

Abstract
Alzheimer's disease (AD) is characterized by neuronal loss in the hippocampus and entorhinal cortex that is manifested by progressive memory impairment and cognitive decline. Autosomal-dominant, familial forms of AD (FAD) are caused by mutations in genes encoding amyloid precursor protein, presenilin-1 (PS1), and presenilin 2. Although it is established that expression of mutant PS1 variants leads to increased production of highly fibrillogenic amyloidβ42 (Aβ42) peptides that deposit in the brains of patients with AD, the mechanism(s) by which Aβ deposition and expression of mutant genes induce lamina- and region-specific vulnerability of neuronal populations is not known. We have examined the hypothesis that expression of transgene-encoded FAD-linked mutant PS1 variants in entorhinal cortex neurons exacerbates the vulnerability of these cells to lesion-induced neuronal loss. To test this notion, we transected the perforant pathway (PP) of transgenic mice harboring either wild-type human PS1 (PS1HWT) or the FAD-linked mutant PS1ΔE9 variant and examined neuronal survival in layer II of the entorhinal cortex (ECL2). Remarkably, PP transections lead to marked reductions in the numbers of ECL2 neurons in the ECL2 of mice expressing mutant PS1, compared with ECL2 neurons in PP-lesioned PS1HWT mice. Finally, and in contrast to studies in nontransgenic mice and in mice expressing PS1HWT, ECL2 neurons that express mutant PS1 and the calcium binding protein calbindin-D28k in ECL2 are also susceptible to lesion-induced neuronal loss. We conclude that expression of FAD-linked mutant PS1 variants enhances the vulnerability of neurons in the entorhinal cortex to PP lesion-induced cytotoxicity.
Keywords: Alzheimer's disease, neurodegeneration, presenilin, perforant pathway, entorhinal cortex, neuronal death
Introduction
Alzheimer's disease (AD) is characterized by progressive memory loss and cognitive deterioration (for review, see Price et al., 1998b). These behavioral impairments are attributed to neuropathological lesions within specific regions in the brain, including the mediotemporal lobe (e.g., hippocampal formation, entorhinal cortex, and association cortices) (Braak and Braak, 1985, 1996; Hyman and Gomez-Isla, 1994). In the entorhinal cortex (EC), a massive loss of the large clusters of layer II (ECL2) stellate cells and a decrease in superficial parts of layer III and the large multipolar neurons of layer IV takes place in early stages of the disease (Hyman et al., 1986; Gomez-Isla et al., 1996; Braak and Braak, 1997). These cellular alterations are accompanied by amyloid deposits composed of amyloid β (Aβ) peptides and neurofibrillary tangles (Hyman et al., 1984, 1986; Jellinger et al., 1991; Braak and Braak, 1996; Price et al., 1998a).
Familial, early onset forms of AD (FAD) are caused by mutations in genes encoding amyloid precursor proteins (APP), presenilin 1 (PS1), and presenilin 2 (PS2), and expression of FAD-linked mutant APP and PS elevates the production, and subsequent deposition, of highly fibrillogenic Aβ42 peptides (for review, see Selkoe, 2001). Emerging evidence has accrued to suggest that expression of PS variants alters intracellular calcium homeostasis (for review, see Leissring et al., 2000; Yoo et al., 2000; Mattson et al., 2001), which may also be associated with altered glutamate uptake, leading to neurotoxicity (Yang and Cook, 2004; Yang et al., 2004).
Glutamatergic stellate neurons from ECL2 give rise to the main excitatory input to the outer two-thirds of the molecular layer of the fascia dentata, via the perforant pathway (PP) (Steward, 1976; Steward and Scoville, 1976; Steward and Vinsant, 1978; Witter et al., 2000) (for review, see Freund and Buzsaki, 1996), and previous studies have shown that disruption of the PP induces neuronal loss in layer II of the entorhinal cortex (Peterson et al., 1994). It has been proposed that axotomy of glutamatergic ECL2 neurons leads to a transitory increase in local extracellular glutamate concentrations that in turn allows influx of calcium ions that ultimately cause cytotoxicity (Coyle and Puttfarcken, 1993). Notably, ECL2 neurons that express both NMDA receptor 1 and the calcium binding protein calbindin-D28k appear to be protected after perforant pathway lesions (Peterson et al., 1994, 1996), suggesting that these cells can buffer the cytotoxic effects of calcium overload (Peterson et al., 1996).
In this report, we test the hypothesis that expression of transgene-encoded FAD-linked mutant PS1 variants in ECL2 neurons exacerbates the vulnerability of these cells to PP lesion-induced toxicity. Remarkably, transgenic mice harboring the FAD-linked PS1ΔE9 variant subject to PP lesions exhibit dramatic reductions in numbers of calbindin-positive and calbindin-negative neurons in the ECL2, compared with neuron numbers in the ECL2 of PP-lesioned mice harboring wild-type human PS1 (PS1HWT). These results suggest that expression of FAD-linked PS1 enhances the vulnerability of ECL2 neurons to PP lesion-induced toxicity that cannot be suppressed by expression of the calcium buffering protein calbindin-D28k
Materials and Methods
Mice. Fourteen male transgenic mice, six mice harboring FAD-linked human wild-type PS1 and eight mice harboring FAD-linked human PS1ΔE9, were used in this study. Male transgenic mice harboring FAD-linked PS1HWT [line S8-4 (Thinakaran et al., 1996)] or human PS1ΔE9 [line S9 (Lee et al., 1997)] were used at 4–5 months of age. All mice were heterozygous for the transgene. The background strain for these mice is [C3H/HeJ × C57BL/6J F3] × C57BL/6J n1 (Lee et al., 1997).
