

Abstract
Do continuous subthreshold oscillations in membrane potential within an electrically coupled network depend on gap junctional coupling? For the inferior olive (IO), modeling and developmental studies suggested that the answer is yes, although physiological studies of connexin36 knock-out mice lacking electrical coupling suggested that the answer is no. Here we addressed the question differently by using a lentivirus-based vector to express, in the IO of adult rats, a single amino acid mutation of connexin36 that disrupts the intracellular trafficking of wild-type connexin36 and blocks gap junctional coupling. Confocal microscopy of green fluorescence protein-labeled dendrites revealed that the mutant connexin36 prevented wild-type connexin36 from being expressed in dendritic spines of IO neurons. Intracellular recordings from lentivirally transduced IO networks revealed that robust and continuous subthreshold oscillations require gap junctional coupling of IO neuron somata within 40 μm of one another. Topological studies indicated that the minimal coupled network for supporting such oscillations may be confined to the dendritic arbor of a single IO neuron. Occasionally, genetically uncoupled IO neurons showed transient oscillations; however, these were not sustained longer than 3 s and were 69% slower and 71% smaller than the oscillations of normal IO neurons, a finding replicated with carbenoxolone, a pharmacological antagonist of gap junctions. The experiments provided the first direct evidence that gap junctional coupling between neurons, specifically mediated by connexin36, allows a continuous network oscillation to emerge from a population of weak and episodic single-cell oscillators. The findings are discussed in the context of the importance of gap junctions for cerebellar rhythms involved in movement.
Keywords: gene transfer, gap junction, rhythm, mutant, cerebellum, rat
Introduction
Continuous, subthreshold oscillations (STOs) in membrane potential figure prominently in current views of brain function because they bring neurons close to the threshold for generating spikes in an orderly and time-varying manner. Substantial work has suggested that specific bandwidths of brain rhythm are underlain by electrical synapses that may permit continuous STOs among neuronal ensembles (Whittington and Traub, 2003; Hestrin and Galarreta, 2005; Sohl et al., 2005). Recently, the issue has been studied intensively in the inferior olive (IO) (Long et al., 2002; DeZeeuw et al., 2003; Placantonakis et al., 2004a,b; Leznik and Llinás, 2005), a precerebellar structure that expresses the highest density of electrical synapses in adult brain (Llinás et al., 1974; Sotelo et al., 1974) and in which neurons maintained in vitro show continuous STOs (Benardo and Foster, 1986; Llinás and Yarom, 1986). The fundamental basis of IO activity is significant both as a unique model of synaptic organization and because of its important role in movement (Llinás, 1974; Welsh et al., 1995; Welsh and Llinás, 1997).
Support for a necessary role of electrical synapses for continuous STOs in the IO has remained indirect, primarily through correlative physiology and mathematical modeling, and considerable uncertainty remains. The strongest support is that STOs in the IO emerge when gap junctions develop (Bleasel and Pettigrew, 1992). One model suggested that electrical synapses enable continuous STOs by coupling neurons that would otherwise act as episodic oscillators based on their intrinsic properties (Yarom, 1991). Another model suggested that electrical synapses could produce continuous STOs among electrically stable neurons when a heterogeneous set of ionic channels among coupled neurons support STOs (Manor et al., 1997). However, IO neurons from mice lacking connexin36 (Cx36), the protein that forms neuronal gap junctions (Condorelli et al., 1998), were electrically uncoupled but showed continuous STOs as robust as wild types (Long et al., 2002; DeZeeuw et al., 2003). Additional study (DeZeeuw et al., 2003) found alterations in the electrical properties of IO neurons of Cx36-null mice that caused them to fire hyper-rhythmically, perhaps confounding the knock-out model. Other studies with nonspecific pharmacological blockers of gap junctions showed that continuous STOs in IO neurons in vitro were not blocked and sometimes strengthened (Leznik and Llinás, 2005), but IO rhythmicity was reduced in vivo (Blenkinsop and Lang, 2006).
This experiment tested the hypothesis that gap junctional coupling is necessary for STOs with a genetic blocker of Cx36 delivered specifically to the IO. We used a lentiviral vector (LV) to express a single amino acid mutation of Cx36 that has dominant-negative effect on wild-type Cx36 and that does not affect the active electrical properties of IO neurons (Placantonakis et al., 2004b). This Cx36 mutant (mutCx36) blocked cytosolic coupling in culture by disrupting Cx36 trafficking (Placantonakis et al., 2002), blocked cytosolic coupling among IO neurons (Placantonakis et al., 2004b), and desynchronized tremor (Placantonakis et al., 2004b). With the LV technique, here we show that continuous STOs in the IO require Cx36-mediated coupling.
Parts of this work have been published previously in abstract form (Placantonakis et al., 2004a).
Materials and Methods
Recombinant LVs and implantation.
The LV system (Cell Genesys, Foster City, CA) described by Placantonakis et al. (2004b) was used. A recombinant LV (mutCx36-GFP-LV) expressed a dominant-negative Cx36 mutation under the control of the phosphoglycerate kinase (PGK) promoter and green fluorescent protein (GFP) under the control of the murine stem cell virus promoter. The mutation of Cx36 was a single amino acid substitution of serine for cysteine at position 231 (C231S). A control vector (GFP-LV) expressed only GFP under the control of the PGK promoter. GFP-LV allowed the effects of LV and GFP to be determined in the absence of the mutant Cx36 protein and aided the localization of Cx36. Estimated titers were 4.4 × 108 transducing units/ml for GFP-LV and 4.5 × 108 transducing units/ml for mutCx36-GFP-LV. LVs were injected into the IO of 41 rats (4–10 weeks old) stereotaxically via the retropharyngeal approach under anesthesia (ketamine, 100 mg/kg, i.p.; xylazine, 10 mg/kg, i.p.). Survival time was 1–2 weeks.
Brainstem slice preparation, intracellular recordings, and dye coupling.
Parasagittal brainstem slices (350 μm) were cut in 4°C modified artificial CSF (ACSF) that contained 252 mm sucrose, 5 mm KCl, 1.25 mm NaH2PO4, 3.5 mm MgSO4, 0.5 mm CaCl2, 10 mm glucose, and 26 mm NaHCO3, aerated with 95% O2/5% CO2 to pH 7.4. Slices were placed in a holding chamber containing room-temperature ACSF in which sucrose was replaced with 126 mm NaCl, and CaCl2 and MgSO4 were changed to 2 mm. Intracellular recordings (n = 86) were performed in a submersion chamber (33–35°C) in current-clamp mode with borosilicate glass microelectrodes (direct current resistance, 80–120 MΩ). Only neurons with membrane potential negative to −50 mV were analyzed. Electrodes were targeted to transduced areas by visualizing GFP.
