

Abstract
In Alzheimer's disease (AD), there is a significant loss of locus ceruleus (LC) noradrenergic neurons. However, functional and anatomical evidence indicates that the remaining noradrenergic neurons may be compensating for the loss. Because the noradrenergic system plays an important role in learning and memory, it is important to determine whether compensation occurs in noradrenergic neurons in the LC and hippocampus of subjects with AD or a related dementing disorder, dementia with Lewy bodies (DLB). We observed profound neuronal loss in the LC in AD and DLB subjects with three major changes in the noradrenergic system consistent with compensation: (1) an increase in tyrosine hydroxylase (TH) mRNA expression in the remaining neurons; (2) sprouting of dendrites into peri-LC dendritic zone, as determined by α2-adrenoreceptors (ARs) and norepinephrine transporter binding sites; and (3) sprouting of axonal projections to the hippocampus as determined by α2-ARs. In AD and DLB subjects, the postsynaptic α1-ARs were normal to elevated. Expression of α1A- and α2A-AR mRNA in the hippocampus of AD and DLB subjects were not altered, but expression of α1D- and α2C-AR mRNA was significantly reduced in the hippocampus of AD and DLB subjects. Therefore, in AD and DLB subjects, there is compensation occurring in the remaining noradrenergic neurons, but there does appear to be a loss of specific AR in the hippocampus. Because changes in these noradrenergic markers in AD versus DLB subjects were similar (except neuronal loss and the increase in TH mRNA were somewhat greater in DLB subjects), the presence of Lewy bodies in addition to plaques and tangles in DLB subjects does not appear to further affect the noradrenergic compensatory changes.
Keywords: norepinephrine, Alzheimer's disease, in situ hybridization, adrenoreceptors, locus ceruleus, hippocampus
Introduction
In Alzheimer's disease (AD), a substantial loss of noradrenergic cell bodies has been documented in postmortem brain tissue in the locus ceruleus (LC), the major source of noradrenergic projections to the whole brain (Mann et al., 1980; Tomlinson et al., 1981; Bondareff et al., 1982; Marcyniuk et al., 1986; Chan-Palay and Asan, 1989; German et al., 1992). However, studies in patients with AD and several postmortem brain tissue studies suggest that the surviving noradrenergic neurons may partially compensate for LC neuronal loss in AD. CSF concentrations of norepinephrine (NE) and its metabolites do not differ in the early stages of AD from those of healthy older individuals and may increase as AD progresses (Mann et al., 1981; Gottfries et al., 1983; Raskind et al., 1984; Tohgi et al., 1992; Elrod et al., 1997).
The content of NE in terminal regions such as the neocortex and hippocampus in postmortem AD subjects is reduced, but the reduction does not correspond to the degree of neuronal loss in the LC (Adolfsson et al., 1979; Mann et al., 1981; Tomlinson et al., 1981; Palmer et al., 1987; Reinikainen et al., 1988; Hoogendijk et al., 1999). A similar result was observed when the synthesizing enzymes for NE were measured in forebrain regions (Cross et al., 1981; Perry et al., 1981; Palmer et al., 1987; Russo-Neustadt et al., 1998). Recently, our laboratory demonstrated a significant increase in the mRNA expression for the rate limiting enzyme tyrosine hydroxylase (TH) in the LC of AD subjects (Szot et al., 2000), suggesting that the remaining neurons in the LC are compensating for the loss of noradrenergic neurons of AD subjects.
Because the noradrenergic system has been shown to play a major role in learning and memory (Gibbs and Summers, 2002), it is important to determine whether compensatory changes are occurring in the noradrenergic nervous system in AD subjects. In addition, we have evaluated a related dementing disorder, dementia with Lewy bodies (DLB). The approach was twofold: (1) to examine the noradrenergic nervous system in the LC and then (2) to examine the noradrenergic system in the hippocampus, another region critical for learning and memory (Morris et al., 2003). In the LC, we measured the expression of TH mRNA and binding sites to the α2-adrenoreceptor (AR) autoreceptor and NE transporter (NET) that are localized to the dendrites of these neurons. In the hippocampus, we measured the α1- and α2-AR binding sites, as well as the mRNA for each of the subtypes for each of the α1- and α2-AR receptors in the LC (α2A-AR) and hippocampus (α1A-, α1D, α2A-, and α2C-AR). The results of these studies indicate that the remaining noradrenergic neurons in AD and DLB subjects show changes in the LC and at terminal projection regions that are consistent with compensation for neuronal loss.
Materials and Methods
Subjects. All postmortem tissue was obtained from the Alzheimer's Disease Research Center, where permission for use of tissue in scientific experiments was obtained. AD is characterized by the insidious onset and gradual progression of impaired memory, language, and executive function. Psychosis, agitation, and other behavioral disturbances characteristically appear late in the disease course. DLB, which accounts for ∼20% of patients with late-life dementia, presents early in its course with psychotic symptoms such as visual hallucinations and with fluctuating cognition and pronounced attentional deficits and often with bradykinesia and increased muscle tone (McKeith et al., 1996; Ballard et al., 1999; Barber et al., 2001). The AD subjects used in these studies met the National Institute on Aging Reagan criteria for AD (Braak stage IV/C or higher with no vascular dementia, frontotemporal dementia, or Lewy body pathology) (McKhann et al., 1984) (Table 1). DLB subjects used in these studies met the same neuropathological diagnostic criteria (Table 1) for AD plus had the presence of Lewy body pathology in the brainstem and limbic regions as confirmed by α-synuclein immunohistochemistry. Medical records of control subjects indicated no clinical, neurological, or psychiatric illness or evidence of cognitive or functional decline and had no obvious neuropathology during autopsy. Subjects who had been diagnosed with depression before death or had taken any antidepressant medication were excluded because depression and antidepressant therapy can result in changes in brain noradrenergic system (Lacroix et al., 1991; Bauer and Tejani-Butt, 1992; Harro and Oreland, 2001; Ordway et al., 2003). Subjects with a history of alcohol or drug abuse were also excluded. Table 1 contains pathology information as well as age, postmortem delay (PMD), and sex for each of the subjects. Hippocampus was studied in the following: 17 nondemented age-comparable control subjects, with an age range of 38–90 years (mean ± SEM, 71.4 ± 3.5 years), seven males and 10 females with an average PMD of 8.5 ± 0.9 h; 15 AD subjects with an age range of 37–94 years (mean ± SEM, 68.6 ± 4.4 years), six males and nine females with an average PMD of 7.4 ± 0.9 h; and 22 DLB subjects with an age range of 63–98 years (mean ± SEM, 79.5 ± 1.6 years), 16 males and six females with an average PMD of 8.0 ± 0.7 h. Because the LC was not always obtained at the time of autopsy, the number of subjects for control and DLB groups was 15, and they are indicated in Table 1.
Table 1.
Specifics on neuropathology, age, PMD, sex, and whether LC was available for each subject
|
Subject |
Braak |
PMD (h) |
Age (years) |
Sex |
LC |
|---|---|---|---|---|---|
| Control | NA | 13 | 38 | F | No |
| Control | I-A | 16 | 52 | F | Yes |
| Control | I-O | 10 | 55 | M | Yes |
| Control | I-A | 10 | 55 | M | Yes |
| Control | II-B | 15 | 60 | F | Yes |
| Control | O-A | 12 | 69 | M | Yes |
| Control | ?-B | 6 | 70 | F | Yes |
| Control | I-B | 11 | 73 | M | Yes |
| Control | NA | 8 | 77 | F | Yes |
| Control | I-O | 4 | 78 | M | Yes |
| Control | II-B | 5 | 78 | M | No |
| Control | I-O | 9 | 80 | F | Yes |
| Control | II-B | 5 | 84 | F | Yes |
| Control | II-B | 6 | 84 | M | Yes |
| Control | II-O | 5 | 85 | F | Yes |
| Control | III-C | 5 | 85 | F | Yes |
| Control | II-C | 5 | 90 | F | Yes |
| AD | V-C | 7 | 37 | M | Yes |
| AD | V-C | 16 | 44 | F | Yes |
| AD | V-C | 6 | 45 | F | Yes |
| AD | VI-C | 4 | 55 | M | Yes |
| AD | V-C | 6 | 61 | F | Yes |
| AD | NA | 14 | 66 | M | Yes |
| AD | VI-C | 6 | 70 | F | Yes |
| AD | VI-C | 4 | 74 | M | Yes |
| AD | V-C | 4 | 74 | F | Yes |
| AD | VI-C | 4 | 75 | F | Yes |
| AD | V-C | 10 | 80 | F | Yes |
| AD | VI-C | 10 | 81 | M | Yes |
| AD | V-C | 7 | 83 | M | Yes |
| AD | V-C | 7 | 90 | F | Yes |
| AD | IV-C | 6 | 94 | F | Yes |
| DLB | V-C | 8 | 63 | M | Yes |
| DLB | V-C | 5 | 67 | M | Yes |
| DLB | VI-C | 12 | 68 | M | Yes |
| DLB | IV-C | 17 | 70 | M | Yes |
| DLB | V-C | 5 | 75 | M | Yes |
| DLB | V-C | 15 | 75 | M | Yes |
| DLB | V-C | 6 | 77 | F | Yes |
| DLB | V-C | 7 | 78 | M | Yes |
| DLB | IV-C | 7 | 79 | M | Yes |
| DLB | V-C | 9 | 79 | M | Yes |
| DLB | V-C | 10 | 79 | M | Yes |
| DLB | V-C | 5 | 80 | M | No |
| DLB | VI-C | 7 | 81 | F | Yes |
| DLB | V-C | 9 | 82 | F | No |
| DLB | VI-C | 10 | 82 | M | No |
| DLB | V-C | 6 | 83 | M | Yes |
| DLB | V-C | 4 | 84 | M | No |
| DLB | V-C | 4 | 85 | M | No |
| DLB | V-C | 5 | 85 | F | Yes |
| DLB | V-C | 12 | 86 | M | No |
| DLB | VI-C | 6 | 88 | F | Yes |
| DLB |
VI-C |
8 |
98 |
F |
No |
NA, Not available.
Tissue. Left hemispheric hippocampal tissue was accessed at autopsy using a protocol that provided the complete hippocampus (unilaterally) in snap-frozen blocks. The fresh medial temporal tissue block for each individual was dissected into 1-cm-thick coronal blocks, snap frozen in liquid nitrogen-cooled isopentane, and stored at –70°C. Serial coronal dorsal hippocampal sections (20 μm) at the level of the lateral geniculate were cut on a cryostat, thaw mounted onto FisherSuper frost slides, and stored at –70°C for each individual.
LC tissue was accessed at autopsy using a protocol that provided the complete LC (bilaterally) in a snap-frozen block of caudal midbrain and rostral brainstem. The block included the region between the third nerve in the midbrain to the trigeminal nerve in the pons. The block was dissected into three horizontal blocks, frozen in liquid nitrogen-cooled isopentane, and stored at –70°C. Serial horizontal sections (20 μm) from the three blocks were cut on a cryostat, thaw mounted onto FisherSuper frost slides, and stored at –70°C. The rostral-to-caudal distance of the LC was determined for each case by examining sections every 500 μm stained with thionin. The rostral (or 0%) was defined as the beginning of the trochlear nucleus, and the caudal pole (100%) ended at the rostral level of the trigeminal motor nucleus (Hoogendijk et al., 1999). After the rostral-to-caudal distance of the LC was determined for each case, sections were systematically taken to include the 30, 50, and 70% levels of the LC because the rostral portion of the LC innervates forebrain structures such as the hippocampus, whereas the caudal portion of the LC innervates hindbrain structures such as the cerebellum and spinal cord (Fallon and Loughlin, 1982; Loughlin et al., 1982).
