

Abstract
Hyperpolarization-activated cyclic nucleotide-gated ion channels (h-channels; Ih; HCN) modulate intrinsic excitability in hippocampal and neocortical pyramidal neurons, among others. Whereas Ih mediated by the HCN2 isoform is regulated by cAMP, there is little known about kinase modulation of Ih, especially for the HCN1 isoform predominant in pyramidal neurons. We used a computational method to identify a novel kinase modulator of h-channels, p38 mitogen-activated protein kinase (p38 MAPK). Inhibition of p38 MAPK in hippocampal pyramidal neurons caused a ∼25 mV hyperpolarization of Ih voltage-dependent activation. This downregulation of Ih produced hyperpolarization of resting potential, along with increased input resistance and temporal summation of excitatory inputs. Activation of p38 MAPK caused a ∼11 mV depolarizing shift in Ih activation, along with depolarized resting potential, and decreased input resistance and temporal summation. Inhibition of related MAPKs, ERK1/2 (extracellular signal-related kinase 1/2) and JNK (c-Jun N-terminal kinase), produced no effect on Ih. These results show that p38 MAPK is a strong modulator of h-channel biophysical properties and may deserve additional exploration as a link between altered Ih and pathological conditions such as epilepsy.
Keywords: HCN, Ih, kinase, dendrite, PLP, hippocampus, epilepsy
Introduction
Hyperpolarization-activated cyclic nucleotide-gated ion channels (HCN; h-channels) regulate excitability in a number of brain regions, including hippocampus, cortex, and thalamus. In CA1 hippocampal and neocortical pyramidal neurons, the voltage-gated current mediated by h-channels (Ih) has a nonuniform subcellular distribution, with a high density in the apical dendrites, and a low density at the soma. Dendritic Ih modifies the signal integration properties of pyramidal neurons by decreasing dendritic input resistance, temporal summation, and length constant, thus reducing neuronal firing from excitatory synaptic stimulation (Magee, 1998, 1999). Increasing evidence implicates Ih in activity-dependent changes in neuronal excitability (Beaumont et al., 2002; van Welie et al., 2004), in learning and memory (Nolan et al., 2004), and in pathological states of hyperexcitability such as epilepsy or neuropathic pain (K. Chen et al., 2001; Chaplan et al., 2003; Shah et al., 2004), and motivates efforts to understand how Ih is modulated at a molecular level.
The best characterized modulator of Ih is cAMP, which increases Ih by a protein kinase A-independent interaction with the HCN molecule (Wainger et al., 2001), shifting Ih voltage-dependent activation in a depolarized direction (Pedarzani and Storm, 1995). The HCN2 isoform accounts for most of the action of cAMP on neuronal Ih; the HCN1 isoform, which is predominant in hippocampus and neocortex, is minimally modulated by cAMP (S. Chen et al., 2001). Protein kinases have been implicated in the modulation of a number of voltage-gated ion channels, and there is some evidence of kinase action on HCN, particularly for protein tyrosine kinases on HCN2 (Chang et al., 1991; Yu et al., 2004; Zong et al., 2005), with one report of HCN1 downregulation in brainstem neurons via TrkB activation (Thoby-Brisson et al., 2003). However, study of exogenously expressed HCN suggests that other important modulators remain to be determined: when both HCN1 and HCN2 channels are excised from the cell, Ih voltage-dependent activation shifts 30–40 mV in a hyperpolarized direction, independent of cAMP activity (S. Chen et al., 2001).
In this study, we sought to identify novel kinase modulators of h-channels using prediction of kinase phosphorylation recognition sequences on the HCN1 and HCN2 molecules. This identified p38 mitogen-activated protein kinase (p38 MAPK) as having a high likelihood of potentially interacting with HCN. In fact, pharmacological blockade of p38 MAPK activity significantly downregulated Ih in hippocampal neurons, with a ∼25 mV hyperpolarizing shift in voltage-dependent activation, causing concomitant hyperpolarization of neuronal resting potential and increases in neuronal input resistance and temporal summation. Activation of p38 MAPK produced an ∼11 mV depolarizing shift in Ih activation, as well as depolarization of resting potential, and reduced input resistance and temporal summation. Blockade of two related MAPKs, extracellular signal-related kinase types 1/2 (ERK1/2) and c-Jun N-terminal kinase (JNK), had no effect on Ih. These results show that p38 MAPK is a strong modulator of Ih activity in hippocampal pyramidal neurons, and may be a critical constitutive factor maintaining the normal properties of neuronal h-channels.
Materials and Methods
Hippocampal slices (400 μm) were prepared from 6- to 10-week-old male Sprague Dawley rats using standard procedures (Poolos and Johnston, 1999). Animal protocols were approved by the Institutional Animal Care and Use Committee at the University of Washington. Neurons were visualized with differential interference contrast microscopy using a Zeiss (Oberkochen, Germany) Axioskop. Recordings were performed at 30−32°C. The extracellular recording solution contained the following (in mm): 125 NaCl, 25 NaHCO3, 10 dextrose, 2.5 KCl, 2 CaCl2, 2 MgCl2, and 1.25 NaH2PO4. The whole-cell recording pipette solution contained the following (in mm): 120 KMeSO4, 20 KCl, 10 HEPES, 4 Na2-ATP, 2 MgCl2, 0.3 Tris-GTP, 0.2 EGTA, pH 7.3, with KOH. For cell-attached patch recordings, the pipette solution contained the following (in mm): 120 KCl, 20 tetraethylammonium-Cl, 10 HEPES, 5 4-aminopyridine, 2 CaCl2, 1 MgCl2, 1 BaCl2, pH 7.4, with KOH. Recorded neurons had resting potentials between −55 and −78 mV.
Whole-cell current-clamp recordings were made using a Dagan (Minneapolis, MN) BVC-700 amplifier, were sampled at 10 kHz, and filtered at 2 kHz. Patch recordings used a Molecular Devices (Foster City, CA) AxoPatch 200B amplifier, and were sampled at 2 kHz and filtered at 500 Hz. Data acquisition used custom software written for the Igor Pro 4.09 analysis environment (Wavemetrics, Lake Oswego, OR). Current injections simulating EPSPs were modeled by an α function of the following form: I = Imax(αt)(1 − e−αt), where α = 1.67. Temporal summation was measured as the ratio of fifth response amplitude (measured from baseline) to the first response amplitude. Voltage-dependent activation of Ih was measured by normalizing the peak tail current amplitudes to a −40 mV voltage command after steady-state activation by various command voltages. This current–voltage relationship was fitted with a Boltzmann function to calculate the half-maximal activation voltage (V1/2) and slope factor. Activation time constants were estimated by fitting Ih traces obtained near V1/2 with a double exponential curve of the form: y0 + A1exp(−x/τ1) + A2exp(−x/τ2), where τ1 and τ2 are the fast and slow time constants, respectively. Curve fitting used the Levenberg–Marquardt algorithm to minimize the χ2 error statistic (Igor Pro 4.09), and all curve fits used for analysis converged properly. Maximal Ih current density was calculated using an estimate of patch area derived from pipette resistance (Sakmann and Neher, 1995). Group data are expressed as mean ± SEM. Statistical significance was calculated using an unpaired two-tailed Student’s t test except where indicated for multiple comparisons, where a one-way ANOVA with Tukey’s post hoc test was used.
