

Abstract
The amygdala is one of several brain regions suspected to be pathological in autism. Previously, we found that young children with autism have a larger amygdala than typically developing children. Past qualitative observations of the autistic brain suggest increased cell density in some nuclei of the postmortem autistic amygdala. In this first, quantitative stereological study of the autistic brain, we counted and measured neurons in several amygdala subdivisions of 9 autism male brains and 10 age-matched male control brains. Cases with comorbid seizure disorder were excluded from the study. The amygdaloid complex was outlined on coronal sections then partitioned into five reliably defined subdivisions: (1) lateral nucleus, (2) basal nucleus, (3) accessory basal nucleus, (4) central nucleus, and (5) remaining nuclei. There is no difference in overall volume of the amygdala or in individual subdivisions. There are also no changes in cell size. However, there are significantly fewer neurons in the autistic amygdala overall and in its lateral nucleus. In conjunction with the findings from previous magnetic resonance imaging studies, the autistic amygdala appears to undergo an abnormal pattern of postnatal development that includes early enlargement and ultimately a reduced number of neurons. It will be important to determine in future studies whether neuron loss in the amygdala is a consistent characteristic of autism and whether cell loss occurs in other brain regions as well.
Keywords: autism, neuropathology, stereology, neuronal density, medial temporal lobe, neuroanatomy, amygdaloid complex
Introduction
Autism is a lifelong neurodevelopmental disorder characterized by impairments in social interaction, deficits in communication, and a restricted repertoire of interests. The cause(s) of autism remains unknown and associated neuropathology has not been clearly established. Given the complexity of symptoms seen in autism, it is probable that several brain areas are dysfunctional. The amygdala, which is involved in the production and recognition of emotions such as fear, has been consistently implicated in the pathophysiology of autism (Baron-Cohen et al., 2000). Structural magnetic resonance imaging (MRI) studies indicate an abnormal growth pattern in the amygdala marked by precocious enlargement (Sparks et al., 2002; Schumann et al., 2004).
Bauman and Kemper (1985, 1994) were the first to report abnormalities in the amygdala in postmortem autism cases. Serial histological sections stained for cell bodies through the amygdala of a 29-year-old male with autism and comorbid seizure disorder were viewed side-by-side with sections from a 25-year-old control male. Density measures were taken in the central part of each amygdaloid nucleus (Bauman and Kemper, 1985). Increased cell-packing density and reduced cell sizes were observed in the central, medial, and cortical nuclei in the autism case. Kemper and Bauman (1993) extended their initial case study with qualitative analyses of five additional cases of autism (four males and one female; ages 9, 10, 12, 22, and 28) and confirmed their initial observations. Kemper and Bauman speculated that the patches of small, densely packed neurons were symptomatic of arrested development in portions of the amygdala. The results are complicated, however, by the fact that four of the six cases also had a seizure disorder. It is well established that epilepsy alone can affect the structure of the amygdala (Pitkanen et al., 1998). More recent postmortem studies of the autistic brain have only superficially evaluated amygdala pathology (Bailey et al., 1998).
Bauman and Kemper (1985, 1994) described changes in neuronal density, or neurons per unit volume, in portions of the autistic amygdala. Recent studies, however, have raised methodological concerns about the interpretation of density measurements as an indication of neuropathology. Haug et al. (1994), for example, found that the process of tissue fixation results in differential shrinkage of brains at different ages; shrinkage was inversely proportional to age. The implication of this finding is that differences in the density of neurons may reflect changes in the volume of the tissue rather than changes in total cell number. The only way to unambiguously interpret pathological changes in neuron number is to actually count a representative sample of neurons in a defined volume (West et al., 1991). The goal of the current study was to measure neuron number, regional volume, and mean neuronal cross-sectional area in the entire amygdala and in individual amygdaloid nuclei. We have analyzed postmortem brains from 9 male cases of autism and compared these with postmortem brains from 10 typically developing age-matched male controls using systematic stereological techniques. None of the cases analyzed in this study had comorbid epilepsy during life.
