

Abstract
The DNA damage response is a network of signaling pathways that affects many aspects of cellular metabolism after the induction of DNA damage. The primary transducer of the cellular response to the double-strand break, a highly cytotoxic DNA lesion, is the nuclear protein kinase ataxia telangiectasia (A-T) mutated (ATM), which phosphorylates numerous effectors that play key roles in the damage response pathways. Loss or inactivation of ATM leads to A-T, an autosomal recessive disorder characterized by neuronal degeneration, particularly the loss of cerebellar granule and Purkinje cells, immunodeficiency, genomic instability, radiosensitivity, and cancer predisposition. The reason for the cerebellar degeneration in A-T is not clear. It has been ascribed by several investigators to cytoplasmic functions of ATM that may not be relevant to the DNA damage response. We set out to examine the subcellular localization of ATM and characterize the ATM-mediated damage response in mouse cerebellar neurons. We found that ATM is essentially nuclear in these cells and that various readouts of the ATM-mediated damage response are similar to those seen in commonly used cell lines. These include the autophosphorylation of ATM, which marks its activation, and phosphorylation of several of its downstream substrates. Importantly, all of these responses are detected in the nuclei of granule and Purkinje cells, suggesting that nuclear ATM functions in these cells similar to other cell types. These results support the notion that the cerebellar degeneration in A-T patients results from defective DNA damage response.
Keywords: ataxia-telangiectasia, DNA damage response, ATM, cerebellum, Purkinje cells, cerebellar granule neurons
Introduction
Living cells are continuously exposed to exogenous and endogenous agents that damage their DNA. The many types of DNA lesions that are induced by these insults are rapidly detected, and an intricate web of signaling pathways, the DNA damage response, is activated. Hallmarks of this response are the activation of cell-cycle checkpoints and the appropriate DNA repair pathways, which lead to cellular survival or, in certain contexts, initiation of apoptotic programs. The DNA damage response is a hierarchical process that is executed through a series of steps. DNA lesions are detected by sensor proteins that recognize the lesions or chromatin alterations that follow DNA damage, and transducer molecules convey the damage signal to downstream effectors. It is this relay system from transducers to effectors that enables a single DNA lesion to modulate numerous pathways. One of the most cytotoxic DNA lesions is the double-strand break (DSB). Even a few DSBs can quickly set in motion a rapidly mounting, vigorous DNA damage response (Matsuura et al., 1998; Varon et al., 1998; van Gent et al., 2001; D'Amours and Jackson, 2002; Bassing and Alt, 2004).
The transducers of the DNA damage response belong to a conserved family of proteins that contain a phosphatidylinositol 3-kinase-like domain, most of which possess serine/threonine kinase activity (Abraham, 2001, 2004; Durocher and Jackson, 2001; Khanna et al., 2001; Shiloh, 2001; Shiloh and Kastan, 2001). Among the members of this family are ataxia-telangiectasia (A-T) mutated (ATM) and the DNA-dependent protein kinase (DNA-PK), which respond primarily to DSBs that are involved in the nonhomologous end-joining pathway of DSB repair, and ataxia telangiectasia Rad 3-related (ATR), which responds primarily to UV damage and stalled replication forks but also shares with ATM substrates in the DSB response pathway. After DSB induction, ATM is rapidly activated in a process of autophosphorylation and conversion of inert ATM homodimers or multimers to active monomers (Bakkenist and Kastan, 2003). Active ATM then phosphorylates a large array of substrates, each of which in turn modulates a damage response pathway. ATM was identified as the product of a gene mutated in the human genetic disease A-T. A-T is characterized by progressive neuronal degeneration, primarily loss of Purkinje and granule cells in the cerebellum, which leads to general motor dysfunction, immunodeficiency, genome instability, extreme radiosensitivity, and high incidence of lymphoreticular malignancies (Lavin and Shiloh, 1997; Abramova et al., 2000; Chun and Gatti, 2004).
The reason for the neuronal degeneration in A-T has been the subject of debate. Several studies claimed that Atm resides in the cytoplasm in cerebellar neurons and attributed the loss of these cells in A-T patients to a deficiency of a cytoplasmic function of ATM (Oka and Takashima, 1998; Kuljis et al., 1999; Barlow et al., 2000). Here we show that Atm is predominantly nuclear in these cells, and all readouts of the Atm-dependent DNA damage response in cerebellar neurons are similar to those seen in commonly used cell lines.
Materials and Methods
Mice.
Atm+/− mice (Barlow et al., 1996) were a generous gift from Dr. Anthony Wynshaw-Boris (University of California, San Diego, CA). Offspring of these mice were genotyped using PCR-based assays based on mouse-tail DNA prepared with the GenElute Mammalian Genomic DNA Miniprep kit (Sigma, St. Louis, MO).
Mice were irradiated using a 6 MV x-ray machine with x-ray doses of 20 Gy. Mice were killed at different time points after ionizing radiation (IR), and the brains were dissected, fixed in 4% formaldehyde in 0.1 m PBS for 24 h, and exposed to sucrose gradient (7, 14, and 21%) for at least 12 h. Brains were frozen in JUNG tissue freezing medium (Leica, Wetzlar, Germany) using dry ice and hexane, after which 12 μm sagittal sections were prepared for immunohistochemical analysis.
Cerebellar granule neurons.
Cultures highly enriched for granule neurons were established from cerebella of 8-d-old mice (Atm+/+ and Atm−/−) as described previously (Nardi et al., 1997).
Organotypic slice cultures from the mouse cerebellum.
Organotypic slice cultures have better preservation of neuronal structure and cell–cell interaction than dissociated cultures. Cultures were prepared according to Noraberg et al. (1999) and Inamura et al. (2000) from 12-d-old wild-type (WT) and Atm−/− mice. Brain slices 400-μm-thick were mounted on Millicell-CM, PICM 030 50 (sterilized culture plate insert), 0.4 μm, 25 mm (Millipore, Bedford, MA) membrane.
Immunocytochemistry.
Cultures were fixed with 4% paraformaldehyde after permeabilization with 0.5% Triton X-100 (for dissociated cerebellar granule cells) or 1% Triton X-100 (for organotypic culture). Subsequently, slides were incubated with blocking solution (1% BSA and 10% serum for dissociated cerebellar granule cells, and 1% BSA, 10% serum, and 0.25% Triton X-100 for organotypic culture) and incubated overnight with the following primary antibodies: mouse anti-calbindin D28K monoclonal antibody (Sigma), rabbit anti-calbindin D28K polyclonal antibody (Chemicon, Temecula, CA), rabbit anti-phosphorylated pSQ/pTQ polyclonal antibody (Cell Signaling Technology, Beverly, MA), rabbit anti-phospho-serine 1981 (pS1981) of ATM (Bethyl Laboratories, Montgomery, TX), mouse anti-phosphorylated H2AX (a variant of histone 2) (γH2AX) monoclonal antibody (Upstate Biotechnology, Waltham, MA), and mouse anti-ATM monoclonal antibody (5C2; a gift from Prof. Eva Lee, University of Texas, San Antonio, TX). Sytox blue (Invitrogen, Carlsbad, CA) was used to visualize cell nuclei (supplemental Fig. S1, available at www.jneurosci.org as supplemental material). Immunofluorescence was visualized using a Zeiss (Oberkochen, Germany) LSM-510 confocal microscope.