Perforant pathway transection. Perforant pathway lesion have been described previously (Lazarov et al., 2002). Briefly, mice were placed on a Kopf stereotaxic apparatus (David Kopf Instruments, Tujunga, CA) after anesthesia. An opening in the scalp at coordinates (in mm): anteroposterior, –4.6; mediolateral, –3.3; dorsoventral, –4.5 to –3.5 (from dura), with the nose bar set at 3.5, was performed using electric drill, and the dura was exposed. A wire knife assembly was used for precision perforant pathway lesion. The assembly was set to the ventral extent of the lesion (–4.5 mm). After penetration, the knife was extended to form a hook that extends medially for 2 mm, the assembly was raised to the dorsal extent of the lesion (3.5 mm), and the knife was retracted, reextended, and again lowered to the ventral extent. The knife hook was raised and lowered five times to verify complete transection of the perforant pathway; each retraction and reextension was performed to ensure that any flexible axons stretched by the knife hook were cut. To verify the accuracy of the lesion, crystals of the fluorescent dye Fluoro-Ruby (Invitrogen, San Diego, CA) were dissolved in saline and introduced into the injury site concomitantly with knife penetration.
Histology and immunohistochemistry. Two weeks after perforant pathway transection, mice were perfused transcardially under deep anesthesia with a saline solution, followed by fixative solution composed of 4% paraformaldehyde and 0.1% glutaraldehyde in 0.1 m phosphate buffer. The brains were removed and kept in fixative overnight, followed by equilibration in 30% sucrose solution. Brains were sectioned horizontally (50 μm) using a frozen-stage equipped microtome and then placed at –20°C in cryoprotectant solution (glycerol and ethylene glycol-based PBS). Every sixth tissue section was subject to immunocytochemistry, as described below, and analyzed by confocal microscopy. Free-floating horizontal brain sections (50 μm) were rinsed three times for 10 min each in Tris-buffered saline (TBS), blocked using blocking–permeabilizing solution (5% donkey serum and 0.25% Triton X-100) for 1–3 h, and incubated with mouse anti-neuronal-specific nuclear protein (anti-NeuN; 1:500; mAB377; Chemicon, Temecula, CA), rabbit anti-calbindin-D28k (1:1000; CB38; Swant, Bellinzona, Switzerland) for 72 h at 4°C. Tissues were then rinsed in blocking solution for 2 h. Sections were then incubated with secondary antibodies (1:250; Jackson ImmunoResearch, West Grove, PA) at room temperature for 2 h. Sections were then washed three times for 10 min each in TBS and mounted on gelatin-coated slides using polyvinyl alcohol–1,4-diazabicyclo-[2.2.2]octane (Sigma, St. Louis, MO) mounting solution (Peterson, 2004).
Imaging and design-based stereology. Brain sections were visualized and imaged using an Olympus (Melville, NY) Fluoview confocal laser scanning microscope. Images at each wavelength were collected separately using a separate and specific excitation filter. Image collection settings were approximately equivalent for all specimens and were taken and recorded using a Fluoview 2.1 program. Collection of samples for stereology was performed using the StereoInvestigator software (MicroBrightField, Colchester, VT). For quantitative design-based stereology analysis, Z series of 20 μm depth were collected from every sixth section, with 2 μm intervals between images. ECL2 neurons were sampled for three-dimensional numerical density of neurons by optical dissector (Peterson et al., 1994). ECL2 volume was estimated using the Cavalieri procedure (Peterson and Jones, 1993; Peterson et al., 1994). ECL2 boundaries used were according to a previous tracer study (Peterson et al., 1994).
Results
Perforant pathway lesion induces neuronal loss in ECL2 of transgenic mice
To examine the impact of perforant pathway lesions on neuronal survival in ECL2, we used transgenic mice harboring mouse prion promoter-driven transgenes that express either human wild-type PS1 (n = 3) (Thinakaran et al., 1996) or the FAD-linked ΔE9 variant (n = 3) (Lee et al., 1997). The expression of each transgene occurs in an ubiquitous manner and in all cells of the CNS (Borchelt et al., 1996). More importantly, we have demonstrated that overexpression of either wild-type or mutant human PS1 transgenes leads to “replacement” of endogenous mouse PS1 with human transgene-encoded PS1 polypeptides (Thinakaran et al., 1996; Lee et al., 1997).
