

Abstract
Neural stem cells (NSCs) offer special therapeutic prospects because they can be isolated from the CNS, expanded ex vivo, and re-implanted into diseased CNS where they not only migrate and differentiate according to cues from host tissue but also appear to be capable of affecting host cells. In nervous (nr) mutant mice Purkinje neuron (PN) mitochondria become abnormal by the second postnatal week, and a majority of PNs die in the fourth to fifth weeks. We previously identified in nr cerebellum a 10-fold increase in tissue plasminogen activator (tPA) as a key component of the mechanism causing nr PN death. Here we report that undifferentiated wild-type murine NSCs, when transplanted into the newborn nr cerebellar cortex, do not replace host PNs but contact imperiled PNs and support their mitochondrial function, dendritic growth, and synaptogenesis, subsequently leading to the rescue of host PNs and restoration of motor coordination. This protection of nr PNs also is verified by an in vitro organotypic slice model in which nr cerebellar slices are cocultured with NSCs. Most importantly, the integrated NSCs in young nr cerebellum rectify excessive tPA mRNA and protein to close to normal levels and protect the mitochondrial voltage-dependent anion channel and neurotrophins, downstream targets of the tPA/plasmin proteolytic system. This report demonstrates for the first time that NSCs can rescue imperiled host neurons by rectifying their gene expression, elevating somatic stem cell therapeutic potential beyond solely cell replacement strategy.
Keywords: neural stem cell, Purkinje neuron degeneration, cell rescue, tissue plasminogen activator, mitochondria, neurotrophin
Introduction
Neural stem cells (NSCs) are defined operationally by their ability to self-renew and differentiate into cells of glial and neuronal lineages and to populate developing or diseased CNS regions (Parker et al., 2005). In previous experiments we observed that mouse (Rosario et al., 1997) and human (Flax et al., 1998) NSCs could repopulate diseased sites in mutant mouse cerebellum by shifting the differentiation fate of their progeny to compensate for a particular missing neural cell type. However, a new concept is emerging that “communication” between injured host brain and NSCs is not unidirectional but, rather, a genuine “dialogue” whereby transplanted NSCs not only migrate and differentiate according to cues from developing or diseased host tissue but also appear to be capable of affecting host cells (Teng et al., 2002; Marconi et al., 2003; Imitola et al., 2004a,b).
Progressive degeneration of Purkinje neurons (PNs) is prominent in many human and mouse diseases (Sarna and Hawkes, 2003). The nervous (nr) autosomal recessive mutation, with its unique mitochondrial phenotype, was the first described inherited mouse model of PN degeneration (Sidman and Green, 1970). Between postnatal day 21 (P21) and P35 a majority of nr PNs degenerate. Motor hyperactivity and ataxia become evident early in the course of PN degeneration. Retinal photoreceptor cells also die at P11–P19 and more slowly after P90 (LaVail et al., 1993). The nr gene was mapped to a 1.4 centimorgan (cM) region of mouse chromosome 8 between the D8Rck1 and D8Mit3 markers (De Jager et al., 1998). This region now is known to contain >10 candidate genes, including tissue plasminogen activator (tPA), a key component in the tPA/plasmin proteolytic system. We recently found that a 10-fold increased tPA in nr cerebellum appears to trigger nr PN death mechanisms, either directly or via downstream targets: voltage-dependent anion channel (VDAC) and selected neurotrophins such as brain-derived neurotrophic factor (BDNF) and neurotrophin 3 (NT3) (Li et al., 2006).
In the present study we examine two fundamental questions. (1) Can NSCs mediate host cell rescue and functional recovery, perhaps a more tractable therapeutic strategy for complex neurodegenerative processes than the more conventionally voiced goal of neuronal replacement? (2) Does NSC-mediated rescue entail a reciprocal interplay between donor and host cells wherein NSCs exert their influence by actually altering gene expression and signaling pathways in host cells? Our study, via demonstrating NSC-mediated restoration of tPA and downstream targets in nr cerebellum, provides new insights into the interplay between exogenous NSCs and imperiled host neurons at levels of cell, organelle, and molecule. This under-appreciated strategy by which stem cells may play a therapeutic role not by cell and gene replacement but rather by restoring molecular homeostasis and rescuing host cells also may apply to other neurodegenerative disorders.
Materials and Methods
Animals.
The nr mutation originally occurred in the BALB/cGr strain (Sidman and Green, 1970) and has been maintained by us congenic with the almost identical BALB/cByJ strain for >12 backcross generations. Progeny used in the present study were obtained from either heterozygous × heterozygous or homozygous (female) × heterozygous (male) crosses. The nr homozygotes were identified in segregating litters by behavioral criteria (hyperactivity and ataxia) after P21. For young (P14–P16) mice the PN mitochondria were checked morphologically, and the nr mutants were recognized by the distinctively enlarged and rounded mitochondrial shapes (Sidman and Green, 1970; Landis, 1973). Mice that had received neonatal NSC transplants and might have retained normal behavior also were classified at adulthood by the retinal phenotype (LaVail et al., 1993). The use of animals in the present study was in accordance with National Institutes of Health-approved institutional guidelines established by Harvard Medical School. All efforts were made to minimize the number of animals used and their suffering.
NSC culture and transplantation.
Murine C17.2 NSCs were maintained, propagated, and transplanted as previously detailed by Snyder et al. (1992) and Marconi et al. (2003). Briefly, NSCs were grown in DMEM supplemented with 10% fetal bovine serum, 5% horse serum, and 2 mm glutamine (Invitrogen, Carlsbad, CA) and were maintained as monolayers at 37°C in 95% air/5% CO2. The cultures were split (1:10) weekly into fresh medium. These NSCs constitutively express lacZ, typically remaining stable and undiminished for at least 4 months. To assess differentiation, we grew cells on coverslips, and we pharmacologically imposed cell cycle arrest by 1 μm mitomycin C (Sigma, St. Louis, MO). Specific antibodies against progenitor and immature/mature neural cell markers then were used to identify the patterns of differentiation of NSCs. In some experiments the conditioned medium from cerebellar slice cultures (see below) was incorporated into the mitomycin C-treated NSC cultures to induce more mature neural differentiation.
Undifferentiated NSCs being prepared for transplantation were trypsinized from 90% confluent flasks and suspended in HBSS (Invitrogen) at 5 × 104 cells/μl. The cerebellum of a cryoanesthetized newborn mouse pup (P0–P7) was identified by transillumination of the head, and NSCs were administered via a glass micropipette. The micropipette was inserted through skin and skull, and 105 NSCs in 2 μl of HBSS or HBSS alone (sham control) was injected into superficial levels of the cerebellar vermis and bilateral hemispheres.
Cerebellar organotypic slice cultures.
The method for culturing organotypic slices has been described previously (Samoilova et al., 2003). Whole cerebellum of P9 mouse pups was removed aseptically and immersed in ice-cold dissecting medium; sagittal slices (400 μm thick) were obtained with a tissue chopper. The slices then were transferred onto sterilized porous membrane inserts (Millipore, Bedford, MA) in 30 mm dishes containing 1 ml of culture medium. The culture medium was composed of 50% MEM with glutamine, 25% HBSS, and 25% horse serum with 20 mm HEPES buffer and 6.5 mg/ml d-glucose (Invitrogen). Slice cultures were kept at 35°C in 5% CO2 for 17 d in vitro (17 DIV), and the medium was exchanged (50% of volume) three times per week. Some slices were cocultured with NSCs (105 cells deposited directly onto the slice). Various specific antibodies were used to assess PN survival and cerebellar cytoarchitecture within whole-mount slices.
For quantification of PN survival the slices were classified, according to the number of compact groups of 100 PNs, into four types (Marin-Teva et al., 2004) (also see Results). More than 20 slices were scored for each treatment, and the percentage of slices belonging to each type was determined. The slices of nr (homozygous) pups with few surviving PNs were distinguishable from the slices of unaffected (heterozygous or wild-type, wt) pups. Mendelian distribution of the subgroups was obtained.
Histological analysis of cerebellum and neural retina.
