

Abstract
Proneurotrophins bind with high affinity to p75 neurotrophin receptor (p75NTR) and lack the capacity to bind Trk receptors, suggesting that proneurotrophins can elicit apoptosis via p75NTR even in cells expressing survival-promoting Trk receptors. In the CNS, basal forebrain (BF) neurons are particularly vulnerable to degeneration in Alzheimer’s disease, and are among the few populations of brain neurons that express p75NTR throughout life. These neurons also express Trk receptors and may be concomitantly exposed to both proneurotrophins and mature neurotrophins during development, disease, or after injury. We investigated the interaction of mature and proneurotrophin signaling in these CNS neurons. Kainic acid-induced seizures elicited production of pro-NGF by BF astrocytes before caspase activation in p75NTR-positive BF neurons, demonstrating local production of proneurotrophins under pathological conditions and suggesting apoptotic signaling in vivo. Mechanisms of proneurotrophin-induced death were analyzed in cultured BF neurons, and required both p75NTR and its coreceptor sortilin. Surprisingly, exposure to both mature neurotrophins and proneurotrophins demonstrated that Trk phosphorylation did not prevent pro-NGF-induced apoptosis via p75NTR. However, activation of PI3K (phosphatidylinositol 3-kinase)/Akt and MEK (mitogen-activated protein kinase kinase)/Erk pathways prevented pro-NGF-induced apoptosis, revealing a novel critical checkpoint in survival versus apoptotic signaling downstream of Trk activation, and suggesting that pro-NGF blocks survival signaling by preventing Akt and Erk activation. This study shows that proneurotrophins are produced in the brain under pathological conditions, and can elicit apoptosis of BF neurons even when Trk receptors are activated.
Keywords: neurotrophins, p75 neurotrophin receptor, apoptosis, signal transduction, basal forebrain, Trk
Introduction
Neurotrophins influence multiple aspects of neuronal function, from facilitation of survival and differentiation to regulation of synaptic activity to induction of apoptosis. Specific effects depend on whether signaling is activated via a member of the Trk receptor tyrosine kinase family or the p75 neurotrophin receptor (p75NTR). The role of Trk receptors in mediating neuronal survival, differentiation, and synaptic function has been well defined, and the signaling pathways have been extensively studied (Friedman and Greene, 1999; Kaplan and Miller, 2000; Patapoutian and Reichardt, 2001). In contrast, the function of p75NTR remains enigmatic, although it participates in multiple cellular activities apparently by functioning as a coreceptor with different partners (Hempstead, 2002; Roux and Barker, 2002; Chao, 2003; Barker, 2004). In the absence of a coexpressed Trk receptor, p75NTR can mediate cell death in neuronal (Rabizadeh et al., 1993; Cotrina et al., 2000; Friedman, 2000) and non-neuronal cells (Casaccia-Bonnefil et al., 1996). p75NTR can be activated selectively by proneurotrophins (Lee et al., 2001), which are potent inducers of p75NTR-mediated cell death (Beattie et al., 2002; Harrington et al., 2004). High-affinity binding of proneurotrophins to p75NTR is attributable to concomitant interaction with sortilin, a member of the Vps10p family of receptors (Nykjaer et al., 2004).
Basal forebrain cholinergic neurons (BFCN) express p75NTR and all three Trk receptors, and are among the few CNS neuronal populations that express p75NTR throughout life. Mature neurotrophins support BFCN survival in culture (Hefti et al., 1985; Alderson et al., 1990; Friedman et al., 1993), and in vivo (Fischer et al., 1987; Kromer, 1987; Klein et al., 1994; Smeyne et al., 1994). The role of p75NTR in BFCN survival has been controversial; however, analysis of two different p75−/− mice indicates an increase in the number of BFCN (van der Zee et al., 1996; Yeo et al., 1997; Peterson et al., 1999; Greferath et al., 2000; Naumann et al., 2002), suggesting that p75NTR may mediate developmental death of these neurons. However, direct demonstration that p75NTR can mediate death of BFCN has been lacking. Because proneurotrophins bind p75NTR selectively without activating Trk receptors, we investigated whether pro-NGF can activate p75NTR signaling in cultured basal forebrain (BF) neurons and induce apoptosis, even with concomitant Trk signaling. Previous studies suggested that Trk activation protected sympathetic neurons from proneurotrophin-induced apoptosis (Lee et al., 2001; Teng et al., 2005); however, it is not known whether this is also true for CNS neurons. We investigated the consequences for signaling and survival of BF neurons exposed simultaneously to both mature and proneurotrophins. We further investigated whether pro-NGF is produced in the basal forebrain under pathological conditions in vivo, potentially leading to death of these neurons. Pro-NGF has been shown to induce death of oligodendrocytes after spinal cord injury (Beattie et al., 2002) and cortical neurons after lesion (Harrington et al., 2004). Pro-NGF is also abundant in the Alzheimer brain (Fahnestock et al., 2001; Peng et al., 2004), raising the possibility that this form of NGF may contribute to death of BF neurons via activation of p75NTR-mediated apoptosis.
Materials and Methods
BDNF was a gift from C. F. Ibáñez (Karolinska Institute, Stockholm, Sweden), and NGF was generously provided by Genentech (San Francisco, CA). Neurotensin was purchased from Bachem (Torrance, CA). Anti-p75 (IgG 192) was purchased from Chemicon (Temecula, CA); other anti-p75 antisera were generously provided by M. V. Chao (Skirball Institute, New York University, New York, New York), L. F. Reichardt (University of California, San Francisco, CA), and B. Carter (Vanderbilt University, Nashville, TN); rabbit anti-TrkBin antiserum was from David Kaplan (Toronto, Ontario, Canada); and chicken anti-TrkBin was purchased from Promega (Madison, WI). Anti-sortilin (anti-neurotensin receptor 3) antibodies were from Alpha Diagnostics (San Antonio, TX) (polyclonal) and from BD Biosciences (Mountain View, CA) (monoclonal). Antibodies to cleaved caspase-3, phospho-Trk (P-Trk), phospho-c-Jun N-terminal kinase (P-JNK), JNK, P-Akt, Akt, P-Erk, and Erk, as well as the phosphatidylinositol 3-kinase (PI3K) inhibitor 2-morpholin-4-yl-8-phenyl-chromen-4-one (LY294002) and the Mek1 inhibitor 2-(2-amino-3-methoxy-phenyl)chromen-4-one (PD98059) were from Cell Signaling Technologies (Beverly, MA). Eagle’s MEM, Ham’s F12, and penicillin–streptomycin were purchased from Invitrogen (Gaithersburg, MD). Polylysine, glucose, insulin, putrescine, progesterone, transferrin, and selenium were obtained from Sigma (St. Louis, MO). Secondary antibodies used for immunostaining were Alexa 488 and Alexa 594 anti-rabbit and anti-mouse antibodies purchased from Invitrogen.
Generation of viral vectors and recombinant murine pro-NGF.
Generation of the furin-resistant pro-NGF was performed as described previously (Lee et al. 2001). Briefly, the cDNA encoding mouse NGF was amplified by reverse transcriptase (RT)-PCR and bidirectionally sequenced. Using PCR-based mutagenesis, six histidine (His) residues were added at the COOH terminus and point mutation of residues KR (amino acids 120 and 121) and residues RR (amino acids 239 and 240) to AA was performed.
Recombinant baculoviral expression vectors were generated encoding the furin-resistant His-tagged pro-NGF using the Bac-to-Bac baculovirus expression system (Invitrogen). Baculoviral stocks were amplified and propagated using Spodoptera frugiperda (Sf9) cells cultured in Sf-900 II serum-free medium for 72–96 h. All baculovirus expression system-related reagents and cells were from Invitrogen.
S. frugiperda (Sf9) cells were infected with a multiplicity of infection (MOI) of 2 and cultured in Sf-900 II serum-free medium for 66 h. The medium was then harvested, and proteinase inhibitors were added (1 mm PMSF, 10 μg/ml leupeptin, and 1 μg/ml aprotonin). Media were subjected to tangential flow dialysis (Minimate TFF Capsule; Pall Corporation, East Hills, NY) against PBS, followed by purification in a HisTrap HP 1 ml column (Amersham Biosciences, Uppsala, Sweden) and eluted with 20 mm Na-phosphate, 0.5 m NaCl, and 1 m imidazole, pH 6.0. Recombinant proteins were dialyzed against PBS and stored at −80°C until use.
Pro-BDNF was purified from the media of 293 cells expressing the pro-BDNF plasmid as described previously (Teng et al., 2005).
Neuronal cultures.
Pregnant rats were killed by exposure to CO2 and soaked in 80% ethanol for 10 min. Rat fetuses were removed at embryonic d 16 (E16) under sterile conditions and kept in PBS on ice. The E16 medial septal area was dissected, dissociated by trituration in serum-free medium, plated on polylysine (0.1 mg/ml)-coated tissue culture wells or plastic Lab-Tek slide wells, and maintained in a serum-free environment (Friedman et al., 1993; Farinelli et al., 1998). Medium consists of a 1:1 mixture of Eagle’s MEM and Ham’s F12 supplemented with glucose (6 mg/ml), putrescine (60 μm), progesterone (20 nm), transferrin (100 μg/ml), selenium (30 nm), penicillin (0.5 U/ml), and streptomycin (0.5 μg/ml). In all experiments, neurons were cultured for 4–5 d before treatment. Cultures maintained under these conditions contained <2% glial cells, confirmed by staining for glial markers.