Immunoblotting analysis.
Tissues or cultured cells were washed with ice-cold PBS and homogenized in ice-cold homogenization buffer [120 mm NaCl, 50 mm Tris buffer, pH 7.4, 1 mm EDTA, 0.5% NP-40, and 1:50 phosphatase inhibitor cocktail (I and II; Sigma) and 1:100 protease inhibitor cocktail for total cerebellum; 150 mm NaCl, 50 mm HEPES, pH 7.4, 10% glycerol, 1% Triton X-100, 1 mm EDTA, 1.5 mm MgCl2, 1 mm EGTA, and 1:50 phosphatase inhibitor cocktail (I and II; Sigma) and 1:100 protease inhibitor cocktail for cultured neurons]. Protein concentration was determined according to Bradford (1976) using BSA as standard. Blots were prepared as described by Harlow and Lane (1988) using 7.5% polyacrylamide gels. Each lane was loaded with 50 μg of protein extract, and, after electrophoresis, the proteins were transferred to an Immobilon polyvinylidene difluoride membrane (Millipore) for at least 12 h at 200 mA. Blots were stained with Ponceau to monitor protein amounts. Membranes were then probed with the following antibodies: mouse anti-tubulin monoclonal antibody (Sigma), rabbit anti-phospho-Ser15 of p53 (Cell Signaling Technology), rabbit anti-p53 (DO1 plus 421), and rabbit anti-phospho-Ser957 of SMC1 (a member of the structural maintenance of chromosome family) (Novus Biologicals, Littleton, CO).
Results
Atm is predominantly nuclear in mature postmitotic neurons
Contrary to its predominantly nuclear localization in proliferating cells, ATM was reported previously to be cytoplasmic in cerebellar human and murine neurons (Oka and Takashima, 1998; Kuljis et al., 1999; Barlow et al., 2000). This is a central issue in A-T and ATM research. To reexamine the subcellular localization of Atm in cerebellar neurons, organotypic cultures derived from WT and Atm−/− mice were reacted with the anti-Atm antibody (5C2; a gift from Prof. Eva Lee) and analyzed by confocal microscopy. The ATM specificity of this antibody in immunostaining was shown recently (Biton et al., 2006) and analyzed by confocal microscopy. Cerebellar neurons in our study exhibited predominantly nuclear staining with this antibody, which was not observed in Atm−/− cells, attesting to the specificity of the antibody (Fig. 1A). This result suggests nuclear localization of Atm in both cerebellar and Purkinje cells. However, close examination of Purkinje cells in these cultures revealed, in addition to the strong nuclear staining, small cytoplasmic immunoreactivity (Fig. 1B), suggesting a minor cytoplasmic fraction of Atm in these cells.
Figure 1.

ATM localization in cerebellar organotypic cultures. A, ATM in Purkinje neuron and in surrounding cells resides in the molecular layer of the cerebellum. In Purkinje neuron, the ATM immunoreactivity is located mainly in the nucleus (white arrow); however, cytoplasmic traces of ATM can be seen. The molecular layer contains several types of neurons such as the basket and satellite neurons. B, This image clearly demonstrates that ATM is predominantly nuclear in these cells. The cultures were fixed and reacted with anti-ATM antibodies (5C2). Scale bars, 20 μm.
Activation of nuclear Atm by DNA damage in mature cerebellar neurons
To monitor ATM activation in cerebellar neurons, organotypic cultures were X-irradiated and subsequently reacted with a specific phospho-antibody against phosphorylated S1981 of human ATM. Autophosphorylation of human ATM on this residue plays a major role in ATM activation and tags activated ATM (Bakkenist and Kastan, 2003). This serine residue is at position 1987 in murine ATM, but the flanking amino acid sequences are identical in both species (Pecker et al., 1996); hence, the site is conserved. To determine whether the antibody against autophosphorylated human ATM detects the autophosphorylation of murine ATM, cerebellar cultures from WT and Atm−/− mice were exposed to 200 ng/ml of the radiomimetic drug neocarzinostatin (NCS) for 30 min, and cellular extracts were subjected to immunoblotting analysis with the antibodies against pS1981 and ATM. The α-pS1981 antibody recognized a damage-induced band that corresponded in size to ATM and was not observed in Atm-deficient cells (Fig. 2A), suggesting that it recognized autophosphorylated ATM. We then used this antibody for immunostaining analysis of irradiated cerebellar cultures. Ionizing radiation induced a dose-dependent nuclear staining in WT mice but not in Atm knock-out mice (Fig. 2B). Temporal analysis showed that the signal appeared shortly after DSB induction and decayed within several hours (Fig. 2C). Interestingly, at certain doses and time points, this immunoreactivity was observed in distinct nuclear foci (Fig. 2C,D). These results suggest that, in cerebellar cells, nuclear ATM undergoes autophosphorylation in response to DNA damage, probably indicating its activation similar to this process in proliferating cells. The same analysis performed in cerebellar sagittal sections obtained from irradiated mice showed that ATM was autophosphorylated in the nuclei of cerebellar neurons in vivo (Fig. 3). Thus, in all sources of cerebellar neurons (cultured cerebellar granules, organotypic cultures, and in vivo brain sections), ATM was autophosphorylated in the nuclei after DNA damage.
Figure 2.
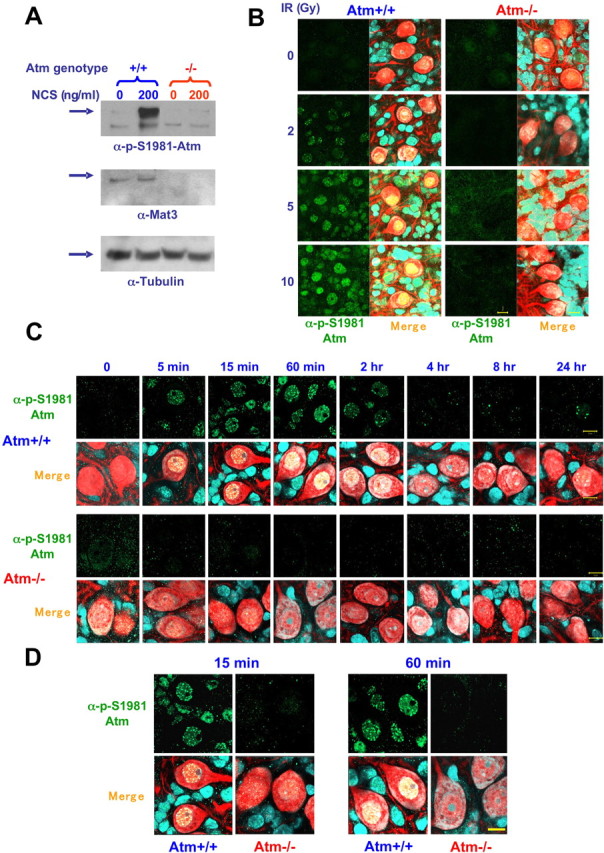
Atm activation after DNA damage. A, Total protein extracts were isolated from Atm−/− and Atm+/+ mice cerebellar granule neurons untreated and treated with NCS (200 ng/ml, 30 min), and immunoblots were reacted with anti-pS1981 ATM antibody (1:1000) and then with anti-ATM antibody (Mat3 [anti-ATM antibody that was prepared against a short sequence (peptide) of murine ATM], 1:1000). This monoclonal antibody was prepared against peptide derived from mouse Atm. To verify equal loading, the blot was stripped and reacted with anti-tubulin antibody. As expected, anti-ATM antibody immunoreacts only with extracts isolated from Atm+/+ cerebella. Note that anti-pS1981 ATM antibody reacts only with activated Atm. B, Dose-dependent activation of Atm. Cerebellar organotypic cultures (day 12 in primary culture) were exposed to various doses of IR (0, 2, 5, and 10 Gy) for 30 min, fixed, and treated with anti-pS1981 ATM antibody (Bethyl Laboratories, Montgomery, TX). Purkinje cells were identified using anti calbindin-28 antibody (red). Note that only the nuclei of Atm+/+ cerebellar neurons that were treated with IR are labeled, suggesting that DNA damage activates Atm in the nuclei of the various cerebellar cells and Purkinje cells. Left panels show the images of fluorescent anti-pS1981 ATM antibody (green), and the right panels are merged images of the fluorescent anti-pS1981 ATM antibody (green), anti-calbindin-28 antibody (red), and Sytox blue. C, Time course of Atm activation in response to IR. Cerebellar organotypic cultures were exposed to 10 Gy, and, at various time points after irradiation, the cultures were fixed and treated with anti-pS1981 ATM antibody. Purkinje cells were identified using anti calbindin-28 antibody (red). Note that Atm is activated as early as 5 min after irradiation and remains active for 2 h. D, Evidence at higher magnification of nuclear foci formed by activated ATM. Merge, α-pS1981–ATM plus calbindin-28 plus Sytox blue. Scale bars, 10 μm.