TH mRNA. Tissue preparation and labeling of the TH oligonucleotide probes was performed as described previously for oligonucleotide labeling (Szot et al., 1997). TH mRNA expression was measured only in the LC, the main loci for noradrenergic innervation throughout the CNS. For each subject, three consecutive slides at the 30, 50, and 70% levels of the LC were labeled with the TH mRNA oligonucleotide probe. The TH probe consisted of three separate oligonucleotide probes to the following nucleotides of the published human sequence (O'Malley et al., 1987) (GenBank accession number X05290): 326–377, 520–571, and 1309–1360. The oligonucleotide probes were 3′ end labeled with [33P]dATP (PerkinElmer, Boston, MA) using terminal deoxyribonucleotidyl transferase (Invitrogen, Piscataway, NJ). The TH probe contained 2.0 × 106 cpm/50 μl and was washed as described in detail in previously published work with oligonucleotides (Szot et al., 1997). Slides were apposed to film (Eastman Kodak, Rochester, NY) for 4 d at room temperature, and then the slides were coated with NTB2 Nuclear Track Emulsion (undiluted) (Eastman Kodak) and stored at –20°C for 14 d. Film and slides were developed as described previously (Szot et al., 1997).
Quantitation of TH mRNA expression was similar to that performed by Szot et al. (2000). The number of cells that achieved labeling threefold higher than background was counted bilaterally on the three slides at each level of the LC for each subject and expressed as TH-positive labeled cell ± SEM. Data were analyzed with ANOVA, followed by a post hoc Fisher's test; statistical significance was taken at p < 0.05. The density of TH mRNA expression per cell was performed measuring the amount of silver grains over the cell bodies of labeled neurons that were threefold higher than background under 20× dark-field illumination with a side-mounted light using the MicroComputer Imaging Device system (MCID) (Imaging Research, St. Catharines, Ontario, Canada). Therefore, all labeled neurons that were counted as positive labeled were also quantitated for the amount of TH mRNA expression per cell. The data are expressed as the average of grains per cell ± SEM bilaterally at each level of the LC for each subject group. Statistical analysis was performed as described above.
α2A-AR mRNA. Tissue preparation and labeling of the α2A-AR oligonucleotide probes was performed as described previously for oligonucleotide labeling (Szot et al., 1997). α2A-AR mRNA was measured in the LC at the three different levels of the LC as described for TH mRNA and in the dorsal hippocampus. Three consecutive slides were used for each level of the LC and in the dorsal hippocampus. The α2A-AR probe consisted of three separate oligonucleotide probes to the following nucleotides of the published human sequence (Kobilka et al., 1987; Fraser et al., 1989): 951–1001, 1071–1121, and 1303–1353. The α2A-AR probe contained 1.3 × 106 cpm/50 μl for the hippocampus and 0.8 × 106 cpm/50 μl for the LC. Slides were apposed to film (Eastman Kodak) for 7 d at room temperature for hippocampus, whereas slides containing the LC were coated with NTB2 Nuclear Track Emulsion (undiluted) (Eastman Kodak) and stored at –20°C for 14 d. Film and slides were developed as described previously (Szot et al., 1997).
Quantitation of α2A-AR mRNA expression in the LC was identical to the method used to quantitate TH mRNA expression. In the hippocampus, optical density (OD) measurements were taken from labeling observed on film using MCID as described previously (Szot et al., 1997). ODs were obtained from films using MCID. Separate OD measurements were made over the three sections for each subject. Background OD was subtracted from each image. The data are expressed as the average OD ± SEM for each region for each subject group. Statistical analysis was performed as described above.
α2C-AR mRNA. Tissue preparation and labeling of the α2C-AR oligonucleotide probes was performed as described previously for oligonucleotide labeling (Szot et al., 1997). α2C-AR mRNA was measured in the dorsal hippocampus. Preliminary work in the LC showed α2C-AR mRNA levels to be undetectable. Three consecutive slides of the dorsal hippocampus were used. The α2C-AR probe consisted of a single oligonucleotide probe to the following nucleotides of the published human sequence (Lomasney et al., 1990): 875–925. The α2C-AR probe contained 0.16 × 106 cpm/50 μl for the hippocampus. Slides were apposed to film (Eastman Kodak) for 17 h at room temperature. Film was developed and analyzed as described previously (Szot et al., 1997). α2C-AR mRNA expression in the hippocampus was measured as OD using MCID as described for α2A-AR mRNA in the hippocampus. The data are expressed as the average OD ± SEM for each region for each subject group. Statistical analysis was performed as described above.
α1A-AR mRNA. Tissue preparation and labeling of the α1A-AR oligonucleotide probes was performed as described previously (Szot et al., 2005). α1A-AR mRNA was measured in the dorsal hippocampus. Three consecutive slides of the dorsal hippocampus were used. The α1A-AR probe consisted of three separate oligonucleotide probes to the following nucleotides of the published human sequence (Schwinn et al., 1990; Hirasawa et al., 1993): 1–45, 1102–1156, and 1435–1483. The α1A-AR probe contained 1.4 × 106 cpm/50 μl for the hippocampus. Slides were apposed to film (Eastman Kodak) for 4 d at room temperature. Film was developed and analyzed as described previously (Szot et al., 1997). α1A-AR mRNA expression in the hippocampus was measured as OD using MCID. The data are expressed as the average OD ± SEM for each region for each subject group. Statistical analysis was performed as described above.
α1D-AR mRNA. Tissue preparation and labeling of the α1D-AR oligonucleotide probes was performed as described previously (Szot et al., 2005). α1D-AR mRNA was measured in the dorsal hippocampus. Three consecutive slides of the dorsal hippocampus were used. The α1D-AR probe consisted of three separate oligonucleotide probes to the following nucleotides of the published human sequence (Weinberg et al., 1994; Schwinn et al., 1995): 587–635, 990–1038, and 1668–1716. The α1D-AR probe contained 0.4 × 106 cpm/50 μl for the hippocampus. Slides were apposed to film (Eastman Kodak) for 7 d at room temperature. Film was developed and analyzed as described previously (Szot et al., 1997). α1D-AR mRNA expression in the hippocampus was measured as OD using MCID. The data are expressed as the average OD ± SEM for each region for each subject group. Statistical analysis was performed as described above.
3H-Prazosin and 125I-HEAT binding (α1-AR). α1-AR binding sites were measured in the hippocampus with two different radiolabeled compounds, 3H-prazosin and 125I-HEAT [(±)-β-[125I-iodo-4-hydroxyphenyl]-ethyl-aminomethyl-tetralone] (PerkinElmer). 3H-Prazosin binding was performed as described previously (Szot et al., 2005). For each subject, four consecutive slides, each containing a section of the dorsal hippocampus was run: three slides for total binding and the fourth for nonspecific binding. Briefly, slides were thawed at room temperature for 10 min, and then 400 μl/slide of incubation buffer (∼0.2 nm 3H-prazosin in 50 mm Tris buffer with 1 mm EDTA, pH 7.4) was placed over the tissue. Slides were incubated for 40 min at room temperature and then washed twice for 2 min in ice-cold 50 mm Tris buffer, pH 7.4, dipped in ice-cold distilled water to remove the salts, and then rapidly dried under a stream of cool air. Nonspecific binding was defined in the presence of 10 μm phentolamine. Slides and 3H standards were apposed to Biomax MR film (Eastman Kodak) for 8 weeks. Films were developed and analyzed as described previously (Szot et al., 1997). Density measurements (microcuries per gram) were determined using MCID unilaterally in the dorsal hippocampus, and the values are expressed as a mean (microcuries per gram) ± SEM for each subject group. Specific binding was obtained by taking the total average value minus nonspecific value in the same region. Specific binding for 3H-prazosin constituted ∼90% of total binding. Statistical analysis was performed as described above.
Receptor binding with 125I-HEAT was performed according to Homma et al. (2000). For each subject, four consecutive slides, each containing a section of the dorsal hippocampus, was run: three slides for total binding and the fourth for nonspecific binding. Slides were thawed as described above, and then 400 μl/slide of incubation buffer (∼100 pm 125I-HEAT in 50 mm Tris buffer with 150 mm NaCl and 5 mm EDTA, pH 7.4) was placed over the tissue. Slides were incubated for 60 min at room temperature and then washed as described above for 3H-prazosin. Nonspecific binding was defined in the presence of 10 μm phentolamine. Slides were apposed to Biomax MS film (Eastman Kodak) for 4 h. Films were developed as described previously (Szot et al., 1997). Autoradiograms were quantified using MCID and expressed as OD. Specific binding was determined as described above. Specific binding for 125I-HEAT constituted ∼30% of total binding.
3H-Nisoxetine binding (NET). NET binding sites were measured in the LC with 3H-nisoxetine (American Radiolabeled Chemicals, St. Louis, MO) according to Bauer and Tejani-Butt (1992). Preliminary work indicated the hippocampus to not have detectable levels of NET binding sites. From each subject, four consecutive slides from the 30, 50, and 70% of the LC were run: three slides for total binding and the fourth for nonspecific binding. Slides were thawed as described above, and then 400 μl/slide of incubation buffer (∼3 nm 3H-nisoxetine in 50 mm Tris buffer with 300 mm NaCl and 5 mm KCl, pH 7.7) was placed over the tissue. Slides were incubated for 90 min at room temperature and then washed as described above for 3H-prazosin. Nonspecific binding was defined in the presence of 1 μm mazindol. Slides were apposed to Biomax MR film (Eastman Kodak) for 8 weeks. Films were developed as described previously (Szot et al., 1997). Density measurements (microcuries per gram) were determined as described above using 3H standards and MCID. Specific binding was obtained by taking the total average value minus nonspecific value in the same region. Specific binding for 3H-nisoxetine constituted 60–70% of total binding. Statistical analysis was performed as described above.
3H-RX821002 binding (α2-AR). α2-AR binding sites were measured in the LC and hippocampus with 3H-RX821002 (PerkinElmer) according to Happe et al. (2004). For each subject, four consecutive slides from the 30, 50, and 70% of the LC and four consecutive sections from the dorsal hippocampus were run as described above. Slides were thawed as described above, and then 400 μl/slide of incubation buffer (∼2nm 3H-RX821002 in 50 mm NaPO4 buffer, pH 7.4) was placed over the tissue. Slides were incubated for 45 min at room temperature and then washed twice for 2 min in ice-cold 50 mm NaPO4 buffer, pH 7.4, dipped in ice-cold distilled water to remove the salts, and then rapidly dried under a stream of cool air. Nonspecific binding was defined in the presence of 10 μm rauwolscine. Slides were apposed to Biomax MR film (Eastman Kodak) for 8 weeks. Films were developed as described previously (Szot et al., 1997). Density measurements and statistical analysis were obtained as described above for 3H-prazosin. Specific binding for 3H-RX821002 constituted ∼90% of total binding.
