

Abstract
Mutations in the C terminus of rhodopsin disrupt a rod outer segment localization signal, causing rhodopsin mislocalization and aggressive forms of retinitis pigmentosa (RP). Studies of cultured photoreceptors suggest that activated mislocalized rhodopsin can cause cell death via inappropriate G-protein-coupled signaling. To determine whether this pathway occurs in vivo, we developed a transgenic Xenopus laevis model of RP based on the class I rhodopsin mutation Q344Ter (Q350Ter in X. laevis). We used a second mutation, K296R, to block the ability of rhodopsin to bind chromophore and activate transducin. We compared the effects of expression of both mutants on X. laevis retinas alone and in combination. K296R did not significantly alter the cellular distribution of rhodopsin and did not induce retinal degeneration. Q350Ter caused rhodopsin mislocalization and induced an RP-like degeneration, including loss of rods and development of sprouts or neurites in some remaining rods, but did not affect the distribution of endogenous rhodopsin. The double mutant K296R/Q350Ter caused a similar degeneration and neurite outgrowth. In addition, we found no protective effects of dark rearing in these animals. Our results demonstrate that the degenerative effects of mislocalized rhodopsin are not mediated by the activated form of rhodopsin and therefore do not proceed via conventional G-protein-coupled signaling.
Keywords: rhodopsin, G-protein-coupled receptors, retinitis pigmentosa, retina, photoreceptors, transgenics
Introduction
Retinitis pigmentosa (RP) is a genetically heterogeneous disorder involving rod photoreceptor death and loss of vision. Approximately 30% of autosomal-dominant RP is caused by mutations in Rhodopsin (Sohocki et al., 2001), over 100 of which have been identified (Berson et al., 2002). Although several animal models exist, researchers have yet to elucidate the pathways leading from mutation to rod death. Understanding these pathways is crucial for the development of RP therapies.
Rhodopsin is a G-protein-coupled receptor and the photo-sensitive pigment of rods. It is synthesized in the cell body [inner segment (IS)] and transported to the disk membranes of a specialized organelle called the outer segment (OS). The chromophore of rhodopsin, 11-cis-retinal, is linked to lysine 296. After absorption of a photon, 11-cis-retinal isomerizes to all-trans retinal, converting rhodopsin to an active conformation and initiating the phototransduction cascade.
Disease-causing mutations are categorized by their effects on rhodopsin structure and function when expressed in cultured cells. Class I mutants are expressed at normal levels, fold correctly, bind 11-cis-retinal, and are transported to the plasma membrane (Sung et al., 1991; Kaushal and Khorana, 1994). Class II mutants are misfolded, bind 11-cis-retinal poorly or not at all, and are retained in the endoplasmic reticulum. The C-terminal eight amino acids, which constitute an OS localization signal (Tam et al., 2000) and an ADP ribosylation factor 4 (arf-4) binding site (Deretic et al., 2005), are a hot spot for class I mutations. In rods, these mutants are not only transported to the OS but also mislocalize to the IS plasma membrane and synapse, where rhodopsin is normally present only in small quantities (Sung et al., 1994; Green et al., 2000; Tam et al., 2000; Moritz et al., 2001).
This loss of polarized rhodopsin distribution leads to rod death, but the mechanism of this pathogenic effect remains unclear. Studies by Alfinito and Townes-Anderson (2002) suggest that the mechanism involves adenylate cyclase activation via a G-protein-coupled cascade initiated by mislocalized rhodopsin. In dissociated salamander retina cultures, OS and IS membranes fuse, resulting in redistribution of rhodopsin to the IS plasma membrane; subsequent cell death can be inhibited by protecting the cultures from light, by chemicals that block G-protein activation (pertussis toxin), and by inhibitors of adenylate cyclase (SQ22536).
To determine whether this mechanism functions in vivo, we generated transgenic Xenopus laevis expressing a representative class I rhodopsin mutation, Q350Ter (analogous to Q344Ter in human rhodopsin), in which the C-terminal five amino acids were truncated. We introduced Q350Ter in the context of an epitope-tagged rhodopsin (eRho) as well as in combination with the mutation K296R. K296R stabilizes the inactive conformation of rhodopsin, thus preventing G-protein activation. The effects of both mutations on rod viability alone and in combination were analyzed. eRhoQ350Ter was mislocalized in rods and caused synaptic sprouting and retinal degeneration; however, blocking eRhoQ350Ter activation (i.e., eRhoK296R/Q350Ter) did not rescue these effects. Our results do not support rhodopsin activation as the initiating mechanism of photoreceptor degeneration caused by class I rhodopsin mutants.
Materials and Methods
Molecular biology. Point mutations M13F, P23H, K296R, and Q350Ter were introduced into the full-length X. laevis rhodopsin cDNA (obtained by PCR from retina RNA) using the method by Nelson and Long (1989). Double and triple mutants were constructed using a combination of standard subcloning procedures and additional rounds of site-directed mutagenesis. The 1D4 C-terminal epitope tag VSKTETSQVAPA was introduced by PCR. Rhodopsin–green fluorescent protein (GFP) fusion constructs were also constructed by PCR, using the vector peGFP-N1 (BD Biosciences, San Jose, CA). All final constructs were sequenced.
For expression in COS cells, full-length 1D4-tagged rhodopsin cDNAs were subcloned into the vector pMT3. For expression in transgenic X. laevis rods, the full-length rhodopsin cDNAs (without 1D4 tags) were cloned into XOP0.8-eGFP-N1, a vector based on peGFP-N1, in which we replaced the cytomegalovirus promoter with 0.8 kb of the Xenopus opsin promoter (Batni et al., 1996). Rhodopsin cDNAs were cloned into EcoR1 and NotI sites, completely replacing the enhanced GFP (eGFP) cDNA. For transgenesis, plasmids were linearized with Fse1.
