < 0.01) (Fig. 2C), indicating a loss of functional synapses. Furthermore, the amplitude of mEPSCs was decreased in shLUCI-expressing neurons to 11.7 ± 1.3 pA from 18.3 ± 1.2 pA in controls (n = 11–13; p < 0.01) (Fig. 2C).
Figure 2.
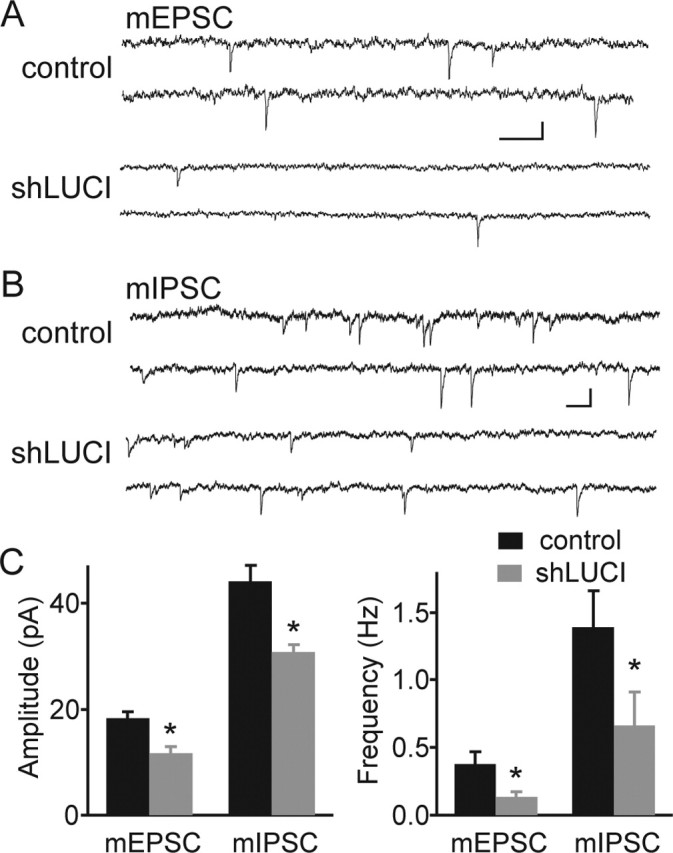
Frequency and amplitude of mEPSCs and mIPSCs are reduced by shLUCI. A, B, Representative traces from control (top) and shLUCI-transfected (bottom) pyramidal neurons displaying mEPSCs (A) and mIPSCs (B). Calibration: A, 10 pA and 500 ms; B, 20 pA and 200 ms. C, Summary of the amplitude (left) and frequency (right) of mEPSCs and mIPSCs in control (black bars) and shLUCI-expressing (gray bars) neurons (n = 11–15). ∗p < 0.05 compared with control.
To determine whether the loss of synaptic inputs to shRNA neurons was specific to excitatory synapses, inhibitory miniature synaptic currents (mIPSCs) were measured. For these experiments, bicuculline was replaced by antagonists of ionotropic glutamate receptors (20 μm NBQX and 50 μm APV). The frequency and the amplitude of mIPSCs were decreased in neurons expressing shLUCI (control: 1.4 ± 0.3 Hz and 44 ± 3 pA, n = 15; shLUCI: 0.66 ± 0.25 Hz and 31 ± 1 pA, n = 14; p < 0.05) (Fig. 2C).
Sequence specificity of the shRNA actions
To understand whether the observed changes are specific to shLUCI or whether they represent a general response to shRNA, we examined the effects on neuronal morphology of two sequences targeting MORF4L1, a protein involved in chromatin regulation (Bertram et al., 1999; Yang, 2004). Previous studies identified shRNA sequences that are equally effective at reducing MORF4L1 levels, but differentially induce an interferon-responsive gene in fibroblasts (Bridge et al., 2003): shMORA activates the interferon-like response, whereas shMORB does not. At 7 DPT, neurons expressing shMORA displayed reduced dendritic complexity, whereas neurons expressing shMORB had normal numbers of dendritic branches (control: 37 ± 3.2, n = 12; shMORA: 23 ± 3, n = 10; shMORB: 34 ± 2.6, n = 10) (Fig. 3B). This indicates that retraction of dendrites depends on the nucleotide sequence of the shRNA and is independent of the targeted mRNA. Analysis of the spine density showed that neurons expressing shMORA had reduced density of dendritic spines (0.1 ± 0.02 μm−1; n = 9; p < 0.01) compared with EGFP control neurons (0.63 ± 0.04 μm−1; n = 11) and that expression of shMORB had no effect on spine density (0.51 ± 0.03 μm−1; n = 7), supporting that loss of MORF4L1 is not responsible for the morphological perturbations induced by shMORA (Fig. 3B, middle).