To induce neuronal loss in the entorhinal cortex, the perforant path of mice harboring either PS1HWT or the PS1ΔE9 variant were transected using a stereotaxically coordinated knife lesion, as described previously (Lazarov et al., 2002). The identification of surviving cells was determined by confocal immunofluorescence microscopy (Fig. 1). To verify lesion accuracy, we introduced the fluorescent dye Fluoro-Ruby into the lesion site at the time of the lesion (for details, see Materials and Methods; for image, see Fig. 1). Mice were allowed to survive for 2 weeks. Animals demonstrating retrograde labeling of ECL2 neurons were determined to have an appropriately placed lesion and were included for subsequent analysis. To quantify the number of neurons in layer II of the entorhinal cortex, serial brain sections immunolabeled for NeuN and calbindin-D28k were analyzed and traced by confocal microscopy (for details, see Materials and Methods; for image, see Fig. 1). Neuronal survival in ECL2 was evaluated using unbiased stereology-based analysis.
Figure 1.

Design-based stereological analysis of neuronal survival in the entorhinal cortex of adult transgenic mice. Representative low-power confocal images of horizontal brain sections of sham and perforant-pathway-lesioned PS1HWT and PS1ΔE9 mice used for stereological analysis of the number of neurons in ECL2. The figure is composed of four panels: top left, brain section of a sham mouse harboring FAD-linked PS1HWT; top right, brain section of a mouse harboring FAD-linked PS1HWT 2 weeks after PP lesion; bottom left, brain section of a sham mouse harboring FAD-linked PS1ΔE9; bottom right, brain section of a mouse harboring FAD-linked PS1ΔE9 2 weeks after PP lesion. Each brain section is immunostained with antibodies raised against NeuN (left top image in each panel) and calbindin-D28k (right top image in each panel) using multiple fluorophore-conjugated secondary antibodies. Fluoro-Ruby [cyanine 3 (Cy3) fluorophore; left bottom image in each panel] can be detected in the lesioned mice (top right, PS1HWT; bottom right, PS1ΔE9). The number of calbindin-expressing neurons was quantified using combined image stacks of NeuN and calbindin (bottom right image in each panel).
Fewer neurons in ECL2 of PS1ΔE9 transgenic mice
To establish that neuronal loss in the ECL2 was the result of the PP lesion and was independent of nonspecific trauma, eight mice (three mice harboring FAD-linked PS1HWT and five mice harboring FAD-linked PS1ΔE9) were anesthetized and subjected to the same surgical procedure as the lesioned mice but without knife penetration and perturbation of the perforant pathway. NeuN+ neurons in the ECL2 of mice harboring either PS1HWT or PS1ΔE9 of sham-operated animals were evaluated after 2 weeks (Figs. 1, 2) and quantified (Fig. 3). Surprisingly, this analysis revealed that the number of NeuN+ neurons in ECL2 in PS1ΔE9 transgenic mice was only 78% of the number of neurons in the ECL2 of PS1HWT mice (Fig. 2, compare NeuN images of sham PS1HWT and sham PS1ΔE9; for quantification, see Fig. 3A). We asked whether the numbers of calbindin-D28k-positive cells were altered in the ECL2 of sham PS1ΔE9, compared with sham-operated PS1HWT mice. Here again, we observed that calbindin-D28k-positive cells in ECL2 of PS1ΔE9 mice were 73% of the cell numbers in PS1HWT ECL2 (Fig. 2, compare calbindin images of sham PS1HWT and sham PS1ΔE9; for quantification, see Fig. 3B). To examine whether the calbindin-D28k-expressing cells were neuronal, we quantified the number of cells that coexpressed NeuN and calbindin-D28k in the ECL2 of sham-operated mice. The analysis revealed that only 71% of ECL2 cells coexpress calbindin-D28k and NeuN in PS1ΔE9 compared with PS1HWT mice (Fig. 2, compare NeuN and calbindin images of sham PS1HWT and sham PS1ΔE9; for quantification, see Fig. 3C). However, differences between sham PS1HWT and sham PS1ΔE9 were not statistically significant (ANOVA; NeuN, p = 0.1128; calbindin-D28k, p = 0.2474; NeuN+ calbindin-D28k+, p = 0.2692).
Figure 2.