Figure 3.

Atm activation after exposure of whole mice to 20 Gy IR. Brain sections were isolated from untreated and irradiated 1-month-old (Atm+/+ and Atm−/−) mice. The brains were fixed, and frozen sections were prepared. The sections treated with anti-pS1981) ATM antibody (1:2000; Bethyl Laboratories). Purkinje cells were visualized using anti-calbindin-28 antibody. The panels show a merged picture [calbindin D28K antibody (green) and anti-pS1981 ATM antibody (red)]. Scale bar, 20 μm. PCL, Purkinje cell layer.
DSB responses in cerebellar neurons
Formation of γH2AX foci
Phosphorylation of histone H2AX on Ser139 at DSB sites is one of the earliest responses to this DNA lesion and is crucial to anchoring additional damage-response proteins to these sites (Fernandez-Capetillo et al., 2004; Lee et al., 2005). γH2AX can be detected by the corresponding phospho-specific antibody in prominent nuclear foci that mark the damage sites. H2AX phosphorylation has shown variable degrees of ATM dependency in different studies (Burma et al., 2001; Girard et al., 2002; Friesner et al., 2005). After ionizing radiation, we observed in cerebellar organotypic cultures a dose-dependent increase in γH2AX nuclear foci in most cells (Fig. 4A). Interestingly, fewer and considerably smaller γH2AX foci were observed in Purkinje cells compared with the other types of cells surrounding them (Fig. 4B,C). Temporal analysis of the formation and disappearance of γH2AX foci demonstrated different time courses in WT and Atm-deficient cerebellar cells: their emergence and subsequent disappearance were considerably delayed in Atm−/− cells. Because the persistence of γH2AX foci is thought to denote unrepaired DSBs, this difference probably represents a delay in DSB sealing in Atm−/− cells.
Figure 4.
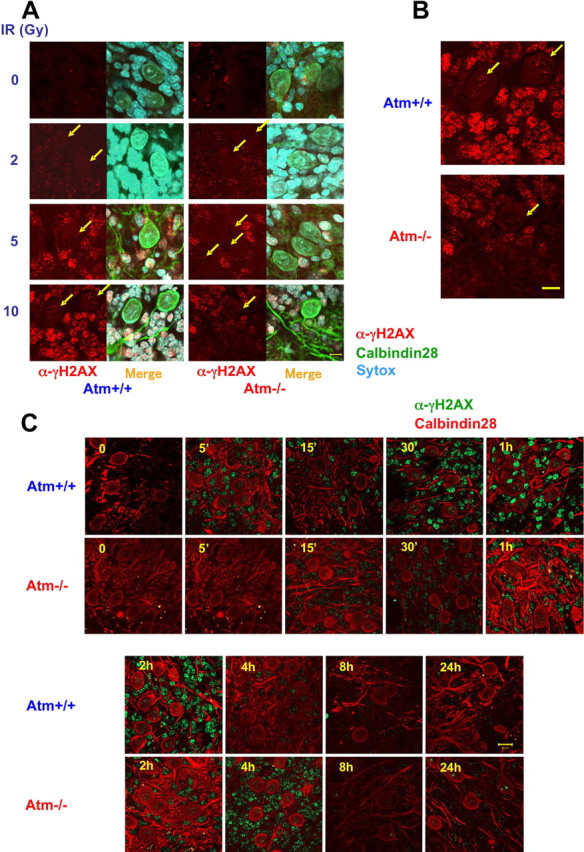
Partial ATM dependence of γH2AX focus formation after DNA damage in cerebellar organotypic cultures. One of the early events in the DSB response is histone H2AX phosphorylation on Ser139 and foci formation of γH2AX at the region of the damage sites. Purkinje cells were identified using anti-calbindin-D28K antibody (green in A, red in C). A, Organotypic cultures were treated with various doses of IR, and, 30 min later, the cultures were fixed and reacted with an anti-γH2AX antibody. γH2AX (in red) can be seen in Atm+/+ as well in Atm−/− cells after treatment with various doses of IR. Merge, α-γH2AX plus calbindin-28 plus Sytox blue. B, Higher magnification of Atm+/+ and Atm−/− cultures that were irradiated with 10 Gy and fixed after 30 min show smaller size of foci in Purkinje cells in contrast to surrounding cells, which have distinct and large foci. Scale bar, 10 μm. C, Organotypic cultures were exposed to 10 Gy, and, at various time points after irradiation, the cultures were fixed and treated with anti-γH2AX antibody (green). γH2AX foci are formed in Atm+/+ cells as early as 5 min after irradiation and last up to 2 h after damage. In contrast, Atm-deficient cells are able to efficiently form γH2AX foci only later (starting at 30 min). Scale bar, 10 μm.
Activated Atm and γH2AX foci are colocalized
A fraction of activated ATM tightly binds to DSB sites (Andegeko et al., 2001; Uziel et al., 2003). Coimmunostaining of irradiated cerebellar organotypic cultures with anti-γH2AX and anti-pS1981/ATM antibodies demonstrated that the foci of γH2AX and activated Atm were indeed colocalized, and the joint foci could be observed as early as 2 min after irradiation. Importantly, activated ATM was detected throughout the nucleus and not only at damage sites (Fig. 5).
Figure 5.
