

Abstract
Hypercapnia evokes a strong cardiorespiratory response including gasping and a pronounced bradycardia; however, the mechanism responsible for these survival responses initiated in the brainstem is unknown. To examine the effects of hypercapnia on the central cardiorespiratory network, we used an in vitro medullary slice that allows simultaneous examination of rhythmic respiratory-related activity and inhibitory synaptic neurotransmission to cardioinhibitory vagal neurons (CVNs). Hypercapnia differentially modulated inhibitory neurotransmission to CVNs; whereas hypercapnia selectively depressed spontaneous glycinergic IPSCs in CVNs without altering respiratory-related increases in glycinergic neurotransmission, it decreased both spontaneous and inspiratory-associated GABAergic IPSCs. Because maternal smoking is the highest risk factor for sudden infant death syndrome (SIDS) and prenatal nicotine exposure is proposed to be the link between maternal smoking and SIDS, we examined the cardiorespiratory responses to hypercapnia in animals exposed to nicotine in the prenatal and perinatal period. In animals exposed to prenatal nicotine, hypercapnia evoked an exaggerated depression of GABAergic IPSCs in CVNs with no significant change in glycinergic neurotransmission. Hypercapnia altered inhibitory neurotransmission to CVNs at both presynaptic and postsynaptic sites. Although the results obtained in this study in vitro cannot be extrapolated with certainty to in vivo responses, the results of this study provide a likely neurochemical mechanism for hypercapnia-evoked bradycardia and the dysregulation of this response with exposure to prenatal nicotine, creating a higher risk for SIDS.
Keywords: ambiguus, SIDS, GABA, glycine, prenatal nicotine, hypercapnia
Introduction
The cardiovascular and respiratory systems are highly integrated, with many physiological challenges requiring finely coordinated responses from both networks. The most ubiquitous cardiorespiratory interaction is respiratory sinus arrhythmia. The heart rate increases during inspiration and decreases during expiration, which maintains the optimal diffusion gradient for oxygen in the lungs (Loewy and Spyer, 1990; Taylor et al., 1999). In addition, respiratory challenges, such as hypoxia or hypercapnia, evoke strong coordinated cardiorespiratory responses. Hypoxia transforms respiration from eupnea to gasping, characterized by a transient increase, followed by a subsequent decrease in respiratory frequency (Guntheroth and Kawabori, 1975; Gozal et al., 2002). In addition, hypoxia and hypercapnia evoke an initial tachycardia, followed by a bradycardia mediated primarily by increases in parasympathetic activity (Taylor and Butler, 1982; Schuen et al., 1997). These responses normally act to increase survival; however, an exaggeration of these responses has been implicated in sudden infant death syndrome (SIDS). Infants at increased risk for SIDS exhibit delayed arousal and experience a greater bradycardia during hypercapnic challenge than control infants (van der Hal et al., 1985; Edner et al., 2002). Furthermore, infants who die of SIDS typically experience a severe bradycardia that precedes or is accompanied by a centrally mediated life-threatening apnea and gasping (Meny et al., 1994; Nachmanoff et al., 1998; Poets et al., 1999; Fewell et al., 2001).
Smoking during pregnancy is a major risk factor for SIDS (Malloy et al., 1988; Bulterys et al., 1990; Haglund and Cnattingius, 1990; Schoendorf and Kiely, 1992), and this risk increases proportionally with the number of cigarettes smoked (Haglund and Cnattingius, 1990). Although cigarettes contain a variety of chemicals, nicotine has been proposed to be the link between maternal smoking and SIDS (Slotkin et al., 1997; Nachmanoff et al., 1998; Bamford and Carroll, 1999; St-John and Leiter, 1999). In animal models of SIDS, prenatal nicotine exposure augments the parasympathetic and diminishes the sympathetic control of heart rate (Slotkin et al., 1997, 1999). Rats exposed to nicotine prenatally have a lower basal heart rate than unexposed animals (Fewell et al., 2001), and neonatal rats exposed to nicotine in utero show reduced tolerance for hypoxia caused by impaired heart rate control (Slotkin et al., 1997). Furthermore, human infants of smoking mothers also exhibit exaggerated heart rate responses to hypoxia (Sovik et al., 2001).
An essential component of heart rate control originates in the parasympathetic cardioinhibitory vagal neurons (CVNs) of the nucleus ambiguus (Mendelowitz, 1999). CVNs are inherently silent and require synaptic neurotransmission to determine their activity (Mendelowitz, 1996). CVNs receive inhibitory GABAergic and glycinergic neurotransmission, and both inhibitory pathways are facilitated by nicotinic acetylcholine receptors (Neff et al., 2003; Wang et al., 2003). However, although the activity of both GABAergic and glycinergic neurotransmission are increased by inspiratory activity, only GABAergic synaptic inputs are mediated by α4β2 nicotinic acetylcholine receptors during inspiration (Neff et al., 2003). This study tests whether hypercapnia modulates central cardiorespiratory interactions, tests whether prenatal nicotine alters these responses, and determines the site(s) of action of hypercapnia.
Materials and Methods
Animal preparation. Sprague Dawley rat pups (postnatal days 1–3; Hill-top, Scottsdale, PA) were anesthetized with ketamine/xylazine (87/13 mg/kg) and exposed to hypothermia to slow the heart. The heart was exposed by a right thoractomy, and the retrograde fluorescent tracer X-rhodamine-5 (and 6)-isothiocyanate (Molecular Probes, Eugene, OR) was injected into the fat pads at the base of the heart. After 24–48 h of recovery, pups were anesthetized with halothane and killed by cervical dislocation, and the hindbrain was rapidly removed and placed in cold physiological saline solution (in mm: 140 NaCl, 5 KCl, 2 CaCl2, 5 glucose, and 10 HEPES, bubbled with 100% O2, pH 7.4). The medulla was removed carefully, preserving the hypoglossal nerve rootlets. The medulla was mounted on a cutting block and placed in a vibrating blade microtome (Leica, Nussloch, Germany). Serial coronal sections of the medulla were sliced until the inferior olive and the nucleus ambiguus were visualized on the rostral surface of the tissue. A single thick slice of the medulla (800 μm) was made that included CVNs, the rostral hypoglossal nucleus and rootlets, and the pre-Botzinger complex. This slice was placed in a recording chamber that allowed perfusion (4 ml/min) with artificial CSF (aCSF) (in mm: 125 NaCl, 3 KCl, 2 CaCl2, 26 NaHCO3, 5 glucose, and 5 HEPES, equilibrated with 95% O2/5% CO2, pH 7.35–7.4). All animal procedures were performed with the approval of the Animal Care and Use Committee of The George Washington University in accordance with the recommendations of the panel on euthanasia of the American Veterinary Medical Association and the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Recording respiratory network activity. The thick medullary slice generates rhythmic respiratory-related motor discharge in hypoglossal cranial nerves. Spontaneous respiratory-related activity was recorded by monitoring motoneuron population activity from hypoglossal nerve rootlets using a suction electrode. Hypoglossal rootlet activity was amplified 50,000 times, filtered (10–300 Hz bandpass; CWE, Ardmore, PA), and electronically integrated (τ = 50 ms; CWE).
