

Abstract
Alix/apoptosis-linked gene-2 (ALG-2)-interacting protein X is an adaptor protein involved in the regulation of the endolysosomal system through binding to endophilins and to endosomal sorting complexes required for transport (ESCRT) proteins, TSG101 and CHMP4b. It was first characterized as an interactor of ALG-2, a calcium-binding protein necessary for cell death, and several observations suggest a role for Alix in controlling cell death. We used electroporation in the chick embryo to test whether overexpressed wild-type or mutated Alix proteins influence cell death in vivo. We show that Alix overexpression is sufficient to induce cell death of neuroepithelial cells. This effect is strictly dependent on its capacity to bind to ALG-2. On the other hand, expression of Alix mutants lacking the ALG-2 or the CHMP4b binding sites prevents early programmed cell death in cervical motoneurons at day 4.5 of chick embryo development. This protection afforded by Alix mutants was abolished after deletion of the TSG101, but not of the endophilin, binding sites. Our results suggest that the interaction of the ALG-2/Alix complex with ESCRT proteins is necessary for naturally occurring death of motoneurons. Therefore, Alix represents a molecular link between the endolysosomal system and the cell death machinery.
Keywords: Alix/AIP1, ALG-2, spinal motoneurons, programmed cell death, ESCRT, embryo
Introduction
Endocytosis and autophagy can be increased massively during neuronal death in the embryo and in the adult, suggesting that activation of the endolysosomal system is instrumental for some types of cell demise. The endolysosomal system is composed of a series of intracellular compartments within which endocytosed molecules and redundant cellular material are hydrolyzed (Pillay et al., 2002). Endocytosed material tends to flow vectorially through the system from early endosomes to late endosomes and into lysosomes, whereas autophagy provides an alternative entry point. On their way toward degradation, ubiquitinated transmembrane proteins trigger the sequential formation of protein hetero-oligomeric complexes called endosomal sorting complexes required for transport (ESCRT I, II, and III), which allow their incorporation into vesicles budding inside intermediate endosomes called the multivesicular bodies (MVB) (Katzmann et al., 2002; Raiborg et al., 2003). Thereafter, the vesicles and their cargoes will be hydrolyzed after fusion of the MVB with the lysosome. Another key regulator of the budding of the MVB limiting membrane is Alix/apoptosis-linked gene-2 (ALG-2)-interacting protein X (AIP1) (Matsuo et al., 2004). This function may be related to the capacity of the protein to bind to lysobisphosphatidic acid and endophilins, both known to regulate the curvature of lipid bilayers (Ringstad et al., 1997; Schmidt et al., 1999; Huttner and Schmidt, 2000; Chatellard-Causse et al., 2002) and to TSG101 and CHMP4b, from ESCRT I and ESCRT III, respectively (Martin-Serrano et al., 2003; Strack et al., 2003; von Schwedler et al., 2003).
Alix was first characterized as an interactor of ALG-2 (Vito et al., 1996, 1999; Missotten et al., 1999), a small Ca2+ binding protein, the expression of which has been shown to be necessary for death to occur (Vito et al., 1996). In adult rats, we have demonstrated recently an early increase of Alix expression in regions of kainate-induced neurodegeneration, as well as in the degenerating striatum in a model of Huntington's disease (Blum et al., 2004; Hemming et al., 2004). This upregulation of Alix may be the cause of cell death, because Alix overexpression induces apoptosis of cultured cerebellar granule cells. On the other hand, expression of the C-terminal half of the protein (Alix-CT), known to block the ESCRT machinery, ensures survival of the same neurons (Trioulier et al., 2004).
Here, we used electroporation in the chick embryo to test whether the expression of wild-type or mutated forms of Alix modulates cell death in vivo. We observed that Alix overexpression killed neuroepithelial cells, whereas Alix-CT expression blocked naturally occurring cell death (NOCD) in motoneurons. Deletion of the ALG-2 binding site abrogated Alix-CT protection and switched full-length Alix from being a death inducer to become an inhibitor of NOCD. We also found that Alix-CT and AlixΔALG-2 death-blocking activities were abolished by removal of the four amino acids necessary for TSG101/ESCRT-I binding, thereby strongly suggesting that the proteins need to bind the ESCRT proteins to inhibit cell death. Therefore, our results show that proteins controlling the endolysosomal system are central in NOCD and suggest that the Alix/ALG-2 complex mediates cell death by recruiting this system.
Materials and Methods
In ovo electroporation. Fertilized Isa Brown eggs (obtained from Société Française de Production avicole, St. Marcellin, France) were incubated at 38°C in a humidified atmosphere for 2.5 d until reaching Hamburger Hamilton (HH) stage 16. Solutions of plasmid DNA were resuspended in 0.05% fast green, 0.33% carboxymethylcellulose in HBSS at a final concentration of 2 μg/μl. After injection into the lumen of the closed neural tube, plasmid DNA was electroporated into cells on one side of the neural tube by electrodes flanking the embryo. Square-wave currents (six 50 ms pulses of 30–50 V) were generated using an Intracept TSS10 (Intracell, Shepreth, UK) electroporator connected to platinum electrodes.
DNA constructs. Alix mouse cDNAs, wild type or mutants (Fig. 1), and enhanced green fluorescent protein (EGFP) cDNA were inserted blunt into the blunted EcoRI site of the pCAGGS expression vector, a kind gift from Tsuyoshi Momose (Nara Institute of Science and Technology, Japan). The vector uses a chick β-actin promoter and a Rous sarcoma virus enhancer. Alix-CT mutants, AlixΔALG-2, and AlixΔALG-2ΔTSG101 were all cloned in fusion with EGFP protein.
Figure 1.

Schematic representation of Alix and its interactors. The mutant proteins used and the effects on cell death are described. Amino acid positions or deletions are indicated. Vertical stripes, Bro1 domain; oblique stripes, proline-rich domain.
Western blotting. To compare chicken and rodent ALG-2, chicken extracts were prepared from whole HH stage 24 embryos by homogenization in 100 μl of 2× loading buffer. Baby hamster kidney (BHK) cells were transfected to overexpress mouse ALG-2 by JetPEI (Polyplus; Ozyme, St. Quentin-en-Yvelines, France) according to the manufacturer's instructions. Lysates were prepared in the following buffer [150 mm NaCl, 50 mm Tris, pH 8.0, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS, and 2× proteases inhibitor mixture (Roche, Meylan, France)] and loaded onto a 12% SDS-polyacrylamide gel before transfer to a nylon membrane (Immobilon P; Millipore, Bedford, MA). ALG-2 was detected with a polyclonal antibody (Swant, Bellinzona, Switzerland) followed by incubation with goat anti-rabbit horseradish peroxidase-conjugated secondary antibody and revealed by chemoluminescence.
