

Abstract
Filamentous tau inclusions in neurons and glia are neuropathological hallmarks of tauopathies. The discovery of microtubule-associated protein tau gene mutations that are pathogenic for a heterogenous group of neurodegenerative disorders, called frontotemporal dementia and parkinsonism linked to chromosome-17 (FTDP-17), directly implicate tau abnormalities in the onset/progression of disease. Although the role of tau pathology in neurons in disease pathogenesis is well accepted, the contribution of glial pathology is essentially unknown. We recently generated a transgenic (Tg) mouse model of tau pathology in astrocytes by expressing the human tau protein under the control of the glial fibrillary acidic protein (GFAP) promoter. Both wild-type and FTDP-17 mutant GFAP/tau Tg animals manifest an age-dependent accumulation of tau inclusions in astrocytes that resembles the pathology observed in human tauopathies. We further demonstrate that both strains of Tg mice manifest compromised motor function that correlates with altered expression of the glial glutamate-aspartate transporter and occurs before the development of tau pathology. Subsequently, the Tg mice manifest additional deficits in neuromuscular strength that correlates with reduced expression of glutamate transporter-1 (GLT-1) and occurs concurrent with tau inclusion pathology. Reduced GLT-1 expression was associated with a progressive decrease in sodium-dependent glutamate transport capacity. Reductions in GLT-1 expression were also observed in corticobasal degeneration, a tauopathy with prominent pathology in astrocytes. Less robust changes were observed in Alzheimer's disease in which neuronal tau pathology predominates. Thus, these Tg mice recapitulate features of astrocytic pathology observed in tauopathies and implicate a role for altered astrocyte function in the pathogenesis of these disorders.
Keywords: tau protein, astrocytes, tauopathy, glutamate transporter, transgenic mice, neurodegenerative disease
Introduction
Filamentous inclusions composed of the microtubule-associated protein tau (MAPT) are defining pathological hallmarks of neurodegenerative diseases collectively referred to as tauopathies (Lee et al., 2001). The discovery of mutations in the MAPT gene in frontotemporal dementia and parkinsonism linked to chromosome 17 (FTDP-17) provided confirmation of the central role of tau abnormalities in the pathogenesis of tauopathies (Hutton et al., 1998; Poorkaj et al., 1998; Spillantini et al., 1998). In many of these diseases, tau pathology occurs not only in neurons but also in glia (Feany and Dickson, 1995; Komori, 1999). This contrasts with the normal CNS distribution, wherein tau is expressed in axons and at only low levels in oligodendrocytes and astrocytes (Shin et al., 1991; LoPresti et al., 1995). Whereas the role of tau pathology in neurons is well accepted, the role of the glial pathology in neurodegeneration is essentially unknown.
To study the contribution of astrocytic tau pathology to disease pathogenesis, we developed a transgenic (Tg) model of tau pathology restricted to astrocytes in which the human MAPT gene is expressed under the control of the glial fibrillary acidic protein (GFAP) promoter (Forman et al., 2005). The Tg mice manifest an age-dependent accumulation of tau pathology in astrocytes that recapitulate the histochemical and biochemical features of pathology observed in human tauopathies. Before the development of tau inclusions in astrocytes, there was a redistribution of the GFAP intermediate filament network resembling the reactive astrocytosis observed in many pathological conditions of the CNS.
Historically, the significance of the astrocyte in CNS function has been underappreciated (Volterra and Steinhauser, 2004). However, recent studies demonstrated that astrocytes contribute to a variety of functions, including synapse formation and plasticity and the regulation of synaptic glutamate homeostasis (Vesce et al., 1999). The latter process is critical to CNS function because glutamate is the major excitatory neurotransmitter. However, at elevated extracellular concentrations, glutamate is neurotoxic (Choi et al., 1987). Levels of synaptic glutamate are thus tightly regulated by five sodium-dependent glutamate transporters present on astrocytes and neurons: the astrocyte-specific glutamate transporters GLT-1 (glutamate transporter-1) and GLAST (glutamate-aspartate transporter), and the neuron-specific transporters EAAC1 (excitatory amino acid carrier 1), EAAT4, expressed predominantly in Purkinje cells, and EAAT5, expressed in retinal photoreceptor and bipolar cells (Furuta et al., 1997; Danbolt, 2001). GLT-1 plays a critical role throughout the CNS, accounting for up to 95% of glutamate uptake (Rothstein et al., 1996; Tanaka et al., 1997). Furthermore, in neurodegenerative diseases such as Alzheimer's disease (AD) and amyotropic lateral sclerosis (ALS), astrocyte-mediated glutamate transport is implicated in disease pathogenesis (Maragakis and Rothstein, 2001).
In the present study, we further analyzed the GFAP/tau wild type (WT) Tg mice as well as a second related Tg line that expresses MAPT with the FTDP-17 mutation P301L. These Tg animals manifest motor deficits before the development of tau pathology that correlates with alterations in the expression and function of glial glutamate transporters. The in vivo relevance was grounded in the finding that similar alterations in GLT-1 were identified in brains of patients with corticobasal degeneration (CBD), a tauopathy with robust astrocytic pathology.
Materials and Methods
Generation of GFAP/tau Tg mice
GFAP/tauWT were generated as described previously, and line 4 was used in all experiments in the current study (Forman et al., 2005). For GFAP/tauP301L Tg mice, cDNA constructs containing the T34 (4R/1N) human tau isoform (Goedert and Jakes, 1990) was subcloned into the eukaryotic expression vector pGfa2 expression vector at the BamHI restriction site that uses the astrocyte-specific GFAP promoter (Brenner and Messing, 1996; Forman et al., 2005). The P301L mutation was introduced into the MAPT gene by site-directed mutagenesis (QuickChange; Stratagene, La Jolla, CA). Linearized constructs were microinjected into fertilized C57BL/6 × C3H (B6C3/F1) mouse embryos and then implanted into pseudopregnant females. Genomic DNA was isolated from mouse tails of pups with the Puregene DNA isolation kit (Gentra Systems, Minneapolis, MN), and potential founders were identified by Southern blotting with 32P-labeled T34 cDNA probes. Candidate founder lines were backcrossed to non-Tg mice to establish germ-line transmission. Human tau heterozygous mice were subsequently bred to homozygosity, and non-Tg littermates were inbred and maintained as control mice.
Histochemical and immunohistochemical analysis
GFAP/tau Tg and non-Tg mice were lethally anesthetized by intraperitoneal injection of ketamine hydrochloride (100 mg/kg) and xylazine (10 mg/kg) and perfused intracardially with PBS, followed by 10% neutral buffered Formalin in isotonic saline in accordance with protocols approved by the University of Pennsylvania. The brains and spinal cords were removed and fixed for an additional 24 h, processed by sequential dehydration in ethanol, paraffin embedded, and cut into 6-μm-thick sections. For immunohistochemical analysis, sections were rehydrated and endogenous peroxidase blocked by incubation with methanol/hydrogen peroxide. Sections were blocked with 2% fetal bovine serum in 50 mm Tris, pH 7.4, 150 mm NaCl, and incubated with primary antibody overnight at 4°C. Primary antibodies included GLT-1, GLAST, OT12 (Forman et al., 2005), AT8 (Goedert et al., 1993), and PHF-1 (Greenberg and Davies, 1990). The polyclonal anti-GLT-1 and anti-GLAST antibodies, raised against peptide sequences from C termini of these transporters, were generously provided by Dr. Rothstein (Johns Hopkins University, Baltimore, MD) (Rothstein et al., 1994). Subsequently, sections were incubated with horseradish peroxidase-conjugated anti-Ig antibodies (Vector Laboratories, Burlingame, CA), followed by visualization with the avidin-biotin peroxidase method with 3,3′-diaminobenzidine as chromogen (Vectastain ABC kit; Vector Laboratories). The sections were viewed with an Olympus Optical (Melville, NY) PX51 microscope equipped with bright-field and fluorescent light sources, and images were obtained using a ProGres C14 Jenoptik camera (Laser Optik Systeme, Mainz, Germany). De-identified paraffin blocks of human brain tissue from AD (n = 6), CBD (n = 6), and normal (n = 6) patients were obtained from the brain bank at the Center for Neurodegenerative Disease Research at the University of Pennsylvania. Formalin-fixed brain tissue with a postmortem interval of <8 h was used to minimize oxidative changes to the glutamate transporters (Beckstrom et al., 1999). Paraffin-embedded tissue from frontal (midfrontal gyrus) and occipital (calcarine cortex) lobes were processed and stained as described above.