Results
None of the noradrenergic parameters measured in this study correlated to the age or sex of the subject or to the PMD. However, we observed significant effects of disease status on noradrenergic parameters as described below.
TH mRNA expression is elevated in the remaining neurons in the LC of AD and DLB subjects
Figure 1 shows the dark-field photomicrographs of TH mRNA expression over cell bodies in the LC of control (A), AD (B), and DLB (C) subjects as defined by cresyl violet staining and the presence of melanin. Quantitation of the number of TH mRNA-positive cell bodies in AD and DLB indicated a significant reduction from control subjects at the 30, 50, and 70% levels of the LC (Fig. 1D). The loss of TH-positive labeled neurons in AD is similar to DLB subjects at the 30 and 50% levels of the LC; however, at the 70% level of the LC, DLB subjects had a significantly greater loss of TH mRNA-positive neurons than AD subjects. In contrast, there was a significant increase in the amount of TH mRNA expression per cell body in all three levels of the LC in both AD and DLB subjects (Fig. 1E). At the 30 and 70% levels of the LC, the amount of TH mRNA expression per cell body was the same between AD and DLB subjects, but, at the 50% level of the LC, DLB subjects had an even greater increase in TH mRNA expression per cell compared with the AD subjects.
Figure 1.
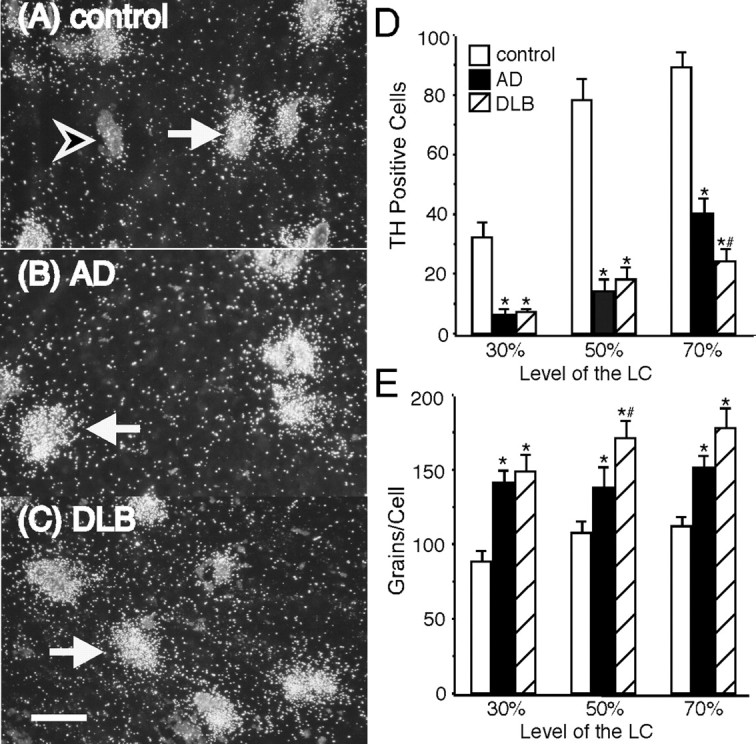
TH mRNA expression in the LC of control(A)(n=15), AD(B)(n=15), and DLB (C)(n=15) subjects at the 50% level of the LC. The number of TH mRNA-positive labeled neurons (D) and the amount of TH mRNA expression per cell (E) in the LC at the 30, 50, and 70% levels of control, AD, and DLB subjects. Filled arrows indicate a labeled neuron in each group, and the unfilled arrow indicates melanin staining without TH mRNA expression. *Significant difference compared with control subjects. #Significant difference compared with AD subjects. Scale bar, 100 μm. Data are represented as mean ± SEM.
A comparison of the number of TH-positive labeled cells to the Clinical Dementia Ratings of AD and DLB subjects showed a significant negative correlation to exist at the 30, 50, and 70% levels of the LC (r =–0.653, p < 0.001; r =–0.571, p < 0.01; r = –0.588, p < 0.01, respectively). This indicates that the loss of TH-positive labeled cells in the LC correlated to the loss of cognitive function in AD and DLB.
A significant negative correlation also existed between the number of TH-positive cells to the amount of TH mRNA expression per cell body at each of the three different levels of the LC (30% level, r = –0.391, p < 0.01; 50% level, r = –0.325, p < 0.05; 70% level, r =–0.463, p < 0.01), indicating that the subjects with the fewest number of TH-positive cells in the LC had the greatest amount of TH mRNA expression per cell. Therefore, the remaining noradrenergic neurons in the LC of AD and DLB subjects appear to be compensating by increasing the expression of the rate-limiting enzyme in the synthesis of NE, and this occurs throughout the LC.
NET binding sites are reduced over LC cell body region but normal over the peri-LC dendritic zone and caudal dorsal raphe
NET is localized to noradrenergic neurons, and its function is to remove released NE from the synapse. NET binding sites, as measured by 3H-nisoxetine, were localized to the LC cell body region and the peri-LC dendritic zone, as well as the caudal dorsal raphe region (cDR) (Fig. 2A–C). Quantitation of NET binding sites over the cell body plus peri-LC dendritic zone indicated a significant decrease in NET binding sites at the 50 and 70% levels of the LC in AD and DLB subjects compared with control subjects (data not shown). The loss of NET binding sites in AD and DLB at these levels of the LC were similar. To determine whether the loss of NET binding sites was specific to just the cell body region, density measurements were taken specifically over the cell body region (Fig. 2A–C, gray shaded area) and the peri-LC dendritic zone (Fig. 2A–C, cross-hatched shaded area). Quantification of NET binding sites over the LC cell body region showed a significant reduction in the AD and DLB subjects compared with control subjects at the 50 and 70% levels of the LC (Fig. 2D). Again the loss of NET binding sites over the LC cell body region is similar between AD and DLB subjects. This would suggest that neuronal number in the LC could contribute to the loss of NET binding sites. Correlation studies partially support this theory. At the 30% level in the LC, a significant correlation was observed between NET binding sites over cell body region and the number of TH-positive labeled cells in control, AD, and DLB subjects (r = 0.666, p < 0.01; r = 0.64, p < 0.05; r = 0.715, p < 0.01, respectively). However, at the 50% level of the LC, only the AD and DLB had a significant correlation to TH-positive labeled neurons (r = 0.772, p < 0.01; r = 0.834, p < 0.01, respectively), and, at the 70% level of the LC, none of the groups displayed a correlation to TH-positive labeled neurons. These data suggests that the loss of noradrenergic neurons in the LC in AD and DLB may account for the loss of NET binding sites at the 30 and 50% levels of the LC but not at the 70% level of the LC. However, it is unclear what contributes to this loss of NET binding sites.
Figure 2.
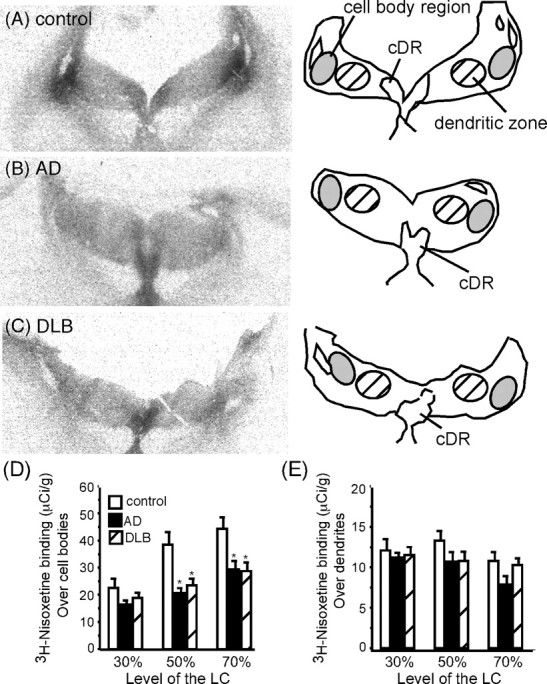
NET binding sites in the LC and cDR of control (A)(n = 15), AD (B)(n = 15), and DLB (C) (n = 15) subjects at the 50% level. The left side of the figure is the autoradiograms of 3H-nisoxetine binding in the LC and cDR. The right side outlines the area labeled by 3H-nisoxetine in the LC, the gray shaded area indicates where the LC cell bodies are located, and the cross-hatched area indicates where the peri-LC dendritic zone is located. Quantification of NET binding sites over the LC cell body region (D;gray shaded area on A–C) and quantification of NET over the peri-LC dendritic area (E; cross-hatched area on A–C) are shown. *Significant difference compared with control subjects. Data are represented as mean ± SEM.
Quantitation of NET binding sites over the peri-LC dendritic zone was not statistically different between the three subject groups (Fig. 2E), despite the loss of TH-positive labeled neurons. This suggests that the remaining noradrenergic neurons in AD and DLB subjects may be demonstrating dendritic sprouting as a means to maintain connections in the peri-LC dendritic area that would have been lost with the reduction of noradrenergic neurons. Axonal sprouting from the remaining neurons also appears to have occurred because the amount of NET binding sites in the cDR in AD and DLB subjects were not different from controls (Fig. 3).
Figure 3.

Quantification of NET binding sites in the cDR at the 50% level of the LC in control (n = 15), AD (n = 15), and DLB (n = 15) subjects. Autoradiographic images can be seen in Fig. 2A–C. Data are represented as mean ± SEM.
α2-AR binding sites in AD and DLB subjects are normal at the 30% level of the LC to modestly reduced at the 50 and 70% levels of the LC
The localization of α2-AR binding sites, as measured by the α2 -AR antagonist 3H-RX821002 (Fig. 4A–C), were similar to that observed with 3H-nisoxetine (Fig. 2A–C). However, unlike 3H-nisoxetine in the LC, 3H-RX821002 was not statistically greater over the cell body region compared with the peri-LC dendritic zone (data not shown). Quantitation of α2-AR binding sites over cell body region plus peri-LC dendritic area (Fig. 4A–C, dashed circular area) indicated a significant reduction in AD and DLB subjects at the 50% level of the LC and in DLB subjects alone at the 70% level of the LC compared with control subjects (Fig. 4D). The loss of α2-AR binding sites in the LC between AD and DLB subjects was not statistically different at any level of the LC. α2-AR binding sites in control, AD, or DLB subjects at the three different levels of the LC did not correlate to TH-positive labeled neurons in the LC, as observed with NET binding sites in the cell body region. This indicates that the loss of TH-positive neurons in the LC of AD and DLB was not responsible for the reduction in binding sites. This was evident at the 30% level of the LC in which there was a tremendous loss of TH-positive neurons in the LC (∼80% loss), but the amount of α2-AR binding was the same between the three subjects groups. At the 50 and 70% levels of the LC, the loss of TH-positive labeled neurons was between 50 and 80%, but the loss of α2-AR binding sites in the LC at the 50 and 70% levels of the LC is ∼18%.
Figure 4.
