

Abstract
Axonal degeneration occurs in many neurodegenerative diseases and after traumatic injury and is a self-destructive program independent from programmed cell death. Previous studies demonstrated that overexpression of nicotinamide mononucleotide adenylyltransferase 1 (Nmnat1) or exogenous application of nicotinamide adenine dinucleotide (NAD) can protect axons of cultured dorsal root ganglion (DRG) neurons from degeneration caused by mechanical or neurotoxic injury. In mammalian cells, NAD can be synthesized from multiple precursors, including tryptophan, nicotinic acid, nicotinamide, and nicotinamide riboside (NmR), via multiple enzymatic steps. To determine whether other components of these NAD biosynthetic pathways are capable of delaying axonal degeneration, we overexpressed each of the enzymes involved in each pathway and/or exogenously administered their respective substrates in DRG cultures and assessed their capacity to protect axons after axotomy. Among the enzymes tested, Nmnat1 had the strongest protective effects, whereas nicotinamide phosphoribosyl transferase and nicotinic acid phosphoribosyl transferase showed moderate protective activity in the presence of their substrates. Strong axonal protection was also provided by Nmnat3, which is predominantly located in mitochondria, and an Nmnat1 mutant localized to the cytoplasm, indicating that the subcellular location of NAD production is not crucial for protective activity. In addition, we showed that exogenous application of the NAD precursors that are the substrates of these enzymes, including nicotinic acid mononucleotide, nicotinamide mononucleotide, and NmR, can also delay axonal degeneration. These results indicate that stimulation of NAD biosynthetic pathways via a variety of interventions may be useful in preventing or delaying axonal degeneration.
Keywords: axotomy, neuropathology, neuroprotection, dorsal root ganglion, nucleus, GFP
Introduction
Axonal degeneration is observed after physical damage to axons and in a variety of neuropathological conditions, including diabetic neuropathies, demyelinating diseases, and neurodegenerative diseases such as Alzheimer’s disease, amyotrophic lateral sclerosis, and Parkinson’s disease. In these diseases, the degeneration of axons often precedes the death of neuronal cell bodies and likely contributes significantly to the development of clinical symptoms (Raff et al., 2002). Axonal degeneration appears to be a unique self-destructive process that is linked to, but distinct from, apoptosis (Coleman, 2005). These new insights suggest that axonal degeneration may act upstream of neuronal cell death and thereby represents a potential therapeutic target for prevention of certain neurodegenerative conditions.
The active nature of axonal degeneration was originally brought to light by the discovery of a spontaneous dominant mutation in mice called Wallerian degeneration slow (wlds). wlds mice have significantly delayed axonal degeneration in both central and peripheral axons after nerve transection (Lunn et al., 1989; Perry et al., 1991). The wlds mutation comprises an 85 kb tandem triplication that results in overexpression of a chimeric nuclear protein called Wlds (Conforti et al., 2000). This protein consists of the N-terminal 70 amino acids of a ubiquitin ligase, Ufd2a (ubiquitin fusion degradation protein 2a), fused to full-length nicotinamide mononucleotide adenylyltransferase 1 (Nmnat1), an enzyme in the nicotinamide adenine dinucleotide (NAD) salvage pathway (Mack et al., 2001; Wang et al., 2001). Using an in vitro Wallerian degeneration assay, we and others have shown previously that the Nmnat1 portion of the Wlds protein is sufficient to protect axons after mechanical or chemical injury (Araki et al., 2004; Wang et al., 2005). Mutations in the Wlds protein or Nmnat1 that render them enzymatically inactive also abolish their ability to delay axonal degeneration (Araki et al., 2004). Furthermore, the wlds phenotype can be mimicked by treating neurons with NAD itself, suggesting that it is the biosynthesis of NAD by Nmnat1 that leads to axonal protection.
Despite the fact that expression of Nmnat1 is sufficient to save axons in vitro (Araki et al., 2004; Wang et al., 2005), there remains a question as to the exact mechanism of this effect. Others have suggested that the N-terminal Ufd2a portion of the Wlds protein is critical, and that the NAD biosynthetic function is not relevant to axonal protection (Conforti et al., 2006). In this study, we manipulated each of the steps in the NAD biosynthetic pathway and found that overexpression of nuclear or non-nuclear forms of Nmnat provide strong protection against axonal degeneration. Nicotinamide phosphoribosyl transferase (NmPRT) and nicotinic acid (Na) phosphoribosyl transferase (NaPRT), which convert the precursor molecules nicotinamide and nicotinic acid, to the respective mononucleotides, also provide axonal protection. We also found that exogenous application of nicotinic acid mononucleotide (NaMN), nicotinamide mononucleotide (NMN), and nicotinamide riboside (NmR) all promoted axonal protection to a similar extent as NAD itself. These results show that stimulating the activity of the NAD biosynthetic pathway at multiple steps provides protection against axonal degeneration after axotomy. Furthermore, they supply evidence that the Wlds effect is mediated via alteration of the NAD biosynthetic pathway and present new avenues for potential therapeutic agents to slow axonal degeneration in diseases of the nervous system.
Materials and Methods
Construction of expression plasmids and mutagenesis.