Biochemical analysis of glutamate transporters
The cerebral hemispheres, brainstem, and spinal cord were dissected from lethally anesthetized GFAP/tau Tg and control mice using protocols approved by the University of Pennsylvania. Tau expression and solubility studies were performed as described previously (Forman et al., 2005). To assess glial glutamate transporter expression, the tissue was homogenized at 3 ml/g wet tissue weight in 50 mm Tris-HCl, 150 mm NaCl, 5 mm EDTA, 1% Nonidet P-40, and 1% SDS, pH 7.4, supplemented with protease inhibitors (1 mm PMSF and 100 μg/ml each of pepstatin A, leupeptin, soybean trypsin inhibitor, N-tosyl-l-phenylalanyl chloromethyl ketone, and N-tosyl-lysine chloromethyl ketone) and centrifuged at 14,000 × g. Twenty-five to 40 μg of total protein extract was resolved on 7.5% SDS-polyacrylamide slab gels. Nitrocellulose replicas of the gels were probed with antibodies specific for GLT-1 (1:5000) (Rothstein et al., 1994), GLAST (1:75) (Rothstein et al., 1994), EAAC1 (1:75) (Rothstein et al., 1994), human tau T14 (1:2000) (Kosik et al., 1988), and actin (1:1000) (Bloom et al., 2003). Primary antibodies were detected with horseradish peroxidase-conjugated anti-mouse or anti-rabbit IgG, respectively (Jackson ImmunoResearch, West Grove, PA). Immunoreactive proteins were revealed using ECL chemiluminescence (NEN, Boston, MA). The blots were developed and quantified using the Fujifilm Image Reader LAS-3000 (Fuji Medical Systems, Stamford, CT). Quantitative Western blot analysis was also performed using the antibodies indicated followed by 2 mCi/ml I 125-labeled secondary antibodies (NEN, Boston, MA) as described previously (Ishihara et al., 1999). The radiolabeled blots were exposed to PhosphorImager plates, and the protein bands were visualized and quantified with ImageQuant software (Molecular Dynamics, Sunnyvale CA). To assess GLT-1 expression in human brains, gray matter from the frontal cortex of normal (n = 3), CBD (n = 3), and AD (n = 3) brains was homogenized at 3 ml/g wet tissue weight using the protocol described above. Fifty micrograms of total protein were resolved on 7.5% SDS-PAGE gels and probed with antibodies specific for GLT-1 and actin.
Glutamate uptake assay
Crude synaptosomal preparations and glutamate transport assays were performed as described previously (Robinson et al., 1991). Briefly, the brain regions indicated were dissected and homogenized in 20 vol of ice-cold 0.32 m sucrose at 4°C using a motorized Teflon/glass homogenizer (Wheaton, Millville, NJ) at 400 rpm for seven strokes, followed by centrifugation at 800 × g for 10 min in a Beckman J2-21 centrifuge (Beckman Coulter, Fullerton, CA) with a JA20.1 rotor. The supernatant was removed and centrifuged at 20,000 × g for 20 min. The resulting pellet was resuspended in 40 vol of 0.32 m sucrose and recentrifuged at 20,000 × g for 20 min. This washed pellet (synaptosomal P2 fraction) was resuspended in 50 vol of 0.32 m sucrose, and protein concentration was determined using the Lowry method (Lowry et al., 1951). The synaptosomal P2 preparation was stored on ice until use.
For glutamate transport assays, 50 μl of the synaptosomal P2 preparation was diluted in 500 μl of Krebs'-HEPES buffer (in mm: 5 Tris base, 10 HEPES, 140 NaCl, 2.5 KCl, 1.2 MgCl2, 1.2 K2HPO4, and 10 dextrose), supplemented with 0.5 μm l-glutamate and 0.5 μCi of l-[3H]glutamate that was prewarmed to 37°C for 5 min. The mixtures were incubated for 3 min at 37°C and terminated by the addition of ice-cold choline buffer (in mm: 5 Tris base, 10 HEPES, 140 choline chloride, 2.5 KCl, 1.2 MgCl2, 1.2 K2HPO4, and 10 dextrose). The synaptosomal preparation was filtered using a Brandel (Gaithersburg, MD) cell harvester and washed with ice-cold choline buffer. Radioactivity retained on the filters (i.e., glutamate transported into the P2 synaptosomal preparations) was quantified using a Beckman Coulter (Fullerton, CA) scintillation counter. All data points were performed in duplicate. Sodium-dependent uptake was calculated as the difference between the amount of radioactivity accumulated in the presence and absence of sodium. Sodium-independent uptake assays were performed in parallel in sodium-free choline buffer. Saturation analyses were performed as above in the presence of 0.5-30 μm nonradioactive l-glutamate and 0.5 μCi of l-[3H]glutamate as tracer. Using the assay conditions described above, sodium dependent transport of l-[3H]glutamate is linear for at least 5 min, and <10% of the exogenous substrate is accumulated. Dihydrokainate (DHK)-sensitive sodium-dependant transport was determined by measuring glutamate uptake (0.5 μm l-glutamate) in the presence and absence of 300 μm DHK (Tocris Cookson, Ballwin, MO), a selective inhibitor of GLT-1-mediated glutamate uptake (Arriza et al., 1994; Koch et al., 1999). The velocity was calculated as described previously and expressed as nanomoles per minute per milligram of protein (Robinson et al., 1991). The kinetic constants for transport of l-[3H]glutamate were calculated using a nonlinear iterative computer program (Prism; GraphPad Software, San Diego, CA) designed to fit and graph the following equation: v = (Vmax × s)/(Km + s), where v is the velocity of transport, Vmax is the maximal velocity of transport, s is the glutamate concentration, and Km is the Michaelis constant. The statistical difference between groups was estimated by ANOVA; p values < 0.05 were considered significant. Data are presented as mean ± SEM from four synaptosomal preparations from four mice.
Motor testing
Accelerating rotarod test. Motor function of GFAP/tau Tg and control mice were monitored using the accelerating rotarod (Ugo Basile, Comerio, Italy) and wire-hang test that assess motor performance/coordination and neuromuscular strength, respectively. In the accelerating rotarod test, each mouse was placed on a rotating cylinder, and the cylinder was gradually accelerated over a 5 min period. Latency to fall from the rotarod was recorded. Test animals underwent training for 2 weeks. Subsequently, each animal was tested two times each on 3 consecutive days for 2 weeks with 45 min intertrial intervals between testing trials. Twelve Tg and 14 non-Tg mice per group were tested for each of the time points, with the exception of the 20-month-old mice, which included six to nine animals. Statistical differences between groups were analyzed by one-way ANOVA, followed by Bonferroni's post hoc test. p values < 0.05 were considered to be significant.
Wire-hang test. Mice were placed with their forepaws on a horizontal wire and were allowed to grasp the wire and remain suspended. Each mouse was tested four times over 2 d, with an intertrial interval of 2 h. The total time the mice remained hanging on the wire was recorded as hanging time in seconds. Eleven to 14 mice per group were used for the grip strength test at 6 months of age and six to nine animals per group at 15 months. Statistical differences between groups were analyzed by one-way ANOVA, followed by Bonferroni's post hoc test. p values < 0.05 were considered to be significant.
CSF analysis. Mice were lethally anesthetized by intraperitoneal injection of ketamine hydrochloride (100 mg/kg) and xylazine (10 mg/kg). CSF samples were collected from the cisterna magna using a micropipette, immediately acidified by dilution in an equal volume of 0.4N per-chloric acid containing 5 μm l-α-aminoadipate, as an internal standard, and stored at -80°C until analysis. The amino acids were measured by a precolumn derivatization with o-phthaldialdehyde using an automated injector as described previously (Robinson et al., 1993). After separation by reverse-phase chromatography using a stepped gradient, amino acids were detected by a McPherson (Chelmsford, MA) model FL-750BX fluorescence detector. Peak areas were integrated using Millennium software (Waters/Millipore, Milford, MA). External standards were injected after every fifth specimen, and a standard curve was constructed to calculate the amounts of internal standard and amino acids in each specimen. Glutamate and aspartate levels in CSF were compared by one-way ANOVA, followed by Bonferroni's test.
Results
GFAP/tauP301L Tg mice develop tau pathology in astrocytes similar to GFAP/tauWT but at lower protein expression levels
The GFAP/tauWT Tg mice express the tau protein specifically in astrocytes (Forman et al., 2005). Tau expression in astrocytes was associated with a reactive astrocytic phenotype, characterized by a redistribution of the intermediate filament GFAP. Furthermore, these Tg mice developed an age-dependent accumulation of astrocytic tau pathology that was abnormally phosphorylated, ubiquitinated, and filamentous, similar to that observed in tauopathies. However, there is accumulating evidence that, in Tg mice, FTDP-17 mutations accelerate the formation of tau inclusions in neurons and induce a more severe neurodegenerative phenotype (Götz, 2001). Nonetheless, the mechanisms whereby tau mutations induce more profound accumulations of fibrillary tau inclusions remain poorly understood. To elucidate mechanisms of astrocyte-specific mechanisms of degeneration associated with FTDP-17 mutations, we generated additional GFAP/tau Tg mice expressing the FTDP-17 mutation P301L (GFAP/tauP301L).
Regional tau protein expression from cortex, brainstem, and spinal cord was determined by immunoblot analysis of the soluble protein extract from 2-month-old GFAP/tauWT and GFAP/tauP301L Tg mice. In wild-type and P301L mutant Tg mice, tau protein expression was region dependent, with the highest levels consistently observed in the spinal cord (Fig. 1A). Quantitative Western blot analyses indicated that the GFAP/tauWT Tg mice expressed fourfold more tau than the GFAP/tauP301L (line 5) Tg mice. To further analyze the regional tau expression of the Tg mice, an immunohistochemical analysis was performed on 2-month-old GFAP/tauWT and GFAP/tauP301L mice (Fig. 1B). The pattern of expression of tau in GFAP/tauP301L Tg mice was similar to that of the GFAP/tauWT Tg mice, with robust tau expression observed in spinal cord and brainstem relative to cerebral cortex and subcortical nuclei. Furthermore, as in the GFAP/tauWT Tg mice, the astrocytic expression of P301L tau protein was associated with prominent GFAP immunoreactivity in astrocytes, predominantly within gray matter (Fig. 1C), changes that were detected as early as 2 months of age (data not shown). The GFAP staining resembles the reactive astrocytosis observed in many pathological conditions of the CNS and may reflect a redistribution of the intermediate filament network similar to that described with the overexpression of tau protein in primary rat astrocyte cultures (Yoshiyama et al., 2003).