Mice (P14–P180) were anesthetized deeply with Avertin (intraperitoneally) and perfused through the heart with fresh 4% paraformaldehyde (Sigma). Brains and eyes were dissected out and kept in the same fixative at 4°C. Cerebellar coronal cryostat sections (8–10 μm thick) were stained with hematoxylin/eosin or neutral red for cerebellar histology and with X-gal (Promega, Madison, WI) for NSCs. Additional animals were perfused with 2% paraformaldehyde plus 2.5% glutaraldehyde (Sigma). Cerebellar samples were cut into thin blocks (<1 mm) and postfixed with osmium tetroxide and potassium ferrocyanide solution (Sigma). Then the blocks were dehydrated and embedded in Epon 812 (Electron Microscopy Sciences, Fort Washington, PA). We cut orienting sections at 1 μm and stained them with alkaline toluidine blue to visualize mitochondria in PNs. In addition, glycol methacrylate-embedded 1–3 μm sections from mouse eyes also were stained with toluidine blue for detection of retinal photoreceptor cell degeneration.
Immunofluorescence staining.
Paraformaldehyde-fixed samples from cell cultures, cultured slices, or tissue sections were incubated in blocking solution consisting of 3% goat serum (Vector Laboratories, Burlingame, CA) and 0.3% Triton X-100 in PBS at room temperature for 30 min and then reacted with specific primary antibodies at 4°C for 16–48 h. Each sample was washed three times, for 20 min each, with PBS and stained by secondary antibodies at 4°C overnight. After being washed with PBS, each sample was sealed with mounting medium containing 4′,6-diamidino-2-phenylindole (DAPI; Vector Laboratories) to stain cell nuclei and was visualized by fluorescence microscopy [Nikon Eclipse E600 (Tokyo, Japan) or Zeiss Axiophot (Oberkochen, Germany)] and/or confocal microscopy (Zeiss Axiovert 100M).
Primary antibodies included mouse monoclonal antibodies: β-galactosidase (β-gal; Chemicon, Temecula, CA) for staining NSCs, calbindin-D-28K (Sigma) specific for PNs, cytochrome c oxidase subunit IV (COX IV; Molecular Probes, Eugene, OR) for mitochondrial inner membrane, glial fibrillary acidic protein (GFAP; Chemicon) for astrocytes, nestin (BD Biosciences, San Jose, CA) and vimentin (Sigma) for undifferentiated neural progenitors, myelin 2′,3′-cyclic nucleotide 3′-phosphodiesterase (CNPase; Covance Research Products, Berkeley, CA) for oligodendrocytes, neuron-specific nuclear protein (NeuN; Chemicon) for cerebellar granule cells, neurofilament (NF; pan-neuronal; Sigma) and tubulin βIII (Sigma) for basket cell axons and PN somata/axons, parvalbumin (Chemicon) for stellate and basket cell somata/axons, postsynaptic density protein-95 (PSD-95; Chemicon) for postsynaptic sites, synapsin I (Chemicon) for presynaptic terminals, and tPA (Progen Biotech, Heidelberg, Germany) for tissue plasminogen activator. Primary antibodies also included rabbit polyclonal antibodies: β-gal (Chemicon) for staining NSCs, activated caspase 3 (Sigma) for apoptotic cells, BDNF (Chemicon) for proneurotrophin BDNF (proBDNF) and mature BDNF (mBDNF), NT3 (Chemicon) for neurotrophin 3, CD68 (Santa Cruz Biotechnology, Santa Cruz, CA) for active microglia, somatostatin (Chemicon) for Golgi cells, TOAD/Ulip/CRMP 4 (TUC4; Chemicon) for immature neuronal cells, and VDAC (Abcam, Cambridge, MA) for mitochondrial outer membrane. Also used was rat monoclonal antibody AN2 (a gift from Dr. J. Trotter, Johannes-Gutenberg University of Mainz, Mainz, Germany) for immature glial cells. Secondary antibodies included goat anti-mouse or anti-rabbit IgG (Chemicon) and donkey anti-rat IgG (Jackson ImmunoResearch, West Grove, PA) conjugated with a fluorescent dye (FITC, cyanines 2 or 3, or Texas Red). Individual antibodies were titrated for optimum staining results.
Western blot.
Proteins were extracted from NSC cultures or mouse tissues with mammalian protein extraction reagent (Pierce, Rockford, IL) containing a protease inhibitor mixture (Roche, Mannheim, Germany) and were quantified with a BCA protein assay kit (Pierce). Protein (50 μg) was denatured at 95°C for 3 min, separated with 12% SDS-PAGE (Bio-Rad, Hercules, CA), and transferred from gel to nitrocellulose membrane (Schleicher & Schuell, Keene, NH). The membrane was blocked with 5% fat-free milk at room temperature for 30 min and incubated with primary antibodies at 4°C overnight. After being washed with TBST buffer (Tris-buffered saline plus Tween 20) for four times, 15 min each, the membrane was incubated with horseradish peroxidase-conjugated secondary antibody for 1 h at room temperature, washed, and then reacted with enhanced ECL Western blot detection reagent (Amersham Biosciences, Piscataway, NJ) for 2 min. A high-performance autoradiography film (Amersham Biosciences) was taped onto the Saran-wrapped membrane in a dark room for 2–30 s and then developed to visualize the bound antibody. β-Actin served as a control protein on the same blot to eliminate loading variations.
Reverse transcription-PCR.
Total RNA was isolated from mouse cerebellum and testis with an RNA purification kit (Invitrogen). The quality of purified RNA was detected in agarose gels. Two-step reverse transcription (RT)-PCR was performed to maximize the uniformity of PCR templates for all reactions. Up to 2 μg of total RNA from each sample was reverse-transcribed into the first-strand cDNA in a 20 μl volume with oligo-dT primers and the SuperScript first-strand synthesis system (Invitrogen). PCR amplification of the coding sequence of a certain gene was performed by using gene-specific oligonucleotide primers and ReddyMix PCR Master Mix (ABgene, Surrey, UK). PCR products were verified with agarose gel.
Behavioral test.
At the eighth week after NSC transplantation both nr mutant mice and wt controls with NSC or sham injections were trained, three trials per day for 3 d, to sustain themselves on a rotating rod (at 10, 20, and 30 rpm for the first, second, and third days, respectively; Economex Instruments, Columbus, OH). The apparatus contained four individual cells (each 9.5 cm in width) and a circular rod with a cross-sectional diameter of 3.5 cm covered by knurled Perspex to provide a sufficiently nonslip surface. The rod was suspended at a height of 20 cm. Rotation of the rod was controlled by a quiet synchronous motor driving a ball-bearing drum mechanism. After the training, mouse motor coordination and fatigue resistance were assessed by measuring the time to falling off the rotating rod at 30 rpm with three trials.
Quantification and statistic analysis.
The number and proportion of the specific staining positive and negative cells were counted from cell and slice cultures or from a series of representative sequentially cut tissue sections. Quantity I software was used for the quantitative analyses of cDNA and protein bands on gel or film. Multiple factor analysis was used to estimate various parameters that potentially affect the efficiency of NSC integration in cerebellar parenchyma. Comparison between groups, either paired or unpaired as required, was performed with (1) the Student’s t or t' test (according to results of the homoscedasticity analysis) for the measurement data, (2) the χ2 test for enumeration data, (3) the linear regression analysis for correlation data, and (4) the Ridit analysis for ranked data. Results were presented as the means ± SD, and differences were considered as significant at p < 0.05 or very significant at p < 0.01.