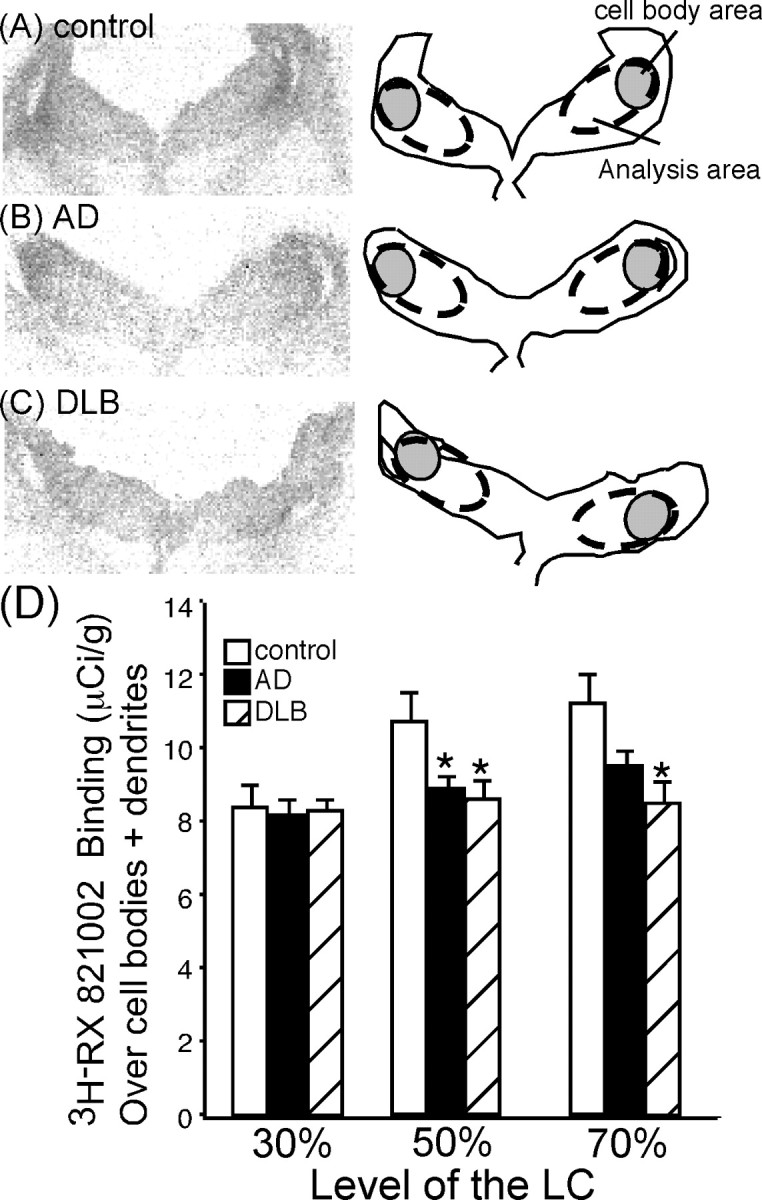
α2-AR binding sites in the LC at the 50% level of control (A)(n = 15), AD (B)(n = 15), and DLB (C) (n = 15) subjects. The left side of the figure is the autoradiograms of 3H-RX821002 binding in the LC. The right side outlines the area labeled by 3H-RX821002 in the LC, and the gray shaded area indicates where the LC cell bodies are located in relation to binding area. D, Quantification of α2-AR binding sites in the LC at the 30, 50, and 70% levels in control, AD, and DLB subjects (dashed circular area). *Significant difference compared with control subjects. Data are represented as mean ± SEM.
The number of positively labeled 2A-AR mRNA neurons in the LC of AD and DLB subjects is reduced, but expression per cell is not different from control subjects
To determine whether a change in mRNA is observed in the remaining neurons to account for the normal to modest reduction in α2-AR binding sites in AD and DLB subjects, in situ hybridization was performed for the α2A-AR mRNA (α2C-AR mRNA was not detected in the LC). α2A-AR mRNA expression in LC neurons would be as the noradrenergic autoreceptor, which is responsible for 3H-RX821002 α2-AR binding observed within the LC and surrounding region. α2A-AR mRNA was expressed at all levels of the LC, but the level of α2A-AR mRNA expression in the LC appeared to be lower (Fig. 5A–C) than that observed for TH mRNA expression (Fig. 1A–C). The number of α2A-AR-positive labeled cell bodies that meet the criteria outlined in Materials and Methods (Fig. 5D) was lower than that observed for TH mRNA expression. For control subjects, the average number of α2A-AR-positive labeled cells at the 30, 50, and 70% levels of the LC were 11, 31, and 31, respectively, whereas TH-positive labeled cells at these levels were 33, 79, and 90, respectively. Therefore, the number of α2A-AR labeled cell bodies was 67, 61, and 66% reduced from TH-positive labeled cells. AD and DBL subjects had a similar reduction of α2A-AR-positive labeled cells in the LC, AD subjects had 57, 53, and 70% reductions, whereas DBL subjects had 63, 68, and 71% reductions at the 30, 50, and 70% levels of the LC.
Figure 5.

α2A-AR mRNA expression in the LC of control (A)(n = 15), AD (B)(n = 15), and DLB (C) (n = 15) subjects at the 50% level of the LC. The number of α2A-AR mRNA-positive labeled neurons (D) and the amount of α2A-AR mRNA expression per cell (E) in the LC at the 30, 50, and 70% levels of control, AD, and DLB subjects are shown. Filled arrows indicate a labeled neuron in each group. *Significant difference compared with control subjects. Scale bar, 100 μm. Data are represented as mean ± SEM.
As observed with the number of TH-positive labeled neurons, both AD and DLB subjects had a significant reduction in the number of α2A-AR-positive labeled cells compared with control subjects at the 30, 50, and 70% levels of the LC (Fig. 5D). The reduction in the number of α2A-AR-labeled neurons was similar to what was observed with TH-positive labeled neurons. However, the amount of α2A-AR mRNA expressed per cell in the remaining neurons in AD and DLB subjects was not significantly different from control subjects (Fig. 5E). There was a tendency for α2A-AR mRNA expression per cell to be higher in AD and DLB subjects, but it did not reach statistical difference.
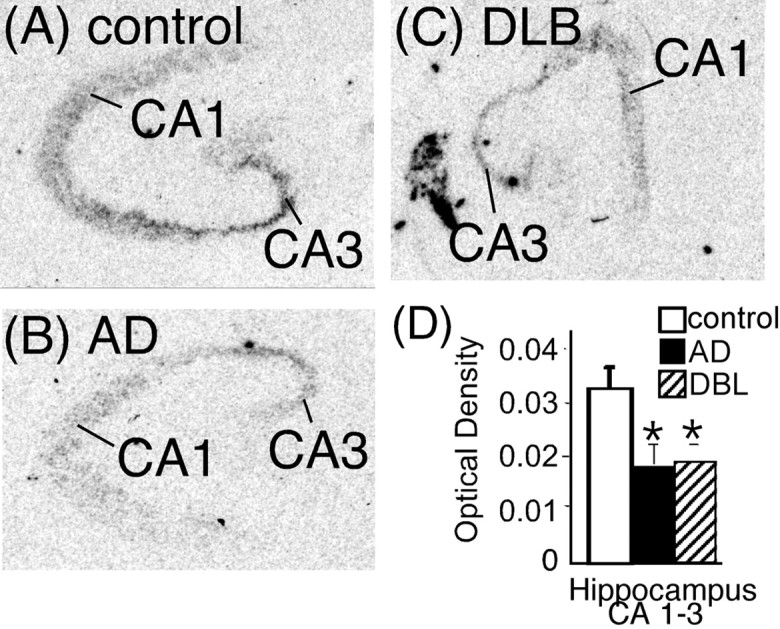
α2-AR binding sites in the hippocampus of AD and DLB are unchanged except for an increase in dentate gyrus granule cell layer
α2-AR binding sites were localized to the dentate gyrus granule cell layer (GCL), stratum lacunosum (SLa), hilus, and pyramidal cell layer of the CA1–CA3 (Fig. 6A–C). This binding pattern was similar to what has been observed previously in the human hippocampus (Pascual et al., 1992; Ordway et al., 1993; Leverenz et al., 2001). α2-AR binding sites are not statistically different between control, AD, and DLB subjects in the SLa, hilus, and pyramidal cell layer, but, in the GCL, AD and DLB subjects had significantly more binding sites than control (Fig. 6D). The increase in α2-AR binding sites in the GCL was the same between AD and DLB subjects. α2-AR binding sites in the GCL of AD and DLB subjects did not correlate to the degree of dementia (Clinical Dementia Rating).
Figure 6.
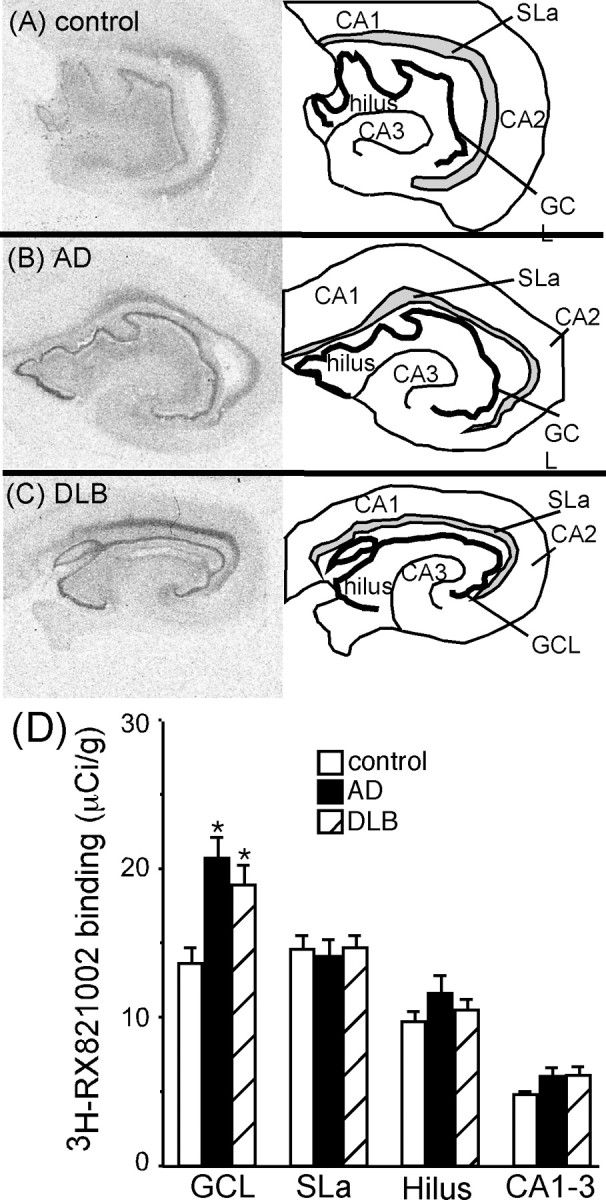
α2-AR binding sites in the dorsal hippocampus of control (A) (n = 16), AD (B) (n = 15), and DLB (C) (n = 21) subjects. The left side of the figure is the autoradiograms of 3H-RX821002 binding in the hippocampus. The right side outlines the areas labeled by 3H-RX821002 in the pyramidal cell layer (CA1–CA3), SLa, dentate gyrus GCL, and hilus. D, Quantification of α2-AR binding sites in the dorsal hippocampus in control, AD, and DLB subjects. *Significant difference compared with control subjects. Data are represented as mean ± SEM.