For infection of BF neurons with adenovirus, cultures were grown for 4 d, treated for 36 h with 50 MOI of adenovirus encoding either myc-tagged activated ras (RasV12) (Mazzoni et al., 1999) or green fluorescent protein (GFP) as control. Neurons were then treated for 5 h with pro-NGF. Expression was monitored by immunostaining the neurons with anti-GFP or anti-myc antibodies. Activation of downstream signaling pathways was determined by immunostaining with anti-P-Akt and anti-P-Erk, and apoptosis was assessed by nuclear morphology with Hoechst labeling.
Neuronal survival assay.
Survival of cultured basal forebrain neurons was assayed by a method we adapted (Farinelli et al., 1998; Maroney et al., 1999; Friedman, 2000), which has been used routinely to assess PC12 cell viability (Rukenstein et al., 1991). After removal of the medium, cultured cells were lysed and intact nuclei were counted using a hemacytometer. Nuclei of dead cells either disintegrate, or if in the process of dying, appear pyknotic and irregularly shaped. In contrast, nuclei of healthy cells are phase bright and have clearly defined limiting membranes. Cell counts were performed in triplicate wells. Statistical significance was determined by ANOVA with Bonferroni or Neuman–Keuls post hoc analysis.
Western blot analysis.
For Western analysis of caspase cleavage, BF cultures were treated for 4 h with vehicle, mature neurotrophins or proneurotrophins, washed with PBS, and harvested in Laemmli sample buffer. Equal amounts of protein were separated by 15% PAGE, transferred to nitrocellulose, and labeled with anti-cleaved caspase-6 or anti-cleaved caspase-3 (1:1000 dilution). These blots were then stripped and reprobed for tubulin. For analysis of different signaling pathways, cells were treated as indicated, washed with PBS, harvested in SDS lysis buffer, and separated by 10% PAGE. Blots were probed with anti-P-JNK, phospho-extracellular signal-regulated kinase (P-ERK), P-Akt, or P-Trk (diluted 1:1000); stripped; and reprobed for JNK, ERK, Akt, or TrkB (1:1000 dilution). All Western blot analyses were performed at least three times with samples from independent experiments.
Seizure induction with kainic acid.
Male Wistar rats (250–275 g) were pretreated for 0.5 h with methyl-scopolamine (1 mg/kg, s.c.; Sigma), and then treated with kainic acid (KA) (10 mg/kg, i.p.). After 2 h of status epilepticus, rats were treated with diazepam (10 mg/kg, i.p.; Henry Schein, Melville, NY) and phenytoin (50 mg/kg, i.p.; Sigma) to stop seizure activity. Additional diazepam was administered as necessary to prevent additional seizures. The p75−/− mice used were the exon III knockouts generated in the Sv129-BALB/c mixed strain (Lee et al., 1992). Control mice were wild-type littermates of the same mixed strain as the knockout mice. These mice were used in preference to the p75−/− mice backcrossed onto the C57BL/6 strain, because C57BL/6 mice are resistant to neuronal loss after seizures (Schauwecker and Steward, 1997). Adult mice (20–25 g) were pretreated for 0.5 h with methyl-scopolomine and with phenytoin (50 mg/kg; Sigma) to prevent mortality associated with tonic seizure, and then injected with 20 mg/kg kainic acid and scored for generalized clonus with loss of righting reflex. Status epilepticus was allowed to proceed for 2 h before treatment with diazepam (10 mg/kg, i.p.). Control animals (rats and mice) received all of the same treatments except that they were injected with saline instead of kainic acid. During recovery, the animals were treated with Hartman’s solution (130 mm NaCl, 4 mm KCl, 3 mm CaCl, 28 mm lactate; 1 ml/100 g) injected subcutaneously until the animals were capable of eating and drinking freely. All animal studies were conducted using the National Institutes of Health guidelines for the ethical treatment of animals and approved by the Rutgers Institutional Animal Care and Use Committee.
Preparation of lysates.
One day after injection with either saline or KA, rats were anesthetized with ketamine/xylazine, and the brains were removed on ice for dissection of the basal forebrain. Tissue was homogenized in Tris buffer with protease inhibitors, and 500 ng of lysate from control or KA-treated animals was added to cultured BF neurons either in the presence of rabbit IgG or blocking antibodies to p75 or sortilin, or after 2 h incubation with 2 μg of anti-pro-NGF.
Immunocytochemistry.
Cultured cells were fixed with 4% paraformaldehyde, blocked for 0.5 h with PBS/5% normal goat serum, and exposed overnight at 4°C to primary antibody in PBS/0.3% Triton. Primary antisera were directed against the following: p75 [192 IgG; Chemicon; 1:1000 dilution; or 9651 (Huber and Chao, 1995; Friedman, 2000); 1:1000 dilution]; the intracellular domain of TrkB (anti-TrkBin) [1:2000 (Allendoerfer et al., 1994); or Promega; 1:1000], indicating the presence of the full-length TrkB receptors; anti-phospho-Trk, anti-phospho-Akt, or anti-cleaved caspase-3 (Cell Signaling Technologies), and anti-sortilin (anti-neurotensin receptor 3; Alpha Diagnostics; 1:1000). Cells were then washed with PBS, exposed to the appropriate secondary antibodies that were coupled to different fluorophores, and highly cross-adsorbed against different species (Alexa 488 and Alexa 594; Invitrogen). Controls included exposing the cells to the appropriate secondary antibodies in the absence of primary antibody, and confirming the specificity of the secondary antibodies to the designated species by ensuring the absence of staining of anti-mouse secondary antibodies to rabbit primary antibodies and vice versa. As an additional control for the pro-NGF immunostaining, the antiserum was preadsorbed with the GST (glutathione S-transferase)-linked propeptide, and staining was eliminated. To identify dying neurons, cells were also labeled with the Hoechst 33342 dye (1 μg/ml; Sigma) and examined by fluorescence microscopy (Stefanis et al., 1999; Troy et al., 2002). Apoptotic nuclei were identified by chromatin condensation and clumping.
Animals were anesthetized with ketamine/xylazine and perfused transcardially with saline followed by 4% paraformaldehyde. The brains were removed, postfixed in 4% paraformaldehyde for 2 h, and cryoprotected in 30% sucrose. Sections (12 μm) were cut on a cryostat (Leica, Nussloch, Germany) and mounted onto charged slides. Sections were blocked in PBS/5% goat serum and permeabilized with PBS/0.3% Triton X-100, and then exposed overnight at 4°C to primary antibodies including the following: anti-p75 (192 IgG; Chemicon; 1:500), anti-cleaved caspase-3 (Cell Signaling Technology; 1:1000), anti-pro-NGF (3 μg/ml) (Beattie et al., 2002), and anti-GFAP (Roche Biosciences, Basel, Switzerland) diluted in PBS/0.3% Triton. Slides were then washed three times in PBS, exposed for 1 h at room temperature to secondary antibodies coupled to the Alexa 488 or 594 fluorophores (1:500 in PBS; Invitrogen), and washed again in PBS. Sections were coverslipped with anti-fading medium (Biomeda, Foster City, CA) and analyzed by fluorescence microscopy (Nikon, Melville, NY). The number of dying neurons in wild-type and p75−/− mice after kainic acid-induced seizures was assessed by labeling with Fluoro-Jade B according to the published protocol (Schmued et al., 1997; Schmued and Hopkins, 2000). Labeled neurons were counted in three different sections in both the medial septum and diagonal band. Epifluorescent images were captured digitally and assembled in Adobe Photoshop.
Results
Pro-NGF elicits death of basal forebrain neurons via p75NTR
Previous studies had suggested that neurotrophins could induce p75NTR-mediated apoptosis only in the absence of Trk signaling (Davey and Davies, 1998; Yoon et al., 1998; Friedman, 2000). Because BF cholinergic neurons coexpress p75NTR with Trk receptors, mature neurotrophins, even at high doses, activate Trk signaling in these cells and do not elicit cell death. However, the recent identification of pro-NGF as a selective ligand for p75NTR that does not bind TrkA, suggested that proneurotrophins might activate apoptosis even in cells that coexpress Trk receptors. E16 rat BF neurons were maintained in culture for 5 d and treated overnight with vehicle, mature (cleaved) NGF, or pro-NGF. Pro-NGF, but not mature NGF, elicited a loss of ∼40% of the BF neurons (Fig. 1). To determine the specificity of the pro-NGF effects, BF neurons were treated with pro-NGF in the presence of blocking antibodies to p75NTR (Huber and Chao, 1995; Friedman, 2000), which prevented pro-NGF-induced loss of BF neurons (Fig. 1). Furthermore, an antibody to NGF, which also binds pro-NGF, prevented loss of BF neurons as well (Fig. 1), whereas control rabbit IgG did not block neuronal loss (data not shown). In addition to pro-NGF, pro-BDNF has similarly been shown to act via p75NTR to induce cell death (Teng et al., 2005). We investigated the effects of pro-BDNF and found that it was as effective as pro-NGF at inducing apoptosis of cultured BF neurons (Fig. 2A,B).
Figure 1.

Pro-NGF elicits loss of BF neurons in culture. BF neurons were grown for 5 d and treated overnight with the following: vehicle (imidazole), mature NGF (100 ng/ml), pro-NGF (1 ng/ml), pro-NGF (1 ng/ml) in the presence of blocking antibodies to NGF (Sigma; which also block pro-NGF; diluted in the medium 1:500) or p75 (9651 (Huber and Chao, 1995; Friedman, 2000); diluted 1:500 in the medium), or the antibodies alone. Data are expressed as mean percentage of control ± SEM in triplicate cultures from five independent experiments (n = 15). The asterisk indicates a value different from control at p < 0.05. Veh, Vehicle; mNGF, mature NGF.