Histological analysis. Chick embryos were collected 24, 48, or 72 h after electroporation and fixed in 4% paraformaldehyde, 0.12 m phosphate buffer, 0.12 mm CaCl2, and 4% sucrose. EGFP expression was visualized by UV illumination using FITC filters. For cryosectioning, samples were incubated in 0.12 m phosphate buffer, containing 15% sucrose for at least 24 h at 4°C. They were embedded in 7.5% gelatin and frozen in nitrogen-chilled isopentane for 1 min at –65°C. Serial sections (8 or 12 μm) were cut with a cryostat and stored at –20°C before use.
Immunofluorescence or immunoperoxidase. Frozen sections were brought to room temperature and saturated in 3% goat serum (GS) in 50 mm Tris, 155 mm NaCl, pH 7.6 (TBS), for 30 min at 37°C. Sections were incubated with the primary antibody for 12–24 h at 4°C. Polyclonal anti-Alix antibody (Chatellard-Causse et al., 2002) was used 1:200 in 1% GS/0.02% saponine in TBS (TBSS). Polyclonal anti-ALG-2 antibody (Swant) was used 1:100 in 1% GS in TBSS. Sections were rinsed five times in TBSS and then treated with the secondary antirabbit Alexa594 antibody (The Jackson Laboratory, Bar Harbor, ME) or a biotinylated goat anti-rabbit secondary antibody (Vector Laboratories, Burlingame, CA), amplified using the ABC kit (Vector Laboratories), and revealed with 3, 3′-diaminobenzidine and nickel intensification. Sections were rinsed in TBS, incubated 30 min at 37°C in Hoechst 33342, 2 μg/ml (Sigma, St. Louis, MO), before being mounted in Mowiol (Calbiochem, La Jolla, CA).
For Islet-1 staining, 8 μm sections were incubated with anti-Islet-1 monoclonal antibody (Developmental Studies Hybridoma Bank, Iowa City, IA) in 1% GS in TBSS and revealed by anti-mouse Alexa594 secondary antibody (The Jackson Laboratory). Islet-1-positive cells were counted on every third section from five embryos (12 sections per embryo).
Terminal deoxynucleotidyl transferase-mediated dUTP-biotin nick end labeling method. To visualize DNA fragmentation, the terminal deoxynucleotidyl transferase-mediated dUTP-biotin nick end labeling (TUNEL) method described by Gavrieli et al. (1992) was used. Sections were permeabilized in a solution composed of 0.1% Triton X-100 in 0.1% citrate buffer, pH 6.0, and then washed in Tris-base saline buffer before incubation with terminal deoxynucleotide transferase (In Situ Cell Death Detection kit; Roche) for 1 h at 37°C in a solution containing TMR red dUTP. Sections were observed using a fluorescence microscope, and positive cells were counted in every third section. Twelve sections per embryo were counted. n = total number of sections analyzed. The average numbers of TUNEL-positive cells in each half of the neural tube were compared statistically with Student's t test as specified in the figure legends.
5-Bromo-2′ deoxyuridine administration and immunohistochemistry. Two hundred microliters of the S-phase marker 5-bromo-2′ deoxyuridine (BrdU; 5 mg/ml) in PBS were dropped onto the vitelline membrane 24 h after electroporation. Embryos were killed 2 h after BrdU administration and then fixed and embedded for cryosectioning.
Immunodetection was performed as described above after pretreatment with 2N HCl for 30 min at 37°C, to denature double-stranded DNA, followed by five rinses in 0.1 m borate buffer, pH 8.5. The sections were saturated and treated with 6 mg/ml of mouse monoclonal antiBrdU antibody (Roche) in 1% GS in TBS for 24 h, 4°C. An anti-mouse Alexa594 antibody (The Jackson Laboratory) was used as secondary antibody. To estimate the number of BrdU-labeled cells, the total fluorescence intensity per unit area was measured in each half of the neural tube in every third section by MetaMorph software (Universal Imaging Corporation, West Chester, PA). The average intensity (arbitrary units) was then calculated for the nonelectroporated or electroporated half of the neural tube of 6–10 embryos, with 12 sections per embryo. The means were statistically compared between control (GFP-transfected) or Alix-transfected embryos using Student's t test.
Results
Alix overexpression in the developing chick spinal cord
Expression plasmids coding for Alix and/or GFP were electroporated into the chick developing neural tube at HH stage 16. Alix was detected with a polyclonal anti-Alix antibody specifically recognizing both chick and mouse Alix (Chatellard-Causse et al., 2002; Fraboulet et al., 2003). In HH stage 21 embryos, 24 h after electroporation of GFP alone, endogenous Alix is mainly detected in distal parts of commissural and motoneuron axons (Fig. 2A,B). After electroporation of the two plasmids, 80% of the cells on the transfected side of the neural tube express both Alix and GFP (Fig. 2C,D). Overexpressed Alix is also detectable in the processes of motoneurons extending out of the ventral neural tube (Fig. 2D,F) and in neural crest derivatives such as the dorsal root ganglia and melanocytes (Fig. 2D).
Figure 2.
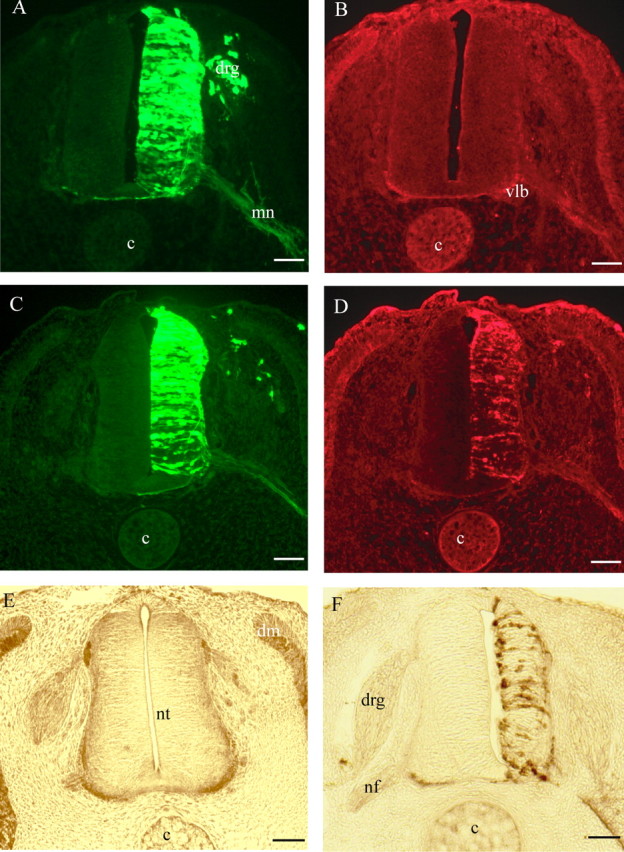
Cross sections of electroporated neural tubes from chick embryos. A–D, HH stage 21 embryos electroporated with the following pCAGGS expression vectors: GFP (A, B) and GFP plus Alix (C, D). A, C, GFP 24 h after electroporation in the neural tissue; GFP is also visible in dorsal root ganglia (drg) and in the axons from motoneurons (mn). Vlb, Ventrolateral border; c, cord. B, D, Immunofluorescence revealing endogenous and overexpressed Alix, respectively, by using polyclonal anti-Alix antibody recognizing endogenous chick and overexpressed mouse Alix. Scale bars, 50 μm. E, F, HH stage 24 embryos, 48 h after electroporation, immunostained with polyclonal anti-Alix revealed by peroxidase. E, Control embryo transfected with GFP pCAGGS plasmid alone. Endogenous Alix is strongly expressed ventrally and laterally along the border of the neural tube (nt). Alix is also expressed in the nerve fibers (nf) and in the dermomyotome (dm). F, Alix-transfected embryo. The neuroepithelium of the electroporated side is clearly reduced in size. Revelation was shorter in F compared with E to avoid saturation of the overexpressed protein and reducing background. Scale bars, 50 μm.