Cell culture and Western blots. COS-7 cells were cultured in DMEM and transfected using the CaPO4 method. Cells were solubilized in PBS plus 1% Triton X-100 after 48 h. Western blots were performed using antibodies B630N, 514-18 (from cell culture supernatant, gift from Dr. P. Hargrave, University of Florida, Gainesville, FL), and anti-GFP (Calbiochem, La Jolla, CA), followed by secondary anti-mouse horse radish peroxidase conjugate (Sigma, St. Louis, MO) and chemiluminescent detection.
Purification of rhodopsin mutants. Mutant pigments were purified from transfected COS cells as described previously (Xie et al., 2003).
Transducin assays. Because two of the rhodopsin mutants used in this work could not be purified in functional form (eRhoK296R does not bind retinal, and eRhoQ350Ter does not contain the 1D4 epitope for immunoaffinity purification of the protein), transducin activation assays were performed using COS cell membranes as a source of the rhodopsin mutants. Transfection of COS cells and preparation of membranes was essentially as described by Robinson et al. (1992). Membranes were assayed as is or after incubation with 11-cis-retinal. All manipulations subsequent to addition of retinal were performed under illumination from dim red lamps. Transducin assays were performed as in the work by Robinson et al. (1992), except that the reaction took place in 10 mm bis-tris-propane buffer, pH 6.7, containing 100 mm NaCl, 5 mm MgCl2, and 0.1 mm EDTA.
Generation of transgenic X. laevis. Transgenic X. laevis were generated using the method by Kroll and Amaya (1996), modified as described previously (Moritz et al., 1999). For each experiment, embryos were derived from multiple egg donors and one sperm donor. We injected similar numbers of eggs from each donor with each transgene construct. Embryos were housed in 4 L tanks in an 18°C incubator on a 12 h light/dark cycle. After 24 h, embryos were exposed to 18 μg/ml G418 for 120 h (Moritz et al., 2002). Dark-reared animals were housed in the same incubator in tanks wrapped in several layers of aluminum foil. At 14 d postfertilization (dpf), surviving X. laevis were killed and one eye was fixed in 4% paraformaldehyde in 0.1 m NaHPO4 buffer, pH 7.4. The contralateral eye was solubilized in 200 μl of a 1:1 mixture of PBS and SDS-PAGE loading buffer, containing 1 mm EDTA and 100 μg/ml PMSF, using a motorized plastic pestle (Kimble/Kontes, Vineland, NJ). Only developmentally normal animals were used in our analysis; animals that were abnormally small or with developmental abnormalities were not used.
Dot blots. Samples of solubilized eyes (2 μl) were diluted 150× in 20 mm NaHPO4 buffer, pH 7.4, and drawn through Immobilon-P membrane (Millipore, Bedford, MA) using a Bio-dot apparatus (Bio-Rad, Hercules, CA). Similar samples derived from nontransgenic eyes [100% wild-type (wt) rhodopsin] or COS cells expressing 1D4-tagged eRho (100% eRho) were applied as controls. Duplicate dot blots were probed with 514-18 and B630N antibodies, followed by IRDye800-CW-conjugated secondary antibody (Rockland, Gilbertsville, PA), and imaged using an Odyssey imaging system (Li-Cor, Lincoln, NE). The ratio of 514-18 to B630N labeling was used to estimate the level of transgenic rhodopsin expression, based on linear extrapolation between the ratios obtained from samples of 100% wt and 100% eRho rhodopsin. Plots of expression level versus log total rhodopsin content were fit to dose–response curves using SigmaPlot (SPSS, Chicago, IL). Because a plateau value of minimum total rhodopsin was not reached, a value of 0.01 was selected for the regression (the first log unit below the lowest data point observed).
Immunolabeling and confocal microscopy. Fixed eyes were infiltrated with 20% sucrose and embedded in OCT medium, frozen, and sectioned and labeled with 2B2 antibody (cell culture supernatant, gift from R. S. Molday, University of British Columbia, Vancouver, British Columbia, Canada) at 1:10 dilution or 11D5 (purified from ascites, gift from D. Deretic, University of New Mexico, Albuquerque, NM) at 1:30,000 dilution, followed by indocarbocyanine-3-conjugated secondary antibody (Jackson ImmunoResearch, West Grove, PA), Alexa-488-conjugated wheat germ agglutinin, and Hoechst 33342 counterstains as described previously (Moritz et al., 2001). For experiments involving 11D5 labeling, at least five 2B2-stained and five 11D5-stained sections were examined for each retina. Sections were imaged using a Zeiss (Oberkochen, Germany) 510 laser scanning confocal microscope equipped with a 10× numerical aperture (N.A.) 0.3 objective and a 40× N.A. 1.2 water immersion objective. Zeiss LSM5 software was used to construct Z-series projections. Figures were assembled using Adobe Photoshop (Adobe Systems, San Jose, CA). Gamma was freely adjusted in images of Hoechst 33342 and wheat germ agglutinin labeling to improve image detail. To preserve linearity, gamma was not adjusted in images of antibody labeling, except where noted in the figure legends.
Results
Epitope tagging of X. laevis rhodopsin
To design an epitope tag for X. laevis rhodopsin that would not interfere with structure and function, we scanned the literature for reports of anti-rhodopsin antibodies with limited cross-reactivity between species that bind regions other than the extreme C terminus. We identified an antibody, 2B2, generated against bovine rhodopsin N-terminal region that did not cross-react with X. laevis rhodopsin (Hicks and Molday, 1986). The region corresponding to the mapped epitope (Molday, 1988) differs by only one amino acid between bovine and X. laevis rhodopsin (F13 vs M13). By introducing the mutation M13F, we conferred 2B2 reactivity to X. laevis rhodopsin. Thus, 2B2 recognizes transgenic epitope-tagged (eRho) but not endogenous (wt) rhodopsin.
