

Abstract
Hypersensitivity to mechanical stimulation is a well documented symptom of neuropathic pain, for which there is currently no effective therapy. Src-family kinases (SFKs) are involved in proliferation and differentiation and in neuronal plasticity, including long-term potentiation, learning, and memory. Here we show that activation of SFKs induced in spinal cord microglia is crucial for mechanical hypersensitivity after peripheral nerve injury. Nerve injury induced a striking increase in SFK phosphorylation in the ipsilateral dorsal horn, and SFKs were activated in hyperactive microglia but not in neurons or astrocytes. Intrathecal administration of the Src-family tyrosine kinase inhibitor 4-amino-5-(4-chlorophenyl)-7-(t-butyl)pyrazolo[3,4-d]pyrimidine (PP2) suppressed nerve injury-induced mechanical hypersensitivity but not heat and cold hypersensitivity. Furthermore, PP2 reversed the activation of extracellular signal-regulated protein kinase (ERK), but not p38 mitogen-activated protein kinase, in spinal microglia. In contrast, there was no change in SFK phosphorylation in primary sensory neurons, and PP2 did not decrease the induction of transient receptor potential ion channel TRPV1 and TRPA1 in sensory neurons. Together, these results demonstrate that SFK activation in spinal microglia contributes to the development of mechanical hypersensitivity through the ERK pathway. Therefore, preventing the activation of the Src/ERK signaling cascade in microglia might provide a fruitful strategy for treating neuropathic pain.
Keywords: Src-family kinase, extracellular signal-regulated protein kinase, p38 mitogen-activated protein kinase, microglia, spinal cord, neuropathic pain
Introduction
Injury to peripheral nerves often produces spontaneous pain, hyperalgesia (increased responsiveness to noxious stimuli), and allodynia (painful responses to normally innocuous stimuli). In contrast to inflammatory pain, the currently available therapeutics for this neuropathic pain is either relatively ineffective or accompanied by substantial side effects. After nerve injury, plastic changes occur in the expression of ion channels, receptors, neuropeptides, and signal transduction-related molecules in the peripheral nervous system and CNS (Scholz and Woolf, 2002; Ji and Strichartz, 2004). In previous studies, much attention has been focused on the primary sensory neurons and their influence on the activity of spinal dorsal horn neurons (Hokfelt et al., 1994; Alvares and Fitzgerald, 1999). However, there is increasing evidence suggesting that glial cells in the spinal cord may also play a role in the pathogenesis of neuropathic pain (Tsuda et al., 2005; Marchand et al., 2005). In fact, microglia and astrocytes are activated in the spinal dorsal horn in several neuropathic pain models. This spinal glial activation is likely involved in the production and release of proinflammatory cytokines and thus increasing pain hypersensitivity (DeLeo and Yezierski, 2001; Watkins et al., 2001a,b; Watkins and Maier, 2003).
Src and other Src-family kinases (SFKs) are expressed widely throughout the mammalian CNS and have been implicated in proliferation and differentiation in the developing CNS (Kuo et al., 1997; Hoffman-Kim et al., 2002). In the developed CNS, there is compelling evidence indicating that these SFKs act as a point of convergence for various signaling pathways and might be crucial for the processes underlying physiological plasticity, including learning and memory, and pathological plasticity, including epilepsy and neurodegeneration (Purcell and Carew, 2003; Salter and Kalia, 2004). Recently, it has been reported that Src is expressed not only in the dorsal root ganglion (DRG) but also in the spinal cord and contributes to the development and maintenance of inflammatory pain hypersensitivity (Igwe, 2003; Guo et al., 2004). However, there has been no study examining SFK activation after nerve injury.
Mitogen-activated protein kinases (MAPKs) play a critical role in intracellular signal transduction and consist of extracellular signal-regulated protein kinase (ERK), p38 MAPK, and c-Jun N-terminal kinase(JNK)/stress-activated protein kinase (SAPK) (Lewis et al., 1998; Widmann et al., 1999). Recent studies have shown that peripheral nerve injury results in MAPK activation in spinal glial cells, and MAPK inhibitors diminish nerve injury-induced mechanical hypersensitivity (Ma and Quirion, 2002; Jin et al., 2003; Tsuda et al., 2004; Zhuang et al., 2005, 2006). We now show that nerve injury induces the activation of SFKs in microglia in the spinal dorsal horn, and this SFK activation increases ERK phosphorylation in spinal microglia and contributes to the development of mechanical hypersensitivity. Our findings point to the potential blockade of the Src/ERK signaling pathway in spinal microglia as a new therapeutic strategy for pain caused by nerve injury.
Materials and Methods
Animals.
A total of 59 male Sprague Dawley rats weighing 200–250 g were used. All procedures were approved by the Hyogo College of Medicine Committee on Animal Research and were performed in accordance with National Institutes of Health guidelines on animal care. Rats that did not receive surgery (n = 7) were used as naive controls for immunohistochemistry, in situ hybridization histochemistry (ISHH), and Western blotting.
Surgical procedures.
All procedures were performed with the rats under pentobarbital anesthesia (50 mg/kg, i.p.). Additional doses of the anesthetics were given as needed. In all rats, no surgery was performed on the right side. Special care was taken to prevent infection and to minimize the influence of inflammation. The hair of the rat's lower back and thigh was shaved, and the skin was sterilized with 0.5% chlorhexidine and covered with clean paper. Sterile operating instruments were used. Rats without surgery (n = 4 for immunohistochemistry, n = 4 for in situ hybridization histochemistry, and n = 3 for Western blotting) were used as naive controls.
To produce an L5 spinal nerve ligation (SNL), a skin incision (3–4 cm) was made in the midline lumbar region (L4–S1). The L6 transverse process was identified, freed of muscular attachments, and partially removed with the help of bone ronguers. This exposed the L5 spinal nerve. The L5 ventral ramus was isolated and freed from the adjacent nerves, and then the L5 spinal nerve was tightly ligated with silk suture and transected distal to the ligature. After surgery, the wound was washed with saline and closed in layers (fascia and skin) with 3-0 silk thread. Animals were allowed to survive for 1, 3, 7, or 14 d after surgery (n = 4 for immunohistochemistry and n = 3 for Western blotting for each time point). In sham-operated rats, the nerve was exposed without ligation.
The intrathecal delivery of 4-amino-5-(4-chlorophenyl)-7-(t-butyl)pyrazolo[3,4-d]pyrimidine (PP2), an Src-family protein tyrosine kinase inhibitor (Calbiochem, La Jolla, CA), or 4-amino-7-phenylpyrazol[3,4-d]pyrimidine (PP3; Calbiochem), a negative control for PP2, was performed as described previously (Fukuoka et al., 2001; Obata et al., 2003). Briefly, a laminectomy of the L5 vertebra was performed under adequate anesthesia with sodium pentobarbital. The dura was cut, and a soft tube (Silascon; outer diameter, 0.64 mm; Kaneka Medix, Osaka, Japan) was inserted into the subarachnoid space of the spinal cord, and the tip of the catheter was implanted at the L4 spinal segmental level. To obtain a sustained drug infusion, an ALZET osmotic pump (7 d pump, 1 μl/hr; Durect, Cupertino, CA) was filled with PP2 (0.005, 0.05, or 0.5 μg/μl) or PP3 (0.5 μg/μl) in 50% DMSO. The catheter of the pump was implanted intrathecally either 3 h before L5 SNL or 7 d after L5 SNL. Normal saline was used as the vehicle control. The lack of effect of DMSO was determined in preliminary experiments, consistent with previous reports (Ji et al., 2002; Obata et al., 2003, 2005). Rats were allowed to survive for up to 7 d after surgery (n = 4 for immunohistochemistry, n = 4 for in situ hybridization histochemistry, and n = 3 for Western blotting).
Behavioral tests.
All rats were tested for mechanical, thermal, and cold hypersensitivity of the plantar surface of the hindpaw 1 d before surgery and 3, 5, or 7 d after surgery. Room temperature and humidity remained stable for all experiments. On each testing day, the rats were brought into the behavior room 1 h before the test session to allow them to habituate to the environment.
Mechanical hypersensitivity was assessed with a Dynamic Plantar Aesthesiometer (Ugo Basile, Comerio, Italy), which is an automated von Frey-type system (Kalmar et al., 2003; Lever et al., 2003). To measure rat hindpaw mechanical thresholds, rats were placed in plastic cages with a wire mesh floor and allowed to acclimate for 15 min before each test session. A paw-flick response was elicited by applying an increasing force (measured in grams) using a plastic filament (0.5 mm in diameter) focused on the middle of the plantar surface of the ipsilateral hindpaw. The force applied was initially below detection threshold and then increased from 1 to 50 g in 1 g steps over 20 s, and then held at 50 g for an additional 10 s. The rate of force increase was 2.5 g/s. The threshold was taken as the force applied to elicit a reflex removal of the hindpaw. This was defined as the mean of three measurements at 1 min intervals. The variability between trials was ∼2 g.
Heat hypersensitivity was tested using the Hargreaves radiant heat apparatus (7370; Ugo Basile). A radiant heat source beneath a glass floor was aimed at the plantar surface of the hindpaw. Three measurements of latency were taken for each hindpaw in each test session. The hindpaws were tested alternately, with intervals between consecutive tests of >5 min. The three measurements of latency per side were averaged.
