

Abstract
M-type (Kv7) potassium channels are closed by Gq/11 G-protein-coupled receptors. Several membrane- or channel-associated molecules have been suggested to contribute to this effect, including depletion of phosphatidylinositol-4,5-bisphosphate (PIP2) and activation of Ca2+/calmodulin and protein kinase C. To facilitate further study of these pathways in intact neurons, we have devised novel membrane-targeted probes that can be applied from the outside of the neuron, by attaching a palmitoyl group to site-directed peptides (“palpeptides”) (cf. Covic et al., 2002a,b). A palpeptide incorporating the 10-residue C terminus of Gαq/11 reduced Gq/11-mediated M-current inhibition in sympathetic neurons by the muscarinic acetylcholine receptor (mAChR) agonist oxotremorine-M but not Go-mediated inhibition of the N-type Ca2+ current by norepinephrine. Instead, the latter was inhibited by the corresponding Go palpeptide. A PIP2 palpeptide, based on the putative PIP2 binding domain of the Kv7.2 channel, inhibited M current (IC50 = ∼1.5 μm) and enhanced inhibition by oxotremorine-M. Inhibition could not be attributed to activation of mAChRs, calcium influx, or block of M channels but was antagonized by intracellular diC8-PIP2 (dioctanoyl-phosphatidylinositol-4,5-bisphosphate), suggesting that it disrupted PIP2–M channel gating. A fluorescently tagged PIP2 palpeptide was highly targeted to the plasma membrane but did not accumulate in the cytoplasm. We suggest that these palpeptides are anchored in the plasma membrane via the palmitoyl group, such that the peptide moiety can interact with target molecules on the inner face of the membrane. The G-protein-replicating palpeptides were sequence specific and probably compete with the receptor for the cognate G-protein. The PIP2 palpeptide was not sequence specific so probably interacts electrostatically with anionic PIP2 head groups.
Keywords: M current; Kv7 channels; KCNQ; muscarinic acetylcholine receptor; palmitoylated peptides; phosphatidylinositol-4,5-bisphosphate; superior cervical ganglion cells; G-protein
Introduction
Potassium M channels are composed of subunits of the Kv7 family, principally Kv7.2 and Kv7.3 (Wang et al., 1998). They assist in regulating the excitability of a variety of neurons (Brown, 1988; Marrion, 1997). M channels can be closed by neurotransmitters and hormones that couple (primarily) to the Gq/G11 family of G-proteins (Hille, 1994; Haley et al., 1998), with a consequential increase in neuronal excitability. A multiplicity of “second-messenger” systems has been suggested to contribute to this inhibition. These include the following: depletion of membrane phosphatidylinositol-4,5-bisphosphate (PIP2) (Suh and Hille, 2002; Ford et al., 2003; Zhang et al., 2003; Suh et al., 2004; Horowitz et al., 2005; Winks et al., 2005); Ca2+ ions (Selyanko and Brown, 1996; Cruzblanca et al., 1998), acting via channel-associated calmodulin (Gamper and Shapiro, 2003); and activation of protein kinase C (Hoshi et al., 2003). These regulators are all thought to act on the proximal (cytoplasmic) C terminus of the channel protein and hence may show complex interactions (Delmas and Brown, 2005).
This poses practical problems in evaluating the relative importance of these (and other) regulatory mechanisms to the physiological responses to different transmitters or hormones. Thus, any drug or other probe that is designed to affect the signaling pathways has to gain access to the intramembrane or intracellular juxtamembrane targets. One common method is to apply these to the cell interior via a patch pipette. However, this is relatively slow, usually irreversible, and, in many neurons, is complicated by the fact that the native M currents “run down” when the cytoplasm is dialysed with an open-tip patch pipette [Brown et al. (1995), their Fig. 9.5]. Alternative approaches such as expression of RNAs or peptide constructs from cDNA plasmids have the disadvantages of irreversibility and possible secondary effects during the time taken for expression.
In the present experiments, we have tried to develop a more dynamic approach to studying intramembrane M-channel regulation that can be applied to intact neurons with minimal disturbance of the intracellular environment and that is rapid enough to be used within the timescale of a normal electrophysiological experiment. We based our approach on that devised by Covic et al. (2002a,b) to perturb receptor–G-protein interaction. This involves the use of peptides that are targeted to, and tethered in, the membrane through an attached fatty acid moiety such as palmitic acid. We call these palmitoylated peptides “palpeptides.” Here we describe results obtained using two sets of such palpeptides: one set directed at the C termini of the α-subunits of the G-proteins Gq/11 and Go, designed to inhibit coupling of the muscarinic acetylcholine receptor (mAChR) to these G-proteins, and the other set based on the putative PIP2 binding site of the Kv7.2 channel (Zhang et al., 2003), designed to occlude PIP2–M channel interaction.
Some of our observations have been reported in abstract form (Robbins and Brown, 2003, 2004).
Materials and Methods
Experiments were conducted using neurons dissociated from rat superior cervical ganglia (SCGs) and cultured in vitro. Sprague Dawley rats (postnatal day 17) were asphyxiated with rising CO2 and decapitated, in accordance with UK Home Office regulations. The SCG was removed and placed in collagenase (500 U ml−1; Sigma, Poole, UK) for 15 min, followed by trypsin (1 mg ml−1; Sigma) for 30 min. Ganglia were triturated with fire-polished glass pipettes, spun down, resuspended, and plated onto either laminin (Sigma)-coated coverslips or 35 mm plastic dishes. Cultures were kept for up to 7 d at 37°C (5% CO2) in L-15 medium supplemented with 10% fetal bovine serum, 2 mm glutamine, 24 mm NaHCO3, 38 mm glucose, 50 U ml−1 penicillin–streptomycin, and 25 ng ml−1 nerve growth factor (Tocris, Avonmouth, UK). Unless indicated otherwise, the materials were from Invitrogen (Paisley, UK).
Peptides were custom-synthesized by ABC (Imperial College London, London, UK) using solid-phase synthesis and purified to >90%. Scrambled versions of the peptides were generated using the program Shuffle Protein located at http://www.bioinformatics.vg/sms/shuffle_protein.html. All peptides used in this study were dissolved in DMSO at 5–10 mm as a stock solution, except when applied via the pipette where they were dissolved in the recording solution. A fluorescein-tagged PIP2 palpeptide (at lysine-8; see Results) was visualized with an epifluorescence microscope fitted with a monochromator (TILL-optoelektronics; TILL Photonics, Graefelfing, Germany), using 475 nm excitation and 535 nm emission. For deconvolution, images were taken in 0.5 μm vertical steps using a high-resolution computer-controlled CCD camera (4880X; Hamamatsu, Bristol, UK) driven with Openlab (Improvision, Coventry, UK) software.
M currents were normally recorded using amphotericin-perforated patch pipettes, and data were acquired and analyzed as described by Hadley et al. (2003). For open-tip pipette recordings, the pipette solution consisted of the following (in mm): 110 K-gluconate, 20 HEPES, 30 KCl, 1 MgCl2, 0.1 EGTA, 2 Na2ATP, and 0.5 Na2GTP, pH 7.2 with NaOH. All currents were recorded in the presence of tetrodotoxin (0.5 μm; Tocris). The transient A current was recorded in the additional presence of linopirdine (10 μm; Sigma) and tetraethylammonium (5 mm; Lancaster, Morecambe, UK); the delayed rectifier current was recorded in the additional presence of linopirdine and 4-aminopyridine (1 mm; Sigma).
Kir2.1 currents were expressed in sympathetic neurons by intranuclear injections of green fluorescent protein (GFP)-Kir2.1 cDNA plasmids using methods described previously (Filippov et al., 1997) and recorded under conditions used for recording M currents.
Calcium channel currents were recorded with 10 mm Ba2+ in the bathing solution and using either amphotericin-perforated patch electrodes with a pipette solution (in mm: 110 Cs acetate, 12 CsCl, 40 HEPES, 3 EGTA, 0.21 CaCl2, 1.25 MgCl2, and 1.25 CsOH) or open-tip pipettes in which the solution was supplemented with 2 NaATP, 0.5 NaGTP, and 20 BAPTA.
Expressed Kv7.2/7.3 or Kv1.2 currents were recorded from cDNA-transfected Chinese hamster ovary (CHO) cells as described by Selyanko et al. (2000).
Amphotericin B, oxotremorine-M, norepinephrine, atropine, pirenzepine, dioctanoyl-phosphatidylinositol-4,5-bisphosphate (diC8-PIP2), and pertussis toxin (PTX) were all obtained from Sigma.
Results
A palpeptide that reduces muscarinic inhibition of M currents by impairing receptor coupling to Gq/11
The first step in receptor-mediated M-current inhibition is the coupling of the activated receptor to the G-protein. Our first experiments were therefore aimed at testing whether we could devise a palpeptide that could selectively disrupt this coupling. To inhibit the M current, we used an mAChR agonist, oxotremorine-M. This inhibition results from stimulation of M1 mAChRs (Marrion et al., 1989; Bernheim et al., 1992) and the consequential activation of the G-proteins Gq and/or G11 (primarily Gq), as deduced from specific antibody injections (Caulfield et al., 1994) and antisense depletion (Haley et al., 1998). We therefore designed a palpeptide replicating the common C-terminal domain of the α-subunits of rat Gq and G11 (palmitoyl-QLNLKEYNLV) (Strathmann and Simon, 1990). This is the domain that forms the primary interaction site of the G-protein with the receptor and confers specificity on G-protein coupling (Bourne, 1997). It is also the same sequence as that previously used as the epitope for generating the antibodies that reduced mAChR-induced M-current suppression (Caulfield et al., 1994). Hence, we reasoned that if such a palpeptide could gain access to the G-protein, it might be able to inhibit receptor–Gq/11 coupling and thereby reduce mAChR-induced M-current inhibition.
