

Abstract
Although p53 is a key modulator of cellular stress responses, the mechanism of p53-mediated apoptosis is ambiguous. p53 can mediate apoptosis in response to death stimuli by transcriptional activation of proapoptotic genes and transcriptional-independent mechanisms. Recent studies have shown that the p53 protein can directly induce permeabilization of the outer mitochondrial membrane by forming a inhibitory complex with a protective Bcl-2 family protein, resulting in cytochrome c release. However, how the mitochondrial p53 pathway mediates neuronal apoptosis after cerebral ischemia remains unclear. We examined the interaction between the mitochondrial p53 pathway and vulnerable hippocampal CA1 neurons in rats using a transient global cerebral ischemia (tGCI) model. Western blot analysis and immunofluorescent staining revealed mitochondrial p53 translocation after tGCI in the hippocampal CA1 neurons. Coimmunoprecipitation revealed that translocated p53 bound to Bcl-XL in the mitochondrial fraction. To examine the effect of a specific p53 inhibitor on the mitochondrial p53 pathway and apoptotic cell death after tGCI, we intravenously administered pifithrin-α (PFT). Mitochondrial p53 translocation and interaction between p53 and Bcl-XL were prevented by treatment with PFT. Moreover, cytochrome c release from mitochondria and subsequent apoptotic CA1 neuronal death were decreased with PFT treatment. These results suggest that the mitochondrial p53 pathway is one of the novel mechanisms mediating delayed death of vulnerable hippocampal CA1 neurons after tGCI.
Keywords: p53, mitochondrial translocation, global cerebral ischemia, apoptosis, hippocampal CA1 neurons, rat
Introduction
The tumor suppressor gene p53 is a central player in cell death, but the mechanism of p53-mediated apoptosis after cell death stimuli remains unclear. Current evidence indicates that p53 encodes a sequence-specific transcription factor that controls the expression of genes whose products mediate apoptosis. These products include Bax (Bcl-2-associated X protein) (Miyashita and Reed, 1995), Noxa (NADPH oxidase activator 1) (Oda et al., 2000a), p53AIP1 (p53 acetate-induced protein 1) (Oda et al., 2000b), and PUMA (p53-upregulated modulator of apoptosis) (Nakano and Vousden, 2001; Yu et al., 2001), which act directly on mitochondria and induce apoptosis. In addition, evidence for transcription-independent p53-mediated apoptosis has been accumulating (Chipuk et al., 2004). In some cell types, a fraction of stabilized p53 rapidly translocates to mitochondria in response to a death stimulus (Marchenko et al., 2000; Sansome et al., 2001; Mihara et al., 2003; Erster et al., 2004). Exogenous p53 was forcibly targeted to mitochondria in p53 null cancer cells, and mitochondrial p53 was shown to be sufficient to launch apoptosis and suppress colony formation in a transcription-independent way (Mihara et al., 2003). Moreover, endogenous mitochondrial p53 forms inhibitory complexes with protective Bcl-XL and Bcl-2 proteins, resulting in cytochrome c release from mitochondria (Mihara et al., 2003). This evidence suggests that p53 can contribute to apoptosis by direct signaling at the mitochondria, thereby amplifying transcription-dependent apoptosis of p53.
Mitochondria are central integrators and transducers for proapoptotic signals in neuronal apoptosis. A major reason for the central role of mitochondria is that these organelles store critical apoptotic activators and effectors of cell death, such as cytochrome c, in their intermembranous space. Permeabilization of mitochondrial membranes causes the release of these activators and triggers apoptotic cell death. Involvement of the mitochondrial pathway in apoptotic cell death after in vivo experimental ischemia has been reported (Fujimura et al., 1999; Sugawara et al., 1999). The transient global cerebral ischemia (tGCI) model has been used to analyze selective vulnerability in the hippocampal CA1 subregion. Neuronal death in the CA1 subregion after tGCI has been shown to occur in a delayed manner (Kirino, 1982). We reported that mitochondrial release of cytochrome c is involved in this delayed neuronal death after tGCI (Sugawara et al., 1999). However, the signaling pathways upstream of the mitochondrial apoptotic mechanism engaged in delayed neuronal death are not fully understood.
Recent findings implicate p53 in neuronal death that occurs in stroke (Crumrine et al., 1994; Li et al., 1994; Tomasevic et al., 1999b; Saito et al., 2005). Many studies have demonstrated that p53 functions as a proapoptotic factor and that it upregulates in apoptotic lesions after an in vivo stroke. However, the precise mechanism of p53 in neuronal injury after stroke remains unknown. Moreover, whether the mitochondrial p53 pathway could affect cytochrome c release and subsequent neuronal cell death after stroke has not been demonstrated. The present study was designed to clarify the role of the mitochondrial p53 pathway after tGCI.
Materials and Methods
Global cerebral ischemia.
Five minutes of tGCI was induced by bilateral common carotid artery occlusion combined with hypotension, using a method described previously with some modifications (Smith et al., 1984; Sugawara et al., 1999). Male Sprague Dawley rats (300–350 g) were anesthetized with 2.0% isoflurane in 70% nitrous oxide and 30% oxygen using a face mask. Rectal temperature was controlled at 37°C during surgery with a homeothermic blanket. The femoral artery was exposed and catheterized with a polyethylene PE-50 catheter to allow continuous recording of arterial blood pressure. A midline neck skin incision was made, and the right jugular vein and both common carotid arteries were exposed. After intravenous injection of 150 IU/kg heparin, blood was quickly withdrawn via the jugular vein. When the mean arterial blood pressure became 30 mmHg, both common carotid arteries were clamped with surgical clips. Blood pressure was maintained at 30 mmHg by withdrawing or infusing blood through the jugular vein during the ischemic period. After 5 min of ischemia, the clips were removed and the blood was reinfused. Sham-operated animals underwent exposure of vessels without blood withdrawal or clamping of carotid arteries. The animals were maintained in an air-conditioned room at 20°C with ad libitum access to food and water before and after surgery. All animals were treated in accordance with Stanford University guidelines, and the animal protocols were approved by the Administrative Panel on Laboratory Animal Care of Stanford University.
Drug treatment.
To examine the effect of a specific p53 inhibitor on the mitochondrial p53 pathway and apoptotic cell death after tGCI, we administered pifithrin-α (PFT) (Sigma, St. Louis, MO) after tGCI. PFT was dissolved in dimethylsulfoxide and PBS. This drug (0.2, 2, or 4 mg/kg in dimethylsulfoxide in PBS) and the vehicle (dimethylsulfoxide in PBS) were injected intravenously via the left jugular vein just after reperfusion as described previously (Leker et al., 2004).
Western blot analysis.
