

Abstract
To study the profile and regulation of apolipoprotein E (apoE) expression in the CNS, we generated mice in which apoE expression can be detected in vivo with unprecedented sensitivity and resolution. cDNA encoding enhanced green fluorescent protein (EGFP) with a stop codon was inserted by gene targeting into the apoE gene locus (EGFPapoE) immediately after the translation initiation site. Insertion of EGFP into one apoE allele provides a real-time location marker of apoE expression in vivo; the remaining allele is sufficient to maintain normal cellular physiology. In heterozygous EGFPapoE mice, EGFP was highly expressed in hepatocytes and peritoneal macrophages. EGFP was also expressed in brain astrocytes; however some astrocytes (∼25%) expressed no EGFP, suggesting that a subset of these cells does not express apoE. EGFP was expressed in <10% of microglia after kainic acid treatment, suggesting that microglia are not a major source of brain apoE. Although hippocampal neurons did not express EGFP under normal conditions, kainic acid treatment induced intense expression of EGFP in injured neurons, demonstrating apoE expression in neurons in response to excitotoxic injury. The neuronal expression was confirmed by in situ hybridization of mouse apoE mRNA and by anti-apoE immunostaining. Smooth muscle cells of large blood vessels and cells surrounding small vessels in the CNS also strongly expressed EGFP, as did cells in the choroid plexus. EGFPapoE reporter mice will be useful for studying the regulation of apoE expression in the CNS and might provide insights into the diverse mechanisms of apoE4-related neurodegeneration.
Keywords: apolipoprotein E, Alzheimer's disease, green fluorescent protein, excitotoxin, knock-in mice, gene regulation
Introduction
The ε4 allele of the gene encoding apolipoprotein E (apoE) has been linked to late-onset familial and sporadic Alzheimer's disease (AD) and has a gene-dose effect on the risk and age of onset of the disease (Corder et al., 1993; Saunders et al., 1993; Roses, 1996; Tang et al., 1998; Romas et al., 2002). Individuals with two copies of the ε4 allele have a 50–90% chance of developing AD by the age of 85, compared with ∼45% for those with one allele (Corder et al., 1993; Farrer et al., 1997) and 20% for the general population (Corder et al., 1993). ApoE is found in amyloid plaques and neurofibrillary tangles (two neuropathological hallmarks of AD) and has been suggested to play important roles in the pathogenesis of these two lesions (Namba et al., 1991; Selkoe, 1991; Wisniewski and Frangione, 1992; Crowther, 1993; Strittmatter et al., 1993a; Roses, 1994; Holtzman et al., 2000; Irizarry et al., 2000; Tanzi and Bertram, 2001).
Initially, apoE was thought to be synthesized in the brain only by astrocytes, oligodendrocytes, and ependymal layer cells (Boyles et al., 1985; Poirier et al., 1991). Although not all studies support the notion (Page et al., 1998; Nishio et al., 2003), increasing evidence suggests that under diverse pathophysiological conditions, CNS neurons also express apoE, albeit at lower levels than astrocytes (Diedrich et al., 1991; Han et al., 1994; Bao et al., 1996; Beffert and Poirier, 1996; Metzger et al., 1996; Xu et al., 1998, 1999a,b). The cellular origin of apoE appears to influence its effects on AD pathology (Huang et al., 2004; Huang, 2006). Astrocyte-derived apoE3 and apoE4 have different effects on the production, deposition, and clearance of Aβ (LaDu et al., 1994; Bales et al., 1999; Holtzman et al., 2000; Irizarry et al., 2000; Ji et al., 2001; Vincent and Smith, 2001; Ye et al., 2005) and on cholesterol efflux (Fagan et al., 1999; Gong et al., 2002). Neuron-derived apoE3 and apoE4 differ in their susceptibility to proteolysis (Huang et al., 2001; Harris et al., 2003; Brecht et al., 2004; Chang et al., 2005) and in their effects on mitochondrial function (Chang et al., 2005), tau phosphorylation (Tesseur et al., 2000a,b; Huang et al., 2001; Harris et al., 2003, 2004a; Brecht et al., 2004), lysosomal leakage (Ji et al., 2002), neurodegeneration (Buttini et al., 1999; Buttini et al., 2002), androgen receptor deficiency (Raber et al., 2002), and cognitive decline (Raber et al., 1998, 2000, 2002). Therefore, a better understanding of the profile and regulation of apoE expression in the CNS is important for unraveling the mechanisms underlying apoE4-related neurodegeneration.
Regulation of apoE expression has been extensively studied in transfected cells (Reardon et al., 1986; Smith et al., 1988; García et al., 1996; Harris et al., 2004b) and in transgenic mice (Shachter et al., 1993; Simonet et al., 1993; Allan et al., 1995, 1997; Shih et al., 2000; Grehan et al., 2001a,b; Zheng et al., 2004) expressing apoE genomic or cDNA constructs. Although these systems have provided valuable information, practical considerations have limited systematic study of apoE expression. The high guanine and cytosine (GC) content of apoE coding sequences requires critical in situ hybridization conditions to limit nonspecific background signals while still providing acceptable sensitivity. Transfected cells are useful for mapping fine structures but have not yielded a fully accurate definition of any cell-specific regulation that reflects in vivo apoE expression. Transgenic models are hampered by variegated expression because of random integration of transgenes into the mouse genome. Enhancers and silencers of nearby endogenous genes can also interfere with transgene expression.
To avoid such limitations, we generated mice that express enhanced green fluorescent protein (EGFP) under control of the endogenous apoE gene promoter and enhancers (EGFPapoE), providing a real-time marker of in vivo apoE expression with unprecedented sensitivity and resolution. EGFP knock-in is a new approach to monitor gene expression in vivo (Aubert et al., 2003; Toyooka et al., 2003). Because it has no signal peptide, EGFP remains intracellular, and surrounding cells are not visible, avoiding confusion as to whether the immunostained apoE in certain cells is produced in situ or taken up through its receptors from the extracellular pool. Insertion of EGFP into one allele of the apoE gene provides a marker of apoE expression in vivo, and the remaining allele maintains normal lipid metabolism and cellular physiology and enables us to confirm the expression of apoE in various cells.
Materials and Methods
Reagents.
Minimum essential medium, Opti-MEM, and fetal bovine serum were obtained from Invitrogen (Rockville, MD). ECL was obtained from Amersham Biosciences (Arlington, IL). Polyclonal rabbit anti-mouse apoE antibody was kindly provided by Dr. Karl Weisgraber (Gladstone Institutes, San Francisco, CA). Kainic acid and monoclonal anti-α-actin were obtained from Sigma (St. Louis, MO). Monoclonal anti-neuron-specific nuclear protein (NeuN) and monoclonal anti-CD11b were from Chemicon (Temecula, CA). Rabbit anti-GFAP was from Dako (Carpinteria, CA). Fluorescein isothiocyanate- and Texas Red-coupled anti-rabbit and anti-mouse were from Vector Laboratories (Burlingame, CA).
Generation of EGFP knock-in mice.
The EGFP-targeting vector was electroporated into embryonic stem (ES) cells (129/Svjae) as described previously (Raffaï et al., 2001), and G418-resistant clones were selected and screened by PCR with primers covering Neo (forward) and 3′ flanking sequence of mouse Apoe (reverse). The DNA sequence recognized by the reverse primer was not included in the vector (Fig. 1A). Therefore, a 1.5 kb fragment could only be amplified from the DNA of the targeted ES cells. Of 200 clones screened, 21 were positive (Fig. 1B).
Figure 1.

Generation and characterization of EGFP-targeted mouse ES cells. A, An EGFP cDNA with start and stop codons and a poly-A sequence (∼1 kb) was inserted into the basic gene-targeting vector used to generate apoE-Arg-61 knock-in mice at the Gladstone Institutes (Raffaï et al., 2001). Homologous recombination between the gene-targeting vector and the Apoe locus in ES cells introduces the EGFP cDNA. A Neo cassette was placed in intron 3. Targeted ES cell clones and mice were identified by PCR with primers and by digestion of genomic DNA with SacI and subsequent Southern blotting with an Apoe 3′ flanking sequence probe, which reveals an expanded 5.3 kb fragment; the wild-type fragment is 3.3 kb. B, PCR screening for ES cell clones with homologous recombination of EGFP revealed a 1.5 kb band in targeted (+/−) ES cells but no band in wild-type (WT) ES cells. C, Southern blotting revealed both 5.3 kb and 3.3 kb DNA fragments in targeted (+/−) ES cells but only a 3.3 kb fragment in wild-type ES cells.