Materials and Methods
This study was approved by the Institutional Review Board of the University of California, Davis. Nineteen postmortem brains, 9 from individuals with autism and 10 from age-matched typically developing controls, were collected for this study. Clinical characteristics are summarized in Table 1. Cases were free of seizure disorder or other major neurological disorders (e.g., schizophrenia, blindness). The diagnosis of autism was confirmed by postmortem use of the Autism Diagnostic Interview–Revised (ADI-R) in seven of the nine cases (Lord et al., 1994). No other clinical information is available for these cases.
Table 1.
Clinical characteristics of postmortem cases
| Age at death (years) | Diagnosis | Postmortem interval (hours) | Hemisphere | Cause of death |
|---|---|---|---|---|
| 10 | Autism | 24 | Right | Choking |
| 11 | Control | 30 | Left | Cardiac arrest |
| 14 | Control | 19 | Right | Drowning |
| 15 | Autism | 3 | Right | Pneumonia |
| 15 | Autism | 12 | Right | Drowning |
| 16 | Autism | 47 | Right | Stopped breathing |
| 17 | Control | 24 | Left | Motor vehicle accident |
| 18 | Control | 20 | Left | Drowning |
| 20 | Autism | 24 | Right | Motor vehicle accident |
| 24 | Control | 19 | Right | Chest gunshot wound |
| 25 | Control | 19 | Left | Drowning |
| 27 | Control | 21 | Right | Hanging |
| 27 | Control | 21 | Left | Cardiac arrest |
| 28 | Autism | 18 | Left | Carbon monoxide |
| 32 | Control | 17 | Left | Arteriosclerosis |
| 35 | Autism | 17 | Left | Pneumonia |
| 36 | Autism | 24 | Left | Cardiac arrest |
| 44 | Autism | 31 | Left | Heart disease |
| 44 | Control | 26 | Left | Pneumonia |
Tissue preparation.
After removal of the brain from the skull, each brain was immersed in 10% buffered formalin for at least 8 weeks before transfer to our laboratory. A proton density-weighted MRI scan was collected on each of the 19 brains with a 1.5 T magnet at the University of California, Davis, Imaging Research Center (Schumann et al., 2001). A 4 cm block containing the entire rostrocaudal extent of the amygdala was cut from one hemisphere and placed into a cryoprotectant solution (10% glycerol in 0.1 m phosphate buffer for 2 d and 20% glycerol for 5 d) in preparation for freezing. The tissue block was frozen with 2-methyl butane (isopentane) and serially sectioned into six series of 50-μm-thick sections and two series of 100-μm-thick sections. One 100 μm series was stained with 0.25% thionin (standard Nissl method) for additional analyses.
Stereological measurements.
All measurements were made using a Nikon Eclipse 600 microscope attached to an Optronics camera with Microfire software, which was connected to a Dell Precision 450 workstation using Stereoinvestigator software (MicroBrightField, Williston, VT). The entire rostrocaudal extent of the human amygdala was defined on every 100 μm Nissl section in which it was present; this was ∼25 sections per case. The amygdala was further partitioned into five subdivisions (Fig. 1): (1) lateral nucleus (∼20 sections), (2) basal nucleus (∼20 sections), (3) accessory basal nucleus (∼20 sections), (4) central nucleus (∼10 sections), and (5) remaining nuclei (∼25 sections). The remaining amygdala nuclei were defined as the nuclei that were enclosed within the outer border of the amygdala after subtracting the lateral, basal, accessory basal, and central nuclei. The remaining nuclei included the anterior cortical nucleus, anterior amygdaloid area, nucleus of the lateral olfactory tract, periamygdaloid cortex (Fig. 1a), medial nucleus, posterior cortical nucleus, amygdalohippocampal area, and intercalated nuclei (Fig. 1c). A detailed protocol for defining the amygdala boundaries has been provided previously (Schumann and Amaral, 2005).
Figure 1.