Figure 2.

Sortilin is required for proneurotrophin-induced death of BF neurons. A, Pro-NGF and pro-BDNF elicit a loss of BF neurons that can be prevented by either anti-p75NTR or anti-sortilin. BF neurons were cultured for 5 d and treated as indicated overnight. Pro-NGF and pro-BDNF were used at 1 ng/ml, and the antibodies were diluted 1:500 in the media. The antibodies alone had no effect on neuronal survival. B, Neurotensin blocks pro-NGF and pro-BDNF-induced neuronal loss in a dose-dependent manner. BF neurons were cultured for 5 d and treated overnight with pro-NGF (1 ng/ml) or pro-BDNF (1 ng/ml) in the presence or absence of 4 or 40 μm neurotensin. For A and B, data are expressed as mean percentage of control ± SEM in triplicate cultures from three independent experiments (n = 9). The asterisks indicate values different from control at p < 0.05. C, Immunostaining demonstrates colocalization of sortilin with p75 on cultured BF neurons. The arrows indicate p75/sortilin-positive apoptotic neurons, and the arrowheads show healthy p75/sortilin-positive neurons. Scale bar, 10 μm.
Role of sortilin in pro-NGF-induced neuronal death
Recent reports have indicated that sortilin functions as a coreceptor with p75NTR for binding and induction of apoptosis by pro-NGF (Nykjaer et al., 2004) and pro-BDNF (Teng et al., 2005). We demonstrated by immunostaining the presence of sortilin in the p75NTR-positive BF neurons (Fig. 2C). To determine whether the coexpressed sortilin was necessary for proneurotrophin-induced apoptosis, neurons were treated overnight with pro-NGF or pro-BDNF in the presence or absence of antibodies to the extracellular domain of sortilin. Anti-sortilin was as effective as anti-p75NTR in preventing proneurotrophin-induced death of BF neurons (Fig. 2A). Moreover, because sortilin was originally identified as a receptor for neurotensin, we used this peptide to compete with pro-NGF for binding to sortilin (Teng et al., 2005). Neurotensin was able to prevent pro-NGF-induced neuronal loss in a dose-dependent manner, 4 μm neurotensin failed to protect the BF neurons against proneurotrophin-induced death, whereas 40 μm neurotensin was protective (Fig. 2B). The ability of neurotensin and anti-sortilin to prevent pro-NGF-induced neuronal death indicates that sortilin, as well as p75NTR, is required for proneurotrophin-induced death of BF neurons. Interestingly, sortilin coexpression was detected in all the p75NTR-positive neurons, both apoptotic and nonapoptotic (Fig. 2C).
Induction of pro-NGF in vivo
Proneurotrophins can elicit death of BF neurons via p75NTR signaling in culture; however, a critical question is whether BF neurons are ever exposed to proneurotrophins in vivo, and whether this could be a mechanism for neuronal death under pathological conditions. To determine whether pro-NGF can be produced in the basal forebrain, adult rats were treated with kainic acid to induce seizures and analyzed 1, 3, and 7 d later by double immunostaining with an antibody that specifically recognizes pro-NGF, but not mature NGF (Beattie et al., 2002) and antibodies to cell type-specific markers. One day after kainic acid-induced seizures, pro-NGF labeling was detected in GFAP-positive cells (Fig. 3), suggesting that pro-NGF is produced in basal forebrain astrocytes after seizures. Moreover, by 3 d after seizures, there was a dramatic increase in neurons coexpressing p75NTR and sortilin (Fig. 4A) and many p75NTR-positive neurons demonstrated immunostaining for cleaved caspase-3 (Fig. 4B), suggesting that production of pro-NGF by basal forebrain astrocytes after seizure may regulate apoptosis of these neurons in vivo as well as in culture.
Figure 3.

Pro-NGF is induced in the BF in vivo by seizures. a–c show sections from a control rat injected with saline, and d–f show sections from a rat injected with kainic acid 1 d after seizures. Sections were labeled for pro-NGF (a, d) and GFAP (b, e). The merged images demonstrate the presence of pro-NGF in astrocytes (f) of KA-treated rats compared with control rats (c). Scale bar, 100 μm.
Figure 4.
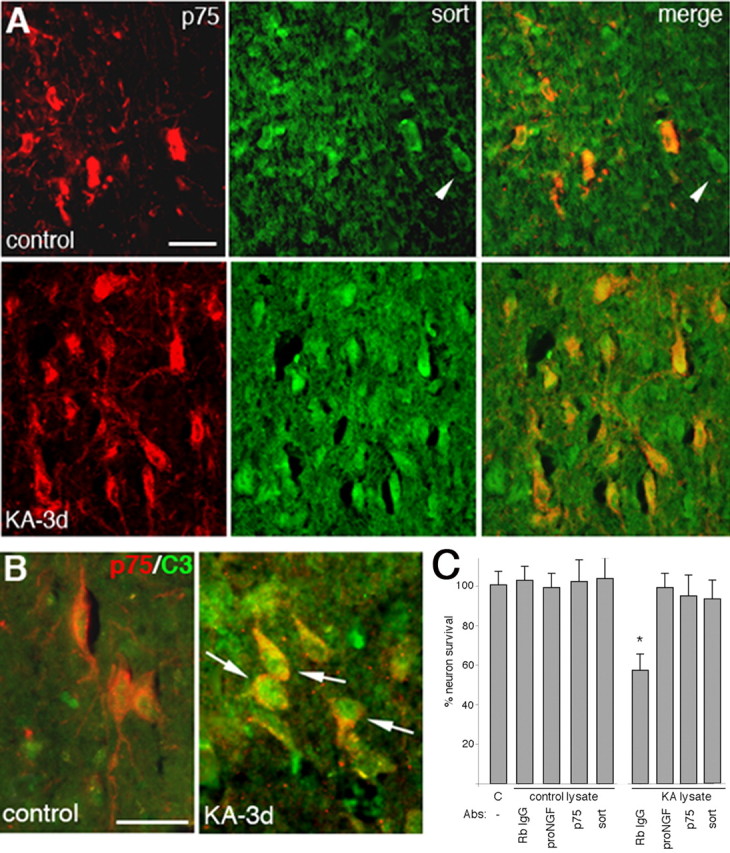
In vivo induction of p75NTR and sortilin in BF neurons after seizures, and activation of caspases in p75NTR-positive neurons. A, Expression of p75NTR (red) and sortilin (green) after kainic acid-induced seizures (bottom row) compared with saline-injected controls (top row). Note that sortilin is expressed in all the p75NTR-positive neurons (orange/yellow) as well as in some neurons that lack p75NTR (arrowheads). Scale bar, 100 μm. B, Rats injected with saline or kainic acid were analyzed after 3 d by immunostaining for p75NTR (red) and cleaved caspase-3 (green). Arrows indicate several p75NTR-positive neurons also labeled for cleaved caspase-3. Scale bar, 25 μm. C, Lysates were prepared from saline (C) or KA-treated rats, incubated with anti-pro-NGF or rabbit (Rb) IgG and added overnight to cultured BF neurons. Lysates from KA-treated, but not control, rats induced a 40% loss of BF neurons that was prevented by preincubation with anti-pro-NGF in three separate experiments with three different sets of lysates. Neuronal death induced by lysates from KA-treated rats was also prevented by the presence of anti-p75NTR (9651) or anti-sortilin. The asterisk indicates a value different from other treatment groups at p < 0.05.
To confirm that the pro-NGF produced in the BF after kainic acid-induced seizures can induce neuronal death via p75 and sortilin, lysates were prepared from control or KA-treated rats and added to cultured BF neurons. BF lysates of kainic acid-treated, but not control, rats induced loss of 40% of cultured BF neurons, which was prevented by the presence of anti-p75 or anti-sortilin, or by preincubating the lysates with anti-pro-NGF (Fig. 4C).
Additional confirmation of the role of p75NTR in mediating neuronal loss in the BF after seizures was obtained by comparing p75−/− mice with wild-type animals. Fluoro-Jade B was used to label dying neurons in the basal forebrain of wild-type or p75−/− mice after KA-induced seizures. In wild-type mice, Fluoro-Jade B labeling after seizures was colocalized with p75NTR expression (Fig. 5A). p75−/− mice showed an ∼65% decrease in the number of dying neurons in the medial septum and an 80% decrease in the diagonal band compared with the wild-type mice 1 d after kainic acid treatment (Fig. 5B), demonstrating a critical role for p75NTR in mediating seizure-induced neuronal death in the basal forebrain.
Figure 5.