The PCR-positive ES cell clones were also screened by Southern blotting. SacI-digested DNA was hybridized with a probe that detects fragments of 5.3 kb (targeted allele) and 3.3 kb (wild-type allele) in targeted ES cells (+/−) but only a 3.3 kb fragment in wild-type cells (Fig. 1A,C). Screening yielded 15 positive ES cell clones (Fig. 1C). Three were microinjected into C57BL/6 blastocysts in the Gladstone Blastocyst Core, yielding >50 chimeric mice harboring EGFP cDNA in the apoE locus. Six male chimeras (>90% brown fur) were crossed with C57BL/6 females to generate heterozygous EGFPapoE mice. Germline transmission resulted in heterozygous F1 EGFPapoE mice (confirmed by PCR and Southern blotting analyses; data not shown) that are 50% C57BL/6. Three additional crosses resulted in F4 mice that are >93% C57BL/6. Heterozygous EGFPapoE mice were then bred to generate homozygotes to maintain the line. For the current study, we used heterozygous EGFPapoE mice, generated by crossing homozygous EGFPapoE and wild-type C57BL/6 mice. Mice were weaned at 21 d of age, housed in a barrier facility at the Gladstone Animal Core with a 12 h light/dark cycle, and fed a chow diet containing 4.5% fat (Ralston Purina, St. Louis, MO).
Northern blotting and quantitative analysis of apoE mRNA.
Total RNA from brains of wild-type and heterozygous EGFPapoE mice was isolated with Triazol (Invitrogen). Total RNA (∼20 μg) was separated by electrophoresis in a 1% agarose gel containing 20% formaldehyde, transferred to Hybond membrane (Amersham Biosciences) and hybridized to a mouse apoE cDNA probe labeled with [32P]dCTP in Ultrahyb solution (Ambion, Austin, TX) at 60°C overnight. The blot was washed in high-stringency buffer (Ambion) at 68°C for 15 min (twice) and exposed to x-ray film for 2–6 h. The blot was then washed with a Strip-EZ buffer (Ambion), rehybridized to a mouse β-actin probe as an internal loading control, and exposed to x-ray film for 2–8 h. The bands of apoE and β-actin mRNAs were scanned, and the ratios of apoE to β-actin were calculated.
Preparation of mouse brain tissues.
Brains from wild-type or heterozygous EGFPapoE mice were collected after a 2 min transcardial perfusion with PBS. One hemibrain from each mouse was homogenized and analyzed for apoE by Western blotting (Huang et al., 2001). In brief, brain tissues were homogenized in ice-cold lysis buffer (50 mm Tris/HCl, pH 8.0, 150 mm NaCl, 4% SDS, 1% Nonidet P-40, 1% sodium deoxycholate, and a mixture of protease and phosphatase inhibitors), placed in a TLA 100.3 rotor, and centrifuged at 35,000 rpm for 30 min at 4°C in an Optima TL ultracentrifuge (Beckman, Fullerton, CA).
Western blotting and quantitative analysis of apoE.
The supernatant (solubilized protein) was subjected to SDS-PAGE and analyzed by Western blotting with a rabbit polyclonal antibody against mouse apoE or a monoclonal antibody against α-actin. The bands of apoE and α-actin from individual mice were scanned, and their intensities were calculated (Huang et al., 2001).
Immunohistochemistry.
The other hemibrain from each mouse was fixed in 3% paraformaldehyde, sectioned, and stained with anti-mouse apoE, anti-NeuN (neuron marker), anti-GFAP (astrocyte marker), anti-α-actin (smooth muscle cell marker), and anti-CD11b (microglial marker) (Buttini et al., 1999; Huang et al., 2001). To block nonspecific reactions, all sections were incubated for 1 h in 10% normal serum from the species that produced the secondary antibodies (Jackson ImmunoResearch, West Grove, PA) in PBS or for 7 min in Superblock (Scytec, Logan, UT), followed by a 1 h incubation in PBS with primary antibodies. Sections were then washed three times in PBS and incubated for 1 h with the corresponding secondary antibodies coupled to Texas Red (Jackson ImmunoResearch). After three washes in PBS, the sections were mounted in VectaShield (Vector Laboratories) and examined for both green (EGFP) and red (other marker staining) channels with a Radiance 2000 laser-scanning confocal system (Bio-Rad, Hercules, CA) mounted on an Optiphot-2 microscope (Nikon, Tokyo, Japan). The images were processed with Photoshop (Adobe Systems, San Jose, CA).
Kainic acid injections.
Kainic acid crosses the blood–brain barrier and induces excitotoxic CNS injury, particularly in the hippocampus and neocortex (Spinler and Cziraky, 1994; Masliah et al., 1997). At 4–6 months of age, heterozygous EGFPapoE mice were injected intraperitoneally with kainic acid (Sigma) dissolved in saline (0.9%) at 25 mg/kg body weight in one dose, as described previously (Buttini et al., 1999). Within ∼15 min, all mice developed seizures. Seizure activity was assessed as described previously (Schauwecker and Steward, 1997). The groups did not differ in the time from injection to seizure onset or in the incidence, intensity, or duration of seizures (data not shown). Mice were killed 1 or 6 d after the injection of kainic acid.
In situ hybridization.
RNA probe complementary to nucleotides 492–783 of mouse apoE mRNA was labeled with [33P]UTP with an RNA transcription kit (Stratagene, La Jolla, CA). The labeled probe was purified through Micro Bio-Spin 30 chromatography columns (Bio-Rad). In situ hybridization was performed as described previously (Grehan et al., 2001b). Briefly, brain paraffin sections (7 μm) were incubated with 20 μg/ml proteinase K (Boehringer Mannheim, Indianapolis, IN) in 50 mm Tris-HCl, pH 8.0, 5 mm EDTA, and 150 mm NaCl for 15 min at room temperature. Proteolytic activity was stopped by immersion for 10 min in 0.2% glycine in PBS. After fixation, acetylation, and dehydration, the sections were incubated for 14–18 h in a humidified chamber at 45°C with labeled probe in a buffer containing 50% formamide, 300 mm NaCl, 20 mm Tris, pH 8.0, 5 mm EDTA, 0.2% polyvinylpyrrolidone, 0.02% Ficoll, 0.02% bovine serum albumin, 10% dextran sulfate, 250 μg/ml sperm DNA, and 0.1 mg/ml tRNA. After two washes at room temperature in 2× SSC and 1.0 mm EDTA for 10 min, the sections were immersed in 20 μg/ml ribonuclease (RNase) A (Sigma) in 500 mm NaCl and 10 mm Tris, pH 8.0, and 10 U/ml T1 RNase (Boehringer Mannheim) for 1 h at 37°C, washed at 60°C in six changes of 0.1× SSC with 1.0 mm EDTA for 4 h, rinsed twice for 10 min each in 0.5× SSC, and dehydrated. For dark-field and bright-field microscopy, the slides were dipped in NTB2 nuclear track emulsion (Eastman Kodak, Rochester, NY), incubated at 4°C for 2–5 d, and developed with D19 developer (Eastman Kodak). The sections were then stained with hematoxylin and eosin (Fisher Scientific, Tustin, CA). After dehydration in a graded series of ethanol (80, 95, and 100%), the slides were rinsed three times in xylene and overlaid with coverslips.
Statistical analysis.
Results are reported as mean ± SD. Differences were evaluated by a t test.
Results
Heterozygous EGFPapoE mice express both EGFP and apoE
As shown by Northern blotting, the average level of brain apoE mRNA in heterozygous EGFPapoE mice was 58% of that in wild-type mice (Fig. 2A), consistent with the expected inactivation of one apoE allele. The average level of apoE protein was 51% of wild-type, as shown by anti-apoE Western blotting (Fig. 2B). Similar Western blotting results were obtained from liver and peritoneal macrophages of heterozygous EGFPapoE mice (data not shown).
Figure 2.

Heterozygous EGFPapoE mice express apoE at ∼50% of the level in wild-type mice. A, Northern blotting analysis of apoE and actin mRNA in brains of wild-type and heterozygous EGFPapoE mice at 5 months of age. B, Western blotting analysis of apoE and actin in brains of wild-type and heterozygous EGFPapoE mice at 5 months of age.
EGFP is expressed in hepatocytes and macrophages
EGFP was expressed at high levels in liver cells (Fig. 3A), the primary source of apoE in both humans and mice. Peritoneal macrophages also expressed EGFP, and the expression was enhanced by cholesterol loading (Fig. 3B), as reported previously (Basu et al., 1981). Thus, in heterozygous EGFPapoE mice, EGFP representing apoE was correctly expressed in hepatocytes and macrophages that normally express apoE.