In sections of HH stage 24 embryos transfected 48 h earlier, we observed that overexpression of Alix reduced the width of the neuroepithelium by ∼25% compared with the untransfected side (Fig. 2F). We could not detect any effect of GFP overexpression under the same conditions (Fig. 2E).
Alix overexpression does not affect cell proliferation in the neural tube
To determine whether the distortion of the epithelium was caused by reduced neuroblast proliferation, we performed BrdU incorporation in HH stage 21 embryos, 24 h after electroporation. Numerous BrdU-positive cells were observed on both sides of the neural tube after transfection with GFP alone (Fig. 3B) or with GFP and Alix (Fig. 3D). There was no significant difference in fluorescence intensity caused by BrdU incorporation between the control and the electroporated half of the neural tube in Alix- or in GFP-electroporated embryos (Fig. 3E).
Figure 3.
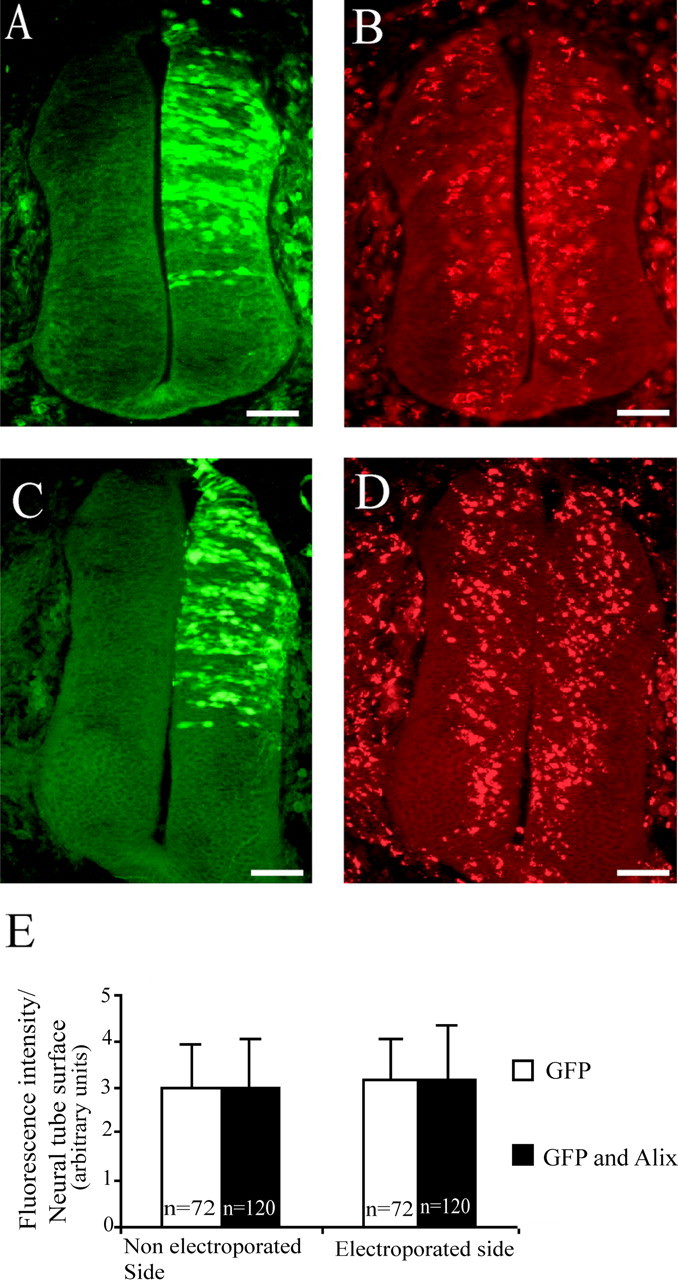
Cross sections of HH stage 21 embryo neural tubes 24 h after electroporation with pCAGGS GFP (A, B) or pCAGGS GFP and pCAGGS Alix (C, D). BrdU was applied to the vitelline membrane for 2 h. A, C, GFP expression 24 h after electroporation. B, D, BrdU immunofluorescence. Scale bars, 50 μm. E, Quantitative analysis of fluorescence intensity per neural tube surface. There was no significant difference between the control and the electroporated half of the neural tube in Alix and GFP or in GFP-only overexpressing embryos. n = number of sections analyzed. Error bars represent SD.
These results demonstrate that the reduced width of the neuroepithelium observed at HH stage 24 is not caused by an effect of Alix on the proliferation of the progenitors.
Alix overexpression leads to a specific increase in the number of apoptotic cells in the transfected neuroepithelium
To determine whether Alix overexpression leads to increased cell death, we looked for changes in nuclear morphology using Hoechst fluorescent staining of chromatin. We clearly observed many nuclei showing marginalization and condensation of chromatin in the Alix-overexpressing neuroepithelia that were almost completely absent in GFP-only transfected embryos (data not shown). The TUNEL method was used to quantify the presence of apoptotic cells in Alix-overexpressing neuroepithelia. Figure 4 shows that the number of TUNEL-positive cells observed in neural tubes 24 h after electroporation was at least three times higher in Alix-overexpressing tissues (Fig. 4C–E,I) than in control tissues at HH stage 21 (Fig. 4A,B,I). Therefore, overexpression of Alix is sufficient to induce cell death in the neuroepithelium.
Figure 4.