Patch-clamp techniques. CVNs in the nucleus ambiguus were identified by the presence of the fluorescent tracer, as described previously (Mendelowitz and Kunze, 1991; Mendelowitz, 1996). Specificity of the cardiac vagal labeling is confirmed by the absence of any labeled neurons in the brainstem when rhodamine is injected into the chest cavity while keeping the pericardial sac intact, or when the injection into the pericardial sac is accompanied by section of the cardiac branch of the vagus nerve (n = 4). In other control experiments (n = 10), intravenous injection of up to 10 mg of rhodamine failed to label any neurons in the medulla, except for rare labeling of neurons in the area postrema, an area with a deficient blood–brain barrier. Although a recent report by Grkovic et al. (2005) suggests there can be a lack of specificity using fluorescent tracers different from the one used in this study, they injected tracers into the cardiac sac via an incision in the abdominal cavity and through the diaphragm, procedures that also greatly increase the probability of erroneous labeling. Slices were viewed with infrared illumination and differential interference optics (Zeiss, Oberkochen, Germany) and under fluorescent illumination with an infrared-sensitive cooled charged-coupled device camera (Photometrics, Tucson, AZ). Neurons containing fluorescent tracer were identified by superimposing the fluorescent and infrared images on a video monitor (Sony, Tokyo, Japan).
Patch pipettes (2.5–4.5 MΩ) were visually guided to the surface of individual CVNs using differential interference optics and infrared illumination (Zeiss). Pipettes were filled with a solution containing (in mm) 150 KCl, 4 MgCl2, 2 EGTA, 2 Na-ATP, and 10 HEPES, pH 7.4. This pipette solution causes the Cl– current induced by the activation of GABA or glycine receptors to be recorded as an inward current. Voltage-clamp recordings were made with Axopatch 200B and pClamp 8 software (Molecular Devices, Union City, CA). All synaptic activity in CVNs was recorded at –80 mV. Only one experiment was performed per preparation.
Focal drug application. Focal drug application was performed using a pneumatic picopump pressure delivery system (WPI, Sarasota, FL). Drugs were ejected from a patch pipette positioned within 30 μmofthe patched CVN. The maximum range of drug application has been determined previously to be 100–120 μm downstream from the drug pipette and considerably less behind the drug pipette (Wang et al., 2002). Glycinergic neurotransmission was isolated by focal application of d-2-amino-5-phosphonovalerate (AP-5; 50 μm), 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX; 50 μm), and gabazine (25 μm) to block NMDA, non-NMDA, and GABA receptors, respectively. GABAergic neurotransmission was isolated by focal application of AP-5, CNQX, and strychnine hydrochloride (1 μm) to block NMDA, non-NMDA, and glycine receptors, respectively. All drugs were obtained from Sigma (St. Louis, MO).
Miniature glycinergic and GABAergic IPSCs (mIPSCs) were isolated with tetrodotoxin (TTX; 1 μm) to block action potential-dependent events. AP-5 (50 μm), CNQX (50 μm), and strychnine hydrochloride (1 μm) or gabazine (25 μm) were applied to block NMDA, non-NMDA, and glycinergic or GABAergic currents, respectively. In a separate set of experiments, glycine (100 μm) or GABA (100 μm) were focally applied to CVNs to evoke glycinergic or GABAergic postsynaptic currents, respectively. Glycine or GABA was puffed for 100 ms every 10 s for 20 min.
Hypercapnia. Rhythmic inspiratory-related activity and the glycinergic or GABAergic IPSCs in a single CVN were recorded simultaneously for 4 min in control aCSF (in mm: 125 NaCl, 3 KCl, 2 CaCl2, 26 NaHCO3, 5 glucose, and 5 HEPES, equilibrated with 95% O2/5% CO2, pH 7.35–7.4, equilibrated with NaOH). Hypercapnia was induced by changing the control perfusate to an identical solution bubbled with 8% CO2/92% O2. After a 1 h equilibration with 8% CO2/92% O2, the pH of this solution was 7.12. Slices were exposed to hypercapnia for 10 min and returned to the original perfusate for 20 min. At the end of each experiment, glycinergic or GABAergic activity was reversibly inhibited using strychnine (1 μm) or gabazine (25 μm), respectively.
Prenatal nicotine exposure. Adult female rats were anesthetized with ketamine/xylazine (87/13 mg/kg, i.p.; Phoenix Pharmaceuticals, St. Joseph, MO) on the third day of gestation and implanted with Alzet osmotic minipumps (Durect, Cupertine, CA) containing (–)nicotine (56.1 mg/ml bacteriostatic saline; Sigma). Osmotic minipumps were chosen to avoid the high plasma nicotine concentrations and subsequent episodic fetal hypoxia–ischemia that can be produced by nicotine injections. Pumps delivered 2.1 mg of nicotine per day, a level approximately equivalent to those that occur in moderate to heavy smokers, for 28 d (Slotkin et al., 1997).
Data analysis. Analysis of spontaneous synaptic currents was performed using MiniAnalysis (version 5.6.12; Synaptosoft, Decatur, GA) with minimal acceptable amplitude at 9 pA for GABAergic events and 12 pA for glycinergic events. The frequency of IPSCs that occurred in CVNs was grouped into 1 s bins and cross-correlated with the onset of inspiratory-related hypoglossal activity. Data were analyzed from all bursts during the last 2 min of the control period, during the last 2 min of hypercapnia, and from 10–12 min during the recovery. These periods were chosen for analysis because during these periods, synaptic activities were consistent and any hypercapnia-evoked changes reached a steady state. Burst duration, frequency, and amplitude were measured using pClamp 8 software (Molecular Devices) and from the filtered (10–300 Hz bandpass) and electronically integrated hypoglossal rootlet activity. Results are presented as mean ± SEM. Statistical comparisons were performed using ANOVA with repeated measures to examine the responses throughout the time course of the experiments and using an unpaired t test to compare the results from control animals with animals that were exposed to nicotine prenatally. Significant difference was set at p < 0.05.
Results
Respiratory activity in the thick medullary slice
As observed in other similar medullary respiratory slice preparations, the frequency of respiratory-related hypoglossal bursts is significantly lower than that of in vivo preparations. This is likely attributable to the reduced temperature of the in vitro preparation and the absence of sensory input to the medulla (Rekling and Feldman, 1998). Hypercapnia evoked a statistically significant (p < 0.01) increase in respiratory burst frequency, followed by a small but not statistically significant depression of respiratory burst frequency (Fig. 1). In addition, hypercapnia significantly depressed (p < 0.01) respiratory burst duration. Hypercapnia induced a small increase in the amplitude of respiratory bursts, but these changes did not reach significance (p > 0.05). The responses in animals exposed to prenatal nicotine closely mimicked and were not significantly different from the responses in unexposed animals (Fig. 1).
Figure 1.