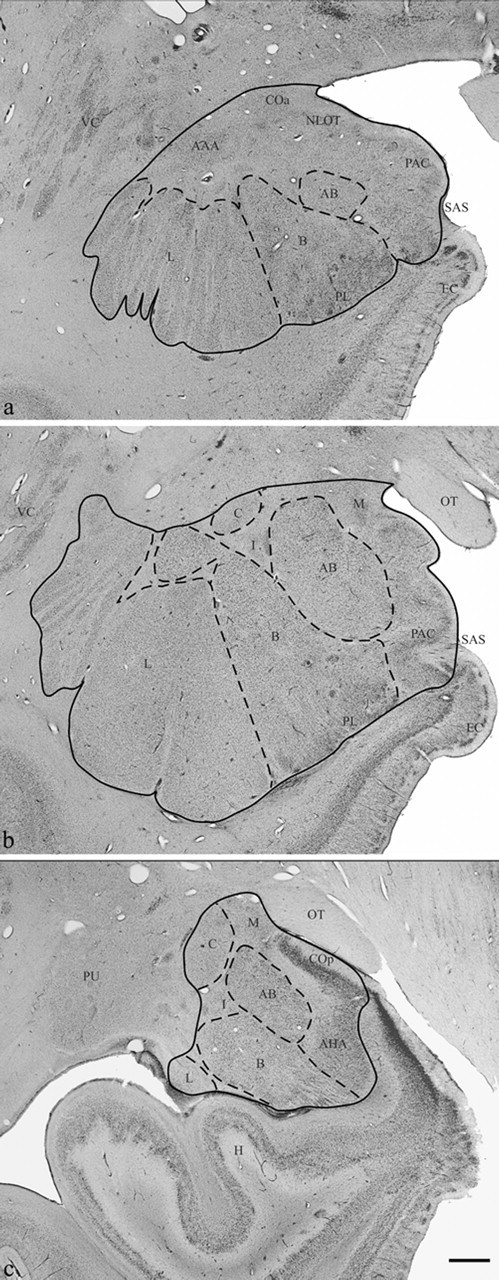
Brightfield photomicrograph of Nissl-stained coronal sections through rostral (a), midrostrocaudal (b), and caudal (c) levels of the amygdala. AAA, Anterior amygdaloid area; AB, accessory basal nucleus; AHA, amygdalohippocampal area; B, basal nucleus; C, central nucleus; COa, anterior cortical nucleus; COp, posterior cortical nucleus; EC, entorhinal cortex; H, hippocampus; I, intercalated nuclei; L, lateral nucleus; M, medial nucleus; NLOT, nucleus of the lateral olfactory tract; OT, optic tract; PAC, periamygdaloid cortex; PL, paralaminar nucleus; PU, putamen; SAS, semiannular sulcus; VC, ventral claustrum. Scale bar, 2 mm.
The postprocessing volume of each region was estimated using the Cavalieri method. This tool was implemented by randomly overlaying a lattice of points on each section and counting the number of points of the lattice located within the region being measured. By knowing the following parameters: the number of points counted within the region, the grid spacing area (0.04 mm2 or 200 μm between points), and the cut thickness (100 μm) of the histological sections, we were able to calculate the volume of each region.
An optical fractionator method was used to estimate the number of neurons in each subdivision of the amygdaloid complex at high magnification with a 100× (1.30 numerical aperture) oil objective (Fig. 2). The optical fractionator method (West et al., 1991) is a three-dimensional probe placed through the reference space, which was used to estimate the number of neurons in each subdivision of the amygdala. This method is independent of volume measurements and thus unaffected by tissue shrinkage. Parameters for applying the optical fractionator method for counting cells in the amygdala nuclei have been described previously (Schumann and Amaral, 2005). Briefly, a neuron was counted if the nucleus associated with that neuron first came into focus within the optical disector counting frame (i.e., a neuron was not counted if the nucleus associated with that neuron came into focus within the guard zone or if the nucleus was touching the left or bottom side of the disector frame). Neurons were distinguished from glia on the basis of size, presence of a visible rim of cytoplasm surrounding the nucleus, and a prominent nucleolus. The optical disector height (thickness) was 9 μm with a 3 μm top and bottom guard zone. The mean section thickness measured at every other counting frame site was used for final calculation of neuron number for each subdivision of the amygdaloid complex. The estimation of neuron number was calculated by the product of the total number of neurons counted at the disectors, the section sampling fraction (number of sections sampled per total sections), the area sampling fraction (area of section sampled per total area), and the thickness sampling fraction (section thickness per disector height).