The coding region of each NAD biosynthetic enzyme was amplified by PCR using Herculase (Stratagene, Garden Grove, CA) from expressed sequence tagged (EST) clones [GenBank accession numbers BC011191 for murine quinolinate phosphoribosyl transferase (QPRT), BQ938564 for murine NaPRT, BC004059 for murine NmPRT, BC038133 for murine Nmnat1, BC005737 for murine Nmnat3, BC016495 for murine nicotinamide riboside kinase 1 (Nrk1), and AI239871 for human Nrk2]. A hexahistidine tag was added at the C terminus of each enzyme. Human glutamine-dependant NAD synthetase (QNS) hexahistidine-tagged cDNA was kindly provided by Nobumasa Hara (Shimane University, Shimane, Japan) (Hara et al., 2003).
To produce the Nmnat1 cytoplasmic mutant (cytNmnat1), the residues corresponding to the nuclear localization site (PGRKRKW located at residues 211–217 to PGAAAAW) were mutated using PCR-mediated site-directed mutagenesis (Ausubel, 2001). A mutant of Nmnat3 (nucNmnat3) localized to the nucleus was generated by adding a nuclear localization signal (KPKKIKTED) derived from human topoisomerase to the C-terminal end of Nmnat3 as described previously (Araki et al., 2004). All of the modified cDNAs encoding these enzymes were cloned into the FCIV lentiviral shuttle vector as described previously (Araki et al., 2004). The integrity of each clone was verified by nucleotide sequence analysis.
NAD biosynthesis substrates.
All substrates for NAD biosynthetic enzymes were purchased from Sigma (Na, N0761; nicotinamide, N0636; NMN, N3501; NaMN, N7764; nicotinic acid adenine dinucleotide (NaAD), N4256; and NAD, N1636). NmR was synthesized from NMN using a phosphatase reaction as described previously (Bieganowski and Brenner, 2004). The conversion of NMN to NmR was monitored using an HPLC reverse-phase column LC-18T (Supelco, Bellefonte, PA) with a buffer containing 50 mm K2HPO4 and 50 mm KH2PO4, pH 7.0. Using a flow rate of 1 ml/min, NmR eluted at 260 ± 10 s and NMN eluted at 150 ± 10 s. The biological activity of NmR was confirmed using yeast strains BY165 and BY278 kindly provided by Charles Brenner (Dartmouth Medical School, Hanover, NH) (Bieganowski and Brenner, 2004).
Quantitation of NAD biosynthetic enzyme expression.
Male Sprague Dawley rats (200–300 g; Harlan Bioproducts, Indianapolis, IN) were anesthetized, and the right sciatic nerve was transected at the hip level using surgical scissors. Dorsal root ganglia (DRGs) from L4 and L5 were collected from three animals at the indicated time points and pooled, and RNA was prepared via Trizol method (Invitrogen, Carlsbad, CA). First-strand cDNA templates were prepared from two independent cDNA syntheses for each sample using 1 μg of RNA template using standard methods. Quantitative reverse transcription (RT)-PCR was performed by monitoring in real time the increase in fluorescence of the SYBR-green dye on a TaqMan 7700 Sequence Detection System (Applied Biosystems, Foster City, CA). The expression levels of glyceraldehye-3-phosphate dehydrogenase was used for normalization of variations between the levels of total cDNA template across different samples. Each RT-PCR quantitation experiment was performed twice using duplicate samples from two independently generated cDNA templates (Araki et al., 2001). All surgical procedures were performed according to National Institute of Health guidelines for care and use of laboratory animals at Washington University.
Cell culture, in vitro axotomy, and quantification of axonal degeneration.
Mouse DRGs dissected from embryonic day 12.5 (E12.5) to E13.5 embryos were cultured as explants on collagen-coated 24-well dishes in DMEM containing 10% FCS and 25 ng/ml nerve growth factor (2.5S; Harlan Bioproducts) as described previously (Araki et al., 2004). Non-neuronal cells were removed from the cultures by adding a mixture of 1 μm 5-fluoro-2′-deoxyuridine and 1 μm uridine to the culture media. Transection of axons was performed at 14–28 d in vitro using an 18 gauge needle to remove the neuronal cell bodies. Lentiviruses expressing NAD biosynthetic enzymes were generated as described previously (Araki et al., 2004). Lentiviruses (104 to 105 total infectious units) were added to each DRG explant, and protein expression from the lentivirus was allowed to accumulate for 4–7 d before axonal transection. Expression of the transgene was confirmed by expression of an enhanced green fluorescent protein (EGFP) reporter that is located downstream of an internal ribosome entry site within the lentiviral vector. For each condition, two DRG explants were used for each experiment, and two to three independent experiments were performed for each condition. Quantitative analysis of axonal degeneration was performed as described previously (Araki et al., 2004). Statistical analysis was performed by Student’s t test.
Monitoring protein expression and localization.
The expression of each NAD biosynthetic enzyme from the respective lentivirus was verified using infected HEK293T cells. Five days after infection, lysates were prepared using buffer containing 20 mm Tris-HCl, pH7.5, 300 mm NaCl, 5 mm EDTA, 0.1% Triton X-100, and protease inhibitor cocktail (Roche, Indianapolis, IN). The expression levels of the hexahistidine-tagged enzymes were analyzed by immunoblotting using anti-hexahistidine tag monoclonal antibody (R & D Systems, Minneapolis, MN). The subcellular localization of each protein was analyzed using HEK293T cells expressing each of the enzymes. The cells were fixed with 4% paraformaldehyde and permeabilized with PBS containing 0.1% Tween 20 (PBS-T). They were then incubated with PBS-T containing 5% BSA for 1 h. Cells were incubated with 1:1000 diluted anti-hexahistidine tag antibody in PBS-T containing 5% BSA for 16 h at 4°C. After incubation with Alexa Fluor 594-conjugated secondary antibody (Invitrogen) in TBS containing 0.1% Tween 20 for 1 h, the proteins were visualized by fluorescence microscopy (Nikon, Melville, NY).