Hypercapnia evokes biphasic changes in central respiratory activity. A, Changing the perfusate from aCSF equilibrated with 95% O2/5% CO2 to aCSF equilibrated with 92% O2/8% CO2 elicited a biphasic change in respiratory activity as burst frequency initially significantly increased and subsequently returned to control levels, as shown in are presentative experiment. B, Respiratory burst frequency (Freq.) significantly increased during hypercapnia in both unexposed (□; p < 0.01; n = 15) and prenatal nicotine-exposed (○; n = 9) animals. C, Hypercapnia significantly depressed burst duration in both unexposed and prenatal nicotine-exposed animals. D, Hypercapnia induced a small, but not statistically significant, rise in burst amplitude (Amp.) in both unexposed and prenatal nicotine-exposed animals. There were no statistically significant differences in the respiratory responses between unexposed and prenatal nicotine-exposed animals. In this and all subsequent figures, *,+p < 0.05 and **,++p < 0.01 in unexposed and prenatal nicotine-treated animals, respectively.
Hypercapnic modulation of glycinergic neurotransmission to CVNs
Unexposed animals
Similar to previous reports (Neff et al., 2003, 2004), glycinergic IPSC frequency in CVNs significantly increased from 9.2 ± 0.6 to 14.6 ± 1.3 Hz during inspiratory bursts (p < 0.01; n = 11 cells), see Figure 2. Hypercapnia evoked a significant decrease in spontaneous glycinergic IPSC frequency (control, 9.2 ± 0.6 Hz; hypercapnia, 6.0 ± 0.5 Hz; p < 0.01; n = 11 cells). However, inspiratory glycinergic IPSC frequency was not significantly altered during hypercapnia (control, 14.6 ± 1.3 Hz; hypercapnia, 13.5 ± 1.5 Hz; p > 0.05; n = 11 cells). The percentage of change in inspiratory/spontaneous glycinergic IPSC frequency was significantly increased from 61.0 ± 8.7% (control) to 133.7 ± 19.6% (hypercapnia) (p < 0.01; n = 11 cells). The amplitude of glycinergic IPSCs in CVNs was not modulated by respiratory activity during control or hypercapnic conditions. However, the amplitude of glycinergic IPSCs was significantly decreased during hypercapnia (control, 69.1 ± 3.2 pA; hypercapnia, 48.6 ± 2.6 pA; p < 0.01; n = 11 cells). Glycinergic neurotransmission returned to control levels during recovery from hypercapnia, and all glycinergic IPSCs were inhibited by the focal application of the glycinergic antagonist strychnine (1 μm).
Figure 2.
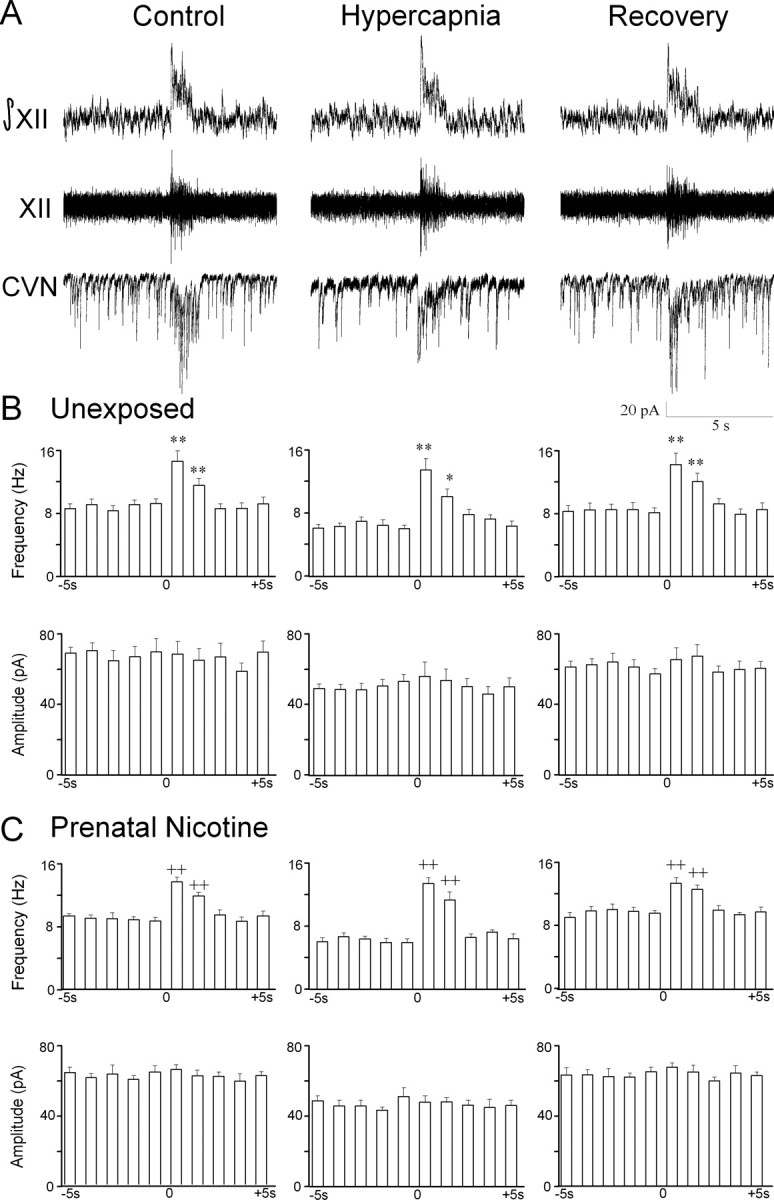
Hypercapnia selectively depresses spontaneous glycinergic IPSC frequency. A, Inspiratory-related bursting activity was recorded from the hypoglossal rootlet (XII) and electronically integrated (XII). Fluorescently identified CVNs were patch clamped in the whole-cell configuration, and glycinergic neurotransmission was isolated by focal application of NMDA, non-NMDA, and GABAA receptor antagonists. The frequency of glycinergic IPSCs significantly increased during inspiratory bursts. Hypercapnia elicited a significant decrease in spontaneous glycinergic IPSCs but did not alter inspiratory-evoked IPSCs. IPSC amplitude was not modulated by inspiratory bursts; however, hypercapnia significantly inhibited glycinergic IPSC amplitude. A typical experiment from an unexposed animal is shown in A, and the average data from 11 cells are shown in B. C, In animals exposed to nicotine prenatally, hypercapnia significantly inhibited the frequency of spontaneous but not inspiratory glycinergic IPSCs. IPSC amplitude was not modulated by inspiratory bursts, although hypercapnia significantly decreased IPSC amplitude. There were no significant differences in unexposed and prenatal nicotine-exposed animals. *p < 0.05; **, ++ p < 0.01.
Prenatal nicotine-exposed animals
Prenatal nicotine exposure did not significantly alter spontaneous or inspiratory glycinergic IPSC frequency under control conditions (p > 0.05; n = 7 cells) (Fig. 2). Furthermore, changes in glycinergic IPSC frequency during hypercapnia were similar to those observed in unexposed animals. Hypercapnia elicited a decrease in spontaneous glycinergic IPSC frequency in CVNs (control, 8.6 ± 0.5 Hz; hypercapnia, 5.6 ± 0.5 Hz; p < 0.01, n = 7 cells). Inspiratory glycinergic IPSC frequency was not altered by hypercapnia (control, 13.5 ± 0.6 Hz; hypercapnia, 12.8 ± 0.7 Hz; p > 0.05; n = 7 cells). Similar to unexposed animals, the percentage of change in inspiratory/spontaneous glycinergic IPSC frequency was significantly increased from 41.7 ± 18.9% (control) to 133.8 ± 11.3% (hypercapnia) (p < 0.05; n = 7 cells). The amplitude of glycinergic IPSCs in CVNs was not modulated by respiratory activity during control of hypercapnic conditions, although the amplitude of glycinergic IPSCs significantly decreased during hypercapnia (control, 64.1 ± 3.7 pA; hypercapnia 50.4 ± 5.0 pA; p < 0.05; n = 7 cells). The changes in glycinergic neurotransmission were reversible and returned to control levels after recovery with the control perfusate. All glycinergic IPSCs were inhibited by the focal application of the glycinergic antagonist strychnine (1 μm).