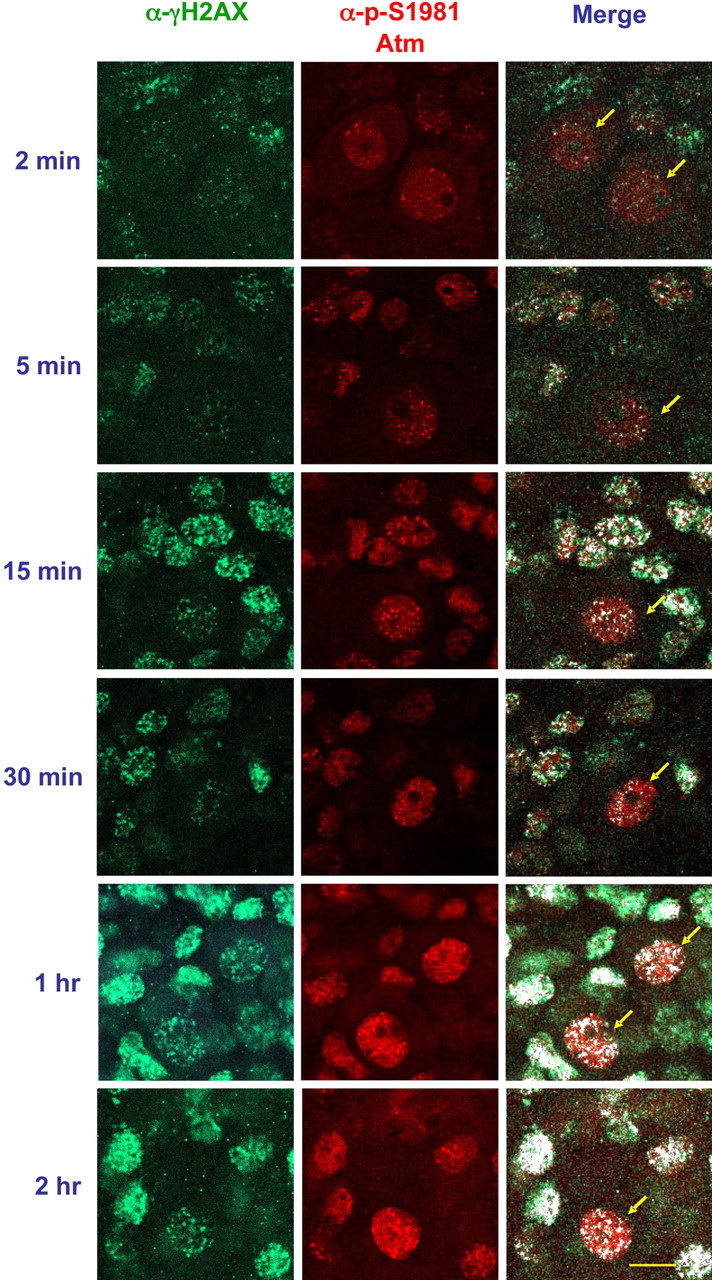
Colocalization of activated ATM and γH2AX in irradiated organotypic cultures. Cerebellar organotypic cultures prepared from Atm+/+ mice were exposed to 10 Gy, and, at various time points thereafter, the cultures were fixed and cotreated with anti-pS1981 ATM antibody (Bethyl Laboratories) (red) and anti- γH2AX (green). The white spots represent colocalization of the two antibodies. Note that ATM is activated throughout the irradiated nucleus, not only at the damage sites. Scale bar, 20 μm.
Nuclear phosphorylation of Atm substrates
In the hierarchy of the DNA damage response, ATM serves as an effector kinase that phosphorylates many substrates in various response pathways. The target sites of ATM are serine or threonine residues followed by glutamine (SQ or TQ motifs) (Kurz and Lees-Miller, 2004). We monitored the phosphorylation of several ATM substrates in cerebellar organotypic cultures after exposure to IR by immunostaining with an antibody that was raised against a collection of peptides containing phosphorylated SQ or TQ motifs. This anti-phospho (SQ/TQ) antibody detects these phosphorylations as a strong nuclear signal (Uziel et al., 2003). The radiation treatment led to marked staining of the nuclei of these cells with a immunofluorescent signal that was ATM dependent and developed over the first hour after irradiation (Fig. 6). In Purkinje cells, this signal exhibited nuclear foci.
Figure 6.
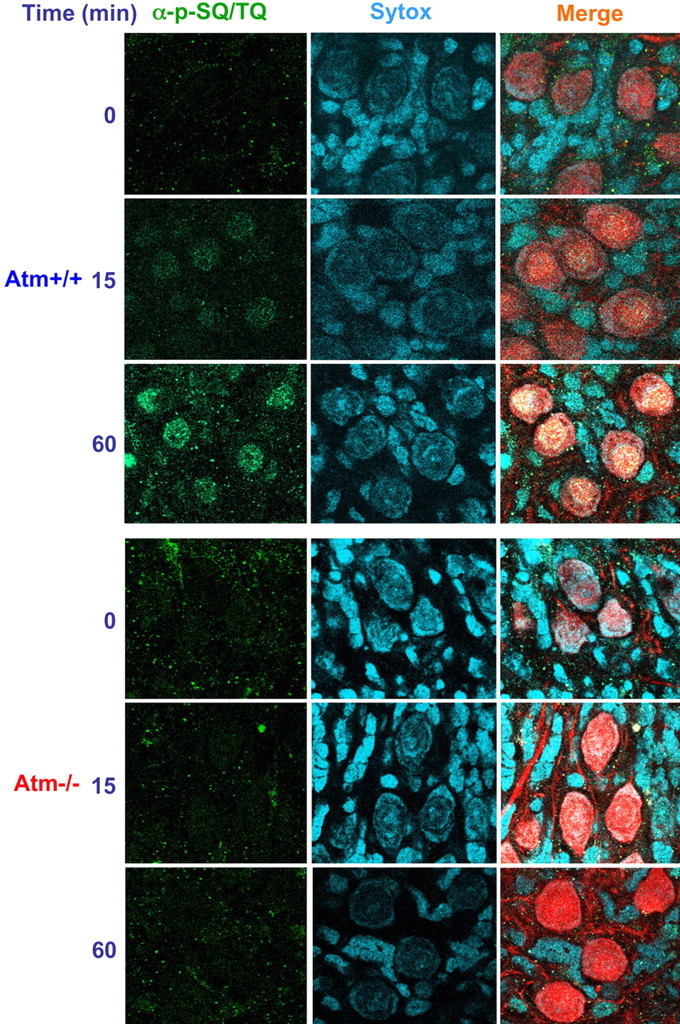
Phosphorylation of Atm substrates (SQ/TQ response) after DNA damage in organotypic culture. Cerebellar organotypic cultures (day 12 in primary culture) prepared from Atm+/+ or Atm−/− mice were exposed to ionizing radiation, fixed, and treated with anti-phosphorylated SQ™Q antibodies. Purkinje cells were identified using anti-calbindin-D28K antibody (red). Cerebellar organotypic cultures were exposed to 10 Gy, and, at various time points (15 and 60 min) after irradiation, the cultures were fixed and reacted with anti-phosphorylated SQ™Q antibodies. Scale bar, 20 μm. Strong nuclear localization of phosphorylated SQ™Q proteins develops as DNA damage increases. Merge, α-p-SQ/TQ plus calbindin28 plus Sytox blue.