Figure 1 indicates that this was indeed the case. Thus, M-current inhibition produced by 1 μm oxotremorine-M (recorded using perforated-patch electrodes) (Fig. 1A) was reduced in a concentration dependent manner within a 5 min application of 3–30 μm of the Gq/11 palpeptide to the bathing solution (Fig. 1B). A scrambled version of this palpeptide (Pal-NLVLNEKLYQ) was ineffective at 30 μm compared with vehicle controls (Fig. 1B). To monitor the time course of palpeptide action, brief (15 s) applications of oxotremorine-M (1 μm) were applied to the cells 2, 10, and 20 min after the addition of either the Gq/11 palpeptide (10 μm) or its scrambled counterpart (Fig. 1C). The inhibition was constant, at ∼50%, in the presence of the scrambled palpeptide but progressively declined with an estimated time constant of ∼15 min in the presence of the Gq/11 palpeptide.
Figure 1.
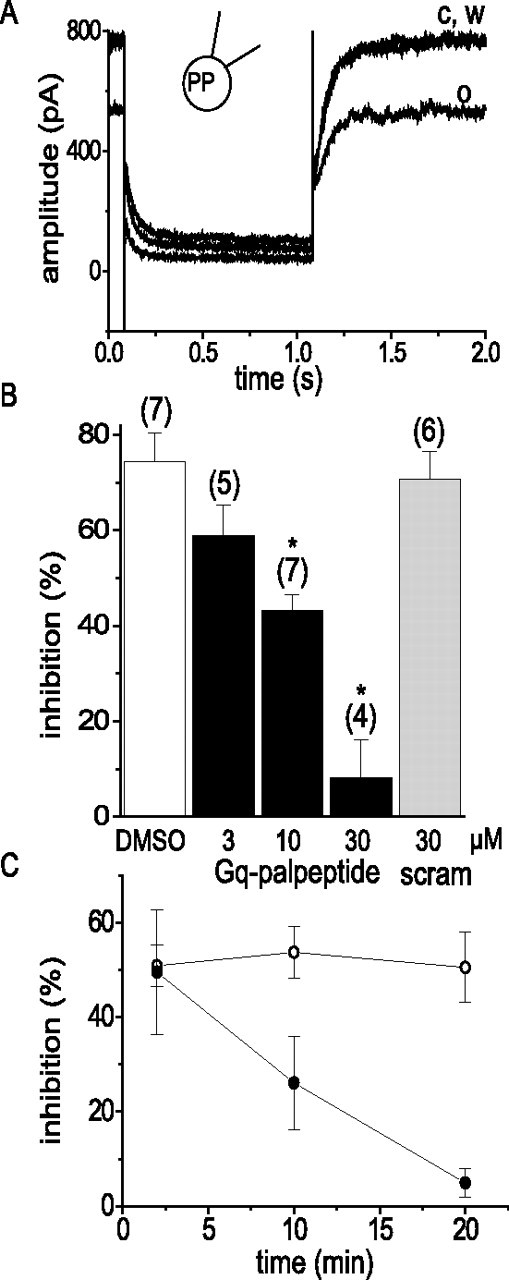
Gq/11 palpeptide reduces muscarinic receptor-mediated inhibition of M current in rat superior cervical sympathetic neurons. A, representative M-current deactivation records obtained using a perforated-patch (PP) electrode before (c) and during (o) bath application of 1 μm oxotremorine-M and after wash out (w). M currents were preactivated by setting the holding potential at −20 mV and deactivated with 1 s hyperpolarizing steps to −50 mV (Adams et al., 1982). B, Pooled data showing mean ± SEM inhibition mediated by 1 μm oxotremorine-M in the presence of vehicle (DMSO), three concentrations (3, 10, and 30 μm) of a palmitoylated decapeptide replicating the C-terminal of Gq/11 (Gq palpeptide), and a palmitolyated scrambled peptide containing the same amino acids (scram; 30 μm). The numbers in parentheses are the number of cells tested. ∗p < 0.01 (ANOVA). C, Time course of Gq/11 palpeptide induced block of muscarinic receptor-mediated M-current inhibition. Oxotremorine-M (1 μm) was applied for 15 s at 2, 10, and 20 min after application of the Gq/11-scrambled palpeptide (10 μm; open circles; n = 5) or the Gq/11 palpeptide (10 μm; filled circles; n = 4).
The Gq/11 palpeptide discriminates between Gq/11 and Go
To assess the G-protein selectivity of the Gq/11 palpeptide, we tested its effect on the inhibition of the calcium current in sympathetic neurons produced by norepinephrine and oxotremorine-M. The former produces a rapid, voltage-dependent inhibition (Hille, 1994) that is mediated primarily by the G-protein Go (Caulfield et al., 1994) and is, accordingly, blocked by PTX (Fig. 2A). The Gq/11 palpeptide had no effect on this inhibition (Fig. 2B). In contrast, when Go-mediated inhibition is blocked by PTX, muscarinic agonists such as oxotremorine-M produce an additional component of slow, voltage-insensitive inhibition (Hille, 1994) that, like M-current inhibition, is induced by stimulation of M1 mAChRs (Bernheim et al., 1992) and is mediated by Gq (Delmas et al., 1998; Haley et al., 2000) (Fig. 2C). This form of inhibition was reduced even more strongly by the Gq/11 palpeptide than was M-current inhibition (Fig. 2D).
Figure 2.

Gq/11 palpeptide reduces muscarinic receptor/Gq-mediated inhibition of calcium current but not adrenoceptor/Go-mediated inhibition. A, Representative calcium currents recorded in whole-cell (WC) patch-clamp mode with 20 mm BAPTA in the pipette solution to isolate Go-mediated inhibition (Beech et al., 1992). Cells were voltage clamped at −70 mV and depolarized with two commands of 200 ms to 0 mV (P1 and P2) interrupted by a 50 ms command to +90 mV. Traces show records in the absence [control (c)] and presence of 10 μm norepinephrine (n). Note that inhibition was voltage sensitive: norepinephrine slowed the P1 current, and the depolarizing prepulse accelerated the P2 current and partly reversed the inhibition. B, Pooled data showing the percentage of inhibition of P1 and P2 currents (mean ± SEM) produced by norepinephrine (NE) in controls (n = 4), in the presence of 1 μm Gq/11 palpeptide (n = 4), and after 24 h pretreatment with 500 ng ml−1 PTX (n = 5). C, Calcium currents recorded in perforated-patch (PP) mode in cells pretreated with PTX, to isolate Gq/11-mediated inhibition. Same voltage protocol as in A. c, Control; o, in the presence of 10 μm oxotremorine-M. Note that (unlike A) inhibition was not voltage sensitive: oxotremorine-M did not slow current activation, and inhibition was not reversed by the depolarizing prepulse. D, Pooled data showing the percentage of oxotremorine-M (oxo-M)-evoked inhibition (mean ± SEM) in PTX-treated control cells (n = 5) and in the presence of 1 μm Gq/11 palpeptide (n = 3). ∗p < 0.05, significantly different from controls.
Thus, a palpeptide replicating the C terminus of Gq/11 appears to be effective in impairing Gq/11-mediated inhibition of both M current and Ca2+ current and preserves the expected selectivity for this class of G-proteins. It should be noted that this is essentially the converse of the approach used by Covic et al. (22217,b) (see Introduction) in which the peptides were based on the complementary G-protein-associating third inner loop of the receptor (in this case, the protease-activating receptors PAR1 and PAR2). These peptides differed from those we used because they were receptor specific rather than G-protein specific. Also, as might be expected, they activated Gq/11 rather than inhibited it (although other homologs could inhibit PAR/Gq activation). We saw no evidence of Gq/11 activation, as reflected by M-current or Ca2+-current inhibition, at concentrations of Gq/11 palpeptide up to 10 μm. Thus, the mean change in M-current amplitude during application of 10 μm Gq/11 palpeptide (+5.0 ± 1.9%; n = 7) was no different from that observed with the vehicle (DMSO, +4.9 ± 1.1%; n = 7). Likewise, 10 μm Gq/11 palpeptide produced no significant change in calcium current amplitude (+1.6 ± 2.3%; n = 7).
A Go palpeptide discriminates between Go and Gq/11
To further test the selectivity of this approach, we synthesized a palmitoylated peptide to mimic the last 10 amino acids of the C terminus of Go1 (ANNLRGCGLY), plus a control scrambled version (CGYGNRLLAN). As above, we used two protocols for recording calcium current. First, to ensure that the inhibition by norepinephrine was dominated by the Go pathway, currents were recorded in whole-cell (open-tip) mode with 20 mm BAPTA in the pipette solution (Beech et al., 1992). Under these conditions, the inhibition was strongly blocked by the Go palpeptide (10 μm) but not by the scrambled palpeptide (Fig. 3A). In contrast, the Go palpeptide did not reduce Gq/11-mediated inhibition by oxotremorine-M (Fig. 3B; compare Fig. 2D). Thus, the Go palpeptide shows the converse Go-Gq/11 specificity to that exhibited by the Gq/11 palpeptide.
Figure 3.