Because 2B2 gave a relatively weak signal on blotting assays, we screened a number of other anti-rhodopsin antibodies for differential reactivity to X. laevis wt and eRho rhodopsins. Using extracts from COS cells expressing rhodopsin amino acids 1–38 fused to the N terminus of GFP, we identified several antibodies that had differential reactivity to eRho. The antibody that gave the strongest signal for eRho on Western blots was 514-18 (Fig. 1), while giving a barely detectable signal for wild-type rhodopsin. This antibody was provided by Dr. Paul Hargrave (personal communication, previously unpublished reagent). The antibody B630N (Adamus et al., 1991) bound wt and eRho equally, and furthermore was unaffected by the RP-causing mutation P23H. B630N and 514-18 are high-affinity antibodies that allowed us to develop very sensitive assays with large linear ranges.
Figure 1.

Effects of M13F on antibody cross-reactivity. Western blots of extracts of COS cells transfected with plasmids designed to express GFP or rhodopsin N1-38–GFP fusion proteins or mock-transfected cells (control) are shown. Blots were probed with antibodies B630N, 514-18, or anti-GFP. Mr, Relative mobility.
Characterization of transduction-deficient rhodopsins
To disrupt rhodopsin signal transduction, we used the mutation K296R (Cohen et al., 1993). Lysine 296 is the site of attachment of the retinal chromophore. In bovine rhodopsin, K296R results in a protein that cannot bind retinal, does not activate transducin, and displays no constitutive activity (Cohen et al., 1993). Because this mutation retains a positive charge, and because mutations at position 296 do not typically result in folding problems, we expected this mutant to retain a native structure. In agreement with the bovine results, K296R, when introduced into eRho, did not form a pigment with retinal (Fig. 2A) and did not activate transducin (Fig. 2B), although wild-type levels of eRhoK296R were established to be present in the COS-cell membrane prep by Western blot analysis using the 1D4 antibody (data not shown).
Figure 2.

Characterization of the effects of rhodopsin mutations on chromophore formation and transducin activation. A, Absorption spectra for mutant rhodopsins purified from COS cell detergent extracts. Spectra are for the mutants eRho (1) and eRhoK296R (2). B, Effect of rhodopsin mutations on ability to activate transducin. Filled symbols, Dark; open symbols, after exposure to light (arrow). Circles, eRho; triangles, eRhoQ350Ter; squares, eRhoK296R. ERhoK296R membranes that were not treated with retinal were also assayed for ability to activate transducin but showed no activity (data not shown).
Effect of K296R on eRhoQ350Ter-induced degeneration
To model RP caused by the human class I mutation Q344Ter, we generated four groups of transgenic X. laevis expressing eRho, eRhoK296R, eRhoQ350Ter, or eRhoK296R/Q350Ter under the control of a 0.8 kb fragment of the X. laevis opsin promoter (Batni et al., 1996).
We conducted a four-way comparison of the effects of the transgenic rhodopsin expression on rod viability using a dot blot assay. At 14 dpf, tadpoles were killed, and one eye was solubilized for dot blots performed with B630N and 514-18 (Fig. 3A). Samples from 22 animals of each type were analyzed as well as control samples of wt rhodopsin (from wild-type retinas) and eRho (heterologously expressed in COS cells). Based on the ratio of 514-18 signal (representing the amount of transgenic rhodopsin) to B630N signal (representing the total amount of rhodopsin), we estimated the expression level of eRho as a percentage of total rhodopsin in each sample (Fig. 3A). High expression of the eRhoQ350Ter and eRhoK296R/Q350Ter rhodopsins correlated with an overall decrease in total rhodopsin and thus retinal degeneration (based on the premise that low total rhodopsin levels indicate absence of rods). Expression of eRho and eRhoK296R did not correlate with a decrease in rhodopsin content, indicating that these rhodopsins did not cause significant rod death (Fig. 3C).
Figure 3.

Dot blot analysis of the effects of rhodopsin expression on total rhodopsin content of transgenic eyes. Dot blots of total eye extract from 22 transgenic animals of each type probed with B630N (total rhodopsin) (A) and 514-18 (eRho rhodopsin) (B) are shown. The positions of four control samples of wt and eRho rhodopsin are indicated on each blot. The ratio of 514-18 signal to B630N signal was used to calculate the percentage of transgenic rhodopsin in each eye. C, Plots of expression level versus total rhodopsin. The log scale data for eRhoQ350Ter and eRhoK296R/Q350Ter were fit to dose–response curves, and eRho and eRhoK296R data were fit to straight lines.
To determine statistical significance, we performed a nonparametric Kruskal–Wallis analysis on the rank-transformed values of the B630N signals shown in Figure 3A (Table 1). Nonparametric statistics were used because of the likelihood that the underlying data distribution was skewed or bimodal rather than normal. The results indicated a highly significant difference between groups. A subsequent multiple-comparisons test performed according to Conover (1999) (Table 2) indicated that eyes from animals expressing Q350Ter rhodopsins (i.e., eRhoQ350Ter and eRhoK296R/Q350Ter) contained significantly lower levels of total rhodopsin relative to eyes expressing non-Q350Ter rhodopsins (i.e., eRho and eRhoK296R). Furthermore, abolishing the ability of rhodopsin to adopt its active conformation (i.e., eRhoK296R/Q350Ter) had no significant effect on the severity of retinal degeneration, as measured by total rhodopsin content. Together, these results indicate that the effects of eRhoQ350Ter were specific to the Q350Ter mutation and not simply rhodopsin overexpression and that the degenerative effects of Q350Ter were not mediated by the active conformation of the mutant rhodopsin.