Fresh brain tissue was removed after 1, 4, 8, 24, and 72 h of reperfusion (n = 4 each). The brain tissue was cut into 1 mm coronal slices using a brain matrix (Zivic Laboratories, Pittsburgh, PA), and the bilateral hippocampus was removed. Then, the hippocampal CA1 subregion was quickly dissected under a microscope and used as a sample. Protein extraction of both the cytosolic and mitochondrial fractions was performed using a multiple centrifugation method as described previously (Fujimura et al., 1999). The tissue was homogenized by gently douncing 30 times in a glass tissue grinder (Wheaton, Millville, NJ) in 7 vol of cold suspension buffer [20 mm HEPES-KOH, pH 7.5, 250 mm sucrose, 10 mm KCl, 1.5 mm MgCl2, 1 mm EDTA, and 1 mm EGTA plus 0.7% protease and phosphatase inhibitor cocktails (Sigma)]. The homogenate was centrifuged at 750 × g for 10 min at 4°C and then at 10,000 × g for 20 min at 4°C. The 10,000 × g pellets were used to obtain the mitochondrial fraction. The supernatant was further centrifuged at 100,000 × g for 60 min at 4°C and was then used for the cytosolic analysis. Protein concentrations were determined by comparison with a known concentration of bovine serum albumin using a kit (Pierce, Rockford, IL). After adding the same volume of Tris–glycine SDS sample buffer (Invitrogen, Carlsbad, CA), we loaded equal amounts of the samples (4–15 μg) per lane. The proteins were separated by SDS-PAGE on a 10–20% Tris–glycine gel (Novex, San Diego, CA) and transferred to a polyvinylidene difluoride membrane (Novex). The primary antibodies were as follows: 1:250 dilution of mouse monoclonal antibody against p53 (554147; BD Biosciences, San Jose, CA), 1:1000 dilution of rabbit polyclonal antibody against Bcl-XL (2762; Cell Signaling Technology, Beverly, MA), 1:1000 dilution of mouse monoclonal antibody against Bcl-2 (610538; BD Biosciences), 1:1000 dilution of rabbit polyclonal antibody against PUMA (4976; Cell Signaling Technology), 1:250 dilution of rabbit polyclonal antibody against Noxa (52–3617; Zymed Laboratories, South San Francisco, CA), 1:1000 dilution of mouse monoclonal antibody against Bax (sc-7480; Santa Cruz Biotechnology, Santa Cruz, CA), 1:1000 dilution of mouse monoclonal antibody against cytochrome c (556433; BD Biosciences), 1:1000 dilution of rabbit polyclonal antibody against caspase-9 (sc-8355; Santa Cruz Biotechnology), 1:10,000 dilution of mouse monoclonal antibody against β-actin (A5441; Sigma), and 1:5000 dilution of mouse monoclonal antibody against cytochrome oxidase (COX) subunit IV (A21348; Invitrogen). Western blots were performed with horseradish peroxidase-conjugated anti-rabbit IgG or anti-mouse IgG (Amersham Biosciences, Buckinghamshire, UK) using enhanced chemiluminescence Western blotting detection reagents (Amersham Biosciences). The film was scanned with a GS-700 imaging densitometer (Bio-Rad, Hercules, CA), and the results were quantified using Multi-Analyst software (Bio-Rad). To confirm the specificity of the antibody, Western blotting with the anti-p53 antibody preabsorbed with the p53 peptide was also performed.
Coimmunoprecipitation.
Sample preparation of mitochondrial fractions was performed as described above in the Western blotting method. The procedure for precipitation was performed as described previously (Saito et al., 2003). Fresh brain tissue was obtained from the hippocampal CA1 subregion after 1, 4, 8, 24, and 72 h of reperfusion (n = 4 each). Protein concentrations were determined by comparison with a known concentration of bovine serum albumin using a kit (Pierce). Thirty micrograms of protein from the mitochondrial fraction were used for coimmunoprecipitation. Whole brain extract was included as a positive control. The protein sample was incubated with 50% slurry of protein G-Sepharose (Amersham Biosciences, Uppsala, Sweden) for 1 h at 4°C, and this mixed sample was centrifuged at 12,000 × g for 1 min. The supernatant was incubated with 2 μg of polyclonal rabbit anti-Bcl-XL antibody (2762; Cell Signaling Technology) or monoclonal anti-mouse Bcl-2 antibody (610538; BD Biosciences) and 20 μl of protein G-Sepharose (50% slurry) for 2 h at 4°C. The negative control was prepared with protein G-Sepharose without an antibody. The 14,000 × g pellets were washed three times and used as the samples bound to each antibody. After adding the same volume of Tris–glycine SDS sample buffer (Invitrogen) to the samples, we boiled these samples to remove the Sepharose beads. After centrifugation at 14,000 × g for 1 min, the supernatant was immunoblotted with a 1:250 dilution of an anti-p53 antibody (554147; BD Biosciences), a 1:1000 dilution of the anti-Bcl-XL antibody (2762; Cell Signaling Technology), or a 1:1000 dilution of the anti-Bcl-2 antibody (610538; BD Biosciences) as described above in the Western blotting method.
Immunofluorescent staining.
The experimental animals were killed after 1, 8, 24, and 72 h of reperfusion. The brains were removed, rapidly frozen in −20°C 2-methylbutane and −50°C dry ice, and stored at −80°C. They were sectioned with a cryostat to a thickness of 20 μm. Frozen brain sections at the level of the hippocampus were placed on slides and fixed by 4% paraformaldehyde in PBS, pH 7.4, for 30 min. After blocking nonspecific binding with 3% bovine serum albumin, the sections were incubated with a rabbit polyclonal anti-p53 antibody (sc-6423; Santa Cruz Biotechnology) at a dilution of 1:50, followed by Texas Red-conjugated anti-rabbit IgG antibody (Jackson ImmunoResearch, West Grove, PA) at a dilution of 1:200. The sections were then covered with Vectashield mounting medium with 4′,6-diamidino-2-phenylindole (DAPI) (Vector Laboratories, Burlingame, CA). To evaluate colocalization of p53 and Bcl-XL, neuron-specific nuclear protein (NeuN), or COX IV, we performed double-immunofluorescent staining. The sections fixed by 4% paraformaldehyde were immunostained with an anti-p53 antibody (sc-6423; Santa Cruz Biotechnology) followed by Texas Red-conjugated anti-rabbit IgG antibody (Jackson ImmunoResearch) as described above. The sections were then incubated with a mouse antibody against NeuN (MAB377X; Chemicon, Temecula, CA) or Bcl-XL (610209; BD Biosciences) at a dilution of 1:50, followed by a fluorescein isothiocyanate-conjugated donkey anti-mouse IgG antibody (Jackson ImmunoResearch) at a dilution of 1:200. For double-immunofluorescent staining of p53 and COX IV, the sections were reacted with a rabbit antibody against COX IV (4844; Cell Signaling Technology) at a dilution of 1:100, followed by fluorescein isothiocyanate-conjugated anti-rabbit monovalent Fab fragments of secondary antibody (Jackson ImmunoResearch) at a dilution of 1:100 for labeling and blocking of COX IV. Then, the sections were incubated with an anti-p53 antibody (sc-6423; Santa Cruz Biotechnology), followed by Texas Red-conjugated anti-rabbit IgG antibody (Jackson ImmunoResearch). The sections were then covered with Vectashield mounting medium with DAPI (Vector Laboratories) and examined under an LSM510 confocal laser scanning microscope (Zeiss, Thornwood, NY).
Histological analysis of hippocampal injury.
The experimental animals were killed after 72 h of reperfusion. The brains were removed, rapidly frozen in −20°C 2-methylbutane and −50°C dry ice, and stored at −80°C. They were sectioned with a cryostat to a thickness of 20 μm and stained with cresyl violet. Neuronal damage in the hippocampus was qualitatively evaluated on the basis of a scoring system of 0 to 4 as described previously (Murakami et al., 1997): grade 0, no damage to neurons in the CA1 subregion; grade 1, scattered ischemic neurons in the CA1 subregion; grade 2, moderate ischemic damage in the CA1 subregion; grade 3, entire pyramidal cells damaged in the CA1 subregion; and grade 4, extensive cell damage in all hippocampal subregions.
In situ labeling of DNA fragmentation.