Figure 2.

Photomicrograph of a Nissl-stained section illustrating the lateral nucleus at 100× (the magnification used for the optical fractionator) from a 15-year-old with autism. Scale bar, 10 μm.
The cross-sectional areas of neurons were also measured using a Nucleator method (Gundersen, 1988). Each time a neuron was counted with the optical fractionator, a grid of six radially extending lines with origin at the nucleus was overlaid onto the neuron. The point at which each line intersected the boundary of the neuron was marked. The mean line length from the nucleus to the border of the neuron was used as the radius to calculate neuronal cross-sectional area.
Statistical analyses.
Two-tailed t tests were performed to identify statistically significant differences between autism and control cases. Comparisons were made of the age of the subjects at death, volume of the total amygdala and amygdala subdivisions, neuron number, neuron cross-sectional area, and neuronal density (neurons per unit volume).
Results
There was no difference in the mean age at death of the autism and control cases (Table 1). There were no group differences in the volume of the total amygdala or of its subdivisions (Table 2). Nor were there differences in the cross-sectional areas of measured neurons. However, there were significantly fewer neurons in the autistic amygdala compared with controls (p = 0.032). While there was a trend for several amygdaloid nuclei to have fewer neurons in the autistic cases, the decrease in the lateral nucleus reached significance (p = 0.029) (Fig. 3). There was also a trend toward fewer neurons in the “remaining nuclei” subdivision in the autism group relative to controls (p = 0.061).
Table 2.
Mean stereological measures ± SD
| Control (n = 10) | Autism (n = 9) | |
|---|---|---|
| Age (years) | 23.9 ± 10 | 24.3 ± 12 |
| Volume (mm3) | ||
| Lateral nucleus | 452 ± 43 | 456 ± 86 |
| Basal nucleus | 343 ± 30 | 334 ± 56 |
| Accessory basal nucleus | 152 ± 16 | 151 ± 27 |
| Central nucleus | 34 ± 6 | 34 ± 8 |
| Remaining nuclei | 399 ± 48 | 396 ± 77 |
| Total amygdala | 1380 ± 114 | 1371 ± 224 |
| Neuron number (×106) | ||
| Lateral nucleus | 4.00 ± 0.40 | 3.47 ± 0.57* |
| Basal nucleus | 3.24 ± 0.52 | 2.88 ± 0.58 |
| Accessory basal nucleus | 1.28 ± 0.20 | 1.10 ± 0.26 |
| Central nucleus | 0.36 ± 0.08 | 0.35 ± 0.05 |
| Remaining nuclei | 3.34 ± 0.53 | 2.94 ± 0.32 |
| Total amygdala | 12.21 ± 1.28 | 10.74 ± 1.47* |
| Neuronal density (no. per cm3) | ||
| Lateral nucleus | 8.98 ± 0.80 | 7.78 ± 1.44* |
| Basal nucleus | 9.38 ± 1.18 | 8.71 ± 1.52 |
| Accessory basal nucleus | 8.61 ± 0.85 | 7.38 ± 1.74 |
| Central nucleus | 10.61 ± 1.56 | 10.58 ± 2.41 |
| Remaining nuclei | 8.22 ± 0.80 | 7.70 ± 1.99 |
| Total amygdala | 8.87 ± 0.53 | 7.99 ± 1.55 |
| Neuronal area (μm2) | ||
| Lateral nucleus | 257 ± 19 | 253 ± 28 |
| Basal nucleus | 265 ± 24 | 268 ± 32 |
| Accessory basal nucleus | 273 ± 21 | 282 ± 25 |
| Central nucleus | 185 ± 15 | 174 ± 12 |
| Remaining nuclei | 211 ± 14 | 216 ± 19 |
| Total amygdala | 238 ± 15 | 239 ± 19 |
*Measurement significantly lower than controls at p < 0.05.
Figure 3.

Number of neurons in five subdivisions of the amygdala of autism (red) and control (blue) brains. The asterisk indicates significant difference (p = 0.03) in neuron number between autism and control lateral nuclei.