Results
Bias control and NSC characterization
For the studies reported here, 176 mouse pups were divided into two groups: 118 pups were transplanted as neonates (between P0 and P7) with murine NSCs, and 58 pups were sham-injected (serving as untreated negative controls). To control against possible bias in animal selection and transplantation procedures, we generated NSC-transplanted and sham-treated mice from two mating schemes: heterozygous males were mated with heterozygous females (for analysis of adult mice older than P30) or with homozygous females (for analysis of preweaning P14–P20 mice). All pups in a given litter were transplanted as newborns by an experimenter blinded to their genotypes. There were no statistical differences between NSC-transplanted and sham-treated mice vis-à-vis age-at-transplantation (P0–P7), sex, and age-at-death (P30–P180; for each comparison, p > 0.05). Of the 74 mice from the NSC-treated group older than P30, 16 were nr homozygous mutants (22%), and of the 40 mice of the same age from the sham-treated control group, nine were nr mutants (23%), both percentages indistinguishable statistically from the Mendelian expectation of 25%. In addition, NSC integration in cerebellar parenchyma was not affected by gender, age-at-transplantation, age-at-death, and phenotype (nr vs wt; in all cases, p > 0.05). Given that the nr gene has not yet been identified and hence could not be used to verify the genotype of a given mouse nor are polymorphisms in closely linked gene markers available because all of the mice are coisogenic BALB/c, homozygous at all genetic loci, another means had to be invoked to control for the possibility that NSC-treated nr mutants might not be distinguishable behaviorally from wt controls because of a good therapeutic response. Therefore, we examined histologically the neural retina in all animals to identify those mice that clearly displayed the nr mutant retinal phenotype (LaVail et al., 1993). Finally, the same stable clone of engraftable, undifferentiated, constitutively lacZ-expressing murine NSCs, prepared for transplantation in an identical manner, was used in all animals.
We next detected NSC differentiation in vitro and in vivo. After cell cycle withdrawal induced by mitomycin C (1 μm), some cultured NSCs differentiated into neurons (8% of NSCs stained with NeuN, NF, or parvalbumin, but none with calbindin), astrocytes (15% stained with GFAP), and oligodendrocytes (1% stained with myelin CNPase), whereas the others remained as progenitor cells (nestin positive or vimentin positive). To establish that the cultured NSCs responded to appropriate cues, we supplemented the mitomycin C-containing culture medium with a conditioned medium from cerebellar slice cultures. In this case, most NSCs lost their progenitor cell markers (nestin negative and vimentin negative) and gained markers for mature neural cells (>40% of NSCs stained with NeuN, NF, parvalbumin, GFAP, or myelin CNPase) or immature neuronal (25% stained with TUC4) or glial (15% stained with AN2) cells. To detect NSC differentiation in vivo, we processed tissue specimens from P14 to P180 mice that had been injected neonatally with NSCs, from neonatally sham-injected mice, and from wild-type controls. Most integrated NSCs were negative for progenitor cell markers (nestin negative and vimentin negative) but were stained positively with immature or mature neural cell markers such as TUC4 (8%), AN2 (48%), NeuN (24%), parvalbumin (3%), and GFAP (1%). None of NSCs was stained with the PN marker (calbindin negative; also see below).
NSC transplantation rescues PNs in nr cerebellum
We examined mutant host cerebellar cytoarchitecture in the specimens from P14 to P180 described in the previous paragraph. At P14 the nr cerebellar laminar architecture was still normal (Fig. 1A). However, although cerebella from adult nr mice (P30, P60, P120, and P180) showed normal parvalbumin-positive interneurons in the molecular layer (ML) and normal NeuN-positive granule cells in the internal granular layer (GL) (Fig. 1B), they also showed severe depletion of calbindin-positive PNs in the lateral hemispheres (Fig. 1C). In addition, the nr mutation activated many endogenous astrocytes (GFAP positive) in adult mouse cerebellar cortex and white matter. The paucity of activated microglial cells (CD68 positive) and the restriction of caspase 3 staining to some granule cells in young (P14–P20) and adult nr cerebella indicated that nr PN death might be independent of either microglial activation or caspase 3-mediated apoptosis. These results were consistent with our previous report (Li et al., 2006).
Figure 1.

NSCs rescued nr cerebellar PNs. A, Neutral red-stained section of young (P14) nr mouse cerebellum displays a normal cortical cytoarchitecture, including the external GL, the ML, the PN layer (PL, between two blue dotted lines), and the internal GL. The right panel is an enlargement of the small green frame in the left panel. B, Cytoarchitecture and neuronal marker expression of adult (P60) nr mouse cerebellum. Cerebellar sections were immunostained with anti-parvalbumin (parv; red) antibody for interneurons in the ML (left), with anti-NeuN (neun; red) antibody for granule cells in the internal GL (right), and with DAPI (dapi; blue) for nuclei. C, PNs in cerebellar vermis and hemispheres of adult (P60) mice. Ca, Cerebellar sections were stained with anti-calbindin (calb; red) antibody for PNs. Representative images show that the PN number obviously was reduced in the cerebellar hemispheres of nr mice and was preserved significantly by NSC transplantation (nr+nsc). Cb, PN numbers in cerebellar vermis and hemispheres of three groups of mice (wt, nr, and nr+nsc). Values represent the means ± SD of PN number (percentage of wt; n = 9; nr vs nr+nsc in hemispheres, p < 0.01). Cc, Western blot shows that the decreased calb in adult nr mouse cerebellum mostly was recovered in nr+nsc cerebellum. D, Host origin of surviving PNs. Da, Cerebellar section of NSC-transplanted nr mouse (P40) was coimmunostained with anti-calb antibody for PNs and anti-β-gal (green) antibody for NSCs. No colocalization of calb and β-gal was found in PNs. Db, Cerebellar section of male NSC-transplanted female nr mice (P40) was stained with DAPI. Barr bodies (yellow arrows) were observed in nuclei of most surviving PNs. E, Relationship between NSC integration and PN survival. Ea, The number of surviving PNs in adult nr+nsc mice (P40–P60) was correlated positively with the number of NSCs integrated in cerebellar parenchyma (red oblique line; r = 0.76; n = 15; p < 0.01), but not with the number of NSCs in the superficial meninges (green oblique line; r = 0.23; n = 15; p > 0.05). Eb, Representative section of adult (P40) nr+nsc mouse cerebellum was stained with X-gal (blue dots). NSCs were counted from three sections of each mouse. Scale bars: A–C, E, 50 μm; Da, 15 μm; Db, 5 μm.
To evaluate the impact of NSC engraftment, we quantified cerebellar PNs on the BALB/cByJ congenic strain, previously undescribed on this genetic background. In the untreated nr cerebellum there was severe depletion of PNs, more pronounced in the lateral hemispheres (>70% PN loss) than in the vermis (∼10% PN loss) (Fig. 1C). However, in the cerebella of adult nr mice that had received NSC implants as neonates, PN number was surprisingly normal in the lateral hemispheres when compared with that in the sham-treated group (p < 0.01) (Fig. 1C), and the neuroinflammatory reactions typically seen in untreated nr cerebellum were also absent (data not shown). We next determined whether these PNs were donor NSCs that had differentiated into new PNs in place of depleted host PNs or were host PNs (somehow protected from cell death in the presence of NSCs). None of the calbindin-positive PNs in the NSC-treated nr mouse cerebellum costained with an antibody to β-gal, a stable marker in the exogenous NSCs (Fig. 1D). β-Gal-expressing cells were indeed abundant, but all were calbindin negative. Furthermore, the surviving PN soma size, width of the ML, dendritic branching, and axon trajectories as visualized with calbindin staining were indistinguishable from normal, a response that could not be attributed reasonably to new PNs derived from NSCs that had downregulated their β-gal marker. As final critical evidence, we scored intranuclear Barr bodies (inactivated X-chromosome in mammalian females) in cerebellar PNs of female adult wt mice and nr mice that had been injected neonatally with male NSCs and found that 243 of 300 PNs (81%) in wt mice and 224 of 300 PNs (75%) in NSC-treated nr mice displayed Barr bodies (n = 3 for each group; p > 0.05) (Fig. 1D). Therefore, these results indicate that the surviving PNs in NSC-transplanted nr mice are “rescued” host cells, not donor NSCs.
The nr mice ± NSCs (i.e., independent of the number of viable PNs) could survive for >18 months. That the surviving PNs in NSC-transplanted nr mice were stable throughout life suggested that NSCs did rescue host PNs rather than merely delaying cell death. Moreover, the number of rescued PNs correlated positively with the number of NSCs integrated into the cerebellar cortical parenchyma (detected by X-gal staining) (r = 0.76; n = 15; p < 0.01), but not with NSCs restricted to the meninges covering the cerebellar surface (r = 0.23; n = 15; p > 0.05) (Fig. 1E). No NSC-injected mice developed tumors.