The sections used for histological analysis of hippocampal injury were used for this study. Frozen brain sections (20 μm thick) at the level of the hippocampus were placed on slides and stained using an in situ technique [terminal deoxynucleotidyl transferase-mediated uridine 5′-triphosphate-biotin nick end labeling (TUNEL) reaction] to detect the DNA-free 3′-OH ends as described previously, with some modification (Sugawara et al., 1999). Briefly, sections were fixed for 30 min in 4% formaldehyde in PBS, pH 7.4. The slides were incubated with NeuroPore (Trevigen, Gaithersburg, MD) for 30 min. They were placed in 1× terminal deoxynucleotidyl transferase buffer (Invitrogen) for 30 min, followed by reaction with terminal deoxynucleotidyl transferase enzyme (Invitrogen) and biotinylated 16-dUTP (Roche Diagnostics, Indianapolis, IN) at 37°C for 60 min. The sections were washed in 2× SSC (150 mol/liter sodium chloride and 15 mol/liter sodium citrate, pH 7.4) for 15 min, followed by washing in PBS two times for 15 min each time. The avidin–biotin technique was applied, and then the nuclei were counterstained with methyl green solution.
Cell death assay.
For quantification of apoptotic-related DNA fragmentation, we used a commercial enzyme immunoassay to determine cytoplasmic histone-associated DNA fragments (Roche Molecular Biochemicals, Mannheim, Germany), which detect apoptotic but not necrotic cell death (Leist et al., 1998). A sample preparation was used as described in the Western blotting method. Fresh brain tissue was taken after 24 and 72 h of reperfusion (n = 4 each) and homogenized with a Teflon homogenizer in 7 vol of ice-cold buffer (50 mm KH2PO4 and 0.1 mm EDTA, pH 7.8). The homogenate was centrifuged at 750 × g for 10 min at 4°C and then at 10,000 × g for 20 min at 4°C. The supernatant was further centrifuged at 100,000 × g for 60 min at 4°C. The resulting supernatant was collected, and the protein concentration was determined. A cytosolic volume containing 20 μg of protein was used for the enzyme-linked immunosorbent assay, following the protocol of the manufacturer.
Cell-counting procedure and statistical analysis.
For quantification of the TUNEL studies, intact pyramidal cells with a distinct nucleus or TUNEL-positive cells were counted in a 1 mm length of the middle portion of the CA1 subfield as described previously (Hayashi et al., 2003a). For evaluation of the results of the Western blot and coimmunoprecipitation studies, the data obtained from each time point (n = 4) were compared with those from the sham-operated group using one-way ANOVA with appropriate post hoc tests (SigmaStat software; Jandel, San Rafael, CA). Comparisons between two groups were achieved with Student’s unpaired t test. The data are expressed as mean ± SD, and significance was accepted with p < 0.05.
Results
Western blot analysis demonstrated mitochondrial translocation of p53 in the CA1 subregion after tGCI
p53 immunoreactivity was evident as a band in both the cytosolic and mitochondrial fractions from the hippocampal CA1 subregion (Fig. 1A). Under normal conditions, cytosolic-dominant distribution of p53 was observed. COX was strongly expressed in the mitochondrial fraction, but virtually no immunoreactivity was seen in the cytosolic fraction. Mitochondrial p53 was increased as early as 1 h after reperfusion, and, 8 and 24 h after reperfusion, a significant increase in mitochondrial p53 was seen compared with the non-ischemic control (Fig. 1B) (p < 0.05). In contrast, cytosolic p53 was decreased as early as 1 h after reperfusion and significantly decreased 8 h after reperfusion compared with the non-ischemic control (Fig. 1C) (p < 0.05). These results suggest that mitochondrial translocation of p53 occurred in the hippocampal CA1 subregion after tGCI.
Figure 1.
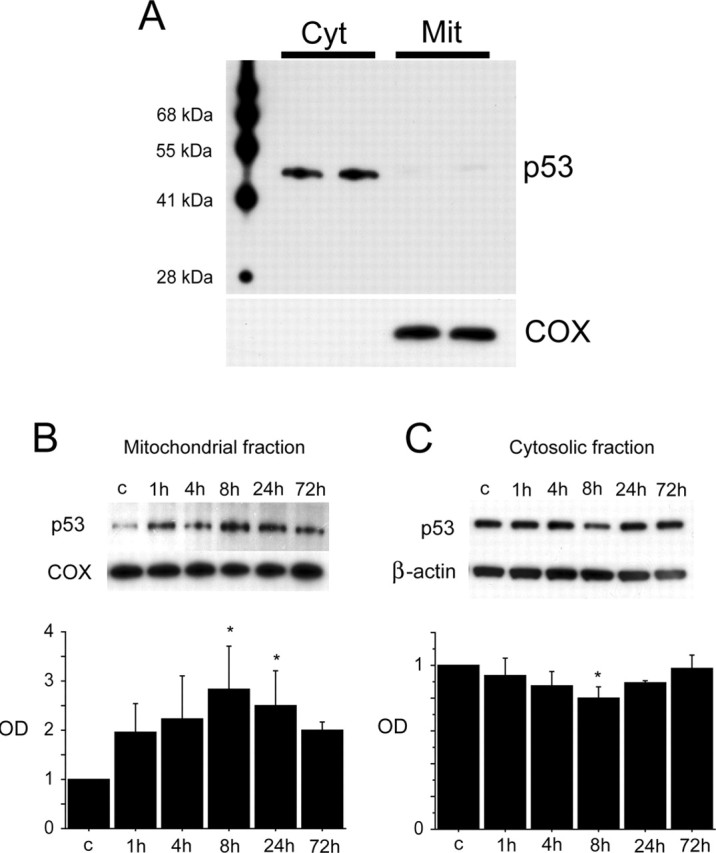
Western blot analysis of p53 in the hippocampal CA1 subregion. A, Western blot analysis of p53 from the cytosolic (Cyt) and mitochondrial (Mit) fractions showed cytosolic-dominant distribution of p53 under normal conditions. p53 immunoreactivity was evident as a single band with a molecular mass of 53 kDa in both the cytosolic and mitochondrial fractions. The results of the COX analysis are shown as an internal mitochondrial control. Molecular weight markers are depicted to the left. B, Western blot analysis of p53 in the mitochondrial fraction from the hippocampal CA1 subregion after tGCI. Expression of mitochondrial p53 showed a gradual increase in a time-dependent manner after tGCI. A quantitative analysis showed that mitochondrial p53 expression increased significantly 8 and 24 h after tGCI (n = 4; *p < 0.05). The results of the COX analysis are shown as an internal mitochondrial control. OD, Optical density. C, Western blot analysis of p53 in the cytosolic fraction from the hippocampal CA1 subregion after tGCI. Expression of cytosolic p53 showed a gradual decrease after tGCI. A quantitative analysis showed that p53 expression was decreased significantly 8 h after tGCI (n = 4; *p < 0.05). Thereafter, it gradually increased. The results of the β-actin analysis are shown as an internal cytosolic control.
Immunofluorescent staining demonstrated that mitochondrial localization of p53 occurred in CA1 neurons after tGCI
Immunofluorescent staining for p53 showed a slight cytosolic immunoreactivity of p53 in both the hippocampal CA1 and CA3 subregions in the non-ischemic brains (Fig. 2). p53 immunoreactivity was gradually increased after reperfusion and peaked 8 h after reperfusion in the hippocampal CA1 subregion. The increased signals of p53 were observed as small particles in the cytosol. Some hippocampal CA1 neurons showed nuclear p53 staining 72 h after reperfusion, when >85% of hippocampal CA1 neurons die (Sugawara et al., 2002). In contrast, p53 immunoreactivity in the CA3 subregion showed no difference among all time points after reperfusion (Fig. 2). Double immunofluorescence for p53 and COX demonstrated that p53 colocalized with COX, which was used as a mitochondrial marker, in the hippocampal CA1 subregion 8 h after tGCI (Fig. 3A–C). Double immunofluorescence for p53 and NeuN demonstrated that p53 expression colocalized with neurons in the hippocampal CA1 subregion 8 h after reperfusion (Fig. 3D–F). These results suggest that mitochondrial localization of p53 occurs in hippocampal CA1 neurons after tGCI.