Table 1.
Kruskal–Wallis analysis of total rhodopsin levels a
|
Group |
Transgene |
n |
Sum of ranks |
Average rank |
|---|---|---|---|---|
| 1 | eRho | 22 | 1276 | 58.0 |
| 2 | eRhoK296R | 22 | 1194 | 54.3 |
| 3 | eRhoQ350Ter | 22 | 849 | 38.6 |
| 4 |
eRhoK296R/Q350Ter |
22 |
596 |
27.1 |
Critical value of test statistic t (α = 0.05), 7.81; value of test statistic t, 20.76 (all groups are not equivalent); p = 0.0001.
Performed as described by Conover (1999).
Table 2.
Multiple comparisons analysis of mean total rhodopsin levels a
|
Comparison |
p |
Conclusion |
|---|---|---|
| Group 1 versus | ||
| 4 | 0.00002 | 1 > 4 |
| 3 | 0.006 | 1 > 3 |
| 2 | 0.6 | Not significantly different |
| Group 2 versus | ||
| 4 | 0.0001 | 2 > 4 |
| 3 | 0.02 | 2 > 3 |
| Group 3 versus | ||
| 4 |
0.10 |
Not significantly different |
Performed as described by Conover (1999).
Effects of dark rearing on Q350Ter-induced degeneration
Although our results show that the active conformation of mutant rhodopsin is not involved in degeneration caused by Q350Ter, the physiological state of light-exposed photoreceptors could indirectly promote Q350Ter-induced degeneration. To determine whether light exposure influenced retinal degeneration, we examined the effects of dark rearing. We generated eRhoQ350Ter-expressing transgenic embryos and divided them into two groups, raised on either a 12 h light/dark cycle or complete darkness from 3 dpf fertilization onwards (i.e., before eye formation). At 14 dpf, we killed the animals and performed a similar dot blot analysis. Dark rearing had no apparent effects on the size or developmental stage of the tadpoles.
We again found that expression of eRhoQ350Ter was correlated with a decrease in total rhodopsin content, likely indicating retinal degeneration. The total rhodopsin content of dark-reared eyes was significantly lower than cyclic reared eyes (Mann–Whitney test, p = 0.008). However, when the data were plotted, it was evident that the reduction in total rhodopsin content of dark-reared eyes occurred primarily at low expression levels; there was no evidence of a protective effect at high expression levels (Fig. 4). The decrease in rhodopsin content was most likely attributable to the effect of dark rearing on the rate of disk synthesis (Besharse et al., 1977). Because new disks are synthesized at a greater rate in cyclic light, dark-reared animals began with a lower complement of total rhodopsin. [We observed a similar effect in nontransgenic animals (data not shown).]
Figure 4.

Effect of dark rearing on total rhodopsin content of eRhoQ350Ter transgenic eyes. Dot blots of total eye extract from 33 animals of each type were probed with B630N (total rhodopsin) and 514-18 (transgenic rhodopsin) as in Figure 3, and plots of expression level versus total rhodopsin were similarly derived from the dot blots and fit to dose–response curves. Note that the greatest difference in rhodopsin content occurs at the lowest expression levels.
Characterization of retinal degeneration
To demonstrate that this reduction in rhodopsin content was caused by RP-like alterations of the retina, we examined contralateral eyes by immunofluorescence microscopy for signs of degeneration. Eyes expressing eRho and eRhoK296R at high levels appeared healthy with abundant photoreceptors and full-length OS (Fig. 5A,B,E). In contrast, eyes expressing eRhoQ350Ter and eRhoK296R/Q350Ter showed a progression ranging from little or no degeneration at low expression levels to absence of rods from large regions of the retina and shortened OS at high expression levels (Fig. 5C, D, F). Cones were present in regions depleted of rods (Fig. 5F).
Figure 5.
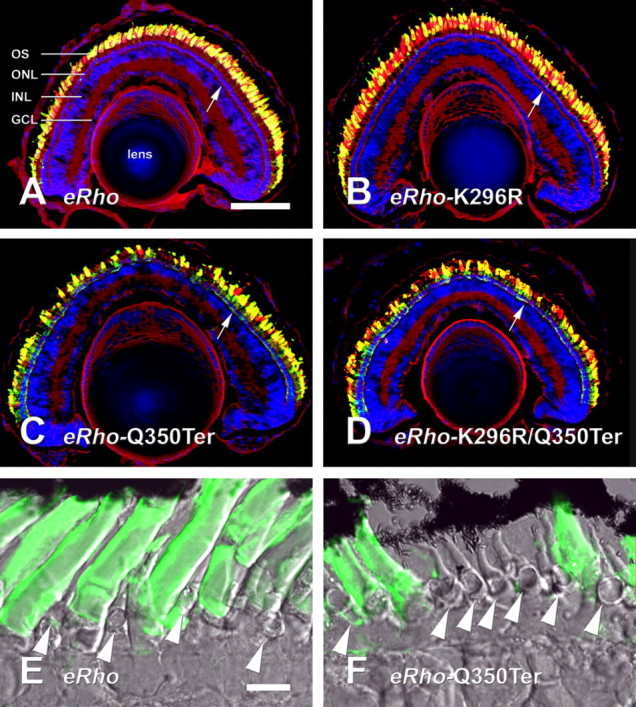
Effects of transgene expression on rod and cone photoreceptor density. A–D, Confocal micrographs of retinas expressing high levels of eRho (A), eRhoK296R (B), eRhoQ350Ter (C), and eRhoK296R/Q350Ter (D) labeled with 2B2 anti-eRho rhodopsin (green), wheat germ agglutinin (red), and Hoechst 33342 nuclear stain (blue). Note the gaps in the photoreceptor layer in C and D. E, Section of retina expressing eRho, with bright-field image superimposed on 2B2 label. F, Similar section expressing eRhoQ350Ter. Note the absence of rods relative to E. Cones are present in the spaces between rods (arrowheads). Mislocalized rhodopsin is present in the rod synapses in C, D, and F (compare regions marked by white arrows to similar regions marked in A, B, and E). Scale bars: (in A) A–D, 100 μm; (in E) E, F, 10 μm. ONL, Outer nuclear layer; INL, inner nuclear layer; GCL, ganglion cell layer.