NSC-rescued PNs retain ability to control motor coordination
To quantify functional recovery, we evaluated motor behavior on a rotarod in four groups of adult mice: (1) wt mice without NSC transplantation (wt; n = 6) or (2) with NSCs (wt+nsc; n = 8) and (3) nr mice without NSCs (nr/nr; n = 6) or (4) with NSCs (nr+nsc; n = 8). A Student’s t' test was used for statistical analysis because of heterogeneity between the groups (F = 10.3 > F0.01 (7, 7, or 5); p < 0.01). NSC engraftment significantly improved the rotarod performance of nr mice (p < 0.05) (Fig. 2A). Linear regression analysis additionally confirmed that the extent to which performance on rotarod improved was correlated positively with the number of rescued PNs (r = 0.83; n = 8; p < 0.05) (Fig. 2B). Similar results were obtained in three independent tests at P60–P120.
Figure 2.

Surviving PNs retained their motor function. A, Results of the rotarod test showed that NSC transplantation did not affect motor function of adult wt mice (wt vs wt+nsc) but significantly restored the motor function of adult nr mice (nr vs nr+nsc). Values represent the means ± SD of time spent on the rod (n = 6–8 for each group of three independent tests; ∗∗p < 0.01). B, The improved motor behavior was correlated positively to the number of surviving PNs in NSC-transplanted nr mouse cerebellum (r = 0.83; n = 8 paired samples; p < 0.01).
PN survival in organotypic slice cultures with and without added NSCs
To begin to dissect the mechanisms underlying NSC-mediated rescue of mutant PNs, we attempted to recreate the phenomenon in vitro by establishing a cerebellar organotypic slice culture system, effective for studying postnatal PN development (Ghoumari et al., 2002; Marin-Teva et al., 2004). The wt cerebellar cortex in slice cultures retained the normal three layers of interneurons, PN somata, and granule cell bodies (Fig. 3A). The slices were evaluated semi-quantitatively for PN number and distribution with the following scale (Marin-Teva et al., 2004): type I, no clusters containing 100 PNs; type II, one cluster of 100–200 PNs; type III, two clusters of 100–200 PNs or one cluster of 200–300 PNs; and type IV, at least three clusters of 100 PNs singly or grouped into one or more clusters totaling 300 or more PNs. In nr cerebellar slice cultures without NSCs, 89, 9, and 2% of slices were classified into type I, type II, and type III, respectively, whereas in cocultures with NSCs the NSCs infiltrated the slice and the PN survival rates shifted to 23% type I, 42% type II, 27% type III, and, now, 8% type IV. Thus the cocultured NSCs in vitro had increased PN numbers significantly in the nr cerebellar slices (p < 0.01) (Fig. 3B). As in the in vivo setting, the restored calbindin-positive PNs in the cocultured slices were not costained with β-gal (Fig. 4B), indicating host origin of the PNs. Not only had the rescue phenomenon been recapitulated in vitro, but we now had a system amenable to dissecting potential underlying mechanisms.
Figure 3.
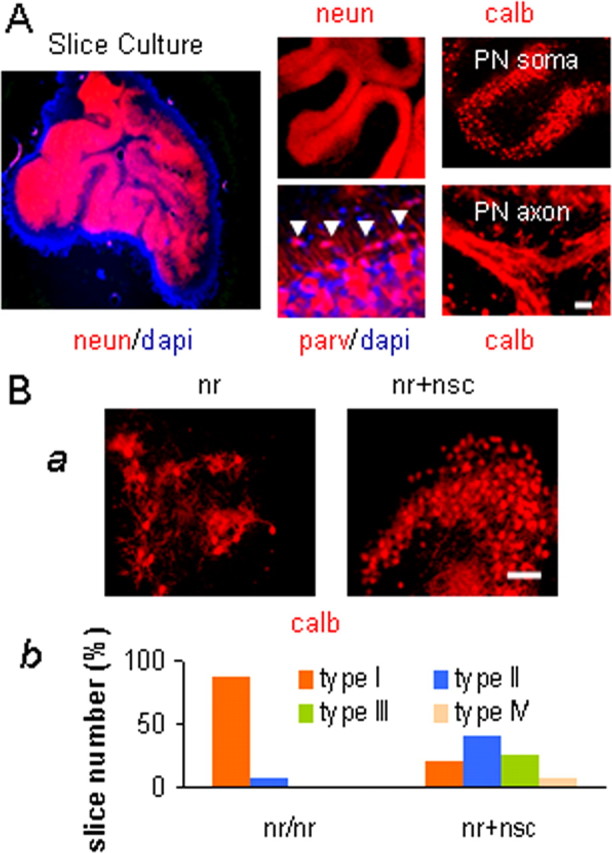
NSCs rescued PNs in nr cerebellar slice cultures. A, Cerebellar slice cultures retained the normal cytoarchitecture of three cortical layers and white matter, i.e., parvalbumin (parv) for the ML, calbindin (calb) for PNs, and NeuN (neun) for the GL. Arrowheads point to basket neurons. B, NSCs rescued PNs in cerebellar slice cultures. Ba, P9 mouse cerebellar slices were cultured for 17 d (P9 + 17 DIV) and stained with anti-calb antibody for PNs. Representative images show that NSCs, when cocultured with cerebellar slices, increased nr PN survival. Bb, Compositions of four types of slices (Marin-Teva et al., 2004) (also see Results) were different between nr cultures and nr+nsc cocultures. For comparison between two kinds of cultures on three independent experiments, p < 0.01. Scale bars: A, B, 50 μm.
Figure 4.

NSCs approached and contacted host PNs. A, NSCs (bgal; green) in vivo, which had been transplanted into the external GL of newborn nr mouse cerebellum, approached (left) and contacted (right) host PNs (calb; red). B, NSCs (bgal; green) in vitro, which had been cocultured with cerebellar slices of nr mice, migrated into the slices and extensively contacted PNs (calb; red). The two right panels are enlargements of the small yellow frame in the left panel. X represents the section images from the x-axis point of view, and Y represents the section images from the y-axis point of view. Both X and Y images show the points of contact between PN (red) and NSC (green). Scale bars: Aa, 30 μm; Ab, 10 μm; B, 25 μm.
NSCs directly contact PNs and protect PN organelle in nr cerebellum
We examined the distribution of the NSCs in cerebellar cortex and direct contacts between donor and host cells. Many transplanted NSCs entered or immediately surrounded the layer of nr PN somata, typically making direct contacts with host PNs (Fig. 4A). The types of intercellular contacts in cocultures of cerebellar slice and NSCs included (1) NSC soma with nr PN soma, (2) NSC soma with nr PN process or nr PN soma with NSC process, and (3) nr PN process with NSC process (Fig. 4B).
To assess whether such contacts are necessary for the rescue of host nr PNs, we placed NSCs under a sterile membrane for which the pores were too small to allow NSCs or their processes to contact host nr PNs in cerebellar slices that were placed on the top side of the membrane. Most PNs in these nr cerebellar slices degenerated, similar to the slices without NSCs as summarized above (Fig. 3B), and most NSCs under the membrane remained nestin positive or vimentin positive. In another experiment the conditioned medium (but no NSCs) from previous cocultures of NSCs and cerebellar slices was added to the culture medium of fresh nr cerebellar cultures but failed to prevent nr PN death. These results indicated that direct intercellular contacts between donor NSCs and host neurons, rather than molecular mediators in the coculture medium, play a role in nr PN protection.
To identify effects of exogenous NSCs on nr PN organelles, we examined histologically the wt and nr PN mitochondria and dendritic and axonal processes in the absence and presence of NSCs. In the absence of NSCs, the young nr cerebellum showed numerous enlarged round mitochondria in 80% of PN somata (Fig. 5A) and significant reductions of >50% in the area occupied by PN dendritic branchlets (Fig. 5B), and the adult nr cerebellum showed reduced numbers of PN axons (Fig. 5C). When NSCs were transplanted into newborn nr cerebella, these subcellular components in nr PNs appeared to be protected. Specifically, in NSC-engrafted preweaning nr cerebella, normal PN mitochondrial morphology was preserved (Fig. 5A), and host PN dendritic branchlets were as elaborate as those in wt mice (Fig. 5B). In neonatally treated nr cerebella examined at adulthood the calbindin immunostaining clearly delineated PN axons with normal trajectories extending through the GL and white matter to synapses on neurons in the cerebellar nuclei (Fig. 5C). These well preserved subcellular features presumably underlie the NSC-mediated retention of normal motor coordination in the nr mutant mice.