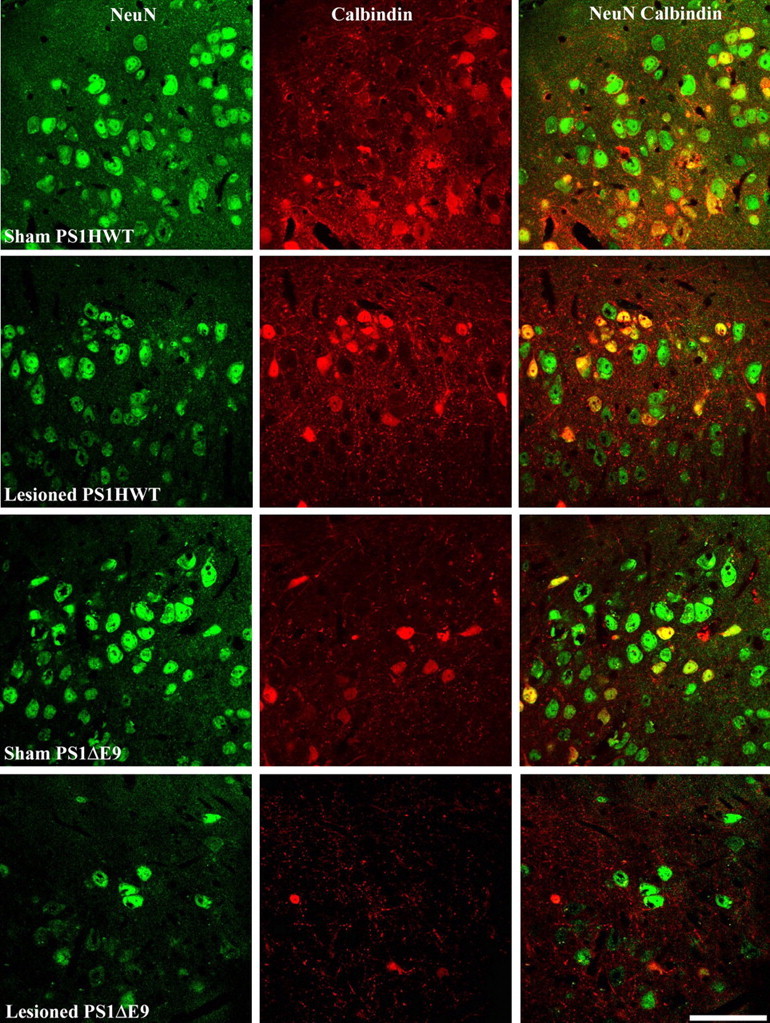
Neuronal survival in ECL2 of sham and lesioned transgenic mice harboring FAD-linked PS1HWT or PS1ΔE9 after transection of the perforant pathway. High-power confocal images of ECL2 neurons as detected in brain sections of sham and lesioned PS1HWT and PS1ΔE9 mice. Top to bottom: sham PS1HWT, lesioned PS1HWT, sham PS1ΔE9, lesioned PS1ΔE9. For each animal, images show NeuN+ (left image column), calbindin-D28k+ (middle image column), and NeuN+ calbindin-D28k+ neurons (right image column) of ECL2. Note the reduced number of neurons in ECL2 of PS1ΔE9 transgenic mice 2 weeks after perforant pathway lesion. Scale bar, 60 μm.
Figure 3.

Reduced survival rate of neurons in ECL2 of PS1ΔE9 transgenic mice 2 weeks after perforant pathway lesion. The number of NeuN+ (A), calbindin-D28k+ (B), and NeuN+ calbindin-D28k+ (C) cells in ECL2 of sham and lesioned PS1HWT and PS1ΔE9 transgenic mice was quantified by unbiased stereology. The number of NeuN+ cells as well as calbindin-D28k + cells was dramatically reduced in ECL2 of PS1ΔE9 compared with ECL2 of PS1HWT 2 weeks after lesion.
Enhanced neuronal loss in ECL2 of PS1ΔE9 transgenic mice after perforant pathway lesion
As expected, we observed neuronal loss in the ECL2 of PS1HWT mice, as well as in PS1ΔE9 transgenic mice 2 weeks after PP lesion (Figs. 2, 3). Transection of the perforant pathway in PS1HWT mice produced a modest reduction in the number of ECL2 neurons (Fig. 2, compare NeuN images of sham PS1HWT and lesioned PS1HWT); quantification revealed that neuronal loss was <10% (Fig. 3A). However, PP transection in PS1ΔE9 mice produced a marked reduction in the number of neurons in ECL2 with 64% survival relative to sham PS1ΔE9 mice (Fig. 2, compare NeuN images of sham and lesioned PS1ΔE9; for quantification, see Fig. 3A). Extent of neuronal survival in the lesioned PS1ΔE9 mice was found to be significantly different from all sham and lesioned groups (ANOVA; p = 0.003 for sham PS1HWT; p = 0.0069 for lesioned PS1HWT; p = 0.034 for sham PS1ΔE9).
Perforant pathway lesions reduce the numbers of calbindin-D28k-expressing neurons in ECL2 of PS1ΔE9 transgenic mice
To examine whether expression of calbindin-D28k might afford protection from PP lesion-induced neuronal death, we quantified the number of calbindin-D28k-expressing cells in sham and PP-lesioned PS1HWT mice. We determined that 96% of calbindin-D28k-expressing cells that survived the lesion in PS1HWT mice are neuronal by virtue of being NeuN positive (Fig. 2, compare calbindin and NeuN calbindin images of sham and lesioned PS1HWT; for quantification, see Fig. 3B,C). Our quantitative analysis revealed that 96.5% of calbindin-D28k-expressing cells survived in PP-lesioned PS1HWT animals, suggesting that any observable neuronal loss in ECL2 cells after the lesion was attributable to cells that did not express calbindin-D28k. In contrast to the data obtained in PS1HWT animals, we observed that of the calbindin-D28k-expressing cells detected in sham PS1ΔE9 mice, only 72% of these survived the PP lesion (Fig. 2, compare albindin images of sham and lesioned PS1ΔE9; for quantification, see Fig. 3B). This analysis further revealed that only 64% of the surviving cells were calbindin-D28k+ NeuN+ cells in the ECL2 of PP-lesioned PS1ΔE9 mice (Fig. 2, compare NeuN calbindin images of sham and lesioned PS1ΔE9; for quantification, see Fig. 3C) (t test, one-tailed; p = 0.033). These results suggest that expression of calbindin-D28k cannot suppress the cell death programs induced in mutant PS1-expressing ECL2 neurons after PP lesions.