Recordings were amplified with an Axoclamp2B amplifier (Molecular Devices, Palo Alto, CA), digitized (5 or 10 kHz sampling), and analyzed with pClamp6 (Molecular Devices), Origin (Microcal Software, Northampton, MA), Datapac 2k2 (Run Technologies, Laguna Hills, CA), Neuroexplorer (Nex Technologies, Littleton, MA), and GB-Stat (Dynamic Microsystems, Silver Spring, MD). Fast Fourier transforms for power spectrum analysis were obtained from 1–60 s records; results are presented as raw power or as percentage of total power. STO amplitude was expressed as the peak-to-peak amplitude. Under the present conditions, 85% of normal IO neurons showed STOs. Measurements of steady-state input resistance were calculated from voltage responses obtained with −0.1 to −0.3 nA current pulses.
Recording electrodes contained 2 m potassium acetate with 4% (w/v) Neurobiotin (Vector Laboratories, Burlingame, CA). At the end of the recording, Neurobiotin was microiontophoresed into the recorded neuron (1–5 nA, 1 s square pulses, 0.5 Hz, 10–40 min); slices were perfused for 2 h after withdrawing the electrode, fixed overnight in 4% Dulbecco's PBS (DPBS)-buffered paraformaldehyde, pH 7.4, and then incubated in blocking solution (1% BSA, 10% goat serum, and 0.4% Triton X-100 in DPBS). Neurobiotin was detected by streptavidin conjugated to Alexa Fluor 568 (Invitrogen, Carlsbad, CA) dissolved at 1–5 μg/ml in DPBS containing 10% blocking solution. Slices were coverslipped on microslides using Prolong Antifade medium (Invitrogen). The number of dye-coupled neurons was defined as (number of labeled neurons/injection − 1). Carbenoxolone (200 μm; Sigma, St. Louis, MO) was introduced into the perfusion solution in 10 experiments for up to 40 min (5 ml/min), of which seven were studied for coupling.
Cx36 immunodetection and microscopy.
LV-implanted rats (n = 8) were perfused with 4% paraformaldehyde, and their brains were postfixed in 30% sucrose. Vibratome (30 μm) sections were treated sequentially with blocking solution and with 2 μg/ml rabbit polyclonal anti-Cx36 antibody (Zymed, San Francisco, CA) in 5% blocking solution. After washing with DPBS, the sections were incubated with 10 μg/ml goat anti-rabbit secondary antibody conjugated to Alexa Fluor 568 and mounted on microslides.
An LSM-510 Meta NLO confocal microscope (Zeiss, Thornwood, NY) was used for fluorescence microscopy. Crosstalk between dual labels was prevented by multitrack scanning using alternating laser excitation and emission filtering. Thick tissue imaging was performed with a 20×/0.75 numerical aperture (NA) air objective or a 63×/1.4 NA oil objective (Zeiss Plan-Neofluars). High-resolution imaging of dendrites for quantitation was performed in 30 μm sections using a 100×/1.3 NA oil objective (100 nm pixel sampling). A 488 nm laser (∼4 mW) excited GFP (500–530 nm emission detected); a 543 nm laser (1 mW) excited Alexa Fluor 568 (565–615 nm emission detected). Emission spectra (380–710 nm) were obtained with a 32-channel spectrophotometer (Zeiss Meta detector). Axial projections of optical section stacks (each 0.4–1 μm thick) were generated as transparencies or surface renders.
The immunogold method was used for the ultrastructural localization of Cx36 as described by Mitchell et al. (2004). Briefly, sections immunolabeled for Cx36 were incubated in 1 nm colloidal-gold goat anti-rabbit IgG (1:50; Electron Microscopy Sciences, Fort Washington, PA); gold particles were amplified using silver intensification (Amersham Biosciences, Arlington Heights, IL). After osmium tetroxide fixation, dehydration, embedding, sectioning (75 nm), and counterstaining, tissue was imaged with a Tecnai 12 electron microscope (FEI, Hillsboro, OR).
Quantitation of Cx36 expression and LV transduction.
Images of Cx36 immunofluorescence were converted to binary by thresholding at 1.6 SDs above the mean luminosity. Thresholded image fields (5000 μm2 total) were analyzed by Scion Image (Scion, Frederick, MD) to identify Cx36 puncta and measure their area. Puncta diameter was measured at its longest extent by profile analysis with 100 nm pixel resolution using Image Examiner software (Zeiss). The diameter of 25 spine heads was quantified the same way from printed electron micrographs. Spine heads were verified by their occurrence in glomeruli and in apposition to one another. The occurrence of Cx36 within and around 20 GFP-expressing dendrites (497 μm2 total) was quantified with a colocalization algorithm that used continuous values of fluorescence intensity (Zeiss Image Examiner). In separate GFP and Cx36 image channels, the eight-bit intensity of each pixel was placed into a cross-correlation matrix divided into quadrants by the median intensities of the channels for the image field; pixels expressing intensities of GFP and Cx36 above both medians were considered to show colocalization. For recording experiments, transduction density (TD) was determined by counting the number of GFP-positive cells in 500,000 μm3 surrounding the soma of each recorded neuron. In thick tissue slices, it was sometimes difficult to unambiguously distinguish neurons from glia using GFP expression alone, and so estimates of TD considered both cell types that are transduced by LVs with high efficiency (84%) (Placantonakis et al., 2004b). In three illustrative examples, transduced neurons could be distinguished from glia by soma shape aided by a systematic difference in GFP fluorescence intensity.
Statistics.
Student's t test and ANOVA were used with significance accepted at p < 0.05 (Winer, 1979). Population measures are presented as the mean ± 1 SEM.
Results
LV-based disruption of Cx36 organization in IO dendrites
Most Cx36 signal in the normal IO presented as puncta in the neuropil (Fig. 1A), but Cx36 immunoreactivity also defined proximal dendrites (Fig. 1A, arrowheads) and neuronal somata (Fig. 1A, asterisk). Confocal imaging of Cx36 in IOs transduced by the control LV (GFP only) revealed that neuropilar puncta lined the perimeter of GFP-expressing dendrites (Fig. 1B–E). Three-dimensional reconstruction of optical sections revealed that pairs of Cx36 puncta often apposed one another (Fig. 1F, arrowheads), linking two dendritic shafts running in parallel (Fig. 1G, arrowhead). The proximity of the Cx36 puncta to dendritic shafts (1.4 ± 0.1 μm; n = 20) and their diameter (0.61 ± 0.02 μm; n = 28) suggested that they defined the heads of dendritic spines, the location of gap junctions between IO neurons (Sotelo et al., 1974). Because GFP did not fill dendritic spines, electron microscopy was used to determine whether Cx36 was present in spine heads. Electron microscopy confirmed that Cx36 was present in dendritic spines within the IO and revealed apposed spine heads containing in-register immunopuncta of Cx36 (Fig. 1H, arrowheads). Ultrastructurally, dendritic spine heads of IO neurons containing Cx36 had a diameter of 1.13 ± 0.08 μm (n = 25), a value that was nearly double the size of the fluorescent Cx36 puncta and indicating that the extradendritic Cx36 seen with the laser microscope did not fully fill the spine head.