Results
Overexpression of multiple NAD biosynthetic enzymes delays axonal degeneration
The expression of Wlds protein or Nmnat1 itself is sufficient to protect axons in an in vitro Wallerian degeneration assay, suggesting that increased NAD production is crucial for axonal protection (Araki et al., 2004). Overexpression of NmPRT, which converts nicotinamide to NMN, increases intracellular NAD concentrations in fibroblasts (Revollo et al., 2004), stimulating us to search for additional manipulations of the NAD biosynthetic pathway that can promote axonal protection.
NAD is recognized as a key molecule for cellular metabolism in organisms ranging from archebacteria to humans. It serves as a cofactor for most dehydrogenases, is important for aerobic and anaerobic ATP generation, and acts as a substrate for proteins such as PARP [poly(ADP-ribose) polymerase] or Sir2 (silent information regulator 2). NAD is synthesized via three major pathways in both prokaryotes and eukaryotes: the de novo, salvage, and Preiss-Handler independent. In the de novo pathway, NAD is synthesized from tryptophan (supplemental Fig. S1, available at www.jneurosci.org as supplemental material), whereas in the salvage pathway, NAD is generated from vitamins, including nicotinic acid and nicotinamide, and, in a recently discovered third route, NAD is derived from NmR. The mammalian homologs for most of the yeast NAD biosynthetic enzymes have been cloned and characterized (supplemental Fig. S1, available at www.jneurosci.org as supplemental material), including NmPRT, QNS, Nrk1/2, and Nmnat1–Nmnat3. For this work, we also identified mammalian homologs of NaPRT, which was present in the database as an EST (BQ938564) having NaPRT conserved domain [National Center for Biotechnology Information (NCBI) conserved domain cd01570.2], and QPRT, which was present in the database as an EST (GenBank accession number BC011191) having a QPRT conserved domain (NCBI clone cd01572.2). All of these enzymes were epitope tagged and cloned into the FCIV lentivirus vector (Araki et al., 2004). HEK293 cells were infected with these viruses, and cell lysates were used to confirm the expression (Fig. 1C). The catalytic activity of each enzyme was confirmed by HPLC (supplemental Fig. S2, available at www.jneurosci.org as supplemental material).
Figure 1.

Axonal protection is mediated by enforced expression of NAD biosynthetic enzymes. In vitro Wallerian degeneration assays were performed using DRG neuronal explant cultures. A, DRG neurons were infected with lentivirus expressing the indicated enzyme (Nmnat1, NmPRT, or EGFP control) 5 d before axotomy. Representative pictures taken at 12, 24, 48, and 72 h after transection are shown. B, In vitro Wallerian degeneration assays were performed and axonal degeneration was quantified as outlined in Materials and Methods. The percentage ± SD of remaining axons relative to pretransection at 12, 24, 48, and 72 h after transection is displayed. *p < 0.0001 indicates a significant difference (n = 6) with EGFP-expressing cells. C, Protein expression of QPRT, NaPRT, NmPRT, Nmnat1, QNS, Nrk1, and Nrk2 by lentivirus was confirmed by immunoblot analysis of HEK293T cells infected with each virus. The enzymatic activity of each clone was confirmed as described in the supplemental data (available at www.jneurosci.org as supplemental material). D, The subcellular localization of the indicated NAD biosynthetic enzymes was examined using immunocytochemistry with an anti-hexahistidine tag antibody of transfected HEK293T cells. The cells were also stained with bis-benzimide to highlight the nucleus to determine the nuclear versus cytoplasmic distribution of each protein. Scale bar, 10 μm.
To test whether expression of any of these NAD biosynthetic enzymes could promote axonal protection, we infected primary cultured DRG neurons with lentiviruses expressing each enzyme and performed in vitro Wallerian degeneration experiments. Consistent with previous reports, axonal degeneration from DRG neurons expressing Nmnat1 was delayed for >72 h after axotomy, whereas axons from DRG neurons expressing EGFP degenerated within 24 h after axotomy (Fig. 1A). Next, we compared the extent of axonal degeneration in DRGs expressing each of the NAD biosynthetic enzymes with those expressing Nmnat1 or EGFP. We found that expression of NmPRT delayed axonal degeneration (Fig. 1A), whereas QPRT, NaPRT, Nrk1, Nrk2, and QNS did not protect against axonal degeneration. We quantified the extent of axonal degeneration by calculating the percentage of remaining axons at the indicated times after transection. This analysis revealed that Nmnat1 and NmPRT resulted in a 10-fold and fivefold increase in the number of axons remaining 48 h after axotomy, respectively (Fig. 1B). The protection mediated by NmPRT was observed only when neurons were cultured in medium containing nicotinamide; thus, these results suggest that increased activity of the NAD biosynthetic pathway stemming from nicotinamide promotes axonal protection in this in vitro Wallerian degeneration assay.