Hypercapnic modulation of GABAergic neurotransmission to CVNs
Unexposed animals
Consistent with previous reports (Neff et al., 2003, 2004), the frequency of GABAergic IPSCs significantly increased during inspiratory bursts (spontaneous, 5.1 ± 0.4 Hz; inspiratory, 8.8 ± 0.8 Hz; p < 0.01; n = 15 cells) (Fig. 3). After exposure to hypercapnia, the frequency of spontaneous GABAergic IPSCs decreased (control, 5.1 ± 0.4 Hz; hypercapnia, 3.8 ± 0.5 Hz; p < 0.01; n = 15 cells). In contrast to the glycinergic responses, inspiratory GABAergic IPSC frequency also decreased during the hypercapnic insult (control, 8.8 ± 0.8 Hz; hypercapnia, 5.8 ± 0.4 Hz; p < 0.01; n = 15 cells). The percentage of change in inspiratory/spontaneous GABAergic IPSC frequency was not significantly altered from 78.3 ± 12.3% (control) to 73.5 ± 13.9% (hypercapnia) (p > 0.05; n = 15 cells).
Figure 3.

Hypercapnia diminishes GABAergic IPSC frequency. GABAergic synaptic activity was isolated by focal application of NMDA, non-NMDA, and glycinergic receptor antagonists. A, As shown for a typical experiment, GABAergic IPSC frequency significantly increased during respiratory bursts. B, After exposure to hypercapnia, both spontaneous and inspiratory GABAergic IPSC frequency was depressed. C, GABAergic IPSC amplitude was not modulated by respiratory bursts; however, hypercapnia diminished IPSC amplitude. *p < 0.05; **p < 0.01.
The amplitude of GABAergic IPSCs in CVNs was not modulated by respiratory activity under control or hypercapnic conditions (Fig. 3). However, the amplitude of GABAergic IPSCs was generally inhibited during hypercapnia (control, 51.5 ± 4.3 pA; hypercapnia, 41.4 ± 3.7 pA; p < 0.05; n = 15 cells). The changes in GABAergic neurotransmission were reversible and returned to control levels after recovery with the control perfusate. All GABAergic IPSCs under these recording conditions were blocked by the application of the GABAA antagonist gabazine.
Prenatal nicotine-exposed animals
As in unexposed animals, the frequency of GABAergic IPSCs in CVNs significantly increased during inspiratory bursts in animals exposed to prenatal nicotine (spontaneous, 5.4 ± 0.8 Hz; inspiratory, 11.6 ± 0.7 Hz; p < 0.01; n = 9 cells) (Fig. 4). However, consistent with previous reports (Neff et al., 2003, 2004), the inspiratory-evoked increase in GABAergic IPSC frequency was dramatically increased relative to unexposed animals (unexposed, 8.8 ± 0.8 Hz; exposed, 11.6 ± 0.7 Hz; p < 0.05). Likewise, prenatal nicotine also significantly increased the percentage of change in inspiratory/spontaneous GABAergic IPSC frequency (unexposed, 78.3 ± 12.3%; exposed, 149.7 ± 37.8%; p < 0.05).
Figure 4.

Prenatal nicotine exposure exaggerates GABAergic IPSC frequency depression during hypercapnia. A, In animals exposed to nicotine prenatally, the inspiratory-evoked increase in GABAergic frequency was significantly increased compared with control animals, as shown in a typical experiment. B, Hypercapnia evoked a significant decrease in both spontaneous and inspiratory GABAergic IPSC amplitude. C, GABAergic IPSC amplitude was not modulated by respiratory bursts; however, hypercapnia diminished IPSC amplitude in animals exposed to nicotine prenatally. +p < 0.05; ++p < 0.01.
Hypercapnia significantly decreased spontaneous IPSC frequency in CVNs in animals exposed to prenatal nicotine (control, 5.4 ± 0.8 Hz; hypercapnia 2.0 ± 0.3 Hz; p < 0.01; n = 9 cells). Inspiratory GABAergic IPSC frequency was also significantly depressed during hypercapnia in animals exposed to prenatal nicotine (control, 11.6 ±0.7 Hz; hypercapnia, 4.6 ± 0.3 Hz; p < 0.01; n = 9 cells). Similar to unexposed animals, the percentage of change in inspiratory/spontaneous GABAergic IPSC frequency was not significantly altered in prenatal nicotine-exposed animals during hypercapnia (control, 149.7 ± 37.8%; hypercapnia, 149.4 ± 24.1%; p > 0.05; n = 9 cells). This ratio, however, remained exaggerated relative to the percentage of change in inspiratory/spontaneous frequency during hypercapnia in unexposed animals (unexposed, 73.5 ± 13.9%; exposed, 149.4 ± 24.1%; p < 0.01).
The amplitude of GABAergic IPSCs was not modulated by respiratory activity under control or hypercapnic conditions in animals exposed to prenatal nicotine. However, the amplitude of GABAergic IPSCs was diminished during hypercapnia (control, 63.2 ± 3.1 pA; hypercapnia, 32. 8 ± 2.1%; p < 0.01). The changes in GABAergic neurotransmission in prenatal nicotine-exposed animals were reversible and returned to control levels after recovery with the control perfusate. All GABAergic IPSCs under these recording conditions were blocked by application of the GABAA antagonist gabazine (25 μm).
Effect of hypercapnia on GABAergic and glycinergic spontaneous mIPSCs
To determine whether hypercapnia modulates the presynaptic release and postsynaptic response of CVNs to inhibitory neurotransmitters, spontaneous mIPSCs were examined. These experiments were performed with the application of TTX (1 μm) to prevent action potential-dependent events. Hypercapnia significantly inhibited GABAergic mIPSC frequency (control, 3.2 ± 0.2 Hz; hypercapnia 2.2 ± 0.1 Hz; p < 0.05; n = 8 cells) (Fig. 5). GABAergic mIPSC amplitude was also significantly decreased during hypercapnia (control, 18.7 ± 1.0 pA; hypercapnia, 13.0 ± 1.4 pA; p < 0.01; n = 8 cells). Similarly, hypercapnia significantly reduced glycinergic mIPSC frequency (control, 2.5 ± 0.2 Hz; hypercapnia, 1.8 ± 0.2 Hz; p < 0.05; n = 7 cells) and amplitude (control, 25.2 ± 1.2 pA; hypercapnia 17.8 ± 1.4 pA; p < 0.01).
Figure 5.