Figure 5.

NSCs protected nr PN mitochondria, dendrites, and axons. A, PNs in young (P16) mouse cerebellar sections (1 μm thick) were stained with alkaline toluidine blue. Numerous round mitochondria (dark puncta) were found in nr PN soma (red dotted ellipse), but not in wt or nr+nsc PN soma (red dotted ellipse). B, A significant reduction was shown in young (P16) nr PN small dendrites (calb; red) but recovered in nr+nsc PN dendrites in the ML. C, The nr+nsc PNs extended long axons (calb; red) through white matter toward the cerebellar nuclei, but they remained compromised in adult (P60) nr cerebellum that held residual PNs. Scale bars: A, 5 μm; B, 30 μm; C, 50 μm.
NSCs rectify excessive tPA and abnormal downstream targets in nr cerebellum
To explore whether the rescue of nr PNs was related to NSC-mediated rectification of excessive tPA in nr cerebellum, we measured tPA protein levels in young and adult mouse cerebella. Increased immunofluorescence intensity of tPA protein in young nr cerebellum remarkably was restored in NSC-treated nr cerebellum (Fig. 6A). Western blot also confirmed the 10-fold increases in tPA protein levels in untreated young and adult nr cerebella (Li et al., 2006) and showed reductions close to normal values after NSC transplantation (p < 0.01) (Fig. 6A). Furthermore, RT-PCR revealed that the rectification of tPA protein levels in young and adult NSC-treated nr cerebella was attributable to normal tPA mRNA expression (Fig. 6B), suggesting that NSC transplantation probably rectified tPA gene transcription in nr cerebella. This finding was reinforced by the use of two pairs of primers designed to detect tPA C- and N-terminal regions (Fig. 6B).
Figure 6.
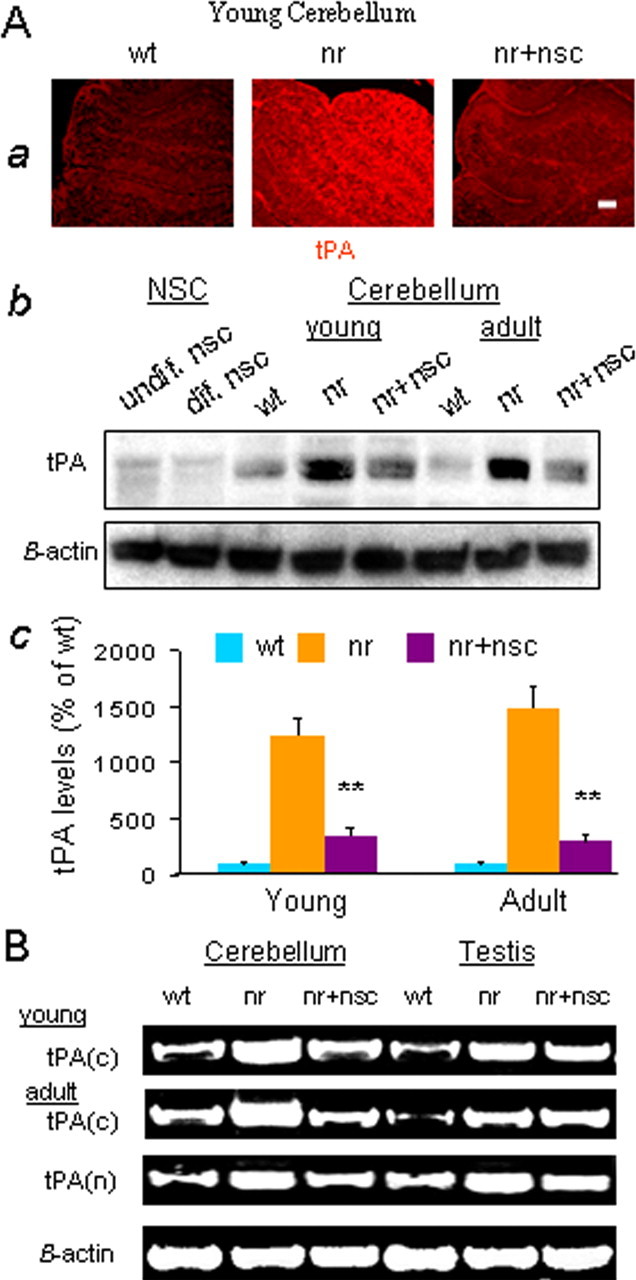
NSCs rectified excessive tPA in nr cerebellum. A, tPA protein levels. Aa, Representative images of young (P16) mouse cerebellum stained with anti-tPA (red) antibody. The tPA level in nr cerebellum was obviously higher than in wt and nr+nsc cerebella. Scale bar, 30 μm. Ab, Levels of tPA protein measured with Western blot were extremely elevated in young (P16) and adult (P120) nr mouse cerebella as compared with those in wt or nr+nsc cerebella. Cultured NSCs in vitro produced very small amounts of tPA protein. Ac, Quantification of tPA protein in young (P16) and adult (P120–P180) cerebella of wt, nr, and nr+nsc mice. Values represent the means ± SD of tPA relative levels (percentage of wt; n = 5). Comparison between nr and nr+nsc, ∗∗p < 0.01. B, tPA(c) and tPA(n) mRNAs were detected by using RT-PCR. Expression of tPA mRNA was upregulated greatly in young (P16) and adult (P120) nr mouse cerebellum and testis, as compared with wt controls. NSC transplantation (nr+nsc) into cerebellum suppressed the increased tPA mRNA in nr cerebellum, but not in nr testis.
Then we examined effects of NSC transplantation into neonatal nr mouse cerebella on a downstream target, VDAC, of the tPA/plasmin proteolytic system (Gonzalez-Gronow et al., 2003; Banerjee and Ghosh, 2004). Distinct isoforms of VDAC encoded by three genes (VDAC1–3) (Sampson et al., 1997) collectively form large voltage-gated pores in the mitochondrial outer membrane (Zizi et al., 1994), which not only act as a pathway for the movement of metabolites like ATP in and out of mitochondria by passive diffusion but also may play a role in synaptic transmission (Jonas, 2004) and apoptotic cell death (Crompton, 1999; Shimizu et al., 1999). We found that alterations of VDAC immunostaining in PN somata and small dendrites of young nr cerebella, which were consistent with the known changes in young nr PN mitochondria (Landis, 1973; Li et al., 2006), were maintained in normal distributions in NSC-treated nr cerebella. Similarly, changes of COX IV in young nr cerebella (Li et al., 2006), a key component of the respiratory chain on mitochondrial inner membrane, also were restored in NSC-treated nr PN dendrites and somata.
We next examined the effects of NSC transplantation on neurotrophins, downstream targets of the tPA/plasmin proteolytic system (Lee et al., 2001; Pang et al., 2004). In young nr cerebella decreased distributions of BDNF and NT3 in the external GL and in PN somata, respectively, were restored effectively by NSC transplantation (Fig. 7A). Because neurotrophins are modulators of PN dendrite growth and synaptogenesis, we additionally detected PSD-95 that is thought to be necessary for rapid formation and remodeling of postsynaptic components (Marrs et al., 2001). Reduced PSD-95 proteins in nr cerebella, either on young PN dendrites receiving inputs from climbing and parallel fibers or on adult PN somata receiving inputs from basket cell axon terminals, were recovered in NSC-treated nr cerebella (Fig. 7A).
Figure 7.