Cross sections of HH stage 21 chick embryo neural tubes electroporated with the following pCAAGS expression vectors: GFP (A, B), GFP plus Alix (C–E), and GFP plus AlixΔALG-2 (F–H). C, F, Immunofluorescence using anti-Alix antibody (A, D, G) expression of GFP 24 h after electroporation. B, E, H, TUNEL labeling. Scale bars, 50 μm. I, Mean numbers ± SD of TUNEL-positive cells in the neural tube of electroporated embryos; n = number of sections analyzed; 12 sections per embryo were counted. **p < 0.001, t test. The number of TUNEL-positive cells is significantly increased in Alix-overexpressing tissues. This effect is abolished when using the AlixΔALG-2 mutant.
The ability of Alix to induce apoptosis is dependent on its interaction with ALG-2
Alix is known to interact with the calcium-binding protein ALG-2. Using an anti-ALG-2 polyclonal antibody that recognizes the chick protein (Fig. 5C), we have revealed the endogenous ALG-2 pattern of expression in HH stage 21 and 24 embryos (Fig. 5A,B). Like Alix, the ALG-2 protein is detected in the nerve fibers of motoneurons and in commissural axons of the ventral part of the neural tube (compare Figs. 5A, 2E). Therefore, the expression profile of each protein is consistent with their possible interaction during embryonic development.
Figure 5.

ALG-2 expression during chick neural tube development. Cross sections of HH stage 21 (A) or 24 (B) chick embryo neural tubes immunostained with polyclonal anti-ALG-2. Endogenous ALG-2 is strongly expressed ventrally in a domain overlapping Alix expression (compare with Fig. 2 B). Scale bars, 50 μm. Polyclonal anti-ALG-2 recognizes a single 19.5 kDa product in chick embryo extracts (chicken), as predicted by the sequence data (GenBank accession numberNM_001030993).Endogenous hamster protein is detected as a single band of 20.5 kDa (BHK + mALG-2). Overexpressed mouse Flag-tagged cDNA is detected as a predominant single band of 23 kDa, as predicted by the amino acid sequence.
We have demonstrated previously that the calcium-dependent ALG-2 binding to Alix requires a 12 amino-acid-long stretch of the Alix C-terminal proline-rich domain (PRD) (Trioulier et al., 2004). AlixΔALG-2, which lacks this region, did not increase the number of TUNEL-positive cells (Fig. 4F–I), thereby showing that apoptosis induced in the chick embryo by Alix is strictly dependent on its ability to bind to ALG-2.
Alix-CT expression protects cervical motoneurons from early cell death
We next asked whether the mechanisms regulated by Alix play a role in NOCD. We tested this in the early phase of cervical motoneuron death, which occurs between HH stage 23 and 25. As seen in Figure 6B, TUNEL staining reveals numerous dying cells in the ventral horn of the cervical segments from HH stage 24 embryos. We tested the effect of the expression of Alix-CT, which lacks the CHMP4b binding region but contains the PRD including the sites of interaction with ALG-2, TSG101, and endophilins (Fig. 1). For this, a construct encoding Alix-CT in fusion with GFP (Fig. 1) was transfected at HH stage 16, and the death profiles were analyzed on cervical sections at HH stage 24. The GFP–Alix-CT protein displayed a vesicular distribution (Fig. 6C), as described previously in cultured cells (Chatellard-Causse et al., 2002; Trioulier et al., 2004). We found that the number of TUNEL-positive motoneurons in the ventral horn was reduced by half by GFP–Alix-CT expression (Figs. 6D, 7A) but not by GFP expression alone (Figs. 6B, 7A). Hoechst staining of the chromatin on the same sections revealed fewer condensed nuclei (data not shown). Therefore, at this developmental step, Alix lacking the CHMP4b binding region is able to block DNA fragmentation and nuclear condensation, which characterize programmed cell death. In contrast, expression of the N-terminal half of Alix, containing the CHMP4b binding region, had no effect on cell survival (data not shown).
Figure 6.

Cross sections of HH stage 24 chick embryo neural tubes electroporated with the following pCAGGS expression vectors: GFP (A, B), GFP–Alix-CT (C, D), GFP–Alix-CTΔALG-2 (E, F), GFP–Alix-CTΔendophilin (G, H), and GFP–Alix-CTΔTSG101 (I, J). A, C, E, G, I, GFP expression 48 h after electroporation. B, D, F, H, J, Corresponding TUNEL labeling. Scale bars, 40 μm.
Figure 7.
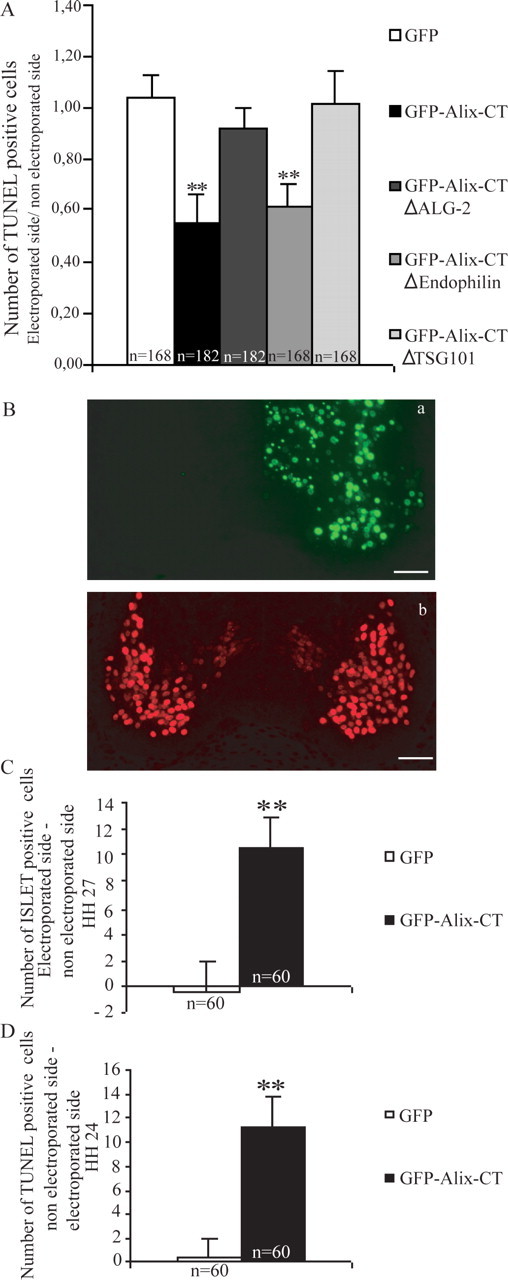
Alix mutants rescue motoneurons from NOCD. A, Ratio of TUNEL-positive cells in the electroporated versus nonelectroporated side of the neural tube. Mean ± SD; n = number of sections analyzed; 12 sections per embryo were counted. **p < 0.001, t test. The number of TUNEL-positive cells was significantly reduced in GFP–Alix-CT- and GFP–Alix-CTΔendophilin-overexpressing embryos. B–D, Evaluation of supernumerary motoneurons on the electroporated side. B, Immunofluorescence staining using an anti-Islet-1 antibody (Bb) on cross section of HH stage 27 chick cervical neural tube electroporated with GFP-Alix-CT (Ba; same section). Scale bars, 40μm. C, Mean numbers ± SD of Islet-1-positive motoneurons supernumerary on HH stage 27 cross sections (B). n = number of sections analyzed; 12 sections were counted per embryo. **p < 0.001, t test. D, Mean numbers ± SD counted after TUNEL staining on HH stage 24 cross sections (see Fig. 6 D).