We used Western blotting analysis to follow the phosphorylation of two well documented ATM substrates: the p53 protein, which is involved in the activation of the cell cycle checkpoints by DNA damage (Kastan et al., 1992; Gottifredi and Prives, 2005), and the cohesin subunit SMC1, whose phosphorylation is required for the intra-S checkpoint (Kim et al., 2002; Yazdi et al., 2002; Niida and Nakanishi, 2005). The target of ATM on human p53 is Ser15 (Banin et al., 1998; Canman et al., 1998), and the equivalent in murine ATM is Ser18 (Xu et al., 1998; Kang et al., 2005). Phosphorylation of Ser18 of p53 in cultured granule cells treated with NCS exhibited a typical time course and ATM dependency (Fig. 7A). Atm+/+ and Atm−/− animals were exposed to 20 Gy, the cerebella were isolated 30 min after IR treatment, and their proteins were extracted and subjected to Western blotting analysis. SMC1 was phosphorylated in an ATM-dependent manner in cerebellar cells (Fig. 7B).
Figure 7.
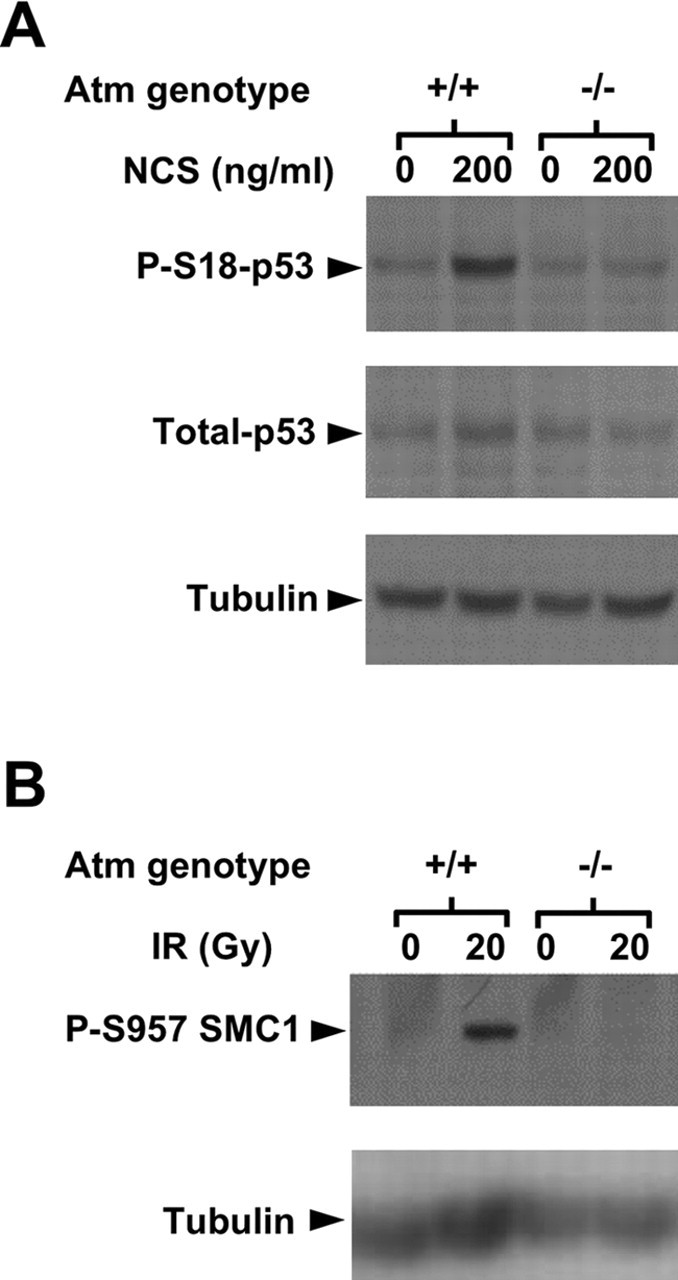
Phosphorylation of specific ATM substrates. A, p53 activation after DNA damage in cultured granule cells. After DNA damage, p53 is activated and stabilized as a result of several posttranslational modifications. Western blot analysis of cultured granule neuronal proteins extracted 30 min after NCS treatment shows that p53 is phosphorylated heavily at Ser18 only in Atm+/+ cells. This phosphorylation lasts 4 h and then declines (data not shown). Western blot analysis of cerebellar protein extracts 30 min after irradiation show that pS957 SMC1 (B) after DNA damage seen only in irradiated Atm+/+ cerebellum.
Discussion
Neuronal degeneration affecting mainly the cerebellum is the major and most disturbing symptom of A-T. Understanding the role of ATM in this tissue is of prime importance for understanding A-T and designing treatment modalities. Our data suggest that, in murine cerebellar neurons, nuclear ATM is activated by DSBs and mediates the cellular response to this lesion, similar to its primary role in proliferating cells. This conclusion is based on our findings: ATM is predominantly nuclear in cerebellar neurons; nuclear ATM autophosphorylates in response to DSB induction and is thus probably activated similarly to its activation in proliferating cells; activated ATM colocalizes with γH2AX foci, suggesting that it binds to DSB sites similarly to activated ATM in proliferating cells; and nuclear ATM-dependent phosphorylation of several ATM substrates is observed in these cells.
A fundamental question with respect to the function of ATM in neuronal cells is its subcellular localization in these cells. Two reports suggested that ATM was cytoplasmic in human and murine cerebellar neurons (Oka and Takashima, 1998; Barlow et al., 2000). In one report by Oka and Takashima (1998), a commercial anti-ATM antibody was used to immunostain human cerebellar sections, but no evidence was provided of the specificity of the antibody to ATM. Such specificity can be established by testing the antibody in immunostaining test against ATM-deficient cells. The vast majority of anti-ATM antibodies tested to date were found to lack ATM specificity in immunostaining (S. Biton, data not shown), but the anti-ATM antibody used in the current study was shown to be specific in this test (Andegeko et al., 2001; Biton et al., 2006). In the study by Barlow et al. (2000) examining ATM localization in murine cerebellar tissues, the antibody used did not react with ATM-deficient cells, thereby fulfilling the basic criterion for Atm specificity. The reason for the discrepancy between our study and that of Barlow et al. is not clear and may reflect differences in experimental protocols. Of note, we did notice a small cytoplasmic fraction of ATM in cultured Purkinje cells and therefore cannot rule out the possibility that ATM resides in these cells in both the nucleus and cytoplasm; nevertheless, the large majority of this protein was nuclear in our cultures.
Unfortunately, we could not detect ATM in frozen cerebellar sections because none of the 10 antibodies that we used displayed specific immunoreactivity in tissue sections (for 5C2 antibody, see supplemental Fig. S2, available at www.jneurosci.org as supplemental material). Collectively, our study clearly demonstrates that ATM is nuclear in cerebellar organotypic cultures, but we cannot generalize this finding to cerebellar frozen sections.