Drugs were applied in the bath and in the recording pipette made either from aqueous stock solutions [N-ethyl-1,2-dimethyl-6-methylimino-N-phenyl-pyrimidin-4-amine hydrochloride (ZD7288); anisomycin] or dissolved in DMSO such that the final DMSO concentration was ≤0.1% [4-[5-(4-fluorophenyl)-2-[4-[(S)-methylsulfinyl]phenyl]-3H-imidazol-4-yl]pyridine (SB203580), 4-(4-fluorophenyl)-2-(4-hydroxyphenyl)-5-(4-pyridyl)1H-imidazole (SB202190), (2Z,3Z)-2,3-bis[amino-(2-aminophenyl)sulfanyl-methylidene]butanedinitrile (U0126), 4-ethyl-2(p-methoxyphenyl)-5-(4′-pyridyl)-1H-imidazole (SB202474), anthra[1,9-cd]pyrazol-6(2H)-one (SP600125)]. SB and SP compounds were obtained from Calbiochem (La Jolla, CA). ZD7288 and U0126 were obtained from Tocris (Balwin, MO). Other chemicals were obtained from Sigma (St. Louis, MO). Recombinant p38α MAPK (activated p38 MAPK; Upstate, Lake Placid, NY) was preactivated by upstream kinase mitogen-activated protein kinase kinase 6 (MKK6) and dissolved in standard whole-cell pipette solution (5 μg/ml). p38α MAPK not preactivated by MKK6 was further inactivated by boiling for 30 min, and then dissolved in pipette solution (inactivated p38 MAPK; 5 μg/ml).
To characterize cellular morphology, neurons were filled via recording micropipettes containing 0.1% biocytin (Invitrogen, Eugene, OR), after which the slices were fixed overnight in 4% paraformaldehyde, blocked overnight in 2% BSA with 0.25% Triton X-100 in PBS, and finally incubated overnight with streptavidin–Alexa 488 (1:200; Invitrogen) with 0.1% Triton X-100 in PBS. Images of the fluorescent neurons were collected in ∼15 planes through the cell with a Nikon (El Segundo, CA) Eclipse TE200/Bio-Rad (Hercules, CA) Radiance 2000 confocal microscope system, and then processed using Image J software (Wayne Rasband, National Institutes of Health, Bethesda, MD) into composite projection micrographs.
Results
We sought to identify novel kinase modulators of Ih using computational means to predict kinase interactions with the HCN molecule. Using the Scansite software tool, we searched the rat HCN1 and HCN2 amino acid sequences (derived from the SWISS-PROT database) for possible kinase phosphorylation sites (Obenauer et al., 2003). Setting our search parameters at the highest level of stringency produced the list of 12 kinases shown in Table 1. Of this list, three kinases had previously described actions on voltage-gated ion channels: protein kinase C (PKC), calcium/calmodulin-dependent protein kinase II (CAMKII), and p38 MAPK (Varga et al., 2004; Chen et al., 2005; Wittmack et al., 2005). Of the entire list, p38 MAPK had the highest score for likely HCN interactions, and was predicted to phosphorylate both HCN1 and HCN2. Thus, we decided to test for actions of p38 MAPK on Ih in hippocampal neurons.
Table 1.
Candidate kinases predicted to phosphorylate HCN1 or HCN2
| Kinase | HCN1 site | HCN1 %ile | HCN2 site | HCN2 %ile |
|---|---|---|---|---|
| p38 MAPK | T778 | 0.004 | T17 | 0.048 |
| p85 SH3 mode 1 | P878 | 0.034 | ||
| PKC μ | S717 | 0.091 | ||
| ATM | S729 | 0.095 | ||
| GSK3 | S680 | 0.114 | S721 | 0.157 |
| CAMKII | S335 | 0.132 | ||
| Akt | S653 | 0.146 | S35 | 0.300 |
| p85 SH3 mode 2 | P752 | 0.018 | ||
| Intersectin SH3 | P662 | 0.040 | ||
| Cdc2 | S742 | 0.053 | ||
| PLCg SH3 | P12 | 0.075 | ||
| Abl SH3 | P723 | 0.091 |
“Site” lists predicted phosphorylation site for each kinase. “%ile” shows ranking of the statistical likelihood of each interaction as a percentage of all possible kinase interactions, in which a smaller percentile indicates a higher ranking. ATM, ataxia-telangiectasia mutated; GSK3, glycogen synthase kinase 3; PLC, phospholipase C; SH3, Src homology 3; Cdc2, cell division cycle 2.
We studied the actions of p38 MAPK on two types of hippocampal neurons: the CA1 pyramidal neuron and a recently described cell type, the pyramidal-like principal neuron (PLP) (Bullis et al., 2004). This latter cell type is a projection neuron whose soma is located in the stratum radiatum, that possesses dendritic morphology quite similar to CA1 hippocampal pyramidal neurons, and is a subset of the previously described class of neurons termed radiatum giant cells (RGCs) (Maccaferri and McBain, 1996; Gulyas et al., 1998; Savic and Sciancalepore, 2001). The main feature distinguishing PLPs from RGCs is the pyramidal neuron-like single apical dendrite that bifurcates at distances >50 μm from a pyramidally shaped soma; in contrast, RGCs typically possess two apical dendrites emanating from an inverted pyramid or shield-shaped soma. An intriguing feature of PLPs is that, although having an apical dendritic morphology similar to CA1 pyramidal neurons, their somatodendritic distribution of Ih is reversed compared with that of CA1 pyramidal cells, with a high density of Ih at the soma but low Ih density in the dendrites that declines with increasing distance from the soma (Bullis et al., 2004). Thus, the high somatic Ih density in PLP neurons provides an opportunity to study Ih in hippocampal neurons using somatic recordings rather than the relatively more difficult dendritic recording techniques needed to study Ih in CA1 pyramidal neurons. In these experiments, we characterized the effects of p38 MAPK inhibition on Ih recorded in both types of neuron.
Examples of CA1 pyramidal and PLP neurons are shown in Figure 1. Representative biocytin fills of the two types showed that PLP neurons have an apical dendritic tree that is extensively arborized, and have a central bifurcation about halfway along the somatodendritic axis. The PLP soma is pyramidal in shape but larger than the CA1 pyramidal soma. The PLP soma also possesses numerous short basilar dendrites and an axon that projects to the alveus. Thus, the morphology of the PLP neuron is similar to that of the CA1 pyramidal neuron.
Figure 1.

Characteristics of Ih in pyramidal and PLP neurons. A, Biocytin-filled image of a typical CA1 hippocampal pyramidal neuron and accompanying traces showing Ih from a dendritic recording. Voltage commands are shown above each trace. The dendritic recording distance was 180 μm. B, Biocytin-filled image of a PLP neuron and sample Ih traces from a somatic recording. PLP neurons showed a similar morphology to CA1 pyramidal neurons, and Ih from somatic PLP recordings demonstrated similar kinetics to that from pyramidal dendrite recordings. C, Voltage-dependent activation of Ih was similar in pyramidal dendrites (circles) and PLP somas (triangles). D, Ih activation time constants were similar for pyramidal dendrites (black bars) and PLP somas (gray bars). Error bars indicate SEM.
The characteristics of Ih recorded in cell-attached patch-clamp mode from pyramidal dendrites and PLP somas were similar. As shown in Figure 1A, Ih evoked by hyperpolarizing voltage commands in pyramidal dendrites was an inward current with a principal fast exponential component and a smaller slow exponential component. This kinetic description is consistent with the predominance of HCN1 over HCN2 expression in CA1 pyramidal neurons, because the HCN1 subtype shows faster kinetics than the HCN2 subtype in exogenous expression systems (Santoro and Tibbs, 1999). Ih recorded in PLP somas showed a similar appearance, with a predominant fast component and a smaller slow component (Fig. 1B). Notably, the density of maximally activated Ih in PLP somas was comparable with that recorded about halfway along the CA1 pyramidal dendrites. The average maximal Ih density evoked by a −150 mV voltage command in pyramidal dendrites was 12.3 ± 3.4 pA/μm2 (n = 12), recorded at a mean dendritic distance from the soma of 156 ± 7.5 μm (n = 12). PLP somas showed an average maximal Ih density of 16.7 ± 2.2 pA/μm2 (n = 11).