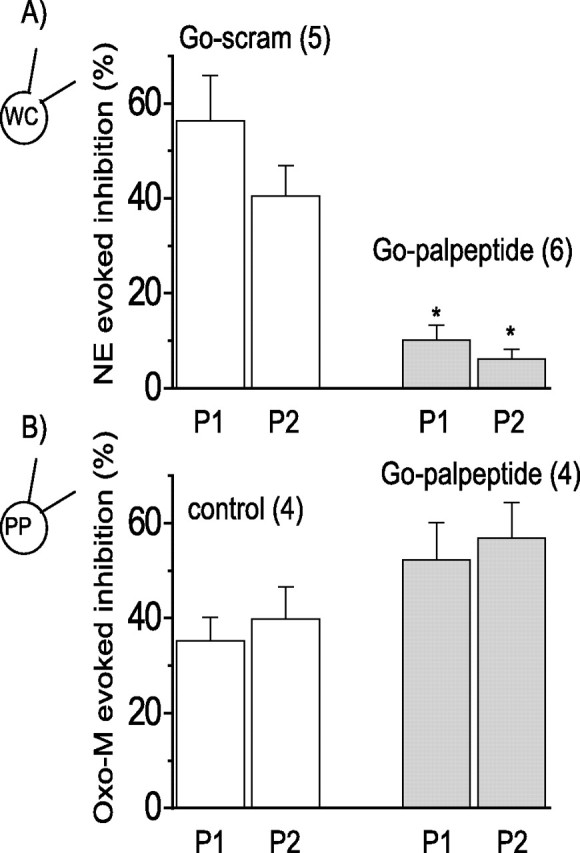
Go palpeptide blocks Go-mediated calcium current inhibition but not Gq/11-mediated inhibition. A, Inhibition of Ca2+ current by norepinephrine (NE; 10 μm) recorded under conditions to isolate the Go mechanism [whole-cell (WC) recording with 20 mm BAPTA] (see Fig. 2A,B). A palmitoylated peptide mimicking the C terminus of Go (Go palpeptide; 10 μm) significantly blocked the inhibition (filled bars; ∗p > 0.01, ANOVA) compared with the inhibition measured in the presence of a palmitoylated scrambled peptide (Go-scram; 10 μm; open bars). The scrambled peptide itself had no significant effect on the Ca2+ current. B, Ca2+ current inhibition by oxotremorine-M (Oxo-M; 10 μm) recorded under conditions that minimize the Go-mediated pathway [perforated-patch (PP) and PTX pretreatment: 500 ng ml−1, 18–24 h] (see Fig. 2C,D). Control inhibition (open bars) was not significantly different from that in the presence of the Go palpeptide (10 μm; filled bars). The numbers in parentheses are the numbers of cells. Error bars indicate SEM.
A PIP2 palpeptide
The G-protein is attached to the membrane through fatty acid groups on the C terminus of the γ-subunit and the N terminus of the α-subunit (Bourne, 1997). It thus projects into the cytoplasm where the C terminus of the α-subunit interacts with the inner loops of the receptor. This seems topographically analogous to the just-submembrane region of the C terminus of the Kv7 channel at which regulators such as PIP2 might act (Delmas and Brown, 2005). Hence, encouraged by the results with our Gq/11 and Go palpeptides, we sought to devise a palpeptide that might have an equivalent effect on PIP2–M channel interaction. We based this peptide (palmitoyl-HRQKHFEKRR) (Table 1) on the region adjacent to histidine-328 in hKv7.2, mutations of which modify the activation of expressed hKv7.2/7.3 currents by diC8-PIP2 (Zhang et al., 2003); this region also has several basic residues that might be expected to interact with the polar head groups of PIP2.
Table 1.
Structure–activity relationship for PIP2-targeted peptides, measured from concentration–response curves for M-current inhibition (see Fig. 5B)
| Peptide | Sequence | IC50(μm) mean ± SEM | Slope mean ± SEM | n |
|---|---|---|---|---|
| PIP2 | HRQKHFEKRR | >>10 | 6 | |
| PIP2K4-fluor (intracellular) | HRQKHFEKRR | 91.70 ± 0.18* | 2.00 ± 0.02 | 4 |
| Pal PIP2 | Pal-HRQKHFEKRR | 1.49 ± 0.22 | 1.39 ± 0.18 | 7 |
| Pal PIP2K8-fluor | Pal-HRQKHFEKRR | 165.32 ± 40.02* | 1.37 ± 0.19 | 6 |
| Pal PIP2H5C | Pal-HRQKCFEKRR | 0.94 ± 0.27 | 1.03 ± 0.14 | 5 |
| Pal PIP2K4M | Pal-HRQMHFEKRR | 0.66 ± 0.14 | 1.01 ± 0.09 | 5 |
| Pal PIP2K8M | Pal-HRQKHFEMRR | 3.20 ± 0.85 | 1.35 ± 0.14 | 5 |
| Pal PIP2K4MK8M | Pal-HRQMHFEMRR | 3.41 ± 0.45 | 1.15 ± 0.16 | 5 |
| Pal PIP2 scram | Pal-HERFKHEQRK | 1.27 ± 0.28 | 1.42 ± 0.36 | 5 |
| Deca-lysine | KKKKKKKKKK | >>10 | 5 | |
| Pal deca-lysine | Pal-KKKKKKKKKK | 0.80 ± 0.24 | 1.67 ± 0.10 | 5 |
| Pal deca-aspartate | Pal-DDDDDDDDDD | >>10 | 6 |
*p < 0.05 (ANOVA), difference from PIP2 palpeptide.
The PIP2 palpeptide is membrane targeted
We first asked whether the PIP2 peptide was taken up into the plasma membrane. To test this, the peptide was tagged with fluorescein at lysine-8. On adding 10 μm of this peptide, fluorescence appeared in the bathing solution and, within a few seconds, became concentrated in the cell membrane (see supplemental video, available at www.jneurosci.org as supplemental material). After washing the peptide out from the bathing solution, the residual fluorescence was restricted to the plasma membrane (Fig. 4) (four experiments). We estimate that the peptide concentrates in the membrane to a level at least 10 times that in the superfusing solution. Importantly, no fluorescence could be detected in the cytoplasm (i.e., the peptide was membrane targeted, not membrane permeant).
Figure 4.
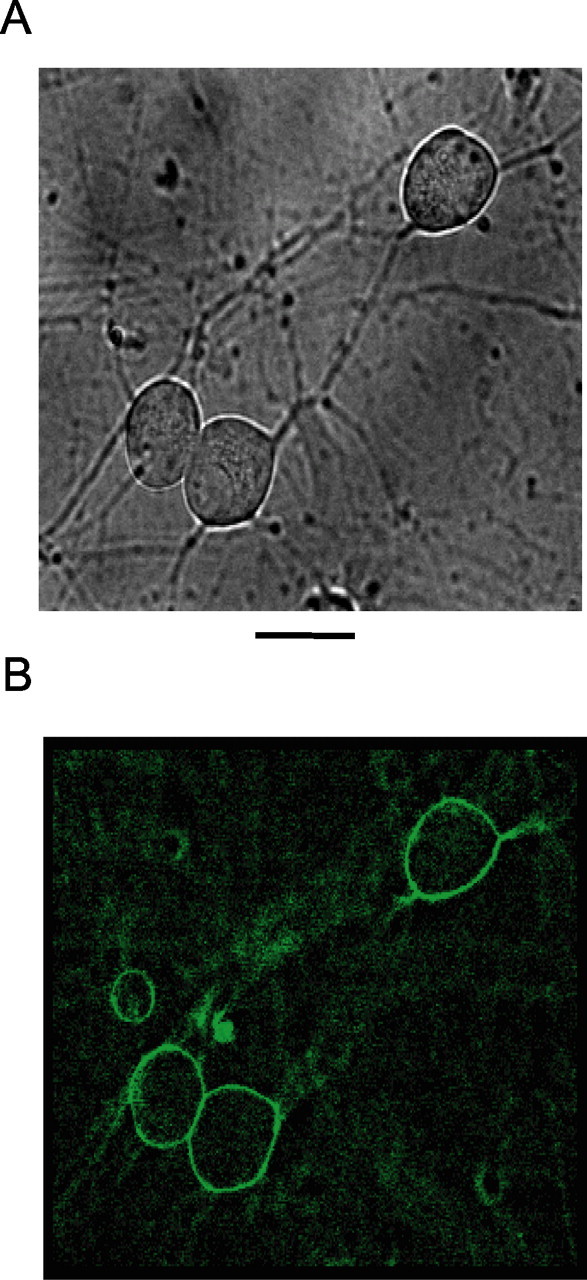
PIP2 palpeptide localizes to the plasma membrane in rat SCG neurons. A, Phase-contrast image of rat sympathetic neurons in culture. B, Deconvolved image of the same cells after live labeling with fluorescein-tagged PIP2 palpeptide (10 μm, applied to the bathing solution) (see Table 1). Excitation wavelength, 475 nm; emission, 535 nm. Scale bar, 25 μm.
PIP2 palpeptide inhibits M current
As shown in Figure 5A, this palpeptide inhibited the M current in a concentration-dependent manner when applied to the bathing fluid, with an IC50 value of 1.5 ± 0.2 μm (Fig. 5B). Inhibition was quite rapid, reaching a peak within 1 min or so, but was rather slow to reverse on washing (Fig. 5C). An equivalent peptide with an unsaturated (palmitoleic, C16:1) fatty acid, with a reduced potency (IC50 = 4.0 ± 0.8 μm; see Table 1), reversed more rapidly (Fig. 5C). In contrast, two other K+ currents recorded from these cells (the fast inactivating A current and the delayed rectifier current) were unaffected by the PIP2 palpeptide up to 10 μm (Fig. 5B).
Figure 5.