Figure 3.
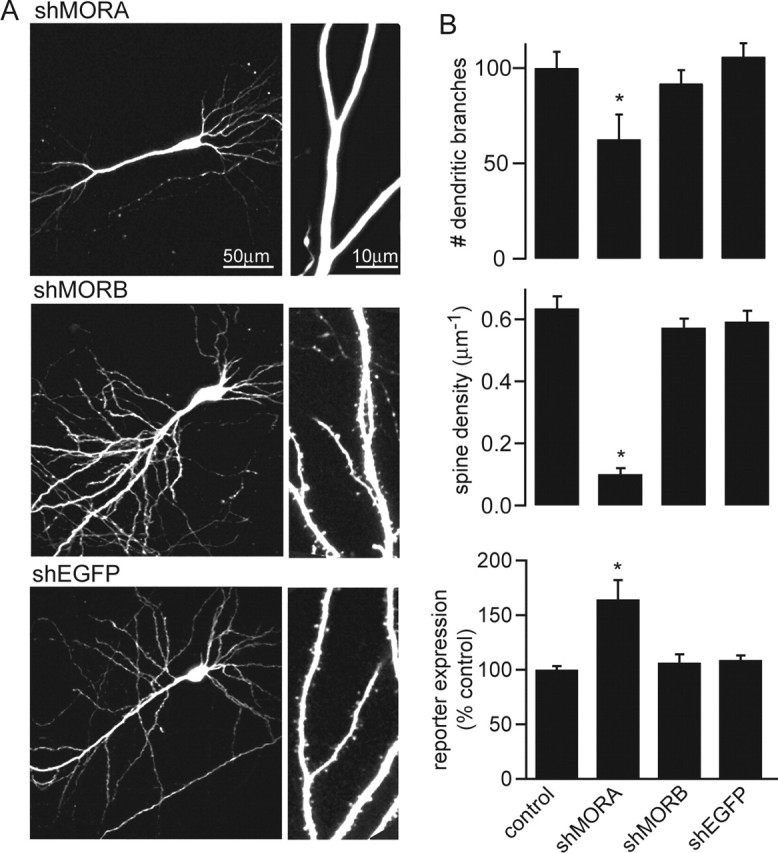
Sequence dependence of the morphological perturbations and the activation of an interferon response. A, Representative whole-cell (left) and apical dendrite (right) images of neurons transfected with shMORA, shMORB, and shEGFP at 7 DPT. B, Top, Summary of the average number of dendritic branches in neurons transfected with the indicated plasmids at 7 DPT (n = 5–12). Middle, Average spine density of neurons transfected with the indicated plasmids and imaged at 7 DPT. Bottom, Relative levels of luciferase activity in dissociated hippocampal neurons at 7 DIV and 24 h after cotransfection with the interferon-inducible reporter (mx2::luciferase) and EGFP (control), shMORA, shMORB, or shEGFP (n = 3–4 independent repetitions). ∗p < 0.05 compared with control.
To further confirm that loss of dendritic spines is not always triggered by robust activation of RNAi, we transfected neurons with EGFP and an shRNA that efficiently targets EGFP (shEGFP) (Shi, 2003). To ensure accurate morphometric analysis despite the reduced green fluorescence, neurons were cotransfected with monomeric red fluorescent protein (mRFP), and red fluorescence was used in the morphological analysis. Expression of shEGFP did not change dendrite complexity (28 ± 2; n = 5) or spine density (0.59 ± 0.03 μm−1; n = 5) compared with control mRFP neurons (26 ± 2.6 and 0.59 ± 0.07 μm−1, n = 7 for branches and spine density, respectively) (Fig. 3B).