Nmnat expression in multiple cellular compartments can promote axonal protection
In wlds mice, the Wlds protein is located within the nucleus and is absent from sciatic nerve and motor neuron terminals (Mack et al., 2001). Nmnat1 is also a nuclear protein; however, the localization of the upstream enzymes in these pathways, including NmPRT, which also promotes axonal protection, has not been well described. We therefore used immunocytochemistry to examine the subcellular localization of each NAD biosynthetic enzyme. HEK293T cells expressing each NAD biosynthetic enzyme were stained with anti-hexahistidine tag antibodies, and the proteins were visualized with the Texas Red-conjugated secondary antibody. As reported previously, Nmnat1 is present in the nucleus. However, QPRT, NaPRT, NmPRT, Nrk1, and Nrk2 are diffusely distributed throughout the cell, and QNS is localized primarily to the cytoplasm (Fig. 1D).
Of the two enzymes that promoted axonal protection, Nmnat1 is predominantly nuclear, and NmPRT is found distributed throughout the cell. These data coupled with the nuclear localization of the Wlds fusion protein suggested that the nuclear production of NAD might be important for the protective function. To test the association between subcellular distribution of the Nmnat enzyme and axonal protection, we mutated the putative Nmnat1 nuclear localization signal (PGRKRKW) located at residues 211–217 to PGAAAAW to generate a cytoplasmic Nmnat1 (designated cytNmnat1). Wild-type and mutant Nmnat1 proteins were expressed in HEK293 cells, and the successful generation of a cytoplasmic Nmnat1 was demonstrated (Fig. 2A). The enzymatic activity of cytNmnat1 was compared with Nmnat1 using proteins affinity purified from transfected HEK293 cell lysates using His-select Nickel Affinity Gel as described in Materials and Methods. We found that the amount of NAD synthesized by the cytNmnat1 was indistinguishable from that produced by wild-type Nmnat1 (supplemental Fig. S3, available at www.jneurosci.org as supplemental material). To test the ability of cytNmnat1 to protect axons, we expressed it in DRG neurons and performed in vitro Wallerian degeneration assays. We found that cytNmnat1 promoted strong axonal protection that was indistinguishable from wild-type Nmnat1 (Fig. 2B,C).
Figure 2.

Nmnat enzymes located in the nucleus, cytoplasm, or mitochondria all promote axonal protection. A, Subcellular localization of Nmnat1, cytNmnat1, Nmnat3, or nucNmnat3 in HEK293T cells. Immunohistochemistry using antibody against the hexahistidine epitope tag was used to detect each protein. The cells were also stained with bis-benzimide. As expected, the cytNmnat1 mutant is located in the cytoplasm, and the nucNmnat3 mutant is located in the nucleus. Scale bar, 10 μm. B, In vitro Wallerian degeneration assay using lentivirus-infected DRG neuronal explant cultures expressing Nmnat1, cytNmnat1, Nmnat3, nucNmnat3, or EGFP control. Representative pictures taken at 12 and 72 h after transection show robust protection against axonal degeneration regardless of the subcellular distribution. C, Quantitative analysis of axonal degeneration in DRG explant cultures expressing Nmnat1, cytNmnat1, Nmnat3, or nucNmnat3 at 12, 24, 48, and 72 h after transection. Nmnat1-, cytNmnat1-, Nmnat3-, and nucNmnat3-expressing cells have a significant difference (p < 0.0001; n = 6) with EGFP-expressing cells at 24, 48, and 72 h after axotomy.
We extended these results by using an Nmnat isoform, Nmnat3, which has comparable enzymatic activity with Nmnat1 (Raffaelli et al., 2002) and is localized within mitochondria (Zhang et al., 2003). We prepared hexahistidine-tagged versions of Nmnat3 and a Nmnat3 mutant in which we added a C-terminal nuclear localization signal (KPKKIKTED) from human topoisomerase I to generate nuclear Nmnat3 (designated nucNmnat3). We expressed wild-type Nmnat3 or nucNmnat3 in HEK293T cells and analyzed their subcellular localization and enzymatic activity. Nmnat3 was distributed outside of the nucleus with bright punctuate staining in the cytoplasm, consistent with a mitochondrial distribution as reported previously, whereas nucNmnat3 was predominantly found in the nucleus as expected (Fig. 2A). The enzymatic activity of Nmnat3 and nucNmnat3 were measured in vitro, and both proteins produced amounts of NAD that were comparable with that of Nmnat1 (supplemental Fig. S3, available at www.jneurosci.org as supplemental material). When these two Nmnat3 proteins were tested in the axonal degeneration assay, we found that both of them afforded protection to a similar extent as Nmnat1 (Fig. 2B,C). Overall, these experiments confirmed the potent axonal protection afforded by Nmnat enzymes and demonstrated that the subcellular site of increased Nmnat enzymatic activity is not important for mediating the delay in axonal degeneration. This would be consistent with the observation that addition of NAD to the culture medium also delays axonal degeneration, suggesting that NAD can diffuse to multiple cellular compartments.
Treatment with NAD precursors can delay axonal degeneration in DRG neurons
Previous work has shown that the addition of NAD to cultured neurons delays axonal degeneration (Araki et al., 2004; Wang et al., 2005). The expression of Nmnat enzymes as well as NmPRT, which ultimately convert NAD precursor substrates to NAD, also promote axonal protection, suggesting that NAD precursors may also be effective in protecting axons. Because some of these precursors may be more stable or enter neurons more efficiently than NAD itself, we tested the ability of many of these substrates to promote axonal protection. These included nicotinamide (the substrate of NmPRT), NMN (the substrate of Nmnat), and nicotinic acid, NmR, NaMN, and NaAD. Each of these substrates was added to cultured DRG neurons 24 h before axotomy at a concentration of 1 mm. We found that NMN, NaMN, and NmR delayed axonal degeneration comparably with NAD, whereas NaAD showed only slight activity and nicotinic acid and nicotinamide had no activity (Fig. 3 A,B). Interestingly, the protective effect of NaMN was equivalent to NMN, suggesting that the reaction converting NaAD to NAD catalyzed by QNS is sufficient to protect axons in the presence of an increased supply of NaAD. However, exogenous application of NaAD was only moderately effective in protecting axons, suggesting that this substrate is unstable or does not enter neurons efficiently (Fig. 3B).