The PIP2 palpeptide selectively inhibits M current. A, Deactivation and reactivation of M current in a rat sympathetic neuron on stepping from −20 to −50 mV for 1 s (see Fig. 1A). Increasing concentrations of PIP2 palpeptide progressively reduced outward M current at −20 mV, and deactivation tail-current amplitudes recorded on stepping to −50 mV. PP, Perforated patch. B, PIP2 palpeptide concentration–inhibition curves against M current (measured from tail currents at −50 mV; filled circles; n = 7), A current (peak current measured at −20 mV from a priming command to −90 mV; open circles; n = 6), and delayed rectifier current (peak current measured at +50 mV from a holding potential of −70 mV; open squares; n = 5). Sigmoid fits were generated by the equation Y = Ymax [Xb/(Kb + Xb)], where K is the IC50 and b is the Hill slope. IC50 = 1.5 ± 0.2 μm; b = 1.4 ± 0.2. C, Time course of M-current inhibition by, and recovery from, the PIP2 palpeptide (3 μm; open circles) and for the equivalent palmitoleic acid-tagged peptide (PIP2 palpeptide; 10 μm; filled circles). The mean ± SEM of relative M-current amplitude was measured every 15 s (n = 4 in each case). The filled bar indicates the application period of the peptides, and the open bar indicates peptide-free wash. D, Concentration–inhibition curve for M-current inhibition by the PIP2 palpeptide in the presence of pirenzepine (0.3 μm; open circles). The sigmoid fit was generated from the equation above: IC50 = 1.33 ± 0.38 μm; b = 1.98 ± 0.41 (n = 5). Control data from B is included for comparison (filled circles). The equivalent nonpalmitoylated peptide had no effect (open triangles; n = 6).
M-current inhibition was not attributable to activation of muscarinic receptors because it was not altered in the presence of the muscarinic receptor antagonist pirenzepine (0.3 μm) (Fig. 5D) or atropine (1 μm; n = 5); and the nonpalmitoylated peptide was inactive on bath application (Fig. 5D). Also, currents generated by coexpression of Kv7.2 and Kv7.3 cDNAs in CHO cells, which are devoid of most neurotransmitter receptors, were equally well inhibited (IC50 = 1.2 μm) by the PIP2 palpeptide (Fig. 6).
Figure 6.
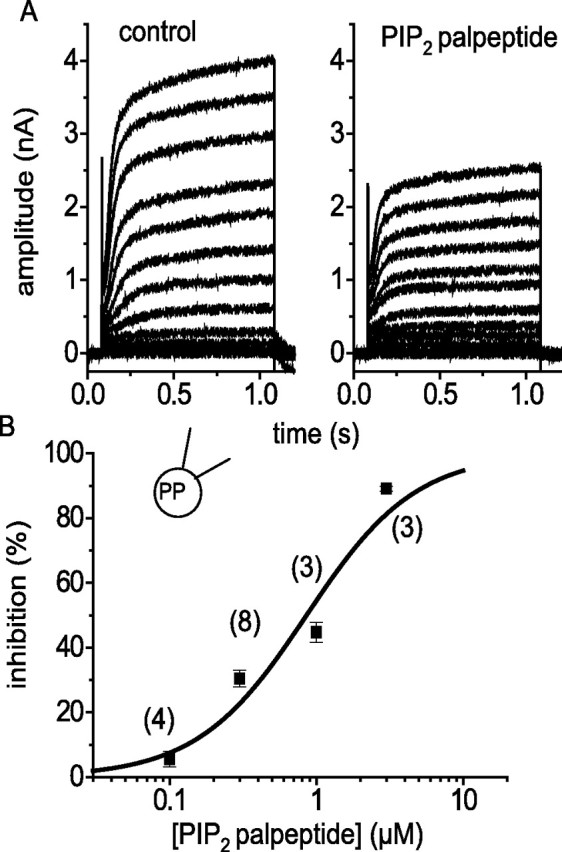
PIP2 palpeptide inhibits Kv7.2/Kv7.3 currents expressed in CHO cells. A, Currents evoked in a CHO cell line expressing Kv7.2 and Kv7.3 cDNAs (Selyanko et al., 2000) in the absence and presence of 1 μm PIP2 palpeptide. Recordings were made as for sympathetic neurons, except that tetrodotoxin was omitted from the bathing medium. Cells were held at −70 mV and stepped sequentially in 10 mV increments to +50 mV for 1 s. B, Concentration–inhibition curve fitted by the Hill equation (see Fig. 5) giving an IC50 of 1.17 μm and a slope of 0.86. The peak current was measured at +10 mV (no leak subtraction). The numbers in parentheses are the number of cells tested at each concentration. PP, Perforated patch.
The PIP2 palpeptide did not require calcium entry to inhibit M current in sympathetic neurons, because the concentration–inhibition curves generated in the absence of external calcium (plus 1 mm EGTA) were comparable to those observed in normal (2.5 mm) Ca2+ solution (IC50 = 0.65 ± 0.21 μm; slope, 1.29 ± 0.20; n = 3). The PIP2 palpeptide remained effective in cells patched with open-tip pipettes containing 10 mm EGTA with no added calcium. Under these conditions, mAChR coupling to M channels is suppressed (Beech et al., 1992), because phospholipase C activation is blocked (Horowitz et al., 2005); this we confirmed in our experiments, because mAChR activation with oxotremorine-M no longer induced membrane-to-cytosol translocation of the GFP-tagged pleckstrin homology (PH) domain of phospholipase Cδ [GFP-PLCδ-PH, a probe for PIP2 hydrolysis (Winks et al., 2005)] (data not shown). This eliminates the possibility that the PIP2 palpeptide inhibited M current by activating phospholipase C.
The potency of the PIP2 palpeptide was not significantly altered by whether the M channels were open or shut. Thus, inhibition by 1 μm PIP2 palpeptide was 37.3 ± 9.9% (n = 4) when the cells were held at −20 mV (M channels open), compared with 27.0 ± 2.0% (n = 4) in cells held at −60 mV (M channels closed) then subsequently depolarized to −20 mV. Also, activation curves for Kv7.2/7.3 currents generated in CHO cells retained the same voltage dependence in the presence of the PIP2 palpeptide (0.1–10 μm; data not shown).
The peptide works from the inside
As noted above (Fig. 5D), a peptide without the palmitoyl group had no effect when applied to the outside of the cell. From the fluorescence observations, we reasoned that the palmitoylated peptide inserted into the membrane and gained access to the (presumed) intracellular submembrane site of action. If so, the nonpalmitoylated peptide might be expected to inhibit M current when applied to the inside of the membrane. To test this, we applied an equivalent fluorescein-tagged but nonpalmitoylated peptide to the interior of the cell via a patch pipette (fluorescently tagged so that we could verify intracellular penetration). This produced a concentration-dependent run down of the M current (Fig. 7). As might be expected from the high membrane partitioning of the palmitoylated peptide (Fig. 4), this required a concentration of the nonpalmitoylated peptide at least 10-fold higher than that of the externally applied palmitoylated peptide to produce an equivalent inhibition.
Figure 7.
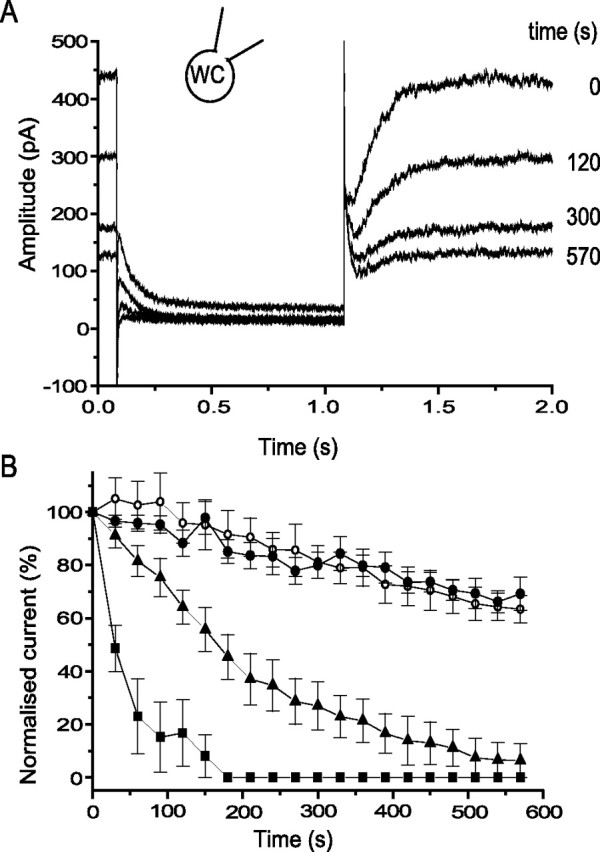
Intracellular application of the nonpalmitoylated PIP2 peptide inhibits M current. A, M-current deactivations evoked by 1 s hyperpolarization command to −50 from −20 mV at the times indicated after membrane breakthrough with a patch pipette containing 100 μm PIP2K4-fluor-peptide (see Table 1). WC, Whole cell. B, Pooled data showing the time course of M current run down in control cells (n = 4; open circles) and in cells dialysed with10 μm (n = 5; filled circles), 100 μm (n = 4; filled triangles), and 300 μm (n = 4; filled squares) PIP2K4-fluor-peptide. Error bars indicate SEM (where larger than the points).
Does the palpeptide affect PIP2–M channel interaction?
Although based on the putative PIP2 binding site of the Kv7.2 channel (Zhang et al., 2003), the palpeptide might close M channels by other mechanisms. If the peptide “competed” for PIP2, one might expect that its inhibitory effect would be reduced by increasing the concentration of PIP2. To test this under the same experimental conditions used in the above experiments, we patched neurons with pipettes (in open-tip configuration) containing 0, 200, or 500 μm of the water-soluble PIP2 analog diC8-PIP2. diC8-PIP2 at 200 μm did not affect the M-current density after a 5 min dialysis (1.63 ± 0.22 pA pF−1 compared with 1.67 ± 0.32 pA pF−1 in controls; n = 6 for both). However, the higher concentration (500 μm) of diC8-PIP2 significantly (p < 0.05, ANOVA) increased M-current density to 2.83 ± 0.39 pA pF−1 (n = 5). This would be expected if the normal sub-unity maximum open probability of M channels (Selyanko and Brown, 1999) was caused by submaximal activation of the constituent Kv7.2/7.3 tetramer at endogenous levels of membrane PIP2 (Li et al., 2005). At both concentrations, diC8-PIP2 produced a significant reduction in the inhibitory effect of oxotremorine-M (Fig. 8A). It also produced a concomitant reduction in the sensitivity of the M current to the PIP2 palpeptide, manifest by a rightward shift of the palpeptide concentration–response curve (Fig. 8B). Although the diC8-PIP2 concentrations seem high, this is a relatively weak activator of these channels (EC50 = 87 μm for activation of expressed Kv7.2/Kv7.3 channels in isolated membrane patches) (Zhang et al., 2003), and high concentrations would be necessary to effectively supplement that of endogenous PIP2 in these neurons (estimated at ∼260 μm as seen by the intracellular fluorescent probe GFP-PLCδ-PH under identical recording conditions) (Winks et al., 2005).