Figure 3.

Expression of EGFP in hepatocytes and peritoneal macrophages in a heterozygous EGFPapoE mouse at 2 months of age. A, Expression of EGFP in hepatocytes as determined by confocal microscopy. B, Peritoneal macrophages were cultured in vitro for 1 or 4 d and analyzed by confocal microscopy. Alternatively, after 4 d of culture, macrophages were incubated with acetylated LDL (AcLDL; 100 μg/ml) for 16 h and analyzed by confocal microscopy.
EGFP is expressed in many, but not all, CNS astrocytes
EGFP was also expressed in CNS astrocytes and was confirmed by anti-GFAP immunofluorescent staining (Fig. 4A–C). Strikingly, EGFP revealed the full cell volume of astrocytes with very fine resolution of cellular processes, whereas anti-GFAP staining highlighted only large processes. Thus, EGFP is better than immunostaining for characterizing cell identity and morphology. Interestingly, some GFAP-positive cells (26 ± 4%) did not express EGFP, suggesting that a subclass of astrocytes might not express apoE, at least under normal conditions.
Figure 4.
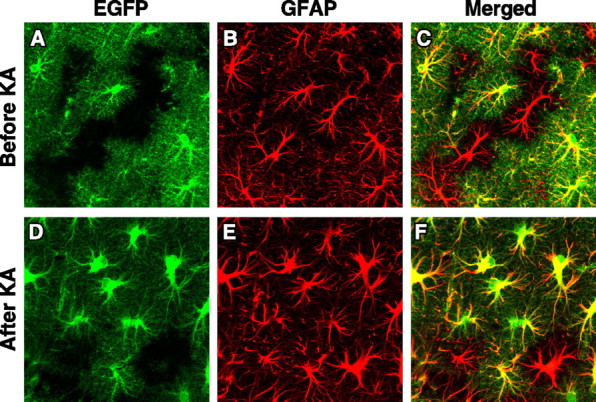
Expression of EGFP in hippocampal astrocytes in heterozygous EGFPapoE mice at 4–6 months of age before (A–C) and 6 d after (D–F) kainic acid (KA) treatment (25 mg/kg body weight). Astrocytes were identified by anti-GFAP immunostaining and confocal microscopy. In the merged image, yellow indicates colocalization of EGFP (green) and GFAP (red).
To determine whether brain insults can induce EGFP-negative astrocytes to express EGFP, we treated heterozygous EGFPapoE mice with kainic acid, which can activate astrocytes, induce gliosis, and increase apoE expression in animal models (Sperk et al., 1983). Hippocampal astrocytes were activated, as indicated by much stronger GFAP staining and enlarged cell bodies and branches; however, 17 ± 3% of astrocytes still did not express EGFP (Fig. 4D–F).
EGFP is expressed in <10% of microglia after kainic acid treatment
EGFP was not expressed in CNS microglia as demonstrated by anti-CD11b, a microglial marker (Chen et al., 2005), immunofluorescent staining (Fig. 5A–C), suggesting that these cells do not express apoE under normal conditions. To determine whether brain insults can induce microglia to express EGFP, we treated heterozygous EGFPapoE mice with kainic acid, which can activate microglia and induce gliosis (Sperk et al., 1983). Hippocampal microglia were activated by kainic acid treatment, as indicated by much stronger CD11b staining and enlarged cell bodies and branches; however, only 6 ± 3% of microglia expressed EGFP (Fig. 5D–F).
Figure 5.
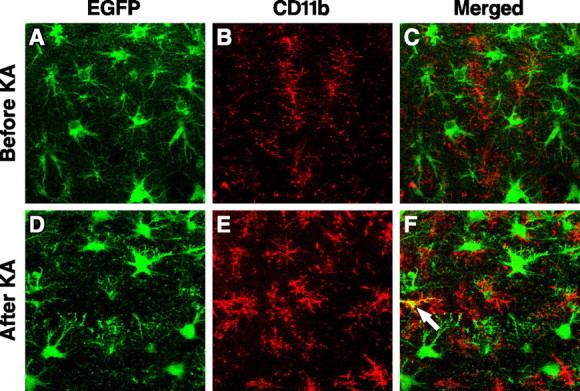
Expression of EGFP in hippocampal microglia in heterozygous EGFPapoE mice at 4–6 months of age before (A–C) and 6 d after (D–F) kainic acid (KA) treatment (25 mg/kg body weight). Microglia were identified by anti-CD11b immunostaining and confocal microscopy. In the merged image, yellow (arrow) indicates colocalization of EGFP (green) and CD11b (red).
EGFP is expressed in hippocampal neurons in response to exitotoxic injury
EGFP was not expressed in hippocampal neurons in heterozygous EGFPapoE mice, as demonstrated by staining for NeuN (a neuronal marker) (Fig. 6A) and GFAP (Fig. 6B). Thus, hippocampal neurons do not express apoE under normal conditions. However, kainic acid treatment induced intense expression of EGFP in injured hippocampal neurons (Fig. 6C–G). Importantly, both EGFP and apoE were only present in injured neurons in the treated mice (Fig. 7A,B,D,E). Neuronal expression of apoE was confirmed by in situ hybridization of mouse apoE mRNA in heterozygous EGFPapoE mice treated with kainic acid (Fig. 7F). ApoE protein and mRNA were undetectable in hippocampal neurons in untreated mice (Fig. 7B,C), in which no neuronal injury was found, as determined by silver staining (Fig. 7A).
Figure 6.
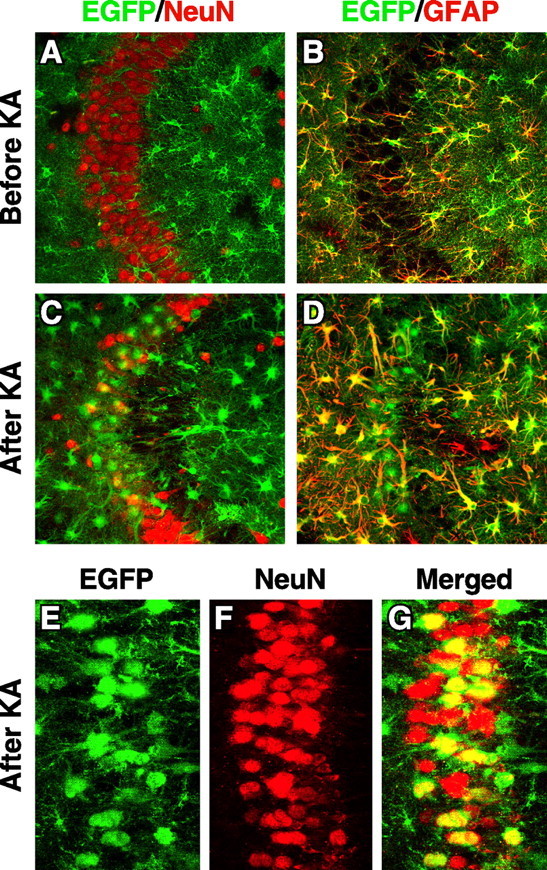
Hippocampal CA3 neurons express EGFP, representing apoE, in response to excitotoxic injury. Heterozygous 5-month-old EGFPapoE mice received peritoneal injections of kainic acid (KA; 25 mg/kg) (C–G), and the brains were collected 1 d (E–G) or 6 d (C, D) later. Untreated age-matched heterozygous EGFPapoE mice served as controls (A, B). Confocal images of immunostained brain sections were collected for EGFP (green) and anti-NeuN (red), a neuronal marker, or anti-GFAP (red), an astrocytic marker. Images in A–D and G are merged, and yellow indicates colocalization.
Figure 7.
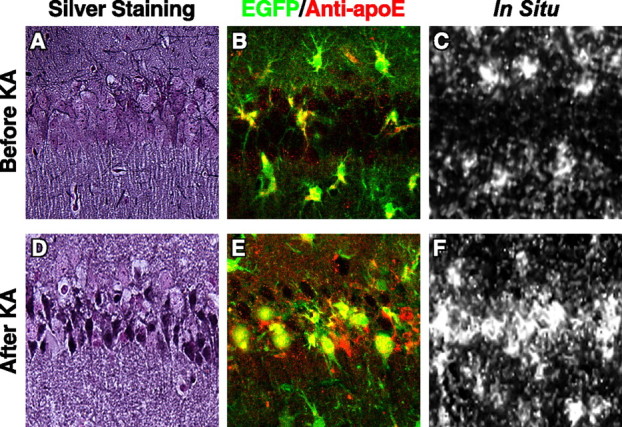
EGFP and apoE protein and mRNA are present only in injured hippocampal neurons in kainic acid-treated mice. Heterozygous 5-month-old EGFPapoE mice received peritoneal injections of kainic acid (KA; 25 mg/kg) (D–F), and the brains were collected 6 d later. Untreated age-matched heterozygous EGFPapoE mice served as controls (A–C). A, D, Gallyas silver staining of the hippocampal CA1 region. B, E, Merged confocal images of EGFP (green) and anti-apoE (red) in the CA1 region. C, F, In situ hybridization of mouse apoE mRNA in the CA1 region.