Figure 3.
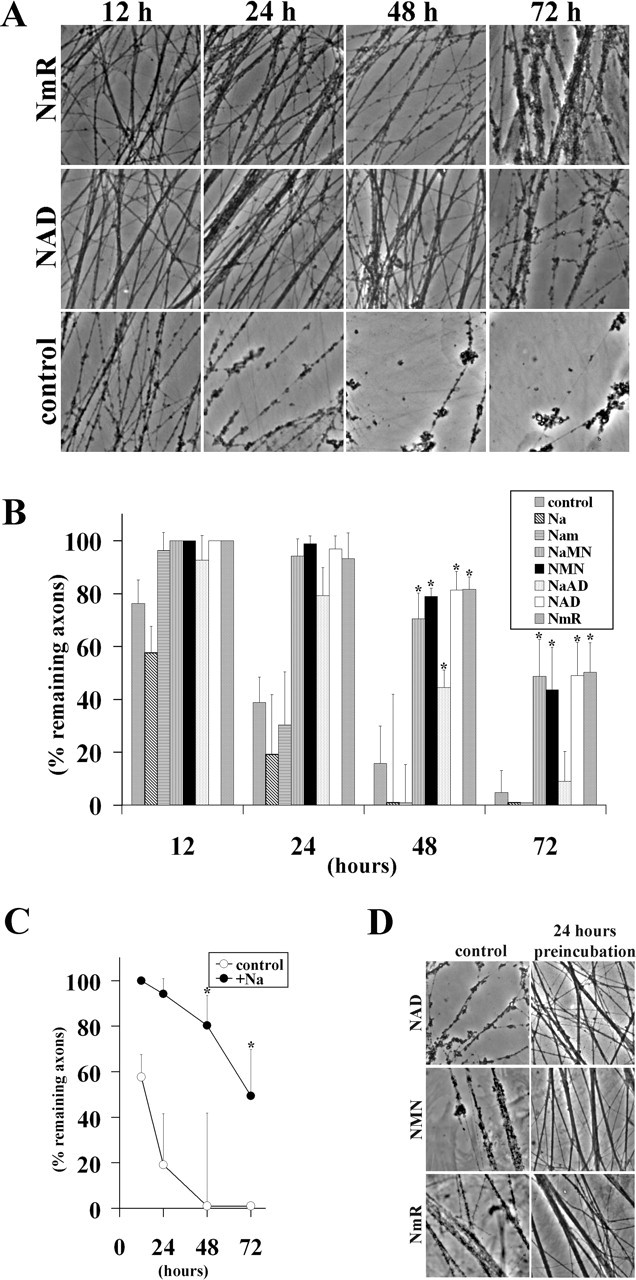
Treatment of cultured DRG neurons with NAD precursors delays axonal degeneration. A, In vitro Wallerian degeneration assay using DRG explant cultures after exogenous application of NAD or NmR. Representative pictures at 12, 24, 48, and 72 h after transection are shown. B, In vitro Wallerian degeneration assay using DRG neuronal explant cultures after exogenous application of Na, nicotinamide (Nam), NaMN, NMN, NaAD, NAD, and NmR. Quantitative analysis of axonal protection at 12, 24, 48, and 72 h after axotomy is shown. *p < 0.002 indicates a significant difference (n = 6) compared with control. C, Axonal degeneration in DRG explants infected with NaPRT-expressing lentivirus is delayed only when 1 mm nicotinic acid is added to the medium 24 h before axotomy. Percentage of remaining axons at 12, 24, 48, and 72 h after transection is shown. *p < 0.01 indicates a significant difference (n = 6) compared with control. D, NAD, NMN, or NmR (1 mm) was added either at the time of transection (control) or 24 h before axonal transection (24 h before incubation). Representative pictures taken 24 h after transection show axonal protection only in cultures in which substrates were added before injury.
We found it surprising that NaPRT was ineffective in protecting axons, whereas NmPRT mediated axonal protection because both NaMN and NMN, the respective products of these two enzymes, were able to delay axonal degeneration. The Wallerian degeneration assays are conducted in culture medium containing nicotinamide but lacking nicotinic acid. This suggested to us that the failure of NaPRT to protect axons could be attributable to insufficient amounts of its substrate, nicotinic acid. We therefore performed experiments in which both substrate and enzyme were increased. For this experiment, DRG neurons were infected with NaPRT lentivirus and cultured in the presence (or absence) of 1 mm nicotinic acid 24 h before axotomy. We found that the combination of increased NaPRT along with 1 mm nicotinic acid effectively delayed axonal degeneration, whereas neither increased enzyme or substrate alone delayed axonal degeneration (Figs. 1B, 3C).