Figure 2.
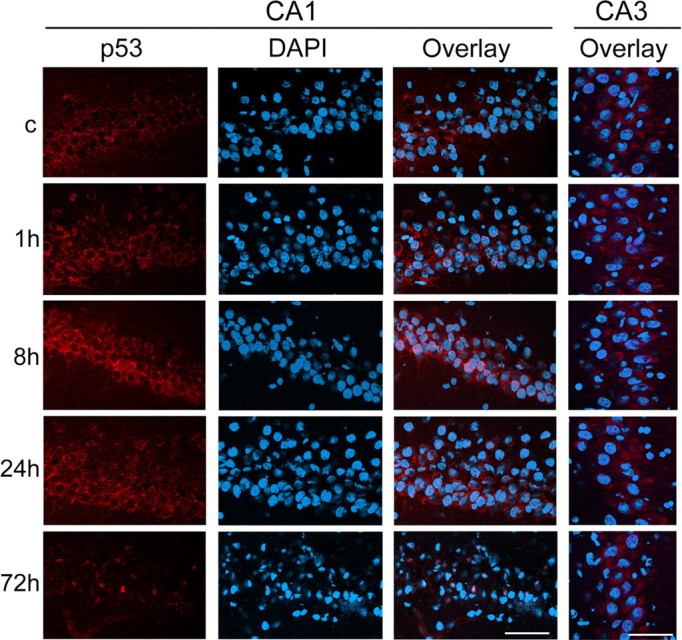
Representative photomicrographs of fluorescent double staining of p53 (red) and DAPI (blue) in the hippocampal CA1 and CA3 subregions after tGCI. p53 staining was indistinct in the non-ischemic brains (c) in both the CA1 and CA3 subregions. Eight hours after reperfusion, it became more intense and had a punctate appearance in the cytosol of the hippocampal CA1 neurons; however, p53 immunoreactivity in the CA3 subregion was no different among all time points after reperfusion. Some hippocampal CA1 cells showed nuclear p53 localization 72 h after reperfusion. Scale bars, 50 μm.
Figure 3.
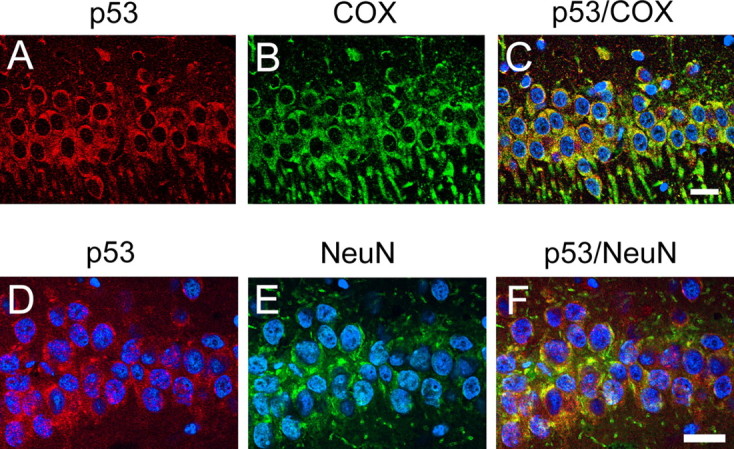
Representative photomicrographs of fluorescent double staining of p53 (red) and COX (green) (A–C) and p53 (red) and NeuN (green) (D–F) in the hippocampal CA1 subregion 8 h after tGCI. Nuclei were counterstained with DAPI (blue). p53 staining was observed in the cytosol (A). COX, which was used as a mitochondrial marker, was also distributed in the cytosol (B). An overlapped image showed that p53 colocalized with COX in the hippocampal CA1 subregion (C). NeuN immunoreactivity (green) showed the distribution of neurons (E). Eight hours after ischemia, p53 was almost exclusively localized in NeuN-positive pyramidal neurons of the hippocampal CA1 subregion (D). An overlapped photo from the same field (F) confirmed the colocalization. Scale bars, 20 μm.
Coimmunoprecipitation demonstrated that direct binding of p53 to Bcl-XL increased in the CA1 subregion after tGCI
To investigate the direct interaction between p53 and Bcl-XL or Bcl-2, we performed coimmunoprecipitation. With Western blot analysis, Bcl-XL and Bcl-2 immunoreactivity was evident as bands of 30 and 26 kDa, respectively, in the mitochondrial fraction from the hippocampal CA1 subregion. There was no significant difference in Bcl-XL or Bcl-2 among all time points after reperfusion (Fig. 4A,C). p53 expression precipitated by Bcl-XL in the mitochondrial fraction increased time dependently, and a significant increase was observed 8 h after reperfusion (Fig. 4B) (p < 0 0.05). In contrast, p53 expression precipitated by Bcl-2 in the mitochondrial fraction was detected as a robust band, and there was no difference among all time points after reperfusion (Fig. 4D). Double immunofluorescence for p53 and Bcl-XL demonstrated that p53 expression colocalized with Bcl-XL in the hippocampal CA1 subregion 8 h after reperfusion (Fig. 4E). These results suggest that direct binding between p53 and Bcl-XL was increased in the hippocampal CA1 subregion after tGCI.
Figure 4.

Direct binding of p53 to Bcl-XL increased in the CA1 subregion after tGCI. A, Western blot analysis of Bcl-XL in the mitochondrial fraction from the hippocampal CA1 subregion after tGCI. Bcl-XL immunoreactivity showed no difference among all time points after tGCI (n = 4 each). The results of the COX analysis are shown as an internal mitochondrial control. c, Control; OD, optical density. B, Coimmunoprecipitation analysis for p53 immunoreactivity precipitated by Bcl-XL in the mitochondrial fraction from the hippocampal CA1 subregion after tGCI. p53 expression gradually increased in a time-dependent manner. Eight hours after tGCI, p53 expression significantly increased (n = 4; *p < 0 0.05). Bcl-XL was used to show equal precipitation. IP, Immunoprecipitation; IB, immunoblotting. C, Western blot analysis of Bcl-2 in the mitochondrial fraction from the hippocampal CA1 subregion after tGCI. Bcl-2 immunoreactivity showed no difference among all time points after tGCI (n = 4 each). The results of the COX analysis are shown as an internal mitochondrial control. D, Coimmunoprecipitation analysis for p53 immunoreactivity precipitated by Bcl-2 in the mitochondrial fraction from the hippocampal CA1 subregion after tGCI. p53 immunoreactivity showed no difference among all time points after tGCI (n = 4 each). Bcl-2 was used to show equal precipitation. E, Representative photomicrographs show fluorescent double staining of p53 (red) and Bcl-XL (green) in the hippocampal CA1 subregion 8 h after tGCI. Nuclei were counterstained with DAPI (blue). An overlapped image demonstrated that p53 expression colocalized with Bcl-XL in the hippocampal CA1 subregion 8 h after reperfusion. Scale bar, 20 μm.