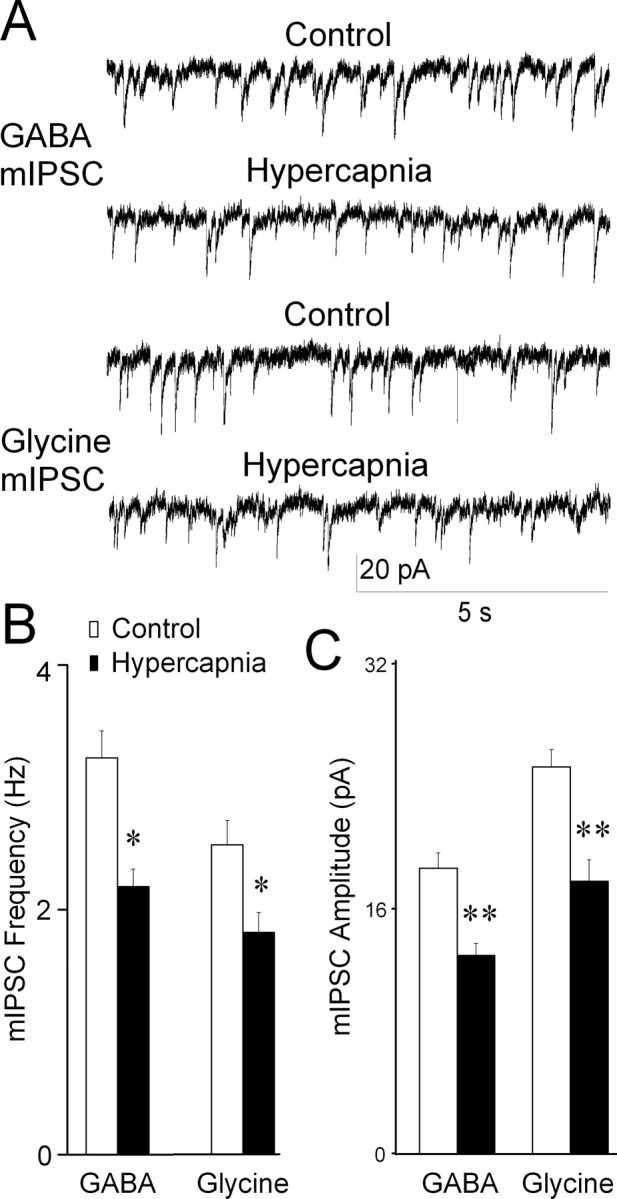
Hypercapnia inhibits GABAergic and glycinergic mIPSCs in CVNs. A, Representative traces showing the effect of hypercapnia on inhibitory mIPSCs. Application of TTX (1 μm), NMDA, non-NMDA, and GABAergic or glycinergic receptor antagonists induces TTX-insensitive, glycinergic, or GABAergic mIPSCs, respectively. Both the frequency and amplitude of GABAergic and glycinergic mIPSCs were significantly reduced after exposure to hypercapnia. B, Average data for seven cells. *p < 0.05; **p < 0.01.
Effect of hypercapnia on postsynaptic responses to exogenous GABA and glycine
To examine whether hypercapnia alters postsynaptic responses of CVNs to exogenous inhibitory neurotransmitters, focal application of GABA or glycine were applied under conditions of synaptic blockade with TTX (1 μm), CNQX (50 μm), AP-5 (50 μm), and strychnine (1 μm) or gabazine (25 μm). Hypercapnia partially inhibited GABA-evoked inward currents (control, 173.8 ± 24.8 pA; hypercapnia, 120.9 ± 16.7 pA; p < 0.01; n = 8 cells) (Fig. 6). Likewise, glycine-evoked inward currents in CVNs were also diminished (control, 395.2 ± 42.1 pA; hypercapnia, 333. 8 ± 33.2 pA; p < 0.05; n = 9 cells).
Figure 6.
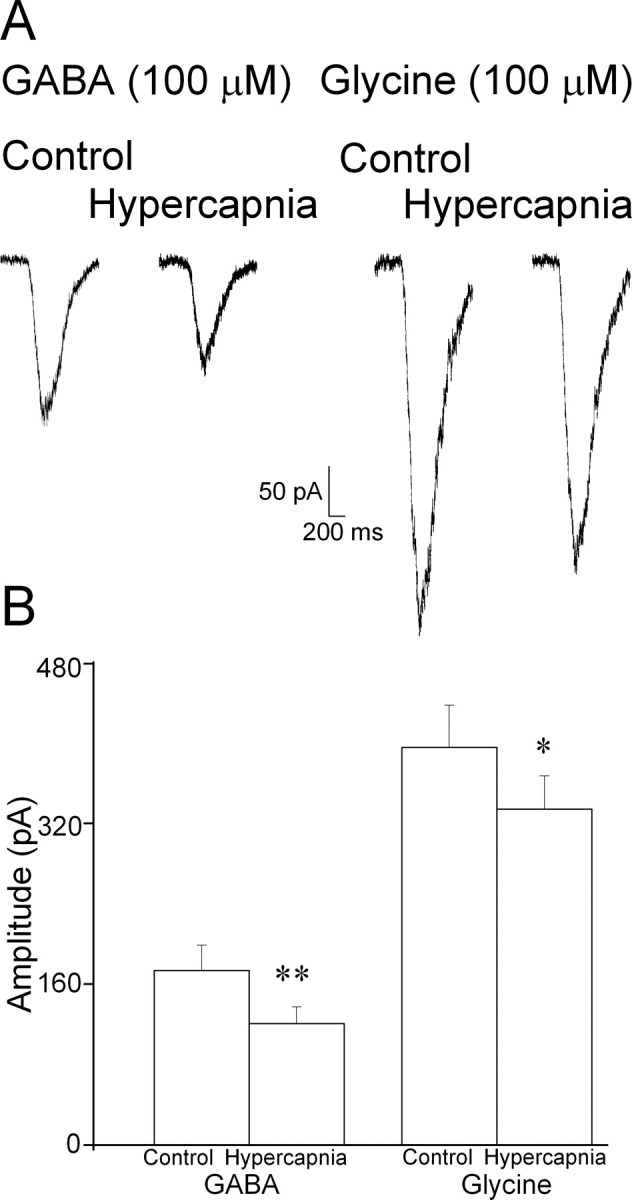
Hypercapnia diminishes CVN postsynaptic responses to inhibitory neurotransmitters. Focal application of GABA (100 μm) or glycine (100 μm) was applied under conditions of synaptic blockade with TTX (1 μm), CNQX (50 μm), AP-5 (50 μm), and strychnine (1 μm) or gabazine (25 μm). A, Representative traces showing the effect of hypercapnia on GABA- and glycine-evoked inward currents in CVNs. As shown in the average data, hypercapnia significantly reduced postsynaptic responses in CVNs to both GABA (n = 8) and glycine (n = 9). *p < 0.05; **p < 0.01.