Localization of transgene products
We examined the subcellular localization of the mutant rhodopsins by immunofluorescence microscopy using antibody 2B2. Large quantities of all transgenic rhodopsins were transported to OS (Figs. 5, 6). IS labeling of transgenic rhodopsin was also detectable in retinas with high-level transgene expression, but the distribution was transgene dependent. In eRho retinas, internal membranes with a distribution consistent with the Golgi apparatus were labeled much more intensely than the plasma membrane. ERhoK296R retinas had a similar pattern of IS label. In eRhoQ350Ter and eRhoK296R/Q350Ter retinas, the most prominent inner segment label was associated with the plasma membrane. These results are consistent with disruption of an OS localization signal by the Q350Ter mutation as described previously (Sung et al., 1994; Tam et al., 2000; Moritz et al., 2001).
Figure 6.

Localization of transgene products in rod photoreceptors. Retinas were labeled with 2B2 anti-eRho rhodopsin (green), wheat germ agglutinin (red), and Hoechst 33342 nuclear stain (blue) Rods are expressing eRho (A), eRhoK296R (B), eRhoQ350Ter (C), or eRhoK296R/Q350Ter (D). Note the strong 2B2 signal from IS plasma membranes in C and D relative to A and B (arrowheads). The asymmetric labeling of OS is an artifact caused by poor penetration of the densely packed disk membranes in fixed tissue. Scale bar, 10 μm. N, Nucleus; S, synapse.
Q350Ter does not affect localization of WT rhodopsin
To determine whether mislocalization of transgenic rhodopsin causes mislocalization of wt rhodopsin, we labeled a number of transgenic retinas with antibody 11D5, which binds the extreme C terminus of rhodopsin (Deretic and Papermaster, 1991) and does not cross-react with the Q350Ter mutant. We did not observe a stronger labeling of IS membranes in retinas expressing high levels of eRhoQ350Ter versus retinas expressing little or no eRhoQ350Ter, although 11D5 was of sufficiently high affinity to detect IS rhodopsin in nontransgenic retinas. We obtained several retinas with dramatically asymmetric expression of eRhoQ350Ter and eRhoK296R/Q350Ter caused by transgene position effects (Moritz et al., 2001) (Fig. 7A). These provided the strongest evidence for a lack of effect on wt rhodopsin localization, because control and transgene-expressing cells were located side by side in the same section. However, 11D5 labeling of IS membranes in these retinas was uniformly low in all rods (Fig. 7B), indicating that the distribution of wt rhodopsin was not significantly affected in cells expressing mislocalizing mutants.
Figure 7.
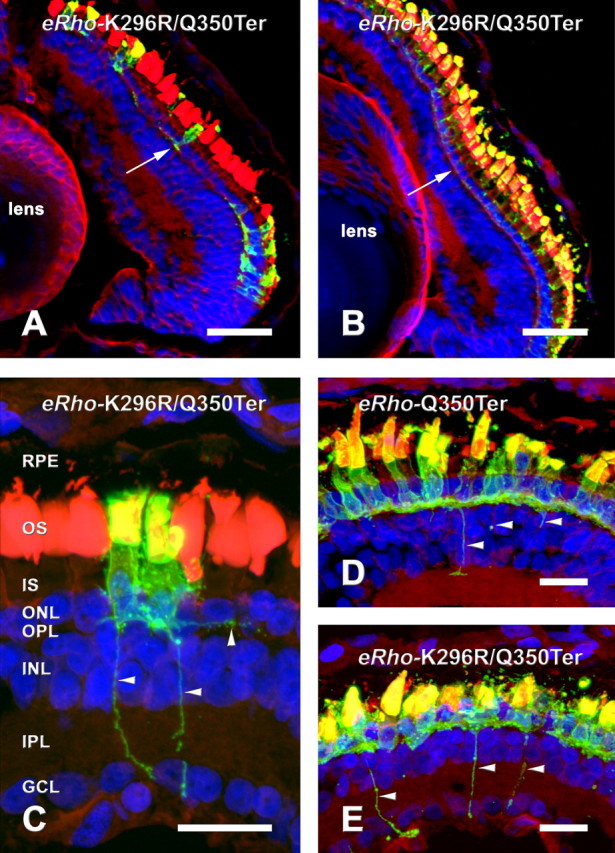
Expression of mislocalized rhodopsin mutants does not cause mislocalization of endogenous rhodopsin and induces formation of sprouts. Retinas were labeled with 2B2 anti-eRho rhodopsin (green) wheat germ agglutinin (red), and Hoechst 33342 nuclear stain (blue). A, B, Retina expressing eRhoK296R/Q350Ter with very nonuniform expression labeled with 2B2 (A) and 11D5 (B). The 11D5 IS label is uniform and relatively faint, with very little label in the synaptic region (compare white arrows in A and B). C, 2B2-labeled section from the same retina shown in B, with horizontal and vertical processes indicated by arrowheads. D, E, ERhoQ350Ter (D) and eRhoK296R/Q350Ter (E) retinas labeled with 2B2 showing several processes (arrowheads). C, D, and E are two-dimensional projections constructed from Z-series of ∼24 confocal images taken at 0.5 μm intervals. Gamma was adjusted in all three channels of C, D, and E to maximize image detail. Scale bars: A, B, 50μm; C–E, 10 μm. RPE, Retinal pigment epithelium; ONL, outer nuclear layer; OPL, outerplexiformlayer; INL, inner nuclear layer; IPL, inner plexiform layer; GCL, ganglion cell layer.