The voltage-dependent activation of Ih was similar in both pyramidal dendrites and PLP somas. As shown in Figure 1C, Ih activation in both cell types was low around −60 mV, and then rapidly increased at more hyperpolarized potentials to plateau around −120 mV. The half-activation voltage (V1/2) in pyramidal dendrites was −86 ± 3.5 mV (n = 12), compared with −89 ± 2.4 mV (n = 11; p > 0.05) in PLP somas. Activation time constants were determined by fitting current traces obtained near V1/2 with double exponential functions. The fast and slow time constants extracted from these functions are shown in Figure 1D and Table 2 and were similar for pyramidal dendrites and PLP somas (p > 0.05 for comparisons of fast and slow time constants, respectively). These data show that Ih, aside from somatodendritic distribution, has equivalent biophysical characteristics in the two cell types, and thus likely results from a similar composition of HCN subunits.
Table 2.
Ih activation time constants measured at V1/2under control conditions and during inhibition of p38 MAPK with SB compounds
| Control | SB | SB vs control | |
|---|---|---|---|
| Pyr. dend. τfast | 54.4 ± 7.99 (n = 11) | 90.0 ± 31.0 (n = 8) | p > 0.05 |
| Pyr. dend. τslow | 664 ± 134 (n = 11) | 1069 ± 441 (n = 8) | p > 0.05 |
| PLP τfast | 63.0 ± 11.4 (n = 11) | 70.2 ± 14.1 (n = 5) | p > 0.05 |
| PLP τslow | 444 ± 49.9 (n = 11) | 1179 ± 297 (n = 5) | p < 0.01 |
All time constants are in milliseconds. Pyr. dend., Pyramidal dendrite.
We then tested whether pharmacological inhibition of p38 MAPK in the two cell types would affect Ih. We used a highly specific cell-permeable inhibitor of p38 MAPK, SB203580 (10 μm) (English and Cobb, 2002). [In a subset of experiments, we also used another highly specific inhibitor, SB202190 (20 μm) with equivalent results.] All kinase inhibitors were delivered via preincubation in the bath for ∼1 h, and in the recording pipette as well, consistent with previous studies that have used a preincubation period to achieve blockade of kinase function in neurons (Yuan et al., 2002; Selcher et al., 2003). Using these drug delivery methods in cell-attached voltage-clamp experiments, we compared Ih in a control group of neurons to that in a group exposed to p38 MAPK inhibitors. As shown in Figure 2A, blockade of p38 MAPK in pyramidal dendrites caused an ∼25 mV hyperpolarizing shift in the voltage-dependent activation of Ih (V1/2 in SB203580: −111 ± 4.7 mV, n = 8; V1/2 in control: −86 ± 3.5, n = 12, p < 0.0005). The slope factor of the activation curve also increased under p38 MAPK blockade (in SB203580: 12 ± 1.6, n = 8; in control: 7.0 ± 0.80, n = 12, p < 0.01). Equivalent results were obtained with inhibition of p38 MAPK in PLP somas (Fig. 2B), with an ∼22 mV hyperpolarizing shift in Ih activation (V1/2 in SB202190: −111 ± 3.6 mV, n = 6; V1/2 in control: −89 ± 2.4 mV, n = 11, p < 0.0001), and an increase in the slope factor (in SB202190: 12 ± 1.7, n = 6; in control: 6.4 ± 0.94, n = 11, p < 0.01). These results appeared to show that blockade of p38 MAPK produced a significant downregulation of Ih voltage-dependent activation that was equivalent in pyramidal neuron dendrites and PLP neuron somas.
Figure 2.

Blockade of p38 MAPK caused a hyperpolarizing shift in Ih activation in both pyramidal dendrites and PLP somas. A, Application of SB203580 (10 μm), a selective inhibitor of p38 MAPK, by bath preincubation and in the recording pipette caused an ∼25 mV hyperpolarizing shift in Ih half-activation voltage (V1/2) in pyramidal dendrites (triangles) compared with control (circles). B, Under similar recording conditions to those in A, blockade of p38 MAPK by SB202190 (20 μm), another p38 MAPK inhibitor, caused a hyperpolarizing shift in V1/2 of similar magnitude in PLP somas to that seen in pyramidal dendrites. C, Preincubation and pipette delivery of U0126 (20 μm), an inhibitor of ERK1/2, had no effect on Ih activation in pyramidal dendrites (triangles) compared with control (circles). D, Preincubation and pipette delivery of SB202474 (10 μm; crosses), an inactive analog of SB203580, and SP600125 (10 μm; triangles), an inhibitor of JNK, caused no significant change in Ih activation in PLP somas compared with control (circles). E, Application of a membrane permeable form of cAMP, 8-Br-cAMP (100 μm; triangles), caused an insignificant depolarizing shift in Ih activation in pyramidal dendrites compared with control (circles). F, Application of anisomycin (20 μm; triangles), an activator of p38 MAPK, caused an ∼11 mV depolarizing shift in Ih activation in PLP somas compared with control (circles). Error bars indicate SEM.
We performed several control experiments to determine whether Ih was similarly regulated by other members of the MAPK family, and whether nonspecific changes in Ih were induced by the delivery of pharmacological agents. First, we tested the effects of blockade of a related MAPK, ERK1/2, which has been shown to regulate the activation of A-type K+ channels (Yuan et al., 2002). We used U0126 (20 μm), a cell-permeable inhibitor of MEK (mitogen-activated protein kinase kinase), the upstream inducer of ERK1/2. Of note, ERK1/2 was not predicted in our computational analysis to recognize a consensus sequence on either HCN1 or HCN2 (Table 1). Delivery of U0126 via similar methods as with p38 MAPK inhibitors caused no significant change in voltage-dependent activation of Ih in pyramidal dendrites (V1/2 in U0126: −82 ± 5.6 mV, n = 4, p > 0.05 compared with control) (Fig. 2C), showing that ERK1/2 does not significantly modulate Ih activation. We then similarly blocked activation of JNK, a third MAPK family member. Treatment of PLP neurons using SP600125 (10 μm), a highly specific antagonist of JNK activation, caused no significant change in Ih activation (V1/2 in SP600125: −87 ± 3.6 mV, n = 4, p > 0.05 compared with control) (Fig. 2D), showing that JNK does not significantly modulate Ih activation. Finally, treatment of PLP neurons with SB202474 (10 μm), an inactive analog of SB203580 and SB202190, caused no change in Ih activation (V1/2 in SB202474: −92 ± 6.1 mV, n = 8, p > 0.05 compared with control) (Fig. 2D), confirming that the effects of SB203580 and SB202190 on Ih activation were not attributable to nonspecific effects of the drug delivery protocol.
We then asked whether activation of p38 MAPK would modulate Ih voltage-dependent activation in a manner opposite to that of p38 MAPK inhibition. The best described activator of Ih, cAMP, produces a depolarizing shift in Ih activation, although this action has been described in cells where HCN2 expression predominates (such as thalamocortical projection neurons), whereas HCN1-mediated Ih is primarily cAMP insensitive (S. Chen et al., 2001). For example, an intracellular level of 1 μm cAMP in thalamocortical neurons was found to maximally shift V1/2 from −91 to −78 mV, a depolarizing shift of 13 mV (Budde et al., 2005). We applied a membrane permeable analog of cAMP, 8-Br-cAMP (100 μm) to pyramidal dendrites (Fig. 2E). This saturating concentration of 8-Br-cAMP caused an insignificant depolarizing shift in Ih activation (V1/2 in 8-Br-cAMP: −82 ± 0.35 mV, n = 3; V1/2 in control: −86 ± 3.5, n = 12; p > 0.05). This insignificant effect of cAMP on pyramidal dendrite Ih is consistent with the predominance of cAMP-insensitive HCN1 expression in pyramidal neurons (Santoro et al., 2000).