Figure 1.

Astrocyte-specific regional human tau expression in GFAP/tauWT and GFAP/tauP301L Tg mice. A, Western blot analysis of soluble tau protein extracted from cortex, brainstem, and spinal cord samples of 2-month-old control (non-Tg), GFAP/tauP301L (PL), and GFAP/tauWT Tg (WT) mice. Ten micrograms of soluble protein extract was resolved by SDS-PAGE and immunoblotted with T14, a monoclonal antibody (mAb) specific for human tau. Both GFAP/tauWT and GFAP/tauP301L Tg lines show highest levels of human tau protein expression in the spinal cord, whereas the lowest levels are detected in the cortex. The GFAP/tauWT Tg mouse expresses approximately fourfold higher levels of human tau protein than GFAP/tauP301L. Molecular weight standards are indicated to the left of each panel. B, Immunohistochemistry was performed with the human tau-specific mAb OT12 (B) on cortex, brainstem, and spinal cords of 2-month-old GFAP/tauWT (WT) and GFAP/tauP301L (PL) Tg mice. Robust tau staining is observed in astrocytes within the gray matter of tau Tg mice. Human tau was not detected in non-Tg mice (data not shown). Insets show tau staining in cells with astrocytic morphology. Scalebar: (in a) a, b, d, e, 200 μm; (inc) c, f, 500 μm; insets, 50 μm. C, Immunohistochemistry for GFAP was performed on the spinal cord of 6 month GFAP/tauWT, GFAP/tauP301L Tg, and non-Tg mice. There is prominent GFAP staining in spinal cord gray matter of GFAP/tauWT and GFAP/tauP301L Tg mice, but only limited GFAP staining is detected in spinal cord gray matter in the non-Tg animals. Scale bar, 100 μm.
To further characterize the GFAP/tau Tg mice, immunostaining was performed on mice from 6 to 24 months of age with antibodies that detect phosphorylation-dependent tau epitopes characteristic of the human tauopathies. The GFAP/tauP301L Tg mice develop astrocytic tau inclusions similar to that described in animals expressing wild-type tau protein (Fig. 2) (Forman et al., 2005). Specifically, there was an age-dependent increase in expression of phosphorylation-dependent tau epitopes. These phosphoepitopes were first detected in brainstem and spinal cord of ∼30% of Tg mice at 12 months of age, and, by 24 months, these epitopes were observed in >95% of animals in brainstem, spinal cord, and subcortical nuclei, especially thalamus. In addition, at 12 months of age, a subset of the tau pathology (as defined by the accumulation of tau phosphoepitopes) was detected with the Gallyas silver impregnation stain. The quantity of Gallyas silver-positive inclusions also increased with age, although the silver stain detected only a subset of the tau pathology revealed by immunohistochemistry with AT8, specific for phosphorylation-dependent tau epitopes (Fig. 2A). Furthermore, there was a progressive accumulation of insoluble, heavily aggregated, and phosphorylated tau protein in the brainstem and spinal cord of GFAP/tauP301L Tg mice (Fig. 2B). There was little insoluble tau in cortex, consistent with low and variable cortical tau pathology detected by immunohistochemistry. Thus, the GFAP/tauP301L Tg mice are phenotypically similar to the Tg mice expressing wild-type protein, except at significantly lower levels of human tau protein expression.
Figure 2.
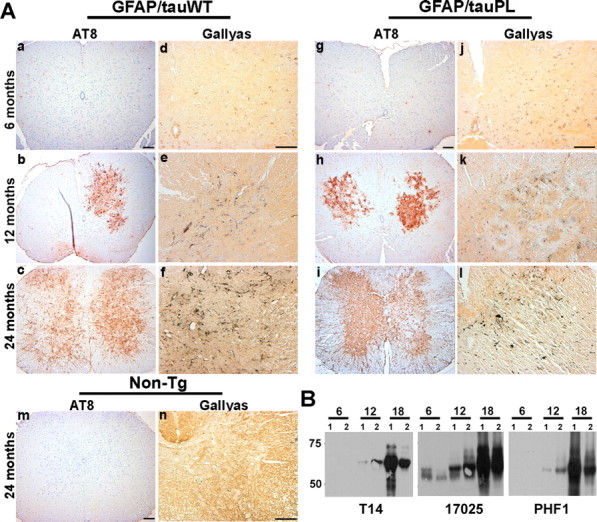
Age-dependent accumulation of pathological tau inclusions in GFAP/tau Tg mice. A, Spinal cord from 6-, 12-, and 24-month-old GFAP/tauWT and GFAP/tauP301L mice were analyzed by immunohistochemistry with AT8, an antibody specific for tau phosphorylated at Ser202 and Thr205 (a-c, g-i, m) and Gallyas silver impregnation stain (d-f, j-l, n). There is an age-dependant accumulation of phosphorylated tau first detected at ∼12 months of age in both lines of tau Tg mice. Only a subset of the AT8-positive pathology is detected with Gallyas silver staining at this age. However, by 24 months, there is robust AT8 immunoreactivity and Gallyas-positive tau pathology in both astrocytic processes and cell soma of GFAP/tauWT and GFAP/tauP301L mice, which is not detected in 24-month-old non-Tg animals (m, n). The distribution and density of tau pathology is similar in the GFAP/tauWT and GFAP/tauP301L Tg mice. Scale bars: (in a) a-c, g-i, m, 100 μm; (in d) d-f, j-l, n, 100 μm. B, Immunoblot analysis of insoluble tau corresponding to 25 mg of starting wet tissue weight extracted from spinal cords of GFAP/tauP301L Tg mice at ages indicated and detected with T14, a human tau-specific mAb, 17025, a polyclonal antibody that detects both human and murine tau, and PHF-1, a mAb specific for tau phosphorylated at Ser396 and Ser404. At each age, two animals were analyzed. Insoluble tau protein is first detected in spinal cord and brainstem (data not shown) at 12 months of age. By 18 months of age, there is extensive insoluble and heavily aggregated tau pathology throughout the brainstem and spinal cord. Some variations in insoluble tau protein was observed between the two animals used at each age.
GFAP/tau Tg mice develop motor impairments before the onset of tau inclusion pathology in astrocytes
To determine whether tau pathology in astrocytes is associated with behavioral deficits, we examined the phenotype of both GFAP/tauWT and GFAP/tauP301L Tg mice. Because both strains of Tg mice manifest robust pathology in spinal cord and brainstem, we performed motor function testing, specifically, the accelerating rotarod and wire-hang test that assess motor performance/coordination and neuromuscular strength, respectively. On the accelerating rotarod, both GFAP/tauWT and GFAP/tauP301L Tg mice showed a decrease in latency to fall relative to non-Tg mice at all ages tested (one-way ANOVA, Bonferroni's post hoc test; p < 0.001) (Fig. 3A). Notably, at 4 months of age, well before the detection of tau phosphoepitopes or silver-positive inclusions, both strains of Tg mice showed significant impairment. Furthermore, starting at 12 months of age, GFAP/tauP301L Tg mice show significant impairment relative to GFAP/tauWT Tg mice (one-way ANOVA, Bonferroni's post hoc test; p < 0.001). In contrast, in the wire-hang test, there was no significant impairment between control and the two strains of tau Tg mice at 6 months of age. However, by 15 months, both GFAP/tauWT and GFAP/tauP301L Tg mice manifest significant deficits in grip strength relative to non-Tg animals (one-way ANOVA, Bonferroni's post hoc test; p < 0.001) (Fig. 3B). Thus, the impaired motor performance observed at 4 months of age in both GFAP/tauWT and GFAP/tauP301L animals could be a result of the redistribution of GFAP observed in the spinal cord in which there is robust tau expression. However, functional consequences of altered GFAP expression are unknown.
Figure 3.