To test whether the reduction in TUNEL-positive cells seen with Alix-CT really turns into a protective effect, we quantified the number of motoneurons at HH stage 27, using an immunostaining for Islet-1, a transcription factor specific for motoneurons (Tsuchida et al. 1994). At this stage, we could not detect any TUNEL-positive cells (data not shown), in good agreement with the previous demonstration that NOCD of motoneurons ends 24 h earlier. In HH stage 27 embryos, transfected at HH stage 16, we found that the side expressing Alix-CT contained on average per section, 10 more Islet-1-positive nuclei than the nonelectroporated side (Fig. 7C). In contrast, no difference could be found in embryos transfected with GFP (Fig. 7C). It is remarkable that the number of 10 supernumerary motoneurons fits almost exactly with the decrease in TUNEL-positive dying cells counted at stage 24 (Fig. 7D). This demonstrates that the reduction in TUNEL positivity at HH stage 24 is a fair estimate of cell survival afforded by Alix-CT.
Deletion of the ALG-2 binding site abrogates the ability of Alix-CT to protect motoneurons from early cell death
Embryos were transfected with an expression vector encoding GFP in fusion with Alix-CT lacking the ALG-2 binding domain (GFP–Alix-CTΔALG-2) (Fig. 1). As illustrated in Figures 6F and 7, TUNEL positivity was significantly higher compared with Alix-CT (Fig. 6D) and was not significantly different from that observed with GFP alone (Fig. 6B). In contrast, deletion of the 14 amino-acid-long stretch of the PRD that is responsible for endophilin binding (Alix-CTΔendophilin) had no effect on Alix-CT activity (Chatellard-Causse et al., 2002) (Figs. 6H, 7). Therefore, the neuroprotective effect of Alix-CT on early death of cervical motoneurons requires the ALG-2 interaction domain.
Full-length Alix deleted of the ALG-2 binding site protects neurons from early cell death
As stated above, AlixΔALG-2 had no proapoptotic activity in the neuroepithelium. We next tested the effect of the mutant on death of motoneurons by using a GFP–AlixΔALG-2, which allowed comparison with GFP–Alix-CT fusion protein. As shown in Figure 8, B and E, AlixΔALG-2 had a similar protecting effect to that of Alix-CT, halving the number of dying motoneurons. Therefore, Alix deleted of its sites of interaction with either ALG-2 or CHMP4b is capable of blocking NOCD.
Figure 8.
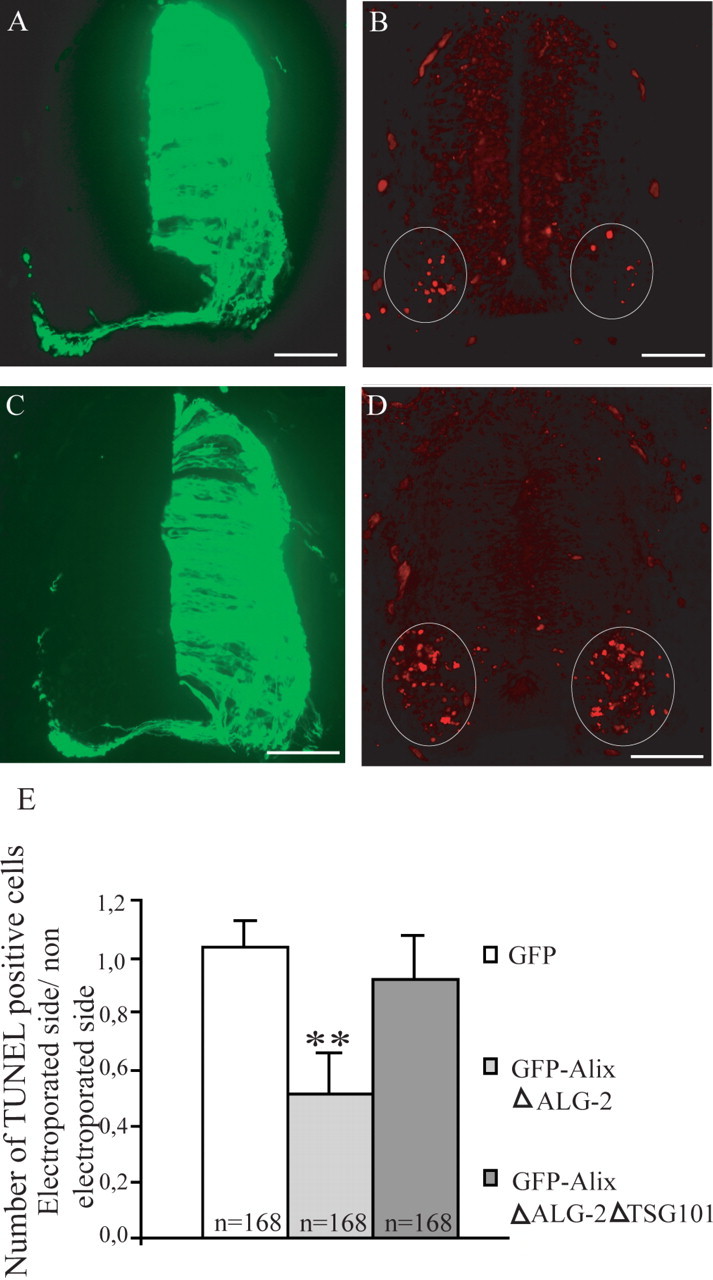
Cross sections of HH stage 21 chick embryo neural tubes electroporated with GFP–AlixΔALG-2 (A, B) and GFP–AlixΔALG-2ΔTSG101 (C, D). A, C, GFP expression 24 h after electroporation. B, D, TUNEL labeling. Scale bars, 45 μm. E, Mean numbers ± SD of TUNEL-positive cells in the neural tube of electroporated embryos, n = number of sections analyzed; 12 sections per embryo were counted. **p < 0.001, t test. The number of TUNEL-positive cells is significantly reduced in GFP–AlixΔALG-2-expressing ventral neural tube. This protection is abolished when using the GFP–AlixΔALG-2ΔTSG101.