Once our data based on the 5C2 antibody and the α-pS1981 antibody placed ATM in the nucleus in murine cerebellar neurons, we monitored the DSB response in these cells using various readouts of this process. The cerebellar organotypic cultures allow close examination of these responses in Purkinje cells and the surrounding neurons. One of the early events at the DSB sites is massive phosphorylation of the tail of histone H2AX on Ser139, which is essential for appropriate initiation of the DSB response and DSB repair (Fernandez-Capetillo et al., 2004). One of the roles of γH2AX is to anchor the damage response Mdc1 (mediator of DNA damage checkpoint protein 1), which is recruited to the damaged site within minutes of DSB induction (Stucki et al., 2005). H2AX phosphorylation is initiated at the break site and rapidly spreads over megabases of genomic DNA; it is therefore evident as prominent nuclear foci (Redon et al., 2002). The vigorous induction of γH2AX foci in cerebellar neurons demonstrates that this cardinal step in the DSB response, which is well documented in proliferating cells (Bekker-Jensen et al., 2006), is performed in these cells as well. It is not surprising that Atm-deficient cerebellar neurons are capable of H2AX phosphorylation but in a later kinetics (Fig. 4), because previous studies showed that other PIKK (phosphoinositide 3-kinase-related kinase) family members such as ATR and DNA-PK also take part in H2AX phosphorylation (Burma et al., 2001; Ward and Chen, 2001; Fernandez-Capetillo et al., 2002; Brown et al., 2003; Celeste et al., 2003; Stiff et al., 2005).
We observed an interesting phenomenon in the cerebellar organotypic cultures: the number and size of γH2AX foci in Purkinje cells were considerably smaller than in the surrounding neurons (Figs. 4, 5). This difference may stem from a unique chromatin organization in Purkinje cells. DNA staining indicates that chromatin in the large nuclei of these cells is less condensed compared with the surrounding neurons (supplemental Fig. S1, available at www.jneurosci.org as supplemental material). This spatial organization may preclude the rapid expansion of H2AX phosphorylation typical of cells with more condensed chromatin. It is possible that changes in chromatin organization at the DSB sites immediately after DSB induction differ in Purkinje cells compared with other cells.
The disappearance of γH2AX foci is thought to represent a reduction in unrepaired DSBs and therefore provides a monitor of their actual repair. We noticed that, even after high IR doses, cerebellar neurons lost the majority of their γH2AX foci within 2 h (Fig. 1C), suggesting a robust repair process. Interestingly, Atm deficiency does not abrogate the repair process altogether but only attenuates it. This is probably attributable to some redundancy between ATM and other PIKK family members, primarily ATR. The relative contribution of ATR to the activation and maintenance of the DSB response may be more significant in murine than in human cerebellum and may explain the lack of cerebellar degeneration in Atm−/− mice (Barlow et al., 1996; Elson et al., 1996; Xu et al., 1996; Chiesa et al., 2000). Support for this notion comes from the work of Frappart et al. (2005), who obtained marked cerebellar degeneration in mice whose Nbs1 (responsible for the nimegen breakage syndrome) protein had been knocked down conditionally in the nervous system. This result can be explained by attenuation of both ATM (Difilippantonio et al., 2005) and ATR (Stiff et al., 2005), both of which require the MRN complex (a complex of MRE11, Rad 50, Nbs1), of which Nbs1 is a subunit, for their activation.
Whereas phosphorylated γH2AX resides solely in nuclear foci, phosphorylated ATM can be detected both in foci and throughout the entire nucleus. Recently Kruhlak et al. (2006) showed that, in response to DSBs, ATM is phosphorylated at the damage sites and spreads rapidly to the nucleus. Our findings thus support the notion that ATM functions similarly in neurons and in proliferating cells (Kruhlak et al., 2006).
We noticed that two ATM substrates, p53 and SMC1, which are involved in activation of the cell cycle checkpoints (Kastan et al., 1992; Niida and Nakanishi, 2005), were phosphorylated in response to DSBs in mature, postmitotic cerebellar neurons. Importantly, these cells did not exhibit even residual DNA synthesis in our cultures, with or without DNA damage (data not shown). This raises questions about the role of these phosphorylations in postmitotic cells. It has been suggested that checkpoint proteins might be involved in DNA repair in these cells. For instance, the Rad24 protein is known to function in the budding yeast as a damage sensor and checkpoint protein (Green et al., 2000). However, Aylon and Kupiec (2004) provided evidence that this protein is also involved in processing DSB ends and in recombination partner choice (Aylon and Kupiec, 2004). Furthermore, p53 was shown to be involved in DNA repair in several model systems: that is, in the re-annealing of single- and double-strand DNA (Oberosler et al., 1993; Bakalkin et al., 1994; Brain and Jenkins, 1994; Jean et al., 1997) (for a detailed review, see Gatz and Wiesmuller, 2006).
In summary, our findings demonstrate that ATM is predominantly nuclear in mature cerebellar neurons and mediates the DSB response similarly to how it functions in this process in proliferating cells. Cerebellar neurons possess a sensitive and vigorous ATM-mediated DSB response, which is probably critical for maintaining the main activity of their genome throughout their long lifespan. In another recent study from our laboratories, similar results were obtained in human neuron-like cells obtained by in vitro neuronal differentiation of neuroblastoma cell lines: in those cells as well, ATM was found to be primarily nuclear and mediate the cellular response to DSBs (Biton et al., 2006). Collectively, both studies strongly suggest that ATM is nuclear in neuronal cells and has similar responsibility in neuronal and proliferating cells. Therefore, attempts to understand the neuronal demise in A-T patients should focus on further exploring the ATM-mediated DNA damage response in the nervous system.
Footnotes
This work was supported by research grants from the Ataxia-Telangiectasia Children's Project (A.B., Y.S.) and the Israel Science Foundation (A.B.). Work in the laboratory of Y.S. is supported also by research grants from the Ataxia-Telangiectasia Medical Research Foundation, The Ataxia-Telangiectasia Medical Research Trust, and National Institutes of Health Grant RO1 NS 31763.