Discussion
AD-associated neuropathology is manifested by disruption of neuronal circuits, intracellular lesions, extracellular lesions (i.e., neurofibrillary tangles and amyloid deposits, respectively), and massive neuronal loss (for review, see Buckner, 2004; Walsh and Selkoe, 2004). Neurons in ECL2 gives rise to the PP, the major source of the excitatory input to the hippocampus. In mild cases of AD, 60% of the neurons in ECL2 are lost, whereas 90% loss occurs in advanced stages of the disease (Gomez-Isla et al., 1996). The mechanism(s) that underlie this massive region-specific cell loss is primarily unknown.
In the present manuscript, we offer several novel insights that impact on our understanding of the role of PS1 and FAD-linked variants on neuronal survival in ECL2. First, we demonstrate that expression of FAD-linked mutant PS1ΔE9 reduces neuronal cell numbers in ECL2 compared with mice expressing wild-type PS1. These findings suggest that expression of the PS1ΔE9 mutant altered developmental cell fate programs that regulate neuronal and non-neuronal cell numbers in ECL2, or alternatively, accelerates neuronal cell loss in the ECL2 during aging.
Second, in mice expressing PS1HWT, we only observed 9% neuronal loss in ECL2 after the PP lesion. This result is somewhat different from the previous demonstration in rat, wherein 30% of ECL2 neurons die at an equivalent time point after partial aspiration lesions of the EC (Peterson et al., 1994). That might result from differences in the lesion approach, which may affect the extent of both primary and secondary damage that each one of the lesions induce. In addition, differential vulnerability of ECL2 neurons to PP lesions might be a species-specific effect or alternatively, might suggest that expression of human PS1 alters the survival of ECL2 neurons. In this regard, recent evidence has suggested a neuroprotective role for human wild-type PS1 (Zhou et al., 2002) and in cell survival pathways (for review, see Niikura et al., 2002).
Finally, and most importantly, we provide unequivocal evidence that expression of the PS1ΔE9 variant enhances the vulnerability of ECL2 neurons to PP lesion-induced neuronal loss. Importantly, neurons that express the calcium binding protein calbindin-D28k are not protected from PP-induced cell death, in sharp contrast to previous studies showing that these neuronal populations are spared in rat ECL2 (Peterson et al., 1994) or mouse ECL2 expressing human wild-type PS1 (this study). Although it is arguable that expression of endogenous mouse PS1 may influence the results we have obtained, it is important to note that endogenous mouse PS1 fails to accumulate in the brains of transgenic mice that express either PS1HWT or PS1ΔE9 (Thinakaran et al., 1996; Lee et al., 1997). The explanation of this protein replacement phenomenon is that overexpression of the human PS1 polypeptides saturates one or more cellular factors that are expressed at limiting levels (Thinakaran et al., 1996, 1997). We now recognize that these saturable factors are nicastrin, APH-1 (anterior pharynx defective), and PEN-2 (presenilin enhancer) (De Strooper, 2003). Hence, it is reasonable to assume that the phenotypes we describe in this study are attributable entirely to the expression of the human transgene-encoded polypeptides in ECL2, without any influence of endogenous mouse PS1.
The mechanism(s) by which FAD-linked mutant PS1 variants enhance neuronal vulnerability is not fully understood, but numerous studies have provided evidence suggesting that expression of mutant PS1 enhances the vulnerability of cultured neurons to toxic insults (Guo et al., 1999a,b; Weihl et al., 1999; Popescu et al., 2001; Terro et al., 2002) and accelerates kainite-induced degeneration of hippocampal neurons in CA1 and CA3 (Guo et al., 1999a). Expression of mutant PS1 variants also perturbs calcium homeostasis (for review, see Mattson and Chan, 2003) (Guo et al., 1996, 1997, 1999a; Leissring et al., 2000; Yoo et al., 2000; Terro et al., 2002).