We showed previously that neurons must be treated with NAD for ∼24 h before axotomy to demonstrate axonal protection (Araki et al., 2004), whereas others have reported that NAD can protect axons when added contemporaneously with the injury (Wang et al., 2005). Because we have now identified multiple substrates capable of delaying axonal degeneration, we tested the time dependency of these substrates. NAD, NMN, or NmR (1 mm each) was added either simultaneously or 24 h before axotomy. At 24 h after transection, there was a significant decrease in the number of degenerating axons when the substrates were added before the injury (Fig. 3D), but no protection was afforded if they were added at the time of injury. Thus, like NAD, both NMN and NmR require a pretreatment period to provide axonal protection. These results strengthen the conclusion that increased NAD synthesis is the crucial step in promoting axonal protection, because a variety of manipulations of NAD biosynthetic pathways, including increased substrate availability, increased enzyme levels, or combinations thereof, delay axonal degeneration.
Expression of enzymes involved in NAD biosynthesis is induced in response to neuronal injury
The impact of NAD biosynthetic activity on axonal protection stimulated us to investigate the expression of the enzymes involved in this pathway in the nervous system during development and after neuronal injury. We surveyed their expression in brain, retina, spinal cord, and DRGs at ages E14, postnatal day 0 (P0), P7, P14, and P21 using quantitative RT-PCR. All enzymes were expressed ubiquitously in the nervous system throughout development and in adulthood, with the exception of Nrk2, whose expression is very low in all tissues examined (data not shown). To determine whether neuronal stress leads to increased expression of these NAD-synthesizing enzymes, we compared their RNA levels in DRGs before injury and at 1, 3, 7, and 14 d after sciatic nerve transection. The expression of most of the enzymes increased twofold to eightfold after injury, whereas Nrk2, which converts NmR to NMN, was induced >20-fold at 14 d after axotomy (Fig. 4). These results suggest that neurons express all NAD biosynthetic enzymes at a basal level and that the pathways that synthesize NAD are activated after neuronal injury to enable increased NAD production for downstream functions associated with the neuronal response to injury.
Figure 4.

Expression of NAD biosynthetic enzymes is regulated by injury in DRG neurons. Quantitative RT-PCR analysis of indicated NAD biosynthesis enzyme mRNA levels in rat DRGs 0, 1, 3, 7, and 14 d after sciatic nerve transection. The expression level was normalized to glyceraldehyde-3-phosphate dehydrogenase and is indicated relative to expression in uninjured DRGs.
Discussion
The mutation in the wlds mutant mouse responsible for the delay in Wallerian degeneration results in the production of a fusion protein composed of a portion of the E3 ubiquitin ligase Ufd2a fused in-frame to the full-length Nmnat1 coding sequence (Conforti et al., 2000). Using an in vitro neuronal culture system, we demonstrated that axonal protection was mediated via the NAD biosynthetic activity of the Nmnat1 portion of the Wlds fusion protein (Araki et al., 2004). Indeed, mutant Wlds or Nmnat1 protein, which harbors a mutation that destroys their NAD-synthesizing activity, fail to save axons, suggesting that an increased supply of NAD is responsible for preventing axonal degeneration. In addition, exogenous application of NAD to cultured neurons before axotomy also delays axonal degeneration, suggesting that NAD may modify a genetic program that results in altered axonal physiology. These studies led us to postulate that manipulation of other enzymatic reactions in the NAD biosynthetic pathway, either through increased expression of the pertinent enzymes or addition of their cognate substrates, might also provide axonal protection. We found that expression of Nmnat1 or Nmnat3 alone could delay axonal degeneration, whereas NmPRT and NaPRT could delay axonal degeneration when high levels of their requisite substrates, nicotinamide or nicotinic acid, were present in the culture medium. The enforced expression of other NAD synthetic enzymes, including QPRT, QNS, Nrk1, and Nrk2, did not promote axonal protection. The addition of multiple NAD precursors, including NaMN, NMN, NaAD, and NmR, gave protection similar to that provided by NAD itself, whereas nicotinic acid and nicotinamide did not. We added NMN or NmR at various times relative to axotomy and found that, like NAD, they afforded protection only if they were administered before axonal injury, suggesting that they protect axons via a similar mechanism. These studies have demonstrated that multiple manipulations that increase activity of the NAD pathway can promote axonal protection, supporting the hypothesis that increased NAD availability is a likely mechanism responsible for the delayed axonal degeneration mediated by the Wlds protein.
The Nmnat enzymes catalyze the final step in NaAD and NAD synthesis by conversion of the mononucleotides NaMN and NMN to NaAD and NAD, respectively. These two mononucleotide compounds can be synthesized from three different pathways: the de novo pathway, the salvage pathway, and a Preiss-Handler independent pathway. NaMN can be synthesized from nicotinic acid via NaPRT (salvage steps) or quinolinic acid (de novo pathway). Our culture medium does not contain nicotinic acid, and production of quinolinic acid through the de novo pathway must be limited because expression of QPRT, which converts quinolinic acid to NaMN, cannot delay axonal degeneration; thus, it appears that NaMN synthesis is relatively low in our assay conditions. Furthermore, although NaAD can promote axonal protection, QNS expression, which converts NaAD to NAD, was not effective, presumably because of low intracellular levels of NaAD secondary to low concentrations of NaMN. These results indicate that the activity of the de novo pathway is insufficient to increase NAD levels high enough to promote axonal protection. Similarly, the intracellular levels of NmR must be low because enforced expression of the Nrk enzymes that convert NmR to NMN do not delay axonal degeneration, whereas addition of NmR itself protects equivalently to NAD administration. It thus appears that the predominant pathway involved in promoting axonal protection under our assay conditions is the salvage pathway that converts nicotinamide, which is present as a major vitamin in the culture medium, to NAD. Consistent with this hypothesis, if nicotinic acid is added to the medium, then enforced expression of NaPRT can also effectively delay axonal degeneration attributable to the increased production of NaMN. The pathways that are most active in vivo under normal versus pathological conditions will dictate which manipulations of NAD production will most effectively alter axonal degeneration dynamics under disease conditions.