We then applied anisomycin, a known activator of p38 MAPK (and JNK), to PLP neurons (Shifrin and Anderson, 1999). Anisomycin (20 μm) caused an ∼11 mV depolarizing shift in Ih activation (V1/2 in anisomycin: −78 ± 2.1 mV, n = 7; V1/2 in control: −89 ± 2.4 mV, n = 11; p < 0.001) (Fig. 2F). Because we previously found that JNK inhibition did not modulate Ih activation, this suggests that anisomycin acts to upregulate Ih via activation of p38 MAPK. Also, because the magnitude of the anisomycin effect on V1/2 was so much larger than the effect of cAMP, it is unlikely that anisomycin is acting through a cAMP-dependent mechanism. Together, these results show that p38 MAPK acts to modulate Ih activation in both depolarizing and hyperpolarizing directions through a broad range depending on whether resting levels of p38 MAPK activation are increased or decreased, respectively. This modulation of Ih activation is not shared by two other members of the MAPK family, ERK1/2 and JNK.
The effects of p38 MAPK inhibition on other biophysical parameters of Ih are shown in Figure 3. Sample Ih traces evoked by command voltages from approximately −30 mV to approximately −150 mV under control and p38 MAPK inhibition conditions from pyramidal dendrite recordings are shown in Figure 3, A and B. As can be seen, inhibition of p38 MAPK significantly reduced Ih evoked by physiological levels of hyperpolarization from rest (e.g., −80 mV). In Figure 3C, the resting potential (RP) change after p38 MAPK inhibition is shown. RP was determined by rupture of the patch at the end of each cell-attached patch recording used to determine voltage-dependent activation in Figure 2, A and B. Inhibition of p38 MAPK produced a hyperpolarization of RP in pyramidal dendrites in which the average RP in control was −60 ± 1.3 mV (n = 12), whereas in the presence of SB203580 (10 μm), RP was −71 ± 3.5 mV (n = 8; p < 0.01). For PLP somas, RP in control was hyperpolarized (−68 ± 3.7 mV; n = 11) compared with pyramidal dendrites under control conditions, and did not hyperpolarize significantly further after treatment with SB202190 (20 μm; RP = −72 ± 2.3 mV; n = 6; p > 0.05 in comparison with PLPs in control). The hyperpolarization of pyramidal dendrite RP after p38 MAPK inhibition is consistent with a hyperpolarizing shift in Ih activation, because less Ih was active at physiological membrane potentials. The relatively hyperpolarized RP of PLPs under control conditions, and the smaller effect of p38 MAPK inhibition on PLP resting potential, may result from the lesser dendritic density of Ih in these cells compared with CA1 pyramidal neurons. Because dendritic surface area constitutes >90% of total neuronal surface area in pyramidal neurons (Fiala and Harris, 1999), it is likely that CA1 pyramidal neurons contain greater total Ih than PLPs. This possibly explains why resting potential in CA1 pyramidal neurons, which is influenced by total neuronal Ih, is more depolarized and more sensitive to Ih downregulation than that of PLP neurons.
Figure 3.

Effects of p38 MAPK blockade on Ih properties. A, Sample Ih traces from a pyramidal dendrite elicited by voltage commands from −30 to −150 mV under control conditions. Tail currents are shown at expanded time scale at right. Traces elicited by a −80 mV command are indicated by an arrowhead. Calibration: 5 pA (1.8 pA for tail currents), 250 ms (12 ms for tail currents). B, Ih traces from pyramidal dendrite using same protocol as in A after p38 MAPK inhibition by SB203580 (10 μm). Traces elicited by a −79 mV command are indicated by an arrowhead. Note reduction of Ih by p38 MAPK inhibition at physiological levels of hyperpolarization. Calibration: 10 pA (5.5 pA for tail currents), 250 ms (12 ms for tail currents). C, Average RP (in millivolts) after patch rupture after voltage-clamp experiments. Pyramidal dendrite (Dend) RP was significantly hyperpolarized after application of SB203580 (10 μm; ∗p < 0.01). PLP soma RP was not significantly hyperpolarized after application of SB202190 (20 μm). D, Maximal Ih density recorded at −150 mV in PLP somas showed no significant change after p38 MAPK inhibition by SB202190 (20 μm). Error bars indicate SEM.
The shift in V1/2 caused by inhibition of p38 MAPK was not associated with a significant decrease in maximal Ih (Fig. 3D). We measured maximal Ih in PLP somas, because in pyramidal dendrites Ih varies with somatodendritic distance, making comparisons more subject to error. In PLP neurons, maximal Ih density in the presence of SB202190 (20 μm) was 12.4 ± 1.35 pA/μm2 (n = 6), not significantly different from that under control conditions (16.7 ± 2.2 pA/μm2; n = 11; p > 0.05). p38 MAPK inhibition did not significantly affect activation time constants measured near V1/2 for either fast or slow components, with the exception of the slow time constant in PLP somas (shown in Table 2). Because the slow component of Ih represents a small minority of the total current, we also measured the slow activation time constant at approximately −150 mV at which the total current amplitude is highest. At −150 mV, the slow activation time constants under control and p38 MAPK inhibition conditions did not differ significantly (control: 601 ± 307 ms, n = 8; SB202190: 690 ± 467 ms, n = 4, p > 0.05). Because of the small amplitudes and large variances in measurements of the slow component of Ih, it is unclear whether the change in this parameter at V1/2 in PLP neurons with p38 MAPK inhibition is of physiological significance.
The results obtained with cell-attached patch voltage-clamp recordings demonstrated that inhibition of p38 MAPK principally caused a decrease in Ih by shifting its voltage-dependent activation in a hyperpolarized direction. We sought confirmation of the effects of Ih downregulation on neuronal excitability by determining whether input resistance (IR) and temporal summation (TS) of EPSP-like voltage transients, two parameters sensitive to the amount of Ih active at rest, were affected by p38 MAPK inhibition. To do so, we used whole-cell current-clamp recordings in pyramidal dendrites and PLP somas.
The effect of p38 MAPK inhibition on IR is shown in Figure 4. We used a KMeSO4-based pipette solution for all current-clamp experiments, based on evidence that this solution does not block Ih as do K-gluconate-based pipette solutions (Velumian et al., 1997). We measured IR as the average of the steady-state voltage response to a set of three hyperpolarizing 100–300 pA current commands. Under control conditions, hyperpolarizing current injections in pyramidal dendrites produced a depolarizing “sag” in the voltage deflection that reduced steady-state IR and was representative of Ih activation (Fig. 4A, Ctrl, arrowhead). To replicate the conditions of kinase inhibition produced in the cell-attached patch-clamp experiments, we preincubated pyramidal neurons in SB203580 (10 μm) for 1 h before obtaining recordings, and then compared these results to a control population of neurons. IR measured in pyramidal dendrites significantly increased from 61 ± 9.1 MΩ (n = 10) under control conditions to 105 ± 13 MΩ (n = 5; p < 0.05) in SB203580, an increase of ∼72% (Fig. 4A, SB). This increase in IR after blockade of p38 MAPK was consistent with downregulation of resting Ih.
Figure 4.