Figure 1.
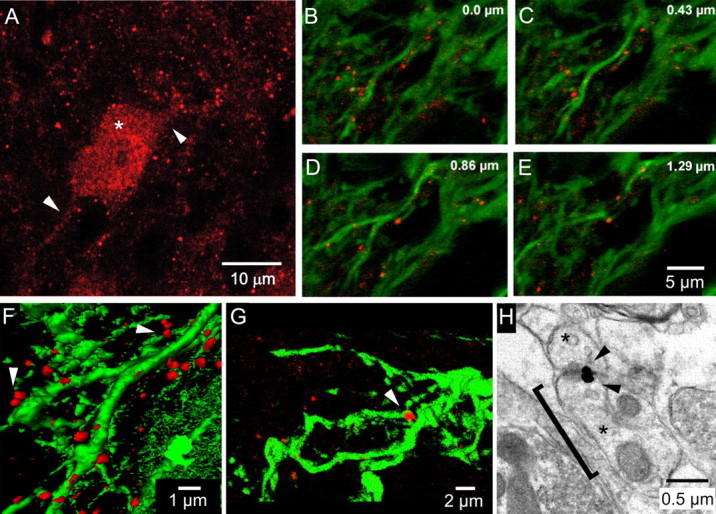
Cx36 in dendritic spines of IO neurons. A, Confocal micrograph of Cx36 immunoreactivity (red) showing Cx36 in the soma of an IO neuron (asterisk), its proximal dendrites (arrowheads), and as puncta in the neuropil. B–E, Sequential optical sections (depth in top right) through a plexus of IO dendrites expressing GFP (green) attributable to GFP-LV and immunolabeled for Cx36 (red). Cx36 puncta sit along the dendritic shafts. F, G, Three-dimensional micrographs demonstrating the spatial relationship of Cx36 puncta to dendritic shafts and in apposition to one another (arrowheads). H, Electron micrograph of two dendritic spines in the IO (asterisks) expressing Cx36 at their apposition (arrowheads), as revealed by nanogold immunolabeling. The bracket indicates 1.3 μm between a dendrite shaft and the apposition.
Transduction of the IO with the LV coding for the mutant Cx36 reduced the punctate expression of Cx36 in the neuropil and induced abnormally large plaques of Cx36 within dendritic shafts (Fig. 2A,B). Whereas 62% of the total Cx36 expression in control tissue had an area corresponding to puncta associated with dendritic shafts (0.2–0.8 μm2) (Fig. 2C), only 17% of the Cx36 expression was in that range after the tissue was transduced by the LV coding for the mutant Cx36 (Fig. 2D). The loss of spine head-sized puncta was offset by the presence of Cx36 plaques exceeding 2.5 μm2 within the GFP-expressing dendritic shafts. Such plaques comprised 50% of the total Cx36 expression after transduction with mutCx36-GFP-LV (Fig. 2D) and were never seen in control IO dendrites. Quantitative analysis indicated that Cx36 was increased within the dendritic shafts after mutCx36-GFP-LV transduction, as indicated by a significant increase in the percentage of pixels containing both Cx36 immunoreactivity and GFP fluorescence (t(18) = −2.95; p < 0.01). Photomicrographs of single IO dendrites demonstrated the significant change in Cx36 expression after transduction with the LV coding for the mutant Cx36 (Fig. 2E,F). The structural data fit with in vitro findings in which the mutant Cx36 prevented wild-type Cx36 from being trafficked to the plasmalemma membrane to form gap junctions (Placantonakis et al., 2002).
Figure 2.

LV-induced changes in dendritic Cx36. A, B, Schematic summary of Cx36 expression in control dendrites transduced by GFP-LV (A) and dendrites transduced by mutCx36-GFP-LV (B). Expressing mutant Cx36 shifts the size and distribution of Cx36 from puncta immediately adjacent to the dendritic shafts to plaques within the dendrites. C, D, Percentage of total Cx36 signal comprised of Cx36 puncta of three size ranges (pie charts) and colocalization cross-correlograms of GFP and Cx36 expression (histograms) for dendrites transduced by GFP-LV (C) and mutCx36-GFP-LV (D). Each dot in the histograms represents a 100 × 100 nm pixel; horizontal and vertical lines indicate the median intensity of Cx36 and GFP fluorescence, respectively. mutCx36-GFP-LV shifts the pixel distribution to the top right, indicating an increase in Cx36 within the dendritic shaft. E, F, Confocal photomicrographs (1 μm optical sections) of Cx36 expression in GFP-expressing dendrites transduced by GFP-LV (E) and mutCx36-GFP-LV (F).
Robust and continuous STOs require local coupling
Intracellular recordings were obtained from neurons in the normal IO (n = 55) and from IOs transduced to express the mutant Cx36 plus GFP (n = 15) or GFP only (n = 6). The organization of gap junctionally coupled networks was assessed by the transcytosolic spread of Neurobiotin (molecular weight of 322.8) after injection into the recorded neuron, as is commonly performed (Goodenough et al., 1996; Rozental et al., 2001; Devor and Yarom, 2002; Placantonakis et al., 2004b; Leznik and Llinás, 2005). Measures of cytosolic coupling assessed by indirect labeling were chosen over measures of electrical coupling using dual recordings because of the unique ability of the former to estimate the full spatial breadth of coupled networks.
STOs from normal IO neurons were continuous and constant in frequency. Among 55 normal IO neurons, the mean STO frequency was 4.8 ± 0.3 Hz (range of 0.5–8.5 Hz). Neurobiotin injected into 14 normal IO neurons showing 9.7 ± 1.4 mV (range of 4–23 mV) STOs at 4.0 ± 0.5 Hz produced indirect labeling in 7.8 ± 2.2 neurons, replicating previous reports (Devor and Yarom, 2002; Leznik and Llinás, 2005). Figure 3A shows a typical example of the coupling and STO of a normal IO neuron.
Figure 3.