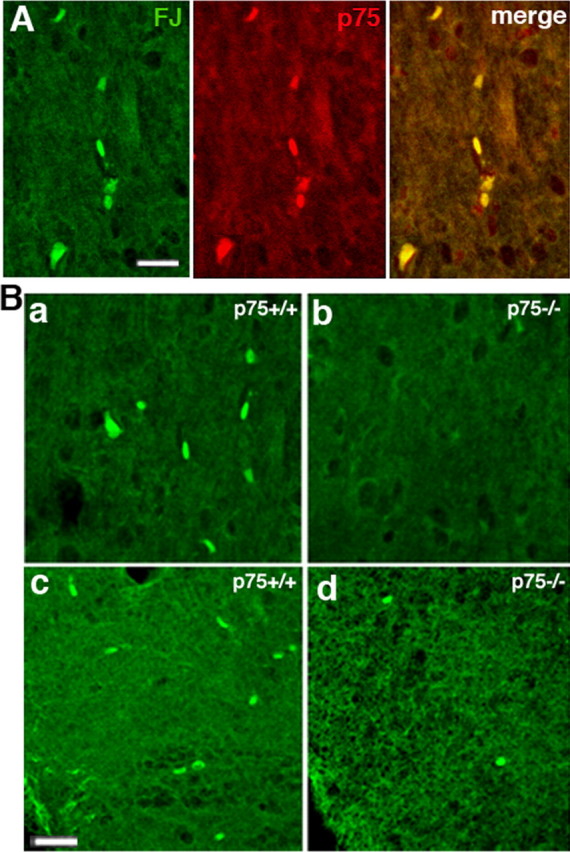
Kainic acid induced fewer dying neurons in the basal forebrain of p75−/− mice than wild-type mice. A, In wild-type mice, kainic acid induced Fluoro-Jade B (FJ) labeling, indicative of dying neurons, in p75+ cells labeled with the 9651 antibody. B, Fluoro-Jade B labeling demonstrated that kainic acid induced death of fewer neurons after 1 d in the p75−/− mice compared with wild-type mice in both the medial septal area (a, b) and diagonal band (c, d). Scale bar, 100 μm.
Signaling pathway activated by pro-NGF
Cultured BF neurons were used to investigate mechanisms governing pro-NGF-induced neuronal apoptosis. Cell death induced via p75NTR activation by mature neurotrophins has been shown to require phosphorylation of JNK (Yoon et al., 1998; Friedman, 2000) and activation of the intrinsic caspase pathway, requiring caspases-9, -6, and -3 (Wang et al., 2001; Troy et al., 2002; Bhakar et al., 2003). To determine whether pro-NGF activated this p75NTR signaling pathway in BF neurons, we examined phosphorylation of JNK and cleavage of caspases. Pro-NGF induced JNK phosphorylation (Fig. 6A) and cleavage of caspase-6 and -3 in these BF neurons (Fig. 6B), indicating that pro-NGF activated the same p75-dependent apoptotic pathway as high doses of mature NGF in other neuronal populations (Troy et al., 2002).
Figure 6.

Pro-NGF-activated signaling in BF neurons. A, BF neurons were cultured for 5 d and treated with pro-NGF for the indicated times. Cells were harvested for Western blot and probed for P-JNK, stripped, and reprobed for total JNK. One representative experiment of three is shown. Numbers indicate the average fold increase from the three experiments in band density compared with control treatment and normalized for total JNK. B, BF neurons were treated with the indicated factors for 4 h and analyzed by Western blot for cleaved caspase-6 (top) and cleaved caspase-3 (bottom). Blots were stripped and reprobed for tubulin. One representative experiment of three is shown. Veh, Vehicle; matNGF, mature NGF.
Pro-NGF can induce death in cells with phosphorylated Trk receptors
To assess whether pro-NGF could elicit apoptosis even in the presence of a survival-promoting factor, cultures were grown from the time of plating in the presence of mature BDNF (10 ng/ml), which facilitates survival of BF neurons (Alderson et al., 1990; Friedman et al., 1993). After 5 d in culture with mature BDNF cells were treated overnight with vehicle, mature NGF, or pro-NGF. Even in the presence of BDNF, pro-NGF, but not mature NGF, elicited a loss of ∼40% of the BF neurons, which was prevented by both the anti-p75 and anti-NGF antibodies (Fig. 7A). Moreover, pretreatment with mature BDNF did not prevent activation of caspase-3 by pro-NGF (Fig. 7B).
Figure 7.
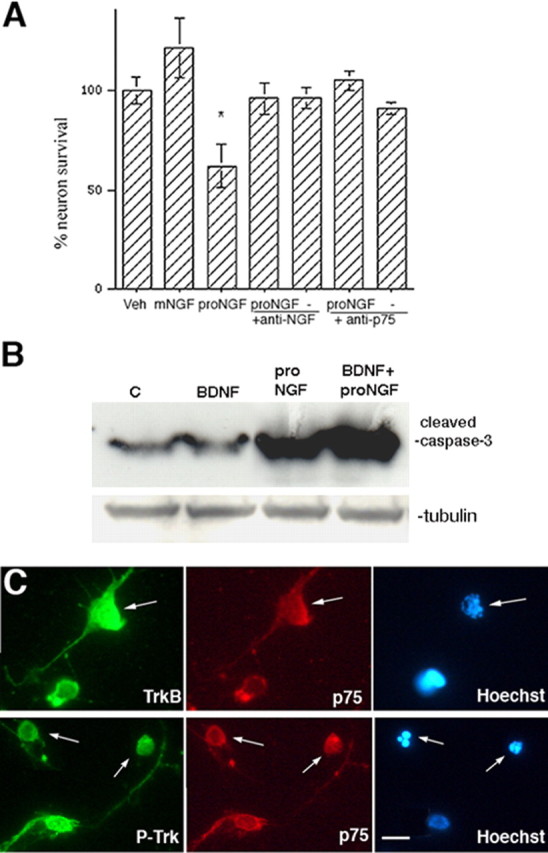
BDNF pretreatment of BF neurons does not protect against pro-NGF-induced apoptosis. A, BF neurons were grown in the presence of BDNF (10 ng/ml) for 5 d and treated overnight with pro-NGF alone, or in the presence of anti-p75NTR or anti-NGF as in Figure 1. Data are expressed as mean percentage of control ± SEM in triplicate cultures from four independent experiments (n = 12). The asterisks indicate values different from control at p < 0.05. Veh, Vehicle; mNGF, mature NGF. B, Western blot demonstrating that cleavage of caspase-3 was induced only by proneurotrophins and was not blocked by pretreatment with mature BDNF. The blot was stripped and reprobed for tubulin. C, Control. C, BF neurons were cultured on slide wells and treated with BDNF and pro-NGF. Neurons were double immunostained for full-length TrkB (anti-TrkBin) (top) or P-Trk (bottom) and p75NTR, and labeled with Hoechst to identify apoptotic nuclei. Scale bar, 10 μm.
Because the basal forebrain is a heterogeneous population of neurons, it was critical to determine which neurons were apoptotic after treatment with pro-NGF. It was possible that, in the presence of BDNF, pro-NGF might induce death only of the neurons expressing TrkA/p75 or TrkC/p75, but not TrkB/p75. To determine whether the neurons that expressed TrkB together with p75NTR were among those that died in response to pro-NGF, neurons were grown on slide wells in the presence of BDNF for 5 d. BDNF was then readded to ensure that the TrkB receptor was activated, and cells were treated for 4 h with vehicle, mature NGF, or pro-NGF. Cells were then fixed and labeled with Hoechst dye to identify apoptotic cells by nuclear morphology, and double immunostained with antibodies to p75NTR (192 IgG) and to either the full-length TrkB (TrkBin) (Allendoerfer et al., 1994; Friedman, 2000) or P-Trk. TrkBin-positive neurons were identified, examined for p75 coexpression, and determined to be either healthy or apoptotic by nuclear morphology. Treatment with pro-NGF resulted in 30–40% of the TrkBin-positive population being apoptotic (Fig. 6C, top; Table 1), whereas in cultures treated with vehicle or mature NGF, only background levels (5–10%) of the TrkBin-positive neurons were apoptotic (Table 1). These data indicate that, even in the presence of BDNF, pro-NGF can induce death of neurons expressing TrkB. Moreover, when neurons were labeled with anti-phospho-Trk, anti-p75NTR, and Hoechst, a similar proportion (∼30%) of neurons with phosphorylated Trk were apoptotic (Fig. 7C, bottom; Table 2), indicating that, in contrast with superior cervical ganglion (SCG) neurons (Lee et al., 2001; Teng et al., 2005), activation of Trk receptors did not prevent the ability of pro-NGF to induce death of BF neurons. Similar results were obtained when the neurons were grown in the presence of NGF or NT3, and then treated with pro-NGF (data not shown).
Table 1.
Pro-NGF induces apoptosis of TrkB+ neurons
| TrkB/p75 | Vehicle | NGF (4 h) | Pro-NGF (4 h) |
|---|---|---|---|
| % Healthy | 96 | 92 | 61 |
| % Apoptotic | 4 | 8 | 39 |
BF neurons were grown on slide wells for 5 d in the presence of BDNF and treated for 4 h with mature NGF or proNGF. The percentage of BF neurons double labeled for TrkB and p75 that were either healthy or apoptotic after the indicated treatments is shown.
Table 2.