Last, we examined whether the sequence dependence of shRNA-triggered spine loss correlates with differential induction of the interferon response pathway. Activation of PKR in response to dsRNA upregulates the transcription of interferon response genes, most of which are mediators of antiviral, antiproliferative activity (Williams, 1999). Mx2 is an interferon-inducible gene that mediates resistance to negative-strand viruses (Asano et al., 2003). A plasmid encoding firefly luciferase under the control of the mx2 promoter (mx2::firefly luciferase) was used to report activation of the interferon response pathway. Dissociated cultured neurons were transfected with mx2::firefly luciferase, CMV::Renilla luciferase, and EGFP, shMORA, shMORB, or shEGFP. The activity of mx2-driven firefly luciferase was measured by chemiluminescence at 24 h and normalized to the activity of CMV-driven Renilla luciferase. shMORA, but not shMORB or shEGFP, activated expression of the reporter relative to EGFP (Fig. 3B, bottom), suggesting that sequence-dependent activation of interferon target genes may underlie the structural and functional perturbations observed in neurons.
Discussion
The activity-dependent formation and regulation of synapses is thought to be the cellular mechanism that enables animals to form memories and learn new behaviors. For this reason, uncovering the signaling cascades that mediate synaptic plasticity is a central goal within the neuroscience community. RNAi is particularly useful in this regard because it allows the low-cost and rapid knockdown of endogenous proteins in postmitotic, differentiated neurons. Furthermore, the use of RNAi circumvents many of the difficulties in interpreting the mechanism of synaptic defects found in knock-out animals that globally lack a protein throughout development. Last, RNAi can be induced sparsely in culture systems and in vivo, allowing for the unambiguous identification of the cell-autonomous role of a protein in regulating synapse structure and function.
Our study shows that expression of shRNAs can trigger severe morphological and functional perturbations in neurons. These include a fast and progressive loss of dendrites, spines, and synapses. These effects are off-target actions of the shRNA because they can be induced by shRNA targeting an exogenous protein without homology to endogenous genes. Our experiments also indicate that activation of the RNAi machinery is neither required nor sufficient for these perturbations because they were caused by shLUCI (which probably does not induce RNAi) and were absent when targeting EGFP, an exogenous protein that was being coexpressed from the vector. Furthermore, only one of two shRNAs known to knockdown the endogenous protein MORF4L1 triggered similar spine and dendrite retraction, indicating that the perturbations are independent of the targeted protein and dependent on the sequence of the shRNA.
Neurons affected by shRNA off-target actions also showed changes in passive membrane properties, including reduced membrane capacitance and increased input resistance. These changes are possibly caused by a reduction of total membrane area that came as a result of loss of dendrite branches and spines. However, despite the increased input resistance that could have occluded a small decrease in the amplitude and frequency of miniature events, we reliably detected reductions on the number and amplitude of mIPSCs and mEPSCs in neurons expressing shLUCI for 1 week. The weakening and loss of excitatory and inhibitory inputs suggests that affected neurons may become postsynaptically silenced and isolated from the remainder of the network.
Spine and dendrite retraction induced by shLUCI required activation of PKR, a member of the cellular antiviral response pathway. Furthermore, the ability of shRNAs to trigger dendrite and spine retraction correlated with activation of an interferon-inducible gene. These observations are in agreement with previous reports that shRNA (Bridge et al., 2003) can activate the interferon response pathway in mammalian cells (de Veer et al., 2005). It is unknown whether activation of the interferon pathway directly mediates the off-target effects of shRNA or is simply an indicator of their presence.
Off-targeting is not an exclusive property of vector-driven RNAi because synthetic siRNAs also potently induce these effects (Sledz et al., 2003; Judge et al., 2005). One potential advantage of using synthetic siRNAs is that chemical modifications, such as incorporation of 2′-O-methyl uridine or guanosine nucleosides into one strand of the siRNA duplex, which have been suggested to diminish the interferon response, can be performed easily (Judge et al., 2006). However, experiments in neurons will be required to directly demonstrate that modified siRNAs diminish off-target actions while retaining the efficiency of silencing of nonmodified siRNAs. Furthermore, synthetic siRNAs are difficult to use in experiments requiring long-lasting silencing of a gene.