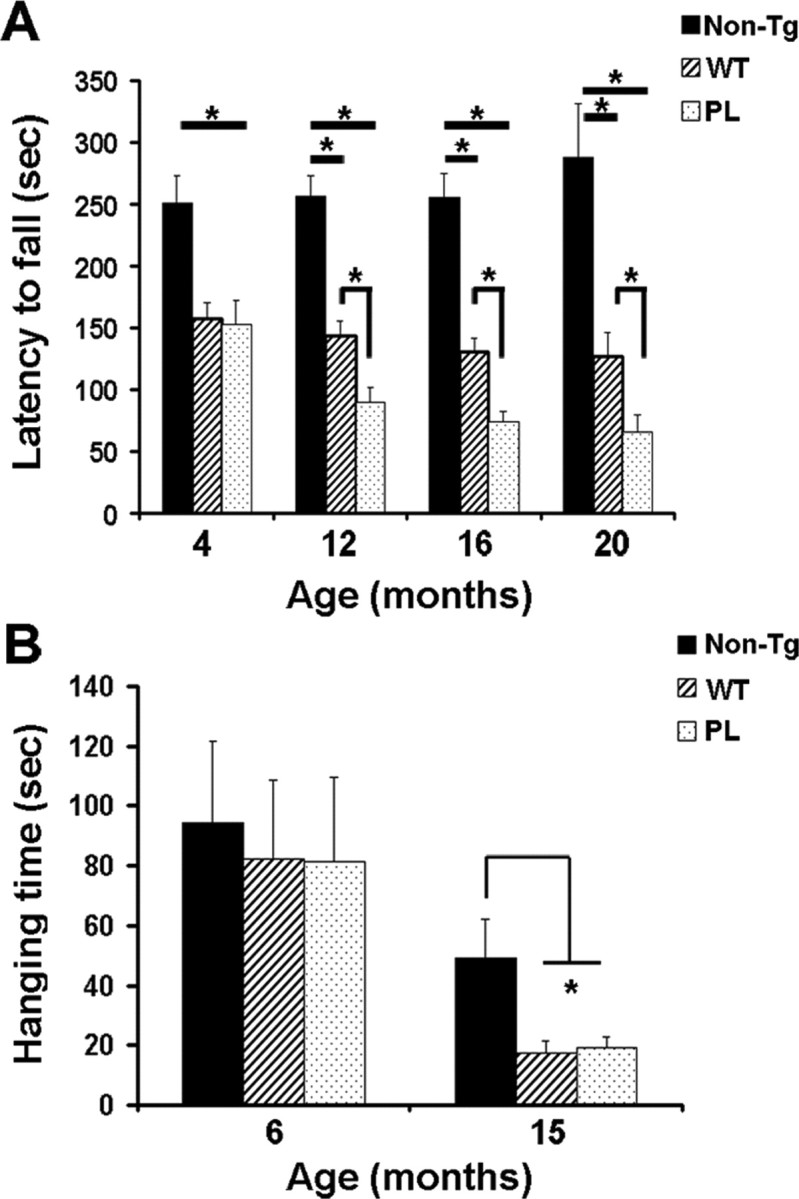
Motor impairment in GFAP/tau Tg mice. A, Both strains of tau Tg and non-Tg mice at the ages indicated were tested on the accelerating rotarod after 2 weeks of training. Latency to fall was recorded for each of 12 trials over 2 weeks. n = 12-14 animals per group at 4 months of age; n = 6-9 animals per group at 12, 16, and 20 months. Impaired performance was observed in both GFAP/tauWT (WT) and GFAP/tauP301L (PL) Tg mice relative to age-matched controls (one-way ANOVA, Bonferroni's post hoc test; *p < 0.001). At 12 months of age, the GFAP/tauP301L Tg mice also manifest impaired performance relative to the GFAP/tauWT Tg animals (one-way ANOVA, Bonferroni's post hoc test; *p < 0.001). Latency to fall, Mean of 12 trials. Error bars indicate SEM. B, GFAP/tau Tg and control mice at the ages indicated were assessed for neuromuscular strength using the wire-hang test. Animals were suspended from a wire, and the latency to fall from the wire was recorded as hanging time in seconds. Each mouse was tested four times over 2 d with an intertrial interval of 2 h. n = 11-14 mice per group at 6 months of age and 6-9 animals per group at 15 months. At 4 months of age, there was no difference in performance between both the strains of tau Tg and non-Tg mice. In contrast, both GFAP/tauWT (WT) and GFAP/tauP301L (PL) mice were significantly impaired at 15 months of age with 65% impairment relative to non-Tg mice (one-way ANOVA, Bonferroni's post hoc test; *p < 0.001). Hanging time, Mean of four trials. Error bars indicate SEM.
Altered expression of GLT-1 and GLAST in GFAP/tauWT and GFAP/tauP301L Tg mice
Glutamate transport is the primary mechanism for the inactivation of synaptically released glutamate (Danbolt, 2001), and the astrocyte-specific glutamate transporter GLT-1 is responsible for up to 95% of glutamate transport (Tanaka et al., 1997). Furthermore, reduction of glial glutamate transporter expression is associated with deficits in motor coordination and neuromuscular strength (Watase et al., 1998). Because the GFAP/tauWT and GFAP/tauP301L Tg mice manifest motor deficits that predated the development of tau pathology, we examined expression of the two glial glutamate transporters in the CNS of both strains of tau Tg and control mice. In 12- and 24-month-old non-Tg mice, immunostaining for both GLT-1 and GLAST revealed a diffuse granular staining pattern throughout the gray matter, consistent with previous observations (Fig. 4A) (Howland et al., 2002). In contrast, in 12-month-old GFAP/tauWT and GFAP/tauP301L Tg animals, there was patchy staining of both GLT-1 (Fig. 4A) and GLAST (data not shown) in the spinal cord and, to a lesser extent, in the brainstem and cortex, suggesting either a redistribution or loss of the transporter proteins. At 24 months of age, there was profound loss of GLT-1 and GLAST immunoreactivity throughout the spinal cord gray matter of Tg animals (Fig. 4A). Similar but less robust changes in GLT-1 and GLAST immunoreactivity were also observed in the brainstem (data not shown) and cortex (supplemental Fig. S1, available at www.jneurosci.org as supplemental material) of the Tg mice compared with non-Tg controls. There were no obvious differences in staining between the GFAP/tauWT and GFAP/tauP301L animals. Thus, the expression of tau in astrocytes leads to an alteration in glial glutamate transporter expression, and the subsequent accumulation of tau aggregates correlates with additional alterations in the expression pattern of these transporters.
Figure 4.
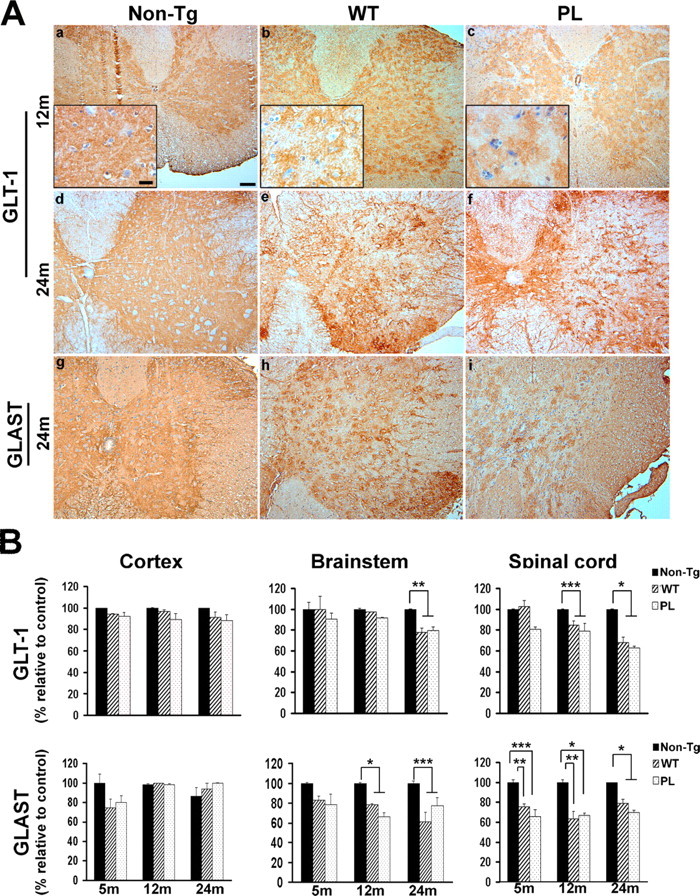
Decreased glial glutamate transporter expression in wild-type and mutant GFAP/tau Tg mice. A, Immunohistochemical analysis was performed on spinal cord sections from 12-month-old (a-c) and 24-month-old (d-i) GFAP/tau Tg and non-Tg mice, as indicated with antibodies to GLT-1 and GLAST. There is altered GLT-1 (a-c) and GLAST (data not shown) immunoreactivity relative to non-Tg animals at 12 months of age. Insets show higher magnification of patchy staining for GLT-1 in both strains of tau Tg mice compared with the diffuse gray matter staining observed in age-matched, non-Tg controls. At 24 months of age, there is progressive loss of both GLT-1 and GLAST immunoreactivity in both GFAP/tauWT and GFAP/tauP301L (PL) Tg animals. Scale bars: a-i, 100 μm; insets, 20 μm. B, Cortex, brainstem, and spinal cord from non-Tg, GFAP/tauWT, and GFAP/tauP301L Tg mice at 5, 12, and 24 months of age were extracted as described in Materials and Methods and immunoblotted with antibodies to GLT-1, GLAST, and actin (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). Immunoblots were quantified with Multigauge version 2.3 software and normalized to actin levels. Data are presented as percentage of non-Tg age-matched control animals. At each time point, four animals per group were analyzed. GLAST expression was significantly decreased in both the brainstem (12 and 24 months) and spinal cord (5, 12, and 24 months) of both the strains of tau Tg mice relative to control animals (one-way ANOVA, Bonferroni's post hoc test; *p < 0.001; **p < 0.01; ***p < 0.05). In contrast, GLT-1 expression was only mildly decreased at 12 months of age in the GFAP/tauWT and GFAP/tauP301L Tg mice. However, at 24 months of age when the GFAP/tauWT and GFAP/tauP301L Tg mice manifest robust tau pathology, there was significant loss of GLT-1 protein expression in both the brainstem and spinal cord (one-way ANOVA, Bonferroni's post hoc test; *p < 0.001; **p < 0.01; ***p < 0.05). Representative immunoblots for human tau (T14), GLT-1, GLAST, EAAC1, and actin from brain regions indicated of 24-month-old animals are shown in supplemental Figure S2 (available at www.jneurosci.org as supplemental material).
