

Abstract
Synaptotagmins comprise a large protein family, of which synaptotagmin 1 (Syt1) is a Ca2+ sensor for fast exocytosis, and its close relative, synaptotagmin 2 (Syt2), is assumed to serve similar functions. Chromaffin cells express Syt1 but not Syt2. We compared secretion from chromaffin cells from Syt1 null mice overexpressing either Syt isoform. High time-resolution capacitance measurement showed that Syt1 null cells lack the exocytotic phase corresponding to the readily-releasable pool (RRP) of vesicles. Comparison with the amperometric signal confirmed that the missing phase of exocytosis consists of catecholamine-containing vesicles. Overexpression of Syt1 rescued the RRP and increased its size above wild-type values, whereas the size of the slowly releasable pool decreased, indicating that the availability of Syt1 regulates the relative size of the two releasable pools. The RRP was also rescued by Syt2 overexpression, but the kinetics of fusion was slightly slower than in cells expressing Syt1. Biochemical experiments showed that Syt2 has a slightly lower Ca2+ affinity for phospholipid binding than Syt1 because of a difference in the C2A domain. These data constitute evidence for the function of Syt1 and Syt2 as alternative, but not identical, calcium-sensors for RRP fusion. By overexpression of Syt1 mutated in the shared PKC/calcium/calmodulin-dependent kinase phosphorylation site, we show that phorbol esters act independently and upstream of Syt1 to regulate the size of the releasable pools. We conclude that exocytosis from mouse chromaffin cells can be modified by the differential expression of Syt isoforms and by Syt abundance but not by phosphorylation of Syt1.
Keywords: chromaffin cell, exocytosis, membrane capacitance, synaptotagmin 1 and 2, phosphorylation, protein kinase C
Introduction
The remarkable heterogeneity and plasticity of neurotransmitter release is made possible by the expression of various protein isoforms and the dynamic regulation of protein function by posttranslational modification. In the present study, we investigated these two aspects of neurotransmitter release as they apply to two different synaptotagmin isoforms and the phosphorylation of synaptotagmin 1 (Syt1). Syt1 is a synaptic-vesicle protein containing two Ca2+- and phospholipid-binding sequences (C2A and C2B) homologous to the C2-region of protein kinase C (Perin et al., 1990; Sutton et al., 1995; Fernandez et al., 2001). Deletion of Syt1 led to impairment of fast, synchronous transmitter release in hippocampal neurons (Geppert et al., 1994). These and other observations suggested Syt1 to be a Ca2+ sensor of synchronized neurotransmitter release, a function that now has been substantiated by several laboratories (for review, see Augustine, 2001; Koh and Bellen, 2003; Yoshihara et al., 2003; Bai and Chapman, 2004; Südhof, 2004). Ca2+ binding to the C2 domains causes Syt1 to bind to phospholipid membranes and the target soluble N-ethylmaleimide-sensitive factor attachment protein receptors (SNAREs) syntaxin and synaptosome-associated protein of 25 kDa (SNAP-25). It is still controversial which of these events actually triggers exocytosis (Zhang et al., 2002; Shin et al., 2003; Bai et al., 2004). The closest relative of Syt1 is synaptotagmin 2 (Syt2), also located to synaptic vesicles, featuring similar overall structure and showing partly complementary expression patterns in the CNS (Geppert et al., 1991; Ullrich et al., 1994; Marquèze et al., 1995). For these reasons, functional redundancy between the two isoforms is widely assumed. The juxtamembrane domain of Syt1 contains a shared phosphorylation site (Thr-112) for PKC and Ca2+/calmodulin-dependent protein kinase II (CaMKII) (Hilfiker et al., 1999), which are both implicated in the regulation of neurotransmitter release (Majewski and Iannazzo, 1998; Turner et al., 1999). Phosphorylation of Syt1 is induced by elevation of [Ca2+]i or by β-phorbol ester treatment in PC12 cells (Hilfiker et al., 1999) and increases binding to syntaxin and SNAP-25 (Verona et al., 2000). The CaMKII/PKC-phosphorylation site is missing in Syt2 as a result of the deletion of eight amino acids, which could have important physiological implications because of the complementary expression pattern. Apart from Syt1, a number of other synaptotagmin isoforms are expressed in neuroendocrine cells, in which they participate in release (e.g., Syt3, Syt7, and Syt9) (Sugita et al., 2001, 2002; Fukuda et al., 2002; Tucker et al., 2003). These isoforms have not been implicated as calcium sensors for synchronized release as measured using electrophysiological techniques; however, they may act during different phases of asynchronous release.
We wanted to investigate the often-cited hypothesis that Syt1 and Syt2 are alternative calcium sensors for fast release and, at the same time, evaluate the role of the Syt1 phosphorylation sites, which might participate in regulatory functions. Because chromaffin cells do not express Syt2 (Geppert et al., 1991; Marquèze et al., 1995), expressing either isoform in Syt1 null chromaffin cells creates a situation in which only one or the other of these two isoforms is present. Similarly, the effect of mutating Syt1 in a phosphorylation site can be investigated without the complication of phosphorylation of wild-type (WT) protein.
Materials and Methods
Preparation of mouse chromaffin cells and solutions. Adrenal glands were removed from newborn Syt1 knock-out (-/-) or control (+/-, +/+) mice, obtained by crossing of heterozygotes (Geppert et al., 1994). The adrenal glands were digested with papain (Worthington, Lakewood, NJ) to obtain isolated chromaffin cells (Sørensen et al., 2003a). Cells were incubated in DMEM (Linaris, Wertheim-Bettingen, Germany) supplemented with penicillin/streptomycin (40,000 U/L and 40 mg/L; Invitrogen, San Diego, CA) and 10 ml/L insulin-transferrin-selenium-X (Invitrogen) at 37°C and 10% CO2 and used within 3 d.
For electrophysiological experiments, the external solution contained the following: 145 mm NaCl, 2.8 mm KCl, 2 mm CaCl2, 1 mm MgCl2, 10 mm HEPES, and 2 g/L d-glucose, pH 7.2, 300 mOsm. The pipette solution used for the flash experiments consisted of the following (in mm): 100 Cs-glutamate, 8 NaCl, 4 CaCl2, 32 HEPES, 2 Mg-ATP, 0.3 GTP, 5 nitrophenyl (NP)-EGTA (supplied by G. Ellis-Davies, Medical College of Philadelphia Hahnemann University, Philadelphia, PA), 0.2 fura-2/0.3 furaptra or 0.4 fura-4F/0.4 furaptra (Invitrogen), pH 7.2 (300 mOsm). Pipette solution for depolarization experiments contained the following (in mm): 70 Cs-glutamate, 30 CsCl, 8 NaCl, 0.125 CaCl2 (free [Ca2+]i was estimated to ∼100 nm), 32 HEPES, 2 Mg-ATP, 0.3 GTP, and 0.2 fura-2/0.3 furaptra, pH 7.2 (300 mOsm). A 200 μm stock solution of phorbol 12-myristate 13-acetate (PMA) (Sigma-Aldrich, Steinheim, Germany) was prepared in DMSO and stored at -20°C until use.
Electrophysiological recordings and data analysis. Whole-cell patch-clamp capacitance, amperometric, and calcium measurements and flash photolysis of caged calcium was performed 8-12 h after infection, as described previously (Nagy et al., 2002). For kinetic analysis, individual capacitance traces were fitted with a triple exponential function:
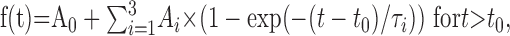 |
where A0 is the capacitance of the cell before the flash, and t0 is the time of the flash. The amplitudes (Ai) and time constants (τi) of the two faster exponentials define the size and release kinetics of the fast and the slow exocytotic burst, respectively. The third exponential corrected for the sustained component but was not used directly. Instead, we subtracted the size of the fast and slow burst from the total amount of secretion during 5 s and calculated the average rate of sustained release. The secretory delay was found by extrapolating the fit function in time until it crossed the preflash membrane capacitance level (Voets, 2000).
All experimental data were analyzed with the program IgorPro 3.16PPC (WaveMetrics, Lake Oswego, OR). Statistical testing of the kinetic parameters was performed by the nonparametric Mann-Whitney U test or by the Kruskal-Wallis test with posttest for comparing three groups. Data are given as the mean ± SEM. Each experimental condition was compared with control cells obtained from the same cell preparations. The rate constants versus calcium and delay versus calcium relationships for the readily-releasable pool (RRP) was calculated using a kinetic scheme described by Sørensen et al. (2003b) (see also Voets, 2000).
Generation of Semliki Forest viruses. The viral vector pSFV1 (Invitrogen) was modified by the introduction of an internal ribosome entry site from polio virus followed by the gene for enhanced green fluorescent protein (EGFP) or enhanced yellow fluorescent protein. The genes coding Syt1 or Syt2 were inserted upstream of the internal ribosome entry site using BamHI and BssHII restriction sites. Syt1 mutants were generated by site-directed mutagenesis, and all constructs were verified by DNA sequencing. Virus production and transfection was performed as described previously (Ashery et al., 1999).
Immunoblot analysis. Overexpression levels of wild-type and mutant Syt1 and Syt2 proteins was estimated after expression in bovine chromaffin cells (Nagy et al., 2002). For quantitation of synaptotagmins in chromaffin cells and mouse cerebellum samples, cell/tissue lysates were analyzed by quantitative immunoblotting using 125I-labeled secondary antibodies and PhosphorImager detection (Molecular Dynamics, Sunnyvale, CA) with vasolin-containing protein (VCP) as internal standards.
Immunostaining. For immunostaining, mouse chromaffin cells were cultured on poly-l-lysine-covered coverslips and infected with SFV as described above. After 8-12 h of expression, the cells were fixed in 3.7% paraformaldehyde in PBS for 1 h at room temperature, permeabilized for 20 min in PBS plus 0.1% Triton X-100, followed by neutralization for 20 min in 50 mm NH4Cl in PBS plus 0.1% Triton X-100. The permeabilized cells were incubated with Syt1 (Ab41.1, 1:100) or Syt2 (A320, 1:200) antibody in the presence of 1% BSA (fraction V) for 2 h, washed four times in PBS, and incubated with cyanine 3 (Cy3)-coupled secondary antibody in the presence of BSA for 1 h. After extensive washing, the cells were imaged on a Leica (Wetzlar, Germany) confocal microscope (TCS SP2) using a PL APO 63×, 1.2 numerical aperture water-immersion confocal scanning objective using 488 nm (for EGFP, emission light collected between 500 and 560 nm) and 543 nm (for Cy3, emission light collected between 563 and 668 nm) excitation. Images were sampled using Leica confocal software and processed using MetaMorph (Universal Imaging Corporation, West Chester, PA).