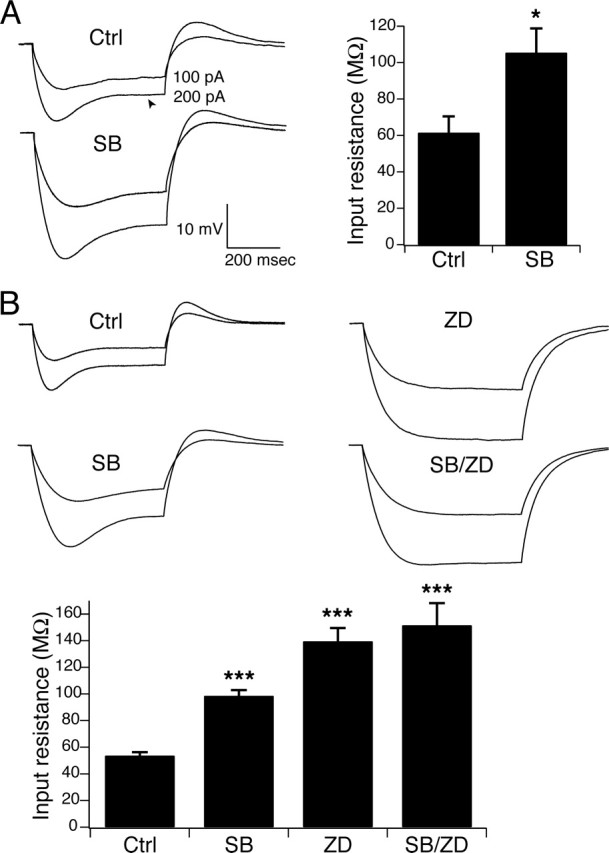
Downregulation of Ih by p38 MAPK inhibition increased IR. A, Sample current-clamp recordings in pyramidal dendrites from separate representative neurons in response to hyperpolarizing current injections of 100 and 200 pA under control conditions and after incubation in SB203580 (10 μm) shows that blockade of p38 MAPK increased IR. The arrowhead indicates depolarizing “sag” in voltage response associated with Ih activation. Resting potential was held at −65 mV by steady-state current injection for all measurements. Group data show that blockade of p38 MAPK by SB203580 caused an ∼72% increase in IR compared with the control population (∗p < 0.05). B, Similar increases in IR were seen during current-clamp recordings in PLP somas after incubation in SB203580. IR increased further when neurons were incubated in ZD7288 (10 μm), a blocker of h-channels, abolishing the depolarizing sag. When PLP neurons were incubated with SB203580 and ZD7288 together, there was no significant difference in IR compared with ZD7288 alone, demonstrating that the effects of SB203580 on IR required intact Ih (∗∗∗p < 0.001 compared with control by one-way ANOVA). Error bars indicate SEM.
Similar results were seen in PLP neurons. Current-clamp recordings at the soma of PLP neurons in response to hyperpolarizing current injections showed similar “sag” of membrane potential reflecting Ih activation (Fig. 4B, Ctrl). Preincubation in SB203580 (10 μm) caused a similar increase in steady-state IR as was seen in pyramidal dendrites (Fig. 4B, SB). IR increased from 53 ± 2.7 MΩ (n = 16) under control conditions to 98 ± 4.3 MΩ (n = 7) in SB203580, an increase of ∼85% (p < 0.001 compared with control by one-way ANOVA). To determine whether this change in IR was specific to Ih, PLP neurons were preincubated in ZD7288 (10 μm), a blocker of Ih. ZD7288 caused an ∼162% increase in IR (139 ± 10 MΩ; n = 10; p < 0.001 compared with control by one-way ANOVA), consistent with complete blockade of Ih (Fig. 4B, ZD). Coapplication of SB203580 and ZD7288 (Fig. 4B, SB/ZD), however, caused no significant additional increase in IR (151 ± 16 MΩ; n = 4; p > 0.05 compared with ZD7288 alone by one-way ANOVA), showing that the IR change produced by SB203580 alone was dependent on intact Ih.
We then tested whether temporal summation of EPSP-like voltage transients would be affected by p38 MAPK inhibition. Ih produces sublinearity in TS which is frequency dependent because of the gradual deactivation of Ih present at rest during the progressive depolarization that occurs during EPSP summation (Magee, 1999). In CA1 pyramidal neuron dendrites, Ih produces maximal sublinearity at ∼20 Hz stimulation (Poolos et al., 2002). In these experiments, we sought evidence for downregulation of Ih in an increase of TS.
Current-clamp recordings were made in pyramidal dendrites and PLP somas under control conditions or after incubation in SB203580. Trains of five α-function current waveforms were injected at 20 Hz to simulate EPSPs. TS was measured as the ratio of the fifth response amplitude to that of the first. Under control conditions in pyramidal dendrites, TS of the fifth response was 154 ± 7.5% (n = 14) of the first response (Fig. 5A, Ctrl). After incubation in SB203580 (10 μm), TS increased to 179 ± 5.8% (n = 5; p < 0.05) (Fig. 5B, SB), an increase of ∼16%. This increased TS was consistent with downregulation of Ih by p38 MAPK inhibition.
Figure 5.

Downregulation of Ih by p38 MAPK inhibition increased TS. A, Representative current-clamp recordings in pyramidal dendrites in response to current injection of 20 Hz α-function waveforms under control conditions (thin trace, Ctrl), after incubation in SB203580 (10 μm; thick trace, SB), or superimposed (Both). TS was measured as the ratio of the peak amplitude of the fifth response to that of the first, and increased after p38 MAPK blockade. Resting potential was held at −65 mV by steady-state current injection for all measurements. Group data showed that incubation in SB203580 caused an ∼16% increase in TS in pyramidal dendrites (∗p < 0.05). B, Similar increases in TS were seen during current-clamp recordings in PLP somas after incubation in SB203580. Sample recordings from separate neurons are shown in response to 20 Hz α-function current injections. Responses in drug (thick traces) are superimposed over control response (thin trace). Group data show that TS increased ∼25% after incubation in SB203580. TS increased further when neurons were incubated in ZD7288 (10 μm). When PLP neurons were incubated with SB203580 and ZD7288 together, there was no significant difference in TS compared with ZD7288 alone, demonstrating that similar to the IR data, the effects of SB203580 on TS required intact Ih (∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001 compared with control by one-way ANOVA). Error bars indicate SEM.
Similar effects of p38 MAPK inhibition on temporal summation were seen in PLP neurons. Current injection of 20 Hz α-function waveforms during PLP somatic current-clamp recordings produced TS, which was similar to that seen in pyramidal dendrites. Under control conditions, TS of the fifth response compared with the first was 145 ± 5.1% (n = 14) (Fig. 5B, Ctrl). After incubation in SB203580 (10 μm), TS was 181 ± 9.0% (n = 6), an increase of ∼25% (p < 0.05 by one-way ANOVA) (Fig. 5B, SB). As in pyramidal dendrites, p38 MAPK inhibition caused an increase in TS, consistent with downregulation of Ih. To determine whether the increase in TS with p38 MAPK inhibition was specific to Ih, we obtained recordings from PLP somas preincubated in ZD7288 (10 μm). Under these conditions, TS was 205 ± 9.3% (n = 10), an increase of ∼41% compared with control (p < 0.001 by one-way ANOVA) (Fig. 5B, ZD). Preincubation in SB203580 and ZD7288 together yielded TS of 198 ± 8.4% (n = 4), which was not significantly different from ZD7288 alone (p > 0.05 by one-way ANOVA) (Fig. 5B, SB/ZD). As with IR measurements, these results showed that the effects of p38 MAPK inhibition on TS were dependent on intact Ih.