Discussion
This study has four major results: (1) the central respiratory network exhibits a biphasic response to hypercapnia; (2) hypercapnia differentially modulates inhibitory neurotransmission to CVNs; whereas hypercapnia selectively depresses spontaneous glycinergic IPSCs in CVNs without altering respiratory-related increases in glycinergic neurotransmission, it decreases both spontaneous and inspiratory GABAergic IPSCs; (3) in animals exposed to prenatal nicotine, hypercapnia evokes an exaggerated depression of GABAergic IPSCs in CVNs with no significant change in glycinergic neurotransmission; and (4) hypercapnia alters inhibitory neurotransmission to CVNs at both presynaptic and postsynaptic locations.
Similar to what is observed both in vivo and in vitro with hypoxia, our results indicate hypercapnia also evokes a biphasic change in central respiratory activity. The central respiratory response was characterized by a transient increased bursting frequency and transformation to a pattern that resembles gasping; respiratory bursts exhibited increased amplitude and decreased duration. This biphasic change was not significantly altered by prenatal nicotine exposure, and the respiratory burst characteristics were nearly indistinguishable from unexposed animals. The effect of prenatal nicotine on respiratory physiology is controversial. Maternal cigarette smoking has been reported to have no effect on ventilatory responses to hypoxia or hypercapnia (Lewis and Bosque, 1995) and diminish the hypoxic ventilatory responses and respiratory drive (Ueda et al., 1999). Similarly, it is reported that prenatal nicotine exposure both does not alter (Bamford et al., 1996) and attenuates the ventilatory response to hypoxia or hypercapnia in rats (Simakajornboon et al., 2004). The results from this study indicate that prenatal nicotine exposure does not alter central respiratory responses to hypercapnia and suggests any potential changes in the ventilatory responses to prenatal nicotine in vivo may depend on changes in the activity of peripheral chemoreceptors rather than within the medulla.
Hypercapnia diminished the frequency of inhibitory glycinergic and GABAergic neurotransmission to CVNs. In addition, the amplitude of glycinergic and GABAergic IPSCs was significantly inhibited during hypercapnia. The suppression of both the frequency and amplitude of inhibitory neurotransmission would enhance excitability of CVNs and increase the probability of firing. These changes are consistent with parasympathetic responses to hypercapnia in in vivo preparations. In both intact and carotid chemo-denervated conscious rats, hypercapnia results in a significant bradycardia that can be blocked by atropine (Oikawa et al., 2005). Similarly, hypercapnia is accompanied by increased vagal outflow to the heart, especially during inspiration in decerebrate cats (Yen et al., 2000). The results from this study indicate the mechanism for these responses is the depression of inhibitory neurotransmission to CVNs during hypercapnia, and the bradycardia evoked by hypercapnia in vivo is likely mediated by disinhibition of CVNs.
Our results are also predictive of the hypercapnia-evoked increase in respiratory sinus arrhythmia observed in vivo (Yasuma et al., 2001; Sasano et al., 2002). Although hypercapnia elicited a uniform depression of GABAergic neurotransmission to CVNs, only the spontaneous glycinergic IPSCs were inhibited, whereas inspiratory-evoked glycinergic neurotransmission to CVNs was preserved. Therefore, the withdrawal of GABAergic neurotransmission during hypercapnia may have a greater role in producing hypercapnia-induced bradycardia. In contrast, sustained inspiratory glycinergic IPSCs in the absence of GABAergic neurotransmission may help maintain respiratory sinus arrhythmia. This may in part account for the increase in respiratory sinus arrhythmia induced by hypercapnia in in vivo studies (Yasuma et al., 2001; Sasano et al., 2002). This is in contrast to hypoxia, which is associated with a decrease in respiratory sinus arrhythmia (Yasuma et al., 2001). Indeed, unlike hypercapnia, both inspiratory-evoked glycinergic and GABAergic inhibitory neurotransmission to CVNs is reduced during exposure to hypoxia in vitro (Neff et al., 2004).
Although hypercapnia significantly reduced the postsynaptic responses of CVNs to glycine, the inspiratory-evoked IPSC frequency increase remained unchanged during hypercapnic insult. Therefore, there may be a separate mechanism that maintains this increased frequency during hypercapnia even when postsynaptic responses are diminished. The maintenance of discrete central cardiorespiratory processes during cardiorespiratory challenge has been described previously. For example, cadmium-insensitive pacemaker neurons within the respiratory network continue to burst throughout hypoxia and maintain respiratory rhythm even after cadmium-sensitive pacemakers have ceased bursting (Pena et al., 2004). Furthermore, during hypoxia, ATP is released within the ventrolateral medulla, where it helps maintain respiratory rhythm during hypoxic slowing (Gourine et al., 2005). A similar mechanism may sustain respiratory sinus arrhythmia during hypercapnia by preserving inspiratory-evoked glycinergic neurotransmission to CVNs.
In addition to inhibiting the presynaptic release of glycine and GABA, we have identified a novel postsynaptic depression of CVN responses to inhibitory neurotransmitters during hypercapnia. The reduced responses to GABA or glycine may result from direct effects of hypercapnia on CVNs. In vivo, carbon dioxide inhalation diminishes the function of GABAA receptors (Sanna et al., 1992; Concas et al., 1993). Furthermore, hypercapnia increases the discharge frequency of medullary inspiratory neurons by reducing the sensitivity of GABAA receptors (Gourine and Spyer, 2001). Hypercapnia may have a similar effect on GABA and glycine receptors in CVNs, reducing postsynaptic responses to inhibitory neurotransmission.
In animals exposed to nicotine prenatally, the effect of hypercapnia on glycinergic IPSC frequency closely mimicked that of unexposed animals. This suggests that pathways that mediate glycinergic neurotransmission to CVNs and its modulation during hypercapnia are insensitive to changes evoked by prenatal nicotine. This is consistent with previous studies that have shown prenatal nicotine exposure altered neither glycinergic IPSC frequency in CVNs nor their modulation by hypoxia (Neff et al., 2004). In contrast, prenatal nicotine significantly exaggerated GABAergic neurotransmission to CVNs and resulted in an exaggerated depression of the frequency of both spontaneous and inspiratory GABAergic IPSCs during hypercapnia. This is consistent with and advances previous studies that have shown prenatal nicotine exaggerated the frequency of GABAergic inputs to CVNs (Neff et al., 2003, 2004). Similar to hypercapnia, prenatal nicotine exposure evokes an exaggerated depression of GABAergic IPSC frequency during hypoxia (Neff et al., 2003, 2004).
The exact mechanism by which prenatal nicotine exposure alters inhibitory GABAergic synaptic neurotransmission to CVNs likely involves changes in α4β2 nicotinic receptors (Neff et al., 2003, 2004). Numerous studies have demonstrated that chronic nicotine exposure causes an exaggerated response to subsequent acute nicotine exposures (Benwell et al., 1995; Balfour et al., 2000; Grottick and Higgins, 2000). Chronic nicotine exposure decreases the number of low-affinity and increases the number and fraction of high-affinity nicotinic receptors (Buisson and Bertrand, 2001). Furthermore, nicotine directly alters the conformation of existing α4β2 receptors, transforming them to a high-affinity state that is easily activated (Vallejo et al., 2005). Because hypercapnia evoked an exaggerated depression of inspiratoryevoked GABAergic IPSCs in cardiac vagal neurons, with no significant change in glycinergic neurotransmission, and similarly, presynaptic α4β2 nicotinic receptors mediate the inspiratory-evoked increase in GABAergic but not glycinergic activity, it is possible hypercapnia specifically inhibits α4β2 nicotinic receptor function.