To determine whether the observed immunohistochemical changes in GLT-1 and GLAST immunoreactivity were associated with reduced protein levels of glutamate transporters, spinal cord, brainstem, and cortex from 5-, 12-, and 24-month-old Tg and control animals were analyzed for GLT-1 and GLAST protein expression. In both strains of tau Tg mice, there was reduced protein expression of both GLAST and GLT-1 in the brainstem and spinal cord that correlated with areas of high regional tau expression (Fig. 4B) (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). Specifically, GLAST protein expression was reduced in the spinal cord (21-37%) and brainstem (21-40%) of both strains of Tg mice as early as 5 months of age, and this reduction did not progress further with aging beyond 12 months. In contrast, only a mild reduction of GLT-1 protein was observed in the spinal cord of 12-month-old GFAP/tau Tg animals. However, at 24 months of age when the GFAP/tauWT and GFAP/tauP301L Tg mice manifest robust tau pathology, there was significant loss of GLT-1 protein expression in both the brainstem (GFAP/tauWT, 22%; GFAP/tauP301L, 21%) and spinal cord (GFAP/tauWT, 32%; GFAP/tauP301L, 38%). Notably, there was no significant difference in glutamate transporter levels between the two strains of tau Tg mice despite significantly lower levels of tau transgene expression in the GFAP/tauP301L animals. Furthermore, in the cerebral hemisphere, in which there is sparse tau pathology, no decrease in GLT-1 and GLAST was detected. In addition, no significant changes in protein expression were observed in the neuronal glutamate transporter EAAC1, thus suggesting a specific loss of the glial glutamate transporters in regions with robust astrocytic tau expression. The reduced GLAST protein expression and impaired performance on the accelerating rotarod test at early ages was consistent with impaired performance of GLAST knock-out mice in a similar assay (Watase et al., 1998). The subsequent loss of GLT-1 protein expression and later development of neuromuscular weakness is also consistent with a previous report showing hindlimb paralysis in a partial GLT-1 knock-out (Tanaka et al., 1997).
Reduced glutamate transport activity in aged tau Tg mice
To determine whether the reduced protein levels of GLT-1 and GLAST lead to reduced glutamate transporter activity, we measured high-affinity sodium-dependent glutamate uptake in crude synaptosomal (P2) preparations from cortex, brainstem, and spinal cord of 12- and 24-month-old GFAP/tauWT and GFAP/tauP301L Tg and control mice. Similar to previous reports, we observed an age-dependent reduction in the velocity of glutamate transport in both strains of Tg mice and non-Tg mice (Fig. 5 and data not shown) (Price et al., 1981). However, in the spinal cord of 12-month-old GFAP/tauWT Tg mice, we observed a 32% reduction in the velocity of glutamate transport relative to control animals (p < 0.01) (Fig. 5A). GFAP/tauP301L mice showed a smaller reduction, but this difference was not significant. Significant changes in glutamate transport were not observed in the brainstem and cortex at this early age. At 24 months of age, both strains of tau Tg mice showed additional decreases in glutamate transport in both spinal cord and brainstem (Fig. 5A). Specifically, glutamate transport from spinal cord preparations of both GFAP/tauWT and GFAP/tauP301L Tg mice was reduced by 60 and 36%, respectively, relative to non-Tg mice. Similarly, glutamate uptake in the brainstem of 24-month-old GFAP/tauWT and GFAP/tauP301L Tg mice was reduced by 46 and 28%, respectively (Fig. 5A). Consistent with the biochemical data above, no significant differences in glutamate uptake were observed in preparations from the cortex. Moreover, there was a 43% decrease in the maximal velocity (Vmax) of glutamate transport in spinal cords of GFAP/tauWT Tg mice at 12 months of age. This was further reduced to 60% at 24 months of age (Fig. 5C,D). However, no significant changes in the affinity for glutamate (Km) were observed at both ages tested (non-Tg, 2.86 ± 0.82 μm; GFAP/tauWT, 2.57 ± 0.25 μm).
Figure 5.

Reduced sodium-dependent glutamate uptake in GFAP/tau Tg mice.A, l-[3H]Glutamate uptake was measured in P2 synaptosomal fractions prepared from cortex (Cx), brainstem (BS), and spinal cord (SC) of 12- and 24-month-old GFAP/tauWT (WT), GFAP/tauP301L (PL), and non-Tg mice. Initial rates of sodium-dependent uptake were expressed as nanomoles per minute per milligram of P2 synaptosomal protein extract. Data are presented as the percentage of glutamate uptake relative to age-matched non-Tg mice. At 12 months of age, there was a significant (32%) reduction in glutamate uptake capacity in the spinal cords of GFAP/tauWT Tg mice relative to the control animals (one-way ANOVA, Bonferroni's post hoc test; *p < 0.001). GFAP/tauP301L showed a smaller reduction (14%), but this was not statistically significant. At 24 months of age, there was an additional reduction in transport capacity in brainstem (WT, 46%; PL, 28%) and spinal cords (WT, 60%; PL, 36%) relative to the control. There were also significant differences in glutamate uptake in synaptosomal preparations from brainstem and spinal cord of 24-month-old GFAP/tauWT and GFAP/tauP301L Tg animals. Data represent mean from four mice analyzed in two independent experiments. Error bars indicate standard SEM (one-way ANOVA, Bonferroni's post hoc test; *p < 0.001; **p < 0.01; ***p < 0.05). B, l-[3H]Glutamate uptake into P2 synaptosomal fractions prepared from spinal cords of 24-month-old GFAP/tauWT, GFAP/tauP301L Tg, and age-matched control animals was measured in the presence and absence of 300 μm DHK, a selective GLT-1 inhibitor. The net sodium-dependent glutamate uptake values are provided that represent the difference in glutamate uptake in the presence and absence of DHK. Error bars represent SEM. C, D, Saturation isotherms for the high-affinity sodium-dependent transport of glutamate in spinal cord P2 synaptosomal fractions of GFAP/tauWT (WT) Tg and non-Tg controls at 12 (C) and 24 (D) months of age. Data represent the mean of four mice performed in two independent experiments. Error bars represent SEM. At 12 months of age, Vmax is reduced 43% relative to the non-Tg controls. By 24 months of age, Vmax is further reduced to 60% (one-way ANOVA, Bonferroni's post hoc test; p < 0.001). In contrast, there was no statistically significant difference in Km between the GFAP/tauWT Tg and non-Tg mice.
To assess the relative contribution of GLT-1 to the sodium-dependent glutamate transport, we measured l-[3H]glutamate uptake in spinal cord P2 preparations in the presence and absence of DHK. DHK inhibits the subtypes of glutamate transporters differentially [i.e., GLT-1, IC50 of 10-50 μm; EAAC1, IC50 of 1.2 mm; GLAST, no inhibition at concentrations up to 10 mm (Garlin et al., 1995; Dowd et al., 1996)]. The effect of DHK on sodium-dependent glutamate uptake was evaluated at a concentration (300 μm) that inhibits GLT-1-mediated activity by up to 90% and EAAC1 by 20% but has no effect on GLAST glutamate transport. DHK inhibited glutamate transport in GFAP/tauWT, GFAP/tauP301L, and non-Tg animals by 75 ± 1.4, 85 ± 3.1, and 90 ± 1.7%, respectively, indicating that the majority of the glutamate uptake activity is mediated by GLT-1. However, consistent with the reduction in GLT-1 expression in aged mice, the component of transport that is DHK sensitive was reduced in GFAP/tauWT and GFAP/tauP301L Tg animals relative to non-Tg mice by 65 and 38%, respectively (Fig. 5B).
Reduced glial glutamate transporters in CBD, a tauopathy with abundant astrocytic tau pathology
Tau protein expression and pathology in astrocytes in our Tg mice models is associated with a functional reduction in the glial glutamate transporters. To determine whether similar alterations are observed in human tauopathies, we immunostained affected and unaffected frontal cortices from six CBD and age-matched control brains, as well as six brains with AD pathology. CBD is a tauopathy with abundant tau pathology in neurons and glia, including astrocytes and oligodendrocytes, whereas the tau pathology in AD primarily localizes to neurons and their processes (Feany and Dickson, 1996). Consistent with published reports, GLT-1 protein was detected diffusely in gray matter of control patients (Rothstein et al., 1995) (Fig. 6A). In contrast, in affected frontal cortex from CBD patients, there was a patchy distribution of GLT-1-immunoreactive protein similar to the 24-month-old GFAP/tauWT and GFAP/tauP301L Tg mice. Unaffected visual cortex from CBD patients showed a pattern of GLT-1 immunoreactivity similar to that of control brains (Fig. 6B). To determine whether the observed immunohistochemical changes in GLT-1 were associated with reduced protein levels, frontal cortices from CBD, AD, and age-matched normal brains were analyzed for GLT-1 protein expression. GLT-1 protein levels were reduced in all CBD cases, and, in two of three CBD brains analyzed, there was a ∼50% reduction in GLT-1 protein levels (supplemental Fig. 3, available at www.jneurosci.org as supplemental material). Alterations in GLT-1 expression have been reported in AD (Li et al., 1997). However, in contrast to the CBD patients, there was only mild reduction in GLT-1 immunoreactivity and no change in GLT-1 protein levels, despite comparable levels of tau pathology. Thus, the widespread astrocytic tau pathology in CBD brain tissue may lead to reduced expression of GLT-1 similar to our GFAP/tau Tg mice.
Figure 6.