Deletion of the TSG101 binding site abrogates the ability of Alix-CT and of AlixΔALG-2 to protect motoneurons from early cell death
Besides binding ALG-2, the proline-rich region of Alix-CT also interacts with TSG101 of ESCRT-I and seems thereby to block the ESCRT machinery. We found that a deletion of four amino acids necessary for Alix binding to TSG101 (von Schwedler et al., 2003) completely prevented Alix-CT from protecting against cell death (Figs. 6J, 7). Similarly, the deletion of the TSG101 binding site from AlixΔALG-2 totally abrogated its rescuing effect (Fig. 8D,E).
Discussion
Here, we show using electroporation of the chick neural tube that overexpression of Alix induces apoptosis. Furthermore, expression of truncated Alix proteins lacking the binding region to CHMP4b of ESCRT-III (Alix-CT) or to ALG-2 (AlixΔALG-2) can block the NOCD of cervical motoneurons. Finally, deletion of four amino acids necessary for these proteins to bind TSG101 of ESCRT-I abolishes their anti-cell-death activity, thereby demonstrating that an intact Alix/ALG-2/ESCRT complex is necessary for neuronal death in vivo.
Electroporation of the neural tube of HH stage 16 embryos allowed us to overexpress Alix in mitotic and postmitotic neuroepithelial cells. Unlike the effect of the Alix human homolog, Hp 95, which has been shown to promote G1 phase arrest in confluent monolayer HeLa cells (Wu et al., 2001), we did not detect any influence of this overexpression on the number of BrdU-incorporating nuclei, thus indicating that Alix does not affect the cell cycle in our model. However, we found that Alix transfection was sufficient to induce apoptosis of the neuroepithelial cells, a result in accordance with our previous observation that enforced Alix expression is sufficient to induce caspase activation and death of cultured cerebellar granule neurons (Trioulier et al., 2004). Alix proapoptotic activity in the neural tube required binding to ALG-2 as a mutant deleted of 12 amino acids within the PRD responsible for ALG-2 binding (Trioulier et al., 2004) was no longer deleterious. Even more interestingly, we found that enforced expression of Alix deleted of its ALG-2 binding site (Alix-ΔALG-2) or of its NH2 terminal half (Alix-CT) protected neurons of the cervical ventral horn from NOCD.
Alix is a main regulator of the endolysosomal system, in particular through binding to a complex called ESCRT, which is necessary for the formation of MVB. MVBs are intermediate endosomes in which membrane proteins en route to the lysosome are sorted into vesicles budding from the endosome-limiting membrane. The vesicles and their cargoes are degraded once the MVB has fused with the lysosome. Several studies in yeast suggest that the sorting of proteins requires the sequential association of three hetero-oligomeric complexes (ESCRT-I, ESCRT-II, and ESCRT-III) on the surface of endosomes (Jiang et al., 2002; Katzmann et al., 2002; Raiborg et al., 2003). TSG101, which is part of ESCRT-I, associates with the four amino acid PSAP (proline, serine, alanine, proline) motif of the Alix PRD domain, whereas CHMP4b, which belongs to ESCRT-III, binds to the first 370 amino acids, the Bro-1 domain, conserved throughout species (Fig. 1) (Martin-Serrano et al., 2003; Strack et al., 2003; von Schwedler et al., 2003; Kim et al., 2005).
Recruitment of Alix to endosomes is known to occur through binding to CHMP4b (Katoh et al., 2003). Several laboratories have shown recently that expression of Alix truncated of its CHMP4b binding site disrupts the ESCRT function in a similar way to Alix downregulation by small interfering RNA (Martin-Serrano et al., 2003). Our finding that Alix mutants lacking either the CHMP4b/ESCRT-III interacting region or the ALG-2 binding site inhibit NOCD suggests that they behave as dominantnegative mutants blocking the formation of an ALG-2/Alix/CHMP4b complex necessary for cell death. This is reinforced by the finding that removal of the ALG-2 binding site on Alix-CT abolished its anti-cell-death activity. A third necessary interactor seems to be TSG101 of ESCRT-I, because deletion of PSAP necessary for its interaction abolished the anti-cell-death activities of both Alix-CT and AlixΔALG-2.
We thus demonstrate for the first time the requirement of an intact ALG-2/Alix/ESCRT complex for neuronal death. One may hypothesize that this complex acts at the level either of the signaling or of the execution of cell death. Alix could regulate signaling endosomes, which are thought to mediate the retrograde communication of neurotrophic factors from axon terminals contacting the target to cell bodies (Howe and Mobley, 2005). However, this is an unlikely scenario, because the cell-death-blocking effect of Alix mutants was observed during the early, short, and synchronized period of NOCD of cervical neurons between HH stages 23 and 26 (Levi-Montalcini, 1950). This NOCD, which affects somatic motoneurons sending axons to the cervical somitic region, is not reduced by neuromuscular blocking or neurotrophic agents (Levi-Montalcini, 1950; Yaginuma et al., 1996). Furthermore, Alix-CTΔendophilin still affords protection, thereby showing that Alix-CT does not need the interaction with endophilins, which are main regulators of tyrosine kinase receptor endocytosis. Another hypothesis is that Alix/ALG-2 may influence cell death by acting on endocytosed death receptors. Indeed, Schneider-Brachert et al. (2004) have demonstrated recently, in lymphoid cell lines, that the initiating caspase 8, which is recruited by the DISC (death-inducing signaling complex) forming around the activated death receptor tumor necrosis factor (TNF) receptor 1 (TNFR1), is only activated after the receptor has been endocytosed, thereby introducing the notion of death inducing signaling endosomes. Interestingly TNFR1 is transiently detected in early developing mouse motoneurons, and TNFα has been shown to commit motoneurons to die later during development (Sedel et al., 2004).
We now need to study whether Alix interacts, directly or indirectly, with caspases for the execution of the death program. A link between ALG-2/Alix and caspases has been suggested in mouse embryonic fibroblasts by the finding that ALG-2 immunoprecipitates with caspase-12 and caspase-9 after stress to the endoplasmic reticulum. In the same report, Rao et al. (2004) showed that Apaf 1–/–cells depleted of ALG-2 were protected against thapsigargin, which induces a rise in cytosolic calcium but not against other endoplasmic reticulum stressors such as tunicamycin or brefeldin. Given the tight calcium-dependent interaction of Alix with ALG-2, our results beg the question of the presence of Alix within caspase-containing complexes and thereby of its direct role in activation of the caspase cascade. If this is the case, Alix/ALG2 would be ideally suited to act as a caspase activation platform regulated by endocytosed receptors.