EGFP is expressed along CNS vessels and in the choroid plexus
EGFP was highly expressed along vessels in the CNS, as indicated by the close proximity or colocalization of EGFP with α-actin (Fig. 8A–C), a smooth muscle cell marker (Deaton et al., 2005). Interestingly, EGFP surrounded smooth muscle cells of small vessels in the hippocampus (Fig. 8A) and cortex (Fig. 8B) but colocalized with smooth muscle cells in large vessels in the cortex (Fig. 8C). In situ hybridization with a probe specific for mouse apoE demonstrated apoE mRNA in cells in or surrounding vessel walls (Fig. 8D). Anti-GFAP immunostaining indicated that the cells expressing EGFP along vessels were negative for GFAP, and thus were not astrocytes (Fig. 8E,F). EGFP was not expressed along veins in the CNS (data not shown). Cells of the choroid plexus also expressed high levels of EGFP (Fig. 9A) and contained apoE mRNA, as shown by in situ hybridization (Fig. 9B).
Figure 8.
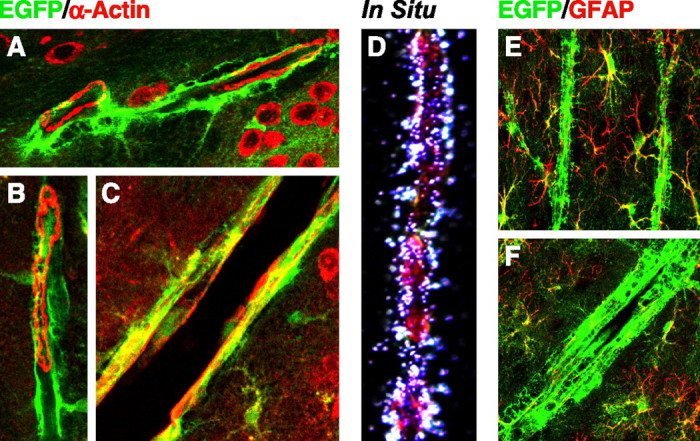
Expression of EGFP in smooth muscle cells in large blood vessels and cells surrounding small blood vessels in brains of heterozygous EGFPapoE mice. A–C, Merged confocal images of EGFP (green) and anti-α-actin (red), a smooth muscle cell marker, were collected from a 5-month-old heterozygous EGFPapoE mouse. A, Small blood vessels in the hippocampus. B, A small blood vessel in the cortex. C, A large blood vessel in the cortex. D, In situ hybridization shows mouse apoE mRNA along the wall of a large blood vessel in the cortex. E, F, Merged confocal images of EGFP (green) and anti-GFAP (red), an astrocyte marker, were collected from a 5-month-old heterozygous EGFPapoE mouse.
Figure 9.

Expression of EGFP in choroid plexus cells of heterozygous EGFPapoE mice. A, Merged confocal image of EGFP (green) and anti-GFAP (red) from a 5-month-old EGFPapoE mouse. B, In situ hybridization revealed mouse apoE mRNA in the choroid plexus.
Discussion
This study of EGFPapoE reporter mice provides insights into the profile and regulation of apoE expression in the CNS. Our findings demonstrate that neurons in the CNS produce apoE in response to injury. They also show that a subclass of astrocytes and most microglia do not express apoE, even after brain insults, and that many types of CNS cells in addition to astrocytes express apoE, including smooth muscle cells in larger blood vessels, cells surrounding small vessels, and cells of the choroid plexus. EGFPapoE reporter mice will be invaluable for studying the regulation of apoE expression in the CNS at different developmental stages or in response to various brain insults and will likely provide additional insights into the roles of apoE in neurobiology and the diverse mechanisms of apoE4-related neurodegeneration.
Our study lays to rest a long-standing controversy concerning the neuronal expression of apoE. Some previous studies showed that at least some neurons express apoE (Diedrich et al., 1991; Poirier et al., 1991; Han et al., 1994; Bao et al., 1996; Metzger et al., 1996; Xu et al., 1996, 1999a,b; Dupont-Wallois et al., 1997; Boschert et al., 1999; Ferreira et al., 2000; Dekroon and Armati, 2001; Hartman et al., 2001; Aoki et al., 2003; Harris et al., 2003); others suggested that they do not (Page et al., 1998; Nishio et al., 2003). More recent studies suggest that neurons might not normally express apoE but do so in response to brain injuries, such as excitotoxic or ischemic injury (Boschert et al., 1999; Aoki et al., 2003). ApoE is also expressed in primary cultured human and rat CNS neurons (Dekroon and Armati, 2001) and in many human neuronal cell lines, including SY-5Y, Kelly, and NT2 cells (Poirier et al., 1991; Dupont-Wallois et al., 1997; Ferreira et al., 2000; Hartman et al., 2001).
In previous studies, anti-apoE immunostaining and in situ hybridization were used to define apoE expression in neurons. Both methods have been criticized for technical or data interpretation limitations. For example, anti-apoE immunostaining can not differentiate whether the apoE in neurons is generated in those cells or taken up from the astrocyte-secreted pool. Likewise, in situ hybridization data are also questioned as false positive, because of the GC-rich nature of the apoE gene. In addition, in situ hybridization might also yield false-negative results because of the poor sensitivity of the technique in detecting low-abundance mRNA in cells. Our EGFPapoE reporter mice avoid those limitations. Thus, our findings conclusively demonstrate that neurons express apoE in response to excitotoxic injury and support the notion that understanding how apoE expression is regulated in neurons is important for unraveling the mechanisms of apoE4-related neurodegenerative disorders.
Surprisingly, ∼20% of hippocampal and cortical astrocytes did not produce apoE, even in response to brain insults. We speculate that apoE-positive and apoE-negative astrocytes play different roles in brain development, neuronal injury and repair, and even some disease processes. Therefore, it would be interesting to know whether aging or neurodegenerative disorders such as AD alter the ratio of those two types of astrocytes.
It has been reported that cultured rat primary microglia and murine microglial cell line express apoE in vitro (Xu et al., 2000; Naidu et al., 2002; Saura et al., 2003; Mori et al., 2004). ApoE protein has also been found in activated microglia in AD brains and in lesioned olfactory bulb of mice by immunocytochemistry (Uchihara et al., 1995; Nathan et al., 2001). We demonstrated in the EGFPapoE reporter mice that microglia do not express apoE under normal conditions, and <10% of microglia produce apoE in response to brain insults. Therefore, microglia are not a major source of brain apoE, at least in mice.
Smooth muscle cells of artery walls in peripheral tissues express apoE (Majack et al., 1988; Moore et al., 2004), and apoE has been detected in cells along vessels in the CNS by anti-apoE immunostaining (Boyles et al., 1985), but the source was unclear. We demonstrated that apoE is produced in smooth muscle cells in large vessels and in cells surrounding small vessels in the CNS. Clearly, those apoE-expressing cells surrounding small vessels are not astrocytes, because they are negative for anti-GFAP immunostaining. Although the cellular identity remains to be determined, apoE generated in these locations may be important in maintaining the integrity and normal function of the blood–brain barrier. In fact, apoE deficiency causes leakage of this structure (Fullerton et al., 2001; Methia et al., 2001). Furthermore, apoE has been found in lesions of cerebral amyloid angiopathy (CAA), and apoE4 is associated with increased severity of CAA in humans and amyloid protein precursor transgenic mice (Schmechel et al., 1993; Greenberg et al., 1995; Fryer et al., 2005).
It has been generally accepted that apoE in CAA is secreted by astrocytes (Fryer et al., 2003, 2005). ApoE4 might deliver more Aβ peptide than apoE3 from the brain parenchyma to vessel walls (Fryer et al., 2005). However, our observation that CNS cells in or around the vessel wall produce copious amounts of apoE raises the possibility that apoE4 generated in this location might retain more Aβ than apoE3, leading to more severe CAA in apoE4 carriers. In line with this hypothesis, CAA in AD brains occurs along the perivascular spaces of small blood vessels and between smooth muscle cells in large vessels (Weller et al., 1998). Interestingly, apoE was not expressed along veins in the CNS of the EGFPapoE mice, and CAA is seldom found along the veins in AD brains (Weller et al., 1998).