Figure 8.
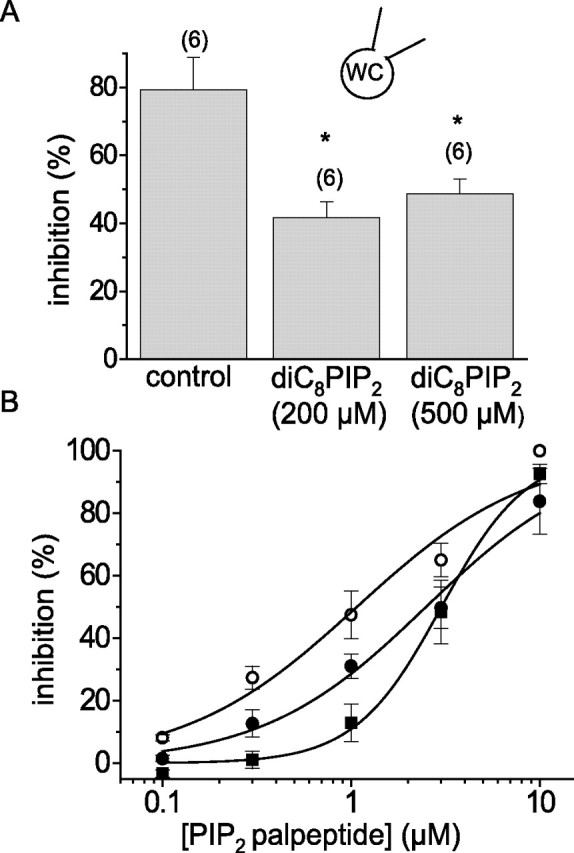
Intracellular diC8-PIP2 reduces the sensitivity of M current to both muscarinic receptor-mediated inhibition and inhibition by the PIP2 palpeptide. A, M-current inhibition by 1 μm oxotremorine-M (%; mean ± SEM) recorded using open-tip patch electrodes with 0, 200, and 500 μm diC8-PIP2 in the recording pipette. The numbers in parentheses indicate the number of cells. ∗p < 0.05 compared with controls. WC, Whole cell. B, Concentration–inhibition curves for PIP2 palpeptide recorded using pipettes containing 0 μm (open circles; n = 6), 200 μm (filled circles; n = 6), and 500 μm (filled squares; n = 5) diC8-PIP2. Solid lines are Hill fits to the mean data giving IC50 values of 1.07 ± 0.24, 2.49 ± 0.28, and 3.01 ± 0.15 μm and slopes of 0.95 ± 0.19, 1.00 ± 0.11, and 1.90 ± 0.18, respectively.
The PIP2 palpeptide sensitizes M current to muscarinic inhibition
Additional evidence that the palpeptide affects PIP2–M channel interaction is provided by its effect on the inhibitory action of oxotremorine-M. Thus, the addition of low concentrations of the PIP2 palpeptide, which of themselves produced minimal M-current inhibition (Fig. 5B), produced a concentration-dependent increase in the sensitivity of the M current to inhibition by oxotremorine-M (Fig. 9): the IC50 value was reduced from 0.84 ± 0.08 μm in the absence of peptide to 0.47 ± 0.07 and 0.22 ± 0.04 μm in the presence of 0.1 and 0.3 μm PIP2 palpeptide, respectively (significantly different from control; p < 0.01, ANOVA; n = 6 for each). Because M-current inhibition by oxotremorine-M probably results primarily from a reduction in available PIP2 (see Introduction), such a sensitization would be expected if the PIP2 palpeptide competed for PIP2 or further reduced PIP2 availability. This arises from the steep and alinear relationship between membrane PIP2 availability and M-channel activity. Thus, our previous calculations (Winks et al., 2005) imply that the 54% inhibition of M current produced by 1 μm oxotremorine-M would need ∼78% depletion of resting membrane PIP2. At 0.3 μm, the PIP2 palpeptide itself produces ∼15% inhibition of M current, implying sequestration of ∼36% of available PIP2. If oxotremorine-M still depleted the residual PIP2 by 78%, the residual M current would now be inhibited by ∼70%, somewhat less than that observed (∼88%), but indicative of the sensitization. [A greater proportion of the residual available PIP2 in the presence of the PIP2 palpeptide might be hydrolyzed by oxotremorine-M, because the amount of PIP2 hydrolyzed at normal levels of PIP2 appeared to be near-saturation as determined from GFP-PLCδPH translocation (Winks et al., 2005).]
Figure 9.

PIP2 palpeptide enhances M-current sensitivity to muscarinic receptor-mediated inhibition. Concentration–inhibition curves for M-current inhibition by oxotremorine-M in control cells (open circles), 0.1 μm PIP2 palpeptide (filled circles), and 0.3 μm PIP2 palpeptide (filled squares; n = 6 in each case). PP, Perforated patch. Error bars indicate SEM.
Peptide homologs
We tested a number of homologs of the PIP2 palpeptide on the ganglionic M current. The results are given in Table 1. First, we mutated lysines to methionines (K4M, K8M, and K4M-K8M) because it had previously been suggested that lysine residues are important for phosphoinositide binding to PH domains (Ferguson et al., 1995). The K4M mutation did not affect inhibitory potency compared with the original palpeptide, but mutations K8M and K4M-K8M significantly reduced potency by twofold or greater. We also changed the 5-histidine to cysteine (H5C) because the equivalent mutation in KCNQ2 reduced PIP2 activation of expressed KCNQ2/3 currents by approximately threefold (Zhang et al., 2003); however, this did not show any reduced potency in our system. Furthermore, a palpeptide composed of the same 10 amino acids, but in random sequence (Table 1, Pal PIP2 scram) was equally potent with the PIP2 palpeptide. Hence, there appeared to be no tight structure–activity relationship for M-current inhibition. Instead, potency seemed to depend more on the number of basic charges. Thus, a deca-lysine palpeptide was at least as potent as the original PIP2 palpeptide, whereas an acidic deca-aspartate palpeptide was totally ineffective at 10 μm.
The effect of the deca-lysine palpeptide suggests that the PIP2 palpeptide, although based on the presumptive PIP2 binding site of the Kv7.2 channel, might not bind in a specific manner to PIP2 but rather might interact electrostatically with the polar head groups of PIP2 to sequester it, in the same manner as long-chain polylysines or some other basic peptides (Gambhir et al., 2004). If so, the PIP2 palpeptide might be expected to affect other ion channels “gated” by PIP2, regardless of the precise amino acid sequence of the PIP2 binding site of the channel. We tested this on two other channels, the endogenous N-type Ca2+ channel and expressed inwardly rectifying K+ channels.
Effect of the PIP2 palpeptide on Ca2+ channels
Recent evidence suggests that the N-type Ca2+ channel is also gated by PIP2 (Wu et al., 2002; Gamper et al., 2005). The PIP2 palpeptide was equally effective in inhibiting currents carried by these channels as those carried by M channels. Thus, at a concentration of 1 μm, the PIP2 palpeptide inhibited the Ca2+ current recorded from sympathetic neurons by 33.7 ± 11.8% (n = 6). However, it did not significantly alter either the “slow” Gq-mediated voltage-independent inhibition produced by 1 μm oxotremorine-M recorded using perforated-patch electrodes (control: 17.1 ± 2.3%, n = 7; plus 1 μm PIP2 palpeptide: 16.2 ± 1.8%, n = 3), or the PTX- and voltage-sensitive (Go-mediated) “fast” inhibition produced by norepinephrine (control: 48.3 ± 6.5, n = 9; plus 1 μm PIP2 palpeptide: 66.1 ± 4.2%, n = 4) (compare Fig. 2 for experimental methods).
Inward rectifier K+ channels
Superior cervical sympathetic neurons do not have prominent native inward rectifier K+ currents, but these can be expressed by cDNA transfection (Ruiz-Velasco and Ikeda, 1998; Filippov et al., 2004). For the present purposes, we opted to express Kir2.1 channels, because these are constitutively active (i.e., do not require a receptor agonist to open them) and are strongly activated by PIP2 (Huang et al., 1998). Figure 10A shows a protocol by which currents through expressed Kir2.1 channels and native M channels could be recorded simultaneously. Unlike M currents, Kir2.1 currents were not inhibited by the PIP2 palpeptide (up to 1 μm) (Fig. 10B). However, coexpression of Kir2.1 reduced the density of the recorded M current from 2.3 ± 0.3 pA/pF (n = 10) to 1.2 ± 0.3 pA/pF (n = 7) (p < 0.05) and strikingly increased the sensitivity of the M current to the PIP2 palpeptide by approximately fivefold (Fig. 10B). All of these effects might be explained by the fact that Kir2.1 is ∼20 times more sensitive to PIP2 than are Kv7.2/Kv7.3 channels (and hence presumably “binds” PIP2 with 20-fold higher affinity): the EC50 value for activation of Kir2.1 channels by diC8-PIP2 is 4.6 μm (Lopes et al., 2002) compared with 87 μm for Kv7.2/7.3 activation (Zhang et al., 2003). Thus, Kir2.1 channels would be much less sensitive than M current to sequestration of a fraction of the endogenous PIP2 by the palpeptide (or competition by the palpeptide for PIP2), whereas overexpression of Kir2.1 channels might themselves compete with the M channels for available PIP2, so reducing M-channel open probability and enhancing sensitivity to further sequestration of PIP2 by the palpeptide. Again, the latter would be predicted from our previous calibrations (Winks et al., 2005): the ∼40% inhibition of M current produced by 1 μm PIP2 palpeptide under normal conditions would correspond to the sequestration of ∼67% of the PIP2 available for channel activation; the ∼48% inhibition produced by Kir2.1 would itself correspond to sequestration of ∼75% of available PIP2; and then a further 67% sequestration of the residual PIP2 by 1 μm PIP2 palpeptide would reduce the residual M current by ∼79% (not far off the observed value of 83%), instead of the 40% seen in the absence of Kir2.1.