To estimate cold sensitivity of the paw, the cold plate test was performed as described previously (Choi et al., 1994; Jasmin et al., 1998). Each rat was placed in a plastic cage with a Peltier cooled cold plate (Neuroscience, Osaka, Japan) kept at a cold temperature (5 ± 0.5°C). After 5 min of adaptation, the numbers of paw lifts within the next 5 min were recorded. Foot lifts associated with locomotion or repositioning of the body were not counted. An assistant, who was unaware of the treatment group, performed all of the behavioral experiments.
Data are expressed as mean ± SD. Differences in changes of values over time of each group were tested using one-way ANOVA, followed by individual post hoc comparisons (Fisher's exact test). One-way ANOVA, followed by individual post hoc comparisons (Fisher's exact test) or pairwise comparisons (t test) were used to assess differences of values between the intrathecal groups. A difference was accepted as significant at p < 0.05.
Immunohistochemistry.
The rats were deeply anesthetized with sodium pentobarbital and perfused transcardially with 1% paraformaldehyde in 0.1 m phosphate buffer (PB), pH 7.4, followed by 4% paraformaldehyde in 0.1 m PB 1, 3, 7, or 14 d after surgery (n = 4 at each time point). After the perfusion, the L5 spinal cord segments and L4/5 DRGs were dissected out and postfixed in the same fixative for 12 h and then replaced with 20% sucrose overnight. Transverse spinal sections (free-floating, 25 μm) and DRG sections (16 μm) were cut in a cryostat and processed for phosphorylated-Src family (Tyr416; p-Src family), p-Src, p-Lck, p-Lyn, p-ERK1/2, p-p38, microtubule-associated protein-2 (MAP2), neuronal-specific nuclear protein (NeuN), glial fibrillary acid protein (GFAP), and OX42 immunohistochemistry according to previously described methods (Noguchi et al., 1995). The polyclonal primary antibody for p-Src family (1:400; Cell Signaling Technology, Beverly, MA), p-Lck (1:400; Cell Signaling Technology), p-Lyn (1:400; Cell Signaling Technology), p-ERK1/2 (1:400; Cell Signaling Technology), p-p38 (1:400; Cell Signaling Technology), GFAP (1:400; DAKO, Glostrup, Denmark) and the monoclonal primary antibody for p-Src (1:400; Calbiochem), MAP2 (1:5000; Sigma, St. Louis, MO), NeuN (neuronal marker, 1:2000; Chemicon, Temecula, CA), and OX42 (CD11b, microglia marker, 1:100; Serotec, Indianapolis, IN) were used for DAB staining.
For the double-immunofluorescent staining for p-Src family and MAP2, NeuN, GFAP, OX42, p-Src, p-ERK1/2, or p-p38, the tyramide signal amplification (TSA; PerkinElmer Life Sciences, Boston, MA) fluorescence procedures (Michael et al., 1997) were used to detect staining for rabbit anti-p-Src family (Tyr416) polyclonal antibody (1:10,000; Cell Signaling Technology). When the two primary antisera raised in rabbits were combined, nonspecific double labeling was not observed. A similar protocol has been used previously (Bennett et al., 1998; Amaya et al., 2000), and the lack of cross-reactivity is thought to be attributable to the fact that the TSA procedure allows the first-series primary antibody to be used at a dilution that is too high to be detected by the second reagent set (Michael et al., 1997). Our data support this explanation. With control single labeling using indirectly labeled immunofluorescence, we were unable to visualize the p-Src family antiserum at the dilutions used for the TSA procedure. Furthermore, the specificity of the labeling with tyramide was assessed by several control experiments: omission of either the primary or secondary antibody and omission of the streptavidin–HRP or the biotinyl–tyramide steps, in four separate experiments, revealed no detectable staining.
In situ hybridization histochemistry.
For the ISHH, the tissue was sectioned (16 μm thick) with a cryostat, thaw-mounted onto Vectabond- (Vector Laboratories, Burlingame, CA) coated slides and stored at −80°C until ready for use. The procedure for ISHH was performed according to previously described methods (Yamanaka et al., 1999; Hashimoto et al., 2001). Briefly, the rat transient receptor potential ion channel TRPA1 and TRPV1 cRNA probe corresponding to nucleotides 302–788 and 149–505, respectively, was prepared. The sections were treated with 10 μg/ml proteinase K in 50 mm Tris-HCl and 5 mm EDTA for 3 min and acetylated with 0.25% acetic anhydride in 0.1 m triethanolamine; then, 35S-labeled RNA probe (5 × 106 cpm/ml) was placed on these sections overnight at 55°C. Hybridized sections were rinsed in 5× SSC and 5 mm DTT for 30 min at 65°C, washed in high-stringency buffer for 30 min at 65°C, and treated with 2 μg/ml RNase A for 30 min at 37°C. Sections were rinsed, dehydrated in an ascending ethanol series, and air dried. For autoradiography, the sections were coated with NTB-3 emulsion (Eastman Kodak, Rochester, NY), diluted 6:4 with distilled water at 45°C, and exposed for 2 weeks in light-tight boxes at 4°C. After development in D19 (Eastman Kodak) and fixation in 24% sodium thiosulfate, the sections were rinsed in distilled water, stained with hematoxylin–eosin, dehydrated in a graded ethanol series, cleared in xylene, and coverslipped.
Measurements of the density of silver grains over randomly selected tissue profiles were performed using the NIH Image program by a blinded assistant, in which only neuronal profiles that contained nuclei were used for quantification. At a magnification of 200× and with bright-field illumination, upper and lower thresholds of gray level density were set such that only silver grains were accurately discriminated from the background in the outlined cell or tissue profile and read by the computer pixel-by-pixel. Subsequently, the area of discriminated pixels was measured and divided by the area of the outlined profile, giving a grain density for each cell or tissue profile. To reduce the risk of biased sampling of the data because of varying emulsion thickness, we used a signal/noise (S/N) ratio for each cell in each tissue section. The S/N ratio of an individual neuron and its cross-sectioned area, which was computed from the outlined profile, was plotted. Based on this scattergram, neurons with a grain density of 10-fold the background level or higher (10 ≤ S/N ratio) were considered positively labeled for TRPA1 and TRPV1 mRNA. At least 1200 neurons from four rats were quantified for each antisense probe in the ISHH study according to previously described methods (Kobayashi et al., 2005). The number of positively labeled DRG neurons was divided by the number of neuronal profiles counted in each DRG.
Data are expressed as mean ± SD. One-way ANOVA, followed by individual post hoc comparisons (Fisher's exact test), was used to assess differences of values between the intrathecal groups. A difference was accepted as significant at p < 0.05.
Western blotting.
Tissue samples from the L5 spinal cord segments and L4/5 DRGs were lysed by homogenizing in 200 μl of lysis buffer containing 20 mm Tris-HCl, pH 8.0, 150 mm NaCl, 1 mm EDTA, 1% Igepal CA-630, 2 mm Na3VO4, 0.5 mm DTT, 1 mm PMSF, 1 μg/ml pepstatin, 5 μg/ml leupeptin, 9 μg/ml aprotinin and 10% glycerol. Lysates were centrifuged at 14,400 × g for 60 min, and the concentration of protein in each sample (supernatant) was determined using the Bio-Rad (Hercules, CA) dye binding. Samples with equal amounts of protein (15 μg) were then separated by 10–20% PAGE, and the resolved proteins were electrotransferred to Hybond-P nitrocellulose (GE Healthcare UK, Little Chalfont, UK). Membranes were incubated with 5% nonfat milk in Tris buffer containing Tween 20 (TBST) (10 mm Tris-HCl, pH 8.0, 150 mm NaCl, and 0.2% Tween 20) for at least 10 min at room temperature and incubated with the polyclonal primary antibody for p-Src family (1:200), total Src (1:1000), p-Lck (1:200), p-Lyn (1:200), p-ERK1/2 (1:1000), total ERK (1:1000), p-p38 (1:200), and total p38 (1:200; all from Cell Signaling Technology) and the monoclonal primary antibody for β-actin (1:1000; Sigma, St. Louis, MO) at 4°C overnight. Membranes were then washed twice with TBST and probed with goat anti-rabbit IgG conjugated with horseradish peroxidase (Vector Laboratories, Burlingame, CA) at room temperature for 2 h. Membranes were finally washed several times with TBST to remove unbound secondary antibodies and visualized using enhanced chemiluminescence (Roche Diagnostics, Indianapolis, IN). Each experiment was repeated at least twice, and in all cases the same results were obtained. The density of specific bands was measured with a computer-assisted imaging analysis system (ATTO Densito graph version 4.02; Atto, Tokyo, Japan) and normalized against a loading control (β-actin). The protein level was expressed as a percentage of the protein level in the normal control ganglia.
Data are expressed as mean ± SD. Differences in changes of values over time of each group were tested using one-way ANOVA, followed by individual post hoc comparisons (Fisher's exact test). One-way ANOVA, followed by individual post hoc comparisons (Fisher's exact test) or pairwise comparisons (t test) were used to assess differences of values between the intrathecal groups. A difference was accepted as significant if p < 0.05.