LV-induced blockade of cytosolic coupling by high transduction. A, A normal IO neuron: cytosolic coupling (i), raw STO record (ii), and STO power spectrum (iii). The recorded neuron (arrow) oscillated at 4 Hz and was dye coupled to 12 neurons (arrowheads). B, GFP (i) and Cx36 (ii) expression in an IO neuron soma and their emission spectra (iii) verifying neuronal transduction by mutCx36-GFP-LV. C, Schematic of the 500,000 μm3 tissue volume used to quantify mutCx36-GFP-LV TD. D, mutCx36-GFP-LV TD exceeding 5 cells/500,000 μm3 significantly decreased cytosolic coupling among IO neurons (black bars) and increased input resistance (gray bars). *p < 0.05 compared with untransduced controls and low-TD. E, F, Intracellular recordings from IO neurons within networks transduced by GFP-LV (E) and mutCx36-GFP-LV (F). Spikes were truncated for clarity in F.
To assess the effect of LVs on STOs and to contrast experiments, it was necessary to determine the density of LV transduction in the network around each recorded neuron. This was performed by counting the number of GFP-expressing cells (Fig. 3B) within 39.5 μm from the recorded soma in three dimensions (500,000 μm3) (Fig. 3C), corresponding to the 40 μm radial distance at which neighboring IO neurons show their greatest coupling coefficients (Devor and Yarom, 2002). The LV transduction density (LV-TD) ranged from 0 to 33 cells/500,000 μm3 (mean, 10.5 ± 3.0). The 15 experiments were divided into two groups based on whether the LV-TD exceeded (high-TD; n = 9) or did not exceed (low-TD; n = 6) 5 transduced cells/500,000 μm3 surrounding the recorded soma. The high-TD group showed significantly (t(13) = 2.5; p < 0.05) less cytosolic coupling (1.9 ± 0.8 indirectly labeled cells) than the low-TD group (9.3 ± 3.4 indirectly labeled cells) and untransduced IO neurons (Fig. 3D). Additionally, direct current injection revealed significantly increased input resistance of neurons in the high-TD group (62 ± 20 MΩ) compared with the control (28 ± 4 MΩ) and low-TD (32 ± 3 MΩ) groups (Fig. 3D). The increase in input resistance indicated a significant reduction in current flow after high LV-TD, consistent with an effective block of gap junction channels. Neurons in the low-TD group were not different from normal IO neurons in values of cytosolic coupling, input resistance, STO amplitude, or STO frequency (all p values >0.05).
The main effect of LV-mediated block of gap junctional coupling on IO neuron physiology was to disrupt the continuity of STOs and reduce their amplitude and frequency until, at the highest values of LV-TD, they were virtually immeasurable with intracellular recording. Whereas IO neurons in the low-TD group showed STOs of 10.4 ± 3.0 mV at 5.6 ± 1.1 Hz, the high-TD group showed STOs of only 3.3 ± 1.0 mV at 2.4 ± 0.5 Hz (for both, t(13) > 2.0; p < 0.05). Plots of STO frequency and amplitude as a function of LV-TD confirmed the significant inverse relationship between LV-TD and the robustness of STOs, indicating that uncoupling of the IO by the LV suppressed STOs in IO neurons.
Of 15 neurons that were recorded in the IO transduced by the LV coding for the mutant Cx36, six neurons showed continuous STOs, whereas nine showed noncontinuous STOs (Fig. 4C). The mean LV-TD for the six neurons showing continuous STOs was 4.5 ± 2.3/500,000 μm3 whereas the LV-TD was significantly greater (t(13) = 2.2; p = 0.05) for neurons that showed noncontinuous STOs (13.7 ± 3.9/500,000 μm3). Only two of nine neurons in the high-TD group showed continuous STOs, whereas four of six in the low-TD group showed continuous STOs. Comparing the five neurons recorded with the highest LV-TD with the four neurons with the lowest TD indicated that an 84% block of coupling reduced STO frequency by 69% and reduced STO amplitude by 71%.
Figure 4.

Suppression of STOs by LV-mediated block of Cx36. A, B, Relationships between mutCx36-GFP-LV transduction density (LV-TD is the number of GFP cells per 500,000 μm3) and STO frequency (A) and STO amplitude (B) for 15 IO neurons. Filled and open circles indicate neurons showing continuous and noncontinuous STOs, respectively. C, Examples of a continuous (top) and noncontinuous (bottom) STO. Spikes are truncated for clarity.
To control for possible effects of LV transduction and GFP expression, the experiment was repeated in six neurons in IOs transduced with GFP-LV to express GFP only. GFP expression by itself did not affect cytosolic coupling (6.5 ± 2.8 indirectly labeled cells) or mean membrane potential (−57.4 ± 4.7 mV). All six neurons showed continuous STOs having a mean amplitude of 8.9 ± 3.4 mV (range of 4–21 mV), a value that was not significantly different than the STO amplitude of nontransduced neurons (t(18) = 0.3; p = 0.8). Thus, the effects of mutCx36-GFP-LV to reduce gap junctional coupling and suppress STOs were specifically attributable to expression of the mutant Cx36 protein.
Changing coupling topology: three illustrative examples
Three examples indicated the necessity of coupling among a group of closely spaced IO neurons for continuous STOs. In the first example, 12 neuronal somata within the dendritic arbor of the recorded neuron were transduced by mutCx36-GFP-LV (Fig. 5A), but coupling was spared to four nontransduced neurons, yet there was no significant STO (Fig. 5B). The four coupled neurons all sat farther than 38 μm (mean, 58 ± 12 μm) from the soma of the recorded neuron (Fig. 5C). Although local transduction density was high, the recorded cell itself was not transduced as indicated by the absence of GFP expression. In the second example (Fig. 5D), 15 neuronal somata within the dendritic arbor of the recorded neuron were transduced by mutCx36-GFP-LV, but coupling was spared to one neuron only 17 μm away (Fig. 5F). Again, there was no significant STO (Fig. 5E). In this experiment, the recorded neuron itself was verified by emission spectroscopy to have been transduced by mutCx36-GFP-LV (Fig. 5F, inset). Notably, an IO neuron only 4 μm away in the same optical plane also expressed GFP but was not dye coupled (Fig. 5F, inset). Both examples showed 2 mV fluctuations in membrane potential that were sometimes sufficiently regular as to appear like small STOs (Fig. 5B,E, asterisks) but that were not sustained longer than 2.2 ± 0.4 s (n = 19). More often, the fluctuations in membrane potential were solitary events (Fig. 5B,E, arrowheads).
Figure 5.