Immunoprecipitations. For assaying the binding of Syt1 or Syt2 to SNARE proteins, tissue lysates from forebrain and spinal cord were used because they are rich in Syt1 and Syt2, respectively. Mouse forebrain and spinal cord were homogenized in 4 and 2 ml of buffer A (10 mm Tris, 1 mm EDTA, and 150 mm NaCl, pH 7.4) containing protease inhibitors. Equal volume of buffer B (10 mm Tris, 1 mm EDTA, 150 mm NaCl, and 2% Triton X-100, pH 7.4) was added, and the homogenate was extracted for 4 h at 4°C. Insoluble material was removed from the extract by centrifugation (40 min at 100,000 × g), and MgCl2 was added to 2 mm final concentration. SNARE proteins were immunoprecipitated using syntaxin monoclonal antibody (HPC-1, when spinal cord samples were used) and polyclonal antibody (U6251, when forebrain samples were used) that were immobilized by protein G-agarose or protein A-agarose beads, respectively. Approximately 6 mg of total protein lysate was used for immunoprecipitation. The binding reactions were incubated overnight at 4°C, and agarose beads were washed six times with 1 ml of the corresponding buffers. Proteins bound to the beads were resuspended in 30 μl of SDS-PAGE sample buffer and were analyzed by SDS-PAGE and immunoblotting using antibodies against different antigens. Antibodies used were as follows: for forebrain samples, Syt1 (Cl41.1), synapsin (Cl10.22), syntaxin 1 (HPC-1), SNAP-25 (SMI 80), and synaptobrevin (Cl 69.1); for spinal cord samples, Syt2 (A320), synapsin (E028), syntaxin (U6251), SNAP-25 (P913), and synaptobrevin (P939).
Phospholipid binding assays. Phospholipid binding to isolated single C2 domains (C2A or C2B) and to the double C2 domain (C2AB) of Syt1 and Syt2 was assessed by a centrifugation assay as described previously (Fernandez et al., 2001; Fernández-Chacón et al., 2002; Shin et al., 2003). Briefly, heavy liposomes consisting of 25% phosphatidylserine (PS) and 75% phosphatidylcholine (PC) were prepared in 50 mm HEPES-NaOH buffer (pH 6.8, 0.1 m NaCl and 4 mm Na2EGTA) containing 0.5 m sucrose. Glutathione S-transferase (GST)-fusion proteins containing the C2 domains (6 μg) were incubated with 100 μg of heavy liposomes (for experiments containing PKC-C2 internal control, 150 μg of heavy liposomes were used) for 10 min at 30°C with different concentrations of free Ca2+ and centrifuged at 13,000 rpm for 10 min. The pellets were washed three times with the corresponding buffer. Bound protein was precipitated, resuspended in 30 ml of 2× SDS sample buffer, and analyzed by SDS-PAGE and Coomassie blue staining. Quantification of binding was performed with NIH ImageJ 1.32j. Data were fitted by the Hill equation f = aCn/(Cn + EC50n), where a is the maximal binding, C is the Ca2+ concentration, and n is the Hill coefficient.
Results
Rescue of synaptotagmin 1 knock-out cells by overexpression of Syt1
It has been shown previously that, in chromaffin cells from Syt1 null animals, the fast phase of the exocytotic burst component is missing (Voets et al., 2001). However, one pitfall of (unconditional) knock-outs is that the phenotype may be secondary to developmental changes. Therefore, knock-out phenotypes should be verified by functional rescue experiments, which, if successful, pave the way for structure-function analysis. To allow such experiments, we recently developed a method to isolate chromaffin cells from single newborn or embryonic mice and keep them in culture for several days, during which time the cells can be transduced with SFV carrying the gene of interest (Sørensen et al., 2003a).
After infection with SFV expressing Syt1 or Syt2, chromaffin cells from Syt1 null mice were subjected to immunostaining and confocal microscopy to investigate the localization of expressed protein. Both isoforms were preferentially targeted to a compartment close to the plasma membrane. Confocal sectioning revealed that the majority of the staining was localized to small, vesicle-like structures in the periphery of the cell, in agreement with the preferential localization of dense-core granules in mouse embryonic chromaffin cells (Voets et al., 2001; Sørensen et al., 2003a). The localization to vesicle-like structures was most clearly observed in cells expressing at moderate levels (Fig. 1A, B), confirming that both Syt isoforms are targeted to vesicles in chromaffin cells. Quantification of the expression level was performed using Western blots of expressing bovine chromaffin cells (Fig. 1C,D), because the amount of protein obtainable from a preparation of mouse chromaffin cells is very low. For comparison with the Syt levels present in brain, samples of protein isolated from the cerebellum were loaded as well. After correction for infection efficiency (20-50% for different constructs), the result showed that Syt1 was expressed approximately fourfold over wild-type levels. Similar levels were found for all Syt1 mutations presented herein. As expected, chromaffin cells did not express Syt2. However, after overexpression, Syt2 was present at approximately five times the level in cerebellum (Fig. 1D, right).
Figure 1.
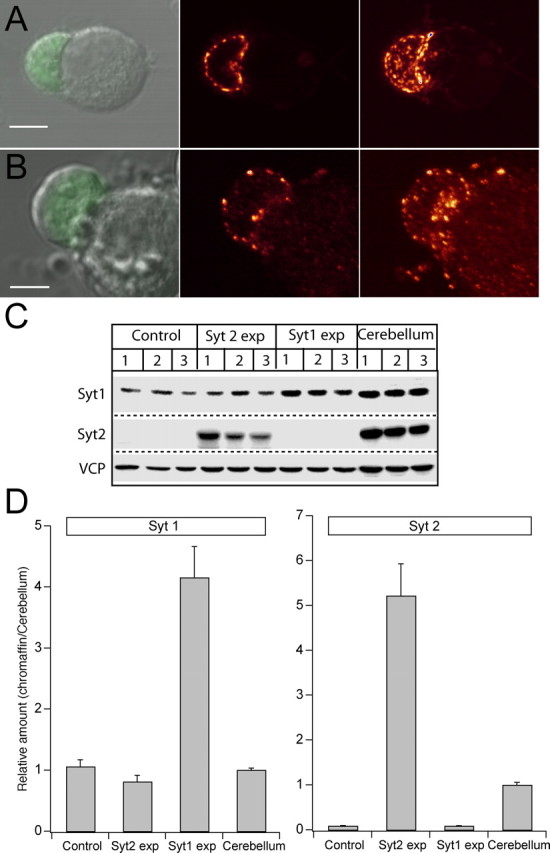
Viral overexpression of synaptotagmin 1 and 2 in chromaffin cells. A, B, Left column, Differential interference contrast image of chromaffin cells from Syt1 null chromaffin cells overlaid with the image obtained in the green channel, indicating EGFP fluorescence. In both cases, the leftmost cell expresses the viral construct at relatively low levels, whereas the right cell does not express. Scale bars, 5 μm. Middle column, Confocal section through the equatorial plane of the cell showing the preferential localization of vesicles to the periphery of the cell. Right column, Projection image of the maximal intensity of a confocal z-stack until the x-y plane. The apparently diffusely located vesicles in the bottom image are attributable to outgrowth of processes by the expressing cell. C, Western blot of lysates from three different preparations of control bovine chromaffin cells, Syt2- and Syt1-expressing chromaffin cells, and cerebellum. VCP was included as an internal standard. D, Quantification of expression level of Syt1 (left) and Syt2 (right) from the Western blot in C, after correction for infection efficiency and normalization to VCP intensity. Overexpression of Syt1 increased the protein concentration to approximately fourfold the value in uninfected bovine chromaffin cells. Chromaffin cells did not express Syt2 before viral infection, which induced expression at approximately five times the level in cerebellum.
Secretion of catecholamines from dense-core vesicles in chromaffin cells was investigated by high time-resolution electrophysiological methods (patch-clamp capacitance measurements and amperometry) combined with simultaneous measurement of [Ca2+]i over a large dynamic range (Rettig and Neher, 2002). Secretion was elicited by flash photolysis of the Ca2+ cage NP-EGTA, which in control cells resulted in secretion consisting of several phases (Fig. 2A, black trace): A rapid so-called burst phase is observed within ∼0.5 s after the flash, followed by a slower sustained phase that is often assumed to represent the refilling of the releasable vesicle pools (Parsons et al., 1995; Ashery et al., 2000). Closer inspection of the capacitance trace revealed the subdivision of the exocytotic burst into a fast [fusion time constant (τfast) ∼20 ms at 20 μm [Ca2+]i] and a slow (τslow ∼200 ms at 20 μm [Ca2+]i) component. These components are believed to result from the fusion of two different but sequentially arranged (Voets et al., 1999) releasable vesicle pools: the RRP and the slowly-releasable pool (SRP), respectively. By fitting a sum of exponentials to the capacitance trace, the size and fusion kinetics of these two pools can be determined simultaneously (see Materials and Methods).
Figure 2.