The concentrations of free cytoplasmic Ca2+ are tightly regulated in neurons and essential for modulating cellular processes, including signal transduction cascades, induction of gene transcription, and neuronal cell death (Ghosh and Greenberg, 1995). The calcium-binding protein calbindin-D28k buffers intracellular calcium (for review, see Baimbridge et al., 1992) and is expressed in the EC in a lamina-specific manner, almost exclusively in ECL2 (Peterson et al., 1994, 1996). It has been suggested that calbindin-D28k plays a critical role in rescuing rat ECL2 neurons from PP-induced cell death (Peterson et al., 1994, 1996) and to block mutant PS1-induced proapoptotic processes (Guo et al., 1998). Indeed, we have demonstrated that the vast majority of calbindin-D28k-expressing ECL2 neurons are spared in PS1HWT transgenic mice after PP lesions (this study). The failure of calbindin-D28k to protect FAD-linked PS1 mutant-expressing ECL2 neurons from PP lesion offer several plausible hypotheses. In view of evidence that expression of mutant PS1 perturbs intracellular calcium homeostasis, it is not inconceivable that excess calcium influx resulting from the perforant path lesion might alter calcium-induced calcium release from intracellular stores to levels that far exceed the buffering capacity of calbindin-D28k, thus overwhelming those calcium-dependent signaling pathways that would normally promote cell survival.
Alternatively, mutant PS1 could enhance PP lesion-induced neuronal loss in ECL2 by pathways that do not involve perturbations of calcium homeostasis. In this regard, it is now well established that expression of FAD-linked mutant PS1 elevates the production of Aβ42 peptides (Price and Sisodia, 1998), and it is conceivable that increased Aβ42 levels and fibrillar intermediates (Hartley et al., 1999; Chromy et al., 2003; Kayed et al., 2003) that compromise neuronal function might enhance the vulnerability of ECL2 neurons to a variety of insults. Future studies to examine the potential role of Aβ42 peptides in this paradigm, by analysis of neuronal vulnerability in the ECL2 of mice that harbor mutant PS1 transgenes on an APP-deficient background, are clearly warranted.
Footnotes
This work was supported by National Institutes of Health Grants AG021494 (S.S.S.) and AG020047 (D.A.P.).
Correspondence should be addressed to Sangram S. Sisodia, Department of Neurobiology, Pharmacology and Physiology, University of Chicago, 947 East 58th Street, Chicago, IL 60637. E-mail: ssisodia@bsd.uchicago.edu.
DOI:10.1523/JNEUROSCI.3961-05.2006
Copyright © 2006 Society for Neuroscience 0270-6474/06/260429-06$15.00/0
References
- Baimbridge KG, Celio MR, Rogers JH (1992) Calcium-binding proteins in the nervous system. Trends Neurosci 15: 303–308. [DOI] [PubMed] [Google Scholar]
- Borchelt DR, Davis J, Fischer M, Lee MK, Slunt HH, Ratovitsky T, Regard J, Copeland NG, Jenkins NA, Sisodia SS, Price DL (1996) A vector for expressing foreign genes in the brains and hearts of transgenic mice. Genet Anal 13: 159–163. [DOI] [PubMed] [Google Scholar]
- Braak H, Braak E (1985) On areas of transition between entorhinal allocortex and temporal isocortex in the human brain. Normal morphology and lamina-specific pathology in Alzheimer's disease. Acta Neuropathol (Berl) 68: 325–332. [DOI] [PubMed] [Google Scholar]
- Braak H, Braak E (1996) Evolution of the neuropathology of Alzheimer's disease. Acta Neurol Scand Suppl 165: 3–12. [DOI] [PubMed] [Google Scholar]
- Braak H, Braak E (1997) Frequency of stages of Alzheimer-related lesions in different age categories. Neurobiol Aging 18: 351–357. [DOI] [PubMed] [Google Scholar]
- Buckner RL (2004) Memory and executive function in aging and AD: multiple factors that cause decline and reserve factors that compensate. Neuron 44: 195–208. [DOI] [PubMed] [Google Scholar]
- Chromy BA, Nowak RJ, Lambert MP, Viola KL, Chang L, Velasco PT, Jones BW, Fernandez SJ, Lacor PN, Horowitz P, Finch CE, Krafft GA, Klein WL (2003) Self-assembly of Abeta(1-42) into globular neurotoxins. Biochemistry 42: 12749–12760. [DOI] [PubMed] [Google Scholar]
- Coyle JT, Puttfarcken P (1993) Oxidative stress, glutamate, and neurodegenerative disorders. Science 262: 689–695. [DOI] [PubMed] [Google Scholar]
- De Strooper B (2003) Aph-1, Pen-2, and Nicastrin with Presenilin generate an active gamma-Secretase complex. Neuron 38: 9–12. [DOI] [PubMed] [Google Scholar]
- Freund TF, Buzsaki G (1996) Interneurons of the hippocampus. Hippocampus 6: 347–470. [DOI] [PubMed] [Google Scholar]