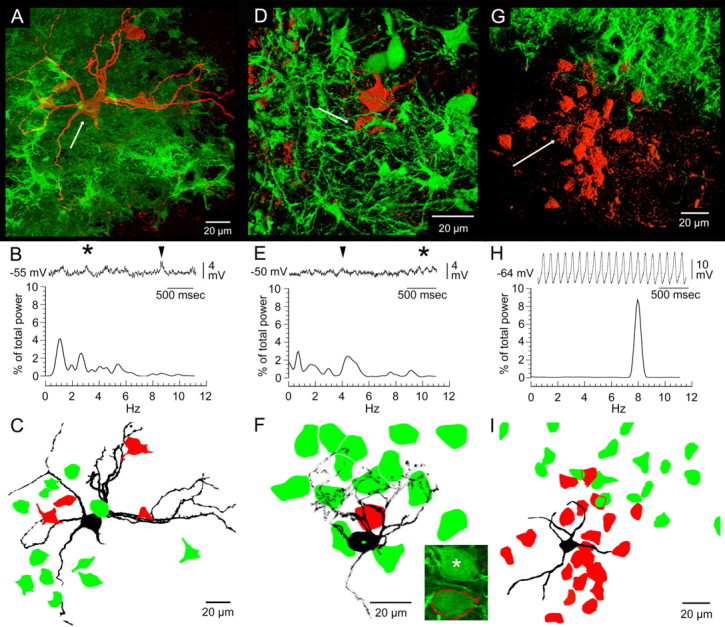
Changes in coupling topology produced by mutant Cx36: effects on STOs. A, D, G, Three-dimensional micrographs of intracellularly recorded IO neurons filled with Neurobiotin (arrows), indirect labeling (red), and GFP expression (green). B, E, H, Three-second epochs of STOs and their frequency spectra obtained from the above neurons indicated by arrows. Asterisks, Transient continuous STOs; arrowheads, single fluctuations. C, F, I, Camera lucida drawings of the above recorded neurons (black), indirectly labeled neurons (red), and GFP-expressing neurons (green). The inset in F shows a 0.5 μm optical section demonstrating that the recorded neuron in D (outlined in red) and a neighboring soma 4 μm away (asterisk) were GFP positive (510 nm emission peak) and uncoupled. The neighboring, uncoupled neuron sat 14 μm directly below the one dye-coupled neuron (red) in the axial thickness of the tissue.
A third example (Fig. 5G) suggested that the minimum network for supporting continuous STOs may not need to extend farther than the dendritic arbor of an IO neuron. Here, a recorded neuron sat 60 μm away from a zone of mutCx36-GFP-LV transduction and expressed a 15 mV, 8 Hz continuous STO (Fig. 5H). There was robust coupling to 21 IO neurons within and immediately outside its dendritic arbor that abutted the edge of a dense transduction zone (Fig. 5I). None of the indirectly labeled neurons at the edge of the transduction zone expressed GFP, and none of the GFP-expressing somata showed indirect labeling. It could be presumed that the indirectly labeled neurons were uncoupled from neurons within the transduction zone and, therefore, that the robust STO was sustained by the local network within the dendritic arbor of the recorded neuron.
Carbenoxolone replicates LV block of Cx36
In 10 of 10 IO neurons, 200 μm carbenoxolone, a pharmacological antagonist of gap junctions, suppressed STOs. The latency to 50% reduction in STO power was 15.2 ± 3.9 min, and the latency to 90% reduction was 26.2 ± 5.5 min (Fig. 6A). The number of indirectly labeled cells during carbenoxolone was 0.4 ± 0.3 (n = 7). Neither the resting membrane potential nor the active electrical properties of IO neurons were affected by 200 μm carbenoxolone (Placantonakis et al., 2004b), reducing the possibility that toxicity caused the suppression. Figure 6B shows a typical experiment.
Figure 6.
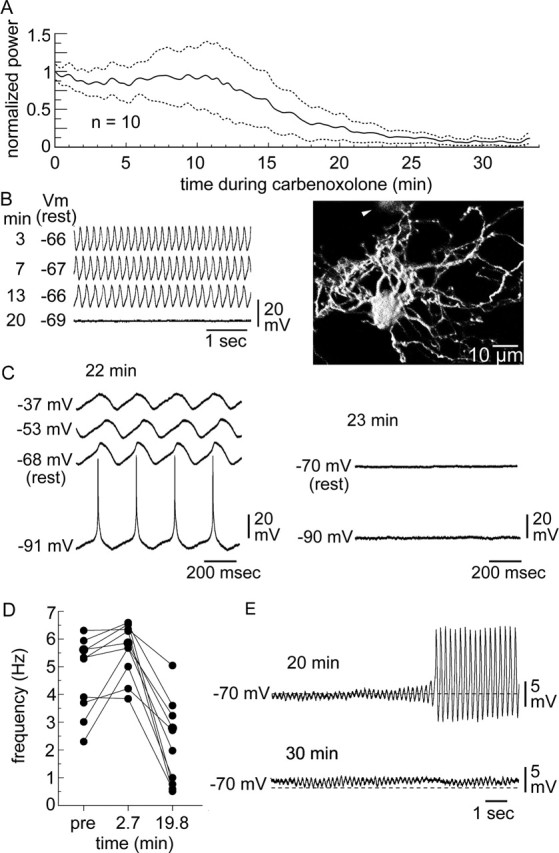
Carbenoxolone replicates LV-mediated block of Cx36. A, Time course of STO suppression by carbenoxolone shown as the mean of peak STO power (solid line) ± 1 SEM (dotted lines). B, An example of STO suppression, showing membrane potential at 3, 7, 13, and 20 min during carbenoxolone (left) and the morphology of the recorded neuron (right) with one indirectly labeled neuron (arrowhead). C, Effect of direct current injection on episodic STOs after 22 min exposure to carbenoxolone (left) and when STOs were blocked by carbenoxolone (right). The occurrence of the STO was voltage insensitive at 22 min and could not be restored by current injection shortly thereafter, after it was blocked by carbenoxolone. D, STO frequency at 2.7 and 19.8 min during carbenoxolone for 10 IO neurons. E, Episodic STOs at 20 min during carbenoxolone showing an STO crescendo from a suppressed state leading to a jump in amplitude (top) and small-amplitude, noncontinuous STOs thereafter (bottom).
Carbenoxolone induced a complex set of effects before maximal suppression of STOs. During the period between 50% reduction in STO power and peak suppression, some neurons showed episodes of normal-amplitude but discontinuous STOs. Direct current injection was used to determine whether the episodic bouts of STOs during carbenoxolone were supported by the coupled network or were single-cell events (Llinás and Yarom, 1986; Long et al., 2002; Leznik and Llinás, 2005). In one noteworthy experiment, the voltage sensitivity of the final episodic STO (at 22 min exposure to carbenoxolone) was tested with direct current injection. This STO occurred over a range of induced voltages (Fig. 6C, left), indicating that electrical coupling within the IO was intact (Llinás and Yarom, 1986) and that carbenoxolone had not yet blocked gap junctions. However, additional exposure to carbenoxolone blocked STOs altogether, and current injection did not reinstate them (Fig. 6C, right). The example provided additional evidence that electrical interactions among coupled IO neurons supported large-amplitude STOs close to the moment when STOs were blocked by carbenoxolone.