Presence of sprouts
In retinas with the highest eRhoQ350Ter and eRhoK296R/Q350Ter expression, 2B2 labeling revealed fine projections that extended from the IS. This occurred in the peripheral retina (where growing amphibian eyes continuously add new rods) as well as surviving central rods. Numerous fine projections gave the cells a hairy appearance (Fig. 7C–E). In some cells, axon-like sprouts were present that projected short distances laterally or into the inner nuclear or ganglion cell layers (Fig. 7C–E).
Discussion
To address the hypothesis that retinal degeneration caused by mutations in the C terminus of rhodopsin is mediated by the active conformation of the mutant rhodopsin, we developed a new model of RP based on primary transgenic X. laevis generated by nuclear transplantation. Our results indicate that activation of rhodopsin mislocalized to the IS plasma membrane and synapse is not the cause of photoreceptor degeneration in our model. The mutation K296R prevented rhodopsin-mediated activation of the heterotrimeric G-protein transducin present in rod IS and OS but did not prevent degeneration caused by Q350Ter. It is important to note that lysine 296 does not comprise part of the G-protein-interacting site of rhodopsin (the cytoplasmic loops), but rather the retinal binding site; K296R prevents transducin activation by inhibition of a global conformational change (Cohen et al., 1993). The mechanism of G-protein activation is highly conserved between G-protein-coupled receptors: rhodopsin/B2-adrenergic receptor chimeras in which the cytoplasmic loops of rhodopsin are substituted with B2-adrenergic receptor sequences can activate Gs (although light-activated rhodopsin does not significantly activate Gs in cultured cells) (Kim et al., 2005). Thus, it is very likely that K296R also prevents activation of heterotrimeric G-proteins other than transducin.
There are several possible explanations for the discrepancy between our results and those obtained with cultured salamander rods (Alfinito and Townes-Anderson, 2002). First, mislocalization of rhodopsin in cultured rods is caused by fusion of OS and IS plasma membranes, probably as a result of mechanical disturbance. Thus, other OS plasma membrane components may also mislocalize to IS, and IS components may be present in the OS. In contrast, other OS proteins (wild-type rhodopsin, peripherin, and the cGMP-gated channel have been investigated) do not delocalize as a result of expression of class I rhodopsin mutants in transgenic animal models (Green et al., 2000). Furthermore, cultured cells may be deprived of soluble factors or cell contacts that could influence cell death mechanisms. However, the apoptotic pathway identified by Alfinito and Townes-Anderson (2002) may be involved in retinal degenerations involving generalized transport defects, such as X-linked RP caused by mutations in RPGR.
An alternative disease mechanism supported by our studies involves crowding effects of large quantities of IS rhodopsin. Delocalized rhodopsin could pack the synaptic or plasma membranes, diluting the effective concentration of normally occurring proteins. Alterations of synaptic protein composition have been documented in transgenic pig retinas expressing P347L rhodopsin (Blackmon et al., 2000). In these animals, rod terminals lost postsynaptic density-95 immunoreactivity before the onset of cell death. Rods also failed to form normal contacts with rod bipolar cells. Synaptic effects are strongly suggested in our study by the phenomenon of sprouting.
We observed projections emanating from the synaptic terminals of transgenic rods, which sometimes extended as far as the ganglion cell layer. These may be analogous to projections observed in retinas of RP patients (sprouts) (Li et al., 1995; Fariss et al., 2000) or neurite outgrowth observed in cultured neurons (Craig and Banker, 1994). The development of these sprouts was also independent of rhodopsin signal transduction, because they occurred in both eRhoQ350Ter and eRhoK296R/Q350Ter retinas. It is possible that the presence of rhodopsin in the synapse causes a defect in axon guidance or synapse formation, forcing rods to seek contacts with unusual cell types. It is not clear whether neurite sprouting contributes to, or is a consequence of, photoreceptor degeneration.
Using our transgenic X. laevis system, we can examine the effects of a wide range of transgene expression levels using immunoassays and microscopy, effectively correlating transgene effects with transgene expression levels. A unique feature of our novel dose–response analysis is the possibility of deriving a Hill coefficient for an activity of a transgene product. Hill coefficients >1 suggest cooperative mechanisms. Unfortunately, our dose–response curves were incomplete, because a lower limit for total rhodopsin content was not met in a significant number of data points. Nevertheless, this lower limit cannot be less than zero, so it is interesting that when we arbitrarily set this value at 10, 1, or 0.1% of total rhodopsin for the regression analysis, Hill coefficient estimates between 4 and 5 for the combined eRhoQ350Ter and eRhoQ350Ter/K296R data points in Figure 3 were obtained in all cases. Similar results were obtained when all cyclic-reared eRhoQ350Ter samples from Figures 3 and 4 were combined in a third set of dot blots (data not shown). An EC50 value of 16% of total rhodopsin was obtained for eRhoQ350Ter, which was also quite independent of the minimum total rhodopsin value used for regression. These values suggest the trigger for cell death involves cooperative action of multiple mutant rhodopsin molecules; combined with the observed synaptic sprouting, our data are consistent with mislocalized rhodopsin disrupting normal synaptic membrane activities through crowding effects.