Results
L5 spinal nerve injury induces an increase in SFK phosphorylation in the ipsilateral dorsal horn
An anti-p-Src family antibody was used to investigate the changes in Src-family phosphorylation. We first performed immunohistochemistry on sections of the L5 spinal cord (Fig. 1A–E). None or very few p-Src family-immunoreactive (IR) cells were found in the spinal dorsal horn of naive control rats (Fig. 1A) and sham-operated rats (Fig. 1E). p-Src family levels started to increase in the ipsilateral dorsal horn at day 1 after L5 SNL (Fig. 1B). Many more p-Src family-IR cells were found in the ipsilateral than the contralateral dorsal horn, predominantly in the superficial dorsal horn (laminas I–III) at day 3 (Fig. 1C) and day 7 (Fig. 1D). Src-family activation after nerve injury was confirmed by Western blot analysis (Fig. 1F). Consistent with the results obtained by immunohistochemistry, nerve injury induced a significant increase in p-Src family levels in the ipsilateral spinal cord. The increase was first evident at day 1 after surgery and continued for 14 d. In contrast, the level of the p-Src family in the contralateral spinal cord was not different from that in naive rats (data not shown).
Figure 1.
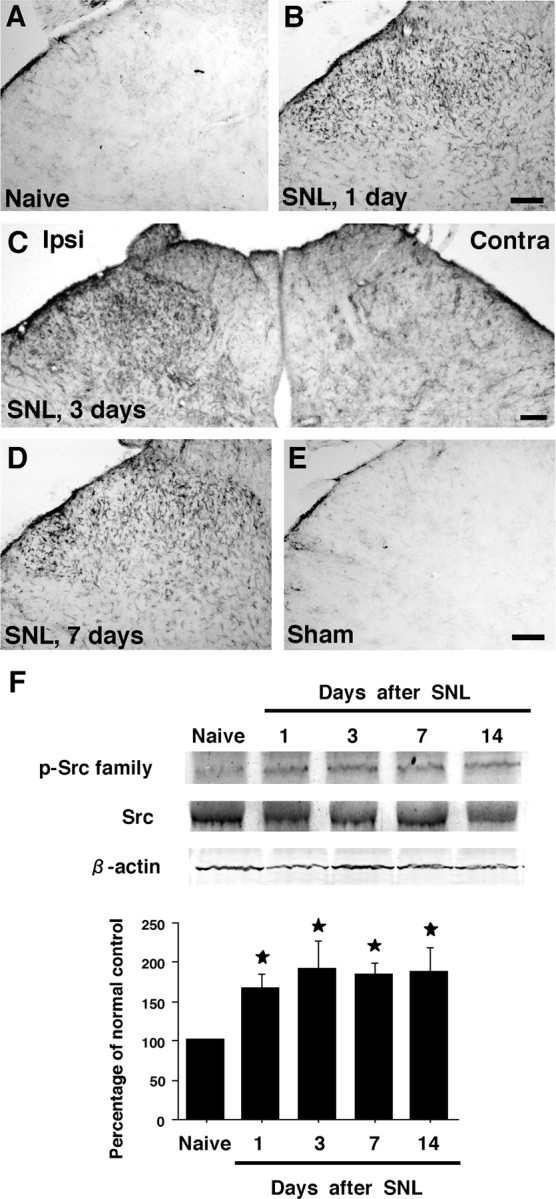
Marked activation of Src family in the spinal dorsal horn after L5 spinal nerve injury. A, E, p-Src family immunostaining in the dorsal horn of naive control rats (A) and sham control rats at day 7 (E). B–D, Immunohistochemistry indicates a substantial increase in the number of p-Src family-IR cells in the ipsilateral (Ipsi) dorsal horn at days 1, 3, and 7 after nerve injury (B, C, and D, respectively). Contra, Contralateral. F, Western blot analysis reveals rapid and persistent Src-family activation in the ipsilateral spinal cord after nerve injury. Quantification of Western blot data is shown at the bottom. Data represent mean ± SD; n = 4 per group. *p < 0.05 compared with the naive control. Scale bar, 100 μm.
To correlate this Src-family activation with the development of neuropathic pain behavior, we examined mechanical pain hypersensitivity. We found significant mechanical hypersensitivity on the ipsilateral hindpaw at day 1 after injury to the L5 nerve, and this pain behavior lasted for >14 d after surgery: the paw withdrawal threshold decreased from 34.2 ± 1.7 g before the injury to 13.8 ± 1.0 g at day 1 and 17.9 ± 1.3 g at day 14. Thus, the level of p-Src family increased markedly in the dorsal horn ipsilateral to the nerve injury with a time course matching that of mechanical hypersensitivity.
L5 spinal nerve injury induces activation of SFKs in spinal microglia
To identify the cell types that expressed the p-Src family after L5 SNL, we performed double immunostaining of the p-Src family with several cell-specific markers: for neurons, MAP2 and NeuN; for astrocytes, GFAP; and for microglia, OX42. We found that the p-Src family did not colocalize with MAP2, NeuN, or GFAP (Fig. 2A–I). Instead, the majority of the p-Src family-IR cells were double labeled with OX42 in the ipsilateral dorsal horn at day 7 after nerve injury, indicating that the Src family is activated in microglia but not in neurons or astrocytes (Fig. 2J–L). Double immunofluorescence indicated that p-Src family was colocalized with OX42 (Fig. 2M–O), but not with MAP2, NeuN, or GFAP (data not shown), in the spinal cord at different times (days 1–14). We also found that nerve injury induced an increase in the number and intensity of OX-42-IR cells in the ipsilateral dorsal horn (Fig. 2K). These OX42-IR cells displayed hypertrophic morphology after injury, a sign of microglial activation. Furthermore, nerve injury induced activation of astrocytes in the ipsilateral dorsal horn, as indicated by increased GFAP expression (Fig. 2H), but these cells did not express the p-Src family. These results indicate that in the dorsal horn after nerve injury, hyperactive microglia are the “main” cell type that expresses the p-Src family.
Figure 2.
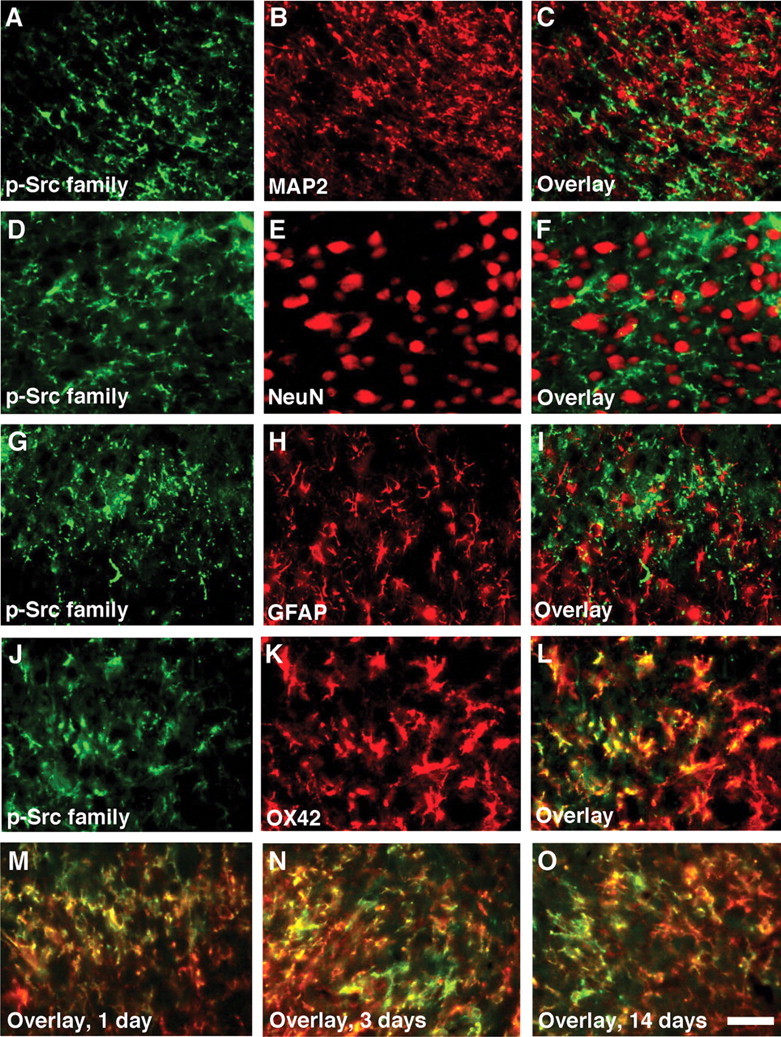
L5 SNL induces Src-family activation in spinal microglia but not in neurons or astrocytes. Immunohistochemical colocalization of green reaction product for p-Src family (A, D, G, J) and red product for MAP2 (B), NeuN (E), GFAP (H), or OX42 (K) in the ipsilateral dorsal horn at day 7 after nerve injury. A–F, Double staining of p-Src family with neuronal markers MAP2 and NeuN shows no colocalization. G–I, Double labeling of p-Src family with GFAP, an astrocyte marker, indicates that Src family is not activated in astrocytes. J–L, Double staining of p-Src family with OX42, a microglia marker, reveals a heavy colocalization in the dorsal horn. M–O, Double immunofluorescence shows that p-Src family (green) is colocalized with OX42 (red) in the superficial dorsal horn (laminas I–III) at days 1, 3, and 14 after nerve injury (M, N, and O, respectively). Two single-stained images were merged. Scale bar, 20 μm.