As reported in a study of the related drug 18β-glycyrrhetinic acid (Leznik and Llinás, 2005), carbenoxolone increased STO frequency from 4.7 ± 0.4 to 5.6 ± 0.3 Hz within a few minutes after its application (Fig. 6D). This effect was noted in nearly all neurons but was most prominent in those that oscillated slowly at 2–3 Hz. However, for all neurons, peak STO frequency was later reduced to 2.2 ± 0.5 Hz at 19.8 ± 2.3 min; thereafter, carbenoxolone abolished STOs altogether in eight experiments.
During the peak suppression induced by 25 min of carbenoxolone, small-amplitude (1.3 ± 0.3 mV), transient (3.6 ± 0.5 s) STOs were observed in two experiments (Fig. 6E). This was a significant (t(18) = 6.4; p < 0.01) decrease in STO amplitude compared with before carbenoxolone (9.6 ± 1.3 mV) and was similar to the effect of mutant Cx36 expression. Such small-amplitude STOs often preceded the large-amplitude bouts at 15–25 min of carbenoxolone (Fig. 6E, top) and persisted when large-amplitude STOs were blocked (Fig. 6E, bottom). Figure 7 compares all of the manipulations, making evident the necessity for gap junctional coupling within the IO for robust STOs.
Figure 7.

Relationship between dye coupling and STO power for effective and ineffective uncoupling agents. High mutCx36-GFP-LV transduction and carbenoxolone significantly reduce gap junctional coupling. The reduction in gap junctional coupling was clearly associated with a significant decrease in the strength of STO (∗p < 0.05 compared with untransduced control).
Discussion
The experiments provide direct evidence that gap junctional coupling mediated by Cx36 is necessary for robust and continuous STOs in IO neurons. Transduction of IO networks to express a mutation of Cx36 with dominant-negative effect on wild-type Cx36 impaired gap junctional coupling and decreased the amplitude, frequency, and continuity of STOs to a point at which STOs were close to immeasurable with the highest uncoupling. Transduction with an LV coding for the mutant Cx36 reduced the expression of Cx36 in neuropilar puncta that appeared to be the sites of dendritic spines, in which gap junctions are formed between IO neurons (Sotelo et al., 1974). The finding that dendritic Cx36 is important for robust STOs was replicated by carbenoxolone, a pharmacological blocker of gap junctions, which also suppressed STO continuity, amplitude, and frequency to the point of abolishing STOs in most cases. Subthreshold rhythmicity and gap junctions have been implicated in many aspects of brain function, especially in motor control attributable to their presence in the IO. Thus, the experiments help elucidate the important issue of the basis of rhythm generation and function in coupled brain networks.
The findings support a model of the IO in which gap junctional coupling mediated by Cx36 allows a network rhythm to emerge from neuronal population that consists of weak, episodic single-cell oscillators (Yarom, 1991). An important finding was that some uncoupled IO neurons were able to emit discontinuous, small-amplitude (∼2 mV) STOs. However, such small STOs in uncoupled IO neurons were slower than normal and could not be sustained for much longer than 2 s. The findings draw attention to the fact that a fundamental basis of IO rhythmicity is the capacity of its constitutive elements for single-cell oscillation (Llinás and Yarom, 1981, 1986; Leznik and Llinás, 2005) but demonstrate a necessary role for Cx36-mediated coupling in a process of mutual reinforcement that allows a continuous and robust STO to emerge as an ensemble property of the IO network.
The present experiments using targeted genetic manipulation of Cx36 in adult rats complement the finding in neonatal rats that STOs within the IO emerge as gap junctions are formed during development (Bourrat and Sotelo, 1983; Bleasel and Pettigrew, 1992). Within the IO, STOs do not occur before postnatal day 4 (P4) when there are no gap junctions, are noncontinuous before P16, grow in amplitude from 3 to 15 mV, increase in frequency from 1 to 7 Hz, and become continuous as the adult complement of gap junctions is reached on day P25 (Bleasel and Pettigrew, 1992). In this experiment, progressively uncoupling the IO in adult rats progressively decreased STO amplitude and frequency to neonatal values and disrupted their continuity (Fig. 4). Thus, genetic blockade of gap junctional coupling returned the IO to a neonatal-like collection of low-amplitude, slow, and noncontinuous single-cell oscillators. The combined evidence indicates that continuous, large-amplitude STOs in the IO require the full adult capacity for gap junctional coupling mediated by Cx36.
Uncoupling the normally developed IO with targeted genetic disruption of Cx36 revealed a more important role for gap junction coupling in spontaneous STOs than was suggested by the study of Cx36 knock-out mice lacking gap junctional coupling, which did not affect spontaneous STOs of IO neurons (Long et al., 2002; DeZeeuw et al., 2003). However, uncoupled IO neurons from Cx36 knock-out mice were abnormally hyper-rhythmic in response to membrane hyperpolarization, a change that was suggested by DeZeeuw et al. (2003) to produce an abnormal capacity for continuous single-cell oscillation. In contrast, LV transduction of IO neurons and expression of the mutant Cx36 protein did not change the electrical properties of IO neurons beyond increasing membrane resistance, as would be expected from closing gap junction channels (Placantonakis et al., 2004b). Moreover, transduction with the control LV lacking the mutant Cx36 coding sequence did not affect STOs, demonstrating that viral or fluorescent protein cytotoxicity was not a factor.
The LV experiments demonstrating that coupling is required for the full expression of STOs in the IO were replicated by pharmacological blockade of gap junctions with carbenoxolone. Carbenoxolone was used as a positive control to verify the effects of the genetic manipulation; the lack of a similar effect would have indicated that the mutant Cx36 blocked STOs through a mechanism unrelated to gap junctions. It is well recognized that there is no specific pharmacological antagonist of gap junctions (Rozental et al., 2001) and that carbenoxolone is not an exception (Rekling et al., 2000; Rouach et al., 2003; Vessey et al., 2004), rendering uncertain any effect of gap-junction-blocking drugs that cannot be verified by a complementary technique. In a recent report (Leznik and Llinás, 2005), the gap junction blocker 18β-glycyrrhetinic acid did not block STOs in the IO but did increase STO frequency while it impaired synchronous firing measured optically. The difference in the two outcomes may relate to the 30% greater potency of carbenoxolone over 18β-glycyrrhetinic acid (Rozental et al., 2001) and our use of twice the concentration. Carbenoxolone did replicate the effect of 18β-glycyrrhetinic acid to transiently increase STO frequency, an effect that may relate to the nonlinear contributions of coupling to STO frequency and that might result from partial uncoupling (Kepler et al., 1990). However, even at a high concentration, carbenoxolone required 26 min to suppress STOs, a latency consistent with the latency of carbenoxolone to block electrical rhythms in other systems for which gap junctions have been implicated, including the locus ceruleus (Ishimatsu and Williams, 1996; Ballantyne et al., 2004), temporal lobe (Konopacki et al., 2004; Kano et al., 2005), and substantia gelatinosa (Asghar et al., 2005). The latency to effect is consistent with the action of carbenoxolone to disaggregate gap junction hemichannels (Goldberg et al., 1996; Guan et al., 1996), which may require more time to reach maximum effect than agents that bind to extracellular sites with high affinity. Alternative explanations are that both carbenoxolone and the mutant Cx36 protein suppressed STOs by effects unrelated to their actions on gap junction proteins and that compensatory changes triggered by the mutant Cx36 protein contributed to STO suppression. It is possible that such effects, if any, are not detectable by analyzing the active membrane properties of IO neurons or their morphology.