Finally, cells in the choroid plexus expressed high levels of apoE. The choroid plexus secretes CSF, expresses many receptors (e.g., apoE receptor-2), secretes numerous molecules (e.g., growth factors), transports nutrients from the blood to CSF, and clears brain metabolites (e.g., Aβ peptides) (Kim et al., 1996; Martel et al., 1997; Serot et al., 2003; Crossgrove et al., 2005; Moir and Tanzi, 2005). ApoE levels in the CSF have not been reported in mice, but in humans are 5–15% of those in plasma (Pitas et al., 1987; Fukumoto et al., 2003). Our results suggest that the choroid plexus is the major source of apoE in CSF. Because the choroid plexus clears brain Aβ peptides by transporting them from CSF to blood (Crossgrove et al., 2005) and because apoE isoforms interact differently with Aβ peptides (Strittmatter et al., 1993b; LaDu et al., 1994, 1995; Ma et al., 1994; Sanan et al., 1994; Wisniewski et al., 1994), apoE generated by the choroid plexus might affect Aβ clearance in an isoform-specific manner. In fact, apoE knock-out mice have higher Aβ levels in CSF than wild-type mice (DeMattos et al., 2004). Furthermore, the function of the choroid plexus declines with aging and in AD patients (Serot et al., 2003), which might lead to decreased clearance of Aβ peptides.
Clearly, apoE is expressed in different types of cells, in addition to astrocytes, in the CNS. We hypothesize that apoE from different cellular sources has distinct roles in both physiological and pathophysiological pathways, including the pathogenesis of AD (Huang et al., 2004; Huang, 2006). For example, apoE4 generated in injured neurons may be involved in mitochondrial dysfunction and neurofibrillary tangle formation (Huang et al., 2001; Harris et al., 2003; Brecht et al., 2004; Chang et al., 2005), apoE4 generated in astrocytes may be primarily responsible for amyloid plaque formation, apoE4 generated in or around blood vessels may be important for CAA formation, and apoE generated in the choroid plexus may participate in the clearance of Aβ peptides. This hypothesis is supported by the early observation that apoE generated locally in the liver is much more efficient than that generated in the periphery in mediating the hepatic clearance of remnant lipoproteins (Mahley and Ji, 1999; Raffaï et al., 2003).
Footnotes
This work was supported in part by National Institutes of Heath Grants P01 AG022074 and R01 HL37063 and a postdoctoral fellowship from the John Douglas French Alzheimer's Foundation. We thank Dr. Karl Weisgraber for critical reading of this manuscript, Karina Fantillo for manuscript preparation, Stephen Ordway and Gary Howard for editorial assistance, John C. W. Carroll and John Hull for graphics, and Chris Goodfellow for photography.
References
- Allan CM, Walker D, Taylor JM (1995). Evolutionary duplication of a hepatic control region in the human apolipoprotein E gene locus. Identification of a second region that confers high level and liver-specific expression of the human apolipoprotein E gene in transgenic mice. J Biol Chem 270:26278–26281. [DOI] [PubMed] [Google Scholar]
- Allan CM, Taylor S, Taylor JM (1997). Two hepatic enhancers, HCR. 1 and HCR. 2, coordinate the liver expression of the entire human apolipoprotein E/C-I/C-IV/C-II gene cluster. J Biol Chem 272:29113–29119. [DOI] [PubMed] [Google Scholar]
- Aoki K, Uchihara T, Sanjo N, Nakamura A, Ikeda K, Tsuchiya K, Wakayama Y (2003). Increased expression of neuronal apolipoprotein E in human brain with cerebral infarction. Stroke 34:875–880. [DOI] [PubMed] [Google Scholar]
- Aubert J, Stavridis MP, Tweedie S, O'Reilly M, Vierlinger K, Li M, Ghazal P, Pratt T, Mason JO, Roy D, Smith A (2003). Screening for mammalian neural genes via fluorescence-activated cell sorter purification of neural precursors from Sox1-gfp knock-in mice. Proc Natl Acad Sci USA 100:11836–11841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bales KR, Verina T, Cummins DJ, Du Y, Dodel RC, Saura J, Fishman CE, DeLong CA, Piccardo P, Petegnief V, Ghetti B, Paul SM (1999). Apolipoprotein E is essential for amyloid deposition in the APPV717F transgenic mouse model of Alzheimer's disease. Proc Natl Acad Sci USA 96:15233–15238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao F, Arai H, Matsushita S, Higuchi S, Sasaki H (1996). Expression of apolipoprotein E in normal and diverse neurodegenerative disease brain. NeuroReport 7:1733–1739. [DOI] [PubMed] [Google Scholar]
- Basu SK, Brown MS, Ho YK, Havel RJ, Goldstein JL (1981). Mouse macrophages synthesize and secrete a protein resembling apolipoprotein E. Proc Natl Acad Sci USA 78:7545–7549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beffert U, Poirier J (1996). Apolipoprotein E, plaques, tangles and cholinergic dysfunction in Alzheimer's disease. Ann NY Acad Sci 777:166–174. [DOI] [PubMed] [Google Scholar]
- Boschert U, Merlo-Pich E, Higgins G, Roses AD, Catsicas S (1999). Apolipoprotein E expression by neurons surviving excitotoxic stress. Neurobiol Dis 6:508–514. [DOI] [PubMed] [Google Scholar]
- Boyles JK, Pitas RE, Wilson E, Mahley RW, Taylor JM (1985). Apolipoprotein E associated with astrocytic glia of the central nervous system and with nonmyelinating glia of the peripheral nervous system. J Clin Invest 76:1501–1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brecht WJ, Harris FM, Chang S, Tesseur I, Yu G-Q, Xu Q, Fish JD, Wyss-Coray T, Buttini M, Mucke L, Mahley RW, Huang Y (2004). Neuron-specific apolipoprotein E4 proteolysis is associated with increased tau phosphorylation in brains of transgenic mice. J Neurosci 24:2527–2534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buttini M, Orth M, Bellosta S, Akeefe H, Pitas RE, Wyss-Coray T, Mucke L, Mahley RW (1999). Expression of human apolipoprotein E3 or E4 in the brains of Apoe–/– mice: isoform-specific effects on neurodegeneration. J Neurosci 19:4867–4880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buttini M, Yu G-Q, Shockley K, Huang Y, Jones B, Masliah E, Mallory M, Yeo T, Longo FM, Mucke L (2002). Modulation of Alzheimer-like synaptic and cholinergic deficits in transgenic mice by human apolipoprotein E depends on isoform, aging, and overexpression of amyloid β peptides but not on plaque formation. J Neurosci 22:10539–10548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang S, Ma TR, Miranda RD, Balestra ME, Mahley RW, Huang Y (2005). Lipid- and receptor-binding regions of apolipoprotein E4 fragments act in concert to cause mitochondrial dysfunction and neurotoxicity. Proc Natl Acad Sci USA 102:18694–18699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Zhou Y, Mueller-Steiner S, Chen LF, Kwon H, Yi S, Mucke L, Gan L (2005). SIRT1 protects against microglia-dependent amyloid-β toxicity through inhibiting NF-κB signaling. J Biol Chem 280:40364–40374. [DOI] [PubMed] [Google Scholar]
- Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, Roses AD, Haines JL, Pericak-Vance MA (1993). Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer's disease in late onset families. Science 261:921–923. [DOI] [PubMed] [Google Scholar]
- Crossgrove JS, Li GJ, Zheng W (2005). The choroid plexus removes β-amyloid from brain cerebrospinal fluid. Exp Biol Med 230:771–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowther RA (1993). Tau protein and paired helical filaments of Alzheimer's disease. Curr Opin Struct Biol 3:202–206. [Google Scholar]
- Deaton RA, Su C, Valencia TG, Grant SR (2005). Transforming growth factor-β1-induced expression of smooth muscle marker genes involves activation of PKN and p38 MAPK. J Biol Chem 280:31172–31181. [DOI] [PubMed] [Google Scholar]