PFT administration decreased histologic CA1 neuronal damage and TUNEL-positive cells after tGCI
To investigate the involvement of p53 in the delayed death of hippocampal CA1 neurons after tGCI, we did an inhibitor study with PFT, a p53 inhibitor. Timing of administration and dose of drug, which were considered to have neuroprotective effects, were decided based on previous studies (Culmsee et al., 2003; Leker et al., 2004). In this study, 2 mg/kg PFT was intravenously administered just after reperfusion. In both the nontreated and vehicle-treated animals, delayed death of hippocampal CA1 neurons was observed 72 h after ischemia, which was consistent with our previous reports using the same tGCI model (Chan et al., 1998; Sugawara et al., 1999). Most of the CA1 neurons in both the nontreated and vehicle-treated animals had shrunken, triangular-shaped, condensed nuclei in the cresyl violet-stained section (Fig. 5C,G,I,M); however, normal features were preserved in the non-ischemic animals and in the PFT-treated animals (Fig. 5A,E,K,O). Most of the morphologically damaged neurons stained with cresyl violet were also positive for TUNEL (Fig. 5B,D,F,H,J,L,N,P). Neuronal damage in the hippocampal CA1 subregion, which was qualitatively evaluated on the basis of a scoring system (Murakami et al., 1997), was significantly decreased in the PFT-treated animals compared with the nontreated and the vehicle-treated animals (Fig. 5Q, *p = 0.0028, **p = 0.0145). In contrast, no significant difference was observed between the nontreated and vehicle-treated animals. Furthermore, the counting studies of TUNEL-positive cells showed that they were significantly decreased in the PFT-treated animals compared with the nontreated and vehicle-treated animals (Fig. 5R, #p = 0.0010, ##p = 0.0026). No significant difference was observed between the nontreated and vehicle-treated animals. These results suggest that intravenous PFT administration might be neuroprotective after tGCI.
Figure 5.

Representative photomicrographs of cresyl violet staining (A, C, E, G, I, K, M, O) and TUNEL staining (B, D, F, H, J, L, N, P), a histological analysis of hippocampal injury (Q), and a cell-counting study of TUNEL-positive cells (R) 72 h after surgery. A–D and I–L show low magnification of the entire hippocampus, and E–H and M–P show high magnification of the hippocampal CA1 subregion. In the non-ischemic animals, no hippocampal CA1 damage was observed (A, B, E, F). Seventy-two hours after ischemia, most of the CA1 neurons in the nontreated (C, G) and vehicle-treated (I, M) animals had shrunken, triangular-shaped, condensed nuclei on the cresyl violet-stained sections. However, the normal features of the nuclei of many neurons were preserved in the PFT-treated animals (K, O). A majority of the damaged neurons became TUNEL-positive (D, H, J, L, N, P). Histological analysis of hippocampal injury (Q) showed a significant decrease in injured CA1 neurons 72 h after tGCI in the PFT-treated animals compared with the non-ischemic and vehicle-treated animals (n = 5; *p = 0.0028, **p = 0.0145). The cell-counting study (R) showed a significant decrease in TUNEL-positive cells in the hippocampal CA1 subregion 72 h after ischemia in the PFT-treated animals compared with the non-ischemic and vehicle-treated animals (n = 5; #p = 0.0010, ##p = 0.0026). c, Control. Scale bar, 50 μm.
Cell death assay demonstrated that DNA fragmentation after tGCI significantly decreased with intravenous administration of PFT
For additional investigation of neuroprotection by PFT against tGCI, we examined apoptotic-related DNA fragmentation after tGCI using a commercial cell death detection kit. In this study, 2 mg/kg PFT was intravenously administered just after reperfusion. As shown in Figure 6A, DNA fragmentation was significantly increased in the hippocampal CA1 subregion 72 h after tGCI compared with the non-ischemic brains in the nontreated animals (Fig. 6A, *p < 0.0001). This result was consistent with the TUNEL-positive cell-counting study. Hence, we examined DNA fragmentation 72 h after reperfusion in the nontreated, vehicle-treated, and PFT-treated animals to assess the protective effect of PFT on DNA damage. As shown in Figure 6B, DNA fragmentation at 72 h of reperfusion was significantly reduced in the hippocampal CA1 subregion of the PFT-treated animals compared with the nontreated and vehicle-treated animals at the same time point (**p = 0.0020, ***p = 0.0066). These results suggest that intravenous PFT administration inhibited DNA fragmentation in the hippocampal CA1 subregion after tGCI.
Figure 6.

DNA fragmentation significantly decreased with administration of PFT. A, Apoptotic-related DNA fragmentation after ischemia was analyzed with a commercial cell death detection kit (n = 4 each). DNA fragmentation increased significantly in the hippocampal CA1 subregion 72 h after tGCI compared with the non-ischemic brains (c) (*p < 0.0001). B, DNA fragmentation in the hippocampal CA1 subregion 72 h after tGCI decreased significantly in the PFT-treated animals compared with the nontreated and vehicle-treated animals (n = 4; **p = 0.0020, ***p = 0.0066).
PFT administration (2 mg/kg) decreased mitochondrial translocation of p53 and direct binding of p53 to Bcl-XL, which resulted in inhibition of cytosolic cytochrome c release and subsequent activation of caspase-9
We next addressed the question of whether PFT plays a role in the mitochondrial or transcriptional p53 pathways after tGCI. We intravenously administered 0.2, 2, or 4 mg/kg PFT just after reperfusion. Western blot analysis showed that mitochondrial p53 expression significantly decreased in the animals treated with 0.2 and 2 mg/kg PFT compared with the vehicle-treated animals 8 h after tGCI (Fig. 7A) (p < 0.05). However, mitochondrial translocation was not inhibited by treatment with 4 mg/kg PFT. PUMA and Noxa, which are transcriptionally controlled by p53 (Oda et al., 2000a; Nakano and Vousden, 2001; Yu et al., 2001), were detected as slight bands in the mitochondrial fraction of the non-ischemic controls. Expression of these proteins became prominent in the vehicle-treated animals 8 h after tGCI (Fig. 7A). Although expression of these proteins showed no statistical difference in the animals treated with 0.2 and 2 mg/kg PFT compared with the vehicle-treated animals, expression was significantly decreased in the rats treated with 4 mg/kg PFT (p < 0.05). Bax is also transcriptionally controlled by p53 (Miyashita and Reed, 1995), but immunoreactivity of Bax showed no prominent changes in the mitochondrial fraction among all groups (Fig. 7A). Bcl-XL revealed no significant changes after tGCI in this study (Fig. 4A), and PFT administration had no effect on the expression of this protein (Fig. 7A). Next, we investigated the role of PFT in protein interaction between p53 and Bcl-XL. Coimmunoprecipitation showed that p53 expression precipitated by Bcl-XL in the rats treated with 2 mg/kg PFT significantly decreased 8 h after tGCI (Fig. 7B) (p < 0.05). To investigate the effect of PFT downstream of the mitochondrial p53 pathway, we examined cytochrome c release to the cytosol and caspase-9 activation. A significant increase in cytosolic cytochrome c was observed 8 and 24 h after reperfusion compared with the non-ischemic brains in the vehicle-treated animals (Fig. 7C, *p < 0.05). A comparison between treatment with the vehicle and 2 mg/kg PFT showed that this protein was less abundant 8 and 24 h after ischemia in the rats treated with 2 mg/kg PFT (#p < 0.05). Next, we examined the activated form of caspase-9, expression of the cleaved p12 subunit, as an initiator of the caspase chain reaction. Expression of cleaved caspase-9 was barely observed in the non-ischemic brains (Fig. 7C), but it gradually increased after reperfusion and showed a significant increase 8 and 24 h after tGCI compared with the non-ischemic brains in the vehicle-treated animals (*p < 0.05). A comparison between treatment with the vehicle and 2 mg/kg PFT showed that this protein was less abundant in the PFT-treated animals 8 and 24 h after ischemia (#p < 0.05, ##p < 0.01). These results indicate that PFT administration (2 mg/kg) inhibits mitochondrial p53 translocation and direct binding between p53 and Bcl-XL in mitochondria, resulting in inhibition of cytosolic cytochrome c release and subsequent activation of caspase-9.
Figure 7.