α2A-AR mRNA expression in the hippocampus of AD and DLB subjects is not different from control subjects
3H-RX821002 binding sites in the human hippocampus represent mainly α2A-ARs (Ordway et al., 1993; Sastre and Garcia-Sevilla, 1994), which are located both presynaptically and postsynaptically. To determine the contribution of postsynaptic α2A-AR to the binding studies in the hippocampus, in situ hybridization to the α2A-AR mRNA was performed. α2A-AR mRNA was expressed at detectable but low levels in the GCL and CA3 region in all subjects (Fig. 7A–C). Quantitation of α2A-AR mRNA in these regions in control, AD, and DLB subjects was not statistically different (Fig. 7D).
Figure 7.
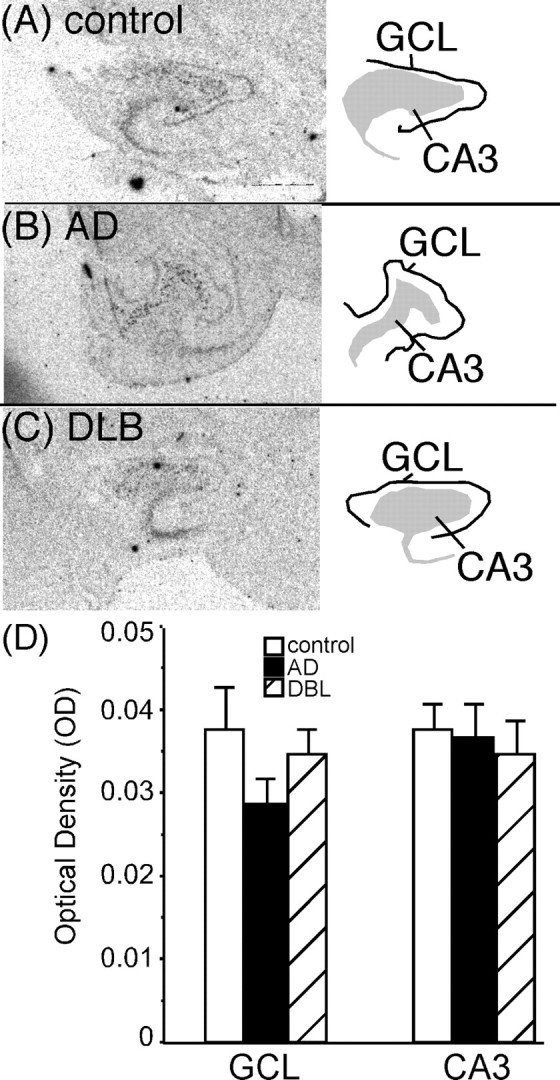
α2A-AR mRNA expression in the dorsal hippocampus of control (A) (n = 16), AD (B)(n = 15), and DLB (C)(n = 22) subjects. The left side of the figure are the autoradiograms of α2A-AR mRNA. The right side outlines the area of α2A-AR mRNA expression in the pyramidal celllayerCA3, hilus, and dentate gyrus GCL.D, Quantification of α2A-AR mRNA expression in the dorsal hippocampus in control, AD, and DLB subjects. Data are represented as mean ± SEM.
α2C-AR mRNA expression in the hippocampus of AD and DLB subjects is significantly reduced in all regions compared with control subjects
α2C-AR mRNA expression in the human hippocampus was observed in the GCL, pyramidal cell layer (CA1–C3), and subiculum (Fig. 8A–C). Quantitation of α2C-AR mRNA in AD and DLB subjects was significantly reduced in all regions of the hippocampus compared with control subjects (Fig. 8D). The reduction in α2C-AR mRNA in the GCL, pyramidal cell layer, and subiculum was comparable between AD and DLB subjects. α2C-AR mRNA expression in these regions did not correlate to the degree of dementia (Clinical Dementia Rating).
Figure 8.
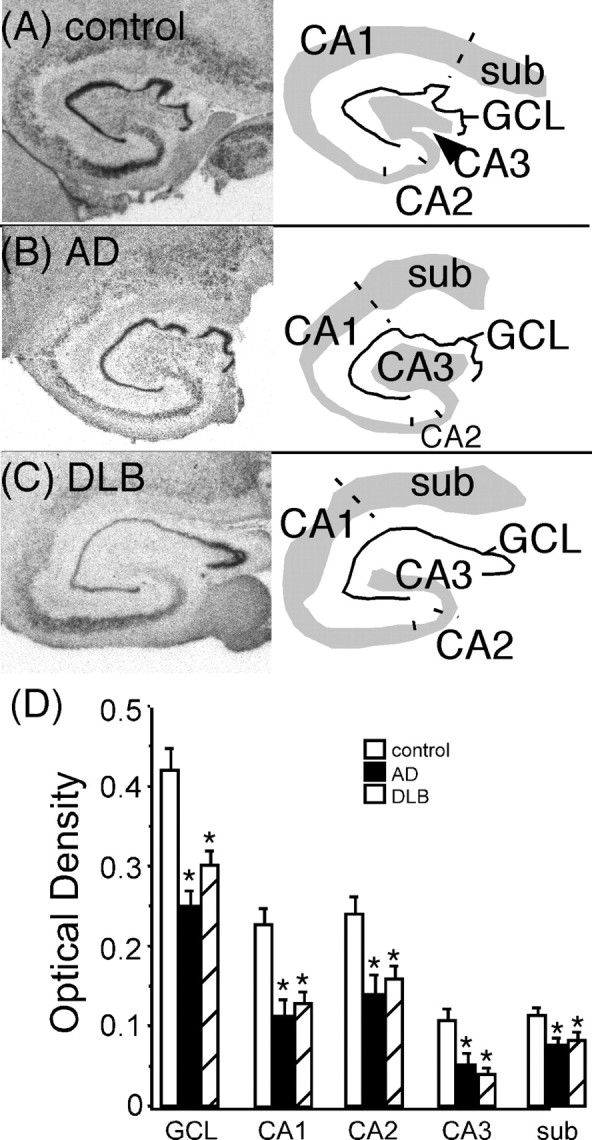
α2C-AR mRNA expression in the dorsal hippocampus of control (A) (n = 16), AD (B)(n = 13), and DLB (C)(n = 22) subjects. The left side of the figure are the autoradiograms of α2C-AR mRNA. The right side outlines the area of α2C-AR mRNA expression in the pyramidal cell layer (CA1–CA3), dentate gyrus GCL, and subiculum (sub). D, Quantification of α2C-AR mRNA expression in the dorsal hippocampus in control, AD, and DLB subjects. *Significant difference compared with control subjects. Data are represented as mean ± SEM.
α1-AR binding sites in the hippocampus are elevated only in the molecular cell layer of the dentate gyrus of AD and DLB subjects
Unlike the α2-AR, the α1-AR is only localized postsynaptically. 3H-Prazosin binding sites in control, AD, and DLB showed a binding pattern (Fig. 9A–C) that was described previously in the human hippocampus (Szot et al., 2005). 3H-Prazosin labeled all three layers of the dentate gyrus (molecular, granular, and hilus) and the stratum lucidum. Quantitation of α1-AR binding sites in all three layers of the dentate gyrus in the three subject groups demonstrated a significant increase in AD and DLB subjects compared with controls (data not shown). On visual inspection of the autoradiograms (Fig. 9A–C), it was apparent that, in AD and DLB subjects, there was a greater amount of binding in the molecular cell layer. To determine whether 3H-prazosin binding sites were elevated in just the molecular cell layer in AD and DLB subjects, binding sites were quantitated over just the molecular cell layer and then over the GCL, hilus, and stratum lucidum (Fig. 9D). A significant increase in 3H-prazosin binding sites in the molecular cell layer was observed in AD and DLB subjects compared with control subjects, whereas in the GCL, hilus, and stratum lucidum, there was no difference between subjects groups. The increase in α1-AR binding sites in the molecular cell layer was not different between AD and DLB subjects (Fig. 9D). This indicates a selective increase in α1-AR binding sites in the hippocampus of AD and DLB subjects without a change in the other areas. α1-AR binding sites in the molecular cell layer of AD and DLB subjects did not correlate to the degree of dementia (Clinical Dementia Rating).
Figure 9.
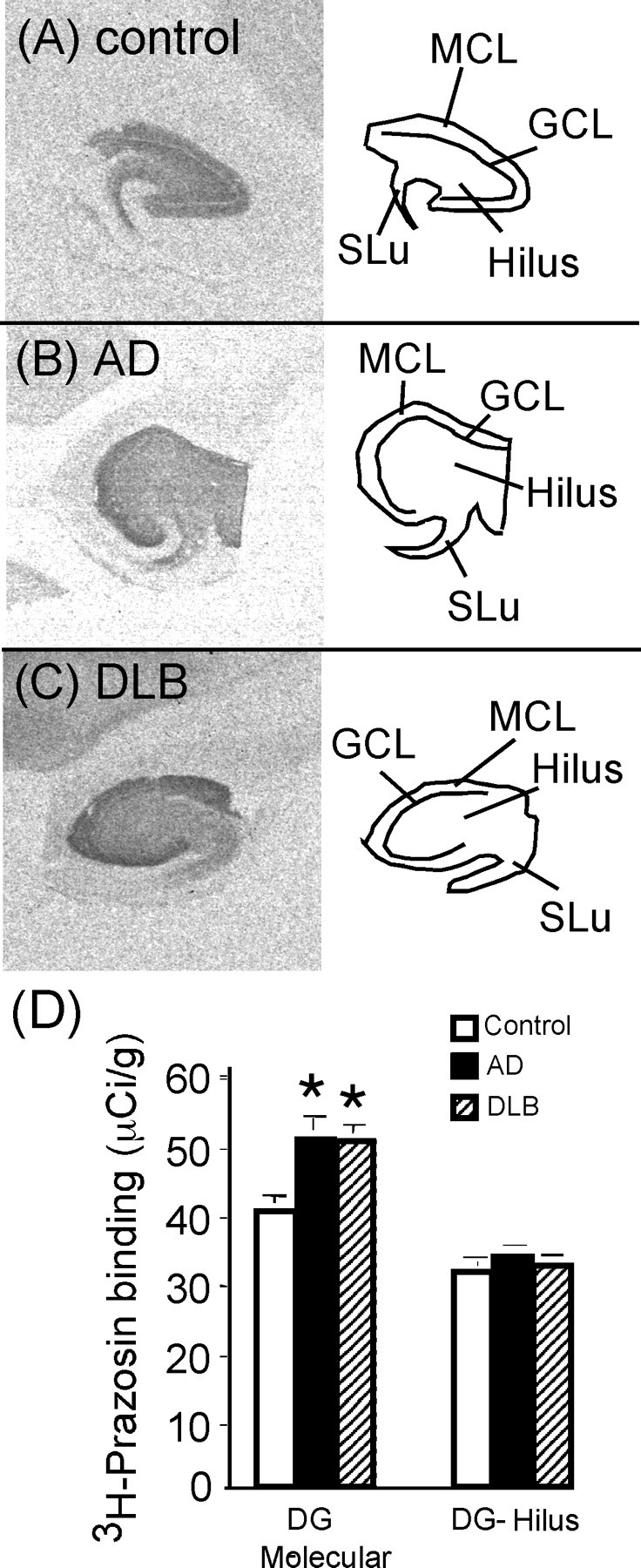
α1-AR binding sites in the dorsal hippocampus of control (A) (n = 17), AD (B) (n = 15), and DLB (C) (n = 22) subjects. The left side of the figure is the autoradiograms of 3H-prazosin binding in the hippocampus. The right side outlines the areas labeled by 3H-prazosin in the dentate gyrus, which is composed of the molecular cell layer (MCL), GCL and hilus, and the stratum lucidum (SLu). D, Quantification of α1-AR binding sites in the dorsal hippocampus in control, AD, and DLB subjects. DG, Dentate gyrus. *Significant difference compared with control subjects. Data are represented as mean ± SEM.