Both the Wlds and Nmnat1 proteins are localized to the nucleus and provide robust axonal protection, whereas NmPRT and NaPRT, which are distributed throughout multiple cellular compartments, mediate moderate protection. These results raise the possibility that NAD production in the nucleus is more effective in mediating axonal protection. Others have also suggested that the nuclear localization may be important for Wlds-mediated axonal protection, possibly by the interaction of valosin-containing-protein (VCP/p97) with the Ufd2a portion of the Wlds fusion protein (Conforti et al., 2006; Laser et al., 2006). However, we found that an Nmnat1 cytoplasmic mutant and the closely related Nmnat3, which is localized to mitochondria, also mediated robust axonal protection. These results suggest that neither Nmnat location nor interaction with VCP is crucial for axonal protection. Instead, it is likely that axonal protection is provided by NAD that is rapidly distributed throughout multiple cellular compartments. Furthermore, the more robust axonal protection mediated by Nmnat compared with NaPRT and NmPRT could result if NAD synthesis in neurons is limited by Nmnat activity.
A recent study from Wang et al. (2005) confirmed the ability of Nmnat1 and NAD to delay axonal degeneration; however, much higher levels of NAD (5 vs 1 mm) were required for protection in those experiments. They also noted that axonal degeneration could be delayed in their system when NAD was added at the time of injury or even slightly after the injury (i.e., no preincubation step was required). In contrast and in accord with our previous study, we find that neurons must be preincubated with NAD or other protective compounds such as NMN or NmR to protect against axonal degeneration. Our observations suggest that the mechanism of axonal protection mediated by preincubation with NAD, NMN, or NmR is different from that observed using postaxotomy treatment. In addition, another recent study claims that both Nmnat1 and NAD are ineffective in preventing axonal degeneration, whereas the Wlds fusion protein mediated protection (Conforti et al., 2006). However, in this report, it was shown that an enzymatically inactive Wlds mutant did not provide axonal protection as well as Wlds, again indicating that NAD production is an important feature of Wlds-mediated axonal protection. These discrepancies may be derived from differences in the neuronal culture conditions such as components in the culture medium, substrates for cell adhesion, or the age of the neurons. All of these parameters, in particular the number of days in vitro, affect intracellular calcium concentrations and requirements for trophic factors (Tong et al., 1996), and it is likely that aspects of Wallerian degeneration are also affected by these factors. In addition, in vivo studies with transgenic mice suggest that Nmnat1 overexpression provides weak axonal protection compared with Wlds (Conforti et al., 2006), whereas dNmnat overexpression in Drosophila olfactory receptor neurons provided strong axonal protection (MacDonald et al., 2006). A detailed characterization of NAD-dependent delay in axonal degeneration in vitro, and eventually in vivo, will help reveal the precise molecular mechanisms of this protective phenomenon.
The expression of many NAD biosynthetic enzymes is altered after axotomy. In particular, the levels of Nrk2 were dramatically increased. This result suggests that Nrk2-dependent NmR conversion to NMN is an important aspect of the neuronal response to stress. The NAD biosynthetic pathway that uses NmR as a substrate is independent from the de novo and salvage pathways, because NmR is found in milk and has been proposed as a vitamin (Bieganowski and Brenner, 2004). In contrast to precursors such as nicotinamide and nicotinic acid, the exogenous application of NmR protects axons from degeneration as well as NAD. This observation suggests that Nrk activity is high enough to convert exogenously applied NmR to NMN in cultured DRG neurons. This would imply that NmR levels in the culture medium are normally very low and would explain why enforced expression of Nrk1 or Nrk2 was ineffective in mediating axonal protection. Because NmR is the only dietary nutrient in the NAD biosynthetic pathway in our study that promotes axonal protection and the enzyme Nrk2, which converts it to NMN, is dramatically induced after axotomy, it may be useful in the treatment of diseases in which axonopathy is a major component of the pathophysiology.
In summary, we found that multiple manipulations that increase activity of the NAD pathway can promote axonal protection, supporting the hypothesis that increased NAD availability is a likely mechanism responsible for the delayed axonal degeneration mediated by the Wlds protein. Additional investigation to apply these findings in vivo will be critical to assess the potential utility of this pathway as a therapeutic target.
Footnotes
This work was supported by the Hope Center for Neurological Disorders, National Institutes of Health Grants AG13730, NS39358, and NS049087, and Muscular Dystrophy Association Grant 3972 (J.M.). We thank Kelli Simburger and Tim Fahrner for experimental assistance, members of the Milbrandt laboratory for their comments on this manuscript, and E. Johnson and S. Imai for helpful discussions. We and Washington University may derive benefit from a licensing agreement with Sirtris Pharmaceuticals, which did not provide any support for this work.