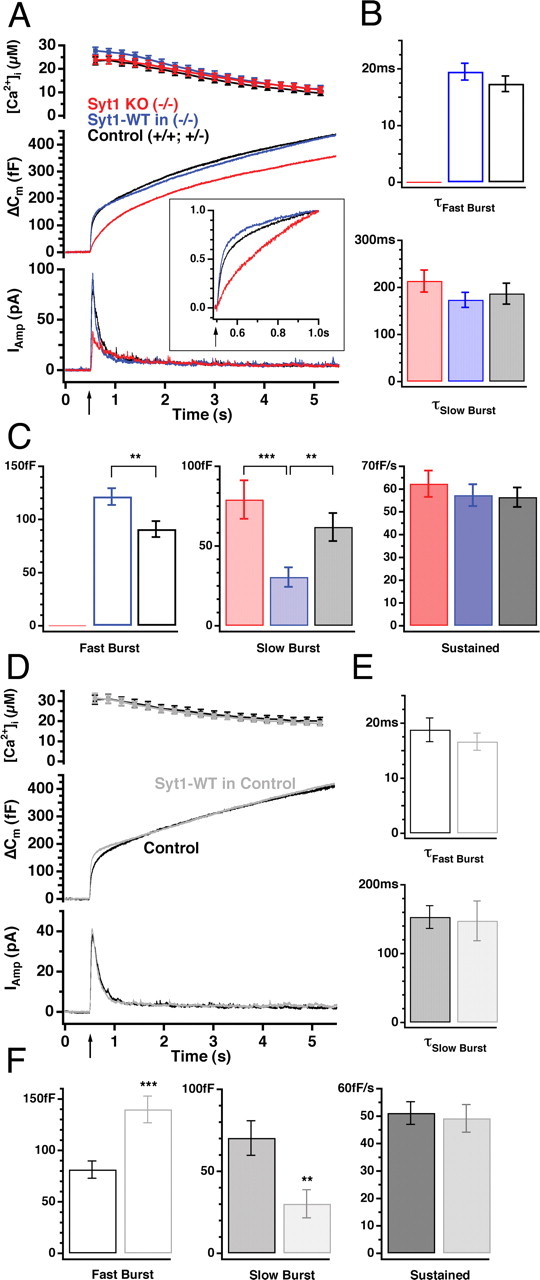
Overexpression of wild-type synaptotagmin 1 results in a larger fast burst and a smaller slow burst of secretion without changing fusion rates. A, Mean calcium concentration (top; error bars represent SEM), capacitance (middle), and amperometric (bottom) responses after a step-like elevation of [Ca2+]i induced by flash photolysis (flash at arrow). Both the capacitance and amperometric traces display a burst-like increase within the first 0.5 s after the flash in the control cells (+/+ and +/-, black; n = 43 cells, N = 8 animals), followed by a slower sustained phase of secretion representing vesicle recruitment (priming) and consecutive fusion. Syt1 null cells lack the fast burst component either measured as cell capacitance or by amperometry (red; n = 41, N = 8), which is reconstituted when cells overexpress wild-type Syt1 (Syt1-WT, blue; n = 42, N = 8). The inset shows the first half-second of the capacitance traces scaled to control level (black). Whereas secretion from the null cells is slowed down (red), overexpression of Syt1 (blue) appears to be faster than control. B, C, The mean ± SEM of kinetic parameters. There was no significant difference between the time constants of both the fast or the slow burst components (B), whereas overexpression of Syt1 in null cells significantly increased the size of the fast burst component and decreased the size of the slow burst component compared with control cells (C). Note that knock-out (KO) cells completely missed the fast burst component, without significant changes in the other kinetic parameters (for color coding, see A). D-F, Overexpression of Syt1-WT in control cells (Syt1-WT, gray; n = 28) also increased fast burst without changing total burst size compared with non-infected control cells (black; n = 28), as seen in the middle capacitance trace (D). Triple-exponential fit revealed that, like in the knock-out cells, overexpression of Syt1 in control cells did not change time constants (E) but led to an increased size of the fast burst accompanied with a decreased slow burst size (F). **p < 0.01; ***p < 0.001.
Flash photolysis of NP-EGTA in isolated chromaffin cells from Syt1 null mice led to secretion lacking the fast burst component without any change in the slow burst and the sustained component (Fig. 2A-C, red traces), in agreement with data from mouse adrenal slices (Voets et al., 2001). We next wanted to test whether overexpression of wild-type Syt1 restores the missing fast burst of secretion. In knock-out cells infected for 8-12 h with SFV carrying the Syt1 WT gene, the fast burst component was completely restored (Fig. 2A-C, blue trace). In fact, exponential fitting showed that the size of the fast burst component (RRP) was on average larger in null cells rescued by overexpression of Syt1 compared with cells from control (+/+ or +/-) littermates (Fig. 2C, left panel). This increase in RRP size was accompanied by a decrease in the size of the SRP (Fig. 2C, middle panel), such that the sum of RRP and SRP size (i.e., the total number of primed vesicles) remained constant (153 ± 9 fF for control cells; 153 ± 12 fF for null cells overexpressing Syt1; p > 0.7).
The increase in RRP size over control values in the rescue experiment may either be attributable to the increased abundance of Syt1 induced by overexpression or else caused by another difference between Syt1 null and control cells. To clarify this question, we overexpressed Syt1 in control cells and observed similar changes as in the rescue experiment: the size of the fast burst component was increased at the expense of the slow burst (Fig. 2D-F). These data show that increased availability of Syt1, as obtained by viral overexpression, changes the equilibrium size relationship between RRP and SRP.
In chromaffin cells, a linear vesicle maturation scheme is postulated (see Discussion), in which vesicles have to go through the SRP state before they can reach the RRP, characterized by maximal fusion rate constants. This linear arrangement was demonstrated by cross-depletion experiments (Voets et al., 1999). The maturation step between the SRP and RRP is assumed reversible (Voets et al., 1999) and Ca2+ independent (Voets, 2000). Within the framework of this model, the simplest interpretation of our results is that the SRP-to-RRP conversion is governed by a rate-limiting step involving the recruitment of Syt1 (the vesicular fast calcium sensor) to the release machinery.
Comparison of capacitance increase and amperometric current in the presence and absence of a fast burst
The above interpretation of Syt1 function only makes sense if the vesicles in the SRP and RRP are identical, except for their fusion rate. However, based on differences between secretion measured with capacitance and amperometry, it has been suggested that the fastest component of the capacitance increase contains a component that is not related to the release of oxidizable material (Oberhauser et al., 1996; Ninomiya et al., 1997; Kasai, 1999) and/or that smaller vesicles (that may or may not contain catecholamines) might contribute to this fast burst component. The study of chromaffin cells from Syt1 null animals, in which the fast burst is selectively eliminated, allows the test of this hypothesis. In Figure 3A is shown the amperometric response from two cells, one Syt1 null cell and one null cell rescued by Syt1 overexpression. Both the amperometric current measurement (Fig. 3A, left ordinate axis) and the integrated signal (right ordinate axis) show that secretion of oxidizable material started after a longer delay and proceeded slower in the cell not expressing Syt1. In Figure 3B, the integrated amperometric signal is compared with the capacitance signal in the same two cells. The integrated amperometric signal is slower than the capacitance signal. This is expected because of the diffusional delay that is characteristic for amperometric recordings (Haller et al., 1998). Thus, a fast phase of both capacitance increase and amperometric current is missing in cells not expressing Syt1, and there is a longer delay between flash and amperometric signal in Syt1 null cells, which is also found in the capacitance responses (Voets et al., 2001 and data not shown). The scaling of the right ordinate axis in Figure 3B was chosen so that the capacitance and amperometric charge agree at late times for the wild-type cell. In the example of Figure 3B, this scaling does not result in a similar agreement for the null trace. However, another characteristic of amperometric recordings is the very large variability, because only a small fraction of the fusing vesicles are detected. When averaging all cells (Fig. 3C), it can be appreciated that the released amperometric charge and the capacitance increase is reduced by the same proportion in the absence of Syt1. A quantitative analysis is presented in Figure 3D: under the assumption that chromaffin granules contain catecholamines at an approximately constant concentration (Albillos et al., 1997; Gong et al., 2003), the third root of the amperometric charge should be proportional to the square root of the capacitance increase (because both quantities are proportional to vesicle radius). This is indeed seen both for the Syt1 null cells and after rescue with Syt1 (Fig. 3D). Moreover, the two regression lines in Figure 3D tightly overlap, which indicates a similar vesicular catecholamine concentration in both cases.
Figure 3.

The fast burst of secretion consists of the fusion of catecholamine-secreting vesicles. A, Example of the amperometric signal after flash photolysis of caged Ca2+ for an Syt1 null cell (gray) and a cell overexpressing Syt1-WT (black). Shown is the amperometric current (left axis) and its time integral (right axis). In the Syt1-overexpressing cell, secretion of oxidizable substance starts sooner and proceeds faster than in the Syt1 null cell. B, Comparison of capacitance increase (left axis) and integrated amperometric current (right axis) for the same two cells as in A. C, Comparison of mean capacitance increase (solid lines, left axis) and mean integrated amperometric current (dotted lines, right axis) for the whole population of Syt1 null cells (gray) and control cells (black). Notice that deletion of Syt1 causes a proportional decrease in capacitance increase and amperometric charge. D, Plot of the third root of the amperometric charge against the square root of the capacitance increase, using the amperometric integral and capacitance change obtained at 1 s after flash photolysis of caged Ca2+. Regression lines through the origin are drawn for both population of vesicles. The tight overlap of the two lines demonstrates similar vesicular catecholamine concentrations in the absence and presence of Syt1. KO, Knock-out.
These data show that the fast phase of the capacitance increase, which is missing in Syt1 null cells, must result from release of vesicles containing the same amount of catecholamines per unit area vesicle membrane as the vesicles that are released during slower phases of secretion. We conclude that, under our experimental conditions, the amperometric signal detects the same population of fusing vesicles as the capacitance signal and that vesicles from the RRP have similar catecholamine content as slower-releasing vesicles, confirming an important assumption behind the linear maturation scheme for secretory vesicles.
However, it is important to note that the consistency between amperometric and capacitance measurements depends on the experimental conditions. Under conditions of lower preflash calcium (<0.3 μm) and/or higher postflash calcium (>80-100 μm) concentrations, the consistency breaks down because of the fusion of non-catecholamine-containing vesicles (Xu et al., 1998; Sørensen, 2004).
Overexpression of synaptotagmin 2 results in slower fast burst than synaptotagmin 1
Next we asked whether the other vesicular synaptotagmin Syt2 could functionally substitute for Syt1. Chromaffin cells express Syt1 but not Syt2 (Fig. 1D) (Geppert et al., 1991; Marquèze et al., 1995); therefore, the Syt1 knock-out chromaffin cells serve as an ideal system for studying the different properties of the two synaptotagmins, without any perturbance caused by the presence of the close relative.
Flash photolysis showed that overexpression of Syt2 restores the missing fast burst component to the same amplitude as Syt1 overexpression (Fig. 4A-C), demonstrating functional rescue of Syt1 knock-out cells by Syt2. However, a closer look at the initial half-second of the response indicates a slower burst when Syt2 is present, whether detected by amperometry or by capacitance measurements (Fig. 4A, inset). Exponential fitting revealed a slightly slower fast burst in cells overexpressing Syt2 (τ1 = 29.8 ± 2.5 ms at 27.7 ± 2.3 μm [Ca2+]I; n = 30) compared with cells overexpressing Syt1 (τ1 = 18.6 ± 2.1 ms at 25.9 ± 2.5 μm [Ca2+]I; n = 29). This difference in fast time constant was statistically significant (p < 0.001). The time constant of the slow burst (τ2 = 201 ± 32 ms for Syt1; τ2 = 272 ± 40 ms for Syt2; p = 0.20) (Fig. 4B), as well as the size of both fast and slow burst components and sustained rate were not significantly different between isoforms (Fig. 3C).