Pro-NGF induces apoptosis of P-Trk+ but not P-Akt+ neurons
| Vehicle | BDNF (15 min) | Pro-NGF (30 min) | BDNF (15 min) + Pro-NGF (30 min) | |
|---|---|---|---|---|
| P-Trk/TrkBa | ||||
| % Healthy | 65/94 | 97/97 | 54/75 | 72/72 |
| % Apoptotic | 0/6 | 0/3 | 8/25 | 28/28 |
| P-Akt /P-Trkb | ||||
| % Healthy | 93/93 | 92/92 | 68/68 | 56/56 |
| % Apoptotic | 0/7 | 0/8 | 0/32 | 0/44 |
| P-Akt /CC3c | No double labeling (see Fig. 9C); 100% of the P-Akt+ cells were healthy and lacked cleaved caspase-3 (CC3) staining, and 100% of the CC3+ were apoptotic and lacked P-Akt labeling. | |||
BF neurons were grown on slide wells for 5 d and treated with BDNF and/or pro-NGF as indicated. Neurons were double labeled with the antibodies indicated in column 1 and scored as healthy or apoptotic based on Hoechst nuclear labeling. Numbers indicate the percentage of neurons labeled with both antibodies out of the percentage of neurons with only the second antibody that were either healthy or apoptotic.
aPercentage of TrkB + neurons that also were labeled for P-Trk that were healthy or apoptotic with the indicated treatments (e.g., in vehicle-treated cultures, 94% of the neurons with TrkB were healthy, 65% of those had P-Trk as a result of the neurons being grown with BDNF; 6% of the TrkB-positive neurons were apoptotic, none had P-Trk.) Note that with BDNF + pro-NGF all the apoptotic neurons with TrkB also had P-Trk indicative of receptor activation.
bPercentage of healthy or apoptotic P-Trk + neurons that were also labeled for P-Akt in the different treatment conditions. Note that none of the apoptotic neurons were labeled for P-Akt (Fig. 9).
CDouble labeling was performed for cleaved caspase-3 and P-Akt; no overlap was observed. Cells were counted in 10 random fields in duplicate wells from three independent experiments.
Activation of signaling pathways by mature versus proneurotrophins
To analyze the different signaling pathways activated by mature and proneurotrophins, BF neurons were grown in culture for 5 d, treated with mature or proneurotrophins for the indicated periods of time, lysed, and analyzed by Western blot for activation of the relevant Trk receptor and different downstream signaling proteins. Mature neurotrophins have been shown to activate the PI3K-Akt and Ras-MAP (mitogen-activated protein) kinase pathways via the Trk receptors (Greene and Kaplan, 1995), whereas cell death induced via p75NTR binding has been shown to require activation of the intrinsic caspase pathway (Wang et al., 2001; Troy et al., 2002). Cultured BF neurons treated for 4 h and analyzed by Western blot for caspase cleavage showed that pro-NGF, but not vehicle or mature NGF or BDNF, induced cleavage of caspase-3 (Figs. 6B, 7B). Conversely, BF neurons treated with mature neurotrophins, but not proneurotrophins, for 15 min showed phosphorylation of Akt and Erk (shown for BDNF treatment) (Fig. 8), indicating that in this heterogeneous population of neurons, only the appropriate signaling pathway was activated by the relevant ligand.
Figure 8.
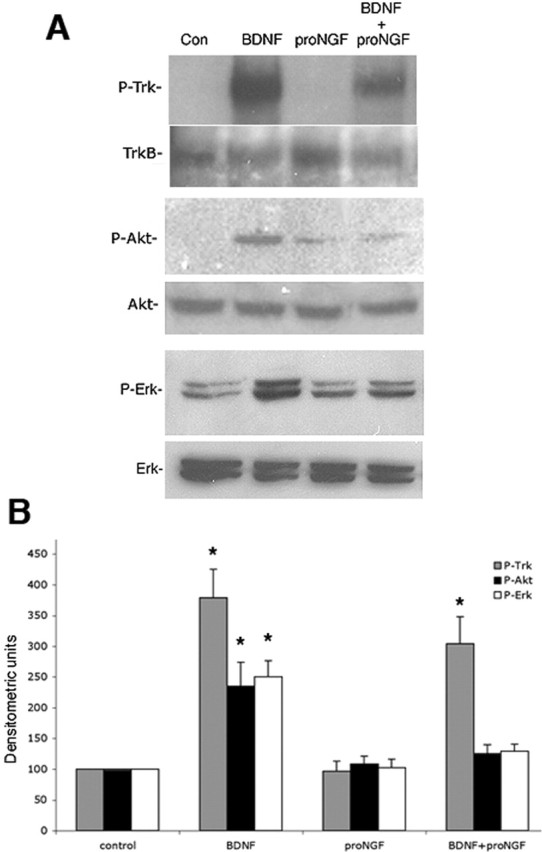
Western blot analysis of distinct signaling pathways activated by mature neurotrophins or proneurotrophins. Cultured BF neurons were treated for 15 min with mature BDNF or pro-NGF or both. A, Cells were analyzed by Western blot for P-Trk, P-Akt, and P-Erk. Blots were then stripped and reprobed for TrkB, total Akt, and total Erk, respectively. Con, Control. B, Quantitation of P-Trk (gray bars), P-Akt (black bars), and P-Erk (open bars) relative to total TrkB, Akt, and Erk, respectively, from three independent experiments. The asterisks indicate values significantly different from control at p < 0.05. Error bars indicate SEM.
To investigate how exposure of BF neurons to both a mature neurotrophin and proneurotrophin would influence activation of signaling pathways and ultimately neuronal survival, cultured BF neurons grown in the presence of BDNF were reexposed to BDNF for 15 min before pro-NGF treatment to ensure activation of TrkB signaling. As shown above, BDNF induction of TrkB phosphorylation did not prevent caspase cleavage and neuronal apoptosis in response to pro-NGF (Fig. 7). Conversely, however, pro-NGF prevented the ability of BDNF to induce phosphorylation of Akt and Erk, but did not inhibit phosphorylation of the TrkB receptor (Fig. 8). Although Trk phosphorylation appeared to be attenuated by the presence of pro-NGF, quantification of multiple blots did not show a significant different between BDNF treatment alone and BDNF plus pro-NGF (Fig. 8B). Activation of signaling pathways was also analyzed at the cellular level by immunostaining and counting the healthy and apoptotic neurons with activated Trk, Akt, Erk, or caspase-3. Quantification of healthy and apoptotic neurons showing activation of Trk, Akt, or caspase-3 under different treatment conditions is shown in Table 2. This analysis showed that many (65%) of the TrkB+ neurons had phosphorylated Trk because the neurons were grown with BDNF (Table 2, P-Trk/TrkB). When BDNF was readded, all the healthy TrkB+ neurons (97% of the total neurons) had P-Trk. Pro-NGF induced apoptosis of 25–30% of the TrkB+ neurons. In the absence of readded BDNF, some of these apoptotic neurons had P-Trk, and with BDNF treatment, all of the TrkB+ neurons had P-Trk whether healthy or apoptotic, indicating that the phosphorylation of the Trk receptor did not prevent apoptotic signaling. With vehicle or BDNF treatment, all of the healthy neurons with P-Trk were also labeled for P-Akt (Table 2, P-Akt/P-Trk) (the number of neurons with P-Akt and P-Trk increased with BDNF treatment, but the percentage of P-Trk+ neurons that also had P-Akt did not change). After pro-NGF treatment, 30–40% of the neurons were apoptotic, even with readdition of BDNF. However, none of the apoptotic neurons had P-Akt, even those with P-Trk (Fig. 9A; Table 2, P-Akt/P-Trk). Because BDNF alone induced phosphorylation of Akt as well as TrkB in 92% of the cells, but BDNF plus pro-NGF induced phosphorylation of Akt only in the 56% of the neurons that were healthy, it suggests that pro-NGF prevented Akt phosphorylation in the apoptotic population. Consistent with this observation, evaluation of P-Akt and cleaved caspase-3 staining clearly demonstrated two separate populations, with cleaved caspase-3 labeling in neurons that had fragmented nuclei and were apoptotic, and P-Akt labeling only in neurons that were not apoptotic and did not have cleaved caspase-3 (Fig. 9C; Table 2, P-Akt/CC3). P-Erk labeling distributed with P-Akt in the nonapoptotic population (Fig. 9B).
Figure 9.

Pro-NGF induces apoptosis of BDNF-treated BF neurons with activated Trk receptors but not with phosphorylated Akt or Erk. A, Neurons were double immunostained for P-Trk and P-Akt and labeled with Hoechst. Anti-P-Akt labeled a subset of the P-Trk-positive neurons that were not apoptotic (arrows). The arrowheads indicate apoptotic neurons that are P-Trk positive and P-Akt negative. B, Neurons were double labeled for P-Akt and P-Erk. P-Erk was present in most but not all of the P-Akt-positive population, which were all healthy. An arrow indicates a P-Akt-positive neuron that lacks P-Erk. An arrowhead indicates an apoptotic cell that had neither P-Akt nor P-Erk. C, Neurons were double labeled for P-Akt and cleaved caspase-3 (C3) and labeled with Hoechst. The arrows indicate healthy neurons, and the arrowheads indicate apoptotic neurons. All of the cleaved caspase-3-positive neurons were apoptotic and lacked P-Akt, whereas all of the P-Akt-positive neurons were healthy and lacked cleaved caspase-3. Scale bar: (in A) A–C, 20 μm.