To rule out the possibility that the selective increase in α1-AR binding sites in the molecular cell layer was a peculiarity of 3H-prazosin, another α1-AR antagonist was used. 125I-HEAT labeled exactly the same regions of the hippocampus as 3H-prazosin (data not shown). Quantitation of 125I-HEAT, similar to 3H-prazosin, demonstrated a selective increase in 125I-HEAT binding sites in the molecular cell layer of AD and DLB subjects compared with control subjects (data not shown). No change was observed in the GCL, hilus, and stratum lucidum in the three subject groups (data not shown).
α1A-AR mRNA expression in the GCL of AD and DLB subjects is not different from control subjects
α1A-AR mRNA expression in the human hippocampus was observed only in the GCL (Szot et al., 2005) (Fig. 10A–C). Quantitation of α1A-AR mRNA expression in AD and DLB subjects in the GCL did not differ from control subjects (Fig. 10D).
Figure 10.

α1A-AR mRNA expression in the dentate gyrus GCL of the dorsal hippocampus of control (A) (n = 17), AD (B) (n = 15), and DLB (C) (n = 22) subjects. D, Quantification of α1A-AR mRNA expression in the dorsal hippocampus in control, AD, and DLB subjects.
α1D-AR mRNA expression in the pyramidal cell layer of AD and DLB subjects is significantly reduced from control subjects
α1D-AR mRNA was expressed only in the pyramidal cell layer as described previously (Szot et al., 2005) (Fig. 11A–C). Quantitation of α1D-AR mRNA expression in the pyramidal cell layer was significantly reduced in AD and DLB subjects compared with control subjects (Fig. 11D). The reduction in α1D-AR mRNA in AD and DLB subjects was similar. The reduction in α1D-AR mRNA expression did not correlate to the degree of dementia (Clinical Dementia Ratings).
Figure 11.
α1D-AR mRNA expression in the pyramidal cell layer (CA1–C3) of the dorsal hippocampus of control (A) (n = 17), AD (B) (n = 15), and DLB (C) (n = 22) subjects. D, Quantification of α1D-AR mRNA expression in the dorsal hippocampus in control, AD, and DLB subjects. *Significant difference compared with control subjects. Data are represented as mean ± SEM.
Discussion
In AD and the similar dementing disorder DLB, there is a significant loss of noradrenergic neurons in the LC at multiple levels. This loss of neurons is documented at the molecular level with mRNA expression for TH, α2A-AR, and NET (Szot et al., 2000). The loss of neurons as measured by these mRNAs is very similar to the documented loss of noradrenergic neurons as measured by morphometric analysis (Marcyniuk et al., 1986; Chan-Palay and Asan, 1989; German et al., 1992). The degree of neuronal loss in AD subjects reported here is similar to our previously published work with a small subject population (Szot et al., 2000), and, as shown by Bondareff et al. (1981) and Matthews et al. (2002), the amount of TH-positive labeled neuronal loss correlates with the degree of cognitive loss. This correlation of noradrenergic neuronal loss to cognitive decline supports the importance of the noradrenergic system in learning and memory (Gibbs and Summers, 2002); however, how this neuronal loss contributes to cognitive loss is unclear at the present time. It needs to be determined whether the compensatory changes in the remaining neurons in the LC and at terminal projection sites such as the hippocampus and cDR contribute to the cognitive decline or could be used pharmacologically to enhance/restore memory.
In the present study, we performed a systematic molecular evaluation of the nature of this compensation. The first compensatory change in noradrenergic neurons is the significant increase in TH mRNA expression (rate-limiting enzyme in the synthesis of NE), which was observed at all levels of the LC. Differences in TH mRNA expression were observed between AD and DLB subjects (no other noradrenergic marker showed a significant difference between AD and DLB subjects). In DLB subjects, there is an even greater loss of TH-positive labeled cell bodies at the 70% level of the LC compared with AD, and, at the 50% level of the LC, DLB subjects have a greater expression of TH mRNA expression per cell. The increase in TH mRNA expression per cell corresponded to the loss of TH-positive labeled cells, i.e., the greater the degree of neuronal loss, the more TH mRNA expression per cell occurred. These studies, obviously, do not indicate whether the amount of NE released in forebrain structures is normal but support previous reports of normal to elevated levels of CSF NE and NE metabolites compared with age-matched control subjects (Mann et al., 1981; Gottfries et al., 1983; Raskind et al., 1984; Tohgi et al., 1992; Elrod et al., 1997).
The second type of compensatory change is observed with the dendritic innervation of the LC neurons to the peri-LC dendritic zone. The LC receives a vast amount of afferent projections, but the majority of these fibers do not go directly to the LC cell body region; rather they innervate the surrounding LC region (Aston-Jones et al., 2004) termed the peri-LC dendritic zone. NET binding sites are localized over the LC cell body region (Bauer and Tejani-Butt, 1992; Tejani-Butt et al., 1993) and over the peri-LC dendritic region, with the greater density over the LC cell body region. The significant loss of NET binding sites over the LC cell body region in AD and DLB can be attributed to the loss of LC neurons, as noted by Tejani-Butt et al. (1993). However, the normal amount of NET binding sites in the peri-LC dendritic zone of AD and DLB subjects suggests that the remaining noradrenergic neurons are sprouting dendritic connections into the surrounding LC region in an attempt to maintain synaptic connections. This is also supported by the findings with the α2-AR autoreceptor. The density of this autoreceptor is reduced in AD and DLB subjects at certain levels of the LC, but the loss is not as extensive as that observed with neuronal number, supporting the hypothesis of dendritic sprouting. The consequence of the loss of this autoreceptor may be increased firing of noradrenergic neurons and release of NE in the LC and at terminal regions (L'Heureux et al., 1986; Van Gaalen et al., 1997; Kawahara et al., 1999).
Expression of NET (Szot et al., 2000) or α2A-AR mRNA in the remaining noradrenergic neurons in AD and DLB subjects is not elevated, so it is unclear what factors are involved in maintaining the normal NET and α2-AR binding sites in the LC. Animal work does not indicate what may occur to the expression of NET or α2A-AR mRNA in LC neurons after lesioning noradrenergic neurons because it has not been studied. One explanation for the lack of a significant change in NET or α2A-AR mRNA expression in the remaining noradrenergic neurons is a change in the stability of the transcript or receptor protein.
The third type of compensatory change in noradrenergic neurons is with axonal projections. The normal amount of NET binding sites in the cDR and normal to elevated α2-ARs in the hippocampus of AD and DLB subjects indicate that remaining noradrenergic neurons are demonstrating axonal sprouting. Several studies have examined α2-ARs in the hippocampus of AD subjects using autoradiography and membrane binding studies with many different radiolabeled ligands. The results have been conflicting: either a significant decrease (Meana et al., 1992; Pascual et al., 1992) or no change or an increase were observed (Leverenz et al., 2001; Matthews et al., 2002).
Because α2-ARs in the hippocampus may be a composite of the autoreceptor and postsynaptic receptors (including heteroreceptors), measurement of postsynaptic α2A- and α2C-AR mRNA expression was performed, and another postsynaptic AR (α1-AR) was measured in the hippocampus. α2A-AR (the subtype responsible for 3H-RX821002 binding sites) (Ordway et al., 1993; Sastre and Garcia-Sevilla, 1994) mRNA expression in the hippocampus of AD and DLB subjects was not different from controls, suggesting that postsynaptic α2A-AR sites are also normal in AD and DLB subjects.
In contrast to α2A-AR mRNA expression, α2C-AR mRNA expression in the hippocampus of AD and DLB subjects was significantly reduced. Because binding studies with 3H-RX821002 do not reflect the changes in this receptor subtype, the functional consequence of this reduced mRNA expression is unclear. Loss of α2C-AR, as observed in transgenic mice (α2C-AR knock-out mice), results in many behavioral changes, including increased stress and startle response, and locomotion (Sallinen et al., 1997). Interestingly, agitation and pacing are common behavioral symptoms in dementia patients. Perhaps the loss of α2C-AR mRNA in AD and DLB subjects in the hippocampus contributes to these behavior symptoms. The loss of α2C-AR mRNA in the GCL or CA3 cannot be attributed to neuronal loss in AD because these regions do not demonstrate any degree of loss (Simic et al., 1997; Bobinski et al., 1998; Fukutani et al., 2000; Price et al., 2001; Rossler et al., 2002), and the expression of α2A-AR mRNA in the same regions was unchanged in AD and DLB.
Postsynaptic α1-AR binding sites in the hippocampus of AD and DLB subjects are normal to elevated, indicating an intact postsynaptic system. Previous α1-AR binding studies in the hippocampus of AD subjects demonstrated either a decrease in binding with 3H-prazosin (Shimohama et al., 1986) or no change with 125I-HEAT (Kalaria, 1989). These previously published studies used a homogenous membrane preparation from the whole hippocampus. This method would not allow the differentiation of the different layers of the hippocampus (i.e., molecular cell layer from dentate gyrus), and measurement would include both membrane and cytoplasmic material. Because the α1-AR protein may be localized in both the membrane and cytoplasm (Mac-Kenzie et al., 2000; Piascik and Perez, 2001; halothorn et al., 2002), it is likely that homogenizing the tissue would affect the number of available receptors for the α1-AR ligand to bind to relative to slice preparation. The increase in α1-AR binding in AD and DLB subjects is not explained by the expression of α1A- or α1D-AR mRNA. Because α1A-AR contributes mainly to 3H-prazosin binding sites in the human hippocampus (Szot et al., 2005), the lack of a change in α1A-AR mRNA in AD and DLB subjects indicate that the increase does not appear to be the result of increased mRNA expression but may represent a change in transcript or protein stability, or sprouting of the dendrites from GCL neurons into the molecular cell layer. Sprouting of GCL neurons has been shown to occur in AD subjects (Cassell and Brown, 1984; Dickson et al., 1994).