Src, Lck, and Lyn are activated in spinal microglia after nerve injury
The Src family of protein tyrosine kinases includes Src, Lck, and Lyn. To investigate which Src-family kinase is activated in spinal microglia after nerve injury, we first examined the phosphorylation of Src using a specific anti-p-Src antibody. We found that L5 SNL increased Src phosphorylation in the ipsilateral dorsal horn, compared with the contralateral side (Fig. 3A). Furthermore, double immunostaining showed that p-Src completely colocalized with the p-Src family (Fig. 3B–D). We next examined the expression of p-Lck and p-Lyn in the spinal cord. Nerve injury also induced the activation of both Lck and Lyn in the ipsilateral dorsal horn (Fig. 3E and F, respectively). We found that p-Lck and p-Lyn were mainly expressed in the lateral side of the superficial dorsal horn. These p-Lck and p-Lyn-IR cells were predominantly microglia, because they coexpressed the microglial marker OX42 (Fig. 3G–I and J–L, respectively). The changes in p-Lck and p-Lyn in the spinal cord were also confirmed by Western blotting (Fig. 4A and B, respectively). Both p-Lck and p-Lyn levels at day 7 after surgery were significantly increased compared with those of naive control rats, consistent with the results obtained by immunohistochemistry (Fig. 3E,F). These results indicate that SFKs are activated mainly in microglia in the dorsal horn after nerve injury, although some p-Lck and p-Lyn-IR cells, as well as p-Src family-IR cells, did not express OX42 (Figs. 2J–O, 3G–L).
Figure 3.
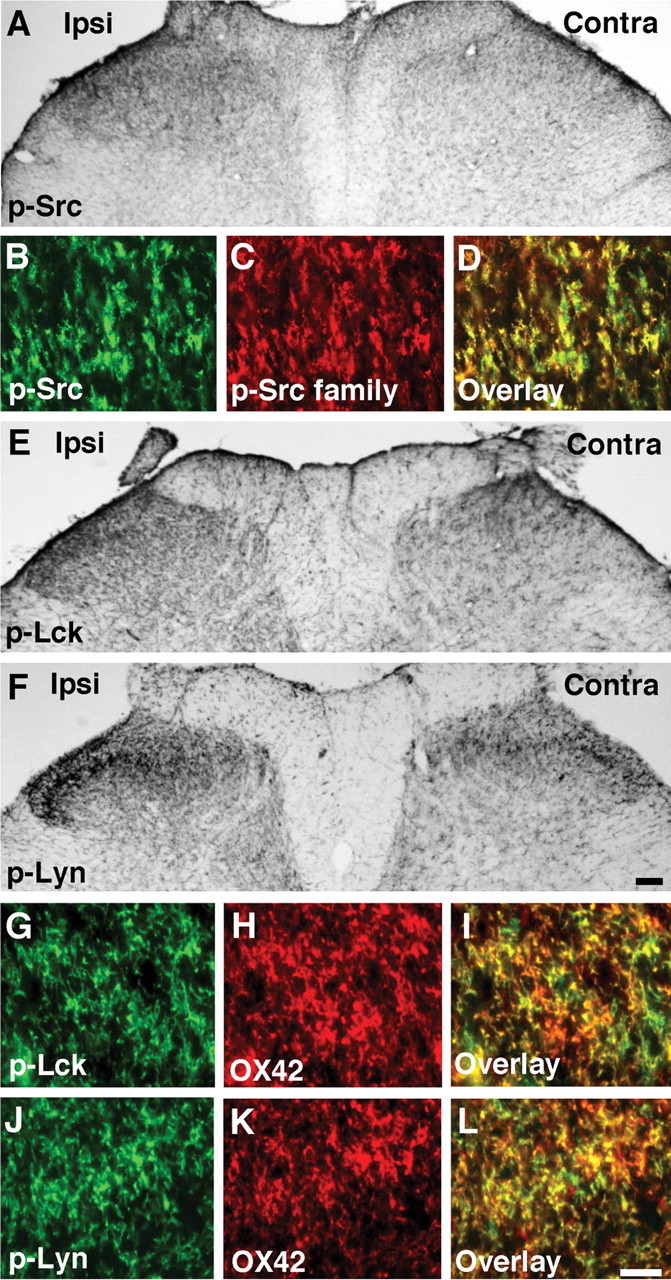
L5 SNL induces the activation of SFKs, such as Src, Lck, and Lyn, in spinal microglia. A, p-Src immunostaining in the ipsilateral (Ipsi) dorsal horn at day 7 after surgery. Contra, Contralateral. B–D, Double staining indicates a complete colocalization (D) between p-Src (B) and p-Src family (C) in the ipsilateral dorsal horn at day 7 after surgery. E, F, p-Lck (E) and p-Lyn (F) immunostaining in the ipsilateral dorsal horn at day 7. G–L, Double labeling for OX42-IR (H, K) and p-Lck-IR (G) or p-Lyn (J) in the ipsilateral dorsal horn at day 7. Scale bars: (in I) A, E, F, 100 μm; (in L) G–L, 20 μm.
Figure 4.

A, B, Western blot analysis reveals activation of Lck (A) and Lyn (B) in the ipsilateral spinal cord at day 7 after L5 SNL. Quantification of Western blot data is shown at right. Data represent mean ± SD; n = 4 per group. *p < 0.05 compared with the naive control.
Inhibition of Src-family activation attenuates nerve injury-induced mechanical hypersensitivity but not heat and cold hypersensitivity
Our results suggest that activation of SFKs in spinal microglia is likely to have an important role in mechanical hypersensitivity after nerve injury. We therefore predicted that suppressing SFK activation in the spinal cord should prevent nerve injury-induced mechanical hypersensitivity. To test this, the Src family of tyrosine kinase inhibitor PP2 or PP3, a negative control for PP2, was delivered intrathecally before L5 SNL and maintained for 7 d via a catheter, the tip of which was implanted at the lumber enlargement. To obtain a sustained drug infusion, the drug was delivered by an osmotic pump connected to a catheter. Intrathecal administration of PP2 (0.5 μg · μl−1 · h−1) or PP3 (0.5 μg · μl−1 · h−1) into naive animals produced no significant changes in basal pain sensitivity (data not shown). We found that intrathecal PP2, but not PP3, significantly reduced the nerve injury-induced mechanical hypersensitivity at days 3, 5, and 7 after surgery (Fig. 5A). However, PP2, as well as PP3, was not able to block the injury-induced thermal and cold hypersensitivity throughout the period we studied (Fig. 5B and C, respectively). Inhibition of Src-family activation by PP2 was confirmed by both immunohistochemistry (Fig. 5D,E) and Western blotting (Fig. 5F). The level of the p-Src family in the vehicle group at day 7 after surgery was significantly greater than that of naive control rats, but this increase in the p-Src family in the ipsilateral dorsal horn was prevented by PP2.
Figure 5.

Effects of the Src family of tyrosine kinase inhibitor PP2 delivered intrathecally on nerve injury-induced neuropathic pain behaviors. A, Mechanical hypersensitivity was determined with a Dynamic Plantar Aesthesiometer at days 3, 5, and 7 after L5 SNL. B, C, Heat and cold hypersensitivity were tested using the plantar test and the cold plate test, respectively. Data represent mean ± SD; n = 8 per group. D, E, p-Src family immunostaining in the ipsilateral dorsal horn in the vehicle group (D) and in the PP2 group (E) at day 7 after nerve injury. F, Western blot analysis reveals inhibition of Src-family activation by PP2. Quantification of Western blot data is shown at right. Data represent mean ± SD; n = 4 per group. *p < 0.05 compared with the naive control. #p < 0.05 compared with vehicle control. Scale bar, 100 μm.
Several reports have shown that inhibition of microglial activation attenuates the development but not existing hypersensitivity in a rat model of neuropathy (Raghavendra et al., 2003). To investigate whether inhibition of Src-family activation would reverse established neuropathic pain, we infused PP2 intrathecally via an osmotic pump 7 d after establishment of L5 SNL-induced mechanical hypersensitivity. This treatment effectively reversed the nerve injury-induced pain hypersensitivity at days 10, 12, and 14 after surgery (Fig. 6).
Figure 6.

Reversal of L5 SNL-induced mechanical hypersensitivity by intrathecal infusion of PP2 via an osmotic pump starting on post-SNL day 7. Data represent mean ± SD; n = 8 per group. #p < 0.05 compared with vehicle control.
Inhibition of Src-family activation reverses the activation of ERK, but not p38 MAPK, in spinal microglia caused by nerve injury
Previous reports have demonstrated that the activation of both ERK and p38 MAPK in microglia in the spinal cord contributes to the generation of neuropathic pain (Jin et al., 2003; Tsuda et al., 2004; Zhuang et al., 2005). To ascertain whether ERK and p38 MAPK phosphorylation in the dorsal horn is regulated by Src-family activation, the levels of p-ERK and p-p38 were compared in the vehicle and PP2 groups using immunohistochemistry (Fig. 7A,B and C,D, respectively). Seven days after nerve injury, numerous p-ERK-IR cells were seen in the ipsilateral dorsal horn in the vehicle group (Fig. 7A). We found that the Src family of tyrosine kinase inhibitor PP2 (0.5 μg · μl−1 · h−1) reversed the nerve injury-induced increase in ERK phosphorylation (Fig. 7B). In contrast, although nerve injury induced a marked increase in the number and intensity of p-p38-IR cells in the ipsilateral dorsal horn, PP2 did not suppress the nerve injury-induced elevation of p-p38 (Fig. 7C,D). These changes in p-ERK and p-p38 in the spinal cord were also confirmed by Western blotting (Fig. 7E and F, respectively). Both p-ERK and p-p38 levels in the vehicle group at day 7 after nerve injury were significantly increased compared with those of naive control rats. Furthermore, we found that PP2 inhibited ERK, but not p38 MAPK activation, consistent with the results obtained by immunohistochemistry (Fig. 7A–D).