However, the sole inhibition of caspase activation cannot explain the protective role of Alix mutants on neuronal death. Indeed, in most cells, caspase inhibition only allows maintenance of an intact nucleus without impairing the dismantling of the cytoplasm, suggesting that caspase-independent pathways are responsible for cytoplasmic destruction. This is the case for cultured cerebellar neurons deprived of potassium, which are not rescued by caspase inhibition but which survive when expressing Alix-CT (Trioulier et al., 2004). This is also true during the first wave of NOCD studied here, because Oppenheim et al. observed that caspase inhibition does not block motoneuron death (Milligan et al., 1995; Yaginuma et al., 1996, 2001; Oppenheim et al., 2001). The Alix-CT cell-death-blocking effect was sustained up to 3 d after transfection and 24 h beyond the expected time of programmed cell death, suggesting that the truncated protein not only blocks caspase-dependent, but also caspase-independent, pathways underlying cell death. These latter remain ill defined, although autophagy has often been designated as a possible way for cytoplasmic destruction (Clarke, 1990). Indeed, numerous autophagic vacuoles accumulate in the cytoplasm of dying cells in which caspases are inhibited, as in the case of cervical neurons treated in situ with caspase inhibitors (Yaginuma et al., 2001). Noteworthy is that Nara et al. (2002) have shown that ESCRT proteins also regulate autophagy, suggesting a close link between MVBs and autophagosomes. Therefore, Alix, as a main regulator of the endolysosomal system and of neuronal death, may represent a good lead to follow to better understand the molecular pathways allowing the caspase-independent destruction of the cytoplasm.
The clear link between the molecular machinery controlling the endolysosomal system and cell death in vivo, which we demonstrate here using Alix, could explain numerous observations of endosomal alterations in a wide range of neurodegenerative diseases, including Alzheimer's disease and several genetic motoneuron diseases, in which mutations occur in genes that regulate endosome function (Nixon, 2005; Reid et al., 2005; Skibinski et al., 2005).
Footnotes
This work was supported in part by the Institut National de la Santé et de la Recherche Médicale, the University Joseph Fourier, a grant from the Association Française contre les Myopathies, and funds from the Association pour la Recherche contre le Cancer. We thank Dr. Karin Sadoul for invaluable help with this manuscript, David Béal for technical support, and Dr. Philippe Mahul for helping with statistics.
Correspondence should be addressed to Rémy Sadoul, Laboratoire Neurodégénérescence et Plasticité, Equipe Mixte INSERM 0108, Pavillon de Neurologie, Centre Hospitalier Universitaire de Grenoble, boite postale 217, 38043 Grenoble Cedex 9, France. E-mail: remy.sadoul@ujf-grenoble.fr.
DOI:10.1523/JNEUROSCI.3069-05.2006
Copyright © 2006 Society for Neuroscience 0270-6474/06/260542-08$15.00/0
S.F. and R.S. contributed equally to this work.
References
- Blum D, Hemming FJ, Galas MC, Torch S, Cuvelier L, Schiffmann SN, Sadoul R (2004) Increased Alix (apoptosis-linked gene-2 interacting protein X) immunoreactivity in the degenerating striatum of rats chronically treated by 3-nitropropionic acid. Neurosci Lett 368: 309–313. [DOI] [PubMed] [Google Scholar]
- Chatellard-Causse C, Blot B, Cristina N, Torch S, Missotten M, Sadoul R (2002) Alix (ALG-2 interacting protein X), a protein involved in apoptosis, binds to endophilins and induces cytoplasmic vacuolization. J Biol Chem 28: 29108–29115. [DOI] [PubMed] [Google Scholar]
- Clarke PG (1990) Developmental cell death: morphological diversity and multiple mechanisms. Anat Embryol (Berl) 181: 195–213. [DOI] [PubMed] [Google Scholar]
- Fraboulet S, Hemming FJ, Mahul AL, Cristina N, Sadoul R (2003) Expression of Alix protein during early avian development. Gene Expr Patterns 3: 139–142. [DOI] [PubMed] [Google Scholar]
- Gavrieli Y, Sherman Y, Ben-Sasson SA (1992) Identification of programmed cell death in situ via specific labeling of nuclear DNA fragmentation. J Cell Biol 119: 493–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemming FJ, Fraboulet S, Blot B, Sadoul R (2004) Early increase of apoptosis-linked gene-2 interacting protein X in areas of kainate-induced neurodegeneration. Neuroscience 123: 887–895. [DOI] [PubMed] [Google Scholar]
- Howe CL, Mobley WC (2005) Long-distance retrograde neurotrophic signaling. Curr Opin Neurobiol 15: 40–48. [DOI] [PubMed] [Google Scholar]
- Huttner WB, Schmidt A (2000) Lipids, lipid modification and lipid-protein interaction in membrane budding and fission–insights from the roles of endophilin A1 and synaptophysin in synaptic vesicle endocytosis. Curr Opin Neurobiol 10: 543–551. [DOI] [PubMed] [Google Scholar]
- Jiang L, Erickson A, Rogers J (2002) Multivesicular bodies: a mechanism to package lytic and storage functions in one organelle? Trends Cell Biol 12: 362–367. [DOI] [PubMed] [Google Scholar]
- Katoh K, Shibata H, Suzuki H, Nara A, Ishidoh K, Kominami E, Yoshimori T, Maki M (2003) The ALG-2-interacting protein Alix associates with CHMP4b, a human homologue of yeast Snf7 that is involved in multivesicular body sorting. J Biol Chem 278: 39104–39113. [DOI] [PubMed] [Google Scholar]
- Katzmann DJ, Odorizzi G, Emr SD (2002) Receptor downregulation and multivesicular-body sorting. Nat Rev Mol Cell Biol 3: 893–905. [DOI] [PubMed] [Google Scholar]
- Kim J, Sitaraman S, Hierro A, Beach BM, Odorizzi G, Hurley JH (2005) Structural basis for endosomal targeting by the Bro1 domain. Dev Cell 8: 937–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levi-Montalcini (1950) The origin and development of the visceral system in the spinal cord of the chick embryo. J Morphol 86: 253–284. [Google Scholar]