The activation of other pathways, in addition to the interferon response pathway, have been associated with the off-target effects of siRNA and shRNA (Persengiev et al., 2004). Furthermore, the off-target actions of several synthetic siRNA have been associated with the presence of homology between the siRNA and the 3′ untranslated region (UTR) of nontargeted genes (Birmingham et al., 2006). Because of the multiple mechanisms by which a siRNA or shRNA can trigger off-target effects, it is often difficult to separate off-target effects from those attributable to loss of the targeted protein. Therefore, we suggest the following guideline in interpreting the results of RNAi-based experiments. First, given that the activation of off-target effects is dependent on the sequence of the shRNA or siRNA, the use of scrambled sequences as a specificity control is insufficient. Second, the interferon response reporter assay described here might provide a generally useful method for rapidly screening shRNAs for activation of at least a subset of off-target effects in neurons. Third, the Smith-Waterman local alignment algorithm can be used to screen for homology between the hexamer or heptamer seed (position 2-7 or 2-8 of the antisense strand) and the 3′UTR of genes and might help prevent off-targeting by shRNA (Birmingham et al., 2006). Last, demonstration of rescue of phenotypic changes by reintroduction of the targeted protein is necessary to conclusively establish that the observed changes result from loss of the targeted protein. Rescue experiments generally require transfection of the shRNA-expressing neuron with a cDNA that encodes the targeted protein but carries silent mutations that render the mRNA insensitive to the shRNA. Moreover, whenever possible, and especially if a rescue experiment cannot be performed, RNAi-based results should be confirmed with the use of other loss-of-function approaches such as dominant-negative mutants or truncations of the targeted protein. In summary, when using RNAi, a combination of controls is required to unambiguously conclude that the loss of the targeted protein is responsible for perturbations of synapse function or structure.
Footnotes
This work was supported by a Searle Scholar Award (B.L.S.) and by National Institutes of Health Training Grant 5T32 NS07484 (V.A.A.). We thank Rochelle Witt and Darko Knutti for helpful technical advice, Adam Carter for the mini analysis algorithms, and the members of the Sabatini laboratory for comments on this manuscript. We thank Dr. Atsushi Asano (mx2 reporter), Darko Knutti (CMV-Renilla luciferase), and Dr. Takenori Takizawa (PKR-K296R) for the gift of plasmids.
References
- Asano A, Jin HK, Watanabe T (2003). Mouse Mx2 gene: organization, mRNA expression and the role of the interferon-response promoter in its regulation. Gene 306:105–113. [DOI] [PubMed] [Google Scholar]
- Banker G, Goslin K (1998). In: Culturing nerve cells Ed 2 Cambridge, MA: MIT.
- Bertram MJ, Berube NG, Hang-Swanson X, Ran Q, Leung JK, Bryce S, Spurgers K, Bick RJ, Baldini A, Ning Y, Clark LJ, Parkinson EK, Barrett JC, Smith JR, Pereira-Smith OM (1999). Identification of a gene that reverses the immortal phenotype of a subset of cells and is a member of a novel family of transcription factor-like genes. Mol Cell Biol 19:1479–1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birmingham A, Anderson EM, Reynolds A, Ilsley-Tyree D, Leake D, Fedorov Y, Baskerville S, Maksimova E, Robinson K, Karpilow J, Marshall WS, Khvorova A (2006). 3′ UTR seed matches, but not overall identity, are associated with RNAi off-targets. Nat Methods 3:199–204. [DOI] [PubMed] [Google Scholar]
- Bridge AJ, Pebernard S, Ducraux A, Nicoulaz AL, Iggo R (2003). Induction of an interferon response by RNAi vectors in mammalian cells. Nat Genet 34:263–264. [DOI] [PubMed] [Google Scholar]
- Chih B, Engelman H, Scheiffele P (2005). Control of excitatory and inhibitory synapse formation by neuroligins. Science 307:1324–1328. [DOI] [PubMed] [Google Scholar]