Figure 7.
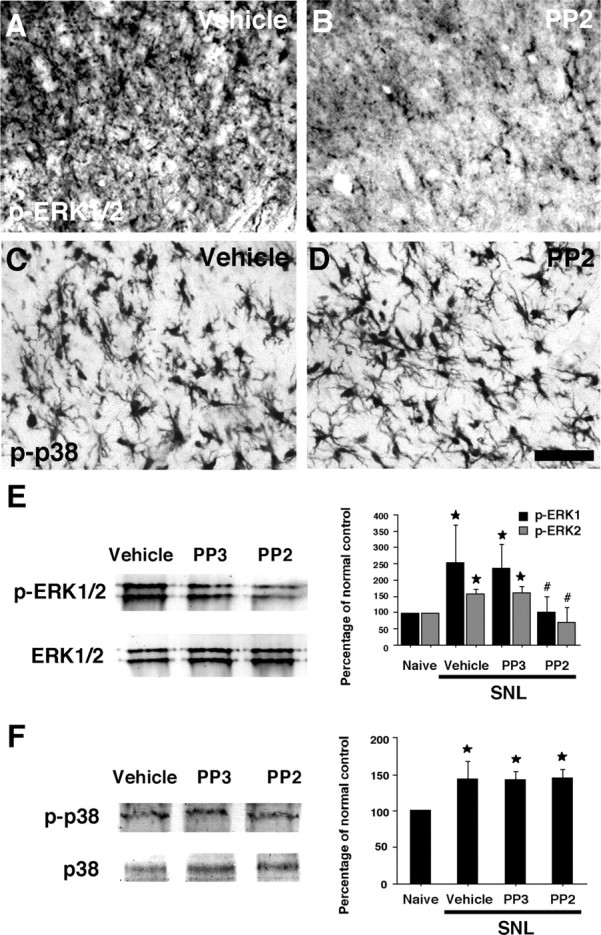
Effects of the Src family of tyrosine kinase inhibitor PP2 delivered intrathecally on nerve injury-induced ERK and p38 MAPK activation in the dorsal horn. A–D, p-ERK (A, B) and p-p38 (C, D) immunostaining in the superficial dorsal horn (laminas I–III) in the vehicle (A, C), and PP2 (B, D) groups at day 7 after L5 SNL. E, F, Western blot analysis indicates that PP2 suppresses the activation of ERK (E), but not p38 MAPK (F) in the ipsilateral spinal cord at day 7. Quantification of Western blot data is shown at right. Data represent mean ± SD; n = 4 per group. *p < 0.05 compared with the naive control. #p < 0.05 compared with vehicle control. Scale bar, 20 μm.
To further examine whether the effects of PP2 on nerve injury-induced mechanical hypersensitivity and ERK activation in the spinal cord are secondary to the inhibition of Src-family activation, we administrated PP2 (0.005, 0.05, or 0.5 μg · μl−1 · h−1) intrathecally to L5 SNL rats. The treatment of PP2 (0.05 μg · μl−1 · h−1) diminished L5 SNL-induced mechanical hypersensitivity only at day 5 after surgery, whereas PP2 (0.005 μg · μl−1 · h−1) application did not block the SNL-induced pain hypersensitivity throughout the 1 week time course of the experiment (Fig. 8A). Furthermore, we found that L5 SNL-induced ERK activation in the spinal cord was not suppressed by PP2 (0.005 or 0.05 μg · μl−1 · h−1) at day 7 after surgery (Fig. 8B).
Figure 8.

Effects of PP2 on nerve injury-induced neuropathic pain behaviors and ERK activation in the dorsal horn. A, Mechanical hypersensitivity was determined with a Dynamic Plantar Aesthesiometer at days 3, 5, and 7 after L5 SNL. Data represent mean ± SD; n = 8 per group. B, Western blot analysis indicates that PP2 suppresses the activation of ERK in the ipsilateral spinal cord at day 7 in a dose-dependent manner. Quantification of Western blot data are shown at right. Data represent mean ± SD; n = 4 per group. *p < 0.05 compared with the naive control. #p < 0.05, ##p < 0.01 compared with vehicle control.
To investigate whether an increase in p-ERK in the ipsilateral dorsal horn after nerve injury is seen in microglia, we performed double labeling for p-ERK and OX42. p-ERK was detected in a subpopulation of OX42-labeled cells at day 3 after nerve injury (Fig. 9A–C), consistent with a previous study (Zhuang et al., 2005). Next, to determine whether the p-Src family-IR cells and p-ERK- or p-p38-IR cells belong to the same subset of spinal microglia, colocalization of the p-Src family with p-ERK or p-p38 was performed (Fig. 9D–I). p-ERK coexpressed with the p-Src family, whereas p-p38- and p-Src family-IR cells were clearly distinguishable. Furthermore, we found that only 33% of p-ERK-IR cells (166 of 519) were p-p38-IR (Fig. 9J–L). These data suggest that the activation of the Src family increases the phosphorylation of ERK, but not p38 MAPK, in a subset of spinal microglia after nerve injury.
Figure 9.
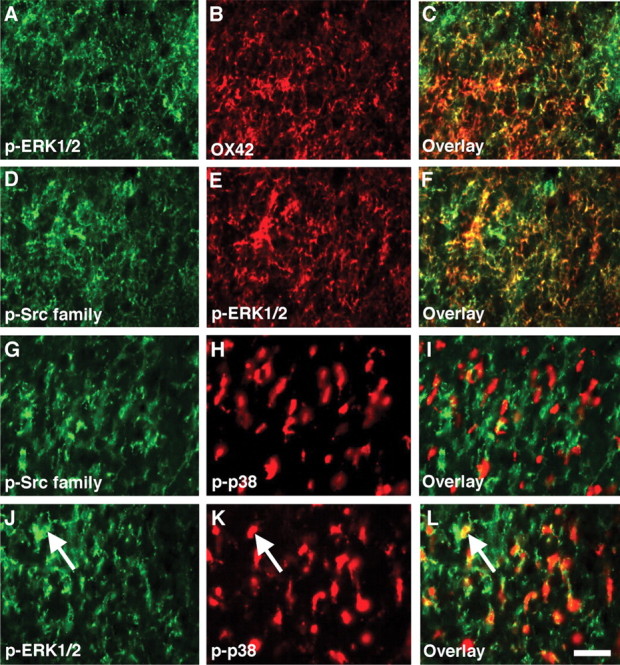
p-ERK, but not p-p38 MAPK, colocalizes with p-Src family in spinal microglia after L5 SNL. A–C, Double staining of p-ERK with OX42 in the ipsilateral dorsal horn at day 3 after nerve injury. p-Src heavily coexpressed with OX42. D–I, Double labeling for p-Src family-IR (D, G) and p-ERK-IR (E) or p-p38 MAPK (H) in the ipsilateral dorsal horn at day 3. The p-Src family-IR cells and p-ERK-IR but not p-p38-IR cells belonged to the same subset of glial cells in the spinal cord. J–L, Double staining of p-ERK with p-p38 MAPK in the ipsilateral dorsal horn at day 3. Note that some ERK-IR cells also expressed p-p38 (arrow). Scale bar, 20 μm.
Inhibition of Src-family activation does not reduce an increase in TRP expression in uninjured primary sensory neurons caused by nerve injury
Recent studies have shown that after L5 SNL, p38 MAPK is activated in undamaged L4 DRG neurons and participates in the development of heat and cold hypersensitivity by regulating transient receptor potential ion channel TRPV1 and TRPA1 expression, respectively (Fukuoka et al., 2001; Obata et al., 2004, 2005). Therefore, we first examined p-Src family immunoreactivity in the uninjured L4 and injured L5 DRG at day 7 after surgery (Fig. 10A–C). We found that nerve injury did not induce an increase in p-Src family expression in the L4/5 DRG neurons. Western blot analysis also revealed no change in p-Src family level (Fig. 10D). Next, we studied TRPV1 and TRPA1 mRNA expression in the L4 DRG using ISHH (Fig. 10E,F and G,H, respectively). Consistent with previous studies (Fukuoka et al., 2001; Obata et al., 2004, 2005), the percentage of both TRPV1 and TRPA1 mRNA-positive neurons significantly increased in the L4 DRG in the vehicle group at day 7 after nerve injury (Fig. 10I and J, respectively). However, we found that the treatment of PP2 did not diminish the nerve injury-induced increase in TRPV1 and TRPA1 expression.
Figure 10.