- Dekroon RM, Armati PJ (2001). Synthesis and processing of apolipoprotein E in human brain cultures. Glia 33:298–305. [DOI] [PubMed] [Google Scholar]
- DeMattos RB, Cirrito JR, Parsadanian M, May PC, O'Dell MA, Taylor JW, Harmony JAK, Aronow BJ, Bales KR, Paul SM, Holtzman DM (2004). ApoE and clusterin cooperatively suppress Aβ levels and deposition: evidence that apoE regulates extracellular Aβ metabolism in vivo. Neuron 41:193–202. [DOI] [PubMed] [Google Scholar]
- Diedrich JF, Minnigan H, Carp RI, Whitaker JN, Race R, Frey W Jr, Haase AT (1991). Neuropathological changes in scrapie and Alzheimer's disease are associated with increased expression of apolipoprotein E and cathepsin D in astrocytes. J Virol 65:4759–4768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupont-Wallois L, Soulié C, Sergeant N, Wavrant-de Wrieze N, Chartier-Harlin M-C, Delacourte A, Caillet-Boudin M-L (1997). ApoE synthesis in human neuroblastoma cells. Neurobiol Dis 4:356–364. [DOI] [PubMed] [Google Scholar]
- Fagan KA, Fouty BW, Tyler RC, Morris KG Jr, Hepler LK, Sato K, LeCras TD, Abman SH, Weinberger HD, Huang PL, McMurtry IF, Rodman DM (1999). The pulmonary circulation of homozygous or heterozygous eNOS-null mice is hyperresponsive to mild hypoxia. J Clin Invest 103:291–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrer LA, Cupples LA, Haines JL, Hyman B, Kukull WA, Mayeux R, Myers RH, Pericak-Vance MA, Risch N, Van Duijn CM (1997). Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta-analysis. J Am Med Assoc 278:1349–1356. [PubMed] [Google Scholar]
- Ferreira S, Dupire M-J, Delacourte A, Najib J, Caillet-Boudin M-L (2000). Synthesis and regulation of apolipoprotein E during the differentiation of human neuronal precursor NT2/D1 cells into postmitotic neurons. Exp Neurol 166:415–421. [DOI] [PubMed] [Google Scholar]
- Fryer JD, Taylor JW, DeMattos RB, Bales KR, Paul SM, Parsadanian M, Holtzman DM (2003). Apolipoprotein E markedly facilitates age-dependent cerebral amyloid angiopathy and spontaneous hemorrhage in amyloid precursor protein transgenic mice. J Neurosci 23:7889–7896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fryer JD, Simmons K, Parsadanian M, Bales KR, Paul SM, Sullivan PM, Holtzman DM (2005). Human apolipoprotein E4 alters the amyloid-β40:42 ratio and promotes the formation of cerebral amyloid angiopathy in an amyloid precursor protein transgenic model. J Neurosci 25:2803–2810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukumoto H, Ingelsson M, Gårevik N, Wahlund L-O, Nukina N, Yaguchi Y, Shibata M, Hyman BT, Rebeck GW, Irizarry MC (2003). APOE ε3/ε4 heterozygotes have an elevated proportion of apolipoprotein E4 in cerebrospinal fluid relative to plasma, independent of Alzheimer's disease diagnosis. Exp Neurol 183:249–253. [DOI] [PubMed] [Google Scholar]
- Fullerton SM, Shirman GA, Strittmatter WJ, Matthew WD (2001). Impairment of the blood–nerve and blood–brain barriers in apolipoprotein E knockout mice. Exp Neurol 169:13–22. [DOI] [PubMed] [Google Scholar]
- García MA, Vázquez J, Giménez C, Valdivieso F, Zafra F (1996). Transcription factor AP-2 regulates human apolipoprotein E gene expression in astrocytoma cells. J Neurosci 16:7550–7556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong J-S, Kobayashi M, Hayashi H, Zou K, Sawamura N, Fujita SC, Yanagisawa K, Michikawa M (2002). Apolipoprotein E (apoE) isoform-dependent lipid release from astrocytes prepared from human apoE3 and apoE4 knock-in mice. J Biol Chem 277:29919–29926. [DOI] [PubMed] [Google Scholar]
- Greenberg SM, Rebeck GW, Vonsattel JPG, Gomez-Isla T, Hyman BT (1995). Apolipoprotein E ε4 and cerebral hemorrhage associated with amyloid angiopathy. Ann Neurol 38:254–259. [DOI] [PubMed] [Google Scholar]
- Grehan S, Allan C, Tse E, Walker D, Taylor JM (2001a). Expression of the apolipoprotein E gene in the skin is controlled by a unique downstream enhancer. J Invest Dermatol 116:77–84. [DOI] [PubMed] [Google Scholar]
- Grehan S, Tse E, Taylor JM (2001b). Two distal downstream enhancers direct expression of the human apolipoprotein E gene to astrocytes in the brain. J Neurosci 21:812–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han S-H, Einstein G, Weisgraber KH, Strittmatter WJ, Saunders AM, Pericak-Vance M, Roses AD, Schmechel DE (1994). Apolipoprotein E is localized to the cytoplasm of human cortical neurons: a light and electron microscopic study. J Neuropathol Exp Neurol 53:535–544. [DOI] [PubMed] [Google Scholar]
- Harris FM, Brecht WJ, Xu Q, Tesseur I, Kekonius L, Wyss-Coray T, Fish JD, Masliah E, Hopkins PC, Scearce-Levie K, Weisgraber KH, Mucke L, Mahley RW, Huang Y (2003). Carboxyl-terminal-truncated apolipoprotein E4 causes Alzheimer's disease-like neurodegeneration and behavioral deficits in transgenic mice. Proc Natl Acad Sci USA 100:10966–10971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris FM, Brecht WJ, Xu Q, Mahley RW, Huang Y (2004a). Increased tau phosphorylation in apolipoprotein E4 transgenic mice is associated with activation of extracellular signal-regulated kinase: modulation by zinc. J Biol Chem 279:44795–44801. [DOI] [PubMed] [Google Scholar]
- Harris FM, Tesseur I, Brecht WJ, Xu Q, Mullendorff K, Chang S, Wyss-Coray T, Mahley RW, Huang Y (2004b). Astroglial regulation of apolipoprotein E expression in neuronal cells. Implications for Alzheimer's disease. J Biol Chem 279:3862–3868. [DOI] [PubMed] [Google Scholar]
- Hartman RE, Wozniak DF, Nardi A, Olney JW, Sartorius L, Holtzman DM (2001). Behavioral phenotyping of GFAP-apoE3 and -apoE4 transgenic mice: apoE4 mice show profound working memory impairments in the absence of Alzheimer's-like neuropathology. Exp Neurol 170:326–344. [DOI] [PubMed] [Google Scholar]
- Holtzman DM, Bales KR, Tenkova T, Fagan AM, Parsadanian M, Sartorius LJ, Mackey B, Olney J, McKeel D, Wozniak D, Paul SM (2000). Apolipoprotein E isoform-dependent amyloid deposition and neuritic degeneration in a mouse model of Alzheimer's disease. Proc Natl Acad Sci USA 97:2892–2897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y (2006). Apolipoprotein E and Alzheimer disease. Neurology 66:S79–S85. [DOI] [PubMed] [Google Scholar]
- Huang Y, Liu XQ, Wyss-Coray T, Brecht WJ, Sanan DA, Mahley RW (2001). Apolipoprotein E fragments present in Alzheimer's disease brains induce neurofibrillary tangle-like intracellular inclusions in neurons. Proc Natl Acad Sci USA 98:8838–8843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Weisgraber KH, Mucke L, Mahley RW (2004). Apolipoprotein E. Diversity of cellular origins, structural and biophysical properties, and effects in Alzheimer's disease. J Mol Neurosci 23:189–204. [DOI] [PubMed] [Google Scholar]
- Irizarry MC, Cheung BS, Rebeck GW, Paul SM, Bales KR, Hyman BT (2000). Apolipoprotein E affects the amount, form, and anatomical distribution of amyloid β-peptide deposition in homozygous APPV717F transgenic mice. Acta Neuropathol 100:451–458. [DOI] [PubMed] [Google Scholar]
- Ji Y, Permanne B, Sigurdsson EM, Holtzman DM, Wisniewski T (2001). Amyloid β40/42 clearance across the blood-brain barrier following intra-ventricular injections in wild-type, apoE knock-out and human apoE3 or E4 expressing transgenic mice. J Alzheimers Dis 3:23–30. [DOI] [PubMed] [Google Scholar]
- Ji Z-S, Miranda RD, Newhouse YM, Weisgraber KH, Huang Y, Mahley RW (2002). Apolipoprotein E4 potentiates amyloid β peptide-induced lysosomal leakage and apoptosis in neuronal cells. J Biol Chem 277:21821–21828. [DOI] [PubMed] [Google Scholar]