- Martin-Serrano J, Yaravoy A, Perez-Caballero D, Bieniasz PD (2003) Divergent retroviral late-budding domains recruit vacuolar protein sorting factors by using alternative adaptor proteins. Proc Natl Acad Sci USA 100: 12414–12419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuo H, Chevallier J, Mayran N, Le Blanc I, Ferguson C, Faure J, Blanc NS, Matile S, Dubochet J, Sadoul R, Parton RG, Vilbois F, Gruenberg J (2004) Role of LBPA and Alix in multivesicular liposome formation and endosome organization. Science 303: 531–534. [DOI] [PubMed] [Google Scholar]
- Milligan CE, Prevette D, Yaginuma H, Homma S, Cardwell C, Fritz LC, Tomaselli KJ, Oppenheim RW, Schwartz LM (1995) Peptide inhibitors of the ICE protease family arrest programmed cell death of motoneurons in vivo and in vitro. Neuron 15: 385–393. [DOI] [PubMed] [Google Scholar]
- Missotten M, Nichols A, Rieger K, Sadoul R (1999) Alix, a novel mouse protein undergoing calcium-dependent interaction with the apoptosis-linked-gene 2 (ALG-2) protein. Cell Death Differ 6: 124–129. [DOI] [PubMed] [Google Scholar]
- Nara A, Mizushima N, Yamamoto A, Kabeya Y, Ohsumi Y, Yoshimori T (2002) SKD1 AAA ATPase-dependent endosomal transport is involved in autolysosome formation. Cell Struct Funct 27: 29–37. [DOI] [PubMed] [Google Scholar]
- Nixon RA (2005) Endosome function and dysfunction in Alzheimer's disease and other neurodegenerative diseases. Neurobiol Aging 26: 373–382. [DOI] [PubMed] [Google Scholar]
- Oppenheim RW, Flavell RA, Vinsant S, Prevette D, Kuan CY, Rakic P (2001) Programmed cell death of developing mammalian neurons after genetic deletion of caspases. J Neurosci 21: 4752–4760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pillay CS, Elliott E, Dennison C (2002) Endolysosomal proteolysis and its regulation. Biochem J 363: 417–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raiborg C, Rusten TE, Stenmark H (2003) Protein sorting into multivesicular endosomes. Curr Opin Cell Biol 15: 446–455. [DOI] [PubMed] [Google Scholar]
- Rao RV, Poksay KS, Castro-Obregon S, Schilling B, Row RH, Del Rio G, Gibson BW, Ellerby HM, Bredesen DE (2004) Molecular components of a cell death pathway activated by endoplasmic reticulum stress. J Biol Chem 279: 177–187. [DOI] [PubMed] [Google Scholar]
- Reid E, Connell J, Edwards TL, Duley S, Brown SE, Sanderson CM (2005) The hereditary sastic paraplegia protein spastin interacts with the ESCRT-III complex-associated endosomal protein CHMP1B. Human Mol Genet 14: 19–38. [DOI] [PubMed] [Google Scholar]
- Ringstad N, Nemoto Y, De Camilli P (1997) The SH3p4/Sh3p8/SH3p13 protein family: binding partners for synaptojanin and dynamin via a Grb2-like Src homology 3 domain. Proc Natl Acad Sci USA 94: 8569–8574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt A, Wolde M, Thiele C, Fest W, Kratzin H, Podtelejnikov AV, Witke W, Huttner WB, Soling HD (1999) Endophilin I mediates synaptic vesicle formation by transfer of arachidonate to lysophosphatidic acid. Nature 401: 133–141. [DOI] [PubMed] [Google Scholar]
- Schneider-Brachert W, Tchikov V, Neumeyer J, Jakob M, Winoto-Morbach S, Held-Feindt J, Heinrich M, Merkel O, Ehrenschwender M, Adam D, Mentlein R, Kabelitz D, Schütze S (2004) Compartmentalization of TNF receptor 1 signaling: internalized TNF receptosomes as death signaling vesicles. Immunity 21: 415–428. [DOI] [PubMed] [Google Scholar]
- Sedel F, Bechade C, Vyas S, Triller A (2004) Macrophage-derived tumor necrosis factor α, an early developmental signal for motoneuron death. J Neurosci 24: 2236–2246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skibinski G, Parkinson NJ, Brown JM, Chakrabarti L, Llyod SL, Hummerich H, Nielsen JE, Hodges JR, Spillantini MG, Thusgaard T, Brandner S, Brun A, Rossor MN, Gade A, Johannsen P, Sorensen SA, Gydesen S, Fisher EMC, Coollinge J (2005) Mutations in the endosomal ESCRTIII-complex subunit CHMP2B in frontotemporal dementia. Nat Genet 37: 806–808. [DOI] [PubMed] [Google Scholar]
- Strack B, Calistri A, Craig S, Popova E, Gottlinger HG (2003) AIP1/ALIX is a binding partner for HIV-1 p6 and EIAV p9 functioning in virus budding. Cell 114: 689–699. [DOI] [PubMed] [Google Scholar]
- Trioulier Y, Torch S, Blot B, Cristina N, Chatellard-Causse C, Verna JM, Sadoul R (2004) Alix, a protein regulating endosomal trafficking, is involved in neuronal death. J Biol Chem 279: 2046–2052. [DOI] [PubMed] [Google Scholar]
- Tsuchida T, Ensini M, Morton SB, Baldassare M, Edlund T, Jessell TM, Pfaff SL (1994) Topographic organization of embryonic motor neurons defined by expression of LIM homeobox genes. Cell 79: 935–943. [DOI] [PubMed] [Google Scholar]
- Vito P, Lacana E, D'Adamio L (1996) Interfering with apoptosis: Ca2+-binding protein ALG-2 and Alzheimer's disease gene ALG-3. Science 271: 521–525. [DOI] [PubMed] [Google Scholar]
- Vito P, Pellegrini L, Guiet C, D'Adamio L (1999) Cloning of AIP1, a novel protein that associates with the apoptosis-linked gene ALG-2 in a Ca2+-dependent reaction. J Biol Chem 274: 1533–1540. [DOI] [PubMed] [Google Scholar]
- von Schwedler UK, Stuchell M, Muller B, Ward DM, Chung HY, Morita E, Wang HE, Davis T, He GP, Cimbora DM, Scott A, Krausslich HG, Kaplan J, Morham SG, Sundquist WI (2003) The protein network of HIV budding. Cell 114: 701–713. [DOI] [PubMed] [Google Scholar]
- Wu Y, Pan S, Che S, He G, Nelman-Gonzalez M, Weil MM, Kuang J (2001) Overexpression of Hp95 induces G1 phase arrest in confluent HeLa cells. Differentiation 67: 139–153. [DOI] [PubMed] [Google Scholar]
- Yaginuma H, Tomita M, Takashita N, McKay SE, Cardwell C, Yin QW, Oppenheim RW (1996) A novel type of programmed neuronal death in the cervical spinal cord of the chick embryo. J Neurosci 16: 3685–3703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaginuma H, Shiraiwa N, Shimada T, Nishiyama K, Hong J, Wang S, Momoi T, Uchiyama Y, Oppenheim RW (2001) Caspase activity is involved in, but is dispensable for, early motoneuron death in the chick embryo cervical spinal cord. Mol Cell Neurosci 18: 168–182. [DOI] [PubMed] [Google Scholar]