α1D-AR mRNA, the other major CNS α1-AR subtype, was significantly reduced in both AD and DLB subjects. The reduction in α1D-AR mRNA cannot be attributed to the cell loss documented in the CA1 in AD (Simic et all., 1997; Bobinski et al., 1998; Fukutani et al., 2000; Price et al., 2001; Rossler et al., 2002) because α2A-AR mRNA expression was unchanged in the same region, in the same subjects. It is possible that these different α-AR subtypes are expressed in different neurons and the neurons expressing α1D-AR mRNA are lost in AD and DLB. The consequence of reduced α1D-AR mRNA expression for protein levels cannot be determined at this time because none of the existing α1-AR ligands bind to this receptor subtype. However, loss of α1D-AR mRNA in pyramidal neurons, especially CA1 neurons, suggests the loss of the modulation of NE on hippocampal pathways. It is interesting to note that α2C-AR mRNA expression is also reduced in these AD and DLB subjects in a similar region. A common factor between these two AR subtypes compared with α1A- and α2A-AR mRNA (which are unchanged in AD and DLB subjects) is that α1D- and α2C-ARs are intracellular receptors, whereas α1A- and α2A-AR are membrane-bound receptors (Daunt et al., 1997; MacKenzie et al., 2000; Piascik and Perez, 2001; Chalothorn et al., 2002).
In conclusion, after the loss of noradrenergic neurons in AD and DLB, three major compensatory changes are observed in the noradrenergic nervous system: (1) an increase per neuron in mRNA for TH, the rate-limiting enzyme in the synthesis of NE; (2) sprouting of dendrites of the remaining LC neurons to the peri-LC dendritic area; and (3) sprouting of LC axons to cDR and hippocampus. Also, other noradrenergic markers are not normal: α1D- and α2C-AR mRNA expression in the hippocampus of AD and DLB subjects are significantly reduced. For all noradrenergic markers, there was no difference between AD and DLB subjects except for minor differences in TH mRNA expression; therefore, the presence of Lewy bodies in addition to plaques and tangles in DLB subjects does not appear to further affect the noradrenergic compensatory changes.
Footnotes
These studies were supported by the Department of Veterans Affairs Research and Development Services, Northwest Network Mental Illness Research, Education, and Clinical Center, and Alzheimer's Disease Research Center.
Correspondence should be addressed to Dr. Patricia Szot, Northwest Network for Mental Illness Research, Education, and Clinical Center (116), Puget Sound Health Care System, 1660 South Columbian Way, Seattle, WA 98108. E-mail: szot@u.washington.edu.
DOI:10.1523/JNEUROSCI.4265-05.2006
Copyright © 2006 Society for Neuroscience 0270-6474/06/260467-12$15.00/0
References
- Adolfsson R, Gottfries CG, Roos BE, Winblad B (1979) Changes in the brain catecholamines in patients with dementia of Alzheimer's type. Br J Psychiatry 135: 216–223. [DOI] [PubMed] [Google Scholar]
- Aston-Jones G, Zhu Y, Card JP (2004) Numerous GABAergic afferents to locus ceruleus in the pericerulear dendritic zone: possible interneuronal pool. J Neurosci 24: 2313–2321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballard C, Holmes C, McKeith I, Neill D, O'Brien J, Cairns N, Lantos P, Perry E, Ince P, Perry R (1999) Psychiatric morbidity in dementia with Lewy bodies: a prospective clinical and neuropathological comparative study with Alzheimer's disease. Am J Psychiatry 156: 1039–1045. [DOI] [PubMed] [Google Scholar]
- Barber R, Panikkar A, McKeith IG (2001) Dementia with Lewy bodies: diagnosis and management. Int J Geriatr Psychiatry 16: S12–S18. [DOI] [PubMed] [Google Scholar]
- Bauer ME, Tejani-Butt SM (1992) Effects of repeated administration of desipramine or electroconvulsive shock on norepinephrine uptake sites measured by [3H] nisoxetine autoradiography. Brain Res 582: 208–214. [DOI] [PubMed] [Google Scholar]
- Bobinski M, de Leon MJ, Tarnawski M, Wegiel J, Bobinski M, Reisberg B, Miller DC, Wisniewski HM (1998) Neuronal and volume loss in CA1 of the hippocampal formation uniquely predicts duration and severity of Alzheimer's disease. Brain Res 805: 267–269. [DOI] [PubMed] [Google Scholar]
- Bondareff W, Mountjoy CQ, Roth M (1981) Selective loss of neurons of origin of adrenergic projection to cerebral cortex (nucleus locus coeruleus) in senile dementia. Lancet 1: 783–784. [DOI] [PubMed] [Google Scholar]
- Bondareff W, Mountjoy CQ, Roth M (1982) Loss of neurons of origin of the adrenergic projection to cerebral cortex (nucleus locus ceruleus) in senile dementia. Neurology 32: 164–168. [DOI] [PubMed] [Google Scholar]
- Cassell MD, Brown MW (1984) The distribution of Timm's stain in the nonsulfide-perfussed human hippocampal formation. J Comp Neurol 222: 461–471. [DOI] [PubMed] [Google Scholar]
- Chalothorn D, McCune DF, Edelmann SE, Garcia-Cazarin ML, Tsujimoto G, Piascik MT (2002) Differences in the cellular localization and agonist-mediated internatization properties of the α1-adrenoceptor subtypes. Mol Pharmacol 61: 1008–1016. [DOI] [PubMed] [Google Scholar]
- Chan-Palay V, Asan E (1989) Alterations in catecholamine neurons of the locus coeruleus in senile dementia of the Alzheimer's type and in Parkinson's disease with and without dementia and depression. J Comp Neurol 287: 373–392. [DOI] [PubMed] [Google Scholar]
- Cross AJ, Crow TJ, Perry EK, Perry RH, Blessed G, Tomlinson BE (1981) Reduced dopamine-beta-hydroxylase activity in Alzheimr's disease. Br Med J (Clin Res Ed) 282: 93–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daunt DA, Hurt C, Hein L, Kallio J, Feng F, Kobilka BK (1997) Subtype-specific intracellular trafficking of α2-adrenrgic receptors. Mol Pharmacol 51: 711–720. [DOI] [PubMed] [Google Scholar]
- Dickson DW, Davies P, Bevona C, Van Hoeven KH, Factor SM, Grober E, Aronson MK, Crystal HA (1994) Hippocampal sclerosis: a common pathological feature of dementia in the very old (> or = 80 years of age) human. Acta Neuropathol 88: 212–221. [DOI] [PubMed] [Google Scholar]
- Elrod R, Peskind ER, DiGiacomo L, Brodkin KL, Veith RC, Raskind MA (1997) Effects of Alzheimer's disease severity on cerebrospinal fluid norepinephrine concentration. Am J Psychiatry 154: 25–30. [DOI] [PubMed] [Google Scholar]
- Fallon JH, Loughlin SE (1982) Monoamine innervation of the forebrain: collateralization. Brain Res Bull 9: 295–307. [DOI] [PubMed] [Google Scholar]
- Fraser CM, Arakawa S, McCombie WR, Venter JC (1989) Cloning, sequence analysis, and permanent expression of a human α2-adrenergic receptor in Chinese hamster ovary cells. Evidence for independent pathways of receptor coupling to adenylate cyclase attenuation and activation. J Biol Chem 264: 11754–11761. [PubMed] [Google Scholar]
- Fukutani Y, Cairns NJ, Shiozawa M, Sasaki K, Sudo S, Isaki K, Lantos PL (2000) Neuronal loss and neurofibrillary degeneration in the hippocampal cortex in late-onset sporadic Alzheimer's disease. Psychiatry Clin Neurosci 54: 523–529. [DOI] [PubMed] [Google Scholar]
- German DC, Manaye KF, White CL, Woodward DJ, McIntire DD, Smith WK, Kalaria RN, Mann DM (1992) Disease-specific patterns of locus coeruleus cell loss. Ann Neurol 32: 667–676. [DOI] [PubMed] [Google Scholar]
- Gibbs ME, Summers RJ (2002) Role of adrenoceptor subtypes in memory consolidation. Prog Neurobiol 67: 345–391. [DOI] [PubMed] [Google Scholar]
- Gottfries CG, Adolfsson R, Aquilonius SM, Carlsson A, Eckernas SA, Nordberg A, Oreland L, Svennerholm L, Wiberg A, Winblad B (1983) Biochemical changes in dementia disorders of Alzheimer's type (AD/SDAT). Neurobiol Aging 4: 261–271. [DOI] [PubMed] [Google Scholar]
- Happe HK, Coulter CL, Gerety ME, Sanders JD, O'Rourke M, Bylund DB, Murrin LC (2004) Alpha-2 adrenergic receptor development in rat CNS: an autoradiographic study. Neuroscience 123: 167–178. [DOI] [PubMed] [Google Scholar]
- Harro J, Oreland L (2001) Depression as a spreading adjustment disorder of monoaminergic neurons: a case for primary implication of the locus coeruleus. Brain Res Rev 38: 79–128. [DOI] [PubMed] [Google Scholar]
- Hirasawa A, Horie K, Tanaka T, Takagaki K, Murai M, Yano J, Tsujimoto G (1993) Cloning, functional expression and tissue distribution of human cDNA for the α1C-adrenergic receptor. Biochem Biophys Res Comm 195: 902–909. [DOI] [PubMed] [Google Scholar]
- Homma N, Hirasawa A, Shibata K, Hashimito K, Tsujimoto G (2000) Both α1A- and α1B-adrenrgic receptor subtypes couple to the transient outward subtype current (ITo) in rat ventricular myocytes. Br J Pharmacol 129: 1113–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoogendijk WJ, Freenstra MG, Botterblom MH, Gilhuis J, Sommer IE, Kamphorst W, Eikelenboom P, Swaab DF (1999) Increased activity of surviving locus ceruleus neurons in Alzheimer's disease. Ann Neurol 45: 82–91. [DOI] [PubMed] [Google Scholar]
- Kalaria RN (1989) Characterization of [125I] HEAT binding to α1-receptors in human brain: assessment in aging and Alzheimer's disease. Brain Res 501: 287–294. [DOI] [PubMed] [Google Scholar]
- Kawahara Y, Kawahara H, Westerink BHC (1999) Tonic regulation of the activity of noradrenrgic neurons in the locus coeruleus of the conscious rat: studies by dual-probe microdialysis. Brain Res 823: 42–48. [DOI] [PubMed] [Google Scholar]
- Kobilka BK, Matsui H, Kobilka TS, Yang-Feng TL, Francke U, Caron MG, Lefkowitz RJ, Regan JW (1987) Cloning, sequencing, and expression of the gene coding for the human platelet α2-adrenergic receptor. Science 238: 650–656. [DOI] [PubMed] [Google Scholar]
- Lacroix D, Blier P, Curet O, de Montigny C (1991) Effects of long term desipramine administration on noradrenergic neurotransmission: electrophysiological studies in the rat brain. J Pharmacol Exp Ther 257: 1081–1091. [PubMed] [Google Scholar]