The morphological data indicate that the C231S mutation of Cx36 prevents wild-type Cx36 from being distributed to dendritic spines, which are the sites of gap junctions in the IO (Sotelo et al., 1974). Similar mutations in connexin32 that occur in Charcot-Marie-Tooth disease (Yum et al., 2002) disrupt connexin trafficking by trapping wild-type connexin32 in intracellular organelles. Similarly, when the C231S mutation of Cx36 was studied in non-neuronal cells, it prevented wild-type Cx36 from being trafficked to the cell membrane by trapping it in the Golgi apparatus and endoplasmic reticulum (Placantonakis et al., 2002), the former being the site of hemichannel assembly (Musil and Goodenough, 1993). The aggregation of Cx36 into plaques within dendritic shafts after transduction with the LV coding for the dominant-negative Cx36 mutant may relate to the projection of the secretory pathway into the dendrites of neurons (Horton and Ehlers, 2003) and suggests that Cx36 is assembled into hemichannels in distal dendrites before being trafficked into spines. Ultimately, the Cx36 mutant expressed in the IO prevented the distribution of Cx36 to the normal sites of gap junctions, blocked cytosolic coupling, and suppressed STOs.
The issue of the relationship between synchrony within coupled neural networks and rhythm generation in the brain is fundamental (Connors and Long, 2004). Within the IO and cerebellum, rhythmic firing and spike synchrony have been implicated in the fine control of skilled movement (Llinás, 1974, 1991; Welsh and Llinás, 1997). During a skilled movement, spike rhythmicity and synchrony within the IO occur conjointly and specific patterns of neuronal synchrony are dynamically regulated in time to relate to specific muscle activations (Welsh et al., 1995; Welsh, 2002). Recently, it was reported that the blockade of neuronal synchrony in vivo with intraolivary injection of carbenoxolone degraded the rhythmic output of the IO (Blenkinsop and Lang, 2006), a finding consistent with our present in vitro data after specific genetic manipulation of Cx36. Our experiment demonstrates further that the enabling of STOs by Cx36-mediated gap junctions within the IO may provide the substrate for entraining spike rhythmicity. More generally, our study indicates that a robust and continuous subthreshold electrical rhythm requires network coupling. Additional in vivo experiments using LV-mediated disruption of Cx36 should be able to elucidate the functional significance of neuronal gap junctional coupling in skilled movement and other aspects of brain function for which electrical synchronization has been implicated.
Footnotes
This work was supported by United States Public Health Service Grants NS31224 and RR016858 (J.P.W.) and DK56465 (H.-P.K.) and by the M. J. Murdock Charitable Trust (S.A.A.).
References
- Asghar AUR, Cilia La Corte PF, LeBeau FEN, Al Dawoud M, Reilly SC, Buhl EH, Whittington MA, King AE (2005). Oscillatory activity within rat substantia gelatinosa in vitro: a role for chemical and electrical neurotransmission. J Physiol (Lond) 562:183–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballantyne D, Andrzejewski M, Muckenhoff K, Scheid P (2004). Rhythms, synchrony and electrical coupling in the locus coeruleus. Respir Physiol Neurobiol 143:199–214. [DOI] [PubMed] [Google Scholar]
- Benardo LS, Foster RE (1986). Oscillatory behavior in inferior olive neurons: mechanism, modulation, cell aggregates. Brain Res Bull 17:773–784. [DOI] [PubMed] [Google Scholar]
- Bleasel AF, Pettigrew AG (1992). Development and properties of spontaneous oscillations of the membrane potential in inferior olivary neurons in the rat. Brain Res Dev Brain Res 65:43–50. [DOI] [PubMed] [Google Scholar]
- Blenkinsop TA, Lang EJ (2006). Block of inferior olive gap junctional coupling decreases Purkinje cell complex spike synchrony and rhythmicity. J Neurosci 26:1739–1748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourrat F, Sotelo C (1983). Postnatal development of the inferior olivary complex in the rat. 1. An electron microscopic study of the medial accessory olive. Dev Brain Res 8:291–310. [DOI] [PubMed] [Google Scholar]
- Condorelli DF, Parenti R, Spinella F, Trovato Salinaro A, Belluardo N, Cardile V, Cicirata F (1998). Cloning of a new gap junction gene (Cx36) highly expressed in mammalian brain neurons. Eur J Neurosci 10:1202–1208. [DOI] [PubMed] [Google Scholar]
- Connors BW, Long MA (2004). Electrical synapses in mammalian brain. Annu Rev Neurosci 27:393–418. [DOI] [PubMed] [Google Scholar]
- Devor A, Yarom Y (2002). Electrotonic coupling in the inferior olivary nucleus revealed by simultaneous double patch recordings. J Neurophysiol 87:3048–3058. [DOI] [PubMed] [Google Scholar]
- DeZeeuw CI, Chorev E, Devor A, Manor Y, Van Der Giessen RS, De Jeu MT, Hoogenraad CC, Bijman J, Ruigrok TJ, French P, Jaarsma D, Kistler WM, Meier C, Petrasch-Parwez E, Dermietzel R, Sohl G, Gueldenagel M, Willicke K, Yarom Y (2003). Deformation of network connectivity in the inferior olive of connexin 36-deficient mice is compensated by morphological and electrophysiological changes at the single neuron level. J Neurosci 23:4700–4711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg GS, Moreno AP, Bechberger JF, Hearn SS, Shivers RR, MacPhee J, Zhang Y-C, Naus CCG (1996). Evidence that disruption of connexon particle arrangements in gap junction plaques is associated with inhibition of gap junctional communication by a glycyrrhetinic acid derivative. Exp Cell Res 222:48–53. [DOI] [PubMed] [Google Scholar]
- Goodenough DA, Goliger JA, Paul DL (1996). Connexins, connexons, and intercellular communication. Annu Rev Biochem 65:475–502. [DOI] [PubMed] [Google Scholar]
- Guan X, Wilson S, Schlender KK, Ruch RJ (1996). Gap-junction disassembly and connexin 43 dephosphorylation induced by 18 beta-glycyrrhetinic acid. Mol Carcinog 16:157–164. [DOI] [PubMed] [Google Scholar]
- Hestrin S, Galarreta M (2005). Electrical synapses define networks of neocortical GABAergic neurons. Trends Neurosci 28:304–309. [DOI] [PubMed] [Google Scholar]
- Horton AC, Ehlers MD (2003). Dual modes of endoplasmic reticulum-to-Golgi transport in dendrites revealed by live-cell imaging. J Neurosci 23:6188–6199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishimatsu M, Williams JT (1996). Synchronous activity in locus coeruleus results from dendritic interactions in pericoerulear regions. J Neurosci 16:5196–5204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kano T, Inaba Y, Avoli M (2005). Periodic oscillatory activity in parahippocampal slices maintained in vitro. Neuroscience 130:1041–1053. [DOI] [PubMed] [Google Scholar]
- Kepler T, Marder E, Abbott L (1990). The effect of electrical coupling on the frequency of model neuronal oscillators. Science 248:82–85. [DOI] [PubMed] [Google Scholar]
- Konopacki J, Kowalczyk T, Golebiewski H (2004). Electrical coupling underlies theta oscillations recorded in hippocampal formation slices. Brain Res 1019:270–274. [DOI] [PubMed] [Google Scholar]
- Leznik E, Llinás R (2005). Role of gap junctions in synchronized neuronal oscillations in the inferior olive. J Neurophysiol 94:2447–2456. [DOI] [PubMed] [Google Scholar]
- Llinás R (1974). Eighteenth Bowditch Lecture. Motor aspects of cerebellar control. Physiologist 17:19–46. [PubMed] [Google Scholar]
- Llinás R (1991). The non-continuous basis of movement execution. In: Motor control: concepts and issues (Humphrey DR, Freund H-J, eds) pp. 223–242. New York: Wiley.