NSCs diminished the effects of excessive tPA on downstream targets. A, Neurotrophins and postsynaptic marker. Aa, Representative images of young (P14) mouse cerebellar sections costained with anti-calb (red) and anti-BDNF (green) antibodies. BDNF, a downstream target of the tPA/plasmin system, existed in the external GL (in rectangle) of wt and nr+nsc cerebella but was reduced greatly in nr cerebellum. Ab, Representative images of young (P14) mouse cerebellar sections costained with anti-calb (red) and anti-NT3 (green) antibodies. NT3, a downstream target of the tPA/plasmin system, existed normally in PN somata of wt and nr+nsc cerebella but was reduced greatly in nr cerebellum. Ac, PSD-95 (red) staining, a postsynaptic dendritic marker, was decreased in the ML and increased in PN somata (parallel lines) of young (P16) nr cerebellum as compared with wt and nr+nsc cerebella. Ad, PSD-95 (red) staining showed many fewer postsynaptic connections (arrowheads) between basket cell axons and PN somata (parallel lines) in adult (P60) nr cerebellum as compared with wt and nr+nsc cerebella. B, NSCs in monolayer cultures obviously expressed foxa2 and gbx2 transcription factors. Ba, Representative images of NSC cultures in undifferentiated phase (undif. nsc) and differentiated phase (dif. nsc). Bb, Results of RT-PCR with the use of two pairs of primers for each factor showed that foxa2 (primer I for 392 bp and primer II for 349 bp) was expressed equally in undifferentiated and differentiated NSCs, whereas gbx2 products (primer I for 462 bp and primer II for 363 bp) were higher in differentiated NSCs than in undifferentiated NSCs. Scale bars: Aa, 30 μm; Ab, 50 μm; Ac, Ad, 15 μm.
NSCs themselves, in proliferation or differentiation phase (Fig. 7B), produced little tPA protein (Fig. 6A) but clearly expressed gastrulation brain homeobox 2 (gbx2) and forkhead box A2 (foxa2) products (Fig. 7B), two crucial transcription factors required for cerebellar development (Li et al., 2002) or neuronal differentiation (Norton et al., 2005), providing a clue to upstream targets regulating tPA gene transcription by which the transplanted NSCs might have rectified tPA expression in host nr cerebellum.
Discussion
NSCs rescue host PNs
In the present study we observe that exogenous NSCs transplanted into a still normal-appearing newborn nr cerebellum rescue host PNs that otherwise would die in the fourth and fifth postnatal weeks (Fig. 1). The NSC-treated nr mice retain near-normal motor coordination, as measured by rotarod performance (Fig. 2). Exogenous NSCs also rescue host nr PNs from death in cerebellar slices in vitro (Fig. 3). In exploring the cellular and molecular mechanisms leading to this NSC-mediated PN rescue, we have observed that NSCs are effective only if integrated into the cerebellar cortical parenchyma where they make direct contact with PNs, not when simply concentrated entirely at the meningeal surface. Likewise, in cerebellar slices in vitro, NSCs laid directly onto the cerebellar slice rescue host PNs, but they fail to do so if separated from the slice by a filter with small pore diameters. Our trials with conditioned medium were also ineffective. Indeed, within the cerebellar parenchyma in vivo the transplanted NSCs concentrate preferentially in proximity to the PN soma layer and, as demonstrated by confocal microscopy, make direct contact with PN somata and dendrites (Fig. 4). PNs rescued by neighboring exogenous NSCs (unlike untreated nr PNs) show normal mitochondria, retain PSD-95 postsynaptic expression, and elaborate normal dendritic branchlets (Fig. 5). Because the nr PN degeneration occurs several weeks after birth (a temporal window for the pathological process to unfold), our results support a critical concept that early stem cell intervention, before the pathophysiological cascade is irreversible, may play a profound role in preventing this abnormal process.
NSCs appear, at least in the case of the nr mutation, to effect their rescue of imperiled PNs by restoring homeostasis to the cell and its milieu, promoting a return toward normal levels of the excessive amounts of tPA that characterize the untreated nr cerebellum (Fig. 6). That this imposition of “molecular homeostasis” may be the basis for this PN rescue is supported by observing that nr PNs could be preserved similarly in organotypic slice cultures in the absence of NSCs but in the presence of an antagonist of a key downstream component in the tPA pathway, PKC, or of an inhibitor of tPA itself (Li et al., 2006). Additional downstream targets of tPA that are altered in nr cerebellum and recovered in NSC-treated nr cerebellum include VDAC and the neurotrophins BDNF and NT3 (Fig. 7), which will be discussed below.
The molecular mechanism of PN rescue involves tPA
The best-known action of tPA is to cleave its substrate, the single-chain enzymatically inactive plasminogen, to the two-chain serine protease, plasmin (Vassalli et al., 1991; Sheehan and Tsirka, 2005). Plasmin can cleave a number of extracellular substrates, including fibrin and laminin (Chen and Strickland, 1997). The potent effect of tPA on fibrin is the basis for its current clinical use as an agent to dissolve acutely formed blood clots in cerebral arteries (Traynelis and Lipton, 2001; Sheehan and Tsirka, 2005). However, tPA also can bind to other receptors and exert profound and varied neurobiological effects. For example, tPA is a presynaptic component, as shown particularly clearly in certain hypothalamic axon terminals (Miyata et al., 2005), and is thought to influence normal synaptogenesis, long-term potentiation, and synaptic alterations in disease states (Madani et al., 1999; Sheehan and Tsirka, 2005). The action of tPA at synapses is based on its cleavage of the NR1 subunit of the NMDA receptor, which probably increases calcium influx (Nicole et al., 2001) and thus accounts for its effect in augmenting the severity of NMDA-mediated excitotoxic lesions. Binding of tPA on the NR2B subunit also may control various forms of seizure activity, particularly well shown in a mouse model of chronic ethanol intake followed by withdrawal-induced seizures (Pawlak et al., 2005). Ion fluxes also are influenced by a direct effect of plasminogen on the mitochondrial outer membrane. Plasminogen contains five binding segments called “kringles,” one of which (kringle 5) specifically can bind brain mitochondrial VDAC to result in partial closure of the VDAC channel (Banerjee and Ghosh, 2004), interfering with mitochondria-related regulation of intracellular calcium and pH (Gonzalez-Gronow et al., 2003). In addition, normal PN development involves a switch from dependence on BDNF for cell genesis to dependence on NT3 for cell maturation (Segal et al., 1995). That the tPA/plasmin proteolytic system can cleave a neurotrophin precursor into the mature neurotrophin is found to be essential for tPA-mediated synaptic plasticity (Pang et al., 2004) and neuronal survival (Lee et al., 2001). It also was found recently that some tPA actions can be counteracted by a naturally produced inhibitor, neuroserpin (Lebeurrier et al., 2005; Galliciotti and Sonderegger, 2006).
With specific reference to mammalian cerebellum, granule cells both secrete tPA and also possess tPA receptors on their surface (Verrall and Seeds, 1989). tPA gene expression is induced during cerebellar development at the stage when granule cells leave the external GL and begin their inward migration (Friedman and Seeds, 1994); this dramatic developmental event is retarded in tPA-deficient mice (Seeds et al., 1999). Lack of tPA activity also impairs cerebellar motor learning (Seeds et al., 2003), and this may be one example of a more general role of tPA in activity-dependent synaptic plasticity (Seeds et al., 1995). Our data correlating high cerebellar tPA with motor impairment in the nr mutant mouse and normal cerebellar tPA with good motor function in NSC-treated nr mouse, coupled with data reported by Seeds et al. (2003), suggest that too high or too low levels of tPA lead to impaired motor function. The same concept is supported by our previous data showing, in the presence of a tPA inhibitor, the increased PN death in wt cerebellar slice cultures (where normal tPA value decreases) in contrast to the decreased PN death in nr slice cultures (where high tPA is restored to near-normal levels) (Li et al., 2006).
Action of tPA on PN survival may not be limited to the nr mutant case. When a double mutant was created from GluRδ2Lc mutant and tPA knock-out mice (GluRδ2Lc/+, tPA−/−), the elimination of tPA was found to delay the cerebellar PN death expected from the dominant GluRδ2Lc/+ mutation (Lu and Tsirka, 2002). A closer examination of tPA and neuroserpin levels as well as other components upstream and downstream from tPA, at various ages in the several mouse mutants affecting PNs and in human PN-selective diseases when possible, clearly is warranted.