Figure 4.
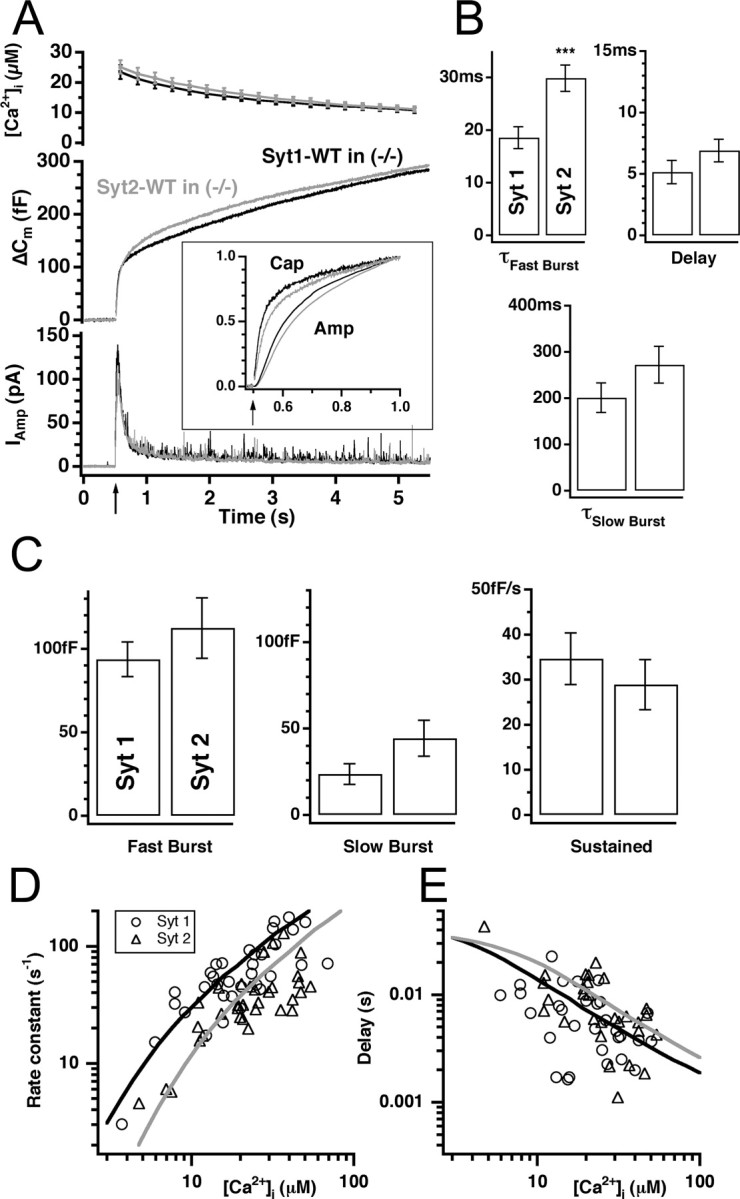
Syt1 and Syt2 are alternative calcium sensors for the RRP. A, The mean [Ca2+]i, capacitance, and amperometric trace from knock-out cells overexpressing either Syt1-WT (black; n = 33) or wild-type synaptotagmin 2 (Syt2-WT, gray; n = 35). The inset shows the first half-second of the capacitance (Cap) and integrated amperometric (Amp) traces scaled to control level. Secretion was slightly slower in the presence of Syt2. B, Kinetic analysis of the individual traces revealed that the time constant of the fast burst component (top left) was significantly increased in cells overexpressing Syt2-WT, whereas the time constant of the slow burst component remained unaffected (bottom). The delay was also slowed down, although not significantly. C, There was no significant difference between the size of both burst components and the sustained rate in cells overexpressing either synaptotagmin isoforms. D, E, Relationship between the postflash [Ca2+]i and the rate constants (1/τ; D) and synaptic delays (E) for the fast component. Cells overexpressing Syt1 display higher rate constants and slightly shorter delay (filled circles) over the examined [Ca2+]i range compared with cells overexpressing Syt2 (open circles). Black (for Syt1-WT) and gray (for Syt2-WT) lines correspond to the best fit with the kinetic scheme described by Sørensen et al. (2003b).
A delay is most often observed between the flash stimulus and the onset of exocytosis. This delay is believed to reflect the binding kinetics of Ca2+ to the Ca2+ sensor of the fusion machinery (Heinemann et al., 1994; Voets, 2000). The exocytotic delay was measured as the time interval between the flash and the time point the exponential fit crosses the preflash membrane capacitance level and was found to be on average a bit longer in cells overexpressing Syt2 (6.9 ± 0.9 ms) than in Syt1-expressing cells (5.2 ± 0.9 ms for Syt1) (Fig. 4B, top right), a difference that was not quite statistically significant (p = 0.11). However, when we increased the number of observations by including results from the second flash stimulation in each cell (when available) into the analysis, the difference in delay became statistically significant (8.8 ± 1 ms for Syt2; 5.5 ± 0.7 ms for Syt1; p < 0.02), whereas the statistical conclusion for all other parameters did not change.
Plotting the rate constant of fast release (1/τ, in which τ is the time constant of fast release) and the secretory delay against the postflash [Ca2+]i allows an inspection of the calcium dependence of the fast release process (Heinemann et al., 1994; Voets, 2000). This plot showed that secretory rates were slower in Syt2-expressing cells over the range of postflash calcium concentrations investigated (Fig. 4D, E; only data from the first stimulation included). In a model of the calcium sensor for exocytosis featuring sequential reversible calcium-binding steps, followed by an irreversible (and calcium-independent) fusion step, such a finding is most easily explained by a decrease in the affinity of the unitary calcium-binding step (Sørensen et al., 2003b). To estimate the difference in affinity, we fitted a model of four sequential cooperative Ca2+-binding steps to the data according to Sørensen et al. (2003b). The result of the fitting is shown as lines in Figure 4D (black line, Syt1, α = 6.8 μm-1s-1, β = 60 s-1, γ = 1328 s-1; gray line, Syt2, α = 4.3 μm-1s-1, β = 76 s-1, γ = 1610 s-1, in which α is the Ca2+-binding rate constant, β is the unbinding rate constant, and γ is the fusion rate constant). The most robust parameter combination of these fits is the KD for Ca2+ binding, estimated as β/α. This value was larger for Syt2 than for Syt1 (Syt1, 60/6.8 = 8.8 μm; Syt2, 76/4.3 = 18 μm).
These data show that Syt1 and Syt2 are alternative, but not identical, calcium sensors for the fast burst of release in chromaffin cells.
Syt2 has a slightly lower Ca2+ affinity for binding to phospholipids than Syt1
Different Syt isoforms deviate in their Ca2+ affinity for binding to phospholipids (Li et al., 1995a,b), which has been proposed to underlie different functions in secretion. For Syt1 and Syt2, the KD for Ca2+-dependent lipid binding to the C2A domain is slightly higher in the case of Syt2, i.e., Syt2 has a lower Ca2+ affinity (Ullrich et al., 1994; Sugita et al., 2002). However, also the C2B domain of synaptotagmins binds to lipids (Fernandez et al., 2001), and the overall lipid-binding depends on both C2 domains (Fernández-Chácon et al., 2002). In a recent study, the kinetics of unbinding of the double C2A-C2B domain from membranes was found to be comparable between Syt1 and Syt2 isoforms (Hui et al., 2005).
We assayed the Ca2+-dependent binding of Syt1 and Syt2 C2 domains to liposomes (25% PS/75% PC). Recombinant C2 fragments were purified as GST-fusion proteins and incubated with liposomes in the presence of different concentrations of free Ca2+. After isolation, the liposomes were analyzed by SDS-PAGE and Coomassie blue staining. We first repeated the experiments using isolated C2A domains and confirmed a lower Ca2+ affinity of phospholipid binding for Syt2 than Syt1 (Fig. 5A) (Ullrich et al., 1994; Sugita et al., 2002). We also compared the Ca2+ affinity of the isolated C2B domains, and here we found the opposite result: the Syt2 C2B domain has a slightly higher affinity for Ca2+-dependent phospholipid binding than the Syt1 C2B domain (Fig. 5B). Finally, using the entire C2A-C2B domain, we found that Syt2 bound liposomes at slightly higher Ca2+ concentrations than the Syt1 C2AB domain (Fig. 5C). Because the difference in Ca2+ affinity was slight, we wanted to rule out that the effect could be caused by interexperimental variability. Therefore, we included the C2 domain of PKCβ in each experiment. After isolation of liposomes, the PKCβ-C2 domain could be distinguished because of its different size. This internal control showed no difference in apparent affinity of PKCβ-C2 in the two experiments shown in Figure 5C. This experiment was performed five times, and the mean binding curve showed a slightly lower affinity for Syt2 than for Syt1 (Fig. 5C). The EC50 for Syt2 was slightly increased (5.5 ± 0.6 μm) compared with Syt1 (4.4 ± 0.5 μm; p < 0.05 by paired t test). Therefore, these experiments lead to the conclusion that the overall Ca2+ affinity for phospholipid binding to the double C2 domain is governed by the combination of both C2A and C2B domains. Overall, we conclude that Syt2 has a slightly lower affinity to phospholipids than Syt1.
Figure 5.