Subject age at death was not correlated with the number of neurons in the total amygdala (Fig. 4) or in any of its subdivisions. When age was included as a covariate, the number of neurons in the total amygdala (p = 0.037) and in the lateral nucleus (p = 0.035) of autism cases remained significantly lower. The density of neurons in the lateral nucleus was lower in the autism group relative to controls (p = 0.049).
Figure 4.

Bivariate scattergram of the number of neurons in the total amygdala of autism (red) and control (blue) brains by age.
Discussion
This is the first study to use systematic, stereological sampling techniques to quantitatively measure the number of neurons in the autistic amygdala. The major finding is that the total amygdala and its lateral nucleus have significantly fewer neurons in the autistic brains compared with age-matched controls.
Before the current study, the amygdala of only seven autism cases had been systematically examined (Bauman and Kemper, 1985; Kemper and Bauman, 1993; Bailey et al., 1998). Bauman and Kemper reported decreased size and increased density of neurons in the medial, central, and cortical nuclei. We did not replicate these findings. There are several fundamental differences between the reports of Bauman and Kemper and the current study. First, the previous studies made qualitative observations of neuronal density and the actual numbers of neurons were not determined. The current study, in contrast, used modern stereological methods to obtain quantitative measurements of true neuron number in the entire amygdala and five reliably defined subdivisions. Second, most of the brains from the autism cases in the previous studies had a comorbid seizure disorder. There is substantial evidence that temporal lobe epilepsy is associated with pathology, including cell loss, of the amygdala (Pitkanen et al., 1998; Vernet et al., 2000). We eliminated this potentially confounding factor by only analyzing postmortem brains from individuals with no history of seizures.
Magnetic resonance imaging studies of typically developing male children indicate that the amygdala undergoes a prolonged postnatal increase in volume and reaches an adult size in late adolescence (Giedd et al., 1996; Giedd, 1997; Schumann et al., 2004). In contrast, the amygdala in autistic boys is at adult size by ∼8 years of age and does not enlarge thereafter (Sparks et al., 2002; Schumann et al., 2004). One possibility to account for the greater initial volume of the autistic amygdala is that there is an exuberant proliferative phase with the generation of a greater number of neurons. This might be reflected in a greater number of neurons in the mature, postmortem brain. We found, however, that there were actually fewer neurons in the amygdala of autistic individuals compared with age-matched controls, although there was no difference in overall volume and neuron size was indistinguishable from controls.
What might account for the observed lower number of neurons in the postmortem, autistic amygdala? Two obvious alternatives are the following: (1) fewer neurons were generated during early development, or (2) a normal or even excessive number of neurons was generated initially but some of these have subsequently degenerated. Unfortunately, we do not currently have data to support or reject either of these possibilities. For example, the early increased size of the autistic amygdala that was identified in MRI studies (Sparks et al., 2002; Schumann et al., 2004) cannot be used as evidence of increased neuronal proliferation because the size difference could be accounted for by an increase in the number of glial cells or fibers rather than differences in neuron number. The resolution of this issue would require similar stereological studies of the amygdala in neonatal autistic subjects. This is currently impractical because the diagnosis of autism is typically made when a child is 3 years of age or older. It is interesting to note, however, that both studies (Sparks et al., 2002; Schumann et al., 2004) performed with young autistic children indicate that the amygdala is larger than normal, whereas studies performed with adolescents (Sparks et al., 2002; Schumann et al., 2004) and young adults (Aylward et al., 1999; Pierce et al., 2001) indicate either no difference or a smaller amygdala. This raises the possibility that the amygdala has a normal or perhaps even increased number of neurons in early postnatal life and that a regressive process takes place at some later time.