Although rhodopsin likely forms dimers and higher-order structures in OS membranes (Fotiadis et al., 2003), expression of eRhoQ350Ter did not cause mislocalization of wt rhodopsin. This is consistent with a mechanism in which the presence of a single localization signal is sufficient to allow transport of a rhodopsin multimer. In this case, our Hill coefficient may represent the number of rhodopsin monomers in a multimer recognized by arf-4. This model predicts that eRhoQ350Ter would only mislocalize at very high expression levels, when formation of mutant: mutant dimers and/or multimers is likely, yet we have observed mislocalization of class I mutants even at low expression levels in this and previous studies (Moritz et al., 2001). Other possible scenarios are that rhodopsin is monomeric when interacting with arf-4, or that Q350Ter mutants do not form multimers. This issue requires additional investigation.
In contrast to the present study, a small increase in endogenous rhodopsin was detected in the IS of transgenic mice expressing an S334Ter rhodopsin truncation mutant, suggesting aberrant wt rhodopsin trafficking (Concepcion et al., 2002). However, the effect was subtle and does not suggest stoichiometric recruitment of wt rhodopsin to the IS by mutant rhodopsin. Minor mislocalization could be explained by a basal rate of misincorporation of wt rhodopsin into post-Golgi carriers destined for IS membranes, combined with an increase in these carriers caused by expression of localization-defective rhodopsin. The absence of mislocalized wt rhodopsin in our study may be species specific, for example because of the greater capacity for rhodopsin transport in frog rods, which have significantly larger OS. Although the larger truncation (15 vs 5 amino acids) could account for the different results, S334Ter expression had no effect on wt rhodopsin distribution in transgenic rats (Green et al., 2000). Because both truncated forms cause retinal degeneration regardless of detectable wt mislocalization (Sung et al., 1994; Chen et al., 1995; Green et al., 2000; the present study), the pathogenic mechanism in all species likely involves the mislocalized truncated rhodopsin.
Because X. laevis retinas contain numerous cones (unlike the retinas of mice and rats), it may be possible to use this system to examine secondary effects of rod death on cone survival, which are responsible for the most debilitating symptoms of RP. Our additional studies will also involve a more detailed examination of the sprouting phenomenon.
Footnotes
This work was supported by the Canadian Institutes of Health Research, The Foundation Fighting Blindness of Canada, the Karl Kirchgessner Foundation, and the Michael Smith Foundation (O.L.M.) and by National Institutes of Health Grant EY07965 (D.D.O.). O.L.M. is a Michael Smith Scholar and W. K. Stell Scholar. We gratefully acknowledge Drives R. S. Molday, P. Hargrave, and D. Deretic for contributing antibody reagents.
Correspondence should be addressed to Orson L. Moritz, Department of Ophthalmology and Visual Sciences, University of British Columbia, 2550 Willow Street, Vancouver, British Columbia, Canada V5Z 3N9. E-mail: olmoritz@interchange.ubc.ca.
DOI:10.1523/JNEUROSCI.3849-05.2006
Copyright © 2006 Society for Neuroscience 0270-6474/06/260203-07$15.00/0
References
- Adamus G, Zam ZS, Arendt A, Palczewski K, McDowell JH, Hargrave PA (1991) Anti-rhodopsin monoclonal antibodies of defined specificity: characterization and application. Vision Res 31: 17–31. [DOI] [PubMed] [Google Scholar]
- Alfinito PD, Townes-Anderson E (2002) Activation of mislocalized opsin kills rod cells: a novel mechanism for rod cell death in retinal disease. Proc Natl Acad Sci USA 99: 5655–5660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batni S, Scalzetti L, Moody SA, Knox BE (1996) Characterization of the Xenopus rhodopsin gene. J Biol Chem 271: 3179–3186. [DOI] [PubMed] [Google Scholar]
- Berson EL, Rosner B, Weigel-DiFranco C, Dryja TP, Sandberg MA (2002) Disease progression in patients with dominant retinitis pigmentosa and rhodopsin mutations. Invest Ophthalmol Vis Sci 43: 3027–3036. [PubMed] [Google Scholar]
- Besharse JC, Hollyfield JG, Rayborn ME (1977) Turnover of rod photoreceptor outer segments. II. Membrane addition and loss in relationship to light. J Cell Biol 75: 507–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackmon SM, Peng YW, Hao Y, Moon SJ, Oliveira LB, Tatebayashi M, Petters RM, Wong F (2000) Early loss of synaptic protein PSD-95 from rod terminals of rhodopsin P347L transgenic porcine retina. Brain Res 885: 53–61. [DOI] [PubMed] [Google Scholar]
- Chen J, Makino CL, Peachey NS, Baylor DA, Simon MI (1995) Mechanisms of rhodopsin inactivation in vivo as revealed by a COOH-terminal truncation mutant. Science 267: 374–377. [DOI] [PubMed] [Google Scholar]
- Cohen GB, Yang T, Robinson PR, Oprian DD (1993) Constitutive activation of opsin: influence of charge at position 134 and size at position 296. Biochemistry 32: 6111–6115. [DOI] [PubMed] [Google Scholar]
- Concepcion F, Mendez A, Chen J (2002) The carboxyl-terminal domain is essential for rhodopsin transport in rod photoreceptors. Vision Res 42: 417–426. [DOI] [PubMed] [Google Scholar]
- Conover WJ (1999) Some methods based on ranks: several independent samples. In: Practical nonparametric statistics, Sec 5.2, Chap 5, Ed 3, pp 288–300. New York: Wiley.