References
- Abraham RT (2001). Cell cycle checkpoint signaling through the ATM and ATR kinases. Genes Dev 15:2177–2196. [DOI] [PubMed] [Google Scholar]
- Abraham RT (2004). PI 3-kinase related kinases: “big” players in stress-induced signaling pathways. DNA Repair (Amst) 3:883–887. [DOI] [PubMed] [Google Scholar]
- Abramova NE, Davies KJ, Crawford DR (2000). Polynucleotide degradation during early stage response to oxidative stress is specific to mitochondria. Free Radic Biol Med 28:281–288. [DOI] [PubMed] [Google Scholar]
- Andegeko Y, Moyal L, Mittelman L, Tsarfaty I, Shiloh Y, Rotman G (2001). Nuclear retention of ATM at sites of DNA double strand breaks. J Biol Chem 276:38224–38230. [DOI] [PubMed] [Google Scholar]
- Aylon Y, Kupiec M (2004). DSB repair: the yeast paradigm. DNA Repair (Amst) 3:797–815. [DOI] [PubMed] [Google Scholar]
- Bakalkin G, Yakovleva T, Selivanova G, Magnusson KP, Szekely L, Kiseleva E, Klein G, Terenius L, Wiman KG (1994). p53 binds single-stranded DNA ends and catalyzes DNA renaturation and strand transfer. Proc Natl Acad Sci USA 91:413–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakkenist CJ, Kastan MB (2003). DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature 421:499–506. [DOI] [PubMed] [Google Scholar]
- Banin S, Moyal L, Shieh S, Taya Y, Anderson CW, Chessa L, Smorodinsky NI, Prives C, Reiss Y, Shiloh Y, Ziv Y (1998). Enhanced phosphorylation of p53 by ATM in response to DNA damage. Science 281:1674–1677. [DOI] [PubMed] [Google Scholar]
- Barlow C, Hirotsune S, Paylor R, Liyanage M, Eckhaus M, Collins F, Shiloh Y, Crawley JN, Ried T, Tagle D, Wynshaw-Boris A (1996). Atm-deficient mice: a paradigm of ataxia telangiectasia. Cell 86:159–171. [DOI] [PubMed] [Google Scholar]
- Barlow C, Ribaut-Barassin C, Zwingman TA, Pope AJ, Brown KD, Owens JW, Larson D, Harrington EA, Haeberle AM, Mariani J, Eckhaus M, Herrup K, Bailly Y, Wynshaw-Boris A (2000). ATM is a cytoplasmic protein in mouse brain required to prevent lysosomal accumulation. Proc Natl Acad Sci USA 97:871–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassing CH, Alt FW (2004). The cellular response to general and programmed DNA double strand breaks. DNA Repair (Amst) 3:781–796. [DOI] [PubMed] [Google Scholar]
- Bekker-Jensen S, Lukas C, Kitagawa R, Melander F, Kastan MB, Bartek J, Lukas J (2006). Spatial organization of the mammalian genome surveillance machinery in response to DNA strand breaks. J Cell Biol 173:195–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biton S, Dar I, Mittelman L, Pereg Y, Barzilai A, Shiloh Y (2006). Nuclear ATM mediates the cellular response to DNA double strand breaks in human neuron-like cells. J Biol Chem 281:17482–17491. [DOI] [PubMed] [Google Scholar]
- Bradford MM (1976). A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254. [DOI] [PubMed] [Google Scholar]
- Brain R, Jenkins JR (1994). Human p53 directs DNA strand reassociation and is photolabelled by 8-azido ATP. Oncogene 9:1775–1780. [PubMed] [Google Scholar]
- Brown KD, Rathi A, Kamath R, Beardsley DI, Zhan Q, Mannino JL, Baskaran R (2003). The mismatch repair system is required for S-phase checkpoint activation. Nat Genet 33:80–84. [DOI] [PubMed] [Google Scholar]
- Burma S, Chen BP, Murphy M, Kurimasa A, Chen DJ (2001). ATM phosphorylates histone H2AX in response to DNA double-strand breaks. J Biol Chem 276:42462–42467. [DOI] [PubMed] [Google Scholar]
- Canman CE, Lim DS, Cimprich KA, Taya Y, Tamai K, Sakaguchi K, Appella E, Kastan MB, Siliciano JD (1998). Activation of the ATM kinase by ionizing radiation and phosphorylation of p53. Science 281:1677–1679. [DOI] [PubMed] [Google Scholar]
- Celeste A, Difilippantonio S, Difilippantonio MJ, Fernandez-Capetillo O, Pilch DR, Sedelnikova OA, Eckhaus M, Ried T, Bonner WM, Nussenzweig A (2003). H2AX haploinsufficiency modifies genomic stability and tumor susceptibility. Cell 114:371–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiesa N, Barlow C, Wynshaw-Boris A, Strata P, Tempia F (2000). Atm-deficient mice Purkinje cells show age-dependent defects in calcium spike bursts and calcium currents. Neuroscience 96:575–583. [DOI] [PubMed] [Google Scholar]
- Chun HH, Gatti RA (2004). Ataxia-telangiectasia, an evolving phenotype. DNA Repair (Amst) 3:1187–1196. [DOI] [PubMed] [Google Scholar]
- D'Amours D, Jackson SP (2002). The Mre11 complex: at the crossroads of DNA repair and checkpoint signalling. Nat Rev Mol Cell Biol 3:317–327. [DOI] [PubMed] [Google Scholar]
- Difilippantonio S, Celeste A, Fernandez-Capetillo O, Chen HT, Martin BR, Laethem FV, Yang YP, Petukhova GV, Eckhaus M, Feigenbaum L, Manova K, Kruhlak M, Camerini-Otero RD, Sharan S, Nussenzweig M, Nussenzweig A (2005). Role of Nbs1 in the activation of the Atm kinase revealed in humanized mouse models. Nat Cell Biol 7:675–685. [DOI] [PubMed] [Google Scholar]
- Durocher D, Jackson SP (2001). DNA-PK, ATM and ATR as sensors of DNA damage: variations on a theme? Curr Opin Cell Biol 13:225–231. [DOI] [PubMed] [Google Scholar]
- Elson A, Wang Y, Daugherty CJ, Morton CC, Zhou F, Campos-Torres J, Leder P (1996). Pleiotropic defects in ataxia-telangiectasia protein-deficient mice. Proc Natl Acad Sci USA 93:13084–13089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Capetillo O, Chen HT, Celeste A, Ward I, Romanienko PJ, Morales JC, Naka K, Xia Z, Camerini-Otero RD, Motoyama N, Carpenter PB, Bonner WM, Chen J, Nussenzweig A (2002). DNA damage-induced G2-M checkpoint activation by histone H2AX and 53BP1. Nat Cell Biol 4:993–997. [DOI] [PubMed] [Google Scholar]
- Fernandez-Capetillo O, Lee A, Nussenzweig M, Nussenzweig A (2004). H2AX: the histone guardian of the genome. DNA Repair (Amst) 3:959–967. [DOI] [PubMed] [Google Scholar]
- Frappart PO, Tong WM, Demuth I, Radovanovic I, Herceg Z, Aguzzi A, Digweed M, Wang ZQ (2005). An essential function for NBS1 in the prevention of ataxia and cerebellar defects. Nat Med 11:538–544. [DOI] [PubMed] [Google Scholar]
- Friesner JD, Liu B, Culligan K, Britt AB (2005). Ionizing radiation-dependent gamma-H2AX focus formation requires ataxia telangiectasia mutated and ataxia telangiectasia mutated and Rad3-related. Mol Biol Cell 16:2566–2576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatz SA, Wiesmuller L (2006). p53 in recombination and repair. Cell Death Differ 13:1003–1016. [DOI] [PubMed] [Google Scholar]
- Girard PM, Riballo E, Begg AC, Waugh A, Jeggo PA (2002). Nbs1 promotes ATM dependent phosphorylation events including those required for G1/S arrest. Oncogene 21:4191–4199. [DOI] [PubMed] [Google Scholar]
- Gottifredi V, Prives C (2005). The S phase checkpoint: when the crowd meets at the fork. Semin Cell Dev Biol 16:355–368. [DOI] [PubMed] [Google Scholar]
- Green CM, Erdjument-Bromage H, Tempst P, Lowndes NF (2000). A novel Rad24 checkpoint protein complex closely related to replication factor C. Curr Biol 10:39–42. [DOI] [PubMed] [Google Scholar]
- Harlow E, Lane D (1988). In: Antibodies: a laboratory manual Cold Spring Harbor, NY: Cold Spring Harbor Laboratory.