Broader implications for stem cell action
The present study strengthens the concept of stem cells serving an underappreciated therapeutic role as “chaperone” cells (Ourednik et al., 2002; Fraidenraich et al., 2004), a notion that likely extends to other neurodegenerative disorders. The fact that NSCs prevented or forestalled host nr PN dysfunction or demise by restoring normal tPA gene expression in the mutant milieu and hence promoted motor behavioral recovery may represent a more tractable therapeutic strategy for this neurogenetic disorder than the more conventionally considered goal of cell replacement. In most neural systems, as in the cerebellum, the prospect of reconstructing the complexity of connections and feedback loops beyond the period of normal neurogenesis seems quite staggering. Preserving extant circuitry clearly would be more approachable. That NSCs effected nr PN rescue so efficiently might not appear so unexpected if one returns to our understanding of the fundamental role that the stem cell plays, first in organogenesis and then in maintaining homeostasis of the organ throughout life. In its first role a given NSC clone yields multiple interacting cell types within the same region, not just the key effector neurons but also the glia that nurture, detoxify, myelinate, and/or consummate the differentiation and refine the synaptic firing of those neurons (Song et al., 2002; Marconi et al., 2003; Christopherson et al., 2005). Similarly, “return-of-function” beyond the classical developmental windows also may require reconstitution of the entire local milieu, a program well suited to the stem cell. In its second role the NSCs may serve to restore equipoise to a disequilibrated milieu not only by fueling cell turnover (Rosario et al., 1997; Flax et al., 1998) but also by regulating gene expression and signaling pathways (Mehler and Gokhan, 2001), by instructing and altering the fate of host cells (Ourednik et al., 2002), and by minimizing impediments to regenerative processes (Park et al., 2002; Teng et al., 2002; Nakamura et al., 2005). Stem cell-mediated cell replacement, therefore, may need to be viewed more broadly to include also providing cells that support and regulate the milieu. Our report provides the first concrete evidence of a stem cell playing a protective role in the rescue of imperiled neurons by rectifying host gene expression.
Footnotes
This work was supported in part by National Institutes of Health Grant CA103056 and the Nancy Lurie Marks Family Foundation to R.L.S. and by the Ataxia-Telangiectasia Children’s Project and Genzyme Corporation (Cambridge, MA) to E.Y.S. We thank Drs. Bela Kosaras, Lili Yu, and Yawen Wang and Bengang Xu for expert assistance and Drs. Jitka and Vaclav Ourednik for helpful insights.
References
- Banerjee J, Ghosh S (2004). Interaction of mitochondrial voltage-dependent anion channel from rat brain with plasminogen protein leads to partial closure of the channel. Biochim Biophys Acta 1663:6–8. [DOI] [PubMed] [Google Scholar]
- Chen ZL, Strickland S (1997). Neuronal death in the hippocampus is promoted by plasmin-catalyzed degradation of laminin. Cell 91:917–925. [DOI] [PubMed] [Google Scholar]
- Christopherson KS, Ullian EM, Stokes CC, Mullowney CE, Hell JW, Agah A, Lawler J, Mosher DF, Bornstein P, Barres BA (2005). Thrombospondins are astrocyte-secreted proteins that promote CNS synaptogenesis. Cell 120:421–433. [DOI] [PubMed] [Google Scholar]
- Crompton M (1999). The mitochondrial permeability transition pore and its role in cell death. Biochem J 341:233–249. [PMC free article] [PubMed] [Google Scholar]
- De Jager PL, Harvey D, Polydorides AD, Zuo J, Heintz N (1998). A high-resolution genetic map of the nervous locus on mouse chromosome 8. Genomics 48:346–353. [DOI] [PubMed] [Google Scholar]
- Flax JD, Aurora S, Yang C, Simonin C, Wills AM, Billinghurst LL, Jendoubi M, Sidman RL, Wolfe JH, Kim SU, Snyder EY (1998). Engraftable human neural stem cells respond to developmental cues, replace neurons, and express foreign genes. Nat Biotechnol 16:1033–1039. [DOI] [PubMed] [Google Scholar]
- Fraidenraich D, Stillwell E, Romero E, Wilkes D, Manova K, Basson CT, Benezra R (2004). Rescue of cardiac defects in Id knockout embryos by injection of embryonic stem cells. Science 306:247–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman GC, Seeds NW (1994). Tissue plasminogen activator expression in the embryonic nervous system. Brain Res Dev Brain Res 81:41–49. [DOI] [PubMed] [Google Scholar]
- Galliciotti G, Sonderegger P (2006). Neuroserpin. Front Biosci 11:33–45. [DOI] [PubMed] [Google Scholar]
- Ghoumari AM, Wehrlé R, De Zeeuw CI, Sotelo C, Dusart I (2002). Inhibition of protein kinase C prevents Purkinje cell death but does not affect axonal regeneration. J Neurosci 22:3531–3542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Gronow M, Kalfa T, Johnson CE, Gawdi G, Pizzo SV (2003). The voltage-dependent anion channel is a receptor for plasminogen kringle 5 on human endothelial cells. J Biol Chem 278:27312–27318. [DOI] [PubMed] [Google Scholar]
- Imitola J, Park KI, Teng YD, Nisim S, Lachyankar M, Ourednik J, Mueller FJ, Yiou R, Atala A, Sidman RL, Tuszynski M, Khoury SJ, Snyder EY (2004a). Stem cells: cross-talk and developmental programs. Philos Trans R Soc Lond B Biol Sci 359:823–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imitola J, Raddassi K, Park KI, Mueller FJ, Nieto M, Teng YD, Frenkel D, Li J, Sidman RL, Walsh CA, Snyder EY, Khoury SJ (2004b). Directed migration of neural stem cells to sites of CNS injury by the stromal cell-derived factor 1α/CXC chemokine receptor 4 pathway. Proc Natl Acad Sci USA 101:18117–18122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonas E (2004). Regulation of synaptic transmission by mitochondrial ion channels. J Bioenerg Biomembr 36:357–361. [DOI] [PubMed] [Google Scholar]
- Landis SC (1973). Changes in neuronal mitochondrial shape in brains of nervous mutant mice. J Hered 64:193–196. [DOI] [PubMed] [Google Scholar]
- LaVail MM, White MP, Gorrin GM, Yasumura D, Porrello KV, Mullen RJ (1993). Retinal degeneration in the nervous mutant mouse I. Light microscopic cytopathology and changes in the interphotoreceptor matrix. J Comp Neurol 333:168–181. [DOI] [PubMed] [Google Scholar]
- Lebeurrier N, Liot G, Lopez-Atalaya JP, Orset C, Fernandez-Monreal M, Sonderegger P, Ali C, Vivien D (2005). The brain-specific tissue-type plasminogen activator inhibitor, neuroserpin, protects neurons against excitotoxicity both in vitro and in vivo Mol Cell Neurosci 30:552–558. [DOI] [PubMed] [Google Scholar]
- Lee R, Kermani P, Teng KK, Hempstead BL (2001). Regulation of cell survival by secreted proneurotrophins. Science 294:1945–1948. [DOI] [PubMed] [Google Scholar]
- Li J, Ma Y, Teng YD, Zheng K, Vartanian TK, Snyder EY, Sidman RL (2006). Purkinje neuron degeneration in nervous (nr) mutant mice is mediated by a metabolic pathway involving excess tissue plasminogen activator. Proc Natl Acad Sci USA 103:7847–7852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li JY, Lao Z, Joyner AL (2002). Changing requirements for Gbx2 in development of the cerebellum and maintenance of the mid/hindbrain organizer. Neuron 36:31–43. [DOI] [PubMed] [Google Scholar]
- Lu W, Tsirka SE (2002). Partial rescue of neural apoptosis in the Lurcher mutant mouse through elimination of tissue plasminogen activator. Development 129:2043–2050. [DOI] [PubMed] [Google Scholar]
- Madani R, Hulo S, Toni N, Madani H, Steimer T, Muller D, Vassalli JD (1999). Enhanced hippocampal long-term potentiation and learning by increased neuronal expression of tissue-type plasminogen activator in transgenic mice. EMBO J 18:3007–3012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marconi MA, Park KI, Teng YD, Ourednik J, Ourednik V, Taylor RM, Marciniak AE, Daadi MM, Rose HL, Lavik EB, Langer R, Auguste KI, Lachyankar M, Freed CR, Redmond DE, Sidman RL, Snyder EY (2003). Neural stem cells: from in vivo to in vitro and back again—practical aspects. In: Stem cell handbook (Sell S, ed.) pp. 191–208. Totowa, NJ: Humana.