- Llinás R, Yarom Y (1981). Electrophysiology of mammalian inferior olivary neurones in vitro. Different types of voltage-dependent ionic conductances. J Physiol (Lond) 315:549–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llinás R, Yarom Y (1986). Oscillatory properties of guinea-pig inferior olivary neurones and their pharmacological modulation: an in vitro study. J Physiol (Lond) 376:163–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llinás R, Baker R, Sotelo C (1974). Electrotonic coupling between neurons in cat inferior olive. J Neurophysiol 37:560–571. [DOI] [PubMed] [Google Scholar]
- Long MA, Deans MR, Paul DL, Connors BW (2002). Rhythmicity without synchrony in the electrically uncoupled inferior olive. J Neurosci 22:10898–10905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manor Y, Rinzel J, Segev I, Yarom Y (1997). Low-amplitude oscillations in the inferior olive: a model based on electrical coupling with heterogeneous channel densities. J Neurophysiol 77:2736–2752. [DOI] [PubMed] [Google Scholar]
- Mitchell JL, Silverman MB, Aicher SA (2004). Rat trigeminal lamina I neurons that project to thalamic or parabrachial nuclei contain the mu-opioid receptor. Neuroscience 128:571–582. [DOI] [PubMed] [Google Scholar]
- Musil LS, Goodenough DA (1993). Multisubunit assembly of an integral plasma membrane channel protein, gap junction connexin43, occurs after exit from the ER. Cell 74:1075–1077. [DOI] [PubMed] [Google Scholar]
- Placantonakis D, Cicirata F, Welsh JP (2002). A dominant negative mutation of neuronal connexin 36 that blocks intercellular permeability. Mol Brain Res 98:15–28. [DOI] [PubMed] [Google Scholar]
- Placantonakis DG, Bukovsky AA, Welsh JP (2004a). Lentiviral expression of a dominant-negative connexin36 mutant in the rat inferior olive disrupts membane potential oscillations. Soc Neurosci Abstr 30:638–8. [Google Scholar]
- Placantonakis DG, Bukovsky AA, Zeng XH, Kiem HP, Welsh JP (2004b). Fundamental role of inferior olive connexin 36 in muscle coherence during tremor. Proc Natl Acad Sci USA 101:7164–7169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rekling JC, Shao XM, Feldman JL (2000). Electrical coupling and excitatory synaptic transmission between rhythmogenic respiratory neurons in the preBötzinger complex. J Neurosci 20: RC113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouach N, Segal M, Koulakoff A, Giaume C, Avignone E (2003). Carbenoxolone blockade of neuronal network activity in culture is not mediated by an action on gap junctions. J Physiol (Lond) 5:729–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozental R, Srinivas M, Spray DC (2001). How to close a gap junction channel. Efficacies and potencies of uncoupling agents. Methods Mol Biol 154:447–476. [DOI] [PubMed] [Google Scholar]
- Sohl G, Maxeiner S, Willecke K (2005). Expression and functions of neuronal gap junctions. Nat Rev Neurosci 6:191–200. [DOI] [PubMed] [Google Scholar]
- Sotelo C, Llinás R, Baker R (1974). Structural study of inferior olivary nucleus of the cat: morphological correlates of electrotonic coupling. J Neurophysiol 37:541–549. [DOI] [PubMed] [Google Scholar]
- Vessey JP, Lalond MR, Mizan HA, Welch NC, Kelly ME, Barnes S (2004). Carbenoxolone inhibition of voltage-gated Ca channels and synaptic transmission in the retina. J Neurophysiol 92:1252–1256. [DOI] [PubMed] [Google Scholar]
- Welsh JP (2002). Functional significance of climbing-fiber synchrony: a population coding and behavioral analysis. Ann NY Acad Sci 978:188–204. [DOI] [PubMed] [Google Scholar]
- Welsh JP, Llinás R (1997). Some organizing principles for the control of movement based on olivocerebellar physiology. Prog Brain Res 11:449–461. [DOI] [PubMed] [Google Scholar]
- Welsh JP, Lang EJ, Sugihara I, Llinás R (1995). Dynamic organization of motor control within the olivocerebellar system. Nature 374:453–457. [DOI] [PubMed] [Google Scholar]
- Whittington MA, Traub RD (2003). Interneuron diversity series: inhibitory interneurons and network oscillations in vitro. Trends Neurosci 26:676–682. [DOI] [PubMed] [Google Scholar]
- In: Statistical principles in experimental design (1979). New York: McGraw-Hill.
- Yarom Y (1991). Rhythmogenesis in a hybrid system: interconnecting an olivary neuron to an analog network of coupled oscillators. Neuroscience 44:263–275. [DOI] [PubMed] [Google Scholar]
- Yum SW, Kloepa KA, Shuman S, Scherer SS (2002). Diverse trafficking abnormalities of connexin32 mutants causing CMTX. Neurobiol Dis 11:43–52. [DOI] [PubMed] [Google Scholar]