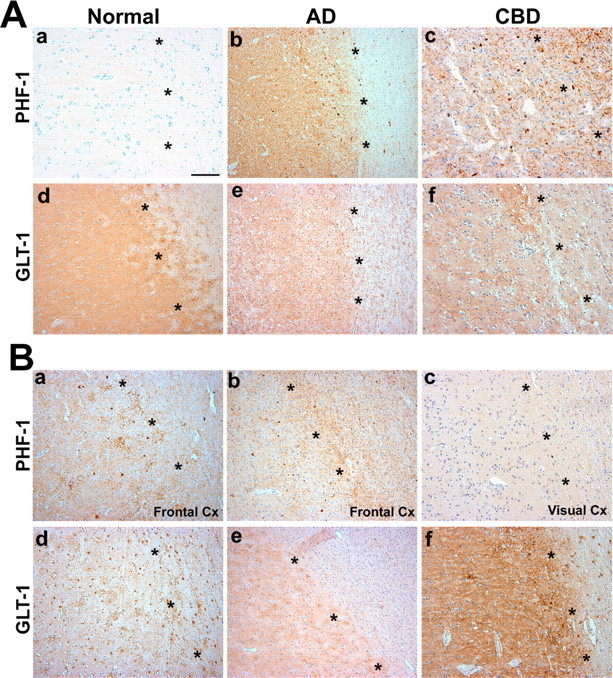
Loss of GLT-1 immunoreactivity in affected cortex of CBD patients. A, Adjacent sections from frontal cortex from patients with CBD (c, f), AD (b, e), and age-matched controls (a, d) were immunostained with PHF-1 (a-c) and GLT-1 (d-f). In cortex from control patients, there is little/no tau pathology (a), and GLT-1 shows diffuse cortical staining (d). In contrast, affected cortex from CBD patients shows robust tau pathology in both gray and white matter (c), with a marked loss of GLT-1 immunoreactivity in cortex (f). Occasional GLT-1-positive astrocytes are observed in the white matter. In frontal cortex from patients with AD, tau pathology is primarily restricted to the neocortex (b), with only mild and variable loss of GLT-1 immunoreactivity (e). B, Adjacent sections of frontal (a, b, d, e) and visual (c, f) cortex (Cx) from additional patients with CBD were immunostained with PHF-1 (a-c) and GLT-1 (d-f). Similar to A, there was robust tau pathology in both gray and white matter of affected cortex from CBD patients (A, B), with a marked loss of GLT-1 immunoreactivity in cortex (d, e). In contrast, in unaffected visual cortex, there is limited tau pathology (c), and, similar to control patients, GLT-1 shows diffuse cortical staining (f). The series of * symbols in each panel demarcate gray (to the left in each photomicrograph) from white matter. Scale bar, 500 μm
Normal CSF glutamate levels in GFAP/tau Tg mice
Glutamate excitotoxicity has been implicated in a variety of disease models, including ischemic reperfusion injury, epilepsy, and neurodegenerative disorders such as ALS (Danbolt, 2001). The mechanism whereby decreased glutamate reuptake into glial cells and/or neurons causes excitotoxicity is likely attributable to the enhanced stimulation of glutamate receptors (Choi et al., 1987). To determine whether the functional decrease in glial glutamate transporters in both strains of tau Tg mice leads to elevated CNS glutamate, we measured CSF glutamate levels in GFAP/tauWT, GFAP/tauP301L Tg, and control mice (Fig. 7). However, we did not observe any statistically significant difference in the CSF glutamate or aspartate levels (Fig. 7). Similarly, there were no differences between the two strains of tau Tg and non-Tg mice in levels of other amino acid neurotransmitters, including methionine, tryptophan, GABA, and asparagine (data not shown). This suggests that the reduction in glial glutamate transporters observed in the spinal cord and brainstem of GFAP/tauWT and GFAP/tauP301L Tg mice is not reflected in CSF because of the dynamic circulation of CSF throughout the subarachnoid space or its production mainly by choroid plexus within the ventricular compartment far removed from brainstem and spinal cord. However, we cannot exclude focal alterations in glutamate levels.
Figure 7.

CSF glutamate (A) and aspartate (B) levels in GFAP/tau Tg mice. CSF samples from cisterna magna of 24-month-old GFAP/tauWT, GFAP/tauP301L Tg, and non-Tg mice were analyzed by HPLC for glutamate and aspartate. No significant differences in concentrations of CSF glutamate and aspartate were detected between both strains of tau Tg and control mice. The amino acids methionine, tryptophan, GABA, and asparagine were also analyzed, but no difference between groups was observed (data not shown). Six animals per group were used for the CSF analyses. Error bars indicate SEM.
Discussion
The role of glial tau pathology in the pathogenesis of tauopathies is poorly understood, and, until recently, it was still debated whether the astrocytic tau pathology is a reactive or a degenerative process (Ikeda et al., 1995; Komori, 1999). However, the astrogliosis observed in tauopathies is associated with neurofibrillary tangle formation and not astrocytic tau pathology (Togo and Dickson, 2002). Moreover, tau expression in astrocytes is normally very low, and thus, the alteration in tau expression likely reflects a disease-specific alteration in tau protein metabolism. To investigate the role of tau pathology specifically in astrocytes, we developed a model of tau pathology in astrocytes that recapitulates key features of astrocytic pathology observed in tauopathies (Forman et al., 2005). These GFAP/tauWT Tg mice manifest an age-dependent accumulation of tau pathology in astrocytes that was abnormally phosphorylated, ubiquitinated, filamentous, and biochemically insoluble.
In the current study, we used these Tg mice and a new Tg line that expresses the P301L FTDP-17 mutation to examine the effect of tau expression and pathology on astrocyte function. Although the GFAP/tauP301L Tg mice expressed fourfold lower levels of tau than the GFAP/tauWT mice, they showed a similar spatial and temporal distribution of tau pathology. Thus, the presence of the P301L mutation did not alter the phenotype of the Tg mice but rather led to a similar pattern of pathology at lower protein concentrations. Despite generating numerous GFAP/tauP301L founder lines, we were unable to successfully breed mice expressing higher levels of mutant tau protein in astrocytes. Both wild-type and mutant GFAP/tau Tg mice express the highest levels of tau protein in the brainstem and spinal cord, and this correlated with the observed distribution of tau pathology. Furthermore, before the development of tau pathology, we observed enhanced GFAP immunoreactivity in gray matter with robust tau expression, morphologically resembling the reactive astrogliosis observed in many CNS pathologies. Furthermore, both strains of Tg mice manifest deficits in motor function before the detection of tau pathology by histochemical (Gallyas-positive inclusions), immunohistochemical (expression of phosphorylation-dependent tau epitopes), or biochemical (accumulation of insoluble tau protein) methodologies. There was also reduced expression of the glial glutamate transporter GLAST, although the functional consequences of this reduction are unclear because the majority of glutamate uptake is mediated by GLT-1. Subsequently, both strains of tau Tg mice develop tau inclusions that were first detected at ∼12 months of age with an age-dependent progression of the quantity and regional extent of this pathology (Forman et al., 2005). After 12 months of age, the GFAP/tauWT and GFAP/tauP301L Tg mice manifested additional deficits in neuromuscular strength that correlated with functional deficits in GLT-1 expression. The in vivo relevance of these findings was grounded in the finding that similar alterations in GLT-1 were identified in affected regions of CBD brains, a tauopathy with robust astrocytic pathology.
Historically, astrocytes were thought to have only a passive function in the CNS, providing nutritional and structural support for neurons. It is now known that astrocytes play a dynamic role in CNS function, including maintenance of the blood-brain barrier, immune modulation, neurogenesis, synaptogenesis, and modulation of synapse function (Ransom et al., 2003). In the latter case, astrocytes modulate synaptic function by the secretion and uptake of neurotransmitters (Haydon, 2001). In the glutaminergic system, astrocytes secrete glutamate in response to activation, modulate glutamate receptor expression, and remove glutamate from the synaptic cleft by glutamate transporters (Araque et al., 2001). This regulation of synaptic glutamate is critical to normal CNS function, and the sodium-dependent glutamate transporters present perisynaptically on astrocytes contribute to the regulation of extracellular glutamate levels. Specifically, studies using antisense oligonucleotides indicated that the bulk of glutamate clearance is dependent on GLT-1 (Maragakis and Rothstein, 2001). The functional relevance of the glutamate transporters was confirmed in animal studies whereby knock-out of GLAST resulted in impaired performance on the accelerating rotarod, whereas partial loss of GLT-1 led to hindlimb paralysis (Tanaka et al., 1997; Watase et al., 1998). Similarly, in the GFAP/tauWT and GFAP/tauP301L Tg mice, we observed an early reduction in GLAST before the development of tau pathology in astrocytes that correlated with impaired motor performance. The subsequent reduction in functional GLT-1 expression coincided with the development of tau pathology in spinal cord and brainstem, as well as neuromuscular weakness. Although no elevations in CSF glutamate or aspartate were detected in the GFAP/tau Tg mice, local alterations in synaptic glutamate levels cannot be excluded.
Disruption in glutamate homeostasis has been implicated in a variety of neurological disorders, including epilepsy, stroke, and multiple sclerosis, and similar perturbations in glutamate regulation may contribute to the pathogenesis of neurodegenerative disease. In a model of Parkinson's disease, disruption of striatal glutamatergic innervation resulted in reduction in both GLT-1 and GLAST protein expression, accompanied by reduced glutamate transport (Ginsberg et al., 1995; Levy et al., 1995). This phenotype was rescued in animals treated with levadopa (Lievens et al., 2000). In a Tg mouse model of AD, the expression of mutant amyloid precursor protein was associated with decreases in Vmax and KD for aspartate uptake accompanied by decreased expression of GLAST and GLT-1 proteins (Masliah et al., 2000). Similar findings were also observed in AD brains, wherein protein levels of GLT-1 were reduced in affected frontal cortices (Li et al., 1997). Moreover, in a Tg model of ALS, the expression of mutant superoxide dismutase 1 (SOD1) caused a 90% reduction in GLT-1 protein expression with associated hindlimb paralysis (Howland et al., 2002). In addition, Trotti et al. (1999) demonstrated that GLT-1 was a target of mutant SOD1-mediated oxidation, which compromised glutamate transporter capacity. In a different Tg mouse model of ALS, increasing the GLT-1 expression delayed loss of muscle strength, prevented loss of motor neurons, and increased survival (Rothstein et al., 2005).