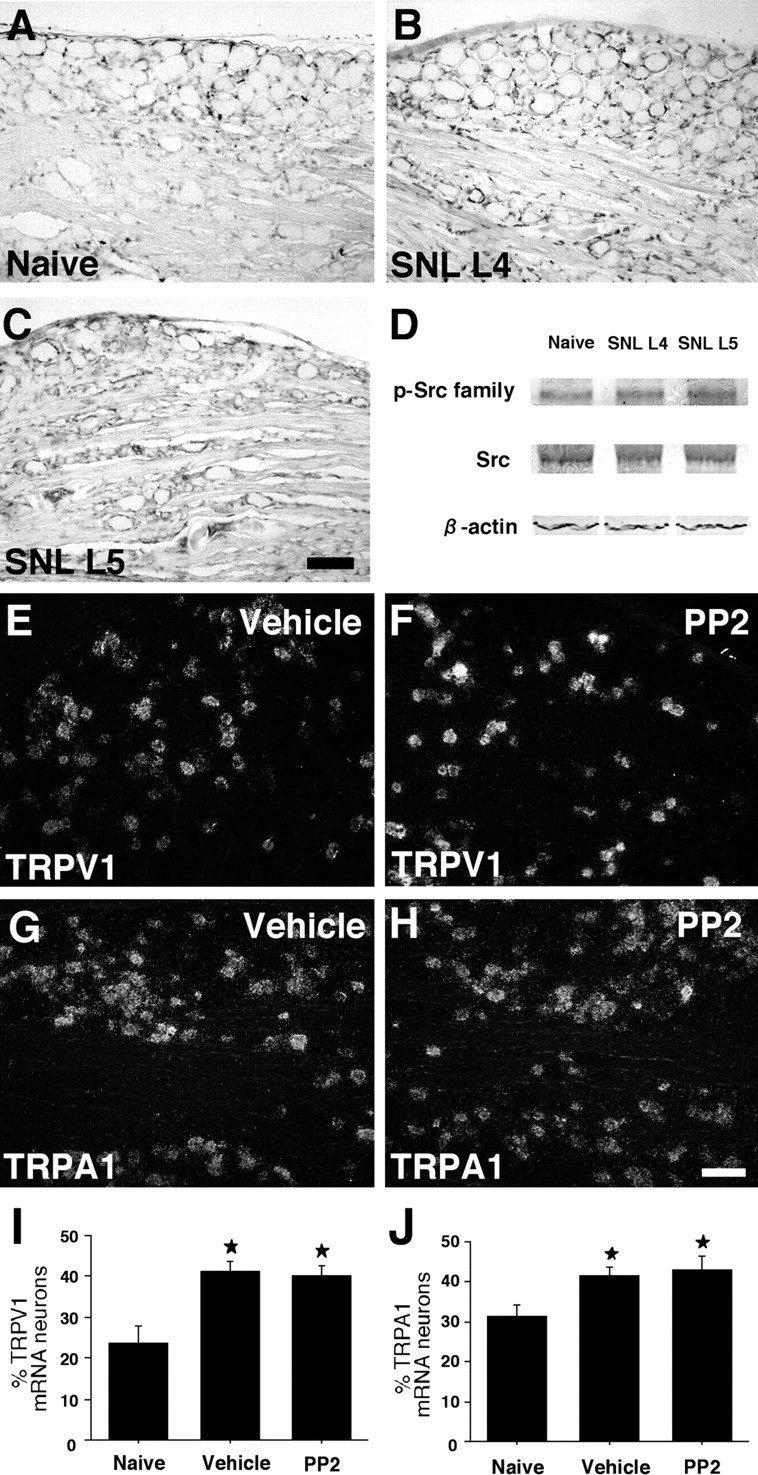
No change in p-Src family expression in primary sensory neurons after L5 SNL. A–C, p-Src family immunostaining in the naive DRG (A) and the ipsilateral L4 (B) and L5 (C) DRG at day 7 after nerve injury. D, Western blot analysis indicates no difference in p-Src family level between the naive DRG and the ipsilateral L4/5 DRG at day 7. E–H, Dark-field photomicrographs of ISHH showing expression of TRPV1 and TRPA1 mRNA in the ipsilateral L4 DRG in the vehicle (E, G) and PP2 (F, H) groups at day 7 after nerve injury. I, J, Quantification of the percentage of TRPV1 (I) and TRPA1 (J) mRNA-positive neurons at day 7. Data represent mean ± SD; n = 4 per group. *p < 0.05 compared with the naive control. Scale bar, 100 μm.
Discussion
The present study demonstrates the following new findings: (1) L5 SNL induced the activation of SFKs including Src, Lck, and Lyn in spinal microglia; (2) PP2 alleviated the nerve injury-induced mechanical hypersensitivity, but not heat and cold hypersensitivity, and prevented the activation of ERK, but not p38 MAPK, in spinal microglia; (3) nerve injury did not increase SFK phosphorylation in the uninjured L4 DRG, and PP2 did not block nerve injury-induced increase in TRPV1 and TRPA1 expression in the L4 DRG.
There is accumulating evidence supporting a role for activated microglia in the pathogenesis of nerve injury-induced pain hypersensitivity. For example, in the spinal dorsal horn, the ATP receptor P2X4 is selectively expressed in activated microglia and contributes to mechanical hypersensitivity after peripheral nerve injury (Tsuda et al., 2003). Furthermore, the chemokine receptor CCR2 is also expressed in spinal microglia, and CCR2-deficient mice do not display mechanical hypersensitivity after nerve injury (Abbadie et al., 2003). In the present study, we found that the phosphorylation of SFKs increased in spinal cord microglia after L5 SNL and that the time course of the activation of SFKs in the spinal cord matched the emergence of enhanced sensitivity to mechanical stimuli. Furthermore, PP2, the Src family of tyrosine kinase inhibitor, diminished this nerve injury-induced mechanical hypersensitivity and ERK activation in spinal microglia. Because the activation of ERK in spinal microglia is involved in the induction and maintenance of mechanical hypersensitivity (Zhuang et al., 2005), these findings suggest that nerve injury induces ERK activation in spinal cord microglia via SFK activation and that this activation of the Src/ERK signaling cascade in spinal microglia contributes to hypersensitivity to mechanical stimulation. However, although the predominant cell type in which SFKs were activated was microglia, we cannot entirely eliminate the possibility that effects of PP2 are on the minority of cells, which are OX42 negative and thus presumed to not be microglia.
In addition to ERK, peripheral nerve injury results in a remarkable increase in p38 phosphorylation, predominantly in spinal microglia (Jin et al., 2003; Tsuda et al., 2004), and this MAPK activation in microglia leads to the production of inflammatory mediators that sensitize dorsal horn neurons in the spinal cord (Ji and Strichartz, 2004; Tsuda et al., 2005; Marchand et al., 2005). For example, MAPK activation appears to regulate the expression of proinflammatory cytokines including interleukin 1β (IL-1β), tumor necrosis factor-α, and IL-6, as well as cyclooxygenase-2 and inducible nitric oxide synthase (Widmann et al., 1999; Koistinaho and Koistinaho, 2002; Ji and Strichartz, 2004). An unexpected finding in the present study was that PP2 did not prevent nerve injury-induced p38 activation in spinal microglia, although PP2 completely attenuated ERK activation. Because either an ERK or p38 inhibitor only partially suppresses nerve injury-induced mechanical hypersensitivity (Jin et al., 2003; Zhuang et al., 2005), it seems likely that in spinal cord microglia, the Src/ERK signaling cascade participates in nerve injury-induced mechanical hypersensitivity, independent of the p38 MAPK pathway. In fact, we found that only 33% of p-ERK-IR cells were p-p38-IR. Thus, we believe that blockade of both Src/ERK and p38 activation simultaneously may provide a more effective means to reduce pain caused by nerve injury.
Five members of the SFKs are now known to be expressed in the mammalian CNS, namely Src, Fyn, Yes, Lck, and Lyn. Recently, it has been demonstrated that Src is expressed in spinal dorsal horn neurons and contributes to inflammatory pain (Guo et al., 2004). We found that after peripheral nerve injury, the cells expressing p-Src, as well as p-Lck and p-Lyn, are microglia rather than neurons or astrocytes in the dorsal horn. Furthermore, a recent report showed that phosphorylation of N-methyl-d-aspartate receptors by Fyn is essential for the maintenance of neuropathic pain (Abe et al., 2005). Considering that nerve injury induces a significant increase in p-Src family expression in spinal microglia, these findings suggest that other SFKs, such as Fyn or Yes, are also activated in microglia and play a role in the generation of mechanical hypersensitivity. In contrast, not only microglia but also astrocytes are activated in the spinal cord after nerve injury, and these activated astrocytes participate in the maintenance of the late phase of neuropathic pain (DeLeo and Yezierski, 2001; Watkins et al., 2001a,b; Watkins and Maier, 2003). Indeed, nerve injury induces ERK and JNK/SAPK activation in spinal astrocytes 2–3 weeks after injury (Ma and Quirion, 2002; Zhuang et al., 2005, 2006). Because microglial responses typically precede astrocyte activation, Src/ERK activation in microglia might have a role in the early establishment of neuropathic pain.
TRPV1, one of the transducer proteins, can generate depolarizing currents in response to noxious thermal stimuli, with an activation temperature of ∼43°C, whereas TRPA1 is activated at ∼17°C, a temperature that is reported as painfully cold by humans (Jordt et al., 2003; Patapoutian et al., 2003; Park et al., 2006). Although the role of TRPA1 in cold transduction is still controversial (Jordt et al., 2004; Bautista et al., 2006), TRPV1 and TRPA1 upregulation in undamaged sensory neurons has been implicated in nerve injury-induced heat and cold hypersensitivity, respectively (Fukuoka et al., 2001; Obata et al., 2004, 2005; Katsura et al., 2006). In the present study also, the levels of TRPV1 and TRPA1 expression increased in the intact L4 DRG after L5 SNL. However, there was no difference in TRP expression between the vehicle and PP2 groups, and PP2 did not block heat and cold hypersensitivity caused by nerve injury. Furthermore, nerve injury did not induce SFK activation in the L4 DRG. Therefore, these findings suggest that SFKs might not be involved in TRPV1 and TRPA1 increase in uninjured sensory neurons, although we cannot deny the possibility that SFKs in DRG neurons may activate these TRP channels by phosphorylation without any modulation at transcription level.