- Ghosh A, Greenberg ME (1995) Calcium signaling in neurons: molecular mechanisms and cellular consequences. Science 268: 239–247. [DOI] [PubMed] [Google Scholar]
- Gomez-Isla T, Price JL, McKeel Jr DW, Morris JC, Growdon JH, Hyman BT (1996) Profound loss of layer II entorhinal cortex neurons occurs in very mild Alzheimer's disease. J Neurosci 16: 4491–4500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Q, Furukawa K, Sopher BL, Pham DG, Xie J, Robinson N, Martin GM, Mattson MP (1996) Alzheimer's PS-1 mutation perturbs calcium homeostasis and sensitizes PC12 cells to death induced by amyloid beta-peptide. NeuroReport 8: 379–383. [DOI] [PubMed] [Google Scholar]
- Guo Q, Sopher BL, Furukawa K, Pham DG, Robinson N, Martin GM, Mattson MP (1997) Alzheimer's presenilin mutation sensitizes neural cells to apoptosis induced by trophic factor withdrawal and amyloid β-peptide: involvement of calcium and oxyradicals. J Neurosci 17: 4212–4222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Q, Christakos S, Robinson N, Mattson MP (1998) Calbindin D28k blocks the proapoptotic actions of mutant presenilin 1: reduced oxidative stress and preserved mitochondrial function. Proc Natl Acad Sci USA 95: 3227–3232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Q, Fu W, Sopher BL, Miller MW, Ware CB, Martin GM, Mattson MP (1999a) Increased vulnerability of hippocampal neurons to excitotoxic necrosis in presenilin-1 mutant knock-in mice. Nat Med 5: 101–106. [DOI] [PubMed] [Google Scholar]
- Guo Q, Fu W, Holtsberg FW, Steiner SM, Mattson MP (1999b) Superoxide mediates the cell-death-enhancing action of presenilin-1 mutations. J Neurosci Res 56: 457–470. [DOI] [PubMed] [Google Scholar]
- Hartley DM, Walsh DM, Ye CP, Diehl T, Vasquez S, Vassilev PM, Teplow DB, Selkoe DJ (1999) Protofibrillar intermediates of amyloid β-protein induce acute electrophysiological changes and progressive neurotoxicity in cortical neurons. J Neurosci 19: 8876–8884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyman BT, Gomez-Isla T (1994) Alzheimer's disease is a laminar, regional, and neural system specific disease, not a global brain disease. Neurobiol Aging 15: 353–354; discussion 379–380. [DOI] [PubMed] [Google Scholar]
- Hyman BT, Van Horsen GW, Damasio AR, Barnes CL (1984) Alzheimer's disease: cell-specific pathology isolates the hippocampal formation. Science 225: 1168–1170. [DOI] [PubMed] [Google Scholar]
- Hyman BT, Van Hoesen GW, Kromer LJ, Damasio AR (1986) Perforant pathway changes and the memory impairment of Alzheimer's disease. Ann Neurol 20: 472–481. [DOI] [PubMed] [Google Scholar]
- Jellinger K, Braak H, Braak E, Fischer P (1991) Alzheimer lesions in the entorhinal region and isocortex in Parkinson's and Alzheimer's diseases. Ann NY Acad Sci 640: 203–209. [DOI] [PubMed] [Google Scholar]
- Kayed R, Head E, Thompson JL, McIntire TM, Milton SC, Cotman CW, Glabe CG (2003) Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science 300: 486–489. [DOI] [PubMed] [Google Scholar]
- Lazarov O, Lee M, Peterson DA, Sisodia SS (2002) Evidence that synaptically released β-amyloid accumulates as extracellular deposits in the hippocampus of transgenic mice. J Neurosci 22: 9785–9793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MK, Borchelt DR, Kim G, Thinakaran G, Slunt HH, Ratovitski T, Martin LJ, Kittur A, Gandy S, Levey AI, Jenkins N, Copeland N, Price DL, Sisodia SS (1997) Hyperaccumulation of FAD-linked presenilin 1 variants in vivo [see comments]. Nat Med 3: 756–760. [DOI] [PubMed] [Google Scholar]
- Leissring MA, Akbari Y, Fanger CM, Cahalan MD, Mattson MP, LaFerla FM (2000) Capacitative calcium entry deficits and elevated luminal calcium content in mutant presenilin-1 knockin mice. J Cell Biol 149: 793–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson MP, Chan SL (2003) Neuronal and glial calcium signaling in Alzheimer's disease. Cell Calcium 34: 385–397. [DOI] [PubMed] [Google Scholar]
- Mattson MP, Chan SL, Camandola S (2001) Presenilin mutations and calcium signaling defects in the nervous and immune systems. BioEssays 23: 733–744. [DOI] [PubMed] [Google Scholar]
- Niikura T, Hashimoto Y, Tajima H, Nishimoto I (2002) Death and survival of neuronal cells exposed to Alzheimer's insults. J Neurosci Res 70: 380–391. [DOI] [PubMed] [Google Scholar]
- Peterson DA (2004) The use of fluorescent probes in cell counting procedures. In: Quantitative methods in neuroscience (Evans SM, Janson AM, Nyengaard JR, eds), pp 85–114. Oxford: Oxford UP.