The mechanism whereby tau expression and pathology leads to reduced expression of glial glutamate transporters in both strains of tau Tg mice is unknown. One possibility is that the selective loss of the GLT-1 and GLAST in astrocytes results from oxidative injury. For example, increased reactive oxygen species caused oxidation of glutamate transporters, resulting in reduced glutamate uptake (Trotti et al., 1998). In addition, in primary rat astrocyte cultures, the overexpression of wild-type and P301L mutant tau caused impairment of microtubule-based plus-end directed transport, resulting in a redistribution of mitochondria from the periphery of cells, although there was no direct evidence of oxidative injury (Yoshiyama et al., 2003). However, tau pathology in neurons may lead to oxidative damage because increased levels of superoxide anion radicals were detected in Tg mice expressing P301L mutant tau in neurons (David et al., 2005). Alternatively, decreased glutamate transporters in the tau Tg mice may be a consequence of neuron degeneration because GLT-1 expression is directly regulated by neuron-astrocyte interactions (Schlag et al., 1998). Thus, the decrease in GLT-1 and GLAST detected in both strains of tau Tg mice and postmortem tissue of CBD patients could result from the loss of neuron-derived factors regulating the expression of these glial glutamate transporters. However, in the GFAP/tau Tg mice, only mild neuron degeneration was observed, and thus the alterations in glial glutamate transporters in the Tg mice are most likely attributable to tau expression and/or pathology in astrocytes (Forman et al., 2005). Alternatively, the accumulation of fibrillar tau inclusions in astrocytes could alter the trafficking of glutamate transporters leading to the observed functional reduction of GLT-1 and GLAST.
In several studies, GLT-1 was demonstrated to account for a large proportion of the glutamate transport capacity in the CNS, and loss of GLT-1 may lead to both a reduction in glutamate uptake capacity and elevated extracellular glutamate (Rothstein et al., 1996). Thus, one study on ALS patients described increased levels of glutamate in the CSF; however, this finding is controversial. In our study, we did not detect an increase in the levels of glutamate in the CSF of GFAP/tauWT and GFAP/tauP301L Tg mice. CSF glutamate levels reflect multiple processes, including glutamate release and uptake, as well as glutamate metabolism. In this regard, decreased activity of glutamate dehydrogenase, as well as a downregulation of GLT-1, was suggested to be the cause of the elevation of CSF and plasma glutamate in ALS patients (Plaitakis, 1990). The lack of increase in the levels of glutamate in the CSF of GFAP/tau Tg mice therefore suggests that the GLT-1 reduction does not significantly modify the total pool of extracellular glutamate. However, focal loss of GLT-1 could alter the local glutamate levels without effecting total CSF glutamate.
In summary, the two strains of tau Tg mice provide the first evidence for compromised astrocyte function as a consequence of tau expression and pathology. The data support the hypothesis that altered glutamate transporter function contributes to the clinical manifestations of tauopathies with astrocytic pathology. The data also raise the possibility that glutamate excitotoxicity contributes to the neurodegeneration characteristic of these diseases. However, additional experiments are necessary to assess the mechanism and functional consequences of these alterations in glutamate transporter function.
Footnotes
This work was supported by National Institutes of Health Grants R01 NS36465, P30 AG10124, P01 AG17586, and P30 26979. V.M.-Y.L. is the John H. Ware Third Chair of Alzheimer's disease research. J.Q.T. is the William Maul Measey-Truman G. Schnabel Jr Professor of Geriatric Medicine and Gerontology. M.S.F. is supported by mentored Clinical Scientist Development Award K08 AG20073 from the National Institute on Aging. We thank Wayland Bergmann for technical help with HPLC analyses.
Correspondence should be addressed to Dr. Mark S. Forman, Department of Pathology and Laboratory Medicine, University of Pennsylvania School of Medicine, 422 Curie Boulevard, 605B Stellar-Chance Building, Philadelphia, PA 19104. E-mail: formanm@mail.med.upenn.edu.
Copyright © 2006 Society for Neuroscience 0270-6474/06/260644-11$15.00/0
References
- Araque A, Carmignoto G, Haydon PG (2001) Dynamic signaling between astrocytes and neurons. Annu Rev Physiol 63: 795-813. [DOI] [PubMed] [Google Scholar]
- Arriza JL, Fairman WA, Wadiche JI, Murdoch GH, Kavanaugh MP, Amara SG (1994) Functional comparisons of three glutamate transporter subtypes cloned from human motor cortex. J Neurosci 14: 5559-5569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckstrom H, Julsrud L, Haugeto O, Dewar D, Graham DI, Lehre KP, Storm-Mathisen J, Danbolt NC (1999) Interindividual differences in the levels of the glutamate transporters GLAST and GLT, but no clear correlation with Alzheimer's disease. J Neurosci Res 55: 218-229. [DOI] [PubMed] [Google Scholar]
- Bloom O, Evergren E, Tomilin N, Kjaerulff O, Low P, Brodin L, Pieribone VA, Greengard P, Shupliakov O (2003) Colocalization of synapsin and actin during synaptic vesicle recycling. J Cell Biol 161: 737-747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner M, Messing A (1996) GFAP transgenic mice. Methods 10: 351-364. [DOI] [PubMed] [Google Scholar]
- Choi DW, Maulucci-Gedde M, Kriegstein AR (1987) Glutamate neurotoxicity in cortical cell culture. J Neurosci 7: 357-368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danbolt NC (2001) Glutamate uptake. Prog Neurobiol 65: 1-105. [DOI] [PubMed] [Google Scholar]
- David DC, Hauptmann S, Scherping I, Schuessel K, Keil U, Rizzu P, Ravid R, Drose S, Brandt U, Muller WE, Eckert A, Gotz J (2005) Proteomic and functional analyses reveal a mitochondrial dysfunction in P301L tau transgenic mice. J Biol Chem 280: 23802-23814. [DOI] [PubMed] [Google Scholar]
- Dowd LA, Coyle AJ, Rothstein JD, Pritchett DB, Robinson MB (1996) Comparison of Na+-dependent glutamate transport activity in synaptosomes, C6 glioma, and Xenopus oocytes expressing excitatory amino acid carrier 1 (EAAC1). Mol Pharmacol 49: 465-473. [PubMed] [Google Scholar]
- Feany MB, Dickson DW (1995) Widespread cytoskeletal pathology characterizes corticobasal degeneration. Am J Pathol 146: 1388-1396. [PMC free article] [PubMed] [Google Scholar]
- Feany MB, Dickson DW (1996) Neurodegenerative disorders with extensive tau pathology: a comparative study and review. Ann Neurol 40: 139-148. [DOI] [PubMed] [Google Scholar]
- Forman MS, Lal D, Zhang B, Dabir DV, Swanson E, Lee VM-Y, Trojanowski JQ (2005) Transgenic mouse model of tau pathology in astrocytes leading to nervous system degeneration. J Neurosci 25: 3539-3550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuta A, Martin LJ, Lin CL, Dykes-Hoberg M, Rothstein JD (1997) Cellular and synaptic localization of the neuronal glutamate transporters excitatory amino acid transporter 3 and 4. Neuroscience 81: 1031-1042. [DOI] [PubMed] [Google Scholar]
- Garlin AB, Sinor AD, Sinor JD, Jee SH, Grinspan JB, Robinson MB (1995) Pharmacology of sodium-dependent high-affinity l-[3H]glutamate transport in glial cultures. J Neurochem 64: 2572-2580. [DOI] [PubMed] [Google Scholar]
- Ginsberg SD, Martin LJ, Rothstein JD (1995) Regional deafferentation down-regulates subtypes of glutamate transporter proteins. J Neurochem 65: 2800-2803. [DOI] [PubMed] [Google Scholar]
- Goedert M, Jakes R (1990) Expression of separate isoforms of human tau protein: correlation with the tau pattern in brain and effects on tubulin polymerization. EMBO J 9: 4225-4230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goedert M, Jakes R, Crowther RA, Six J, Lubke U, Vandermeeren M, Cras P, Trojanowski JQ, Lee VM-Y (1993) The abnormal phosphorylation of tau protein at Ser-202 in Alzheimer disease recapitulates phosphorylation during development. Proc Natl Acad Sci USA 90: 5066-5070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Götz J (2001) Tau and transgenic animal models. Brain Res Rev 35: 266-286. [DOI] [PubMed] [Google Scholar]
- Greenberg SG, Davies P (1990) A preparation of Alzheimer paired helical filaments that displays distinct tau proteins by polyacrylamide gel electrophoresis. Proc Natl Acad Sci USA 87: 5827-5831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haydon PG (2001) GLIA: listening and talking to the synapse. Nat Rev Neurosci 2: 185-193. [DOI] [PubMed] [Google Scholar]