The above results suggest that neurons expressing activated Akt and/or Erk were resistant to pro-NGF-induced cell death. To determine whether activation of the PI3K/Akt and mitogen-activated protein kinase kinase (MEK)/Erk pathways could mediate basal forebrain neuron survival, we asked whether constitutive activation of these pathways would rescue these neurons from pro-NGF-induced cell death. Neurons were infected with a recombinant adenovirus vector encoding myc epitope-tagged, constitutively activated ras (rasV12), which activates both the PI3K/Akt and MEK/Erk pathways in the absence of neurotrophin treatment (Mazzoni et al., 1999). Survival of neurons expressing activated ras was compared with neurons infected with a control adenovirus expressing green fluorescent protein. Neurons infected with the GFP adenovirus showed the same percentage of death after pro-NGF treatment as did uninfected neurons (30%); however, neurons infected with the rasV12 virus were resistant to pro-NGF-induced death (Fig. 10A). The neurons with rasV12 were identified by immunostaining for the myc tag, and these neurons all had phosphorylated Akt and Erk (data not shown). These data suggest that a critical checkpoint in survival versus apoptotic signaling in these CNS neurons is therefore upstream of Akt and Erk but downstream of Trk activation. To confirm the role of the PI3K/Akt and MEK/Erk pathways in regulating survival of these BF neurons, cultures were treated overnight with inhibitors of these pathways while being exposed to pro-NGF. Overnight treatment with the inhibitors alone did not affect basal neuron survival; however, the PI3K inhibitor LY294002 potentiated the effects of pro-NGF, eliciting a greater percentage of cell death than pro-NGF alone (Fig. 10B), suggesting that Akt may be a critical mediator of BF neuron survival. Inhibition of Erk activation with PD98059 did not augment the degree of cell death elicited by pro-NGF, suggesting that the Erk pathway may be less involved in survival regulation than the Akt pathway.
Figure 10.
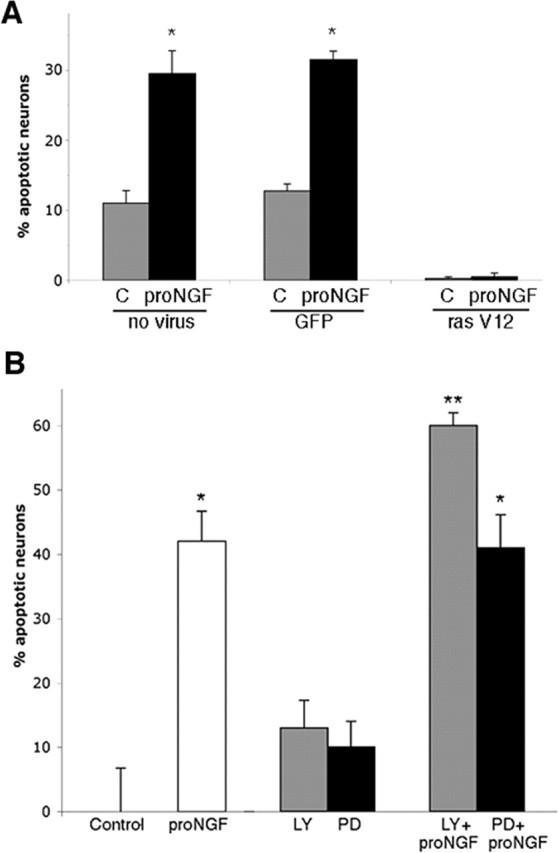
Akt is a critical mediator of BF neuron survival. A, BF neurons were grown in culture for 5 d and infected during the last 36 h with 50 MOI of adenovirus expressing either GFP or constitutively activated ras (ras V12) that was myc tagged. Neurons infected with control virus were identified by GFP labeling, and those infected with the rasV12 virus were identified by immunostaining for the myc tag. Uninfected and infected cultures were treated with pro-NGF for 5 h, fixed, and labeled with Hoechst to identify apoptotic nuclei. Data are expressed as percentage of apoptotic neurons. Triplicate cultures were analyzed in two independent experiments (n = 6). The asterisk indicates a value significantly different from all other groups at p < 0.05. Error bars indicate SEM. C, Control. B, BF neurons were grown for 5 d and treated overnight with the PI3K inhibitor LY294002 (LY) (50 μm) or the Mek1 inhibitor PD98059 (PD) (10 μm) alone or in combination with pro-NGF. Triplicate cultures were analyzed in three independent experiments (n = 9) and expressed as percentage of apoptotic neurons. The asterisk indicates a value significantly different from all other groups at p < 0.05. The double asterisk indicates a value significantly different from pro-NGF alone at p < 0.05. Error bars indicate SEM.
Discussion
Neurotrophins influence multiple cellular functions ranging from neuronal survival and differentiation to induction of apoptosis, depending on which signaling pathways are activated. Trk signaling is critical for regulating neuronal survival, differentiation, and synaptic function, whereas activation of p75NTR can induce cell death. In cells expressing Trk receptors and p75NTR, mature neurotrophins activate signaling of their respective Trk receptors. Thus, an apoptotic response would only be activated by mature neurotrophins in cells expressing p75NTR in the absence of the cognate Trk receptor (Davey and Davies, 1998; Yoon et al., 1998; Friedman, 2000). However, the recent identification of pro-NGF as a selective, high-affinity ligand for p75NTR (Lee et al., 2001) suggested that p75NTR-mediated apoptosis could occur even in neurons expressing Trk receptors, because proneurotrophins would not activate Trk signaling. We investigated this possibility in basal forebrain neurons, which have been extensively studied for effects of mature neurotrophins on neuronal survival and expression of cholinergic traits. These neurons play a critical role in memory and express p75NTR throughout life, which may contribute to their vulnerability to degeneration in Alzheimer’s Disease. In this study, we showed that pro-NGF and pro-BDNF induced apoptosis of BF neurons in culture. Moreover, we demonstrated that pro-NGF was present in BF astrocytes under pathological conditions induced by kainic acid treatment in vivo, and that caspases were activated in p75-expressing BF neurons, suggesting that pro-NGF may induce p75NTR-mediated apoptotic signaling in vivo. This was substantiated by the decrease in neuronal death induced by kainic acid in the basal forebrain of p75−/− mice compared with wild-type animals. BF lysates from KA-treated rats induced loss of cultured BF neurons, which was prevented by antibodies to pro-NGF, p75NTR, and sortilin, confirming the presence of pro-NGF and its apoptotic activity. These observations raised the question of how these neurons would respond if both p75NTR and Trk signaling were activated. Previous studies demonstrated that sympathetic neurons were protected from pro-NGF-induced cell death by Trk activation (Teng et al., 2005). In contrast, our study demonstrated that pro-NGF elicited apoptosis of these central BF neurons even in the presence of mature neurotrophins and phosphorylation of Trk receptors, and revealed a novel checkpoint in survival versus apoptotic signaling downstream of Trk activation.
Role of sortilin in determining apoptosis of BF neurons
We investigated whether coexpression of sortilin with p75NTR was necessary for pro-NGF induction of BF neuronal death. Neurotensin, which can compete with pro-NGF for binding to sortilin (Nykjaer et al., 2004), prevented pro-NGF-induced death of BF neurons in a dose-dependent manner, suggesting that sortilin was a necessary component of the receptor complex. Moreover, treatment with an antibody to the external domain of sortilin was as effective in protecting neurons as the anti-p75 NTR antibody, confirming the role of both receptors in mediating apoptosis. Interestingly, sortilin coexpression was detected in all the p75 NTR-positive neurons, both apoptotic and nonapoptotic, suggesting that although both sortilin and p75 NTR are necessary for proneurotrophin-induced apoptosis, coexpression of these two receptors is not sufficient to determine which neurons die. Whether there is an additional receptor component needed, or whether these receptors may be expressed but not activated or not present on the cell surface in some neurons, requires additional investigation.
Survival versus death signaling
Previous studies suggested that activation of Trk signaling could protect neurons from p75NTR-mediated apoptosis (Yoon et al., 1998). Because BF neurons express p75NTR together with Trk receptors, and mature neurotrophins such as BDNF facilitate survival of BF neurons (Alderson et al., 1990; Friedman et al., 1993), we tested whether maintaining the neurons in the presence of BDNF would abrogate the apoptotic effects of pro-NGF. Surprisingly, when the BF neurons were grown in the presence of BDNF, and then treated with pro-NGF, the proportion of apoptotic neurons was similar to those treated in the absence of BDNF. Double-label immunostaining confirmed that neurons expressing full-length TrkB in addition to p75NTR were apoptotic, even in the presence of BDNF. Moreover, a similar proportion of apoptotic neurons had phosphorylated Trk, indicating that the receptor was activated. Thus, in contrast to SCG neurons (Teng et al., 2005), activation of a Trk receptor did not protect BF neurons from pro-NGF-induced apoptosis.
An investigation of signaling pathways revealed that exposure of BF neurons to mature neurotrophins or proneurotrophins clearly activated distinct pathways, with mature neurotrophins eliciting phosphorylation of Akt and Erk, and proneurotrophins inducing phosphorylation of JNK and cleavage of caspase-6 and -3. However, if a neuron is exposed to both a mature and proneurotrophin, which signal will predominate to influence neuronal survival? Would activation of Trk signaling by mature neurotrophins prevent p75NTR-mediated apoptotic signaling, or would p75NTR signaling interfere with Trk-mediated survival? Pretreatment with a mature neurotrophin (e.g., BDNF) did not prevent pro-NGF-induced caspase cleavage and apoptosis in BF neurons. In contrast, although TrkB phosphorylation was not prevented by pro-NGF, activation of Akt and Erk by BDNF was inhibited by pro-NGF. When examined at the cellular level, 30–40% of the neurons expressing full-length TrkB, or phosphorylated Trk, were apoptotic, indicating that Trk phosphorylation did not prevent pro-NGF binding and signaling via a p75/sortilin complex. All of the healthy, P-Trk-positive neurons showed Akt phosphorylation; however, none of the apoptotic neurons had P-Akt or P-Erk, even those that had P-Trk labeling. The introduction of constitutively activated ras, which induced phosphorylation of both Akt and Erk in the absence of neurotrophin treatment, confirmed the critical role of these pathways in determining neuronal survival, because these neurons were resistant to pro-NGF-induced death. Moreover, the presence of the PI3K inhibitor LY294002 augmented the amount of neuronal death induced by pro-NGF. Thus, pro-NGF seemed to interfere with the prosurvival signaling downstream of Trk receptor activation by blocking Akt phosphorylation, leading to apoptosis.