- Peterson DA, Jones DG (1993) Determination of neuronal number and process surface area in organotypic cultures: a stereological approach. J Neurosci Methods 46: 107–120. [DOI] [PubMed] [Google Scholar]
- Peterson DA, Lucidi-Phillipi CA, Eagle KL, Gage FH (1994) Perforant path damage results in progressive neuronal death and somal atrophy in layer II of entorhinal cortex and functional impairment with increasing post-damage age. J Neurosci 14: 6872–6885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson DA, Lucidi-Phillipi CA, Murphy DP, Ray J, Gage FH (1996) Fibroblast growth factor-2 protects entorhinal layer II glutamatergic neurons from axotomy-induced death. J Neurosci 16: 886–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popescu BO, Cedazo-Minguez A, Popescu LM, Winblad B, Cowburn RF, Ankarcrona M (2001) Caspase cleavage of exon 9 deleted presenilin-1 is an early event in apoptosis induced by calcium ionophore A 23187 in SH-SY5Y neuroblastoma cells. J Neurosci Res 66: 122–134. [DOI] [PubMed] [Google Scholar]
- Price DL, Sisodia SS (1998) Mutant genes in familial Alzheimer's disease and transgenic models. Annu Rev Neurosci 21: 479–505. [DOI] [PubMed] [Google Scholar]
- Price DL, Sisodia SS, Borchelt DR (1998a) Genetic neurodegenerative diseases: the human illness and transgenic models. Science 282: 1079–1083. [DOI] [PubMed] [Google Scholar]
- Price DL, Tanzi RE, Borchelt DR, Sisodia SS (1998b) Alzheimer's disease: genetic studies and transgenic models. Annu Rev Genet 32: 461–493. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ (2001) Alzheimer's disease: genes, proteins, and therapy. Physiol Rev 81: 741–766. [DOI] [PubMed] [Google Scholar]
- Steward O (1976) Reinnervation of dentate gyrus by homologous afferents following entorhinal cortical lesions in adult rats. Science 194: 426–428. [DOI] [PubMed] [Google Scholar]
- Steward O, Scoville SA (1976) Cells of origin of entorhinal cortical afferents to the hippocampus and fascia dentata of the rat. J Comp Neurol 169: 347–370. [DOI] [PubMed] [Google Scholar]
- Steward O, Vinsant SL (1978) Identification of the cells of origin of a central pathway which sprouts following lesions in mature rats. Brain Res 147: 223–243. [DOI] [PubMed] [Google Scholar]
- Terro F, Czech C, Esclaire F, Elyaman W, Yardin C, Baclet MC, Touchet N, Tremp G, Pradier L, Hugon J (2002) Neurons overexpressing mutant presenilin-1 are more sensitive to apoptosis induced by endoplasmic reticulum-Golgi stress. J Neurosci Res 69: 530–539. [DOI] [PubMed] [Google Scholar]
- Thinakaran G, Borchelt DR, Lee MK, Slunt HH, Spitzer L, Kim G, Ratovitsky T, Davenport F, Nordstedt C, Seeger M, Hardy J, Levey AI, Gandy SE, Jenkins NA, Copeland NG, Price DL, Sisodia SS (1996) Endoproteolysis of presenilin 1 and accumulation of processed derivatives in vivo. Neuron 17: 181–190. [DOI] [PubMed] [Google Scholar]
- Thinakaran G, Harris CL, Ratovitski T, Davenport F, Slunt HH, Price DL, Borchelt DR, Sisodia SS (1997) Evidence that levels of presenilins (PS1 and PS2) are coordinately regulated by competition for limiting cellular factors. J Biol Chem 272: 28415–28422. [DOI] [PubMed] [Google Scholar]
- Walsh DM, Selkoe DJ (2004) Deciphering the molecular basis of memory failure in Alzheimer's disease. Neuron 44: 181–193. [DOI] [PubMed] [Google Scholar]
- Weihl CC, Ghadge GD, Kennedy SG, Hay N, Miller RJ, Roos RP (1999) Mutant presenilin-1 induces apoptosis and downregulates Akt/PKB. J Neurosci 19: 5360–5369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witter MP, Naber PA, van Haeften T, Machielsen WC, Rombouts SA, Barkhof F, Scheltens P, Lopes da Silva FH (2000) Cortico-hippocampal communication by way of parallel parahippocampal-subicular pathways. Hippocampus 10: 398–410. [DOI] [PubMed] [Google Scholar]
- Yang Y, Cook DG (2004) Presenilin-1 deficiency impairs glutamate-evoked intracellular calcium responses in neurons. Neuroscience 124: 501–505. [DOI] [PubMed] [Google Scholar]
- Yang Y, Kinney GA, Spain WJ, Breitner JC, Cook DG (2004) Presenilin-1 and intracellular calcium stores regulate neuronal glutamate uptake. J Neurochem 88: 1361–1372. [DOI] [PubMed] [Google Scholar]
- Yoo AS, Cheng I, Chung S, Grenfell TZ, Lee H, Pack-Chung E, Handler M, Shen J, Xia W, Tesco G, Saunders AJ, Ding K, Frosch MP, Tanzi RE, Kim T-W (2000) Presenilin-mediated modulation of capacitative calcium entry. Neuron 27: 561–572. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Zhang W, Easton R, Ray JW, Lampe P, Jiang Z, Brunkan AL, Goate A, Johnson EM, Wu JY (2002) Presenilin-1 protects against neuronal apoptosis caused by its interacting protein PAG. Neurobiol Dis 9: 126–138. [DOI] [PubMed] [Google Scholar]