We then asked whether intracellular perfusion of activated recombinant p38α MAPK would produce opposite effects on neuronal excitability compared with p38 MAPK inhibition. Current-clamp recordings were made in PLP somas using pipette solution that contained either control (KMeSO4-based) solution, or control solution containing inactivated p38α MAPK (5 μg/ml) or activated p38α MAPK (5 μg/ml) (see Materials and Methods). We then measured IR and TS at 15 min intervals. Under control conditions (Fig. 6A,C, Ctrl), IR declined somewhat after 15 min of recording compared with the beginning of the recording (14 ± 3.7% decrease; n = 6). Similarly, IR decreased modestly after 15 min of recording with pipette solution containing inactivated p38α MAPK (7.9 ± 4.1% decrease; n = 6). However, perfusion with activated p38α MAPK caused a significant decrease in IR that was maximal at 15 min (37 ± 7.7% decrease; n = 4; p < 0.01 compared with inactivated p38α MAPK by one-way ANOVA). Associated with decreased IR was a depolarization of resting membrane potential to −48 ± 0.87 mV (n = 4) after 15 min of perfusion. This was significantly depolarized in comparison to RP after 15 min of recording in control solution (−68 ± 1.3 mV; n = 6; p < 0.001 by one-way ANOVA) and inactivated p38α MAPK solution (−66 ± 1.3 mV; n = 6; p < 0.001 by one-way ANOVA).
Figure 6.

Activated p38α MAPK subunits upregulate Ih. A, Representative current-clamp recordings from PLP neurons in response to 200 pA hyperpolarizing current injections measured initially (thin trace, 0 min) or after 15 min of recording in the same neuron (thick trace, 15 min). Resting potential was held at −65 mV by steady-state current injection for all measurements. Under control conditions and with inactivated p38α MAPK added to the pipette solution, IR had decreased minimally after 15 min of recording. With active p38α MAPK added to the pipette solution, IR decreased significantly after 15 min of recording. B, Measurement of TS in PLP neurons in response to 20 Hz α-function current injections and under the same conditions as in A showed that TS changed minimally after 15 min of recording in control and inactivated p38α MAPK conditions. Activated p38α MAPK caused a significant decrease in temporal summation after 15 min of recording. C, D, Group data showing decrease in IR and TS, respectively, after 15 min of recording with activated p38α MAPK compared with control and inactivated p38α MAPK (∗p < 0.05, ∗∗p < 0.01 compared with activated p38α MAPK by one-way ANOVA). Error bars indicate SEM.
Similar changes in temporal summation were seen after perfusion with activated p38α MAPK. As shown in Figure 6, B and D, perfusion of control solution or inactivated p38α MAPK subunits produced small decreases in TS after 15 min of recording (8.6 ± 3.1% decrease, n = 6 in control; 7.1 ± 2.4%, n = 6 in inactivated p38α MAPK). However, application of activated p38α MAPK caused a significant decrease in TS that was maximal at 15 min (24 ± 5.6% decrease; n = 4; p < 0.05 compared with inactivated p38α MAPK by one-way ANOVA). This decrease in temporal summation, along with changes in input resistance and resting potential, was consistent with significant upregulation of Ih by activated p38α MAPK, and was opposite to the changes caused by inhibition of p38 MAPK, confirming the role of p38 MAPK in modulating the voltage-dependent activation of Ih in pyramidal neurons.
Discussion
These results show that p38 MAPK is a novel modulator of h-channels. The actions of p38 MAPK were demonstrated in two hippocampal neuron types with subcellular domains containing high densities of Ih (CA1 pyramidal neuron dendrites and PLP neuron somas), and in both cell types the results of p38 MAPK modulation were similar. Inhibition of p38 MAPK downregulated Ih by shifting its voltage-dependent activation to substantially more hyperpolarized levels. Conversely, activation of p38 MAPK above resting levels produced a depolarizing shift in Ih voltage-dependent activation. This modulation of Ih activation would be expected to produce significant changes in the amount of Ih present at resting membrane potential and thus influence neuronal excitability. Changes in input resistance, temporal summation of subthreshold voltage transients, and resting potential (all parameters sensitive to Ih) produced by modulation of p38 MAPK were both consistent with its action on Ih voltage-dependent activation and dependent on intact Ih. Thus, inhibition of p38 MAPK produced downregulation of Ih, which yielded increased input resistance and temporal summation, and hyperpolarization of resting potential; activation of p38 MAPK caused upregulation of Ih, which in turn diminished input resistance and temporal summation, and depolarized resting potential. Inhibition of related MAPK family members ERK1/2 and JNK produced no effect on Ih activation, showing that the actions of p38 MAPK are specific to that kinase and not a property of all MAPKs nor an artifact of drug delivery methods.
These results provide the first evidence of kinase modulation of Ih in pyramidal neurons. Ih in the CNS has heterogeneous biophysical properties because of differential expression of HCN subunits, mostly HCN1 and HCN2. Hippocampal and neocortical Ih is primarily mediated by HCN1 expression, whereas in thalamus and other subcortical regions, HCN2 expression predominates (Santoro et al., 2000). Most of the evidence of Ih modulation by signaling intermediaries such as cAMP, intracellular Ca2+, and tyrosine kinases has been demonstrated for brain regions in which the HCN2 isoform predominates (Chang et al., 1991; Yu et al., 2004; Zong et al., 2005), with only a single report of kinase action on HCN1-mediated Ih, in brainstem neurons (Thoby-Brisson et al., 2003). cAMP is the best studied modulator of Ih, but the HCN1 isoform is virtually insensitive to cAMP action, consistent with the minimal effect seen here in pyramidal dendrites. Given the predominance of HCN1 expression in hippocampal neurons, it is likely that the actions of p38 MAPK on Ih in these neurons derive mostly from effects on HCN1, although a small contribution from actions on HCN2 cannot be excluded.
The existence of other strong modulators of HCN voltage dependence apart from cAMP had been hypothesized from experiments studying exogenously expressed channels in which excision of membrane patches from the intracellular milieu caused a hyperpolarizing shift in Ih V1/2 of 30–40 mV that was independent of cAMP activity (S. Chen et al., 2001). The ∼25 mV hyperpolarizing shift in V1/2 caused by inhibition of p38 MAPK activity suggests that constitutive (i.e., tonically active) intracellular p38 MAPK activity is a major determinant of normal h-channel properties. A similar phenomenon has been shown for tyrosine kinases in the maintenance of resting CNG (cyclic nucleotide-gated) channel properties (Kramer and Molokanova, 2001), and there is evidence in cell types as diverse as Xenopus oocytes, human embryonic kidney cells, and rat hepatocytes that constitutive p38 MAPK activity regulates a variety of signaling pathways (Feranchak et al., 2001; Hasegawa and Cahill, 2004; Samuvel et al., 2005). The present results, however, do not show whether p38 MAPK phosphorylates specific residues on the HCN protein, leaving open the possibility that downstream effectors of p38 MAPK mediate its actions on Ih. Even if p38 MAPK directly phosphorylates HCN, some examples of kinase/ion channel interaction have been shown to require participation of chaperone proteins, which at present remain poorly defined with regard to HCN (Schrader et al., 2002; Levitan, 2006). Conversely, several antiepileptic drugs shown to upregulate Ih in hippocampal neurons (such as lamotrigine, which shifts V1/2 ∼11 mV in a depolarizing direction) (Poolos et al., 2002; Surges et al., 2003) act via unknown intermediaries. The finding of a novel kinase modulatory pathway offers new lines of investigation for mechanisms of drug action on Ih.
p38 MAPK has multiple actions in neuronal and other tissues. Its initial characterization was as a signaling pathway activated by pathological conditions such as osmotic stress or inflammation mediated by interleukin-1 and tumor necrosis factor (Roux and Blenis, 2004). For this reason, p38 MAPK is an active focus of study in chronic disease such as rheumatoid arthritis and neurogenic pain syndromes (Kumar et al., 2003; Ji, 2004; Saklatvala, 2004). Our understanding of the physiological roles of p38 MAPK in neurons has begun to follow that of its better characterized MAPK sibling, ERK1/2, which in addition to its initial discovery as a mediator of mitogenic stimulation has also been found to modulate a number of ion channels and neural plasticity processes (Yuan et al., 2002; Sweatt, 2004). For example, p38 MAPK has been found to modulate voltage-gated Na+ channels under both resting and pathological conditions (Wittmack et al., 2005; Jin and Gereau, 2006), as well as the aquaporins (Arima et al., 2003), large-conductance calcium-activated K+ channels (Ye et al., 2004), and the serotonin transporter (Samuvel et al., 2005). These demonstrations of constitutive action of p38 MAPK under physiological conditions have mostly lacked identification of upstream activators. However, activation of p38 MAPK by metabotropic glutamate receptors mediates induction of NMDA receptor-independent long-term depression (Bolshakov et al., 2000) and may oppose ERK1/2-dependent long-term potentiation (Thomas and Huganir, 2004). Interestingly, the main subtype of p38 MAPK present in the CNS, p38α, is present at high density in pyramidal neuron dendrites, consistent with its modulation of both Ih and synaptic plasticity (Lee et al., 2000).