PFT administration (2 mg/kg) decreased mitochondrial p53, direct binding of p53 to Bcl-XL, cytosolic cytochrome c release, and subsequent activation of caspase-9. A, Western blot analysis of p53, PUMA, Noxa, Bax, and Bcl-XL in the mitochondrial fraction from the hippocampal CA1 subregion 8 h after tGCI. Mitochondrial p53 expression significantly decreased after treatment with 0.2 and 2 mg/kg PFT compared with vehicle treatment (n = 4; *p < 0.05). Mitochondrial PUMA and Noxa expression significantly decreased in the rats treated with 4 mg/kg PFT compared with the vehicle-treated animals (n = 4; *p < 0.05). Mitochondrial Bax and Bcl-XL showed no significant difference among all groups. The results of the COX analysis are shown as an internal mitochondrial control. OD, Optical density. V, vehicle. B, Coimmunoprecipitation analysis for p53 immunoreactivity precipitated by Bcl-XL in the mitochondrial fraction from the hippocampal CA1 subregion 8 h after tGCI. p53 expression significantly decreased in the animals treated with 2 mg/kg PFT compared with the vehicle-treated animals (n = 4; *p < 0.05). Bcl-XL was used to show equal precipitation. IP, Immunoprecipitation; IB, immunoblotting. C, Western blot analysis of cytochrome c and cleaved caspase-9 in the cytosolic fraction from the hippocampal CA1 subregion 8 h after tGCI. Expression of cytochrome c and cleaved caspase-9 were detected as bands with a molecular mass of 15 and 12 kDa, respectively. In the vehicle-treated animals, cytosolic cytochrome c was significantly increased 8 and 24 h after tGCI (n = 4; *p < 0.05). Cytosolic cytochrome c expression significantly decreased 8 and 24 h after tGCI in the rats treated with 2 mg/kg PFT compared with the vehicle-treated animals (n = 4; #p < 0.05). In the vehicle-treated animals, cleaved caspase-9 expression was significantly increased 8 and 24 h after tGCI (n = 4; *p < 0.05). Cleaved caspase-9 expression significantly decreased 8 and 24 h after tGCI in the animals treated with 2 mg/kg PFT compared with the vehicle-treated animals (n = 4; #p < 0.05, ##p < 0.05). The results of the β-actin analysis are shown as an internal cytosolic control. c, Control.
Discussion
It is well established that the rat tGCI model mimics conditions after transient cardiac arrest and causes selective neuronal death in vulnerable regions, such as hippocampal CA1 pyramidal cells, cerebellar Purkinje cells, and neurons in the third to fifth layers of the cerebral cortex (Kirino, 1982; Pulsinelli et al., 1982). This vulnerability has been attributed to many factors, such as glutamate neurotoxicity, calcium influx, expression of cell suicide genes, activation of apoptotic proteins, mitochondrial dysfunction, endoplasmic reticulum dysfunction, and oxygen free radicals (Abe et al., 1995; Chan, 1996; Sugawara et al., 1999; Hayashi et al., 2003b). Above all, mitochondrial cytochrome c release to the cytosol is considered the critical step toward apoptotic cell death for vulnerable hippocampal CA1 neurons after tGCI (Sugawara et al., 1999). However, upstream signaling of mitochondrial cytochrome c release after tGCI remains unclear. In this study, we demonstrated that mitochondrial p53 translocation and direct inhibitory binding to Bcl-XL induced mitochondrial release of cytochrome c and subsequent activation of caspase-9, resulting in delayed neuronal death of CA1 neurons after ischemia.
Known as the “guardian of the genome,” p53 is the most frequently mutated tumor suppressor in many forms of neoplasia. This enhances its importance as the master regulator of cell proliferation and death. Its regulation is highly complex and involves interactions with MDM2 (transformed mouse 3T3 cell double minute 2), alterations in p53 protein levels, and direct phosphorylation or acetylation (Levine, 1997). Once activated, p53 induces cell cycle arrest or apoptosis under many conditions in which DNA damage poses the risk of malignant transformation. p53 expression induces loss of viability in hippocampal cultures (Jordán et al., 1997), and deletion of the p53 gene confers resistance against excitotoxicity to hippocampal and cortical neurons (Xiang et al., 1996). In p53-dependent neuronal death in vitro, the multidomain Bcl-2 family member Bax has been identified as a major mediator of cell death (Xiang et al., 1998). Other p53-regulated Bcl-2 homology 3 (BH-3) only proteins, such as PUMA, Noxa, and Bid (BH-3 interacting domain death agonist), might also play key roles in neuronal apoptosis (Sax and El-Deiry, 2003). These reports suggest that neurons are clearly susceptible to p53-induced cell death, at least in vitro. However, the role of p53 in neuronal cell death in vivo remains less clear. Deletion of the p53 gene has been associated with neuroprotection after transient focal cerebral ischemia (Crumrine et al., 1994). In contrast, p53 protein levels increased mainly in the ischemia-resistant CA3 subregion after tGCI, suggesting that p53 is not a neuronal cell death marker after cerebral ischemia (Tomasevic et al., 1999b). This inconsistency might depend on the severity of ischemia and types of ischemic models. p53 might primarily serve a protective role by activating DNA repair mechanisms after mild to moderate insults but could become detrimental in response to severe ischemic insults (Tomasevic et al., 1999a). In our study, immunofluorescent staining showed that p53 immunoreactivity increased not in the CA3 neurons but in the CA1 neurons that were destined to die 2–3 d after tGCI. Moreover, p53 inhibition by PFT decreased delayed death of hippocampal CA1 neurons after tGCI. These results strongly suggest the detrimental effect of p53 after tGCI.
A fraction of induced p53 translocates to the mitochondria at the onset of p53-dependent apoptosis but not during p53-independent apoptosis or p53-mediated cell cycle arrest in vitro (Marchenko et al., 2000). Bypassing the nucleus by targeting p53 to mitochondria is still sufficient to launch apoptosis (Marchenko et al., 2000). This evidence suggests the contribution of p53 to apoptosis by direct signaling at the mitochondria in vitro. In the present study, immunofluorescence showed increased p53 signals in the CA1 neurons as small particles in the cytosol and that they colocalized with COX, which was used as a mitochondrial marker. Moreover, Western blot analysis revealed increased p53 immunoreactivity in the mitochondrial fraction and decreased p53 immunoreactivity in the cytosolic fraction after tGCI. These results provide evidence of mitochondrial p53 translocation after cerebral ischemia in vivo. Cytosolic p53 degradation through the MDM2 pathway and the ubiquitin–proteasome system was observed after cerebral ischemia (Saito et al., 2005). These degradation mechanisms need to be considered in regard to cytosolic p53 decreasing other than p53 translocation in the present study. Erster et al. (2004) found that mitochondrial p53 accumulation rapidly occurred in radiosensitive organs such as the thymus, spleen, testis, and brain within 2 h after exposure to γ-irradiation in vivo. They proposed that mitochondrial translocation of p53 triggers a rapid proapoptotic response that jumpstarts and amplifies the slower transcription-dependent response. In our study, Western blot analysis showed that p53 immunoreactivity in the mitochondrial fraction began to increase 1 h after ischemia and significantly increased 8 and 24 h after ischemia. In contrast, nuclear p53 immunoreactivity was first observed 72 h after ischemia in the immunofluorescent study. This result suggests the important role of rapid p53 mitochondrial accumulation in delayed death of CA1 neurons.