The present study demonstrated that ERK was only activated in a fraction of p-Src family-IR cells. Therefore, we also cannot exclude the possibility that by overdosing PP2 (0.5 μg · μl−1 · h−1) the effect is not limited to SFKs and that nonspecific effects contribute to ERK inhibition and the behavioral effects. At this dose, however, PP2 did not reverse nerve injury-induced heat/cold hypersensitivity, as well as p38 activation in spinal microglia. Furthermore, at a low dose (0.005 or 0.05 μg · μl−1 · h−1), PP2 did not decrease not only L5 SNL-induced mechanical hypersensitivity but also ERK activation in the spinal cord. Some reports have shown that activated microglia participate in heat hypersensitivity, as well as mechanical hypersensitivity after nerve injury (Tanga et al., 2005). Nevertheless, our results indicate that SFK activation in spinal microglia has a crucial role in the pathogenesis of mechanical hypersensitivity rather than heat/cold hypersensitivity after peripheral nerve injury, through the ERK pathway. Although it is important to establish how these transduction-related molecules are activated in spinal microglia after nerve injury and how they cause signaling to neurons in the dorsal horn (Tsuda et al., 2005; Marchand et al., 2005), blocking the activation of the Src/ERK signaling cascade in spinal microglia may represent a new approach to effectively treating clinical neuropathic pain.
Footnotes
This work was supported in part by Grants-in-Aid for Scientific Research, and the Open Research Center Grant, Hyogo College of Medicine, both from the Japanese Ministry of Education, Science, and Culture. This work was also supported by a grant from the Japan Health Sciences Foundation. We thank Yuki Obata, Nobumasa Ushio, and Noriko Kusumoto for technical assistance. We thank D. A. Thomas for correcting the English usage.
References
- Abbadie C, Lindia JA, Cumiskey AM, Peterson LB, Mudgett JS, Bayne EK, DeMartino JA, MacIntyre DE, Forrest MJ. Impaired neuropathic pain responses in mice lacking the chemokine receptor CCR2. Proc Natl Acad Sci USA. 2003;100:7947–7952. doi: 10.1073/pnas.1331358100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abe T, Matsumura S, Katano T, Mabuchi T, Takagi K, Xu L, Yamamoto A, Hattori K, Yagi T, Watanabe M, Nakazawa T, Yamamoto T, Mishina M, Nakai Y, Ito S. Fyn kinase-mediated phosphorylation of NMDA receptor NR2B subunit at Tyr1472 is essential for maintenance of neuropathic pain. Eur J Neurosci. 2005;22:1445–1454. doi: 10.1111/j.1460-9568.2005.04340.x. [DOI] [PubMed] [Google Scholar]
- Alvares D, Fitzgerald M. Building blocks of pain: the regulation of key molecules in spinal sensory neurones during development and following peripheral axotomy. Pain. 1999;([Suppl 6]):S71–S85. doi: 10.1016/S0304-3959(99)00140-2. [DOI] [PubMed] [Google Scholar]
- Amaya F, Decosterd I, Samad TA, Plumpton C, Tate S, Mannion RJ, Costigan M, Woolf CJ. Diversity of expression of the sensory neuron-specific TTX-resistant voltage-gated sodium ion channels SNS and SNS2. Mol Cell Neurosci. 2000;15:331–342. doi: 10.1006/mcne.1999.0828. [DOI] [PubMed] [Google Scholar]
- Bautista DM, Jordt SE, Nikai T, Tsuruda PR, Read AJ, Poblete J, Yamoah EN, Basbaum AI, Julius D. TRPA1 mediates the inflammatory actions of environmental irritants and proalgesic agents. Cell. 2006;124:1269–1282. doi: 10.1016/j.cell.2006.02.023. [DOI] [PubMed] [Google Scholar]
- Bennett DL, Michael GJ, Ramachandran N, Munson JB, Averill S, Yan Q, McMahon SB, Priestley JV. A distinct subgroup of small DRG cells express GDNF receptor components and GDNF is protective for these neurons after nerve injury. J Neurosci. 1998;18:3059–3072. doi: 10.1523/JNEUROSCI.18-08-03059.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi Y, Yoon YW, Na HS, Kim SH, Chung JM. Behavioral signs of ongoing pain and cold allodynia in a rat model of neuropathic pain. Pain. 1994;59:369–376. doi: 10.1016/0304-3959(94)90023-X. [DOI] [PubMed] [Google Scholar]
- DeLeo JA, Yezierski RP. The role of neuroinflammation and neuroimmune activation in persistent pain. Pain. 2001;90:1–6. doi: 10.1016/s0304-3959(00)00490-5. [DOI] [PubMed] [Google Scholar]
- Fukuoka T, Kondo E, Dai Y, Hashimoto N, Noguchi K. Brain-derived neurotrophic factor increases in the uninjured dorsal root ganglion neurons in selective spinal nerve ligation model. J Neurosci. 2001;21:4891–4900. doi: 10.1523/JNEUROSCI.21-13-04891.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo W, Wei F, Zou S, Robbins MT, Sugiyo S, Ikeda T, Tu JC, Worley PF, Dubner R, Ren K. Group I metabotropic glutamate receptor NMDA receptor coupling and signaling cascade mediate spinal dorsal horn NMDA receptor 2B tyrosine phosphorylation associated with inflammatory hyperalgesia. J Neurosci. 2004;24:9161–9173. doi: 10.1523/JNEUROSCI.3422-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto N, Yamanaka H, Fukuoka T, Dai Y, Obata K, Mashimo T, Noguchi K. Expression of HGF and cMet in the peripheral nervous system of adult rats following sciatic nerve injury. Neuroreport. 2001;12:1403–1407. doi: 10.1097/00001756-200105250-00022. [DOI] [PubMed] [Google Scholar]
- Hoffman-Kim D, Kerner JA, Chen A, Xu A, Wang TF, Jay DG. pp60(c-src) is a negative regulator of laminin-1-mediated neurite outgrowth in chick sensory neurons. Mol Cell Neurosci. 2002;21:81–93. doi: 10.1006/mcne.2002.1157. [DOI] [PubMed] [Google Scholar]
- Hokfelt T, Zhang X, Wiesenfeld-Hallin Z. Messenger plasticity in primary sensory neurons following axotomy and its functional implications. Trends Neurosci. 1994;17:22–30. doi: 10.1016/0166-2236(94)90031-0. [DOI] [PubMed] [Google Scholar]
- Igwe OJ. c-Src kinase activation regulates preprotachykinin gene expression and substance P secretion in rat sensory ganglia. Eur J Neurosci. 2003;18:1719–1730. doi: 10.1046/j.1460-9568.2003.02878.x. [DOI] [PubMed] [Google Scholar]
- Jasmin L, Kohan L, Franssen M, Janni G, Goff JR. The cold plate as a test of nociceptive behaviors: description and application to the study of chronic neuropathic and inflammatory pain models. Pain. 1998;75:367–382. doi: 10.1016/s0304-3959(98)00017-7. [DOI] [PubMed] [Google Scholar]
- Ji RR, Strichartz G. Cell signaling and the genesis of neuropathic pain. Sci STKE 2004. 2004:reE14. doi: 10.1126/stke.2522004re14. [DOI] [PubMed] [Google Scholar]
- Ji RR, Befort K, Brenner GJ, Woolf CJ. ERK MAP kinase activation in superficial spinal cord neurons induces prodynorphin and NK-1 upregulation and contributes to persistent inflammatory pain hypersensitivity. J Neurosci. 2002;22:478–485. doi: 10.1523/JNEUROSCI.22-02-00478.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin SX, Zhuang ZY, Woolf CJ, Ji RR. p38 mitogen-activated protein kinase is activated after a spinal nerve ligation in spinal cord microglia and dorsal root ganglion neurons and contributes to the generation of neuropathic pain. J Neurosci. 2003;23:4017–4022. doi: 10.1523/JNEUROSCI.23-10-04017.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordt SE, McKemy DD, Julius D. Lessons from peppers and peppermint: the molecular logic of thermosensation. Curr Opin Neurobiol. 2003;13:487–492. doi: 10.1016/s0959-4388(03)00101-6. [DOI] [PubMed] [Google Scholar]
- Jordt SE, Bautista DM, Chuang HH, McKemy DD, Zygmunt PM, Hogestatt ED, Meng ID, Julius D. Mustard oils and cannabinoids excite sensory nerve fibres through the TRP channel ANKTM1. Nature. 2004;427:260–265. doi: 10.1038/nature02282. [DOI] [PubMed] [Google Scholar]
- Kalmar B, Greensmith L, Malcangio M, McMahon SB, Csermely P, Burnstock G. The effect of treatment with BRX-220, a co-inducer of heat shock proteins, on sensory fibers of the rat following peripheral nerve injury. Exp Neurol. 2003;184:636–647. doi: 10.1016/S0014-4886(03)00343-1. [DOI] [PubMed] [Google Scholar]