Although the exact relationship between the Trk and p75NTR signaling pathways in neurons remains to be fully elucidated, our data suggest that proneurotrophins can induce p75NTR-dependent apoptosis, even in neurons with activated Trk receptors. However, once Akt and Erk are activated, those neurons can no longer be induced to undergo apoptosis by pro-NGF. Akt is critical for neurotrophin-mediated survival in many neuronal cell types (Brunet et al., 2001), and is clearly a critical mediator of BF neuron survival as well. However, in these neurons, Trk phosphorylation is not sufficient to prevent apoptosis, the critical point in survival versus death fate determination occurs between Trk activation and phosphorylation of downstream targets, specifically Akt.
Seizure-induced apoptosis of BF neurons in vivo
Although pro-NGF can induce apoptosis of BF neurons in culture, an important consideration is whether BF neurons are ever exposed to proneurotrophins in vivo. We, and others, had previously shown that seizures induce p75NTR-mediated apoptosis of hippocampal neurons (Roux et al., 1999; Troy et al., 2002), and a previous study had indicated that BF neurons undergo p75NTR-mediated apoptosis after kainic acid-induced seizures (Oh et al., 2000). We confirmed the importance of p75NTR in mediating death of BF neurons, demonstrating that p75−/− mice showed a 65% reduction in neuronal death in the medial septum and an 80% reduction in the diagonal band compared with wild-type mice after kainic acid-induced seizures. Adult rats were analyzed by immunostaining for pro-NGF after seizures. One day after KA treatment, pro-NGF immunostaining was colocalized with GFAP in the basal forebrain, indicating the presence of pro-NGF in astrocytes under these pathological conditions. Moreover, by 3 d after seizure, activated caspase-3 was detected in p75NTR-positive neurons, indicating the presence of apoptotic BF neurons as a consequence of the seizures, possibly because of the production of pro-NGF. The apoptotic activity of pro-NGF produced in the BF after seizures was confirmed by the ability of BF lysates from kainic acid-treated rats to induce death of cultured BF neurons, which was prevented by anti-pro-NGF, anti-p75NTR, and anti-sortilin.
Inflammatory stimuli induce NGF mRNA in CNS glial cells in vitro (Friedman et al., 1990; Spranger et al., 1990) and in vivo (DeKosky et al., 1994; Oderfeld-Nowak and Bacia, 1994; Goss et al., 1998); however, which form of the NGF protein is produced has not been addressed. In addition, NGF and BDNF mRNAs are induced by seizure activity in neurons of the hippocampus and other forebrain regions (Gall and Isackson, 1989; Isackson et al., 1991). The upregulation of neurotrophin expression under pathological conditions has traditionally been considered as a potential protective mechanism for CNS neurons. However, from this study as well as others, it is clear that different forms of the neurotrophin protein may have profoundly different consequences, preferentially activating different receptors to induce cell survival or death (Friedman, 2005). In addition to seizures, pro-NGF is elevated in the brains of Alzheimer’s patients (Fahnestock et al., 2001; Peng et al., 2004). With progression of the disease there is a loss of TrkA receptors, but a maintenance of the level of p75NTR (Counts and Mufson, 2005), suggesting that pro-NGF signaling via p75NTR may contribute to neuronal loss in Alzheimer’s disease. Thus, investigating the mechanisms that regulate neurotrophin production, processing, and signaling is critical for understanding the multifunctional roles of these factors in the brain.
Footnotes
*M.V. and W.S. contributed equally to this work.
This work was supported by National Institutes of Health Grants NS42778 (W.J.F.) and NS30687 (B.L.H.) and by the Alzheimer’s Disease Program of the American Health Assistance Foundation (W.J.F.). We thank Jing-Yu Pan, Deepak Srinivasan, and Michael Schmidt for excellent technical assistance.
References
- Alderson RF, Alterman AL, Barde Y-A, Lindsay RM (1990). Brain-derived neurotrophic factor increases survival and differentiated functions of rat septal cholinergic neurons in culture. Neuron 5:297–306. [DOI] [PubMed] [Google Scholar]
- Allendoerfer KL, Cabelli RJ, Escandon E, Kaplan DR, Nikolics K, Shatz CJ (1994). Regulation of neurotrophin receptors during the maturation of the mammalian visual system. J Neurosci 14:1795–1811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker PA (2004). p75NTR is positively promiscuous: novel partners and new insights. Neuron 42:529–533. [DOI] [PubMed] [Google Scholar]
- Beattie MS, Harrington AW, Lee R, Kim JY, Boyce SL, Longo FM, Bresnahan JC, Hempstead BL, Yoon SO (2002). ProNGF induces p75-mediated death of oligodendrocytes following spinal cord injury. Neuron 36:375–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhakar AL, Howell JL, Paul CE, Salehi AH, Becker EB, Said F, Bonni A, Barker PA (2003). Apoptosis induced by p75NTR overexpression requires Jun kinase-dependent phosphorylation of Bad. J Neurosci 23:11373–11381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunet A, Datta SR, Greenberg ME (2001). Transcription-dependent and -independent control of neuronal survival by the PI3K-Akt signaling pathway. Curr Opin Neurobiol 11:297–305. [DOI] [PubMed] [Google Scholar]
- Casaccia-Bonnefil P, Carter BD, Dobrowsky RT, Chao MV (1996). Death of oligodendrocytes mediated by the interaction of nerve growth factor with its receptor p75. Nature 383:716–719. [DOI] [PubMed] [Google Scholar]
- Chao MV (2003). Neurotrophins and their receptors: a convergence point for many signalling pathways. Nat Rev Neurosci 4:299–309. [DOI] [PubMed] [Google Scholar]
- Cotrina ML, Gonzalez-Hoyuela M, Barbas JA, Rodriguez-Tebar A (2000). Programmed cell death in the developing somites is promoted by nerve growth factor via its p75NTR receptor. Dev Biol 228:326–336. [DOI] [PubMed] [Google Scholar]
- Counts SE, Mufson EJ (2005). The role of nerve growth factor receptors in cholinergic basal forebrain degeneration in prodromal Alzheimer disease. J Neuropathol Exp Neurol 64:263–272. [DOI] [PubMed] [Google Scholar]
- Davey F, Davies A (1998). TrkB signalling inhibits p75-mediated apoptosis induced by NGF in embryonic proprioceptive neurons. Curr Biol 115:56–67. [DOI] [PubMed] [Google Scholar]
- DeKosky ST, Goss JR, Miller PD, Styren SD, Kochanek PM, Marion D (1994). Upregulation of nerve growth factor following cortical trauma. Exp Neurol 130:173–177. [DOI] [PubMed] [Google Scholar]
- Fahnestock M, Michalski B, Xu B, Coughlin MD (2001). The precursor pro-nerve growth factor is the predominant form of nerve growth factor in brain and is increased in Alzheimer’s disease. Mol Cell Neurosci 18:210–220. [DOI] [PubMed] [Google Scholar]
- Farinelli SE, Greene LA, Friedman WJ (1998). Neuroprotective actions of dipyridamole on cultured CNS neurons. J Neurosci 18:5112–5123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer W, Wictorin K, Björklund A, Williams LR, Varon S, Gage FH (1987). Amelioration of cholinergic neuron atrophy and spatial memory impairment in aged rats by nerve growth factor. Nature 329:65–68. [DOI] [PubMed] [Google Scholar]
- Friedman WJ (2000). Neurotrophins induce death of hippocampal neurons via the p75 receptor. J Neurosci 20:6340–6346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman WJ (2005). Interactions of interleukin-1 with neurotrophic factors in the central nervous system: beneficial or detrimental? Mol Neurobiol 32:133–144. [DOI] [PubMed] [Google Scholar]
- Friedman WJ, Greene LA (1999). Neurotrophin signaling via Trks and p75. Exp Cell Res 253:131–142. [DOI] [PubMed] [Google Scholar]
- Friedman WJ, Larkfors L, Ayer-LeLievre C, Ebendal T, Olson L, Persson H (1990). Regulation of β-nerve growth factor expression by inflammatory mediators in hippocampal cultures. J Neurosci Res 27:374–382. [DOI] [PubMed] [Google Scholar]
- Friedman WJ, Ibanez CF, Hallbook F, Persson H, Cain LD, Dreyfus CF, Black IB (1993). Differential actions of neurotrophins in the locus coeruleus and basal forebrain. Exp Neurol 119:72–78. [DOI] [PubMed] [Google Scholar]
- Gall CM, Isackson PJ (1989). Limbic seizures increase neuronal production of messenger RNA for nerve growth factor. Science 245:758–761. [DOI] [PubMed] [Google Scholar]