The compound PFT can selectively inhibit p53 transcriptional activity in various cell lines and prevents DNA damage-induced apoptosis (Komarov et al., 1999). PFT protects normal cells, but not the cells from p53-null mice, in culture and in vivo, against death induced by anticancer treatments. In addition, PFT protected neurons against oxygen-glucose deprivation in vitro (Culmsee et al., 2003) and reduced ischemic brain damage in both transient focal cerebral ischemia (Culmsee et al., 2001, 2003; Leker et al., 2004) and tGCI (Culmsee et al., 2003). Neuroprotection with a single dose of PFT was observed up to 3 h after ischemia (Culmsee et al., 2003; Leker et al., 2004). Mechanisms of PFT neuroprotection against cerebral ischemia have been reported to include inhibition of p53 translocation into the nucleus and prevention of its DNA-binding activity (Culmsee et al., 2001; Leker et al., 2004) or preservation of nuclear factor-κB activity (Culmsee et al., 2003). In this study, apoptotic neuronal cell death in the hippocampal CA1 subregion after tGCI was significantly decreased by intravenous administration of 2 mg/kg PFT. Apoptotic cell death was confirmed by TUNEL staining and the cell death assay. Interestingly, the three different doses of PFT produced a U-shaped dose curve in mitochondrial translocation. A high dose of PFT (4 mg/kg) had no effect on mitochondrial p53 translocation after tGCI, but it inhibited upregulation of proapoptotic PUMA and Noxa, which are transcriptional target genes of p53. In contrast, low to moderate doses of PFT (0.2 and 2 mg/kg) selectively inhibited mitochondrial p53 translocation without inhibiting the transcriptional target genes. Moreover, 2 mg/kg PFT inhibited direct binding between p53 and Bcl-XL in mitochondria and cytochrome c release and subsequent caspase-9 activation. Inhibition of mitochondrial p53 translocation and subsequent apoptotic cell death by PFT were also observed in the renal ischemia-reperfusion model (Kelly et al., 2003). Mitochondrial release of cytochrome c is the critical step for delayed neuronal death of hippocampal CA1 neurons in our model of tGCI (Sugawara et al., 1999). The study on PFT administration suggests that mitochondrial p53 translocation and direct interaction between p53 and Bcl-XL might be among the upstream mechanisms of mitochondrial cytochrome c release after cerebral ischemia. Moreover, a reduction in oxidative stress inhibited cytochrome c release and subsequent neuronal death after tGCI in human copper/zinc-superoxide dismutase (SOD1) transgenic rats (Sugawara et al., 2002). We also observed a reduction in mitochondrial p53 translocation after tGCI in SOD1 transgenic rats (supplemental figure, available at www.jneurosci.org as supplemental material). This evidence might support the idea that mitochondrial p53 translocation is a key step to cytochrome c release and subsequent neuronal death after tGCI. In contrast, the role of the transcriptional pathway of p53 needs to be considered. Proapoptotic PUMA and Noxa expression was increased after tGCI and was inhibited by a high dose of PFT (4 mg/kg). A recent study suggested that the transcriptional p53 pathway has a cooperative relationship with the cytosolic p53 pathway in cell death mechanisms (Chipuk et al., 2005). Thus, this cooperative relationship might underlie the delayed neuronal death of hippocampal neurons after tGCI in our study. A more detailed relationship between the mitochondrial and nuclear pathways of p53 in neuronal death needs to be investigated in a future study.
Although our study demonstrated the important role p53 plays in ischemic neuronal death, it is likely that the inhibitor we used may have had an off-target effect. Thus, it is imperative that additional strategies, such as small interfering RNA or p53−/− mice, be used to support our hypothesis. In this regard, a recent study by Yonekura et al. (2006) has demonstrated that hippocampal CA1 neuronal death was significantly reduced in p53−/− mice compared with p53+/+ wild-type mice after global cerebral ischemia. Our rat study has provided additional insights into mechanisms of p53 mitochondrial translocation, p53/Bcl-XL binding, cytochrome c release, and caspase-9 activation that lead to neuronal apoptosis.
In summary, our present study provides evidence of a role for the mitochondrial p53 signaling pathway in the delayed neuronal death of hippocampal CA1 neurons. We found that p53 translocates to mitochondria and directly binds to Bcl-XL after tGCI. Mitochondrial translocation of p53 and protein interaction between p53 and Bcl-XL induce release of cytochrome c from mitochondria to the cytosol, resulting in delayed hippocampal CA1 neuronal death after tGCI.
Footnotes
This work was supported by National Institutes of Health Grants P50 NS14543, R01 NS25372, R01 NS36147, and R01 NS38653. We thank Liza Reola, Bernard Calagui, and Trisha Crandall for technical assistance, Cheryl Christensen for editorial assistance, and Elizabeth Hoyte for figure preparation.
References
- Abe K, Aoki M, Kawagoe J, Yoshida T, Hattori A, Kogure K, Itoyama Y (1995). Ischemic delayed neuronal death. A mitochondrial hypothesis. Stroke 26:1478–1489. [DOI] [PubMed] [Google Scholar]
- Chan PH (1996). Role of oxidants in ischemic brain damage. Stroke 27:1124–1129. [DOI] [PubMed] [Google Scholar]
- Chan PH, Kawase M, Murakami K, Chen SF, Li Y, Calagui B, Reola L, Carlson E, Epstein CJ (1998). Overexpression of SOD1 in transgenic rats protects vulnerable neurons against ischemic damage after global cerebral ischemia and reperfusion. J Neurosci 18:8292–8299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chipuk JE, Kuwana T, Bouchier-Hayes L, Droin NM, Newmeyer DD, Schuler M, Green DR (2004). Direct activation of Bax by p53 mediates mitochondrial membrane permeabilization and apoptosis. Science 303:1010–1014. [DOI] [PubMed] [Google Scholar]
- Chipuk JE, Bouchier-Hayes L, Kuwana T, Newmeyer DD, Green DR (2005). PUMA couples the nuclear and cytoplasmic proapoptotic function of p53. Science 309:1732–1735. [DOI] [PubMed] [Google Scholar]
- Crumrine RC, Thomas AL, Morgan PF (1994). Attenuation of p53 expression protects against focal ischemic damage in transgenic mice. J Cereb Blood Flow Metab 14:887–891. [DOI] [PubMed] [Google Scholar]
- Culmsee C, Zhu X, Yu Q-S, Chan SL, Camandola S, Guo Z, Greig NH, Mattson MP (2001). A synthetic inhibitor of p53 protects neurons against death induced by ischemic and excitotoxic insults, and amyloid β-peptide. J Neurochem 77:220–228. [DOI] [PubMed] [Google Scholar]
- Culmsee C, Siewe J, Junker V, Retiounskaia M, Schwarz S, Camandola S, El-Metainy S, Behnke H, Mattson MP, Krieglstein J (2003). Reciprocal inhibition of p53 and nuclear factor-κB transcriptional activities determines cell survival or death in neurons. J Neurosci 23:8586–8595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erster S, Mihara M, Kim RH, Petrenko O, Moll UM (2004). In vivo mitochondrial p53 translocation triggers a rapid first wave of cell death in response to DNA damage that can precede p53 target gene activation. Mol Cell Biol 24:6728–6741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujimura M, Morita-Fujimura Y, Kawase M, Copin J-C, Calagui B, Epstein CJ, Chan PH (1999). Manganese superoxide dismutase mediates the early release of mitochondrial cytochrome c and subsequent DNA fragmentation after permanent focal cerebral ischemia in mice. J Neurosci 19:3414–3422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi T, Saito A, Okuno S, Ferrand-Drake M, Chan PH (2003a). Induction of GRP78 by ischemic preconditioning reduces endoplasmic reticulum stress and prevents delayed neuronal cell death. J Cereb Blood Flow Metab 23:949–961. [DOI] [PubMed] [Google Scholar]