- Howland DS, Liu J, She Y, Goad B, Maragakis NJ, Kim B, Erickson J, Kulik J, DeVito L, Psaltis G, DeGennaro LJ, Cleveland DW, Rothstein JD (2002) Focal loss of the glutamate transporter EAAT2 in a transgenic rat model of SOD1 mutant-mediated amyotrophic lateral sclerosis (ALS). Proc Natl Acad Sci USA 99: 1604-1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutton M, Lendon CL, Rizzu P, Baker M, Froelich S, Houlden H, Pickering-Brown S, Chakraverty S, Isaacs A, Grover A, Hackett J, Adamson J, Lincoln S, Dickson D, Davies P, Petersen RC, Stevens M, de Graaff E, Wauters E, van Baren J, et al. (1998) Association of missense and 5′-splice-site mutations in tau with the inherited dementia FTDP-17. Nature 393: 702-705. [DOI] [PubMed] [Google Scholar]
- Ikeda K, Akiyama H, Kondo H, Haga C, Tanno E, Tokuda T, Ikeda S (1995) Thorn-shaped astrocytes: possibly secondarily induced tau-positive glial fibrillary tangles. Acta Neuropathol (Berl) 90: 620-625. [DOI] [PubMed] [Google Scholar]
- Ishihara T, Hong M, Zhang B, Nakagawa Y, Lee MK, Trojanowski JQ, Lee VM-Y (1999) Age-dependent emergence and progression of a tauopathy in transgenic mice overexpressing the shortest human tau isoform. Neuron 24: 751-762. [DOI] [PubMed] [Google Scholar]
- Koch HP, Chamberlin AR, Bridges RJ (1999) Nontransportable inhibitors attenuate reversal of glutamate uptake in synaptosomes following a metabolic insult. Mol Pharmacol 55: 1044-1048. [DOI] [PubMed] [Google Scholar]
- Komori T (1999) Tau-positive glial inclusions in progressive supranuclear palsy, corticobasal degeneration and Pick's disease. Brain Pathol 9: 663-679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosik KS, Orecchio LD, Binder L, Trojanowski JQ, Lee VM-Y, Lee G (1988) Epitopes that span the tau molecule are shared with paired helical filaments. Neuron 1: 817-825. [DOI] [PubMed] [Google Scholar]
- Lee VM-Y, Goedert M, Trojanowski JQ (2001) Neurodegenerative tauopathies. Annu Rev Neurosci 24: 1121-1159. [DOI] [PubMed] [Google Scholar]
- Levy LM, Lehre KP, Walaas SI, Storm-Mathisen J, Danbolt NC (1995) Down-regulation of glial glutamate transporters after glutamatergic denervation in the rat brain. Eur J Neurosci 7: 2036-2041. [DOI] [PubMed] [Google Scholar]
- Li S, Mallory M, Alford M, Tanaka S, Masliah E (1997) Glutamate transporter alterations in Alzheimer disease are possibly associated with abnormal APP expression. J Neuropathol Exp Neurol 56: 901-911. [DOI] [PubMed] [Google Scholar]
- Lievens JC, Salin P, Had-Aissouni L, Mahy N, Kerkerian-Le Goff L (2000) Differential effects of corticostriatal and thalamostriatal deafferentation on expression of the glutamate transporter GLT1 in the rat striatum. J Neurochem 74: 909-919. [DOI] [PubMed] [Google Scholar]
- LoPresti P, Szuchet S, Papasozomenos SC, Zinkowski RP, Binder LI (1995) Functional implications for the microtubule-associated protein tau: localization in oligodendrocytes. Proc Natl Acad Sci USA 92: 10369-10373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowry OH, Rosebrough NJ, Farr AL, Randall RJ (1951) Protein measurement with the Folin phenol reagent. J Biol Chem 193: 265-275. [PubMed] [Google Scholar]
- Maragakis NJ, Rothstein JD (2001) Glutamate transporters in neurologic disease. Arch Neurol 58: 365-370. [DOI] [PubMed] [Google Scholar]
- Masliah E, Alford M, Mallory M, Rockenstein E, Moechars D, Van Leuven F (2000) Abnormal glutamate transport function in mutant amyloid precursor protein transgenic mice. Exp Neurol 163: 381-387. [DOI] [PubMed] [Google Scholar]
- Plaitakis A (1990) Glutamate dysfunction and selective motor neuron degeneration in amyotrophic lateral sclerosis: a hypothesis. Ann Neurol 28: 3-8. [DOI] [PubMed] [Google Scholar]
- Poorkaj P, Bird TD, Wijsman E, Nemens E, Garruto RM, Anderson L, Andreadis A, Wiederholt WC, Raskind M, Schellenberg GD (1998) Tau is a candidate gene for chromosome 17 frontotemporal dementia. Ann Neurol 43: 815-825. [DOI] [PubMed] [Google Scholar]
- Price MT, Olney JW, Haft R (1981) Age-related changes in glutamate concentration and synaptosomal glutamate uptake in adult rat striatum. Life Sci 28: 1365-1370. [DOI] [PubMed] [Google Scholar]
- Ransom B, Behar T, Nedergaard M (2003) New roles for astrocytes (stars at last). Trends Neurosci 26: 520-522. [DOI] [PubMed] [Google Scholar]
- Robinson MB, Hunter-Ensor M, Sinor J (1991) Pharmacologically distinct sodium-dependent l-[3H]glutamate transport processes in rat brain. Brain Res 544: 196-202. [DOI] [PubMed] [Google Scholar]
- Robinson MB, Djali S, Buchhalter JR (1993) Inhibition of glutamate uptake with l-trans-pyrrolidine-2,4-dicarboxylate potentiates glutamate toxicity in primary hippocampal cultures. J Neurochem 61: 2099-2103. [DOI] [PubMed] [Google Scholar]
- Rothstein JD, Martin L, Levey AI, Dykes-Hoberg M, Jin L, Wu D, Nash N, Kuncl RW (1994) Localization of neuronal and glial glutamate transporters. Neuron 13: 713-725. [DOI] [PubMed] [Google Scholar]
- Rothstein JD, Van Kammen M, Levey AI, Martin LJ, Kuncl RW (1995) Selective loss of glial glutamate transporter GLT-1 in amyotrophic lateral sclerosis. Ann Neurol 38: 73-84. [DOI] [PubMed] [Google Scholar]
- Rothstein JD, Dykes-Hoberg M, Pardo CA, Bristol LA, Jin L, Kuncl RW, Kanai Y, Hediger MA, Wang Y, Schielke JP, Welty DF (1996) Knockout of glutamate transporters reveals a major role for astroglial transport in excitotoxicity and clearance of glutamate. Neuron 16: 675-686. [DOI] [PubMed] [Google Scholar]
- Rothstein JD, Patel S, Regan MR, Haenggeli C, Huang YH, Bergles DE, Jin L, Dykes HM, Vidensky S, Chung DS, Toan SV, Bruijn LI, Su ZZ, Gupta P, Fisher PB (2005) Beta-lactam antibiotics offer neuroprotection by increasing glutamate transporter expression. Nature 433: 73-77. [DOI] [PubMed] [Google Scholar]
- Schlag BD, Vondrasek JR, Munir M, Kalandadze A, Zelenaia OA, Rothstein JD, Robinson MB (1998) Regulation of the glial Na+-dependent glutamate transporters by cyclic AMP analogs and neurons. Mol Pharmacol 53: 355-369. [DOI] [PubMed] [Google Scholar]
- Shin RW, Iwaki T, Kitamoto T, Tateishi J (1991) Hydrated autoclave pretreatment enhances tau immunoreactivity in formalin-fixed normal and Alzheimer's disease brain tissues. Lab Invest 64: 693-702. [PubMed] [Google Scholar]
- Spillantini MG, Murrell JR, Goedert M, Farlow MR, Klug A, Ghetti B (1998) Mutation in the tau gene in familial multiple system tauopathy with pre-senile dementia. Proc Natl Acad Sci USA 95: 7737-7741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka K, Watase K, Manabe T, Yamada K, Watanabe M, Takahashi K, Iwama H, Nishikawa T, Ichihara N, Kikuchi T, Okuyama S, Kawashima N, Hori S, Takimoto M, Wada K (1997) Epilepsy and exacerbation of brain injury in mice lacking the glutamate transporter GLT-1. Science 276: 1699-1702. [DOI] [PubMed] [Google Scholar]
- Togo T, Dickson DW (2002) Tau accumulation in astrocytes in progressive supranuclear palsy is a degenerative rather than a reactive process. Acta Neuropathol (Berl) 104: 398-402. [DOI] [PubMed] [Google Scholar]
- Trotti D, Danbolt NC, Volterra A (1998) Glutamate transporters are oxidant-vulnerable: a molecular link between oxidative and excitotoxic neurodegeneration? Trends Pharmacol Sci 19: 328-334. [DOI] [PubMed] [Google Scholar]
- Trotti D, Rolfs A, Danbolt NC, Brown Jr RH, Hediger MA (1999) SOD1 mutants linked to amyotrophic lateral sclerosis selectively inactivate a glial glutamate transporter. Nat Neurosci 2: 848. [DOI] [PubMed] [Google Scholar]
- Vesce S, Bezzi P, Volterra A (1999) The active role of astrocytes in synaptic transmission. Cell Mol Life Sci 56: 991-1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volterra A, Steinhauser C (2004) Glial modulation of synaptic transmission in the hippocampus. Glia 47: 249-257. [DOI] [PubMed] [Google Scholar]
- Watase K, Hashimoto K, Kano M, Yamada K, Watanabe M, Inoue Y, Okuyama S, Sakagawa T, Ogawa S, Kawashima N, Hori S, Takimoto M, Wada K, Tanaka K (1998) Motor discoordination and increased susceptibility to cerebellar injury in GLAST mutant mice. Eur J Neurosci 10: 976-988. [DOI] [PubMed] [Google Scholar]
- Yoshiyama Y, Zhang B, Bruce J, Trojanowski JQ, Lee VM-Y (2003) Reduction of detyrosinated microtubules and Golgi fragmentation are linked to tau-induced degeneration in astrocytes. J Neurosci 23: 10662-10671. [DOI] [PMC free article] [PubMed] [Google Scholar]