The alterations in cardiorespiratory responses to hypercapnia after prenatal nicotine exposure may be of clinical significance. Smoking during pregnancy increases the risk of SIDS two to four times (Malloy et al., 1988; Bulterys et al., 1990; Haglund and Cnattingius, 1990; Schoendorf and Kiely, 1992), and nicotine has been proposed to be the link between maternal smoking and SIDS (Slotkin et al., 1997; Nachmanoff et al., 1998; Bamford and Carroll, 1999; St-John and Leiter, 1999). Some have suggested that SIDS may result from a direct alteration of the development of brainstem sites responsible for cardiorespiratory control and arousal attributable to prenatal nicotine exposure (Meny et al., 1994; Slotkin et al., 1997; Nachmanoff et al., 1998; St-John and Leiter, 1999). Furthermore, infants at increased risk for SIDS exhibit delayed arousal and experience a greater bradycardia during hypercapnic challenge than control infants (van der Hal et al., 1985; Edner et al., 2002). The exaggerated depression of GABAergic IPSC frequency in CVNs during hypercapnia observed in the present study would result in a significant reduction in heart rate and provides a possible mechanism by which severe bradycardia is evoked by hypercapnia in SIDS.
In summary, hypercapnia evokes a biphasic response in the central respiratory network. In addition, hypercapnia differentially modulates inhibitory neurotransmission to CVNs, broadly diminishing GABAergic while selectively depressing spontaneous glycinergic neurotransmission. In animals exposed to nicotine prenatally, CVNs receive exaggerated GABAergic synaptic transmission, and hypercapnia evokes an exaggerated depression of GABAergic IPSCs in CVNs. In addition, hypercapnia alters both presynaptic release and postsynaptic responses of CVNs to inhibitory neurotransmitters. Although the results obtained in this study in vitro cannot be extrapolated with certainty to in vivo responses, the results from this study provide a likely neurochemical mechanism for the bradycardia elicited by hypercapnia and the dysregulation of this response by prenatal nicotine and provide a cellular basis for the increased risk of SIDS after prenatal nicotine exposure.
Footnotes
This work was supported by National Institutes of Health Grants 59895 and 72006 to D.M.
Correspondence should be addressed to Dr. David Mendelowitz, Department of Pharmacology and Physiology, The George Washington University, 2300 Eye Street Northwest, Washington, DC20037. E-mail:dmendel@gwu.edu.
DOI:10.1523/JNEUROSCI.4221-05.2006
Copyright © 2006 Society for Neuroscience 0270-6474/06/260021-09$15.00/0
References
- Balfour DJ, Wright AE, Benwell ME, Birrell CE (2000) The putative role of extra-synaptic mesolimbic dopamine in the neurobiology of nicotine dependence. Behav Brain Res 113: 73–83. [DOI] [PubMed] [Google Scholar]
- Bamford O, Carroll J (1999) Dynamic ventilatory responses in rats: normal development and effects of prenatal nicotine exposure. Respir Physiol 117: 29–40. [DOI] [PubMed] [Google Scholar]
- Bamford OS, Schuen JN, Carroll JL (1996) Effect of nicotine exposure on postnatal ventilatory responses to hypoxia and hypercapnia. Respir Physiol 106: 1–11. [DOI] [PubMed] [Google Scholar]
- Benwell ME, Balfour DJ, Birrell CE (1995) Desensitization of the nicotine-induced mesolimbic dopamine responses during constant infusion with nicotine. Br J Pharmacol 114: 454–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buisson B, Bertrand D (2001) Chronic exposure to nicotine upregulates the human α4β2 nicotinic acetylcholine receptor function. J Neurosci 21: 1819–1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulterys MG, Greenland S, Kraus JF (1990) Chronic fetal hypoxia and sudden infant death syndrome: interaction between maternal smoking and low hematocrit during pregnancy. Pediatrics 86: 535–540. [PubMed] [Google Scholar]
- Concas A, Sanna E, Cuccheddu T, Mascia MP, Santoro G, Maciocco E, Biggio G (1993) Carbon dioxide inhalation, stress and anxiogenic drugs reduce the function of GABAA receptor complex in the rat brain. Prog Neuropsychopharmacol Biol Psychiatry 17: 651–661. [DOI] [PubMed] [Google Scholar]
- Edner A, Ericson M, Milerad J, Katz-Salamon M (2002) Abnormal heart rate response to hypercapnia in boys with an apparent life-threatening event. Acta Paediatr 91: 1318–1323. [DOI] [PubMed] [Google Scholar]
- Fewell JE, Smith FG, Vienna KYN (2001) Prenatal exposure to nicotine impairs protective responses of rat pups to hypoxia in an age dependent manner. Respir Physiol 127: 61–73. [DOI] [PubMed] [Google Scholar]
- Gourine AV, Spyer KM (2001) Chemosensitivity of medullary inspiratory neurones: a role for GABAA receptors? NeuroReport 12: 3395–3400. [DOI] [PubMed] [Google Scholar]
- Gourine AV, Llaudet E, Dale N, Spyer KM (2005) Release of ATP in the ventral medulla during hypoxia in rats: role in hypoxic ventilatory response. J Neurosci 25: 1211–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gozal D, Gozal E, Reeves SR, Lipton AJ (2002) Gasping and autoresuscitation in the developing rat: effect of antecedent intermittent hypoxia. J Appl Physiol 92: 1141–1144. [DOI] [PubMed] [Google Scholar]
- Grkovic I, Fernandez K, McAllen RM, Anderson CR (2005) Misidentification of cardiac vagal pre-ganglionic neurons after injections of retrograde tracer into the pericardial space in the rat. Cell Tissue Res 321: 335–340. [DOI] [PubMed] [Google Scholar]
- Grottick AJ, Higgins GA (2000) Effect of subtype selective nicotinic compounds on attention as assessed by the five-choice serial reaction time task. Behav Brain Res 117: 197–208. [DOI] [PubMed] [Google Scholar]
- Guntheroth WG, Kawabori I (1975) Hypoxia apnea and gasping. J Clin Invest 56: 1371–1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haglund B, Cnattingius S (1990) Cigarette smoking as a risk factor for sudden infant death syndrome: a population-based study. Am J Public Health 80: 29–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis KW, Bosque EM (1995) Deficient hypoxia awakening response in infants of smoking mothers: possible relationship to sudden infant death syndrome. J Pediatr 127: 691–699. [DOI] [PubMed] [Google Scholar]
- Loewy AD, Spyer KM, eds (1990) Central regulation of autonomic functions. New York: Oxford UP.