- Hayashi T, Saito A, Okuno S, Ferrand-Drake M, Dodd RL, Nishi T, Maier CM, Kinouchi H, Chan PH (2003b). Oxidative damage to the endoplasmic reticulum is implicated in ischemic neuronal cell death. J Cereb Blood Flow Metab 23:1117–1128. [DOI] [PubMed] [Google Scholar]
- Jordán J, Galindo MF, Prehn JHM, Weichselbaum RR, Beckett M, Ghadge GD, Roos RP, Leiden JM, Miller RJ (1997). p53 expression induces apoptosis in hippocampal pyramidal neuron cultures. J Neurosci 17:1397–1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly KJ, Plotkin Z, Vulgamott SL, Dagher PC (2003). p53 mediates the apoptotic response to GTP depletion after renal ischemia-reperfusion: protective role of a p53 inhibitor. J Am Soc Nephrol 14:128–138. [DOI] [PubMed] [Google Scholar]
- Kirino T (1982). Delayed neuronal death in the gerbil hippocampus following ischemia. Brain Res 239:57–69. [DOI] [PubMed] [Google Scholar]
- Komarov PG, Komarova EA, Kondratov RV, Christov-Tselkov K, Coon JS, Chernov MV, Gudkov AV (1999). A chemical inhibitor of p53 that protects mice from the side effects of cancer therapy. Science 285:1733–1737. [DOI] [PubMed] [Google Scholar]
- Leist M, Kühnle S, Single B, Nicotera P (1998). Differentiation between apoptotic and necrotic cell death by means of the BM cell death detection ELISA or annexin V staining. Biochemica 2:25–28. [Google Scholar]
- Leker RR, Aharonowiz M, Greig NH, Ovadia H (2004). The role of p53-induced apoptosis in cerebral ischemia: effects of the p53 inhibitor pifithrin α. Exp Neurol 187:478–486. [DOI] [PubMed] [Google Scholar]
- Levine AJ (1997). p53, the cellular gatekeeper for growth and division. Cell 88:323–331. [DOI] [PubMed] [Google Scholar]
- Li Y, Chopp M, Zhang ZG, Zaloga C, Niewenhuis L, Gautam S (1994). p53-immunoreactive protein and p53 mRNA expression after transient middle cerebral artery occlusion in rats. Stroke 25:849–855. [DOI] [PubMed] [Google Scholar]
- Marchenko ND, Zaika A, Moll UM (2000). Death signal-induced localization of p53 protein to mitochondria. A potential role in apoptotic signaling. J Biol Chem 275:16202–16212. [DOI] [PubMed] [Google Scholar]
- Mihara M, Erster S, Zaika A, Petrenko O, Chittenden T, Pancoska P, Moll UM (2003). p53 has a direct apoptogenic role at the mitochondria. Mol Cell 11:577–590. [DOI] [PubMed] [Google Scholar]
- Miyashita T, Reed JC (1995). Tumor suppressor p53 is a direct transcriptional activator of the human bax gene. Cell 80:293–299. [DOI] [PubMed] [Google Scholar]
- Murakami K, Kondo T, Epstein CJ, Chan PH (1997). Overexpression of CuZn-superoxide dismutase reduces hippocampal injury after global ischemia in transgenic mice. Stroke [Erratum (1997) 28:2573] 28:1797–1804. [DOI] [PubMed] [Google Scholar]
- Nakano K, Vousden KH (2001). PUMA, a novel proapoptotic gene, is induced by p53. Mol Cell 7:683–694. [DOI] [PubMed] [Google Scholar]
- Oda E, Ohki R, Murasawa H, Nemoto J, Shibue T, Yamashita T, Tokino T, Taniguchi T, Tanaka N (2000a). Noxa, a BH3-only member of the Bcl-2 family and candidate mediator of p53-induced apoptosis. Science 288:1053–1058. [DOI] [PubMed] [Google Scholar]
- Oda K, Arakawa H, Tanaka T, Matsuda K, Tanikawa C, Mori T, Nishimori H, Tamai K, Tokino T, Nakamura Y, Taya Y (2000b). p53AIP1, a potential mediator of p53-dependent apoptosis, and its regulation by Ser-46-phosphorylated p53. Cell 102:849–862. [DOI] [PubMed] [Google Scholar]
- Pulsinelli WA, Brierley JB, Plum F (1982). Temporal profile of neuronal damage in a model of transient forebrain ischemia. Ann Neurol 11:491–498. [DOI] [PubMed] [Google Scholar]
- Saito A, Hayashi T, Okuno S, Ferrand-Drake M, Chan PH (2003). Overexpression of copper/zinc superoxide dismutase in transgenic mice protects against neuronal cell death after transient focal ischemia by blocking activation of the Bad cell death signaling pathway. J Neurosci 23:1710–1718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito A, Hayashi T, Okuno S, Nishi T, Chan PH (2005). Modulation of p53 degradation via MDM2-mediated ubiquitylation and the ubiquitin-proteasome system during reperfusion after stroke: role of oxidative stress. J Cereb Blood Flow Metab 25:267–280. [DOI] [PubMed] [Google Scholar]
- Sansome C, Zaika A, Marchenko ND, Moll UM (2001). Hypoxia death stimulus induces translocation of p53 protein to mitochondria. Detection by immunofluorescence on whole cells. FEBS Lett 488:110–115. [DOI] [PubMed] [Google Scholar]
- Sax JK, El-Deiry WS (2003). p53 downstream targets and chemosensitivity. Cell Death Differ 10:413–417. [DOI] [PubMed] [Google Scholar]
- Smith ML, Bendek G, Dahlgren N, Rosen I, Wieloch T, Siesjö BK (1984). Models for studying long-term recovery following forebrain ischemia in the rat. 2. A 2-vessel occlusion model. Acta Neurol Scand 69:385–401. [DOI] [PubMed] [Google Scholar]
- Sugawara T, Fujimura M, Morita-Fujimura Y, Kawase M, Chan PH (1999). Mitochondrial release of cytochrome c corresponds to the selective vulnerability of hippocampal CA1 neurons in rats after transient global cerebral ischemia. In: J Neurosci 19: pp. RC39(1–6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugawara T, Noshita N, Lewén A, Gasche Y, Ferrand-Drake M, Fujimura M, Morita-Fujimura Y, Chan PH (2002). Overexpression of copper/zinc superoxide dismutase in transgenic rats protects vulnerable neurons against ischemic damage by blocking the mitochondrial pathway of caspase activation. J Neurosci 22:209–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomasevic G, Kamme F, Stubberöd P, Wieloch M, Wieloch T (1999a). The tumor suppressor p53 and its response gene p21WAF1/Cip1 are not markers of neuronal death following transient global cerebral ischemia. Neuroscience 90:781–792. [DOI] [PubMed] [Google Scholar]
- Tomasevic G, Shamloo M, Israeli D, Wieloch T (1999b). Activation of p53 and its target genes p21WAF1/Cip1 and PAG608/Wig-1 in ischemic preconditioning. Mol Brain Res 70:304–313. [DOI] [PubMed] [Google Scholar]
- Xiang H, Hochman DW, Saya H, Fujiwara T, Schwartzkroin PA, Morrison RS (1996). Evidence for p53-mediated modulation of neuronal viability. J Neurosci 16:6753–6765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang H, Kinoshita Y, Knudson CM, Korsmeyer SJ, Schwartzkroin PA, Morrison RS (1998). Bax involvement in p53-mediated neuronal cell death. J Neurosci 18:1363–1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yonekura I, Takai K, Asai A, Kawahara N, Kirino T (2006). p53 potentiates hippocampal neuronal death caused by global ischemia. J Cereb Blood Flow Metab in press. [DOI] [PubMed]
- Yu J, Zhang L, Hwang PM, Kinzler KW, Vogelstein B (2001). PUMA induces the rapid apoptosis of colorectal cancer cells. Mol Cell 7:673–682. [DOI] [PubMed] [Google Scholar]