Phospholipid and SNARE binding by synaptotagmin 1 and 2. A, B, Apparent Ca2+ affinities of synaptotagmin 1 and 2 single C2 domains (as purified GST-fusion proteins). The C2 domains analyzed are the C2A domains of Syt1 and Syt2 (A) and C2B of Syt1 and Syt2 (B). Liposomes composed of 25% PS/75% PC were incubated with the double domains at the Ca2+ concentrations shown (clamped with Ca2+/EGTA buffers). After binding, the samples were centrifuged to sediment the liposomes, and bound protein was analyzed by SDS-PAGE and Coomassie blue staining. Top panels, Representative Coomassie blue-stained gels from single experiments. Bottom panels, Quantification of binding. The data were fitted by the Hill equation. The EC50 values are presented in the figures, together with the number of experiments. C, Apparent Ca2+ affinities of synaptotagmin 1 and 2 double C2A-C2B domains. The C2 domain from PKCβ as a GST-fusion protein was included in each reaction as an internal control. Bottom panel, The EC50 of Syt2 C2AB was 5.5 ± 0.6 μm, which is slightly higher than the EC50 for Syt1 C2AB of 4.4 ± 0.5 μm (paired t test, p < 0.05; n = 5). The EC50 of PKCβ-C2 domains did not vary significantly between the experiments, showing that there was no systematic difference in the calcium concentration between the experiments. D, Immunoprecipitation (IP) using syntaxin antibodies. Protein samples were taken from forebrain, which is rich in Syt1, and spinal cord, which is rich in Syt2. Immunoprecipitation of syntaxin led to coprecipitation of the other SNAREs and synaptotagmin 1 and 2, respectively.
To supplement our finding that Syt1 and Syt2 can substitute for each other in neurotransmitter release, we performed immunoprecipitation of syntaxin 1 from brain extracts of the forebrain and spinal cord, which are rich in Syt1 and Syt2, respectively. These experiments confirmed that both Syts bind to the neuronal SNAREs (Fig. 5D). This observation using native proteins supplements previous findings of equal binding of the two isoforms to syntaxin and SNAP-25 using C2AB domains fused to GST (Rickman et al., 2004a,b).
Mutation of Syt1 at phosphorylation sites leaves secretion unchanged
The difference in secretory rate that we found above correlates with the difference in Ca2+ affinity of Syt1 and Syt2. However, the different secretory rate might also be caused by other differences between the proteins. Notably, Syt1 has a shared PKC/CaMKII phosphorylation site (Thr-112) not present in Syt2. In some secretory systems, β-phorbol ester treatment causes an increase in release rate constant in the absence of a change in readily-releasable pool size (Wu and Wu, 2001; Zhu et al., 2002; Lou et al., 2005). One possibility is that this could be caused by phosphorylation of Syt1 to increase its effectiveness in triggering release. To investigate this possibility, we generated mutations of Syt1 and overexpressed them in Syt1 knock-out chromaffin cells. By replacing Thr-112 with alanine, we created a mutant that might behave as the nonphosphorylated Syt1 (a “nonphosphomimetic” mutant), whereas replacing Thr-112 with the negatively charged aspartate results in a mutant that might act as the constitutively phosphorylated Syt1 (a “phosphomimetic” mutant). Importantly, however, in both mutants, dynamic phosphorylation of Syt1 at this site is no longer possible. Likewise, we created double mutations in the caseine kinase II phosphorylation sites at Thr-125 and Thr-128 (Bennett et al., 1993; Davletov et al., 1993; Hilfiker et al., 1999).
As shown in Figure 6A-D, neither overexpression of the phosphomimetic T112D mutant of Syt1 nor overexpression of the nonphosphomimetic T112A in knock-out cells changed flash-evoked secretion compared with overexpression of wild-type Syt1. Importantly, we did not observe significant differences in the time constant of the fast burst component measured at similar postflash [Ca2+]i (15.7 ± 1.5 ms at 21 ± 2 μm calcium for WT; 16.1 ± 1.8 ms at 26 ± 1 μm for T112D; and 15.5 ± 1.6 ms at 25 ± 3 μm calcium for T112A), which argues against a potential role of this phosphorylation in the regulation of the fusion kinetics of RRP fusion. Likewise, the response to a second flash stimulation was intact in cells overexpressing Syt1 T112A and Syt1 T112D (Fig. 6B). This experiment assays the refilling of the vesicles pools and shows also that this function is intact in the absence of dynamic synaptotagmin phosphorylation.
Figure 6.
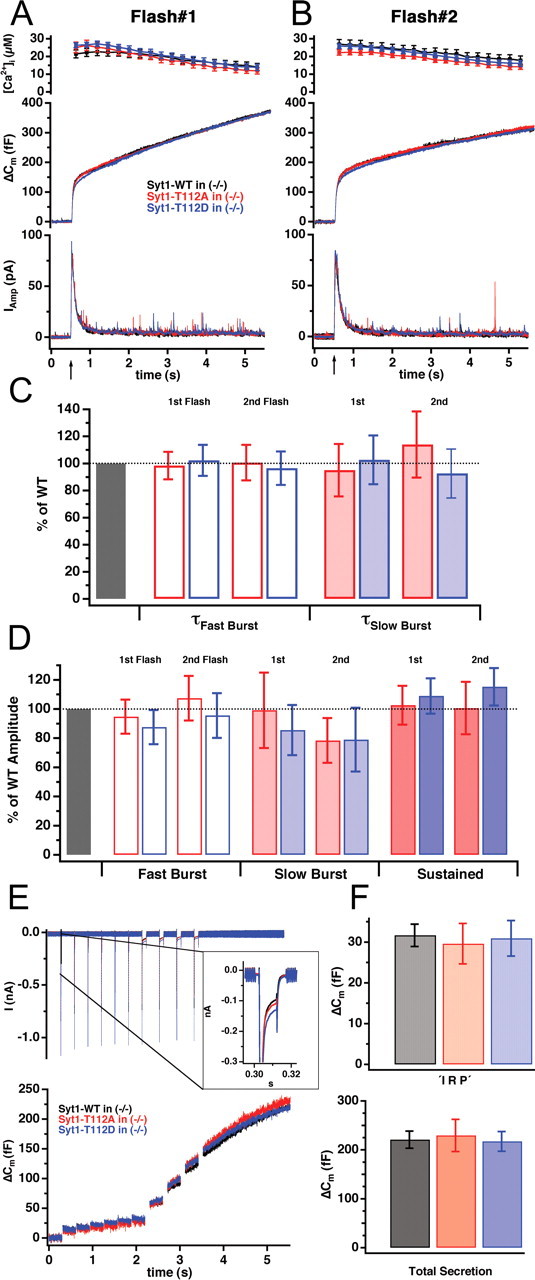
Mutation of Syt1 at the PKC/CaMKII phosphorylation site Thr-112 did not modify secretion. A, Mean capacitance (middle) and amperometric (bottom) responses to the first flash stimulation (arrow) from knock-out cells overexpressing wild-type Syt1 (black; n = 19), the nonphosphomimetic T112A mutant (red; n = 18), or the phosphomimetic T112D mutant (blue; n = 22). B, The corresponding response evoked by a second flash stimulation given ∼80 s after the first stimulus. C, D, Time constants for the fast and slow burst component (C) and amplitudes of the different kinetic components (D) were normalized to the corresponding control data (Syt1-WT overexpression) and displayed as mean ± SEM. No significant difference was found between the kinetic components. E, Voltage protocol (6 10-ms depolarization steps from the resting -70 to +5 mV, followed by 4 100-ms steps) applied to assay depolarization-induced secretion showed that capacitance changes (bottom) in cells overexpressing either Syt1-T112A (red; n = 20) or Syt1-T112D (blue; n = 18) were indistinguishable from Syt1-WT-overexpressing cells (black; n = 20). The top row shows the mean currents measured in voltage clamp; as seen in the inset, the calcium currents were also similar in cells overexpressing different Syt1 mutants. F, No significant difference was found in the secretion evoked by the six 10 ms pulses, which is thought to be the secretion of vesicles located in the vicinity of the calcium channels (top; IRP), as well as in the total secretion (bottom).
Syt1 was proposed to interact with N- and P/Q-type calcium channels (Charvin et al., 1997; Sheng et al., 1997), either of them contributing ∼30% of calcium influx in mouse chromaffin cells (Aldea et al., 2002). This interaction might serve as a molecular link between synaptic vesicles and the calcium channels and in addition might modulate channel function (Seagar et al., 1999). To investigate the functional consequences of Syt1 phosphorylation during exocytosis triggered by calcium influx, we applied a voltage protocol that allows us to distinguish a subpopulation of the RRP, which is located close to calcium channels [immediately releasable pool (IRP)] (Horrigan and Bookman, 1994; Voets et al., 1999). Six 10 ms depolarization steps were applied to selectively release vesicles from the IRP, followed by four 100 ms pulses to evoke secretion of the releasable vesicles located farther from calcium channels as described by Voets et al. (1999). Using this protocol, Voets et al. (2001) showed that the IRP is eliminated in the absence of Syt1. Figure 6, E and F, shows that, although somewhat variable, the amplitude of the calcium currents (87 ± 5 pA for WT; 114 ± 11 pA for T112D; and 98 ± 16 pA for T112A; Kruskal-Wallis three-group comparison, p = 0.11) (Fig. 6E, inset) and the size of IRP were unaffected by overexpression of either phosphorylation mutants. This set of experiments indicates that phosphorylation of Syt1 at Thr-112 does not affect the relative localization of vesicles to calcium channels, nor does it seem to regulate channel activity.
Flash experiments using the double mutations T125A/T128A and T125D/T128D in the caseine kinase phosphorylation sites indicated that both mutants rescued secretion as well as Syt1, indicating also that the caseine kinase phosphorylation site is not required for basal Syt1 function (supplemental Fig. 1, available at www.jneurosci.org as supplemental material).
PKC modulation of catecholamine secretion happens upstream of Syt1 function
The above experiments show that Syt1 mutated at Thr-112 rescues secretion as well as wild-type Syt1. However, the question is whether this also would be the case during conditions in which the appropriate protein kinases were activated. The application of β-phorbol esters leads to strong PKC activation and results in a twofold to threefold increase in secretion from bovine chromaffin cells, which is inhibited by bisindolylmaleimide I (Gillis et al., 1996; Smith et al., 1998). In addition, phorbol esters have other C1 domain-containing targets that can modify secretion, including Munc13 isoforms (Brose and Rosenmund, 2002). We asked whether the phosphorylation of Syt1 plays any role in mediating the effect of β-phorbol esters. Figure 7A-C shows that extracellular application of 100 nm PMA for 3 min leads to a robust increase of secretion in knock-out cells, which mainly was the result of an increased size of the slow burst component. Moreover, overexpression of Syt1-T112A did not prevent the potentiating effect of PMA, which increased secretion to similar extent as in cells overexpressing Syt1-WT (∼2-fold increase of both burst components and ∼1.5-fold increase of the sustained component) (Fig. 7D-G). The time constants of the two burst components were unaffected by PMA (Fig. 7F). Note that, in the presence of PMA, the size of the SRP is as large in Syt1 null cells as the summed size of SRP and RRP in Syt1-expressing cells. Thus, PMA increases the number of primed vesicles, whereas Syt1 is responsible for converting a part of these to the fast-releasing RRP state. Overall, the data show that PMA acts upstream and independently of Syt1.