- Leverenz JB, Miller MA, Dobie DJ, Peskind ER, Raskind MA (2001) Increased alpha 2-adrenergic receptor binding in locus coeruleus projection areas in dementia with Lewy bodies. Neurobiol Aging 22: 555–561. [DOI] [PubMed] [Google Scholar]
- L'Heureux R, Dannis T, Curet O, Scatton B (1986) Measurement of endogenous noradrenaline release in the rat cerebral cortex in vivo by transcortical dialysis: effect of drugs affecting noradrenergic transmission. J Neurochem 46: 1794–1801. [DOI] [PubMed] [Google Scholar]
- Lomasney JW, Lorenz W, Allen LF, King K, Regan JW, Yang-Feng TL, Caron MG, Lefkowitz RJ (1990) Expansion of the α2-adrenergic receptor family: cloning and characterization of a human α2-adrenergic receptor subtype, the gene for which is located on chromosome 2. Proc Natl Acad Sci USA 87: 5094–5098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loughlin SE, Foote SL, Fallon JH (1982) Locus coeruleus projections to cortex: topography, morphology and collateralizations. Brain Res Bull 9: 287–294. [DOI] [PubMed] [Google Scholar]
- MacKenzie JF, Daly CJ, Pediani JD, McGrath JC (2000) Quantitative imaging in live human cells reveals intracellular α1-adrenoreceptor ligand-binding sites. J Pharmacol Exp Ther 294: 434–443. [PubMed] [Google Scholar]
- Mann DM, Lincoln J, Yates PO, Stamp JE, Toper S (1980) Changes in the monoamine containing neurons of the human CNS in senile dementia. Br J Psychiatry 136: 533–541. [DOI] [PubMed] [Google Scholar]
- Mann JJ, Stanley M, Neophytides A, de Leon MJ, Ferris SH, Gershon S (1981) Central amine metabolism in Alzheimer's disease: in vivo relationship to cognitive deficit. Neurobiol Aging 2: 57–60. [DOI] [PubMed] [Google Scholar]
- Marcyniuk B, Mann DM, Yates PO (1986) Loss of nerve cells from locus coeruleus in Alzheimer's disease is topographically arranged. Neurosci Lett 64: 247–252. [DOI] [PubMed] [Google Scholar]
- Matthews KL, Chen CPL-H, Esiri MM, Keene J, Minger SL, Francis PT (2002) Noradrenergic changes, aggressive behavior, and cognition in patients with dementia. Biol Psychiatry 51: 407–416. [DOI] [PubMed] [Google Scholar]
- McKeith IG, Galasko D, Kosaka K, Perry EK, Dickson DW, Hansen LA, Salmon DP, Lowe J, Mirra SS, Byrne FJ, Lennox G, Quinn NP, Edwardson JA, Ince PG, Bergeron C, Burns A, Miller BL, Lovestone S, Collerton D, Jansen EN, et al. (1996) Consensus guidelines for the clinical and pathologic diagnosis of dementia with Lewy bodies (DLB): report of the consortium on DLB international workshop. Neurology 47: 1113–1124. [DOI] [PubMed] [Google Scholar]
- McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlam EM (1984) Clinical diagnosis of Alzheimer's disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer's disease. Neurology 34: 939–944. [DOI] [PubMed] [Google Scholar]
- Meana JJ, Barturen F, Garro MA, Garcia-Sevilla JA, Fontan A, Zarranz JJ (1992) Decreased density of pre-synaptic α2-adrenoceptors in postmortem brains of patients with Alzheimer's disease. J Neurochem 58: 1896–1904. [DOI] [PubMed] [Google Scholar]
- Morris RG, Moser EI, Riedel G, Martin SJ, Sandin J, Day M, O'Carroll C (2003) Elements of a neurobiological theory of the hippocampus: the role of activity-dependent synaptic plasticity in memory. Philos Trans Soc Lond B Biol Sci 358: 773–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Malley KL, Anhalt MJ, Martin BM, Kelsoe JR, Winfield SL, Ginns EJ (1987) Isolation and characterization of the human tyrosine hydroxylase gene: identification of 5′ alternative splice sites responsible for multiple mRNAs. Biochemistry 26: 6910–6914. [DOI] [PubMed] [Google Scholar]
- Ordway GA, Jaconetta SM, Halaris AE (1993) Characterization of subtypes of alpha-2 adrenoreceptors in the human brain. J Pharmacol Exp Ther 264: 967–976. [PubMed] [Google Scholar]
- Ordway GA, Schenk J, Stockmeier CA, May W, Klimek V (2003) Elevated agonist binding to α2-adrenoceptors in the locus coeruleus in major depression. Biol Psychiatry 53: 315–323. [DOI] [PubMed] [Google Scholar]
- Palmer AM, Wilcock GK, Esiri MM, Francis PT, Bowen DM (1987) Monoaminergic innervation of the frontal and temporal lobes in Alzheimer's disease. Brain Res 401: 231–238. [DOI] [PubMed] [Google Scholar]
- Pascual J, Grijalba B, Garcia-Sevilla JA, Zarranz JJ, Pazos A (1992) Loss of high-affinity α2-adrenoceptors in Alzheimer's disease: an autoradiographic study in frontal cortex and hippocampus. Neurosci Lett 142: 36–40. [DOI] [PubMed] [Google Scholar]
- Perry EK, Blessed G, Tomlinson BE, Perry RH, Crow TJ, Cross AJ, Dockray GJ, Dimaline R, Arregui A (1981) Neurochemical activities in human temporal lobe related to aging and Alzheimer-type changes. Neurobiol Aging 2: 251–256. [DOI] [PubMed] [Google Scholar]
- Piascik M, Perez DM (2001) α1-Adrenergic receptors: New insights and directions. J Pharmacol Exp Ther 298: 403–410. [PubMed] [Google Scholar]
- Price JL, Ko AL, Wade MJ, Tsou SK, Mckeel DW, Morris JC (2001) Neuron number in the entorhinal cortex and CA1 in preclinical Alzheimer's disease. Arch Neurol 58: 1395–1402. [DOI] [PubMed] [Google Scholar]
- Raskind MA, Peskind ER, Halter JB, Jimerson DC (1984) Norepinephrine and MHPG levels in CSF and plasma in Alzheimer's disease. Arch Gen Psychiatry 41: 343–346. [DOI] [PubMed] [Google Scholar]
- Reinikainen KJ, Paljarvi L, Huuskonen M, Soininen H, Laakso M, Reikkinen P (1988) A post-mortem study of noradrenergic, serotonergic and GABAergic neurons in Alzheimer's disease. J Neurol Sci 84: 101–116. [DOI] [PubMed] [Google Scholar]
- Rossler M, Zarski R, Bohl J, Ohm TG (2002) Stage-dependent and sector-specific neuronal loss in hippocampus during Alzheimer's disease. Acta Neuropathol 103: 363–369. [DOI] [PubMed] [Google Scholar]
- Russo-Neustadt A, Zomorodian TJ, Cotman CW (1998) Preserved cerebellar tyrosine hydroxylase-immunoreactive neuronal fibers in a behaviorally aggressive subgroup of Alzheimer's disease patients. Neuroscience 87: 55–61. [DOI] [PubMed] [Google Scholar]
- Sallinen J, Link RE, Haapalinna A, Viitamaa T, Kulatunga M, Sjoholm B, MacDonald E, Pelto-Huikko M, Leino T, Barsh GS, Kobilka BK, Scheinin M (1997) Genetic alteration of α2C-adrenoceptor expression in mice: influence on locomotor, hypothermic, and neurochemical effects of dexmedetomidine, a subtype-nonselective α2-adrenoceptor agonist. Mol Pharmacol 51: 36–46. [DOI] [PubMed] [Google Scholar]
- Sastre M, Garcia-Sevilla JA (1994) Alpha 2-adrenoceptor subtypes identified by [3H]RX821002 binding in the human brain: the agonist guanoxabenz does not discriminate different forms of the predominant alpha 2A subtype. J Neurochem 63: 1077–1085. [DOI] [PubMed] [Google Scholar]
- Schwinn DA, Lomasney JW, Lorenz W, Szklut PJ, Fremeau RT, Yang-Feng TL, Caron MG, Lefkowitz RJ, Cotecchis S (1990) Molecular cloning and expression of the cDNA for a novel α1-adrenergic receptor subtype. J Biol Chem 265: 8183–8189. [PubMed] [Google Scholar]
- Schwinn DA, Johnston GI, Page SO, Mosley MJ, Wilson KH, Worman NP, Campbell S, Fidock MD, Furness LM, Parry-Smith DJ, Peter B, Bailey DS (1995) Cloning and pharmacological characterization of human alpha-1 adrenergic receptors: sequence corrections and direct comparison with other species homologues. J Pharmacol Exp Ther 272: 134–142. [PubMed] [Google Scholar]
- Shimohama S, Taniguchi T, Fujiwara M, Kameyama M (1986) Biochemical characterization of α-adrenergic receptors in human brain and changes in Alzheimer-type dementia. J Neurochem 47: 1295–1301. [PubMed] [Google Scholar]
- Simic G, Kostovic I, Winblad B, Bogdanovic N (1997) Volume and number of neurons of the human hippocampal formation in normal aging and Alzheimer's disease. J Comp Neurol 379: 482–494. [DOI] [PubMed] [Google Scholar]
- Szot P, White SS, Veith RC (1997) Effect of pentylenetetrazol on the expression of tyrosine hydroxylase mRNA and norepinephrine and dopamine transporter mRNA. Mol Brain Res 44: 46–54. [DOI] [PubMed] [Google Scholar]
- Szot P, Leverenz JB, Peskind ER, Kiyasu E, Rohde K, Miller MA, Raskind MA (2000) Tyrosine hydroxylase and norepinephrine transporter mRNA expression in the locus coeruleus in Alzheimer's disease. Mol Brain Res 84: 135–140. [DOI] [PubMed] [Google Scholar]
- Szot P, White SS, Greenup JL, Leverenz JB, Peskind ER, Raskind MA (2005) α1-adrenoreceptor in human hippocampus: Binding and receptor subtype mRNA expression. Mol Brain Res 139: 367–371. [DOI] [PubMed] [Google Scholar]
- Tejani-Butt SM, Yang J, Zaffer H (1993) Norepinephrine transporter sites are decreased in the locus coeruleus in Alzheimer's disease. Brain Res 631: 147–150. [DOI] [PubMed] [Google Scholar]
- Tohgi H, Ueno M, Abe T, Takahashi S, Nozaki Y (1992) Concentrations of monoamines and their metabolites in the cerebral spinal fluid from patients with senile dementia of the Alzheimer's type and vascular dementia of the Binswanger's type. J Neurol Transm Park Dis Dement Sect 4: 69–77. [DOI] [PubMed] [Google Scholar]
- Tomlinson BE, Irving D, Blessed G (1981) Cell loss in the locus coeruleus in senile dementia of the Alzheimer's type. J Neurol Sci 49: 419–428. [DOI] [PubMed] [Google Scholar]
- Van Gaalen M, Kawahara H, Kawahara Y, Westerink BHC (1997) The locus coeruleus noradrenrgic system in the rat brain studied by dual-probe microdialysis. Brain Res 763: 56–62. [DOI] [PubMed] [Google Scholar]
- Weinberg DH, Trivedi P, Tan CP, Mitra S, Perkins-Barrow A, Borkowski D, Strader CD, Bayne M (1994) Cloning, expression and characterization of human α adrenergic receptors α1A, α1B and α1C Biochem Biophys Res Commun 201: 1296–1304. [DOI] [PubMed] [Google Scholar]