- Malloy MH, Kleinman JC, Land GH, Schramm WF (1988) The association of maternal smoking with age and cause of infant death. Am J Epidemiol 128: 46–55. [DOI] [PubMed] [Google Scholar]
- Mendelowitz D (1996) Firing properties of identified parasympathetic cardiac neurons in nucleus ambiguus. Am J Physiol 271: H2609–H2614. [DOI] [PubMed] [Google Scholar]
- Mendelowitz D (1999) Advances in parasympathetic control of heart rate and cardiac function. News Physiol Sci 14: 155–161. [DOI] [PubMed] [Google Scholar]
- Mendelowitz D, Kunze DL (1991) Identification and dissociation of cardiovascular neurons from the medulla for patch clamp analysis. Neurosci Lett 132: 217–221. [DOI] [PubMed] [Google Scholar]
- Meny RG, Carroll JL, Carbone MT, Kelly DH (1994) Cardiorespiratory recordings from infants dying suddenly and unexpectedly at home. Pediatrics 93: 44–49. [PubMed] [Google Scholar]
- Nachmanoff DB, Panigrahy A, Filiano JJ, Mandell F, Sleeper LA, Valdes-Dapena M, Krous HF, White WF, Kinney HC (1998) Brainstem 3H-nicotine receptor binding in the sudden infant death syndrome. J Neuropathol Exp Neurol 57: 1018–1025. [DOI] [PubMed] [Google Scholar]
- Neff RA, Wang J, Baxi S, Evans C, Mendelowitz D (2003) Respiratory sinus arrhythmia: endogenous activation of nicotinic receptors mediates respiratory modulation of brainstem cardioinhibitory parasympathetic neurons. Circ Res 93: 565–572. [DOI] [PubMed] [Google Scholar]
- Neff RA, Simmens SJ, Evans C, Mendelowitz D (2004) Prenatal nicotine exposure alters central cardiorespiratory responses to hypoxia in rats: implications for sudden infant death syndrome. J Neurosci 24: 9261–9268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oikawa S, Hirakawa H, Kusakabe T, Nakashima Y, Hayashida Y (2005) Autonomic cardiovascular responses to hypercapnia in conscious rats: the roles of the chemo- and baroreceptors. Auton Neurosci 117: 105–114. [DOI] [PubMed] [Google Scholar]
- Pena F, Parkis MA, Tryba AK, Ramirez JM (2004) Differential contribution of pacemaker properties to the generation of respiratory rhythms during normoxia and hypoxia. Neuron 43: 105–117. [DOI] [PubMed] [Google Scholar]
- Poets CF, Meny RG, Chobanian MR, Bonofiglo RE (1999) Gasping and other cardiorespiratory patterns during sudden infant deaths. Pediatr Res 45: 350–354. [DOI] [PubMed] [Google Scholar]
- Rekling JC, Feldman JL (1998) PreBotzinger complex and pacemaker neurons: hypothesized site and kernel for respiratory rhythm generation. Annu Rev Physiol 60: 385–405. [DOI] [PubMed] [Google Scholar]
- Sanna E, Cuccheddu T, Serra M, Concas A, Biggio G (1992) Carbon dioxide inhalation reduces the function of GABAA receptors in the rat brain. Eur J Pharmacol 216: 457–458. [DOI] [PubMed] [Google Scholar]
- Sasano N, Vesely AE, Hayano J, Sasano H, Somogyi R, Preiss D, Miyasaka K, Katsuya H, Iscoe S, Fisher JA (2002) Direct effect of Pa(CO2) on respiratory sinus arrhythmia in conscious humans. Am J Physiol Heart Circ Physiol 282: H973–H976. [DOI] [PubMed] [Google Scholar]
- Schoendorf KC, Kiely JL (1992) Relationship of sudden infant death syndrome to maternal smoking during and after pregnancy. Pediatrics 90: 905–908. [PubMed] [Google Scholar]
- Schuen JN, Bamford OS, Carroll JL (1997) The cardiorespiratory response to anoxia: normal development and the effect of nicotine. Respir Physiol 109: 231–239. [DOI] [PubMed] [Google Scholar]
- Simakajornboon N, Vlasic V, Li H, Sawnani H (2004) Effect of prenatal nicotine exposure on biphasic hypoxic ventilatory response and protein kinase C expression in caudal brain stem of developing rats. J Appl Physiol 96: 2213–2219. [DOI] [PubMed] [Google Scholar]
- Slotkin TA, Saleh JL, McCook EC, Seidler FJ (1997) Impaired cardiac function during postnatal hypoxia in rats exposed to nicotine prenatally: implications for perinatal morbidity and mortality, and for sudden infant death syndrome. Teratology 55: 177–184. [DOI] [PubMed] [Google Scholar]
- Slotkin TA, Epps TA, Stenger ML, Sawyer KJ, Seidler FJ (1999) Cholinergic receptors in heart and brainstem of rats exposed to nicotine during development: implications for hypoxia tolerance and perinatal mortality. Brain Res Dev Brain Res 113: 1–12. [DOI] [PubMed] [Google Scholar]
- Sovik S, Lossius K, Walloe L (2001) Heart rate response to transient chemoreceptor stimulation in term infants is modified by exposure to maternal smoking. Pediatr Res 49: 558–565. [DOI] [PubMed] [Google Scholar]
- St-John WM, Leiter JC (1999) Maternal nicotine depresses eupneic ventilation of neonatal rats. Neurosci Lett 267: 206–208. [DOI] [PubMed] [Google Scholar]
- Taylor EW, Butler PJ (1982) Nervous control of heart rate: activity in the cardiac vagus of the dogfish. J Appl Physiol 53: 1330–1335. [DOI] [PubMed] [Google Scholar]
- Taylor EW, Jordan D, Coote JH (1999) Central control of the cardiovascular and respiratory systems and their interactions in vertebrates. Physiol Rev 79: 855–916. [DOI] [PubMed] [Google Scholar]
- Ueda Y, Stick SM, Hall G, Sly PD (1999) Control of breathing in infants born to smoking mothers. J Pediatr 135: 226–232. [DOI] [PubMed] [Google Scholar]
- Vallejo YF, Buisson B, Bertrand D, Green WN (2005) Chronic nicotine exposure upregulates nicotinic receptors by a novel mechanism. J Neurosci 25: 5563–5572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Hal AL, Rodriguez AM, Sargent CW, Platzker AC, Keens TG (1985) Hypoxic and hypercapneic arousal responses and prediction of subsequent apnea in apnea of infancy. Pediatrics 75: 848–854. [PubMed] [Google Scholar]
- Wang J, Irnaten M, Venkatesan P, Evans C, Baxi S, Mendelowitz D (2002) Synaptic activation of hypoglossal respiratory motorneurons during inspiration in rats. Neurosci Lett 332: 195–199. [DOI] [PubMed] [Google Scholar]
- Wang J, Wang X, Irnaten M, Venkatesan P, Evans C, Baxi S, Mendelowitz D (2003) Endogenous acetylcholine and nicotine activation enhances GABAergic and glycinergic inputs to cardiac vagal neurons. J Neurophysiol 89: 2473–2481. [DOI] [PubMed] [Google Scholar]
- Yasuma F, Hirai M, Hayano JI (2001) Differential effects of hypoxia and hypercapnia on respiratory sinus arrhythmia in conscious dogs. Jpn Circ J 65: 738–742. [DOI] [PubMed] [Google Scholar]
- Yen CT, Hwang JC, Wu JS (2000) Cardiac and pulmonary vagal neurons receive excitatory chemoreceptor input. Chin J Physiol 43: 9–13. [PubMed] [Google Scholar]