References
- Araki T, Nagarajan R, Milbrandt J (2001). Identification of genes induced in peripheral nerve after injury. Expression profiling and novel gene discovery. J Biol Chem 276:34131–34141. [DOI] [PubMed] [Google Scholar]
- Araki T, Sasaki Y, Milbrandt J (2004). Increased nuclear NAD biosynthesis and SIRT1 activation prevent axonal degeneration. Science 305:1010–1013. [DOI] [PubMed] [Google Scholar]
- Ausubel FM (2001). In: Current protocols in molecular biology Vol 1: Chap 8 New York: Wiley. [Google Scholar]
- Bieganowski P, Brenner C (2004). Discoveries of nicotinamide riboside as a nutrient and conserved NRK genes establish a Preiss-Handler independent route to NAD+ in fungi and humans. Cell 117:495–502. [DOI] [PubMed] [Google Scholar]
- Coleman M (2005). Axon degeneration mechanisms: commonality amid diversity. Nat Rev Neurosci 6:889–898. [DOI] [PubMed] [Google Scholar]
- Conforti L, Tarlton A, Mack TG, Mi W, Buckmaster EA, Wagner D, Perry VH, Coleman MP (2000). A Ufd2/D4Cole1e chimeric protein and overexpression of Rbp7 in the slow Wallerian degeneration (WldS) mouse. Proc Natl Acad Sci USA 97:11377–11382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conforti L, Fang G, Beirowski B, Wang MS, Sorci L, Asress S, Adalbert R, Silva A, Bridge K, Huang XP, Magni G, Glass JD, Coleman MP (2006). NAD(+) and axon degeneration revisited: Nmnat1 cannot substitute for Wld(S) to delay Wallerian degeneration. Cell Death Differ in press. [DOI] [PubMed]
- Hara N, Yamada K, Terashima M, Osago H, Shimoyama M, Tsuchiya M (2003). Molecular identification of human glutamine- and ammonia-dependent NAD synthetases. Carbon-nitrogen hydrolase domain confers glutamine dependency. J Biol Chem 278:10914–10921. [DOI] [PubMed] [Google Scholar]
- Laser H, Conforti L, Morreale G, Mack TG, Heyer M, Haley JE, Wishart TM, Beirowski B, Walker SA, Haase G, Celik A, Adalbert R, Wagner D, Grumme D, Ribchester RR, Plomann M, Coleman MP (2006). The slow Wallerian degeneration protein, WldS, binds directly to VCP/p97 and partially redistributes it within the nucleus. Mol Biol Cell 17:1075–1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lunn ER, Perry VH, Brown MC, Rosen H, Gordon S (1989). Absence of Wallerian degeneration does not hinder regeneration in peripheral nerve. Eur J Neurosci 1:27–33. [DOI] [PubMed] [Google Scholar]
- MacDonald JM, Beach MG, Porpiglia E, Sheehan AE, Watts RJ, Freeman MR (2006). The Drosophila cell corpse engulfment receptor draper mediates glial clearance of severed axons. Neuron 15:869–881. [DOI] [PubMed] [Google Scholar]
- Mack TG, Reiner M, Beirowski B, Mi W, Emanuelli M, Wagner D, Thomson D, Gillingwater T, Court F, Conforti L, Fernando FS, Tarlton A, Andressen C, Addicks K, Magni G, Ribchester RR, Perry VH, Coleman MP (2001). Wallerian degeneration of injured axons and synapses is delayed by a Ube4b/Nmnat chimeric gene. Nat Neurosci 4:1199–1206. [DOI] [PubMed] [Google Scholar]
- Perry VH, Brown MC, Lunn ER (1991). Very slow retrograde and Wallerian degeneration in the CNS of C57BL/Ola Mice. Eur J Neurosci 3:102–105. [DOI] [PubMed] [Google Scholar]
- Raff MC, Whitmore AV, Finn JT (2002). Axonal self-destruction and neurodegeneration. Science 296:868–871. [DOI] [PubMed] [Google Scholar]
- Raffaelli N, Sorci L, Amici A, Emanuelli M, Mazzola F, Magni G (2002). Identification of a novel human nicotinamide mononucleotide adenylyltransferase. Biochem Biophys Res Commun 297:835–840. [DOI] [PubMed] [Google Scholar]
- Revollo JR, Grimm AA, Imai S (2004). The NAD biosynthesis pathway mediated by nicotinamide phosphoribosyltransferase regulates Sir2 activity in mammalian cells. J Biol Chem 279:50754–50763. [DOI] [PubMed] [Google Scholar]
- Tong JX, Eichler ME, Rich KM (1996). Intracellular calcium levels influence apoptosis in mature sensory neurons after trophic factor deprivation. Exp Neurol 138:45–52. [DOI] [PubMed] [Google Scholar]
- Wang J, Zhai Q, Chen Y, Lin E, Gu W, McBurney MW, He Z (2005). A local mechanism mediates NAD-dependent protection of axon degeneration. J Cell Biol 170:349–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang MS, Fang G, Culver DG, Davis AA, Rich MM, Glass JD (2001). The Wlds protein protects against axonal degeneration: a model of gene therapy for peripheral neuropathy. Ann Neurol 50:773–779. [DOI] [PubMed] [Google Scholar]
- Zhang X, Kurnasov OV, Karthikeyan S, Grishin NV, Osterman AL, Zhang H (2003). Structural characterization of a human cytosolic NMN/NaMN adenylyltransferase and implication in human NAD biosynthesis. J Biol Chem 278:13503–13511. [DOI] [PubMed] [Google Scholar]