Figure 7.
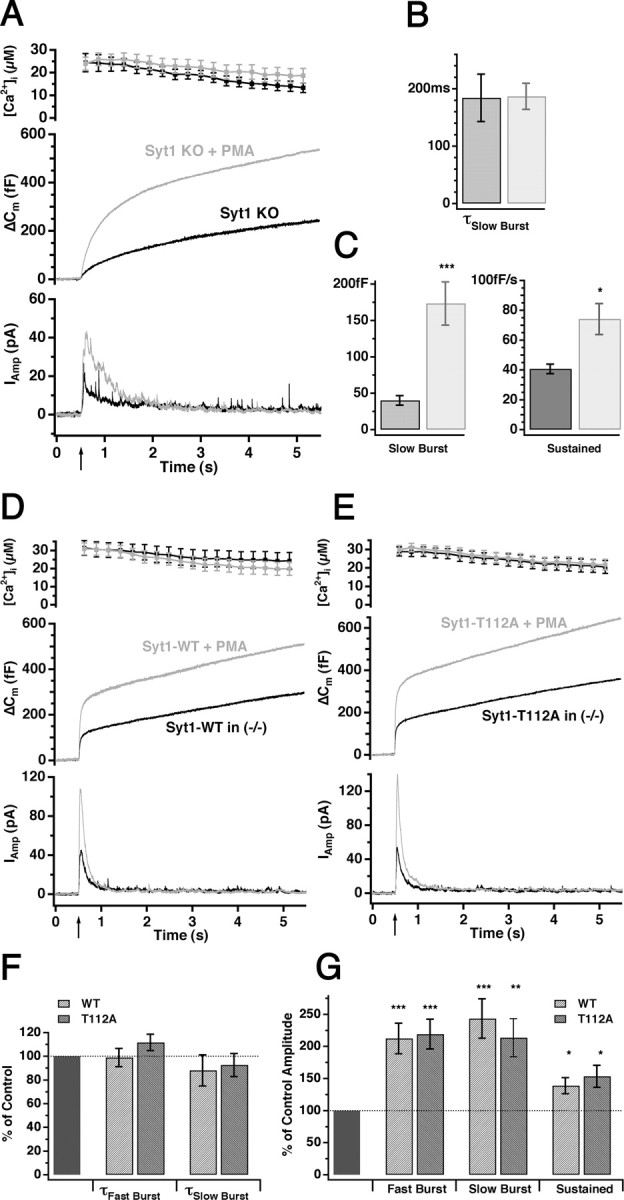
Synaptotagmin 1 is not required for the effect of β phorbol ester in chromaffin cells. A, Superfusion of Syt1 null cells with 100 nm PMA (3 min before giving the flash stimulus) resulted in a massive increase of both the capacitance (middle) and the amperometric (bottom) response (gray; n = 13) compared with nontreated knock-out cells (black; n = 16). B, C, Kinetic analysis showed that PMA (gray bars) significantly increased both the amplitude of the slow burst component (C, left) and the sustained rate (C, right), whereas the time constant of the slow burst component remained unaffected (B). D, E, PMA similarly increased the secretion in both knock-out cells overexpressing wild-type Syt1 (D, gray, n = 19; nontreated cells, black, n = 18) and knock-out cells overexpressing the nonphosphomimetic T112A mutant of Syt1 (E, gray; PMA treated, n = 18; nontreated, black, n = 18). F, G, The kinetic parameters were normalized to the corresponding nontreated parameters and displayed as mean ± SEM. PMA did not change the time constants of either burst component (F) but significantly increased the amplitude of both burst components and the sustained rate (G) in cells overexpressing either wild-type or T112A mutant Syt1. *p < 0.05; **p < 0.01; ***p < 0.001.
Discussion
Overexpression of wild-type vesicular synaptotagmins: the effect of quantity and quality
We used chromaffin cells from Syt1 null mice and demonstrated that viral overexpression of Syt1 or Syt2 rescued the fast burst component of secretion, which is lacking in Syt1 knock-out chromaffin cells. Thus, both isoforms are able to trigger secretion from a readily-releasable vesicle pool.
Despite the overall similarity of secretion driven by either synaptotagmin isoforms, a difference in fusion rate from the RRP was observed, Syt2 being slower than Syt1. In addition, overexpression of Syt1 led to larger RRP size than in control cells, whereas the size of the SRP was reduced in parallel. The fusion rate of the larger RRP in Syt1-overexpressing cells was unchanged (Fig. 2). To understand the significance of these results, we consider the linear model for maturation of large dense-core vesicles in chromaffin cells (Voets et al., 1999; Ashery et al., 2000; Voets, 2000):

where UPP (unprimed pool) is a reserve pool of docked, but unprimed, vesicles that can gain release competence by maturing into the SRP and the RRP. Vesicles in the SRP and the RRP can undergo exocytosis, but the RRP vesicles are characterized by faster fusion kinetics than the SRP vesicles (rslow < rfast). C represents the pool of secreted vesicles, and, importantly, only the final secretion steps (rslow, rfast) are irreversible, whereas the maturation steps are reversible. Note that, in this simplified model, rslow and rfast represent the overall rates of fusion; in a more extensive description, each of these parameters would be modeled by sequential Ca2+ binding to a calcium sensor, followed by an irreversible fusion step (compare with Sørensen, 2004).
The increase in RRP size by Syt1 overexpression and the lack of an RRP in the absence of Syt1 suggests that the equilibrium between RRP and SRP (i.e., k2/k-2) is affected by the availability of Syt1. In a previous study, we found that this step is regulated by cAMP-dependent phosphorylation of an unknown target protein (Nagy et al., 2004), raising the possibility that Syt1 availability might be regulated by cAMP. However, whether this is a physiological mechanism remains unknown. It is also possible to explain the larger size of the RRP after Syt1 overexpression by a lower release rate from the RRP at basal [Ca2+]i, in agreement with the published role of Syt1 in suppressing basal release (Yoshihara and Littleton, 2002). However, the basal [Ca2+]i in our experiments was 400-600 nm, at which concentration the refilling rate dominates over basal release in determining the size of the RRP (Voets, 2000). It should be noted that, in both cases, the reversible model above cannot easily explain the compensatory decrease seen in SRP size during Syt1 overexpression. To account for this finding, the model could be extended by including a maximal upper size for the entire primed vesicle pool (= RRP + SRP size), which could be caused by the existence of a limited resource for priming.
It has been suggested that the two releasable vesicle pools SRP and RRP are attributable to the existence of two alternative conformations of the SNARE complex, loose and tight, respectively (Xu et al., 1999; Voets et al., 2001). In light of our data, we can suggest that Syt1/Syt2 binding to the loose SNARE complex drives the conformational change into a tighter complex. Most likely this is caused by Ca2+-independent binding of Syt to the SNAREs, because this step is usually assumed Ca2+ independent (Voets et al., 1999; Voets, 2000). In a second reaction, Syt might trigger release by Ca2+-dependent binding to phospholipids or SNAREs, which would explain the different abilities of Syt isoforms to tune the fast speed of release.
Biochemically, we found that the KD for Ca2+-dependent lipid binding to the double C2AB domain is higher for Syt2 than for Syt1, although the difference is small. This lower affinity of Syt2 is determined by a combination of the lower affinity of the C2A domain and a slightly higher affinity of the C2B domain, demonstrating that both C2 domains cooperate to determine the overall Ca2+ affinity of phospholipid binding. This difference may underlie the different fusion speeds seen when overexpressing the two isoforms in chromaffin cells. We should remember, however, that the apparent calcium affinity of C2 domains depends critically on the local phospholipid composition. This is also a relevant consideration when comparing the triggering rate between different experimental systems. The brainstem expresses predominantly Syt2 (Geppert et al., 1991; Ullrich et al., 1994; Marquèze et al., 1995), yet triggering rates measured by flash photolysis of caged Ca2+ are faster in the calyx of Held synapse than in chromaffin cells by approximately an order of magnitude (Schneggenburger and Neher, 2000). This could be caused by the different vesicle type, the phospholipid composition, or the different expression of accessory proteins.
How does our work compare with studies of synaptotagmin in central neurons? In autaptic hippocampal neurons, single EPSCs follow a biphasic time course, and, in the absence of Syt1, the balance is shifted in favor of the asynchronous component, whereas the total amount of secretion is only slightly reduced (Shin et al., 2003; Nishiki and Augustine, 2004a). Overexpression of Syt1 in Syt1 null autaptic neurons restored the synchronicity of the EPSCs (Han et al., 2004; Nishiki and Augustine, 2004b). For an immediate consideration, these data are consistent with our rescue data in Figure 2, if one equates synchronized secretion with our RRP and asynchronous secretion with our SRP, as suggested (Nishiki and Augustine, 2004a). However, closer inspection reveals differences, because in chromaffin cells the Syt1 R233Q mutation noticeably slows down fast flash-induced release from the RRP (Sørensen et al., 2003b), whereas in neurons the amplitude of a single EPSC is reduced, but the kinetics of the two components are unaffected (Fernández-Chacón et al., 2001; Han et al., 2004). Likewise, in neurons, no difference in EPSC shape was found when Syt1 nulls were rescued by Syt1 or Syt2 (Stevens and Sullivan, 2003). Therefore, it appears that, in neurons, a kinetic step independent of calcium binding to Syt1 determines the timing of synaptic vesicle fusion from the two pools. Most likely this is the rapid rise and decay of the Ca2+ signal in microdomains around transiently open calcium channels.