- Craig AM, Banker G (1994) Neuronal polarity. Annu Rev Neurosci 17: 267–310. [DOI] [PubMed] [Google Scholar]
- Deretic D, Papermaster DS (1991) Polarized sorting of rhodopsin on post-Golgi membranes in frog retinal photoreceptor cells. J Cell Biol 113: 1281–1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deretic D, Williams AH, Ransom N, Morel V, Hargrave PA, Arendt A (2005) Rhodopsin C terminus, the site of mutations causing retinal disease, regulates trafficking by binding to ADP-ribosylation factor 4 (ARF4). Proc Natl Acad Sci USA 102: 3301–3306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fariss RN, Li ZY, Milam AH (2000) Abnormalities in rod photoreceptors, amacrine cells, and horizontal cells in human retinas with retinitis pigmentosa. Am J Ophthalmol 129: 215–223. [DOI] [PubMed] [Google Scholar]
- Fotiadis D, Liang Y, Filipek S, Saperstein DA, Engel A, Palczewski K (2003) Atomic-force microscopy: rhodopsin dimers in native disc membranes. Nature 421: 127–128. [DOI] [PubMed] [Google Scholar]
- Green ES, Menz MD, LaVail MM, Flannery JG (2000) Characterization of rhodopsin mis-sorting and constitutive activation in a transgenic rat model of retinitis pigmentosa. Invest Ophthalmol Vis Sci 41: 1546–1553. [PubMed] [Google Scholar]
- Hicks D, Molday RS (1986) Differential immunogold-dextran labeling of bovine and frog rod and cone cells using monoclonal antibodies against bovine rhodopsin. Exp Eye Res 42: 55–71. [DOI] [PubMed] [Google Scholar]
- Kaushal S, Khorana HG (1994) Structure and function in rhodopsin. 7. Point mutations associated with autosomal dominant retinitis pigmentosa. Biochemistry 33: 6121–6128. [DOI] [PubMed] [Google Scholar]
- Kim JM, Hwa J, Garriga P, Reeves PJ, RajBhandary UL, Khorana HG (2005) Light-driven activation of beta 2-adrenergic receptor signaling by a chimeric rhodopsin containing the beta 2-adrenergic receptor cytoplasmic loops. Biochemistry 44: 2284–2292. [DOI] [PubMed] [Google Scholar]
- Kroll KL, Amaya E (1996) Transgenic Xenopus embryos from sperm nuclear transplantations reveal FGF signaling requirements during gastrulation. Development 122: 3173–3183. [DOI] [PubMed] [Google Scholar]
- Li ZY, Kljavin IJ, Milam AH (1995) Rod photoreceptor neurite sprouting in retinitis pigmentosa. J Neurosci 15: 5429–5438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molday RS (1988) Monoclonal antibodies to rhodopsin and other proteins of rod outer segments. In: Progress in retinal research, Vol 8 (Osbourne N, Chader J, eds), pp 173–209. Oxford: Pergamon. [Google Scholar]
- Moritz OL, Tam BM, Knox BE, Papermaster DS (1999) Fluorescent photoreceptors of transgenic Xenopus laevis imaged in vivo by two microscopy techniques. Invest Ophthalmol Vis Sci 40: 3276–3280. [PubMed] [Google Scholar]
- Moritz OL, Tam BM, Papermaster DS, Nakayama T (2001) A functional rhodopsin-green fluorescent protein fusion protein localizes correctly in transgenic Xenopus laevis retinal rods and is expressed in a time-dependent pattern. J Biol Chem 276: 28242–28251. [DOI] [PubMed] [Google Scholar]
- Moritz OL, Biddle KE, Tam BM (2002) Selection of transgenic Xenopus laevis using antibiotic resistance. Transgenic Res 11: 315–319. [DOI] [PubMed] [Google Scholar]
- Nelson RM, Long GL (1989) A general method of site-specific mutagenesis using a modification of the Thermus aquaticus polymerase chain reaction. Anal Biochem 180: 147–151. [DOI] [PubMed] [Google Scholar]
- Robinson PR, Cohen GB, Zhukovsky EA, Oprian DD (1992) Constitutively active mutants of rhodopsin. Neuron 9: 719–725. [DOI] [PubMed] [Google Scholar]
- Sohocki MM, Daiger SP, Bowne SJ, Rodriquez JA, Northrup H, Heckenlively JR, Birch DG, Mintz-Hittner H, Ruiz RS, Lewis RA, Saperstein DA, Sullivan LS (2001) Prevalence of mutations causing retinitis pigmentosa and other inherited retinopathies. Hum Mutat 17: 42–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung CH, Schneider BG, Agarwal N, Papermaster DS, Nathans J (1991) Functional heterogeneity of mutant rhodopsins responsible for autosomal dominant retinitis pigmentosa. Proc Natl Acad Sci USA 88: 8840–8844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung CH, Makino C, Baylor D, Nathans J (1994) A rhodopsin gene mutation responsible for autosomal dominant retinitis pigmentosa results in a protein that is defective in localization to the photoreceptor outer segment. J Neurosci 14: 5818–5833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tam BM, Moritz OL, Hurd LB, Papermaster DS (2000) Identification of an outer segment targeting signal in the COOH terminus of rhodopsin using transgenic Xenopus laevis J Cell Biol 151: 1369–1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie G, Gross AK, Oprian DD (2003) An opsin mutant with increased thermal stability. Biochemistry 42: 1995–2001. [DOI] [PubMed] [Google Scholar]