Beyond these examples, it is not yet clear what physiological or pathological roles may be played by p38 MAPK modulation of Ih. Experimental and computational modeling studies show that the influence of Ih on overall excitability in CA1 hippocampal and neocortical pyramidal neurons is inhibitory (Poolos et al., 2002); thus, it would be expected that p38 MAPK activation would reduce overall CA1 pyramidal neuron excitability. Considering the diverse actions that Ih has on neuronal excitability, pacemaker activity, and synaptic plasticity, it is likely that p38 MAPK signaling modulates excitability in other neuron types as well. Furthermore, multiple lines of evidence have implicated both loss of Ih (for review, see Poolos, 2005) and loss of kinase activity in the development of epilepsy (Kurz et al., 2001; Sanchez et al., 2005). Thus, it will be an interesting area for future investigation to determine whether derangement of p38 MAPK signaling plays a role in epileptogenesis.
Footnotes
This work was supported by the National Institutes of Health and GlaxoSmithKline. We thank Terrance Jones for producing the biocytin histology images. Sangwook Jung, Galen Flynn, and Bill Zagotta also provided valuable suggestions.
References
- Arima H, Yamamoto N, Sobue K, Umenishi F, Tada T, Katsuya H, Asai K (2003). Hyperosmolar mannitol simulates expression of aquaporins 4 and 9 through a p38 mitogen-activated protein kinase-dependent pathway in rat astrocytes. J Biol Chem 278:44525–44534. [DOI] [PubMed] [Google Scholar]
- Beaumont V, Zhong N, Froemke RC, Ball RW, Zucker RS (2002). Temporal synaptic tagging by Ih activation and actin: involvement in long-term facilitation and cAMP-induced synaptic enhancement. Neuron 33:601–613. [DOI] [PubMed] [Google Scholar]
- Bolshakov VY, Carboni L, Cobb MH, Siegelbaum SA, Belardetti F (2000). Dual MAP kinase pathways mediate opposing forms of long-term plasticity at CA3-CA1 synapses. Nat Neurosci 3:1107–1112. [DOI] [PubMed] [Google Scholar]
- Budde T, Caputi L, Kanyshkova T, Staak R, Abrahamczik C, Munsch T, Pape HC (2005). Impaired regulation of thalamic pacemaker channels through an imbalance of subunit expression in absence epilepsy. J Neurosci 25:9871–9882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bullis JB, Jones TD, Poolos NP (2004). h-channel distribution in “pyramidal-like” principal neurons in stratum radiatum of hippocampus differs from pyramidal neurons. Soc Neurosci Abstr 29:52.59. [Google Scholar]
- Chang F, Cohen IS, DiFrancesco D, Rosen MR, Tromba C (1991). Effects of protein kinase inhibitors on canine Purkinje fibre pacemaker depolarization and the pacemaker current i(f). J Physiol 440:367–384. Lond. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaplan SR, Guo HQ, Lee DH, Luo L, Liu C, Kuei C, Velumian AA, Butler MP, Brown SM, Dubin AE (2003). Neuronal hyperpolarization-activated pacemaker channels drive neuropathic pain. J Neurosci 23:1169–1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K, Aradi I, Thon N, Eghbal-Ahmadi M, Baram TZ, Soltesz I (2001). Persistently modified h-channels after complex febrile seizures convert the seizure-induced enhancement of inhibition to hyperexcitability. Nat Med 7:331–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Wang J, Siegelbaum SA (2001). Properties of hyperpolarization-activated pacemaker current defined by coassembly of HCN1 and HCN2 subunits and basal modulation by cyclic nucleotide. J Gen Physiol 117:491–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Cantrell AR, Messing RO, Scheuer T, Catterall WA (2005). Specific modulation of Na+ channels in hippocampal neurons by protein kinase Cε. J Neurosci 25:507–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- English JM, Cobb MH (2002). Pharmacological inhibitors of MAPK pathways. Trends Pharmacol Sci 23:40–45. [DOI] [PubMed] [Google Scholar]
- Feranchak AP, Berl T, Capasso J, Wojtaszek PA, Han J, Fitz JG (2001). p38 MAP kinase modulates liver cell volume through inhibition of membrane Na+ permeability. J Clin Invest 108:1495–1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiala JC, Harris KM (1999). Dendrite structure. In: Dendrites (Stuart G, Spruston N, Hausser M, eds) pp. 1–34. Oxford: Oxford UP.
- Gulyas AI, Toth K, McBain CJ, Freund TF (1998). Stratum radiatum giant cells: a type of principal cell in the rat hippocampus. Eur J Neurosci 10:3813–3822. [DOI] [PubMed] [Google Scholar]
- Hasegawa M, Cahill GM (2004). Regulation of the circadian oscillator in Xenopus retinal photoreceptors by protein kinases sensitive to the stress-activated protein kinase inhibitor, SB 203580. J Biol Chem 279:22738–22746. [DOI] [PubMed] [Google Scholar]
- Ji RR (2004). Peripheral and central mechanisms of inflammatory pain, with emphasis on MAP kinases. Curr Drug Targets Inflamm Allergy 3:299–303. [DOI] [PubMed] [Google Scholar]
- Jin X, Gereau RW IV (2006). Acute p38-mediated modulation of tetrodotoxin-resistant sodium channels in mouse sensory neurons by tumor necrosis factor-α. J Neurosci 26:246–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer RH, Molokanova E (2001). Modulation of cyclic-nucleotide-gated channels and regulation of vertebrate phototransduction. J Exp Biol 204:2921–2931. [DOI] [PubMed] [Google Scholar]
- Kumar S, Boehm J, Lee JC (2003). p38 MAP kinases: key signalling molecules as therapeutic targets for inflammatory diseases. Nat Rev Drug Discov 2:717–726. [DOI] [PubMed] [Google Scholar]
- Kurz JE, Sheets D, Parsons JT, Rana A, Delorenzo RJ, Churn SB (2001). A significant increase in both basal and maximal calcineurin activity in the rat pilocarpine model of status epilepticus. J Neurochem 78:304–315. [DOI] [PubMed] [Google Scholar]
- Lee SH, Park J, Che Y, Han PL, Lee JK (2000). Constitutive activity and differential localization of p38α and p38β MAPKs in adult mouse brain. J Neurosci Res 60:623–631. [DOI] [PubMed] [Google Scholar]
- Levitan IB (2006). Signaling protein complexes associated with neuronal ion channels. Nat Neurosci 9:305–310. [DOI] [PubMed] [Google Scholar]
- Maccaferri G, McBain CJ (1996). Long-term potentiation in distinct subtypes of hippocampal nonpyramidal neurons. J Neurosci 16:5334–5343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magee JC (1998). Dendritic hyperpolarization-activated currents modify the integrative properties of hippocampal CA1 pyramidal neurons. J Neurosci 18:7613–7624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magee JC (1999). Dendritic Ih normalizes temporal summation in hippocampal CA1 neurons. Nat Neurosci 2:508–514. [DOI] [PubMed] [Google Scholar]
- Nolan MF, Malleret G, Dudman JT, Buhl DL, Santoro B, Gibbs E, Vronskaya S, Buzsaki G, Siegelbaum SA, Kandel ER, Morozov A (2004). A behavioral role for dendritic integration: HCN1 channels constrain spatial memory and plasticity at inputs to distal dendrites of CA1 pyramidal neurons. Cell 119:719–732. [DOI] [PubMed] [Google Scholar]
- Obenauer JC, Cantley LC, Yaffe MB (2003). Scansite 2.0: proteome-wide prediction of cell signaling interactions using short sequence motifs. Nucleic Acids Res 31:3635–3641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedarzani P, Storm JF (1995). Protein kinase A-independent modulation of ion channels in the brain by cyclic AMP. Proc Natl Acad Sci USA 92:11716–11720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poolos NP (2005). The h-channel: a potential channelopathy in epilepsy? Epilepsy Behav 7:51–56. [DOI] [PubMed] [Google Scholar]
- Poolos NP, Johnston D (1999). Calcium-activated potassium conductances contribute to action potential repolarization at the soma but not the dendrites of hippocampal CA1 pyramidal neurons. J Neurosci 19:5205–5212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poolos NP, Migliore M, Johnston D (2002). Pharmacological upregulation of h-channels reduces the excitability of pyramidal neuron dendrites. Nat Neurosci 5:767–774. [DOI] [PubMed] [Google Scholar]
- Roux PP, Blenis J (2004). ERK and p38 MAPK-activated protein kinases: a family of protein kinases with diverse biological functions. Microbiol Mol Biol Rev 68:320–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saklatvala J (2004). The p38 MAP kinase pathway as a therapeutic target in inflammatory disease. Curr Opin Pharmacol 4:372–377. [DOI] [PubMed] [Google Scholar]
- Sakmann B, Neher E (1995). Geometric parameters of pipettes and membrane patches. In: Single-channel recording (Sakmann B, Neher E, eds) pp. 637–650. New York: Plenum.