- Marin-Teva JL, Dusart I, Colin C, Gervais A, van Rooijen N, Mallat M (2004). Microglia promote the death of developing Purkinje cells. Neuron 41:535–547. [DOI] [PubMed] [Google Scholar]
- Marrs GS, Green SH, Dailey ME (2001). Rapid formation and remodeling of postsynaptic densities in developing dendrites. Nat Neurosci 4:1006–1013. [DOI] [PubMed] [Google Scholar]
- Mehler MF, Gokhan S (2001). Developmental mechanisms in the pathogenesis of neurodegenerative diseases. Prog Neurobiol 63:337–363. [DOI] [PubMed] [Google Scholar]
- Miyata S, Nakatani Y, Hayashi N, Nakashima T (2005). Matrix-degrading enzymes tissue plasminogen activator and matrix metalloprotease-3 in the hypothalamo-neurohypophysial system. Brain Res 1058:1–9. [DOI] [PubMed] [Google Scholar]
- Nakamura M, Okano H, Toyama Y, Dai HN, Finn TP, Bregman BS (2005). Transplantation of embryonic spinal cord-derived neurospheres support growth of supraspinal projections and functional recovery after spinal cord injury in the neonatal rat. J Neurosci Res 15:457–468. [DOI] [PubMed] [Google Scholar]
- Nicole O, Docagne F, Ali C, Margaill I, Carmeliet P, MacKenzie ET, Vivien D, Buisson A (2001). The proteolytic activity of tissue-plasminogen activator enhances NMDA receptor-mediated signaling. Nat Med 7:59–64. [DOI] [PubMed] [Google Scholar]
- Norton WH, Mangoli M, Lele Z, Pogoda HM, Diamond B, Mercurio S, Russell C, Teraoka H, Stickney HL, Rauch GJ, Heisenberg CP, Houart C, Schilling TF, Frohnhoefer HG, Rastegar S, Neumann CJ, Gardiner RM, Strahle U, Geisler R, Rees M (2005). Monorail/Foxa2 regulates floorplate differentiation and specification of oligodendrocytes, serotonergic raphe neurones, and cranial motoneurones. Development 132:645–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ourednik J, Ourednik V, Lynch WP, Schachner M, Snyder EY (2002). Neural stem cells display an inherent mechanism for rescuing dysfunctional neurons. Nat Biotechnol 20:1103–1110. [DOI] [PubMed] [Google Scholar]
- Pang PT, Teng HK, Zaitsev E, Woo NT, Sakata K, Zhen S, Teng KK, Yung WH, Hempstead BL, Lu B (2004). Cleavage of proBDNF by tPA/plasmin is essential for long-term hippocampal plasticity. Science 306:487–491. [DOI] [PubMed] [Google Scholar]
- Park KI, Teng YD, Snyder EY (2002). The injured brain interacts reciprocally with neural stem cells supported by scaffolds to reconstitute lost tissue. Nat Biotechnol 20:1111–1117. [DOI] [PubMed] [Google Scholar]
- Parker MA, Anderson JK, Corliss DA, Abraria VE, Sidman RL, Park KI, Teng YD, Cotanche DA, Snyder EY (2005). Expression profile of an operationally-defined neural stem cell clone. Exp Neurol 194:320–332. [DOI] [PubMed] [Google Scholar]
- Pawlak R, Melchor JP, Matys T, Skrzypiec AE, Strickland S (2005). Ethanol-withdrawal seizures are controlled by tissue plasminogen activator via modulation of NR2B-containing NMDA receptors. Proc Natl Acad Sci USA 102:443–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosario CM, Yandava BD, Kosaras B, Zurakowski D, Sidman RL, Snyder EY (1997). Differentiation of engrafted multipotent neural progenitors towards replacement of missing granule neurons in meander tail cerebellum may help determine the locus of mutant gene action. Development 124:4213–4224. [DOI] [PubMed] [Google Scholar]
- Samoilova M, Li J, Pelletier MR, Wentlandt K, Adamchik Y, Naus CC, Carlen PL (2003). Epileptiform activity in hippocampal slice cultures exposed chronically to bicuculline: increased gap junctional function and expression. J Neurochem 86:687–699. [DOI] [PubMed] [Google Scholar]
- Sampson MJ, Lovell RS, Craigen WJ (1997). The murine voltage-dependent anion channel gene family. Conserved structure and function. J Biol Chem 272:18966–18973. [DOI] [PubMed] [Google Scholar]
- Sarna JR, Hawkes R (2003). Patterned Purkinje cell death in the cerebellum. Prog Neurobiol 70:473–507. [DOI] [PubMed] [Google Scholar]
- Seeds NW, Williams BL, Bickford PC (1995). Tissue plasminogen activator induction in Purkinje neurons after cerebellar motor learning. Science 270:1992–1994. [DOI] [PubMed] [Google Scholar]
- Seeds NW, Basham ME, Haffke SP (1999). Neuronal migration is retarded in mice lacking the tissue plasminogen activator gene. Proc Natl Acad Sci USA 96:14118–14123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeds NW, Basham ME, Ferguson JE (2003). Absence of tissue plasminogen activator gene or activity impairs mouse cerebellar motor learning. J Neurosci 23:7368–7375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segal RA, Pomeroy SL, Stiles CD (1995). Axonal growth and fasciculation linked to differential expression of BDNF and NT3 receptors in developing cerebellar granule cells. J Neurosci 15:4970–4981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheehan JJ, Tsirka SE (2005). Fibrin-modifying serine proteases thrombin, tPA, and plasmin in ischemic stroke: a review. Glia 50:340–350. [DOI] [PubMed] [Google Scholar]
- Shimizu S, Narita M, Tsujimoto Y (1999). Bcl-2 family proteins regulate the release of apoptogenic cytochrome c by the mitochondrial channel VDAC. Nature 399:483–487. [DOI] [PubMed] [Google Scholar]
- Sidman RL, Green MC (1970). “Nervous,” a new mutant mouse with cerebellar disease. In: Les mutants pathologiques chez l’animal. In: Leur interpret pour la recherche biomedicale (Sabourdy M, ed.) pp. 69–79. Paris: Centre National de la Recherche Scientifique.
- Snyder EY, Deitcher DL, Walsh C, Arnold-Aldea S, Hartwieg EA, Cepko CL (1992). Multipotent neural cell lines can engraft and participate in development of mouse cerebellum. Cell 68:33–51. [DOI] [PubMed] [Google Scholar]
- Song H, Stevens CF, Gage FH (2002). Astroglia induce neurogenesis from adult neural stem cells. Nature 417:39–44. [DOI] [PubMed] [Google Scholar]
- Teng YD, Lavik EB, Qu X, Ourednik J, Zurakowski D, Langer R, Snyder EY (2002). Functional recovery following traumatic spinal cord injury mediated by a unique polymer scaffold seeded with neural stem cells. Proc Natl Acad Sci USA 99:3024–3029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traynelis SF, Lipton SA (2001). Is tissue plasminogen activator a threat to neurons? Nat Med 7:17–18. [DOI] [PubMed] [Google Scholar]
- Vassalli JD, Sappino AP, Belin D (1991). The plasminogen activator/plasmin system. J Clin Invest 88:1067–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verrall S, Seeds NW (1989). Characterization of 125I-tissue plasminogen activator binding to cerebellar granule neurons. J Cell Biol 109:265–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zizi M, Forte M, Blachly-Dyson E, Colombini M (1994). NADH regulates the gating of VDAC, the mitochondrial outer membrane channel. J Biol Chem 269:1614–1616. [PubMed] [Google Scholar]