Figure 10.
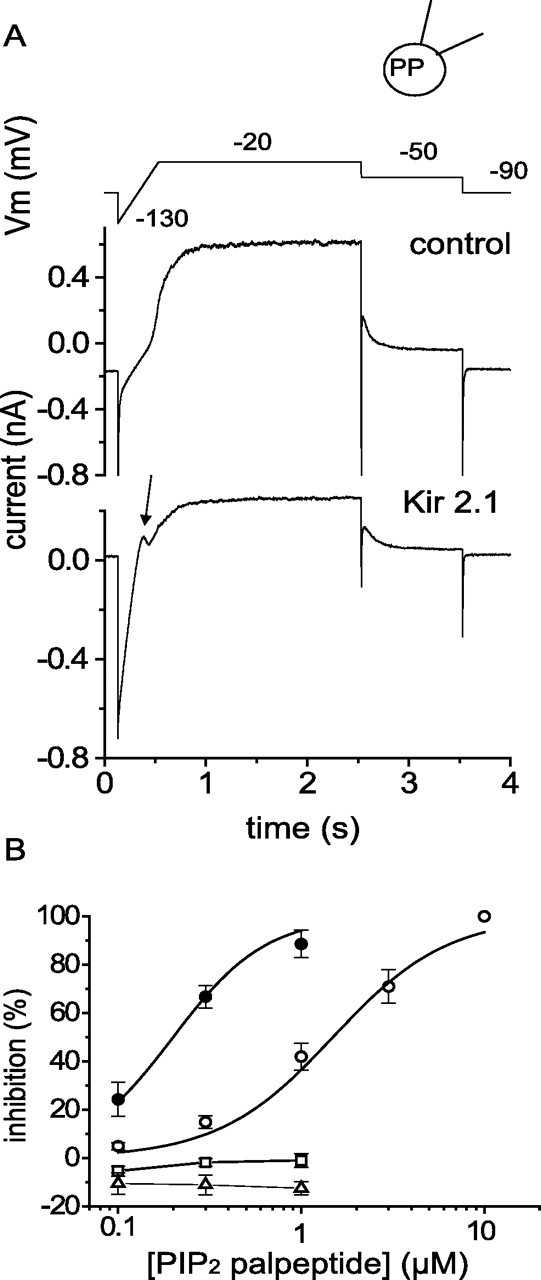
Expression of Kir2.1-GFP reduces M-current amplitude and increases M-current sensitivity to PIP2 palpeptide. A, Current traces recorded in perforated-patch mode generated by the voltage commands shown in the top trace in a control neuron (middle trace) and in a Kir2.1-GFP-expressing neuron (bottom trace). In the latter, note the increased inward current after the first hyperpolarizing step to −130 mV and characteristic outward notch current (arrow) as the Kir current declines during depolarization. PP, Perforated patch. B, Concentration–inhibition curves for M-current inhibition by the PIP2 palpeptide in Kir2.1-GFP-transformed cells (filled circles; n = 3) and for inhibition of outward Kir current (open squares; n = 3,) and inward Kir current (open triangles; n = 3). The data for M-current inhibition by PIP2 palpeptide in control cells shown in Figure 5B are included for comparison (open circles; n = 7). Error bars indicate SEM.
The role of the fatty acid
The palmitoyl group is required for partitioning and anchoring of the palpeptide into the membrane. This is indicated by the concentration and retention of fluorescence in the membrane shown in Figure 4. In contrast, the fluorescently tagged but nonpalmitoylated peptide used in the intracellular dialysis experiments (Fig. 7) showed no such membrane accumulation. We were interested to know how the activity of the peptide might be affected by the nature of the attached fatty acid. We have not yet explored this systematically but have noted two pertinent points. First, substitution of the unsaturated (C16:1) palmitoleic acid for the saturated (C16:0) palmitic acid increased the IC50 value of the PIP2 palpeptide in inhibiting the M current from 1.49 ± 0.22 μm (n = 7) to 4.0 ± 0.82 μm (n = 6). As shown in Figure 5B, it also accelerated the reversibility of the inhibition. Presumably the “kinked” unsaturated methylene chain occupies a larger volume and does not insert so tightly into the membrane phospholipid bilayer. Second, a PIP2 peptide with a longer-chain (C24:0) lignoceric acid moiety was totally ineffective at concentrations up to 10 μm. There may be several reasons for this, but one possibility is that it fails to present the peptide moiety at an appropriate distance from the inner side of the membrane to interact with the PIP2 head groups (see Discussion).
A palmitoylated peptide cannot access the potassium channel pore
We presume that palpeptides are anchored in the membrane through the palmitoyl group and that the peptide domains of the Gq/11, Go, and PIP2 palpeptides project into the cytoplasm to a sufficient extent as to interact with the muscarinic receptor or with the polar head groups of PIP2 because these are at the inner face of the membrane (see Discussion). The question arises whether a membrane-attached palpeptide might project sufficiently far into the cytoplasm to interact (for example) with the aqueous pore of an ion channel. To test this, we constructed a peptide (ShB) replicating the first 20 amino acids of the Shaker inactivating domain (Zagotta et al., 1990) and tested this on currents generated by expressed Kv1.2 channels (which lack the fast-inactivating N-terminal ball-peptide). As shown in Figure 11A, intracellular dialysis of 0.5–1 mg ml−1 (224–448 μm) of the nonpalmitoylated version (MAAVAGLYGLGEDRQHRKKQ-CONH2) of this peptide via the patch pipette rapidly induced fast inactivation of the previously noninactivating current. In contrast, no such inactivation was induced by extracellular application of up to 100 μm of an N-terminal palmitoylated version of this peptide (ShB palpeptide) (Fig. 11B).
Figure 11.

ShB peptide induces inactivation of Kv1.2 currents when applied intracellularly, but an ShB palpeptide does not when applied extracellularly. A, Currents recorded from CHO cells expressing Kv1.2 cDNA using open-tip patch electrodes. Top trace, Currents generated by a series of 1.5 s voltage steps from −70 to +70 mV (10 mV increments). Note that currents do not inactivate. Middle trace, Currents generated by a step to +70 mV recorded at the times indicated after patching with a pipette containing 448 μm (1 mg ml−1) ShB peptide. Bottom trace, Series of currents generated as in the top trace but after 10 min equilibration with the ShB peptide. WC, Whole cell. B, Kv1.2 current recorded using perforated-patch (PP) electrodes in the absence and presence of increasing concentrations (micromolars) of ShB palpeptide added to the bathing solution. The bottom panel shows the percentage of inactivation of the Kv1.2 current after 1.5 s depolarization (measured between the two vertical lines on top panel) in the presence of increasing concentrations of externally applied ShB palpeptide and (right block) 448 μm intracellular nonpalmitoylated ShB peptide. Error bars (where applicable) are means ± SEMs of the number of cells in parentheses. C, Control; IC, intracellular nonpalmitoylated ShB peptide.
Discussion
In these experiments, we designed two sets of membrane-targeted probes to perturb signal transduction in the mAChR–M current inhibitory pathway in intact neurons. The first probe was a palmitoylated peptide based on the C terminus of the α-subunit of Gq/11 (the “Gq/11 palpeptide”), designed to inhibit receptor coupling to Gq. This was essentially a complementary peptide to those designed by Covic et al. (22217,b) to replicate the cognate cytoplasmic domain of another Gq-coupled receptor. However, unlike the latter, the Gq/11 palpeptide did not activate Gq and thereby inhibit M current. Instead, it reduced the M-current inhibition produced by mAChR stimulation, just like an antibody directed against the same C-terminal sequence (Caulfield et al., 1994). This was specifically directed against receptor coupling to Gq/11 in so far that, whereas it also suppressed M1 mAChR-induced Gq/11-mediated Ca2+ current inhibition, it did not affect Go-mediated adrenergic inhibition. Conversely, the latter effect, but not the former, was selectively inhibited by another palpeptide containing the C-terminal sequence of Go. Thus, these lipidated peptides essentially replicate the selective inhibitory effects of Gq/11 or Go antibodies raised using the same sequences as epitopes (Caulfield et al., 1994). We hypothesize that they bind to the complementary sequences in the inner loops of the cognate receptors and therefore compete against the relevant G-protein α-subunit C terminus for receptor binding.