- Samuvel DJ, Jayanthi LD, Bhat NR, Ramamoorthy S (2005). A role for p38 mitogen-activated protein kinase in the regulation of the serotonin transporter: evidence for distinct cellular mechanisms involved in transporter surface expression. J Neurosci 25:29–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez RM, Dai W, Levada RE, Lippman JJ, Jensen FE (2005). AMPA/kainate receptor-mediated downregulation of GABAergic synaptic transmission by calcineurin after seizures in the developing rat brain. J Neurosci 25:3442–3451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santoro B, Tibbs GR (1999). The HCN gene family: molecular basis of the hyperpolarization-activated pacemaker channels. Ann NY Acad Sci 868:741–764. [DOI] [PubMed] [Google Scholar]
- Santoro B, Chen S, Luthi A, Pavlidis P, Shumyatsky GP, Tibbs GR, Siegelbaum SA (2000). Molecular and functional heterogeneity of hyperpolarization-activated pacemaker channels in the mouse CNS. J Neurosci 20:5264–5275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savic N, Sciancalepore M (2001). Electrophysiological characterization of “giant” cells in stratum radiatum of the CA3 hippocampal region. J Neurophysiol 85:1998–2007. [DOI] [PubMed] [Google Scholar]
- Schrader LA, Anderson AE, Mayne A, Pfaffinger PJ, Sweatt JD (2002). PKA modulation of Kv4.2-encoded A-type potassium channels requires formation of a supramolecular complex. J Neurosci 22:10123–10133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selcher JC, Weeber EJ, Christian J, Nekrasova T, Landreth GE, Sweatt JD (2003). A role for ERK MAP kinase in physiologic temporal integration in hippocampal area CA1. Learn Mem 10:26–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah M, Anderson A, Leung V, Lin X, Johnston D (2004). Seizure-induced plasticity of h channels in entorhinal cortical layer III pyramidal neurons. Neuron 44:495–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shifrin VI, Anderson P (1999). Trichothecene mycotoxins trigger a ribotoxic stress response that activates c-Jun N-terminal kinase and p38 mitogen-activated protein kinase and induces apoptosis. J Biol Chem 274:13985–13992. [DOI] [PubMed] [Google Scholar]
- Surges R, Freiman TM, Feuerstein TJ (2003). Gabapentin increases the hyperpolarization-activated cation current Ih in rat CA1 pyramidal cells. Epilepsia 44:150–156. [DOI] [PubMed] [Google Scholar]
- Sweatt JD (2004). Mitogen-activated protein kinases in synaptic plasticity and memory. Curr Opin Neurobiol 14:311–317. [DOI] [PubMed] [Google Scholar]
- Thoby-Brisson M, Cauli B, Champagnat J, Fortin G, Katz DM (2003). Expression of functional tyrosine kinase B receptors by rhythmically active respiratory neurons in the pre-Botzinger complex of neonatal mice. J Neurosci 23:7685–7689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas GM, Huganir RL (2004). MAPK cascade signalling and synaptic plasticity. Nat Rev Neurosci 5:173–183. [DOI] [PubMed] [Google Scholar]
- van Welie I, van Hooft JA, Wadman WJ (2004). Homeostatic scaling of neuronal excitability by synaptic modulation of somatic hyperpolarization-activated Ih channels. Proc Natl Acad Sci USA 101:5123–5128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varga AW, Yuan LL, Anderson AE, Schrader LA, Wu GY, Gatchel JR, Johnston D, Sweatt JD (2004). Calcium-calmodulin-dependent kinase II modulates Kv4.2 channel expression and upregulates neuronal A-type potassium currents. J Neurosci 24:3643–3654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velumian AA, Zhang L, Pennefather P, Carlen PL (1997). Reversible inhibition of IK, IAHP, Ih and ICa currents by internally applied gluconate in rat hippocampal pyramidal neurones. Pflügers Arch 433:343–350. [DOI] [PubMed] [Google Scholar]
- Wainger BJ, DeGennaro M, Santoro B, Siegelbaum SA, Tibbs GR (2001). Molecular mechanism of cAMP modulation of HCN pacemaker channels. Nature 411:805–810. [DOI] [PubMed] [Google Scholar]
- Wittmack EK, Rush AM, Hudmon A, Waxman SG, Dib-Hajj SD (2005). Voltage-gated sodium channel Nav1.6 is modulated by p38 mitogen-activated protein kinase. J Neurosci 25:6621–6630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye CP, Yano S, Tfelt-Hansen J, MacLeod RJ, Ren X, Terwilliger E, Brown EM, Chattopadhyay N (2004). Regulation of a Ca2+-activated K+ channel by calcium-sensing receptor involves p38 MAP kinase. J Neurosci Res 75:491–498. [DOI] [PubMed] [Google Scholar]
- Yu HG, Lu Z, Pan Z, Cohen IS (2004). Tyrosine kinase inhibition differentially regulates heterologously expressed HCN channels. Pflügers Arch 447:392–400. [DOI] [PubMed] [Google Scholar]
- Yuan LL, Adams JP, Swank M, Sweatt JD, Johnston D (2002). Protein kinase modulation of dendritic K+ channels in hippocampus involves a mitogen-activated protein kinase pathway. J Neurosci 22:4860–4868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zong X, Eckert C, Yuan H, Wahl-Schott C, Abicht H, Fang L, Li R, Mistrik P, Gerstner A, Much B, Baumann L, Michalakis S, Zeng R, Chen Z, Biel M (2005). A novel mechanism of modulation of hyperpolarization-activated cyclic nucleotide-gated channels by SRC kinase. J Biol Chem 280:34224–34232. [DOI] [PubMed] [Google Scholar]