- Goss JR, O’Malley ME, Zou L, Styren SD, Kochanek PM, DeKoskey ST (1998). Astrocytes are the major source of nerve growth factor upregulation following traumatic brain injury in the rat. Exp Neurol 149:301–309. [DOI] [PubMed] [Google Scholar]
- Greene LA, Kaplan DR (1995). Early events in neurotrophin signaling via Trk and p75 receptors. Curr Top Neurobiol 5:579–587. [DOI] [PubMed] [Google Scholar]
- Greferath U, Bennie A, Kourakis A, Bartlett PF, Murphy M, Barrett GL (2000). Enlarged cholinergic forebrain neurons and improved spatial learning in p75 knockout mice. Eur J Neurosci 12:885–893. [DOI] [PubMed] [Google Scholar]
- Harrington A, Leiner B, Blechschmitt C, Arevalo J, Lee R, Morl K, Meyer M, Hempstead B, Yoon S, Giehl K (2004). Secreted proNGF is a pathophysiological death-inducing ligand after adult CNS injury. Proc Natl Acad Sci USA 101:6226–6230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hefti F, Hartikka J, Eckenstein F, Gnahn H, Heumann R, Schwab M (1985). Nerve growth factor increases choline acetyltransferase but not survival or fiber outgrowth of cultured fetal septal cholinergic neurons. Neuroscience 14:55–68. [DOI] [PubMed] [Google Scholar]
- Hempstead BL (2002). The many faces of p75NTR. Curr Opin Neurobiol 12:260–267. [DOI] [PubMed] [Google Scholar]
- Huber LJ, Chao MV (1995). Mesenchymal and neuronal cell expression of the p75 neurotrophin receptor are distinguished during morphogenesis of transgenic animals. Dev Biol 167:227–238. [DOI] [PubMed] [Google Scholar]
- Isackson PJ, Huntsman MM, Murray KD, Gall CM (1991). BDNF mRNA expression is increased in adult rat forebrain after limbic seizures: temporal patterns of induction distinct from NGF. Neuron 6:937–948. [DOI] [PubMed] [Google Scholar]
- Kaplan DR, Miller FD (2000). Neurotrophin signal transduction in the nervous system. Curr Opin Neurobiol 10:381–391. [DOI] [PubMed] [Google Scholar]
- Klein R, Silos-Santiago I, Smeyne RJ, Lira SA, Brambilla R, Bryant S, Zhang L, Snider W, Barbacid M (1994). Disruption of the neurotrophin-3 receptor gene trkC eliminates 1a muscle afferents and results in abnormal movements. Nature 368:249–251. [DOI] [PubMed] [Google Scholar]
- Kromer LF (1987). Nerve growth factor treatment after brain injury prevents neuronal death. Science 235:214–216. [DOI] [PubMed] [Google Scholar]
- Lee KF, Li E, Huber LJ, Landis SC, Sharpe AH, Chao MV, Jaenisch R (1992). Targeted mutation of the gene encoding the low affinity NGF receptor p75 leads to deficits in the peripheral sensory nervous system. Cell 69:737–749. [DOI] [PubMed] [Google Scholar]
- Lee R, Kermani P, Teng KK, Hempstead BL (2001). Regulation of cell survival by secreted proneurotrophins. Science 294:1945–1948. [DOI] [PubMed] [Google Scholar]
- Maroney AC, Finn JP, Bozyczko-Coyne D, O’Kane TM, Neff NT, Tolkovsky AM, Park DS, Yan CY, Troy CM, Greene LA (1999). CEP-1347 (KT7515), an inhibitor of JNK activation, rescues sympathetic neurons and neuronally differentiated PC12 cells from death evoked by three distinct insults. J Neurochem 73:1901–1912. [PubMed] [Google Scholar]
- Mazzoni IE, Said FA, Aloyz R, Miller FD, Kaplan D (1999). Ras regulates sympathetic neuron survival by suppressing the p53-mediated cell death pathway. J Neurosci 19:9716–9727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naumann T, Casademunt E, Hollerbach E, Hofmann J, Dechant G, Frotscher M, Barde YA (2002). Complete deletion of the neurotrophin receptor p75NTR leads to long-lasting increases in the number of basal forebrain cholinergic neurons. J Neurosci 22:2409–2418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nykjaer A, Lee R, Teng K, Jansen P, Madsen P, Nielsen M, Jacobsen C, Kliemannel M, Schwarz E, Willnow T, Hempstead B, Petersen C (2004). Sortilin is essential for proNGF-induced neuronal cell death. Nature 427:843–848. [DOI] [PubMed] [Google Scholar]
- Oderfeld-Nowak B, Bacia A (1994). Expression of astroglial nerve growth factor in damaged brain. Acta Neurobiol Exp 54:73–80. [PubMed] [Google Scholar]
- Oh JD, Charisathian K, Chase TN, Butcher LL (2000). Overexpression of neurotrophin receptor p75 contributes to the excitotoxin-induced cholinergic neuronal death in rat basal forebrain. Brain Res 853:174–185. [DOI] [PubMed] [Google Scholar]
- Patapoutian A, Reichardt LF (2001). Trk receptors: mediators of neurotrophin action. Curr Opin Neurobiol 11:272–280. [DOI] [PubMed] [Google Scholar]
- Peng S, Wuu J, Mufson EJ, Fahnestock M (2004). Increased proNGF levels in subjects with mild cognitive impairment and mild Alzheimer disease. J Neuropathol Exp Neurol 63:641–649. [DOI] [PubMed] [Google Scholar]
- Peterson DA, Dickinson-Anson HA, Leppert JT, Lee KF, Gage FH (1999). Central neuronal loss and behavioral impairment in mice lacking neurotrophin receptor p75. J Comp Neurol 404:1–20. [DOI] [PubMed] [Google Scholar]
- Rabizadeh S, Oh J, Zhong L, Yang J, Bitler CM, Butcher LL, Bredesen DE (1993). Induction of apoptosis by the low-affinity NGF receptor. Science 261:345–358. [DOI] [PubMed] [Google Scholar]
- Roux P, Barker P (2002). Neurotrophin signaling through the p75 neurotrophin receptor. Prog Neurobiol 67:203–233. [DOI] [PubMed] [Google Scholar]
- Roux PP, Colicos MA, Barker PA, Kennedy TE (1999). p75 neurotrophin receptor expression is induced in apoptotic neurons after seizure. J Neurosci 19:6887–6896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rukenstein A, Rydel RE, Greene LA (1991). Multiple agents rescue PC12 cells from serum-free cell death by translation- and transcription-independent mechanisms. J Neurosci 11:2552–2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schauwecker PE, Steward O (1997). Genetic determinants of susceptibility to excitotoxic cell death: Implications for gene targeting approaches. Proc Natl Acad Sci USA 94:4103–4108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmued LC, Hopkins KJ (2000). Fluoro-Jade B: a high affinity fluorescent marker for the localization of neuronal degeneration. Brain Res 874:123–130. [DOI] [PubMed] [Google Scholar]
- Schmued LC, Albertson C, Slikker W Jr (1997). Fluoro-Jade: a novel fluorochrome for the sensitive and reliable histochemical localization of neuronal degeneration. Brain Res 751:37–46. [DOI] [PubMed] [Google Scholar]
- Smeyne RJ, Klein R, Schnapp A, Long LK, Bryant S, Lewin A, Lira SA, Barbacid M (1994). Severe sensory and sympathetic neuropathies in mice carrying a disrupted trk/NGF receptor gene. Nature 368:246–249. [DOI] [PubMed] [Google Scholar]
- Spranger M, Lindholm D, Bandtlow C, Heumann R, Gnahn H, Naher-Noe M, Thoenen H (1990). Regulation of nerve growth factor (NGF) synthesis in the rat central nervous system: comparison between the effects of interleukin-1 and various growth factors in astrocyte cultures and in vivo. Eur J Neurosci 2:69–76. [DOI] [PubMed] [Google Scholar]
- Stefanis L, Park DS, Friedman WJ, Greene LA (1999). Caspase-dependent and independent death of camptothecin-treated embryonic cortical neurons. J Neurosci 19:6235–6247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng HK, Teng KK, Lee R, Wright S, Tevar S, Almeida RD, Kermani P, Torkin R, Chen ZY, Lee FS, Kraemer RT, Nykjaer A, Hempstead BL (2005). ProBDNF induces neuronal apoptosis via activation of a receptor complex of p75NTR and sortilin. J Neurosci 25:5455–5463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Troy CM, Friedman JE, Friedman WJ (2002). Mechanisms of p75-mediated death of hippocampal neurons: role of caspases. J Biol Chem 277:34295–34302. [DOI] [PubMed] [Google Scholar]
- van der Zee CEE, Ross GM, Riopelle RJ, Hagg T (1996). Survival of cholinergic forebrain neurons in developing p75 NGFR-deficient mice. Science 274:1729–1732. [DOI] [PubMed] [Google Scholar]
- Wang X, Bauer JH, Li Y, Shao Z, Zetoune FS, Cattaneo E, Vincenz C (2001). Characterization of a p75NTR apoptotic signaling pathway using a novel cellular model. J Biol Chem 276:33812–33820. [DOI] [PubMed] [Google Scholar]
- Yeo TT, Chua-Couzens J, Butcher LL, Bredesen DE, Cooper JD, Valletta JS, Mobley WC, Longo FM (1997). Absence of p75NTR causes increased basal forebrain cholinergic neuron size, choline acetyltransferase activity, and target innervation. J Neurosci 17:7594–7605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon SO, Casaccia-Bonnefil P, Carter B, Chao MV (1998). Competitive signaling between TrkA and p75 nerve growth factor receptors determines cell survival. J Neurosci 18:3273–3281. [DOI] [PMC free article] [PubMed] [Google Scholar]