Another attempt at comparing the systems would be to equate the readily-releasable vesicle pool in hippocampal neurons, evaluated by hyperosmotic sucrose application (Rosenmund and Stevens, 1996), with the chromaffin cell RRP. During overexpression of Syt1 in hippocampal neurons, the release probability (= EPSC charge/“sucrose pool” charge) increased, whereas the size of the sucrose pool remained constant (Han et al., 2004). Although both datasets indicate a gain-of-function effect, the interpretation of the neuronal data are exactly the opposite of our finding that Syt1 overexpression increased the size of the RRP, without changing the release rate constant. These differences may be explained by a different correspondence between vesicle pools. For instance, the sucrose pool might be kinetically heterogeneous, so that action potentials can draw only on one subpool, as suggested recently (Moulder and Mennerick, 2005). Hence, the sucrose pool may correspond to the pools denoted RRP and SRP (and maybe even UPP) in chromaffin cells. In this case, an increase in the size of the RRP subpool at the expense of the SRP would indeed be interpreted as an increase in release probability when using sucrose application. The advantage of our measurements is that, by applying a strong stimulus, we empty the primed pools completely, allowing the clear distinction of different vesicle pool sizes and their release rates. In conclusion, we suggest that synaptotagmin abundance regulates releasable pool sizes rather than release probability.
Does phosphorylation of synaptotagmin 1 have a physiological function?
Phorbol ester treatment leads to different effects on secretion in different systems: in chromaffin cells, an increase in releasable pool size is observed, whereas the release rate constants from the pools are unaffected (Gillis et al., 1996; Smith et al., 1998; Yang et al., 2002; this study). However, in pituitary gonadotrophs and in the calyx of Held, an increase in release rate constant is seen in the absence of a change in readily-releasable pool size (Wu and Wu, 2001; Zhu et al., 2002; Lou et al., 2005). Using rescue of null cells with Syt1 constructs mutated in phosphorylation sites, we did not find any modification of secretion in the presence or absence of phorbol esters (Figs. 6, 7). Furthermore, even in Syt1 null cells, phorbol esters increased secretion. These data show that phorbol esters act independently and upstream of synaptotagmin to regulate the releasable vesicle pool size in mouse chromaffin cells.
Conclusion
We conclude that phorbol esters act upstream of synaptotagmin to regulate the size of the primed vesicle pool (SRP + RRP size), whereas both Syt1 and Syt2 have the ability to shift primed vesicles into the high-releasable state (RRP). The calcium dependence of fusion from the RRP varies in parallel with the Ca2+ dependence of lipid binding of Syt1/Syt2. All of these observations are consistent with the idea that Syt1 and Syt2 are alternative (but non-identical) Ca2+ sensors for fast secretion.
Footnotes
This work was supported by the grants from the Deutsche Forschungsgemeinschaft (Sonderforschungsbereich 523/TP4, So708 1-1, and Graduirtenkolleg 521), Korean Science and Engineering Foundation, and Deutscher Akademischer Austauschdienst. We thank Ina Herfort and Dirk Reuter for expert technical assistance. We also thank Ralf B. Nehring for help and Reinhard Jahn for providing antibody against synaptotagmin 1.
Correspondence should be addressed to Jakob B. Sørensen, Max-Planck-Institute for Biophysical Chemistry, Am Fassberg 11, 37077 Göttingen, Germany. E-mail: jsoeren@gwdg.de.
G. Nagy's present address: National Institute of Neurosurgery, H-1145 Budapest, Hungary.
J. H. Kim's present address: Vollum Institute, Oregon Health and Science University, Portland, OR 97239.
Copyright © 2006 Society for Neuroscience 0270-6474/06/260632-12$15.00/0
References
- Albillos A, Dernick G, Horstmann H, Almers W, Alvarez de Toledo G, Lindau M (1997) The exocytotic event in chromaffin cells revealed by patch amperometry. Nature 389: 509-512. [DOI] [PubMed] [Google Scholar]
- Aldea M, Jun K, Shin HS, Andres-Mateos E, Solis-Garrido LM, Montiel C, Garcia AG, Albillos A (2002) A perforated patch-clamp study of calcium currents and exocytosis in chromaffin cells of wild-type and α1A knockout mice. J Neurochem 81: 911-921. [DOI] [PubMed] [Google Scholar]
- Ashery U, Betz A, Xu T, Brose N, Rettig J (1999) An efficient method for infection of adrenal chromaffin cells using the Semliki Forest virus gene expression system. Eur J Cell Biol 78: 525-532. [DOI] [PubMed] [Google Scholar]
- Ashery U, Varoqueaux F, Voets T, Betz A, Thakur P, Koch H, Neher E, Brose N, Rettig J (2000) Munc 13-1 acts as a priming factor for large densecore vesicles in bovine chromaffin cells. EMBO J 19: 3586-3596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Augustine GJ (2001) How does calcium trigger neurotransmitter release? Curr Opin Neurobiol 11: 320-326. [DOI] [PubMed] [Google Scholar]
- Bai J, Chapman ER (2004) The C2 domains of synaptotagmin—partners in exocytosis. Trends Biochem Sci 29: 143-151. [DOI] [PubMed] [Google Scholar]
- Bai J, Wang CT, Richard DA, Jackson MB, Chapman ER (2004) Fusion pore dynamics are regulated by synaptotagmin·t-SNARE interactions. Neuron 41: 929-942. [DOI] [PubMed] [Google Scholar]
- Bennett MK, Miller KG, Scheller RH (1993) Casein kinase II phosphorylates the synaptic vesicle protein p65. J Neurosci 13: 1701-1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brose N, Rosenmund C (2002) Move over protein kinase C, you've got company: alternative cellular effectors of diacylglycerol and phorbol esters. J Cell Sci 115: 4399-4411. [DOI] [PubMed] [Google Scholar]
- Charvin N, Lévêque C, Walker D, Berton F, Raymond C, Kataoka M, Shoji-Kasai Y, Takahashi M, De Waard M, Seagar MJ (1997) Direct interaction of the calcium sensor protein synaptotagmin I with a cytoplasmic domain of the α1A subunit of the P/Q-type calcium channel. EMBO J 16: 4591-4596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davletov B, Sontag JM, Hata Y, Petrenko AG, Fykse EM, Jahn R, Südhof TC (1993) Phosphorylation of synaptotagmin I by casein kinase II. J Biol Chem 268: 6816-6822. [PubMed] [Google Scholar]
- Fernandez I, Arac D, Ubach J, Gerber SH, Shin OH, Gao Y, Anderson RGW, Südhof TC, Rizo J (2001) Three-dimensional structure of the synaptotagmin C2B-domain: synaptotagmin 1 as a phospholipids binding machine. Neuron 32: 1057-1069. [DOI] [PubMed] [Google Scholar]
- Fernández-Chacón R, Königstorfer A, Gerber SH, García J, Matos MF, Stevens CF, Brose N, Rizo J, Rosenmund C, Südhof TC (2001) Synaptotagmin I functions as a calcium regulator of release probability. Nature 410: 41-49. [DOI] [PubMed] [Google Scholar]
- Fernández-Chácon R, Shin O, Königstorger A, Matos MF, Meyer AC, Garcia J, Gerber SH, Rizo J, Südhof TC, Rosenmund C (2002) Structure/function analysis of Ca2+ binding to the C2A domain of synaptotagmin 1. J Neurosci 22: 8438-8446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuda M, Kowalchyk JA, Zhang X, Martin TFJ, Mikoshiba K (2002) Synaptotagmin IX regulates Ca2+-dependent secretion in PC12 cells. J Biol Chem 277: 4601-4604. [DOI] [PubMed] [Google Scholar]
- Geppert M, Archer III BT, Südhof TC (1991) Synaptotagmin II. A novel differentially distributed form of synaptotagmin. J Biol Chem 266: 13548-13552. [PubMed] [Google Scholar]
- Geppert M, Goda Y, Hammer RE, Li, Rosahl TW, Stevens CF, Südhof TC (1994) Synaptotagmin I: a major Ca2+ sensor for transmitter release at a central synapse. Cell 79: 717-727. [DOI] [PubMed] [Google Scholar]
- Gillis KD, Möβner R, Neher E (1996) Protein kinase C enhances exocytosis from chromaffin cells by increasing the size of the readily releasable pool of secretory granules. Neuron 16: 1209-1220. [DOI] [PubMed] [Google Scholar]
- Gong LW, Hafez I, Alvarez de Toledo G, Lindau M (2003) Secretory vesicles membrane area is regulated in tandem with quantal size in chromaffin cells. J Neurosci 23: 7917-7921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haller M, Heinemann C, Chow RH, Heidelberger R, Neher E (1998) Comparison of secretory responses as measured by membrane capacitance and by amperometry. Biophys J 74: 2100-2113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han W, Rhee J-S, Maximov A, Lao Y, Mashimo T, Rosenmund C, Südhof TC (2004) N-glycosylation is essential for vesicular targeting of synaptotagmin 1. Neuron 41: 85-99. [DOI] [PubMed] [Google Scholar]
- Heinemann C, Chow RH, Neher E, Zucker RS (1994) Kinetics of the secretory response in bovine chromaffin cells following flash photolysis of caged Ca2+ Biophys J 67: 2546-2557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilfiker S, Pieribone VA, Nordstedt C, Greengard P, Czernik AJ (1999) Regulation of synaptotagmin I phosphorylation by multiple protein kinases. J Neurochem 73: 921-932. [DOI] [PubMed] [Google Scholar]
- Horrigan FT, Bookman RJ (1994) Releasable pools and the kinetics of exocytosis in adrenal chromaffin cells. Neuron 13: 1119-1129. [DOI] [PubMed] [Google Scholar]
- Hui E, Bai J, Wang P, Sugimori M, Llinas RR, Chapman ER (2005) Three distinct kinetic groupings of the synaptotagmin family: candidate sensors for rapid and delayed exocytosis. Proc Natl Acad Sci USA 102: 5210-5214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasai H (1999) Comparative biology of Ca2+-dependent exocytosis: implications of kinetic diversity for secretory function. Trends Neurosci 22: 88-93. [DOI] [PubMed] [Google Scholar]
- Koh TW, Bellen HJ (2003) Synaptotagmin I, a Ca2+ sensor for neurotransmitter release. Trends Neurosci 26: 413-422. [DOI] [PubMed] [Google Scholar]