The second type of peptide was designed to inhibit PIP2“gating” of M channels and was based on the putative PIP2-binding sequence of hKv7.2 (Zhang et al., 2003). As predicted (see Introduction), this inhibited M currents. Of course, this might occur through a multiplicity of mechanisms; some, at least, we can exclude. First, it did not activate the muscarinic receptors because its effect was not prevented by atropine or pirenzepine. Second, it is unlikely to have interacted with other Gq-coupled receptors or Gq-mediated phospholipase C activation because it was equally effective on Kv7.2/7.3 channels expressed in CHO cells, which have few such endogenous receptors, and also because it was effective when intracellular Ca2+ was buffered by high concentrations of a calcium chelator that disrupts Gq/phospholipase C-mediated M-current inhibition (Beech et al., 1992; Horowitz et al., 2005). Finally, it is also unlikely to have blocked the M channels directly, either from the outside or from the inside, because an externally applied nonpalmitoylated peptide was ineffective, and, as indicated by the tests with the Shaker palpeptide, a membrane-attached palpeptide cannot access a potassium-channel pore from the inside.
Some positive evidence for an interaction with PIP2 is provided by the apparently competitive antagonism of the effect of the palpeptide on M current by intracellular diC8-PIP2 (Fig. 8). The potentiation of the inhibitory effect of oxotremorine-M by low concentrations of the PIP2 palpeptide would also accord with this modus operandi, because mAChR-induced inhibition seems to result primarily from depletion of membrane PIP2 (see Introduction) so would be enhanced if some fraction of available PIP2 was already sequestered.
Thus, the effect of the PIP2 palpeptide observed in these experiments accords with the currently prevalent view that the activity of native M channels is dependent on available membrane PIP2 levels, and that M-current inhibition by mAChR stimulation results primarily from Gq/phospholipase C-catalyzed depletion of PIP2. However, a rather surprising observation was the effect of overexpressing Kir2.1 channels in both reducing M-current density and sensitizing the current to PIP2 palpeptide inhibition. This we attribute to competitive sequestration of PIP2 by the Kir2.1 channels. If this interpretation is correct, it suggests that the pool of PIP2 available to the M channels, or PIP2 mobility, may be rather more restricted that previously assumed, because the density of PIP2 molecules in the inner leaflet of the membrane (estimated at several thousand molecules per square micrometer) (Xu et al., 2003; Suh et al., 2004) exceeds the probable membrane density of M channels by approximately three orders of magnitude.
Although based on a putative PIP2 binding domain in the Kv7.2 channel protein (Zhang et al., 2003), the PIP2 palpeptide did not show any “tight” structure–activity relationship as an M-current inhibitor. Thus (and unlike the G-protein palpeptides), a scrambled peptide containing the same amino acids was as effective as the original PIP2 palpeptide. Rather, activity among a restricted series of peptides seemed to depend on the presence or absence of basic residues and (to an extent) on the number of such residues. This suggests that these palpeptides do not bind in a structurally specific manner to PIP2 Instead, like many other basic peptides (Gambhir et al., 2004; McLaughlin and Murray, 2004), they probably interact electrostatically with the polar head groups of PIP2 and thereby “sequester” PIP2 in such a manner as to hinder its interaction with the Kv7.2 channel. Hence, they do not compete specifically against Kv7.2 channels for PIP2 but can also potentially inhibit other channels gated by PIP2. Thus, we found that the PIP2 palpeptide also inhibited PIP2-regulated N-type Ca2+ currents (Gamper et al., 2005): although the PIP2 binding site on these channels has not been identified, it seems unlikely to be identical to that in Kv7.2 channels. On the other hand, it was ineffective on PIP2-gated Kir2.1 channels. This we attribute to the fact that these channels bind PIP2 with an affinity ∼20-fold higher than that of Kv7.2 channels (Lopes et al., 2002; cf. Zhang et al., 2003). The equivalent affinity of the N-type Ca2+ channel for PIP2 has not been determined but is probably similar to that of native M channels, because oxotremorine-M inhibits both channels with virtually identical concentration dependence (Haley et al., 2000). Therefore, the effectiveness of these PIP2 palpeptides in disrupting PIP2 channel interaction will depend on the affinity of the channel for PIP2. In agreement with this, the PIP2 palpeptide did not reduce oxotremorine-M-induced M-current inhibition (i.e., it did not antagonize phospholipase C-catalyzed PIP2 hydrolysis. This would accord with the high affinity of PIP2 for the PH domain of phospholipase C (dissociation constant, 1–2 μm) (Hirose et al., 1999).
We emphasize that these palpeptides are membrane targeted, not membrane permeant. Thus, the fluorescently labeled derivative clearly concentrated within the membrane, with very little (if any) accumulation within the cytosol (Fig. 4). Presumably, the palmitoyl group anchors the peptide within the inner leaflet of the membrane, with the hydrophobic peptide moiety projecting from the inner face of the membrane into the cytosol (Kuliopulos and Covic, 2003). In the case of the Gq/11 palpeptide, this would closely replicate the natural position at which the G-protein interacts with the receptor because the native G-protein α-subunit is itself anchored in the membrane (in part, at least) by a palmitoyl group (Wedegaertner et al., 1993). In this position, the basic PIP2 palpeptide can also interact electrostatically with the polar head groups of PIP2, like the myristoylated MARCKS protein (McLaughlin et al., 2002). Adherence of the peptide domain to within a few angstroms of the membrane is indicated by the fact that a ShB palpeptide was unable to block the pore the of the Kv1.2 channel, which requires access from the cytoplasm through a side-portal above the cytoplasmic T1 domain (Long et al., 2005). This further illustrates the point that these peptides do not penetrate in high concentrations into the cytoplasm itself, because an intracellularly applied nonpalmitoylated ShB peptide was very effective.
Compared with other methods of investigating ion channel regulation, such as antibody injection or minigene expression, these palpeptides have some clear practical advantages: they are easy to use in intact cells, are rapid in effect and (to an extent) reversible, and therefore can be used as a membrane-targeted drug within the lifetime of a conventional experiment. Reasonable specificity may be anticipated in designing peptides to disrupt protein–protein interaction. Thus, the G-protein-based peptides show similar subtype selectivity to antibodies generated using the same sequences as epitopes, although the former are much more convenient to use and give more reproducible results: indeed, the Gq/11 palpeptide has recently been used in cerebellar slices to disrupt metabotropic glutamate receptor coupling to transient receptor potential channels (Duguid and Smart, 2005). Not surprisingly, the peptides directed against PIP2 show less specificity in terms of the requisite amino acid sequence because they are only required to neutralize charges on the phospholipid head groups: any higher specificity required for high-affinity binding of PIP2 to a particular ion channel presumably requires a more complex structure that a short peptide is unlikely to replicate. Nevertheless, they appear effective in sequestering PIP2 and are more convenient to use in intact cells than, for example, PIP2 antibodies. One constraint is that they might well neutralize charges on other phosphatidylinositol phosphates (or possibly other anionic membrane constituents). However, this is also true for some PIP2 antibodies and so is unlikely to severely limit their use in evaluating the role of PIP2 as an ion channel regulator because the latter is the most abundant of the dynamically regulated membrane phospholipids (McLaughlin and Murray, 2004).
Thus, lipidated peptides of this type should be helpful in probing other forms of ion channel regulation (or indeed regulation of other membrane proteins) that depend on interactions within or just subjacent to the cell membrane. By altering the nature of the fatty acid, it may even be possible to target the peptides to different membrane microenvironments.
Footnotes
This work was supported by the United Kingdom Medical Research Council and by the European Union Framework 6 program. We thank J. Reilly for cell culture, Dr. A. Filippov for the cDNA nuclear injections, and Dr. M. Leyland (Department of Cell Physiology and Pharmacology, University of Leicester, Leicester, UK) for the Kir2.1-GFP cDNA.
References
- Adams PR, Brown DA, Constanti A (1982). M-currents and other potassium currents in bullfrog sympathetic neurones. J Physiol (Lond) 330:537–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beech DJ, Bernheim L, Mathie A, Hille B (1992). Intracellular Ca2+ buffers disrupt muscarinic suppression of Ca2+ current and M current in rat sympathetic neurons. Proc Natl Acad Sci USA 88:652–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernheim L, Mathie A, Hille B (1992). Characterization of muscarinic receptor subtypes inhibiting Ca2+ current and m current in rat sympathetic neurons. Proc Natl Acad Sci USA 89:9544–9548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourne HR (1997). How receptors talk to trimeric G proteins. Curr Opin Cell Biol 9:134–142. [DOI] [PubMed] [Google Scholar]
- Brown DA (1988). M currents. In: Ion channels (Narahashi T, ed.) pp. 55–99. New York: Plenum. [DOI] [PubMed]
- Brown DA, Buckley NJ, Caulfield MP, Duffy SM, Jones S, Lamas J, Marsh SJ, Robbins J, Selyanko AA (1995). Coupling of muscarinic acetylcholine receptors to neural ion channels: closure of K+ channels. In: Molecular mechanisms of acetylcholine receptor function (Wess J, ed.) pp. 165–182. Austin, TX: RG Landes Company.