- de Veer MJ, Sledz CA, Williams BR (2005). Detection of foreign RNA: implications for RNAi. Immunol Cell Biol 83:224–228. [DOI] [PubMed] [Google Scholar]
- Dunah AW, Hueske E, Wyszynski M, Hoogenraad CC, Jaworski J, Pak DT, Simonetta A, Liu G, Sheng M (2005). LAR receptor protein tyrosine phosphatases in the development and maintenance of excitatory synapses. Nat Neurosci 8:458–467. [DOI] [PubMed] [Google Scholar]
- Elbashir SM, Harborth J, Lendeckel W, Yalcin A, Weber K, Tuschl T (2001). Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature 411:494–498. [DOI] [PubMed] [Google Scholar]
- Fire A, Xu S, Montgomery MK, Kostas SA, Driver SE, Mello CC (1998). Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans Nature 391:806–811. [DOI] [PubMed] [Google Scholar]
- Hannon GJ (2002). RNA interference. Nature 418:244–251. [DOI] [PubMed] [Google Scholar]
- Hering H, Sheng M (2003). Activity-dependent redistribution and essential role of cortactin in dendritic spine morphogenesis. J Neurosci 23:11759–11769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Judge AD, Sood V, Shaw JR, Fang D, McClintock K, MacLachlan I (2005). Sequence-dependent stimulation of the mammalian innate immune response by synthetic siRNA. Nat Biotechnol 23:457–462. [DOI] [PubMed] [Google Scholar]
- Judge AD, Bola G, Lee AC, MacLachlan I (2006). Design of noninflammatory synthetic siRNA mediating potent gene silencing in vivo Mol Ther 13:494–505. [DOI] [PubMed] [Google Scholar]
- Lo DC, McAllister AK, Katz LC (1994). Neuronal transfection in brain slices using particle-mediated gene transfer. Neuron 13:1263–1268. [DOI] [PubMed] [Google Scholar]
- Persengiev SP, Zhu X, Green MR (2004). Nonspecific, concentration-dependent stimulation and repression of mammalian gene expression by small interfering RNAs (siRNAs). RNA 10:12–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuel CE (2001). Antiviral actions of interferons. Clin Microbiol Rev 14:778–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schratt GM, Tuebing F, Nigh EA, Kane CG, Sabatini ME, Kiebler M, Greenberg ME (2006). A brain-specific microRNA regulates dendritic spine development. Nature 439:283–289. [DOI] [PubMed] [Google Scholar]
- Shi Y (2003). Mammalian RNAi for the masses. Trends Genet 19:9–12. [DOI] [PubMed] [Google Scholar]
- Sledz CA, Williams BR (2004). RNA interference and double-stranded-RNA-activated pathways. Biochem Soc Trans 32:952–956. [DOI] [PubMed] [Google Scholar]
- Sledz CA, Holko M, de Veer MJ, Silverman RH, Williams BR (2003). Activation of the interferon system by short-interfering RNAs. Nat Cell Biol 5:834–839. [DOI] [PubMed] [Google Scholar]
- Stoppini L, Buchs PA, Muller D (1991). A simple method for organotypic cultures of nervous tissue. J Neurosci Methods 37:173–182. [DOI] [PubMed] [Google Scholar]
- Sui G, Soohoo C, Affar el B, Gay F, Shi Y, Forrester WC, Shi Y (2002). A DNA vector-based RNAi technology to suppress gene expression in mammalian cells. Proc Natl Acad Sci USA 99:5515–5520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takizawa T, Tatematsu C, Nakanishi Y (2002). Double-stranded RNA-activated protein kinase interacts with apoptosis signal-regulating kinase 1. Implications for apoptosis signaling pathways. Eur J Biochem 269:6126–6132. [DOI] [PubMed] [Google Scholar]
- Tavazoie SF, Alvarez VA, Ridenour DA, Kwiatkowski DJ, Sabatini BL (2005). Regulation of neuronal morphology and function by the tumor suppressors Tsc1 and Tsc2. Nat Neurosci 8:1727–1734. [DOI] [PubMed] [Google Scholar]
- Vo N, Klein ME, Varlamova O, Keller DM, Yamamoto T, Goodman RH, Impey S (2005). A cAMP-response element binding protein-induced microRNA regulates neuronal morphogenesis. Proc Natl Acad Sci USA 102:16426–16431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams BR (1999). PKR: a sentinel kinase for cellular stress. Oncogene 18:6112–6120. [DOI] [PubMed] [Google Scholar]
- Yang XJ (2004). The diverse superfamily of lysine acetyltransferases and their roles in leukemia and other diseases. Nucleic Acids Res 32:959–976. [DOI] [PMC free article] [PubMed] [Google Scholar]