- Li C, Davletov BA, Südhof TC (1995a) Distinct Ca2+ and Sr2+ binding properties of synaptotagmins. Definition of candidate Ca2+ sensors for the fast and slow components of neurotransmitter release. J Biol Chem 270: 24898-24902. [DOI] [PubMed] [Google Scholar]
- Li C, Ullrich B, Zhang JZ, Anderson RG, Brose N, Südhof TC (1995b) Ca2+-dependent and -independent activities of neural and non-neural synaptotagmins. Nature 375: 594-599. [DOI] [PubMed] [Google Scholar]
- Lou X, Scheuss V, Schneggenburger R (2005) Allosteric modulation of the presynaptic Ca2+ sensor for vesicle fusion. Nature 435: 497-501. [DOI] [PubMed] [Google Scholar]
- Majewski H, Iannazzo L (1998) Protein kinase C: a physiological mediator of enhanced transmitter output. Prog Neurobiol 55: 463-475. [DOI] [PubMed] [Google Scholar]
- Marquèze B, Boudier JA, Mizuta M, Inagaki N, Seino S, Seagar M (1995) Cellular localization of synaptotagmin I, II, and III mRNAs in the central nervous system and pituitary and adrenal glands of the rat. J Neurosci 15: 4906-4917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moulder KL, Mennerick S (2005) Reluctant vesicles contribute to the total readily releasable pool in glutamatergic hippocampal neurons. J Neurosci 25: 3842-3850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy G, Matti U, Nehring RB, Binz T, Rettig J, Neher E, Sørensen JB (2002) Protein kinase C-dependent phosphorylation of synaptosome-associated protein of 25 kDa at Ser187 potentiates vesicle recruitment. J Neurosci 22: 9278-9286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy G, Reim K, Matti U, Brose N, Binz T, Rettig J, Neher E, Sørensen JB (2004) Regulation of releasable vesicle pool sizes by protein kinase A-dependent phosphorylation of SNAP-25. Neuron 41: 417-429. [DOI] [PubMed] [Google Scholar]
- Ninomiya Y, Kishimoto T, Yamazawa T, Ikeda H, Miyashita Y, Kasai H (1997) Kinetic diversity in the fusion of exocytotic vesicles. EMBO J 16: 929-934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishiki T, Augustine GJ (2004a) Synaptotagmin I synchronizes transmitter release in mouse hippocampal neurons. J Neurosci 24: 6127-6232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishiki T, Augustine GJ (2004b) Dual roles of the C2B domain of synaptotagmin I in synchronizing Ca2+-dependent neurotransmitter release. J Neurosci 24: 8542-8550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberhauser AF, Robinson IM, Fernandez JM (1996) Simultaneous capacitance and amperometric measurements of exocytosis: a comparison. Biophys J 71: 1131-1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons TD, Coorssen JR, Horstmann H, Almers W (1995) Docked granules, the exocytotic burst, and the need for ATP hydrolysis in endocrine cells. Neuron 15: 1085-1096. [DOI] [PubMed] [Google Scholar]
- Perin MS, Fried VA, Mignery GA, Jahn R, Südhof TC (1990) Phospholipid binding by a synaptic vesicle protein homologous to the regulatory region of protein kinase C. Nature 345: 260-263. [DOI] [PubMed] [Google Scholar]
- Rettig J, Neher E (2002) Emerging roles of presynaptic proteins in Ca2+-triggered exocytosis. Science 298: 781-785. [DOI] [PubMed] [Google Scholar]
- Rickman C, Archer DA, Meunier FA, Craxton M, Fukuda M, Burgoyne RD, Davletov B (2004a) Synaptotagmin interaction with the syntaxin/SNAP-25 dimer is mediated by an evolutionarily conserved motif and is sensitive to inositol hexakisphosphate. J Biol Chem 279: 12574-12579. [DOI] [PubMed] [Google Scholar]
- Rickman C, Craxton M, Osborne S, Davletov B (2004b) Comparative analysis of tandem C2 domains from the mammalian synaptotagmin family. Biochem J 378: 681-686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenmund C, Stevens CF (1996) Definition of the readily releasable pool of vesicles at hippocampal synapses. Neuron 16: 1197-1207. [DOI] [PubMed] [Google Scholar]
- Schneggenburger R, Neher E (2000) Intracellular calcium dependence of transmitter release rates at a fast central synapse. Nature 406: 889-893. [DOI] [PubMed] [Google Scholar]
- Seagar M, Leveque C, Charvin N, Marqueze B, Martin-Moutot N, Boudier JA, Boudier JL, Shoji-Kasai Y, Sato K, Takahashi M (1999) Interactions between proteins implicated in exocytosis and voltage-gated calcium channels. Philos Trans R Soc Lond B Biol Sci 354: 289-297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng ZH, Yokoyama CT, Catterall WA (1997) Interaction of the synprint site of N-type Ca2+ channels with the C2B domain of synaptotagmin I. Proc Natl Acad Sci USA 94: 5405-5410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin OH, Rhee JS, Tang J, Sugita S, Rosenmund C, Sudhof TC (2003) Sr2+ binding to the Ca2+ binding site of the synaptotagmin 1 C2B domain triggers fast exocytosis without stimulating SNARE interactions. Neuron 37: 99-108. [DOI] [PubMed] [Google Scholar]
- Smith C, Moser T, Xu T, Neher E (1998) Cytosolic Ca2+ acts by two separate pathways to modulate the supply of release-competent vesicles in chromaffin cells. Neuron 20: 1243-1253. [DOI] [PubMed] [Google Scholar]
- Sørensen JB (2004) Formation, stabilisation and fusion of the readily releasable pool of secretory vesicles. Pflügers Arch 448: 347-362. [DOI] [PubMed] [Google Scholar]
- Sørensen JB, Nagy G, Varoqueaux F, Nehring RB, Brose N, Wilson MC, Neher E (2003a) Differential control of the releasable vesicle pools by SNAP-25 splice variants and SNAP-23. Cell 114: 75-86. [DOI] [PubMed] [Google Scholar]
- Sørensen JB, Fernández-Chacón R, Südhof TC, Neher E (2003b) Examining synaptotagmin 1 function in dense core vesicle exocytosis under direct control of Ca2+ J Gen Physiol 122: 265-276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens CF, Sullivan JM (2003) The synaptotagmin C2A domain is part of the calcium sensor controlling fast synaptic transmission. Neuron 39: 299-308. [DOI] [PubMed] [Google Scholar]
- Südhof TC (2004) The synaptic vesicle cycle. Annu Rev Neurosci 27: 509-547. [DOI] [PubMed] [Google Scholar]
- Sugita S, Han W, Butz S, Liu X, Fernandez-Chacon R, Lao Y, Sudhof TC (2001) Synaptotagmin VII as a plasma membrane Ca2+ sensor in exocytosis. Neuron 30: 459-473. [DOI] [PubMed] [Google Scholar]
- Sugita S, Shin O-H, Han W, Lao Y, Südhof TC (2002) Synaptotagmins form a hierarchy of exocytotic Ca2+ sensors with distinct Ca2+ affinities. EMBO J 21: 270-280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutton RB, Davletov BA, Berghuis AM, Südhof TC, Sprang SR (1995) Structure of the first C2 domain of synaptotagmin I: a novel Ca2+/phospholipid-binding fold. Cell 80: 929-938. [DOI] [PubMed] [Google Scholar]
- Tucker WC, Edwardson JM, Bai J, Kim HJ, Martin TF, Chapman ER (2003) Identification of synaptotagmin effectors via acute inhibition of secretion from cracked PC12 cells. J Cell Biol 162: 199-209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner KM, Burgoyne RD, Morgan A (1999) Protein phosphorylation and the regulation of synaptic membrane traffic. Trends Neurosci 22: 459-464. [DOI] [PubMed] [Google Scholar]
- Ullrich B, Li C, Zhang JZ, McMahon H, Anderwon RGW, Geppert M, Südhof TC (1994) Functional properties of multiple synaptotagmins in brain. Neuron 13: 1281-1291. [DOI] [PubMed] [Google Scholar]
- Verona M, Zanotti S, Schäfer T, Racagni G, Popoli M (2000) Changes of synaptotagmin interaction with t-SNARE proteins in vitro after calcium/calmodulin-dependent phosphorylation. J Neurochem 74: 209-221. [DOI] [PubMed] [Google Scholar]
- Voets T (2000) Dissection of three Ca2+-dependent steps leading to secretion in chromaffin cells from mouse adrenal slices. Neuron 28: 537-545. [DOI] [PubMed] [Google Scholar]
- Voets T, Neher E, Moser T (1999) Mechanisms underlying phasic and sustained secretion in chromaffin cells from mouse adrenal slices. Neuron 23: 607-615. [DOI] [PubMed] [Google Scholar]
- Voets T, Moser T, Lund PE, Chow RH, Geppert M, Südhof TC, Neher E (2001) Intracellular calcium dependence of large dense-core vesicle exocytosis in the absence of synaptotagmin I. Proc Natl Acad Sci USA 98: 11680-11685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu XS, Wu LG (2001) Protein kinase C increases the apparent affinity of the release machinery to Ca2+ by enhancing the release machinery downstream of the Ca2+ sensor. J Neurosci 21: 7928-7936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu T, Binz T, Niemann H, Neher E (1998) Multiple kinetic components of exocytosis distinguished by neurotoxin sensitivity. Nat Neurosci 1: 192-200. [DOI] [PubMed] [Google Scholar]
- Xu T, Rammner B, Margittai M, Artalejo AR, Neher E (1999) Inhibition of SNARE complex assembly differentially affects kinetic components of exocytosis. Cell 99: 713-722. [DOI] [PubMed] [Google Scholar]
- Yang Y, Udayansankar S, Dunning J, Chen P, Gillis KD (2002) A highly Ca2+-sensitive pool of vesicles is regulated by protein kinase C in adrenal chromaffin cells. Proc Natl Acad Sci USA 99: 17060-17065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshihara M, Littleton JT (2002) Synaptotagmin I functions as a calcium sensor to synchronize neurotransmitter release. Neuron 36: 897-908. [DOI] [PubMed] [Google Scholar]
- Yoshihara M, Adolfsen B, Littleton JT (2003) Is synaptotagmin the calcium sensor? Curr Opin Neurobiol 13: 315-323. [DOI] [PubMed] [Google Scholar]
- Zhang X, Kim-Miller MJ, Fukuda M, Kowalchyk JA, Martin TFJ (2002) Ca2+-dependent synaptotagmin binding to SNAP-25 is essential for Ca2+-triggered exocytosis. Neuron 34: 599-611. [DOI] [PubMed] [Google Scholar]
- Zhu H, Hille B, Xu T (2002) Sensitization of regulated exocytosis by protein kinase C. Proc Natl Acad Sci USA 99: 17055-17059. [DOI] [PMC free article] [PubMed] [Google Scholar]