Is there a unifying hypothesis that might account for the findings of an increased volume of the autistic amygdala in infancy and a smaller number of neurons in the mature brain? Although quite speculative at present, one intriguing hypothesis relates to the concept of “allostatic load” (McEwen, 2003, 2004). As espoused by McEwen, stress promotes adaptation (allostasis) through a variety of mechanisms such as the release of stress hormones. However, persistent stressors can lead to damage to multiple body systems (allostatic overload). This hypothesis is based on several premises. One is that precocious growth of brain regions such as the frontal lobe and amygdala is a characteristic biological feature of autism (Redcay and Courchesne, 2005) perhaps through a fundamental defect in regulatory mechanisms of ontogenesis. Precocious growth of the amygdala may lead to early and excessive functional activity. A second consideration is the abundance of evidence from animal (LeDoux, 2000; Davis et al., 2003) and human (Adolphs et al., 1994, 1995; Buchel and Dolan, 2000) studies that implicate the amygdala in the detection of danger and the production of fear and anxiety. One component of the response to fear is the regulation by the amygdala of both autonomic activity and corticotropin and cortisol production through its descending connections from the central nucleus (Schulkin et al., 1994). In other words, the greater the output from the amygdala, the larger the stress response. Interestingly, the amygdala in children with an anxiety disorder is both larger (De Bellis et al., 2000) and more active (Thomas et al., 2001) than that of age-matched controls. Even in older autistic individuals, Dalton et al. (2005) have recently demonstrated that the amygdala is more active when provoked by images of faces, a stimulus category that is typically avoided by people with autism. This is consistent with the literature indicating that anxiety is a common comorbid feature of autism (Muris et al., 1998).
Thus, one possibility is that a biological defect inherent to autism leads to the production of a larger and more active amygdala. The more active amygdala produces a heightened level of fear and anxiety typical of autism (Muris et al., 1998) as well as a heightened and chronic stress response. Over time, the heightened stress response could possibly have damaging effects leading to the loss of neurons and a smaller amygdala. This scenario is not without precedent, because in depression, early stages are associated with amygdala enlargement (Frodl et al., 2002), whereas long-term depression is associated with atrophy of the amygdala (Sheline et al., 1998). One caveat to this interpretation of our data is the inherent heterogeneity of the human brain and the substantial variability within the diagnosis of autism. Although there was a significant reduction in the number of neurons in the autistic amygdala as a group, not all individuals with autism had fewer neurons in the amygdala relative to their age-matched typically developing counterparts. Our data are consistent with the view that there may be many causes of autism and many neurological sequelae. It will be important to perform similar quantitative analyses on larger numbers of postmortem brains to determine whether amygdaloid neuron loss is a consistent and important pathological characteristic of autism.
To conclude, this is the first postmortem study to provide quantitative evidence that the amygdala is structurally abnormal in autism. In conjunction with the findings from our MRI studies (Schumann et al., 2004), the amygdala appears to undergo an abnormal pattern of development that includes early enlargement and a decreased number of neurons in adulthood. No study to date has used modern stereological techniques in any area of the autistic brain to determine whether there are differences in neuron number. Therefore, it is unclear whether the finding of decreased neuron numbers in the autistic amygdala is idiosyncratic for this brain region or a more generalized feature of the autistic brain that will eventually be observed in other regions. As mentioned above, many areas of the brain appear to undergo abnormal overgrowth early in development, including the frontal lobe and amygdala (Redcay and Courchesne, 2005). Unfortunately, sparse tissue availability has made postmortem studies difficult to perform. Nonetheless, future stereological studies performed in other brain regions will define the generality of the findings reported here in the autistic brain.
Footnotes
This work was supported by the National Alliance for Autism Research, National Institutes of Health Grant MH41479, and the M.I.N.D. Institute. We thank Gwyndolen Harburg and John Sylvain for their technical assistance and Dr. Jane Pickett at the Autism Tissue Program for facilitating tissue collection. Tissue samples were provided by the National Institute of Child Health and Human Development Brain and Tissue Bank for Developmental Disorders at the University of Miami from director Dr. Carol Petito; the Harvard Brain Tissue Resource Center from director Dr. Francine Benes; the University of California, Davis, School of Medicine from Dr. Claudia Greco; and the University of California, San Diego, from Dr. Eric Courchesne. We also thank Dr. Richie Davidson for helpful conversations leading to the hypothesis proposed at the end of this paper. We are deeply indebted to the families of our donors who have made this study possible.