- Kim D-H, Iijima H, Goto K, Sakai J, Ishii H, Kim H-J, Suzuki H, Kondo H, Saeki S, Yamamoto T (1996). Human apolipoprotein E receptor 2. A novel lipoprotein receptor of the low density lipoprotein receptor family predominantly expressed in brain. J Biol Chem 271:8373–8380. [DOI] [PubMed] [Google Scholar]
- LaDu MJ, Falduto MT, Manelli AM, Reardon CA, Getz GS, Frail DE (1994). Isoform-specific binding of apolipoprotein E to β-amyloid. J Biol Chem 269:23403–23406. [PubMed] [Google Scholar]
- LaDu MJ, Pederson TM, Frail DE, Reardon CA, Getz GS, Falduto MT (1995). Purification of apolipoprotein E attenuates isoform-specific binding to β-amyloid. J Biol Chem 270:9039–9042. [DOI] [PubMed] [Google Scholar]
- Ma J, Yee A, Brewer HB Jr, Das S, Potter H (1994). Amyloid-associated proteins α1-antichymotrypsin and apolipoprotein E promote assembly of Alzheimer β-protein into filaments. Nature 372:92–94. [DOI] [PubMed] [Google Scholar]
- Mahley RW, Ji Z-S (1999). Remnant lipoprotein metabolism: key pathways involving cell-surface heparan sulfate proteoglycans and apolipoprotein E. J Lipid Res 40:1–16. [PubMed] [Google Scholar]
- Majack RA, Castle CK, Goodman LV, Weisgraber KH, Mahley RW, Shooter EM, Gebicke-Haerter PJ (1988). Expression of apolipoprotein E by cultured vascular smooth muscle cells is controlled by growth state. J Cell Biol 107:1207–1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martel CL, Mackic JB, Matsubara E, Governale S, Miguel C, Miao W, McComb JG, Frangione B, Ghiso J, Zlokovic BV (1997). Isoform-specific effects of apolipoproteins E2, E3, and E4 on cerebral capillary sequestration and blood–brain barrier transport of circulating Alzheimer's amyloid β. J Neurochem 69:1995–2004. [DOI] [PubMed] [Google Scholar]
- Masliah E, Westland CE, Rockenstein EM, Abraham CR, Mallory M, Veinberg I, Sheldon E, Mucke L (1997). Amyloid precursor proteins protect neurons of transgenic mice against acute and chronic excitotoxic injuries in vivo. Neuroscience 78:135–146. [DOI] [PubMed] [Google Scholar]
- Methia N, André P, Hafezi-Moghadam A, Economopoulos M, Thomas KL, Wagner DD (2001). ApoE deficiency compromises the blood brain barrier especially after injury. Mol Med 7:810–815. [PMC free article] [PubMed] [Google Scholar]
- Metzger RE, LaDu MJ, Pan JB, Getz GS, Frail DE, Falduto MT (1996). Neurons of the human frontal cortex display apolipoprotein E immunoreactivity: implications for Alzheimer's disease. J Neuropathol Exp Neurol 55:372–380. [DOI] [PubMed] [Google Scholar]
- Moir RD, Tanzi RE (2005). LRP-mediated clearance of Aβ is inhibited by KPI-containing isoforms of APP. Curr Alzheimer Res 2:269–273. [DOI] [PubMed] [Google Scholar]
- Moore ZWQ, Zhu B, Kuhel DG, Hui DY (2004). Vascular apolipoprotein E expression and recruitment from circulation to modulate smooth muscle cell response to endothelial denudation. Am J Pathol 164:2109–2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori K, Yokoyama A, Yang L, Yang L, Maeda N, Mitsuda N, Tanaka J (2004). l-Serine-mediated release of apolipoprotein E and lipids from microglial cells. Exp Neurol 185:220–231. [DOI] [PubMed] [Google Scholar]
- Naidu A, Xu Q, Catalano R, Cordell B (2002). Secretion of apolipoprotein E by brain glia requires protein prenylation and is suppressed by statins. Brain Res 958:100–111. [DOI] [PubMed] [Google Scholar]
- Namba Y, Tomonaga M, Kawasaki H, Otomo E, Ikeda K (1991). Apolipoprotein E immunoreactivity in cerebral amyloid deposits and neurofibrillary tangles in Alzheimer's disease and kuru plaque amyloid in Creutzfeldt–Jakob disease. Brain Res 541:163–166. [DOI] [PubMed] [Google Scholar]
- Nathan BP, Nisar R, Randall S, Short J, Sherrow M, Wong GK, Struble RG (2001). Apolipoprotein E is upregulated in olfactory bulb glia following peripheral receptor lesion in mice. Exp Neurol 172:128–136. [DOI] [PubMed] [Google Scholar]
- Nishio M, Kohmura E, Yuguchi T, Nakajima Y, Fujinaka T, Akiyama C, Iwata A, Yoshimine T (2003). Neuronal apolipoprotein E is not synthesized in neuron after focal ischemia in rat brain. Neurol Res 25:390–394. [DOI] [PubMed] [Google Scholar]
- Page KJ, Hollister RD, Hyman BT (1998). Dissociation of apolipoprotein and apolipoprotein receptor response to lesion in the rat brain: An in situ hybridization study. Neuroscience 85:1161–1171. [DOI] [PubMed] [Google Scholar]
- Pitas RE, Boyles JK, Lee SH, Hui D, Weisgraber KH (1987). Lipoproteins and their receptors in the central nervous system. Characterization of the lipoproteins in cerebrospinal fluid and identification of apolipoprotein B,E(LDL) receptors in the brain. J Biol Chem 262:14352–14360. [PubMed] [Google Scholar]
- Poirier J, Hess M, May PC, Finch CE (1991). Astrocytic apolipoprotein E mRNA and GFAP mRNA in hippocampus after entorhinal cortex lesioning. Mol Brain Res 11:97–106. [DOI] [PubMed] [Google Scholar]
- Raber J, Wong D, Buttini M, Orth M, Bellosta S, Pitas RE, Mahley RW, Mucke L (1998). Isoform-specific effects of human apolipoprotein E on brain function revealed in ApoE knockout mice: increased susceptibility of females. Proc Natl Acad Sci USA 95:10914–10919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raber J, Wong D, Yu G-Q, Buttini M, Mahley RW, Pitas RE, Mucke L (2000). Apolipoprotein E and cognitive performance. Nature 404:352–354. [DOI] [PubMed] [Google Scholar]
- Raber J, Bongers G, LeFevour A, Buttini M, Mucke L (2002). Androgens protect against apolipoprotein E4-induced cognitive deficits. J Neurosci 22:5204–5209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raffaï RL, Weisgraber KH (2002). Hypomorphic apolipoprotein E mice. A new model of conditional gene repair to examine apolipoprotein E-mediated metabolism. J Biol Chem 277:11064–11068. [DOI] [PubMed] [Google Scholar]
- Raffaï RL, Dong L-M, Farese RV Jr, Weisgraber KH (2001). Introduction of human apolipoprotein E4 “domain interaction” into mouse apolipoprotein E. Proc Natl Acad Sci USA 98:11587–11591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raffaï RL, Hasty AH, Wang Y, Mettler SE, Sanan DA, Linton MF, Fazio S, Weisgraber KH (2003). Hepatocyte-derived apoE is more effective than non-hepatocyte-derived apoE in remnant lipoprotein clearance. J Biol Chem 278:11670–11675. [DOI] [PubMed] [Google Scholar]
- Reardon CA, Lau Y-F, Paik Y-K, Weisgraber KH, Mahley RW, Taylor JM (1986). Expression of the human apolipoprotein E gene in cultured mammalian cells. J Biol Chem 261:9858–9864. [PubMed] [Google Scholar]
- Romas SN, Santana V, Williamson J, Ciappa A, Lee JH, Rondon HZ, Estevez P, Lantigua R, Medrano M, Torres M, Stern Y, Tycko B, Mayeux R (2002). Familial Alzheimer disease among Caribbean Hispanics. A reexamination of its association with APOE. Arch Neurol 59:87–91. [DOI] [PubMed] [Google Scholar]
- Roses AD (1994). The Alzheimer diseases. Curr Neurol 14:111–141. [Google Scholar]
- Roses AD (1996). Apolipoprotein E alleles as risk factors in Alzheimer's disease. Annu Rev Med 47:387–400. [DOI] [PubMed] [Google Scholar]
- Sanan DA, Weisgraber KH, Russell SJ, Mahley RW, Huang D, Saunders A, Schmechel D, Wisniewski T, Frangione B, Roses AD, Strittmatter WJ (1994). Apolipoprotein E associates with β amyloid peptide of Alzheimer's disease to form novel monofibrils. Isoform apoE4 associates more efficiently than apoE3. J Clin Invest 94:860–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunders AM, Strittmatter WJ, Schmechel D, St George-Hyslop PH, Pericak-Vance MA, Joo SH, Rosi BL, Gusella JF, Crapper-MacLachlan DR, Alberts MJ, Hulette C, Crain B, Goldgaber D, Roses AD (1993). Association of apolipoprotein E allele ε4 with late-onset familial and sporadic Alzheimer's disease. Neurology 43:1467–1472. [DOI] [PubMed] [Google Scholar]