- Inamura N, Araki T, Enokido Y, Nishio C, Aizawa S, Hatanaka H (2000). Role of p53 in DNA strand break-induced apoptosis in organotypic slice culture from the mouse cerebellum. J Neurosci Res 60:450–457. [DOI] [PubMed] [Google Scholar]
- Jean D, Gendron D, Delbecchi L, Bourgaux P (1997). p53-mediated DNA renaturation can mimic strand exchange. Nucleic Acids Res 25:4004–4012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang J, Ferguson D, Song H, Bassing C, Eckersdorff M, Alt FW, Xu Y (2005). Functional interaction of H2AX, NBS1, and p53 in ATM-dependent DNA damage responses and tumor suppression. Mol Cell Biol 25:661–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kastan MB, Zhan Q, el-Deiry WS, Carrier F, Jacks T, Walsh WV, Plunkett BS, Vogelstein B, Fornace AJ Jr (1992). A mammalian cell cycle checkpoint pathway utilizing p53 and GADD45 is defective in ataxia-telangiectasia. Cell 71:587–597. [DOI] [PubMed] [Google Scholar]
- Khanna KK, Lavin MF, Jackson SP, Mulhern TD (2001). ATM, a central controller of cellular responses to DNA damage. Cell Death Differ 8:1052–1065. [DOI] [PubMed] [Google Scholar]
- Kim ST, Xu B, Kastan MB (2002). Involvement of the cohesin protein, Smc1, in Atm-dependent and independent responses to DNA damage. Genes Dev 16:560–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruhlak MJ, Celeste A, Dellaire G, Fernandez-Capetillo O, Muller WG, McNally JG, Bazett-Jones DP, Nussenzweig A (2006). Changes in chromatin structure and mobility in living cells at sites of DNA double-strand breaks. J Cell Biol 172:823–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuljis RO, Chen G, Lee EY, Aguila MC, Xu Y (1999). ATM immunolocalization in mouse neuronal endosomes: implications for ataxia-telangiectasia. Brain Res 842:351–358. [DOI] [PubMed] [Google Scholar]
- Kurz EU, Lees-Miller SP (2004). DNA damage-induced activation of ATM and ATM-dependent signaling pathways. DNA Repair (Amst) 3:889–900. [DOI] [PubMed] [Google Scholar]
- Lavin MF, Shiloh Y (1997). The genetic defect in ataxia-telangiectasia. Annu Rev Immunol 15:177–202. [DOI] [PubMed] [Google Scholar]
- Lee MS, Edwards RA, Thede GL, Glover JN (2005). Structure of the BRCT repeat domain of MDC1 and its specificity for the free COOH-terminal end of the gamma-H2AX histone tail. J Biol Chem 280:32053–32056. [DOI] [PubMed] [Google Scholar]
- Matsuura S, Tauchi H, Nakamura A, Kondo N, Sakamoto S, Endo S, Smeets D, Solder B, Belohradsky BH, Der Kaloustian VM, Oshimura M, Isomura M, Nakamura Y, Komatsu K (1998). Positional cloning of the gene for Nijmegen breakage syndrome. Nat Genet 19:179–181. [DOI] [PubMed] [Google Scholar]
- Nardi N, Avidan G, Daily D, Zilkha-Falb R, Barzilai A (1997). Biochemical and temporal analysis of events associated with apoptosis induced by lowering the extracellular potassium concentration in mouse cerebellar granule neurons. J Neurochem 68:750–759. [DOI] [PubMed] [Google Scholar]
- Niida H, Nakanishi M (2005). DNA damage checkpoints in mammals. Mutagenesis 21:3–9. [DOI] [PubMed] [Google Scholar]
- Noraberg J, Kristensen BW, Zimmer J (1999). Markers for neuronal degeneration in organotypic slice cultures. Brain Res Brain Res Protoc 3:278–290. [DOI] [PubMed] [Google Scholar]
- Oberosler P, Hloch P, Ramsperger U, Stahl H (1993). p53-catalyzed annealing of complementary single-stranded nucleic acids. EMBO J 12:2389–2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oka A, Takashima S (1998). Expression of the ataxia-telangiectasia gene (ATM) product in human cerebellar neurons during development. Neurosci Lett 252:195–198. [DOI] [PubMed] [Google Scholar]
- Pecker I, Avraham KB, Gilbert DJ, Savitsky K, Rotman G, Harnik R, Fukao T, Schrock E, Hirotsune S, Tagle DA, Collins FS, Wynshaw-Boris A, Ried T, Copeland NG, Jenkins NA, Shiloh Y, Ziv Y (1996). Identification and chromosomal localization of Atm, the mouse homolog of the ataxia-telangiectasia gene. Genomics 35:39–45. [DOI] [PubMed] [Google Scholar]
- Redon C, Pilch D, Rogakou E, Sedelnikova O, Newrock K, Bonner W (2002). Histone H2A variants H2AX and H2AZ. Curr Opin Genet Dev 12:162–169. [DOI] [PubMed] [Google Scholar]
- Shiloh Y (2001). ATM and ATR: networking cellular responses to DNA damage. Curr Opin Genet Dev 11:71–77. [DOI] [PubMed] [Google Scholar]
- Shiloh Y, Kastan MB (2001). ATM: genome stability, neuronal development, and cancer cross paths. Adv Cancer Res 83:209–254. [DOI] [PubMed] [Google Scholar]
- Stiff T, Reis C, Alderton GK, Woodbine L, O'Driscoll M, Jeggo PA (2005). Nbs1 is required for ATR-dependent phosphorylation events. EMBO J 24:199–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stucki M, Clapperton JA, Mohammad D, Yaffe MB, Smerdon SJ, Jackson SP (2005). MDC1 directly binds phosphorylated histone H2AX to regulate cellular responses to DNA double-strand breaks. Cell 123:1213–1226. [DOI] [PubMed] [Google Scholar]
- Uziel T, Lerenthal Y, Moyal L, Andegeko Y, Mittelman L, Shiloh Y (2003). Requirement of the MRN complex for ATM activation by DNA damage. EMBO J 22:5612–5621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Gent DC, Hoeijmakers JH, Kanaar R (2001). Chromosomal stability and the DNA double-stranded break connection. Nat Rev Genet 2:196–206. [DOI] [PubMed] [Google Scholar]
- Varon R, Vissinga C, Platzer M, Cerosaletti KM, Chrzanowska KH, Saar K, Beckmann G, Seemanova E, Cooper PR, Nowak NJ, Stumm M, Weemaes CM, Gatti RA, Wilson RK, Digweed M, Rosenthal A, Sperling K, Concannon P, Reis A (1998). Nibrin, a novel DNA double-strand break repair protein, is mutated in Nijmegen breakage syndrome. Cell 93:467–476. [DOI] [PubMed] [Google Scholar]
- Ward IM, Chen J (2001). Histone H2AX is phosphorylated in an ATR-dependent manner in response to replicational stress. J Biol Chem 276:47759–47762. [DOI] [PubMed] [Google Scholar]
- Xu Y, Ashley T, Brainerd EE, Bronson RT, Meyn MS, Baltimore D (1996). Targeted disruption of ATM leads to growth retardation, chromosomal fragmentation during meiosis, immune defects, and thymic lymphoma. Genes Dev 10:2411–2422. [DOI] [PubMed] [Google Scholar]
- Xu Y, Yang EM, Brugarolas J, Jacks T, Baltimore D (1998). Involvement of p53 and p21 in cellular defects and tumorigenesis in Atm−/− mice. Mol Cell Biol 18:4385–4390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yazdi PT, Wang Y, Zhao S, Patel N, Lee EY, Qin J (2002). SMC1 is a downstream effector in the ATM/NBS1 branch of the human S-phase checkpoint. Genes Dev 16:571–582. [DOI] [PMC free article] [PubMed] [Google Scholar]