- Katsura H, Obata K, Mizushima T, Yamanaka H, Kobayashi K, Dai Y, Fukuoka T, Tokunaga A, Sakagami M, Noguchi K. Antisense knock down of TRPA1, but not TRPM8, alleviates cold hyperalgesia after spinal nerve ligation in rats. Exp Neurol. 2006;200:112–123. doi: 10.1016/j.expneurol.2006.01.031. [DOI] [PubMed] [Google Scholar]
- Kobayashi K, Fukuoka T, Yamanaka H, Dai Y, Obata K, Tokunaga A, Noguchi K. Differential expression patterns of mRNAs for P2X receptor subunits in neurochemically characterized dorsal root ganglion neurons in the rat. J Comp Neurol. 2005;481:377–390. doi: 10.1002/cne.20393. [DOI] [PubMed] [Google Scholar]
- Koistinaho M, Koistinaho J. Role of p38 and p44/42 mitogen-activated protein kinases in microglia. Glia. 2002;40:175–183. doi: 10.1002/glia.10151. [DOI] [PubMed] [Google Scholar]
- Kuo WL, Chung KC, Rosner MR. Differentiation of central nervous system neuronal cells by fibroblast-derived growth factor requires at least two signaling pathways: roles for Ras and Src. Mol Cell Biol. 1997;17:4633–4643. doi: 10.1128/mcb.17.8.4633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lever I, Cunningham J, Grist J, Yip PK, Malcangio M. Release of BDNF and GABA in the dorsal horn of neuropathic rats. Eur J Neurosci. 2003;18:1169–1174. doi: 10.1046/j.1460-9568.2003.02848.x. [DOI] [PubMed] [Google Scholar]
- Lewis TS, Shapiro PS, Ahn NG. Signal transduction through MAP kinase cascades. Adv Cancer Res. 1998;74:49–139. doi: 10.1016/s0065-230x(08)60765-4. [DOI] [PubMed] [Google Scholar]
- Ma W, Quirion R. Partial sciatic nerve ligation induces increase in the phosphorylation of extracellular signal-regulated kinase (ERK) and c-Jun N-terminal kinase (JNK) in astrocytes in the lumbar spinal dorsal horn and the gracile nucleus. Pain. 2002;99:175–184. doi: 10.1016/s0304-3959(02)00097-0. [DOI] [PubMed] [Google Scholar]
- Marchand F, Perretti M, McMahon SB. Role of the immune system in chronic pain. Nat Rev Neurosci. 2005;6:521–532. doi: 10.1038/nrn1700. [DOI] [PubMed] [Google Scholar]
- Michael GJ, Averill S, Nitkunan A, Rattray M, Bennett DL, Yan Q, Priestley JV. Nerve growth factor treatment increases brain-derived neurotrophic factor selectively in TrkA-expressing dorsal root ganglion cells and in their central terminations within the spinal cord. J Neurosci. 1997;17:8476–8490. doi: 10.1523/JNEUROSCI.17-21-08476.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noguchi K, Kawai Y, Fukuoka T, Senba E, Miki K. Substance P induced by peripheral nerve injury in primary afferent sensory neurons and its effect on dorsal column nucleus neurons. J Neurosci. 1995;15:7633–7643. doi: 10.1523/JNEUROSCI.15-11-07633.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obata K, Yamanaka H, Dai Y, Tachibana T, Fukuoka T, Tokunaga A, Yoshikawa H, Noguchi K. Differential activation of extracellular signal-regulated protein kinase in primary afferent neurons regulates brain-derived neurotrophic factor expression after peripheral inflammation and nerve injury. J Neurosci. 2003;23:4117–4126. doi: 10.1523/JNEUROSCI.23-10-04117.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obata K, Yamanaka H, Kobayashi K, Dai Y, Mizushima T, Katsura H, Fukuoka T, Tokunaga A, Noguchi K. Role of mitogen-activated protein kinase activation in injured and intact primary afferent neurons for mechanical and heat hypersensitivity after spinal nerve ligation. J Neurosci. 2004;24:10211–10222. doi: 10.1523/JNEUROSCI.3388-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obata K, Katsura H, Mizushima T, Yamanaka H, Kobayashi K, Dai Y, Fukuoka T, Tokunaga A, Tominaga M, Noguchi K. TRPA1 induced in sensory neurons contributes to cold hyperalgesia after inflammation and nerve injury. J Clin Invest. 2005;115:2393–2401. doi: 10.1172/JCI25437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park CK, Kim MS, Fang Z, Li HY, Jung SJ, Choi SY, Lee SJ, Park K, Kim JS, Oh SB. Functional expression of thermo-transient receptor potential channels in dental primary afferent neurons: implication for tooth pain. J Biol Chem. 2006;281:17304–17311. doi: 10.1074/jbc.M511072200. [DOI] [PubMed] [Google Scholar]
- Patapoutian A, Peier AM, Story GM, Viswanath V. ThermoTRP channels and beyond: mechanisms of temperature sensation. Nat Rev Neurosci. 2003;4:529–539. doi: 10.1038/nrn1141. [DOI] [PubMed] [Google Scholar]
- Purcell AL, Carew TJ. Tyrosine kinases, synaptic plasticity and memory: insights from vertebrates and invertebrates. Trends Neurosci. 2003;26:625–630. doi: 10.1016/j.tins.2003.09.005. [DOI] [PubMed] [Google Scholar]
- Raghavendra V, Tanga F, DeLeo JA. Inhibition of microglial activation attenuates the development but not existing hypersensitivity in a rat model of neuropathy. J Pharmacol Exp Ther. 2003;306:624–630. doi: 10.1124/jpet.103.052407. [DOI] [PubMed] [Google Scholar]
- Salter MW, Kalia LV. Src kinases: a hub for NMDA receptor regulation. Nat Rev Neurosci. 2004;5:317–328. doi: 10.1038/nrn1368. [DOI] [PubMed] [Google Scholar]
- Scholz J, Woolf CJ. Can we conquer pain? Nat Neurosci. 2002;5([Suppl]):1062–1067. doi: 10.1038/nn942. [DOI] [PubMed] [Google Scholar]
- Tanga FY, Nutile-McMenemy N, DeLeo JA. The CNS role of Toll-like receptor 4 in innate neuroimmunity and painful neuropathy. Proc Natl Acad Sci USA. 2005;102:5856–5861. doi: 10.1073/pnas.0501634102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuda M, Shigemoto-Mogami Y, Koizumi S, Mizokoshi A, Kohsaka S, Salter MW, Inoue K. P2X4 receptors induced in spinal microglia gate tactile allodynia after nerve injury. Nature. 2003;424:778–783. doi: 10.1038/nature01786. [DOI] [PubMed] [Google Scholar]
- Tsuda M, Mizokoshi A, Shigemoto-Mogami Y, Koizumi S, Inoue K. Activation of p38 mitogen-activated protein kinase in spinal hyperactive microglia contributes to pain hypersensitivity following peripheral nerve injury. Glia. 2004;45:89–95. doi: 10.1002/glia.10308. [DOI] [PubMed] [Google Scholar]
- Tsuda M, Inoue K, Salter MW. Neuropathic pain and spinal microglia: a big problem from molecules in “small” glia. Trends Neurosci. 2005;28:101–107. doi: 10.1016/j.tins.2004.12.002. [DOI] [PubMed] [Google Scholar]
- Watkins LR, Maier SF. Glia: a novel drug discovery target for clinical pain. Nat Rev Drug Discov. 2003;2:973–985. doi: 10.1038/nrd1251. [DOI] [PubMed] [Google Scholar]
- Watkins LR, Milligan ED, Maier SF. Spinal cord glia: new players in pain. Pain. 2001a;93:201–205. doi: 10.1016/S0304-3959(01)00359-1. [DOI] [PubMed] [Google Scholar]
- Watkins LR, Milligan ED, Maier SF. Glial activation: a driving force for pathological pain. Trends Neurosci. 2001b;24:450–455. doi: 10.1016/s0166-2236(00)01854-3. [DOI] [PubMed] [Google Scholar]
- Widmann C, Gibson S, Jarpe MB, Johnson GL. Mitogen-activated protein kinase: conservation of a three-kinase module from yeast to human. Physiol Rev. 1999;79:143–180. doi: 10.1152/physrev.1999.79.1.143. [DOI] [PubMed] [Google Scholar]
- Yamanaka H, He X, Matsumoto K, Shiosaka S, Yoshida S. Protease M/neurosin mRNA is expressed in mature oligodendrocytes. Brain Res Mol Brain Res. 1999;71:217–224. doi: 10.1016/s0169-328x(99)00187-4. [DOI] [PubMed] [Google Scholar]
- Zhuang ZY, Gerner P, Woolf CJ, Ji RR. ERK is sequentially activated in neurons, microglia, and astrocytes by spinal nerve ligation and contributes to mechanical allodynia in this neuropathic pain model. Pain. 2005;114:149–159. doi: 10.1016/j.pain.2004.12.022. [DOI] [PubMed] [Google Scholar]
- Zhuang ZY, Wen YR, Zhang DR, Borsello T, Bonny C, Strichartz GR, Decosterd I, Ji RR. A peptide c-Jun N-terminal kinase (JNK) inhibitor blocks mechanical allodynia after spinal nerve ligation: respective roles of JNK activation in primary sensory neurons and spinal astrocytes for neuropathic pain development and maintenance. J Neurosci. 2006;26:3551–3560. doi: 10.1523/JNEUROSCI.5290-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]