- Caulfield MP, Jones S, Vallis Y, Buckley NJ, Kim G-D, Milligan G, Brown DA (1994). Muscarinic M-current inhibition via Gαq/11 and α-adrenoceptor inhibition of Ca2+ current via Gαo in rat sympathetic neurones. J Physiol (Lond) 477:415–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Covic L, Gresser AL, Talavera J, Swift S, Kuliopulos A (22217). Activation and inhibition of G protein-coupled receptors by cell-penetrating membrane-tethered peptides. Proc Natl Acad Sci USA 99:643–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Covic L, Misra M, Badar J, Singh C, Kuliopulos A (2002b). Pepducin-based intervention of thrombin-receptor signaling and systemic platelet activation. Nat Med 8:1161–1165. [DOI] [PubMed] [Google Scholar]
- Cruzblanca H, Koh DS, Hille B (1998). Bradykinin inhibits M current via phospholipase C and Ca2+ release from IP3-sensitive Ca2+ stores in rat sympathetic neurons. Proc Natl Acad Sci USA 95:7151–7156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delmas P, Brown DA (2005). Pathways modulating neural KCNQ/M (Kv7) potassium channels. Nat Rev Neurosci 6:850–862. [DOI] [PubMed] [Google Scholar]
- Delmas P, Abogadie FC, Dayrell M, Haley JE, Milligan G, Caulfield MP, Brown DA, Buckley NJ (1998). G-proteins and G-protein subunits mediating cholinergic inhibition of N-type calcium currents in sympathetic neurons. Eur J Neurosci 10:1654–1666. [DOI] [PubMed] [Google Scholar]
- Duguid IC, Smart TG (2005). Retrograde mGluR1 activation enhances endocannabinoid release from cerebellar Purkinje cells. Soc Neurosci Abstr 31:41–3. [Google Scholar]
- Ferguson KM, Lemmon MA, Schlessinger J, Sigler PB (1995). Structure of the high affinity complex of inositol trisphosphate with a phospholipase C pleckstrin homology domain. Cell 83:1037–1046. [DOI] [PubMed] [Google Scholar]
- Filippov AK, Webb TE, Barnard EA, Brown DA (1997). Inhibition by heterologously-expressed P2Y2 nucleotide receptors of N-type calcium currents in rat sympathetic neurones. Br J Pharmacol 121:849–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filippov AK, Fernandez-Fernandez JM, Marsh SJ, Simon J, Barnard EA, Brown DA (2004). Activation and inhibition of neuronal G protein-gated inwardly rectifying K+ channels by P2Y nucleotide receptors. Mol Pharmacol 66:468–477. [DOI] [PubMed] [Google Scholar]
- Ford CP, Stemkowski PL, Light PE, Smith PA (2003). Experiments to test the role of phosphatidylinositol 4,5-bisphosphate in neurotransmitter-induced M-channel closure in bullfrog sympathetic neurons. J Neurosci 23:4931–4941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gambhir A, Hangyas-Mihalyne G, Zaitseva I, Cafioso DS, Wang J, Murray D, Pnetyala SN, Smith SO, MacLaughlin S (2004). Electrostatic sequestration of PIP2 on phospholipid membranes by basic/aromatic regions of proteins. Biophys J 86:2188–2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamper N, Shapiro MS (2003). Calmodulin mediates Ca2+-dependent modulation of M-type K+ channels. J Gen Physiol 122:17–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamper N, Reznikov V, Yamada Y, Yang J, Shapiro MS (2005). Phosphatidylinositol 4,5-bisphosphate signals underlie receptor-specific Gq/11-mediated modulation of N-type Ca2+ channels. J Neurosci 24:10980–10992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadley JK, Passmore GM, Tatulian L, Al-Qatari M, Ye F, Wickenden AD, Brown DA (2003). Stoichiometry of expressed KCNQ2/KCNQ3 potassium channels and subunit composition of native ganglionic M channels deduced from block by tetraethylammonium. J Neurosci 23:5012–5019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haley JE, Abogadie FC, Delmas P, Dayrell M, Vallis Y, Milligan G, Caulfield MP, Brown DA, Buckley NJ (1998). The α subunit of Gq contributes to muscarinic inhibition of the M-type potassium current in sympathetic neurons. J Neurosci 18:4521–4531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haley JE, Delmas P, Offermans S, Abogadie FC, Simon MI, Buckley NJ, Brown DA (2000). Muscarinic inhibition of calcium current and M current in Gαq-deficient mice. J Neurosci 20:3973–3979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hille B (1994). Modulation of ion-channel function by G-protein-coupled receptors. Trends Neurosci 17:531–536. [DOI] [PubMed] [Google Scholar]
- Hirose K, Kadowski S, Tanabe M, Takeshima H, Iino M (1999). Spatiotemporal dynamics of inositol 1,4,5-trisphosphate that underlies complex Ca2+ mobilization patterns. Science 284:1527–1530. [DOI] [PubMed] [Google Scholar]
- Horowitz LF, Hirdes W, Suh BC, Hilgemann DW, Mackie K, Hille B (2005). Phospholipase C in living cells: activation, inhibition, Ca2+ requirement, and regulation of M current. J Gen Physiol 126:243–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshi N, Zhang JS, Omaki M, Takeuchi T, Yokoyama S, Wanaverbecq N, Langeberg LK, Yoneda Y, Scott JD, Brown DA, Higashida H (2003). AKAP150 signaling promotes suppression of the M-current by muscarinic agonists. Nat Neurosci 6:564–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang CL, Feng S, Hilgemann DW (1998). Direct activation of inward rectifier potassium channels by PIP2 and its stabilization by Gβγ. Nature 391:803–806. [DOI] [PubMed] [Google Scholar]
- Kuliopulos A, Covic L (2003). Blocking receptors on the inside: pepducin-based interventions of PAR signaling and thrombosis. Life Sci 74:255–262. [DOI] [PubMed] [Google Scholar]
- Li Y, Gamper N, Hilgemann DW, Shapiro MS (2005). Regulation of Kv7 (KCNQ) K+ channel open probability by phosphatidylinositol 4,5-bisphosphate. J Neurosci 25:9825–9835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long SB, Campbell EB, McKinnon R (2005). Crystal structure of a mammalian voltage-dependent Shaker family K+ channel. Science 309:897–903. [DOI] [PubMed] [Google Scholar]
- Lopes CM, Zhang H, Rohacs T, Jin T, Yang J, Logothetis DE (2002). Alterations in conserved Kir channel-PIP2 interactions underlie channelopathies. Neuron 34:933–944. [DOI] [PubMed] [Google Scholar]
- Marrion NV (1997). Control of M-current. Annu Rev Physiol 59:483–504. [DOI] [PubMed] [Google Scholar]
- Marrion NV, Smart TG, Marsh SJ, Brown DA (1989). Muscarinic suppression of the M-current in the rat sympathetic ganglion is mediated by receptors of the M1-subtype. Br J Pharmacol 98:557–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin S, Murray D (2004). Plasma membrane phosphoinositide organization by protein electrostatics. Nature 438:605–611. [DOI] [PubMed] [Google Scholar]
- McLaughlin S, Wang J, Gambhir A, Murray D (2002). PIP2 and proteins: interactions, organization and information flow. Annu Rev Biophys Biomol Struct 31:151–175. [DOI] [PubMed] [Google Scholar]
- Robbins J, Brown DA (2003). Investigation of M-current transduction mechanisms using palmitoylated peptides. Soc Neurosci Abstr 29:545–1. [Google Scholar]
- Robbins J, Brown DA (2004). A membrane-targetted palmitylated peptide replicating a phosphatidylinositol-4,5-bisphosphate binding site inhibits IK(M) and modulates receptor-mediated inhibition. J Physiol (Lond) 557P:C43. [Google Scholar]
- Ruiz-Velasco V, Ikeda SR (1998). Heterologous expression and coupling of G protein-gated inwardly rectifying K+ channels in adult rat sympathetic neurons. J Physiol (Lond) 513:761–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selyanko AA, Brown DA (1996). Intracellular calcium directly inhibits potassium M channels in excised membrane patches from rat sympathetic neurons. Neuron 16:151–162. [DOI] [PubMed] [Google Scholar]
- Selyanko AA, Brown DA (1999). M-channel gating and simulation. Biophys J 77:701–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selyanko AA, Hadley JK, Wood IC, Abogadie FC, Jentsch TJ, Brown DA (2000). Inhibition of KCNQ1–4 channels expressed in mammalian cells via M1 muscarinic acetylcholine receptors. J Physiol (Lond) 522:349–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strathmann M, Simon MI (1990). G protein diversity: a distinct class of α-subunits is present in vertebrates and invertebrates. Proc Natl Acad Sci USA 87:9113–9117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suh B-C, Hille B (2002). Recovery from muscarinic modulation of M current requires phosphatidylinositol 4,5-bisphosphate synthesis. Neuron 35:507–520. [DOI] [PubMed] [Google Scholar]
- Suh BC, Horowitz LF, Hirdes W, Mackie K, Hille B (2004). Regulation of KCNQ2/KCNQ3 current by G protein cycling: the kinetics of receptor-mediated signaling by Gq. J Gen Physiol 123:663–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang HS, Pan Z, Shi W, Brown BS, Wymore RS, Dixon JE, McKinnon D (1998). KCNQ2 and KCNQ3 potassium channel subunits: molecular correlates of the M-channel. Science 282:1890–1893. [DOI] [PubMed] [Google Scholar]
- Wedegaertner PB, Chu DH, Wilson PT, Levis MJ, Bourne HR (1993). Palmitoylation is required for signaling functions and membrane attachment of Gq alpha and Gs alpha. J Biol Chem 268:25001–25008. [PubMed] [Google Scholar]
- Winks JS, Hughes S, Filippov AK, Tatulian L, Abogadie FC, Brown DA, Marsh SJ (2005). Relationship between membrane phosphatidylinositol-4,5-bisphosphate and receptor-mediated inhibition of native neuronal M channels. J Neurosci 25:3400–3413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu L, Bauer CS, Zhen XG, Xie C, Yang J (2002). Dual regulation of voltage-gated calcium channels by PtdIns(4,5)P2. Nature 419:947–952. [DOI] [PubMed] [Google Scholar]
- Xu C, Watras J, Loew LM (2003). Kinetic analysis of receptor-activated phosphoinositide turnover. J Cell Biol 161:779–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zagotta WN, Hoshi T, Aldrich RW (1990). Restoration of inactivation in mutants of Shaker potassium channels by a peptide derived from ShB. Science 250:568–571. [DOI] [PubMed] [Google Scholar]
- Zhang H, Cracium LC, Mirshahi T, Rohács T, Lopes CMB, Jin T, Logothetis DE (2003). PIP2 activates KCNQ channels, and its hydrolysis underlies receptor-mediated inhibition of M currents. Neuron 37:963–975. [DOI] [PubMed] [Google Scholar]