- Saura J, Petegnief V, Wu X, Liang Y, Paul SM (2003). Microglial apolipoprotein E and astroglial apolipoprotein J expression in vitro: opposite effects of lipopolysaccharide. J Neurochem 85:1455–1467. [DOI] [PubMed] [Google Scholar]
- Schauwecker PE, Steward O (1997). Genetic determinants of susceptibility to excitotoxic cell death: implications for gene targeting approaches. Proc Natl Acad Sci USA 94:4103–4108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmechel DE, Saunders AM, Strittmatter WJ, Crain BJ, Hulette CM, Joo SH, Pericak-Vance MA, Goldgaber D, Roses AD (1993). Increased amyloid β-peptide deposition in cerebral cortex as a consequence of apolipoprotein E genotype in late-onset Alzheimer disease. Proc Natl Acad Sci USA 90:9649–9653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selkoe DJ (1991). The molecular pathology of Alzheimer's disease. Neuron 6:487–498. [DOI] [PubMed] [Google Scholar]
- Serot J-M., Béné M-C., Faure GC. (2003). Choroid plexus, aging of the brain, and Alzheimer's disease. Front Biosci 8:s515–s521. [DOI] [PubMed] [Google Scholar]
- Shachter NS, Zhu Y, Walsh A, Breslow JL, Smith JD (1993). Localization of a liver-specific enhancer in the apolipoprotein E/C-I/C-II gene locus. J Lipid Res 34:1699–1707. [PubMed] [Google Scholar]
- Shih S-J, Allan C, Grehan S, Tse E, Moran C, Taylor JM (2000). Duplicated downstream enhancers control expression of the human apolipoprotein E gene in macrophages and adipose tissue. J Biol Chem 275:31567–31572. [DOI] [PubMed] [Google Scholar]
- Simonet WS, Bucay N, Lauer SJ, Taylor JM (1993). A far-downstream hepatocyte-specific control region directs expression of the linked human apolipoprotein E and C-I genes in transgenic mice. J Biol Chem 268:8221–8229. [PubMed] [Google Scholar]
- Smith JD, Melián A, Leff T, Breslow JL (1988). Expression of the human apolipoprotein E gene is regulated by multiple positive and negative elements. J Biol Chem 263:8300–8308. [PubMed] [Google Scholar]
- Sperk G, Lassmann H, Baran H, Kish SJ, Seitelberger F, Hornykiewicz O (1983). Kainic acid induced seizures: neurochemical and histopathological changes. Neuroscience 10:1301–1315. [DOI] [PubMed] [Google Scholar]
- Spinler SA, Cziraky MJ (1994). Lipoprotein(a): physiologic function, association with atherosclerosis, and effects of lipid-lowering drug therapy. Ann Pharmacother 28:343–351. [DOI] [PubMed] [Google Scholar]
- Strittmatter WJ, Saunders AM, Schmechel D, Pericak-Vance M, Enghild J, Salvesen GS, Roses AD (1993a). Apolipoprotein E: high-avidity binding to β-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc Natl Acad Sci USA 90:1977–1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strittmatter WJ, Weisgraber KH, Huang DY, Dong L-M, Salvesen GS, Pericak-Vance M, Schmechel D, Saunders AM, Goldgaber D, Roses AD (1993b). Binding of human apolipoprotein E to synthetic amyloid β peptide: isoform-specific effects and implications for late-onset Alzheimer disease. Proc Natl Acad Sci USA 90:8098–8102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang M-X, Stern Y, Marder K, Bell K, Gurland B, Lantigua R, Andrews H, Feng L, Tycko B, Mayeux R (1998). The APOE-ε4 allele and the risk of Alzheimer disease among African Americans, whites, and Hispanics. J Am Med Assoc 279:751–755. [DOI] [PubMed] [Google Scholar]
- Tanzi RE, Bertram L (2001). New frontiers in Alzheimer's disease genetics. Neuron 32:181–184. [DOI] [PubMed] [Google Scholar]
- Tesseur I, Van Dorpe J, Bruynseels K, Bronfman F, Sciot R, Van Lommel A, Van Leuven F (2000a). Prominent axonopathy and disruption of axonal transport in transgenic mice expressing human apolipoprotein E4 in neurons of brain and spinal cord. Am J Pathol 157:1495–1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tesseur I, Van Dorpe J, Spittaels K, Van den Haute C, Moechars D, Van Leuven F (2000b). Expression of human apolipoprotein E4 in neurons causes hyperphosphorylation of protein tau in the brains of transgenic mice. Am J Pathol 156:951–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyooka Y, Tsunekawa N, Akasu R, Noce T (2003). Embryonic stem cells can form germ cells in vitro Proc Natl Acad Sci USA 100:11457–11462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchihara T, Duyckaerts C, He Y, Kobayashi K, Seilhean D, Amouyel P, Hauw J-J (1995). ApoE immunoreactivity and microglial cells in Alzheimer's disease brain. Neurosci Lett 195:5–8. [DOI] [PubMed] [Google Scholar]
- Vincent B, Smith JD (2001). Astrocytes down-regulate neuronal β-amyloid precursor protein expression and modify its processing in an apolipoprotein E isoform-specific manner. Eur J Neurosci 14:256–266. [DOI] [PubMed] [Google Scholar]
- Weller RO, Massey A, Newman TA, Hutchings M, Kuo Y-M, Roher AE (1998). Cerebral amyloid angiopathy. Am J Pathol 153:725–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wisniewski T, Frangione B (1992). Apolipoprotein E: a pathological chaperone protein in patients with cerebral and systemic amyloid. Neurosci Lett 135:235–238. [DOI] [PubMed] [Google Scholar]
- Wisniewski T, Castaño EM, Golabek A, Vogel T, Frangione B (1994). Acceleration of Alzheimer's fibril formation by apolipoprotein E in vitro. Am J Pathol 145:1030–1035. [PMC free article] [PubMed] [Google Scholar]
- Xu P-T, Schmechel D, Rothrock-Christian T, Burkhart DS, Qiu H-L, Popko B, Sullivan P, Maeda N, Saunders AM, Roses AD, Gilbert JR (1996). Human apolipoprotein E2, E3, and E4 isoform-specific transgenic mice: human-like pattern of glial and neuronal immunoreactivity in central nervous system not observed in wild-type mice. Neurobiol Dis 3:229–245. [DOI] [PubMed] [Google Scholar]
- Xu P-T, Gilbert JR, Qiu H-L, Rothrock-Christian T, Settles DL, Roses AD, Schmechel DE (1998). Regionally specific neuronal expression of human APOE gene in transgenic mice. Neurosci Lett 246:65–68. [DOI] [PubMed] [Google Scholar]
- Xu P-T, Gilbert JR, Qiu H-L, Ervin J, Rothrock-Christian TR, Hulette C, Schmechel DE (1999a). Specific regional transcription of apolipoprotein E in human brain neurons. Am J Pathol 154:601–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu P-T, Schmechel D, Qiu H-L, Herbstreith M, Rothrock-Christian T, Eyster M, Roses AD, Gilbert JR (1999b). Sialylated human apolipoprotein E (apoEs) is preferentially associated with neuron-enriched cultures from APOE transgenic mice. Neurobiol Dis 6:63–75. [DOI] [PubMed] [Google Scholar]
- Xu Q, Li Y, Cyras C, Sanan DA, Cordell B (2000). Isolation and characterization of apolipoproteins from murine microglia. Identification of a low density lipoprotein-like apolipoprotein J-rich but E-poor spherical particle. J Biol Chem 275:31770–31777. [DOI] [PubMed] [Google Scholar]
- Ye S, Huang Y, Müllendorff K, Dong L, Giedt G, Meng EC, Cohen FE, Kuntz ID, Weisgraber KH, Mahley RW (2005). Apolipoprotein (apo) E4 enhances amyloid β peptide production in cultured neuronal cells: apoE structure as a potential therapeutic target. Proc Natl Acad Sci USA 102:18700–18705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng P, Pennacchio LA, Le Goff W, Rubin EM, Smith JD (2004). Identification of a novel enhancer of brain expression near the apoE gene cluster by comparative genomics. Biochim Biophys Acta 1676:41–50. [DOI] [PubMed] [Google Scholar]