References
- Adolphs R, Tranel D, Damasio H, Damasio A (1994). Impaired recognition of emotion in facial expressions following bilateral damage to the human amygdala. Nature 372:669–672. [DOI] [PubMed] [Google Scholar]
- Adolphs R, Tranel D, Damasio H, Damasio AR (1995). Fear and the human amygdala. J Neurosci 15:5879–5891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aylward EH, Minshew NJ, Goldstein G, Honeycutt NA, Augustine AM, Yates KO, Barta PE, Pearlson GD (1999). MRI volumes of amygdala and hippocampus in non-mentally retarded autistic adolescents and adults. Neurology 53:2145–2150. [DOI] [PubMed] [Google Scholar]
- Bailey A, Luthert P, Dean A, Harding B, Janota I, Montgomery M, Rutter M, Lantos P (1998). A clinicopathological study of autism. Brain 121:889–905. [DOI] [PubMed] [Google Scholar]
- Baron-Cohen S, Ring HA, Bullmore ET, Wheelwright S, Ashwin C, Williams SC (2000). The amygdala theory of autism. Neurosci Biobehav Rev 24:355–364. [DOI] [PubMed] [Google Scholar]
- Bauman M, Kemper TL (1985). Histoanatomic observations of the brain in early infantile autism. Neurology 35:866–874. [DOI] [PubMed] [Google Scholar]
- Bauman M, Kemper TL (1994). Neuroanatomic observations of the brain in autism. In: The neurobiology of autism (Bauman M, Kemper TL, eds) pp. 119–145. Baltimore: Johns Hopkins UP.
- Buchel C, Dolan RJ (2000). Classical fear conditioning in functional neuroimaging. Curr Opin Neurobiol 10:219–223. [DOI] [PubMed] [Google Scholar]
- Dalton KM, Nacewicz BM, Johnstone T, Schaefer HS, Gernsbacher MA, Goldsmith HH, Alexander AL, Davidson RJ (2005). Gaze fixation and the neural circuitry of face processing in autism. Nat Neurosci 8:519–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis M, Walker DL, Myers KM (2003). Role of the amygdala in fear extinction measured with potentiated startle. Ann NY Acad Sci 985:218–232. [DOI] [PubMed] [Google Scholar]
- De Bellis MD, Casey BJ, Dahl RE, Birmaher B, Williamson DE, Thomas KM, Axelson DA, Frustaci K, Boring AM, Hall J, Ryan ND (2000). A pilot study of amygdala volumes in pediatric generalized anxiety disorder. Biol Psychiatry 48:51–57. [DOI] [PubMed] [Google Scholar]
- Frodl T, Meisenzahl E, Zetzsche T, Bottlender R, Born C, Groll C, Jager M, Leinsinger G, Hahn K, Moller HJ (2002). Enlargement of the amygdala in patients with a first episode of major depression. Biol Psychiatry 51:708–714. [DOI] [PubMed] [Google Scholar]
- Giedd JN (1997). Normal development. In: Child and adolescent psychiatric clinics of North America neuroimaging (Peterson BS, ed.) pp. 265–282. Philadelphia: Saunders.
- Giedd JN, Vaituzis AC, Hamburger SD, Lange N, Rajapakse JC, Kaysen D, Vauss YC, Rapoport JL (1996). Quantitative MRI of the temporal lobe, amygdala, and hippocampus in normal human development: ages 4–18 years. J Comp Neurol 366:223–230. [DOI] [PubMed] [Google Scholar]
- Gundersen HJ (1988). The nucleator. J Microsc 151:3–21. [DOI] [PubMed] [Google Scholar]
- Haug JS, Goldner CM, Yazlovitskaya EM, Voziyan PA, Melnykovych G (1994). Directed cell killing (apoptosis) in human lymphoblastoid cells incubated in the presence of farnesol: effect of phosphatidylcholine. Biochim Biophys Acta 1223:133–140. [DOI] [PubMed] [Google Scholar]
- Kemper TL, Bauman ML (1993). The contribution of neuropathologic studies to the understanding of autism. Neurol Clin 11:175–187. [PubMed] [Google Scholar]
- LeDoux JE (2000). Emotion circuits in the brain. Annu Rev Neurosci 23:155–184. [DOI] [PubMed] [Google Scholar]
- Lord C, Rutter M, Le Couteur A (1994). Autism Diagnostic Interview–Revised: a revised version of a diagnostic interview for caregivers of individuals with possible pervasive developmental disorders. J Autism Dev Disord 24:659–685. [DOI] [PubMed] [Google Scholar]
- McEwen BS (2003). Mood disorders and allostatic load. Biol Psychiatry 54:200–207. [DOI] [PubMed] [Google Scholar]
- McEwen BS (2004). Protection and damage from acute and chronic stress: allostasis and allostatic overload and relevance to the pathophysiology of psychiatric disorders. Ann NY Acad Sci 1032:1–7. [DOI] [PubMed] [Google Scholar]
- Muris P, Steerneman P, Merckelbach H, Holdrinet I, Meesters C (1998). Comorbid anxiety symptoms in children with pervasive developmental disorders. J Anxiety Disord 12:387–393. [DOI] [PubMed] [Google Scholar]
- Pierce K, Muller RA, Ambrose J, Allen G, Courchesne E (2001). Face processing occurs outside the fusiform “face area” in autism: evidence from functional MRI. Brain 124:2059–2073. [DOI] [PubMed] [Google Scholar]
- Pitkanen A, Tuunanen J, Kalviainen R, Partanen K, Salmenpera T (1998). Amygdala damage in experimental and human temporal lobe epilepsy. Epilepsy Res 32:233–253. [DOI] [PubMed] [Google Scholar]
- Redcay E, Courchesne E (2005). When is the brain enlarged in autism? A meta-analysis of all brain size reports. Biol Psychiatry 58:1–9. [DOI] [PubMed] [Google Scholar]
- Schulkin J, McEwen BS, Gold PW (1994). Allostasis, amygdala, and anticipatory angst. Neurosci Biobehav Rev 18:385–396. [DOI] [PubMed] [Google Scholar]
- Schumann CM, Amaral DG (2005). Stereological estimation of the number of neurons in the human amygdaloid complex. J Comp Neurol 491:320–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumann CM, Buonocore MH, Amaral DG (2001). Magnetic resonance imaging of the post-mortem autistic brain. J Autism Dev Disord 31:561–568. [DOI] [PubMed] [Google Scholar]
- Schumann CM, Hamstra J, Goodlin-Jones BL, Lotspeich LJ, Kwon H, Buonocore MH, Lammers CR, Reiss AL, Amaral DG (2004). The amygdala is enlarged in children but not adolescents with autism; the hippocampus is enlarged at all ages. J Neurosci 24:6392–6401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheline YI, Gado MH, Price JL (1998). Amygdala core nuclei volumes are decreased in recurrent major depression. NeuroReport 9:2023–2028. [DOI] [PubMed] [Google Scholar]
- Sparks BF, Friedman SD, Shaw DW, Aylward EH, Echelard D, Artru AA, Maravilla KR, Giedd JN, Munson J, Dawson G, Dager SR (2002). Brain structural abnormalities in young children with autism spectrum disorder. Neurology 59:184–192. [DOI] [PubMed] [Google Scholar]
- Thomas KM, Drevets WC, Dahl RE, Ryan ND, Birmaher B, Eccard CH, Axelson D, Whalen PJ, Casey BJ (2001). Amygdala response to fearful faces in anxious and depressed children. Arch Gen Psychiatry 58:1057–1063. [DOI] [PubMed] [Google Scholar]
- Vernet O, Farmer JP, Montes JL, Villemure JG, Meagher-Villemure K (2000). Dysgenetic mesial temporal sclerosis: an unrecognized entity. Childs Nerv Syst 16:719–723. [DOI] [PubMed] [Google Scholar]
- West MJ, Slomianka L, Gundersen HJ (1991). Unbiased stereological estimation of the total number of neurons in the subdivisions of the rat hippocampus using the optical fractionator. Anat Rec 231:482–497. [DOI] [PubMed] [Google Scholar]